 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTime-dependent photoemission from droplets: influence of size and charge on the photophysics near the surface†
Loren
Ban
 *,
Hanchao
Tang
,
Bruce L.
Yoder
and
Ruth
Signorell
*
*,
Hanchao
Tang
,
Bruce L.
Yoder
and
Ruth
Signorell
*
Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog Weg 2, CH-8093 Zürich, Switzerland. E-mail: lban@ethz.ch; rsignorell@ethz.ch
First published on 4th May 2022
Abstract
Photoemission from submicrometer droplets containing a mixture of dioctyl phthalate and dioctyl sebacate was investigated by femtosecond and nanosecond photoionization. Photoelectron spectra recorded after ionization with single 4.7 eV femtosecond or nanosecond laser pulses showed marked differences between the two cases. These differences were attributed to ionization of long-lived states which only occurred within the duration of the nanosecond pulse. The tentative assignment of the long-lived states to dioctyl phthalate triplet states is discussed. A nanosecond–femtosecond pump–probe scheme using 4.7 eV (pump) and 3.1 eV (probe) pulses was used to investigate the decay dynamics of these long-lived states. The dynamics showed an accelerated decay rate at higher dioctyl phthalate concentrations. Furthermore, the dependence of the decay dynamics on droplet size and charge was investigated. The decay of the long-lived states was found to be faster in smaller droplets as well as in neutral droplets compared with both positively and negatively charged droplets. Possible mechanisms to explain these observations and the dominance of contributions from the droplets surface are discussed.
Introduction
Photoelectron spectroscopy (PES) of droplets and particles has been established as a versatile spectroscopic method to investigate aerosol samples of nanometer and micrometer size.1–6 Due to their large surface-to-volume ratio, studies of aerosol interfaces are of great importance to understand their physics and chemistry.7,8 In the context of atmospheric aerosol studies, PES has been used to investigate heterogeneous reactivity at droplet surfaces.9–11 This was done by using high-energy radiation (in the soft X-ray and near-edge X-ray regimes) which yields surface-sensitive information due to short experimental probing depths limited to a few nanometers by inelastic electron scattering. As a result, it was shown that chemical gradients are formed at the droplet surface as a result of diffusion-limited mixing in the droplet, therefore altering the chemical kinetics compared to bulk systems.9,12Droplet PES also offers insight into fundamental properties of the condensed phase such as electron transport scattering13–18 or the electronic structure.6,19–22 In a recent publication,18 we illustrated how a charged droplet–vacuum interface influences such properties. For example, the study of low-energy electron escape from charged droplet–vacuum interfaces can provide detailed information on the shape of the interface potential. Quantum effects make low-energy electrons particularly sensitive to the height and width of the interface potential.18 Furthermore, a charged interface leads to characteristic shifts in the measured photoelectron spectra which need to be taken into account for precise determination of electronic structure in droplet PES.18
In this work, we extend our previous study of the droplet–vacuum interface and examine its influence on the photophysics near the surface. As a model system, we chose bis(2-ethylhexyl) phthalate (DEHP) dissolved in bis(2-ethylhexyl) sebacate (DEHS) droplets, where both components are liquid and their low volatility offers good control over the interface properties. The study was motivated by the observation that resonant photoionization of DEHP droplets with 4.7 eV light leads to significantly different photoelectron spectra depending on whether nanosecond or femtosecond laser pulses were used. To get at the root of this phenomenon, we performed nanosecond–femtosecond pump–probe experiments and show that following excitation at 4.7 eV, DEHP survives in long-lived states. Measurements of the electron yield decay dynamics showed these long-lived states to decay on a microsecond timescale. The dynamics turned out to depend on the DEHP concentration as well as on the droplet’s size (500 nm and 1200 nm in diameter) and charge state (neutral and charge states of up to about ±20e). These results suggest that surface effects could play a role in the decay of the long-lived states. Although still at an early stage, this work illustrates the prospects of droplet PES for time-resolved studies of photoinduced processes in submicrometer systems where surface effects can lead to enhanced reactivity.
Experimental and theoretical methods
The experimental setup used in this work is shown in Fig. 1. It consists of an air-side configuration used to prepare the aerosol sample, and a droplet photoelectron spectrometer. Here we briefly describe the key elements. Further details can be found in the ESI (Section 1).†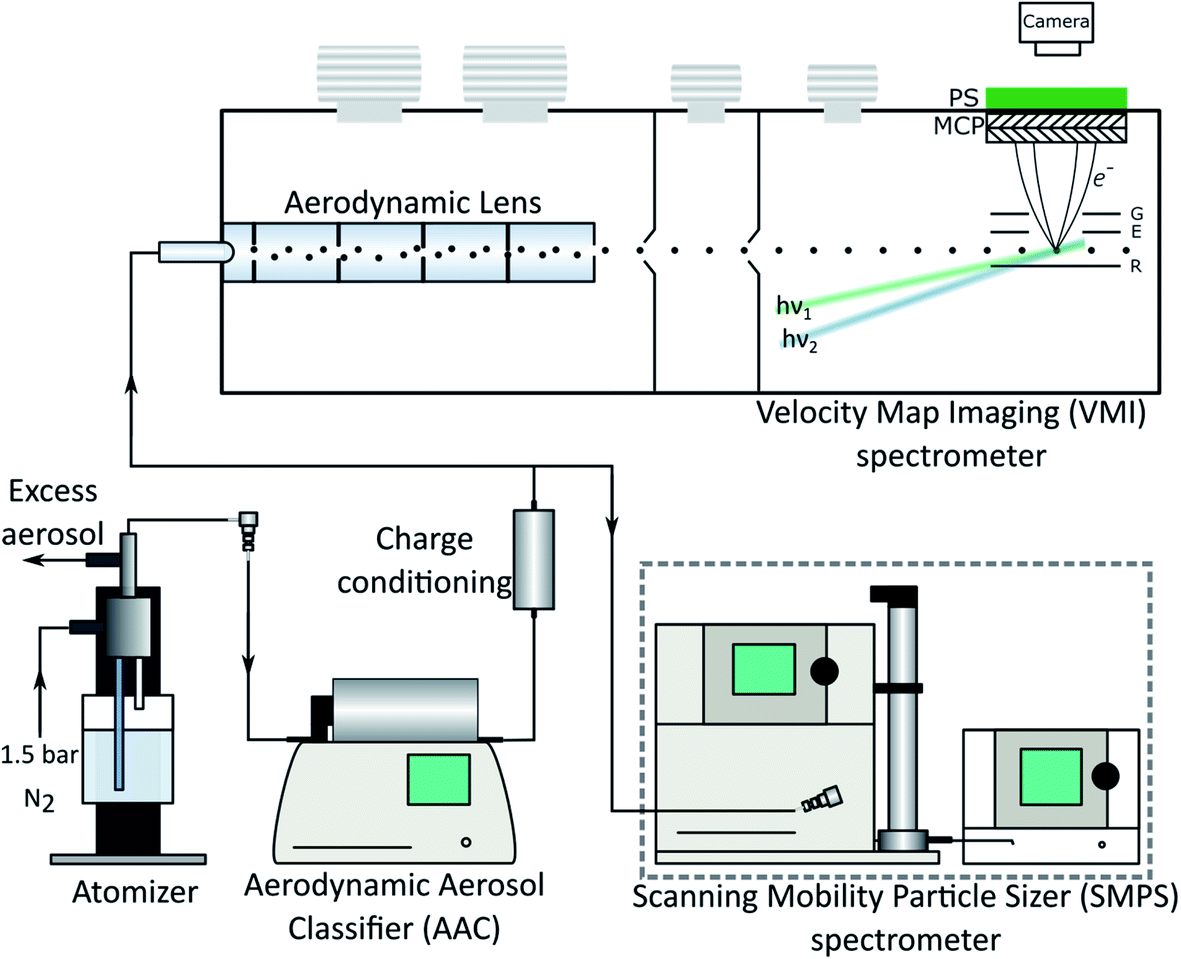 | ||
| Fig. 1 Sketch of the experimental setup consisting of the aerosol generation part (atomizer, AAC, charge conditioning), photoelectron spectrometer (top) and in-parallel particle sizing by the SMPS. The droplets were transferred to vacuum via the aerodynamic inlet device and ionized by the laser pulses (hν1,2) in between the repeller (R) and extractor (E) plates. Photoelectrons were detected in a velocity map imaging configuration (R, E and ground G plates) by the imaging detector (microchannel plates MCP, phosphor screen PS, camera). | ||
Aerosol sample preparation
The aerosol is prepared by atomization of liquid bis(2-ethylhexyl) phthalate (DEHP) and bis(2-ethylhexyl) sebacate (DEHS) mixtures, followed by size-selection in an Aerodynamic Aerosol Classifier (AAC) based on their aerodynamic diameter Da.23 A neutral aerosol sample is generated by removing charged droplets from the aerosol flow with an electrostatic precipitator (ESP). Positively and negatively charged aerosol samples are generated by replacing the ESP with a unipolar diffusion charger.18 The aerosol flow is then directed to the photoelectron spectrometer. A Scanning Mobility Particle Sizer (SMPS) monitors droplet size and charge in parallel to the photoionization experiment, as shown in Fig. 1.Typical droplet size and charge distributions obtained from the SMPS are shown in Fig. 2 and 3. The size-selected (Da = 500 nm) sample shows a single peak with a full-width at half-maximum of about 90 nm. The corresponding charge distributions for positively and negatively charged samples are shown in Fig. 3. The average droplet charge states of +15e, +20e and −17e are obtained, with a standard deviation of a few charge units.
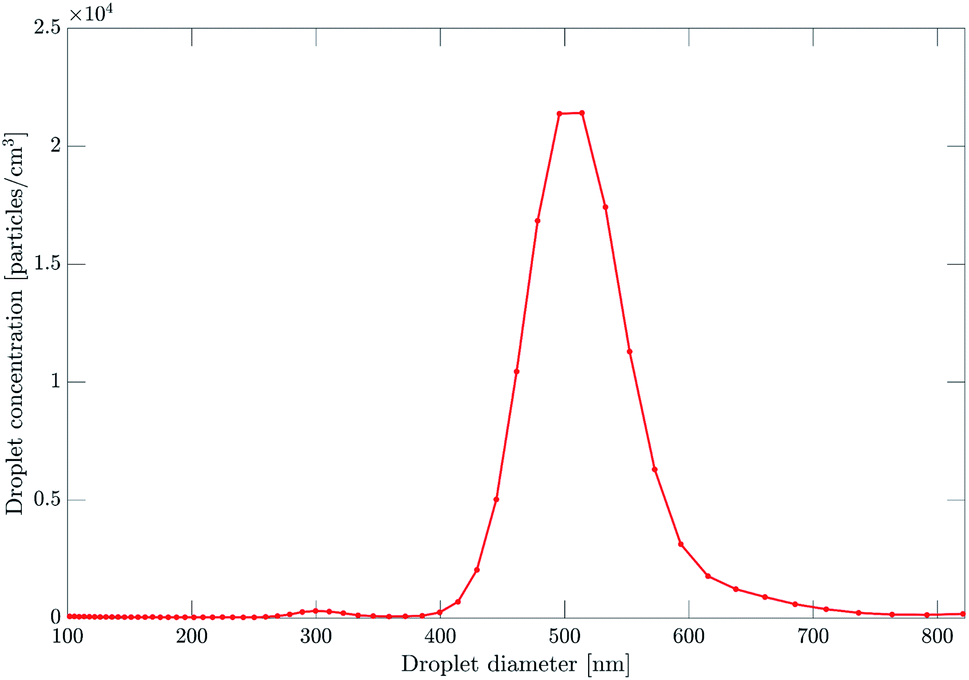 | ||
| Fig. 2 Droplet size distribution of the DEHP/DEHS aerosol size-selected at 500 nm by the AAC. The distribution shows a peak corresponding to particles of 500 nm in diameter with a full width at half-maximum of about 90 nm. | ||
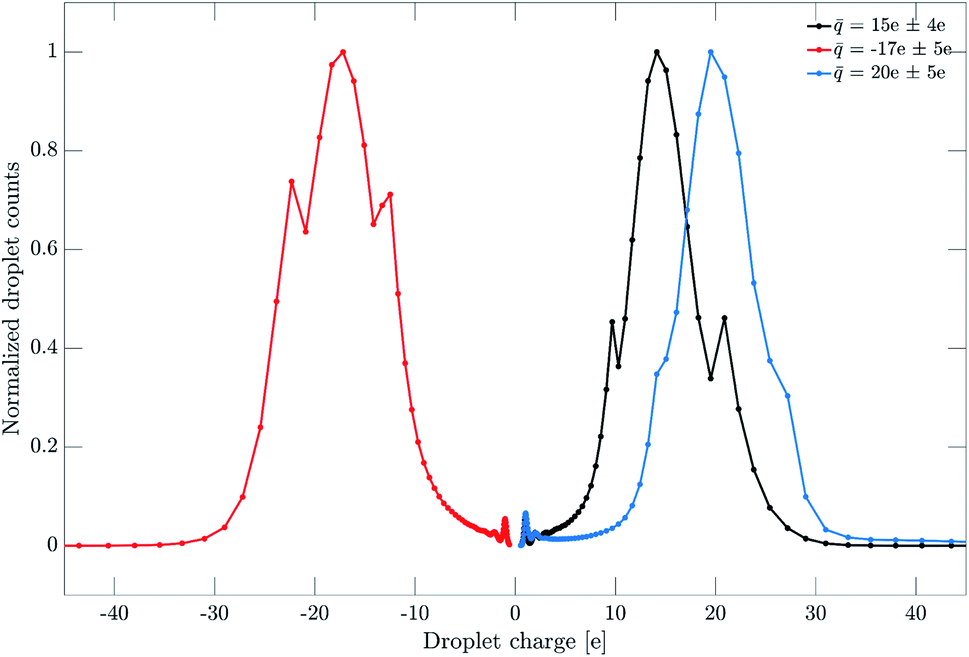 | ||
| Fig. 3 Charge distributions of droplets with diameter of 500 nm after passing through the unipolar diffusion charger at positive and negative polarity. The average charge states (of elementary charge units) are shown in the legend. These charge states correspond to the measurements presented in the last section of the paper. | ||
Photoelectron spectrometer
The second part of the aerosol flow enters the photoelectron spectrometer through an aerodynamic lens (ADL), whose design is based on ref. 24. The ADL transmits a wide range of particle diameters in the submicrometer range and is similar to those used in our previous studies.13,16,18,25 The droplet beam is intersected by femtosecond (hν1) and nanosecond (hν2) laser pulses in the photoelectron spectrometer. The femtosecond pulses have photon energies of either 3.1 eV or 4.7 eV and nominal durations of <70 fs, with peak intensities of ∼1–35 GW cm−2. The nanosecond pulses have a photon energy of 4.7 eV, a pulse duration of ∼7 ns and peak intensities of ∼100 kW cm−2. For pump–probe experiments performed during this work, the instrument response function (IRF) is well approximated by the temporal profile of the nanosecond pulse.The photoelectron spectrometer is operated in a Velocity Map Imaging (VMI) configuration, with two different detection modes employed in this work. In the first part of the paper, we use the camera to record angle-resolved photoelectron images. Photoelectron kinetic energy (eKE) spectra are retrieved by image reconstruction along the laser propagation axis1 with MEVIR26 and background-corrected by subtraction of an image collected in the absence of the droplet beam (with laser beams on). In the second part of the paper, we decoupled the electron signal directly from the imaging detector and recorded electron time-of-flight (eTOF) spectra to retrieve total photoelectron yields. Recording photoelectron yields requires the collection of fewer laser shots to achieve a reasonable signal-to-noise ratio and was therefore used to perform pump–probe studies over a wide range of time delays (see below).
DFT calculations
To interpret the experimental data, we need information on the electronic energy levels relevant for our system. To the best of our knowledge, no detailed calculations exist in the literature for either liquid or gaseous DEHP. As a basis for our analysis, we performed density functional theory (DFT) calculations on a model system to obtain information on the electronic level structure that could be related to the experimental data. The chosen model system was a gas-phase dibutyl phthalate (DBP) molecule whose electronic structure should be similar to DEHP27 (Fig. 4) with the advantage of providing a smaller and thus a more tractable system for quantum chemical calculations. The calculations were further simplified by constraining the conformational space that can be covered by the aliphatic chains. To estimate the vertical ionization energies (VIE) and adiabatic ionization energies (AIE) in the liquid droplet, we performed additional calculations using a polarizable continuum model. The calculations were compared with the limited information on phthalates (see above) and other similar aromatic systems, for which more detailed information is available in the literature (i.e. benzene and benzoic acid). Details of the calculations and the comparisons can be found in the ESI (Section 2).†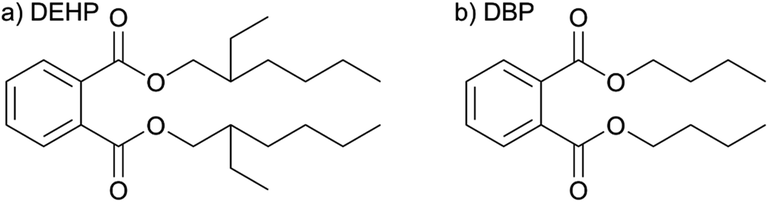 | ||
| Fig. 4 Chemical structures of DEHP (a) and DBP (b). | ||
Results and discussion – neutral droplets
Single-pulse multiphoton ionization at 4.7 eV
In Fig. 5 we compare photoelectron images obtained by single-pulse photoionization of DEHP droplets with nanosecond (panel a) and femtosecond (panel b) pulses. Neutral droplets with a diameter of 500 nm and a DEHP concentration of 1.5 M were used. The signal in the photoelectron spectra originates from resonant multiphoton ionization (REMPI) of DEHP (see below). The solvent, DEHS, does not significantly contribute to the signal since it has no aromatic moiety and non-resonant multiphoton ionization is inefficient at the laser intensities used. This was confirmed by additional measurements of pure DEHS droplets at equal laser intensities.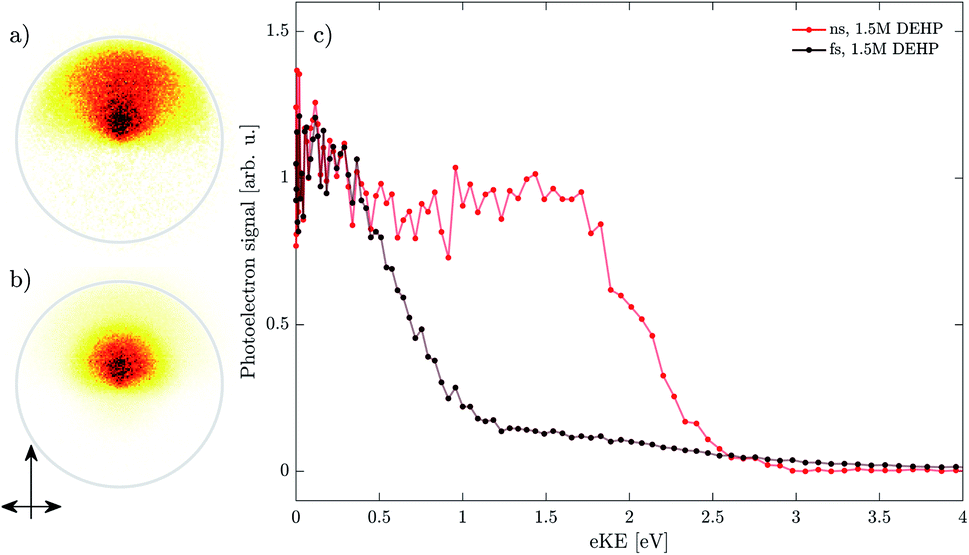 | ||
| Fig. 5 Droplet photoelectron images generated by nanosecond (a) and femtosecond (b) ionization of neutral DEHP/DEHS droplets of 500 nm diameter and DEHP concentration of 1.5 M. The gray circles indicate an eKE of 2.4 eV. Light propagation (vertical) and polarization (horizontal) vectors are indicated in panel (b). Corresponding photoelectron spectra are shown in panel (c). The spectra are normalized to their respective signal at eKE ∼ 0.2 eV. | ||
The images are asymmetric along the laser propagation axis, with signal found on the image half away from the incident laser direction (single-headed arrow in Fig. 5). This is a characteristic feature of VMI PES of weakly-absorbing droplets that show so-called nanofocusing.1,5,28 A clear difference between the ns and fs images can be seen, with the signal in the nanosecond case extending towards larger radii, i.e., higher electron kinetic energies.
For a more quantitative comparison, the corresponding photoelectron spectra are shown in Fig. 5c. The spectra are normalized to their respective signal at eKE ∼ 0.2 eV. The nanosecond spectrum (red) shows two bands that extend up to electron kinetic energies (eKE) of ∼3 eV. The spectrum is in good agreement with our previous study on pure DEHP droplets.18 By contrast, the femtosecond spectrum (black) shows about one order of magnitude lower signal at eKE > 1 eV (relative to the signal at 0.2 eV) and the signal seems to extend to higher eKE (see difference at ∼3 eV).
Gaussian component fitting of femto and nano spectra
To illustrate the differences more clearly, Fig. 6 shows the same spectra on a logarithmic y-axis. The femtosecond spectrum (dots in Fig. 6b) does indeed extend to significantly higher eKE than the nanosecond spectrum (dots in Fig. 6a). We analyzed the spectra in terms of a multi-component Gaussian fit to the experimental data. Rather than focusing on the peak maxima we compare high kinetic energy onsets, which we defined as eKEσ = eKE0 + 3σ, where eKE0 is the Gaussian peak maximum and σ its standard deviation. Difficulties in using the peak maxima for the determination of vertical ionization energies (VIE) in condensed phase photoelectron spectroscopy have been previously assigned to fast excited-state dynamics29,30 and inelastic electron scattering.31,32 At the low eKE values measured in this work, inelastic electron scattering can additionally distort the peak shape away from a Gaussian, leading to an asymmetric tail toward lower eKEs.14 Effects of electron scattering can in principle be accounted for by using a scattering model,14 but scattering cross sections are not available for DEHP/DEHS mixtures.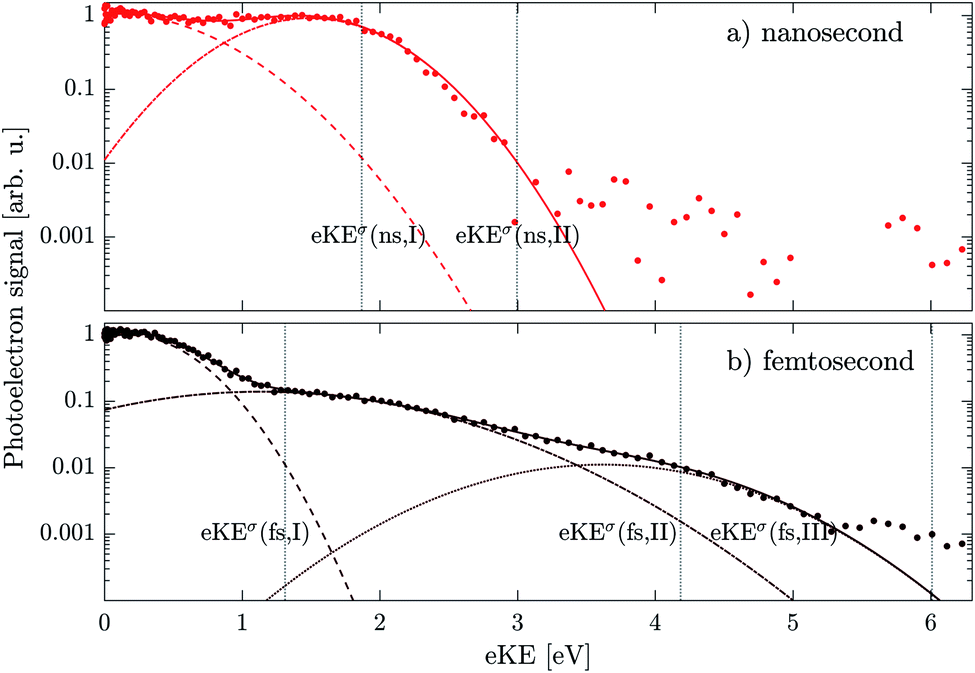 | ||
| Fig. 6 Droplet photoelectron spectra (circles) from Fig. 5 shown on a logarithmic y-scale. The spectra are fitted by a multi-component Gaussian function (full-lines, components shown as dashed, dashed-dotted and dotted lines). Panel (a) shows nanosecond and panel (b) femtosecond ionization. Individual onset values are indicated by vertical gray dotted lines. | ||
Using eKE onsets (to be compared with the AIE) instead of peak maxima (to be compared with the VIE) circumvents some of these issues. The eKE onsets determined from the multi-component fits are qualitatively compared with the calculated ionization energies (IE, Table SI1).†
The resulting fits to the data are shown as full lines in Fig. 6, with dashed lines representing individual components. The nanosecond case (Fig. 6a) is well fitted by 2 component bands with onsets of eKEσ(ns,I) = 1.9 eV and eKEσ(ns,II) = 3.0 eV and similar peak amplitudes. In the femtosecond case (Fig. 6b), 3 components were necessary to fit the data with eKEσ(fs,I) = 1.3 eV, eKEσ(fs,II) = 4.2 eV and eKEσ(fs,III) = 6.0 eV. The first component (fs,I) has about one and two orders of magnitude higher amplitude than components two (fs,II) and three (fs,III), respectively.
Band assignment and a simple energy diagram
We base our band assignment on the DFT calculations described in the ESI (Section 2).†Fig. 7 shows a simplified electronic energy diagram of a gas phase DBP molecule, which is assumed to be similar to a gas phase DEHP molecule. It includes vertical excitation energies of the singlet state manifold. The lowest excited state (S1) lies at 3.2 eV above the ground state (S0). Higher excited states (Sn) lie in the range of ∼4.7 eV and enable resonant excitation at the photon energies used here. The gas phase VIE is calculated for a vertical transition to the lowest ionic state (D0) and equals 9.0 eV. The condensed phase VIE is estimated by the transition to the ionic state Dsolv0 and equals 8.1 eV. A condensed phase shift of 0.9 eV is reasonable considering the values determined for gas phase and condensed phase benzene.33 We only account for the shift in the ionization energies, since energy levels of the neutral DEHP molecule will only be slightly shifted by the weakly polar (ε ∼ 4)34 solvent DEHS. The corresponding AIE equals 8.6 eV and 7.8 eV for the gas and condensed phase, respectively.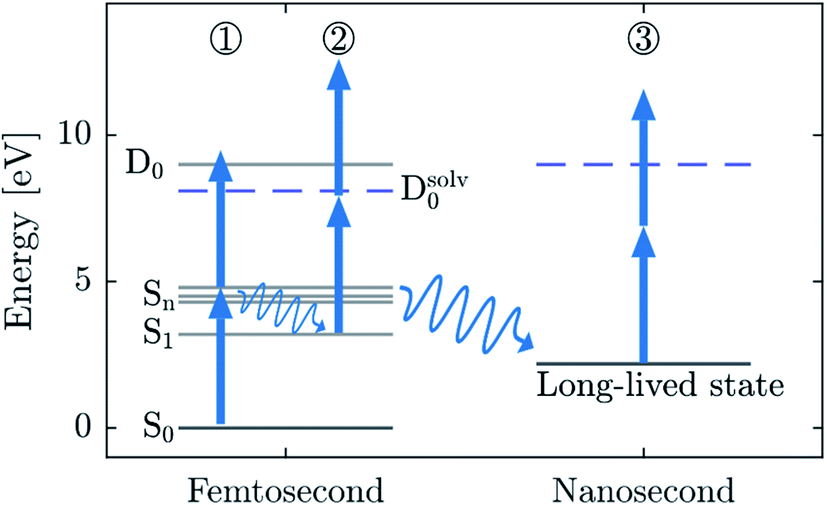 | ||
| Fig. 7 A sketch of an energy level diagram of a DEHP molecule indicating 3 different processes that could explain the bands observed in the femtosecond (pathway 1 and 2) and nanosecond (pathway 1 and 3) spectra. The horizontal axis indicates the timescale at which these processes take place. Arrows indicate the 4.7 eV laser pulses. We suggest a 2-photon ionization process of the long-lived states on a nanosecond timescale (process 3). | ||
First, we consider the 1 + 1 REMPI process indicated by (1) in Fig. 7. Absorption of the first 4.7 eV photon leads to resonant excitation (blue arrow). Absorption of an additional photon is enough to reach the ionic state manifold (D0, Dsolv0) completing the 1 + 1 REMPI (1). The calculated condensed phase AIE (see ESI, Table SI1)† yields an eKE onset value of 1.6 eV in reasonable agreement with the experimentally determined eKEσ(ns,I) of 1.9 eV and eKEσ(fs,I) of 1.3 eV. Therefore, we assign these bands to a 1 + 1 REMPI from the ground state. The difference in the onsets of ∼0.6 eV might be ascribed to the overlap of the (ns,I) band with the asymmetric low eKE tail of inelastically scattered electrons of the strong (ns,II) band. The fit accounts for this by increasing the width of the lower eKE Gaussian component, thereby shifting eKEσ(ns,I) to correspondingly larger values. This shift is virtually absent in the fs spectrum because the (fs,II) band is much weaker than the (fs,I) band.
On the premise that ionization requires a minimum of 2 photons (process (1)), the difference between the ns and fs spectra could arise from the presence of higher-order (1 + n) REMPI. These processes are much more probable in the fs case due to the shorter pulse duration and, in turn, significantly higher peak intensities (GW cm−2 in the fs case, kW cm−2 in the ns case). Since bands originating from (1 + n) REMPI would be shifted to higher eKE by integer multiples of the photon energy, we can assign the eKEσ(fs,III) band at 6.0 eV to 1 + 2 REMPI (eKEσ(fs,III) − eKEσ(fs,I) = 4.7 eV). Significantly weaker amplitude of the 1 + 2 REMPI band is reasonable since it includes an additional non-resonant step which has a significantly lower cross section.
However, bands eKEσ(ns,II) at 3 eV and eKEσ(fs,II) at 4.2 eV cannot be explained in this way. Their eKE is too high to be explained by the 1 + 1 REMPI and too low to be explained by the 1 + 2 REMPI. This indicates that the excited-state dynamics in the Sn manifold taking place within the pulse duration plays a role.29,30 The effect of this dynamics will be very different in the ns case and the fs case. Only ultrafast (<70 fs) relaxation processes are expected to play a role in the fs case, while slower processes such as intersystem crossing can occur within the duration of the ns pulse. In both cases, all the states generated during these dynamics can absorb additional photons and be ionized. This picture is illustrated by the “ladder” and “ladder switching” models.35 We propose that the bands eKEσ(ns,II) and eKEσ(fs,II) originate from distinct molecular states formed as a result of the dynamics initiated by single-photon excitation. These intermediate states are ionized by additional photons within the duration of the same ns or fs pulse.
The (fs,II) band can be assigned to 2-photon ionization of the S1 state upon fast internal conversion of the Sn manifold, designated as process (2) in Fig. 7. The calculated values of the condensed phase AIE and the S1 energy lead to an estimate of the eKE onset value of 4.8 eV, in reasonable agreement with the experimental onset eKEσ(fs,II) = 4.2 eV. Part of the difference might be ascribed to geometry relaxation in the S1 state not accounted for in our DFT calculations. Our assignment implies an internal conversion of the Sn manifold to S1 state on a sub-100 fs timescale, which is not unlikely considering literature on similar systems.36–38
The long duration of the ns pulse (∼7 ns), by contrast, also allows for slow relaxation processes to take place (e.g. intersystem crossing, photofragmentation and solvated electron formation). For the ns band we determined an onset eKEσ(ns,II) = 3.0 eV which would lead to IE = 6.4 eV (2-photon ionization) or IE = 1.7 eV (1-photon ionization). The IE of 1.7 eV is rather low and could only be explained by weakly bound molecular states such as anions formed by electron attachment after 2-photon ionization by process (1). It might also be conceivable, that weakly-bound solvated electrons are formed by localization of a photoelectron generated in process (1) – analogous to the above-mentioned electron attachment. The low-polarity of the solvent DEHS (ε ∼ 4),34 however, makes these options appear unlikely. In addition, solvated electrons were not reported in previous studies on similar systems.39 One might also think of anionic species formed along the fragmentation pathway of DEHP. However, DEHP fragments formed by UV dissociation which are reported in the literature include only neutral and cationic species which are unlikely to have such a low IE.40–42
Discarding major contributions from such 2 + 1 processes, i.e., process (1) followed by single photon ionization, we are left with the possibility of a 1 + 2 process. We propose to assign the (ns,II) band to the 2-photon ionization of long-lived states formed after photoexcitation to the Sn manifold, as indicated by process (3) in Fig. 7. The strong contribution of the (ns,II) band to the spectrum indicates that the efficiency of this ionization channel is comparable to the direct (1 + 1) REMPI channel (process (1)). This implies that the long-lived states are formed with a high probability and that it has a comparable ionization cross section to the ground state.
Concentration dependence of single-pulse multiphoton ionization at 4.7 eV
Fig. 8 shows photoelectron images and the corresponding spectra for nanosecond ionization of pure DEHP (panel c and open circles) and of DEHP in different concentrations in DEHS (panel b and full circles for 1.5 M, panel a and crosses for 0.5 M). Images and spectra collected upon femtosecond ionization are shown in Fig. 9 for a similar range of DEHP concentrations (panel a and open circles for 1.5 M, panel b and full circles for 0.5 M, panel c and crosses for 0.1 M). All spectra are normalized to their signal at eKE ≈ 0.2 eV (see Fig. 5).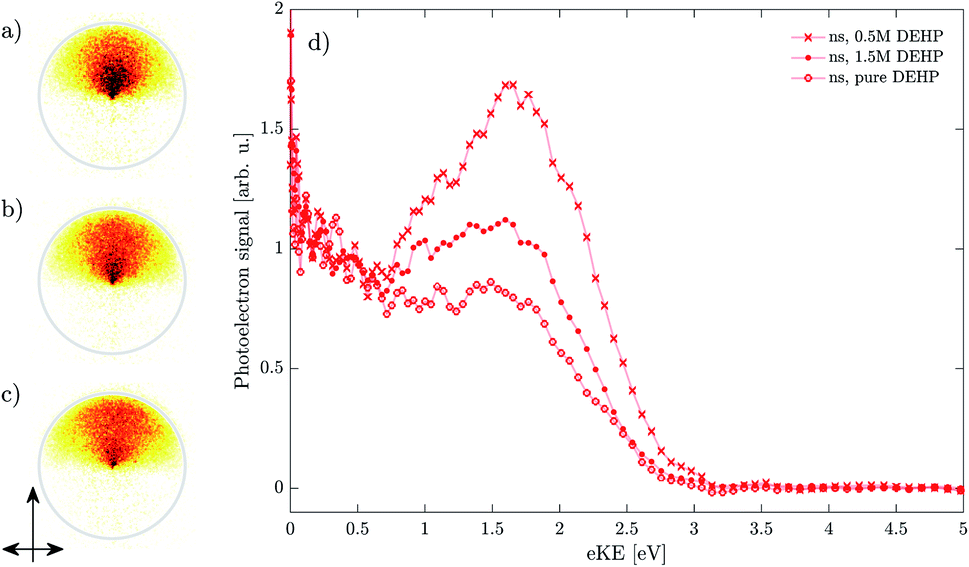 | ||
| Fig. 8 Droplet photoelectron images ((a) 0.5 M DEHP, (b) 1.5 M DEHP, (c) pure DEHP) and spectra (d) from nanosecond ionization of neutral droplets with varying DEHP concentration. The single-headed arrow denotes the laser propagation direction and the double-headed arrow denotes the linear polarization of the electric field. The spectra are normalized to their respective signal at eKE ∼ 0.2 eV and smoothed by a 3-point moving average. | ||
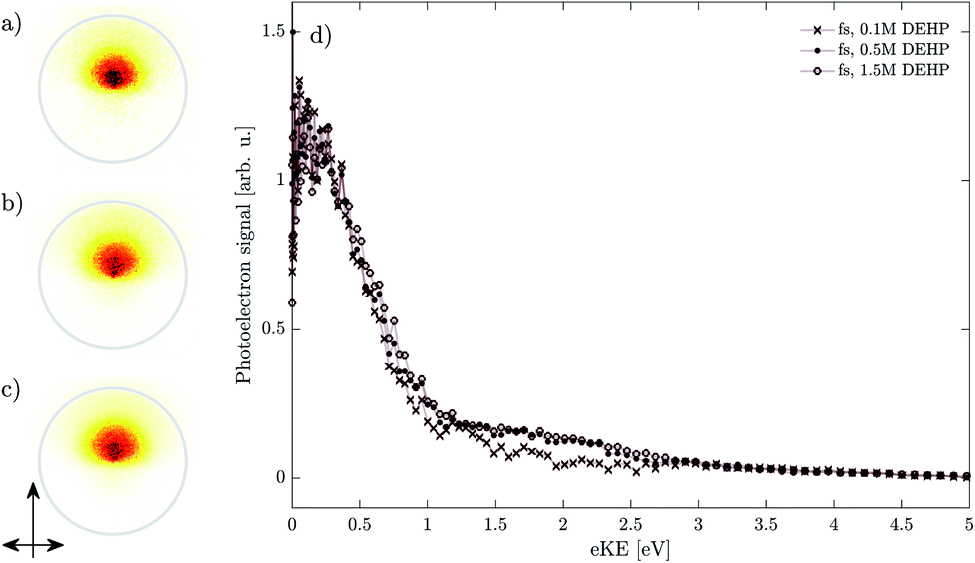 | ||
| Fig. 9 Droplet photoelectron images ((a) 1.5 M DEHP, (b) 0.5 M DEHP, (c) 0.1 M DEHP) and spectra (d) from femtosecond ionization of neutral droplets with varying DEHP concentration. The single-headed arrow denotes the laser propagation direction and the double-headed arrow denotes the linear polarization of the electric field. The spectra are normalized to their respective signal at eKE ∼ 0.2 eV. | ||
In the ns case, the intensity of the (ns,II) band relative to that of the (ns,I) band increases with decreasing concentration. By contrast, the fs spectra show similar relative band intensities for all concentrations. This illustrates that the (fs,II) and (fs,III) bands are only minor spectral components whose relative contributions do not depend on the concentration of DEHP. This is consistent with the proposed origin of these bands in fast unimolecular relaxation (internal conversion) and higher order ionization.
The pronounced concentration dependence in the ns case (Fig. 8) suggests that the contribution of the (ns,II) band is sensitive to the surrounding DEHP molecules. The relative contribution of the (ns,II) band decreases at higher DEHP concentrations. This behavior suggests that the long-lived states are efficiently quenched at higher concentrations by additional DEHP molecules. While this simple mechanism qualitatively explains the data, we note that a complex interplay with additional mechanisms (e.g. bimolecular association to form a DEHP dimer) could also be consistent with the experimentally observed concentration dependence (see below).
Nanosecond–femtosecond pump–probe VMI
To verify the assignment of the (ns,II) band to long-lived states formed upon photoexcitation, we turned to pump–probe studies. We used nanosecond 4.7 eV pulses to photo-excite (pump) the system followed by a femtosecond pulse, also at 4.7 eV, as a probe, with the pump–probe delay set to Δt = 30 ns. To determine whether the (ns,II) band also appears in the ns–fs pump–probe spectrum, the signal contributions from the single pulse excitations have to be removed from the VMI. This was achieved by subtracting an image obtained at Δt = −20 μs from the image at Δt = 30 ns. The image at long negative time delay serves as a background that contains only the respective single-pulse contributions (which do not depend on the time delay) but no pump–probe signal.Fig. 10 compares the resulting pump–probe spectrum (black) with the single-pulse nanosecond case (red). The pump–probe spectrum (black) contains a clear contribution from a high-energy band that agrees well with the (ns,II) band of the single-pulse nanosecond experiment (red). This supports the assignment presented above, indicating that the long-lived states are generated during the ns pump pulse (7 ns) and persist for at least 30 ns before they are ionized by the fs-probe. The VIE and the eKE onset are in good agreement with the single-pulse nanosecond spectrum (red). While we cannot exclude that the long-lived states undergo further transformations after the ns-pump pulse, these transformations do not seem to affect their photoelectron spectrum significantly. Therefore, we conclude that the differences in photoionization by femtosecond and by nanosecond pulses (i.e. the (ns,II) band) are due to strong contributions from long-lived states formed during the nanosecond pulse.
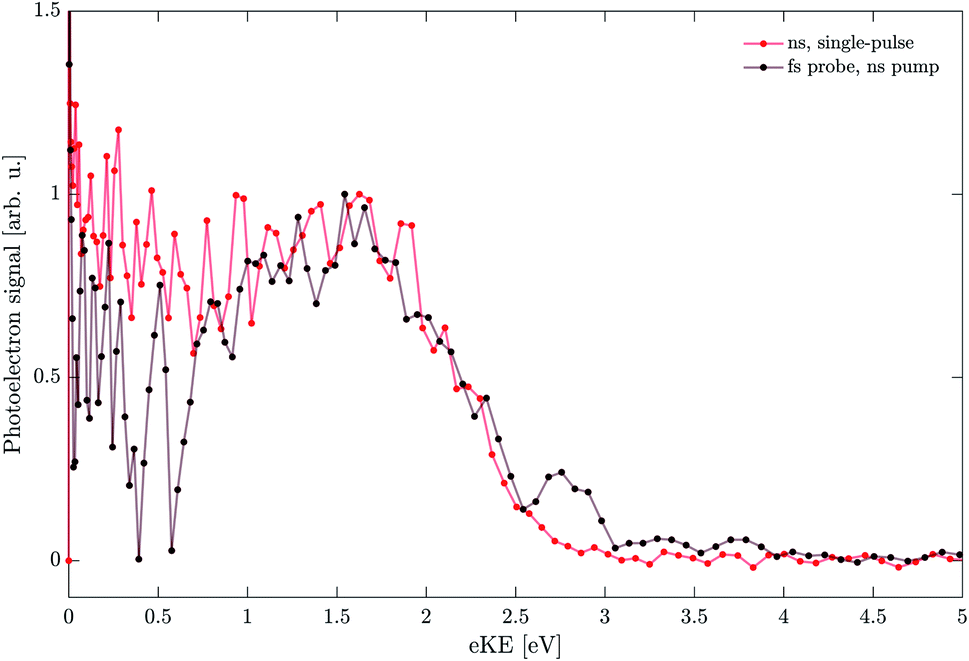 | ||
| Fig. 10 Comparison of the nanosecond single-pulse photoelectron spectrum (red) with the pump–probe spectrum (black). Nanosecond (4.7 eV) pulses were used as a pump and femtosecond (4.7 eV) pulses as a probe, delayed by ∼30 ns. The spectra are normalized to their respective signal at eKE ∼ 1.75 eV. | ||
A weak feature in the pump–probe spectrum at eKE ∼ 2.8 eV might be attributed to an artefact of the image reconstruction or the background subtraction. At low kinetic energies (eKE < 0.7 eV) the strong single-pulse femtosecond contribution (Fig. 5) in the background image causes significant scatter in the pump–probe signal after background subtraction so that meaningful analysis of this eKE range is currently impossible.
Total electron yield decay dynamics
To complement the photoelectron spectra, we performed pump–probe electron yield measurements to gain insight into the decay dynamics of the long-lived states. We used neutral droplets with a diameter of 500 nm composed of varying DEHP concentrations in DEHS. Nanosecond 4.7 eV pulses were used as a pump and time-delayed femtosecond 3.1 eV pulses as a probe. The photon energy of the fs-probe pulse was lowered so as to produce almost no single-pulse background. In this way, more reliable background subtraction and better signal-to-noise ratios were achieved, enabling measurements over a longer pump–probe delay range. We assumed that the same long-lived states are probed with both 4.7 eV and 3.1 eV pulses, which seems reasonable since 2-photon ionization is also feasible at lower photon energy (see Section 2 in the ESI).† Time-dependent total electron yields Y(Δt) were extracted from the eTOF traces after correcting for the single-pulse background from the nanosecond and femtosecond pulses (see ESI, Section 3).†
Fig. 11 shows the decay dynamics of the electron yield Y(Δt) for t > 10 ns (just outside of the instrument response function) as a function of the DEHP concentration. All data are normalized to the value Y(Δt = 10 ns) of the measurement at the highest concentration (1.5 M). The experimental data (circles, shaded areas are the estimated signal uncertainties) show that the electron yield systematically increases with increasing DEHP concentration. As such this is no surprise as the number of absorbing DEHP molecules in the probe volume increases. However, this dependence is complicated by additional fast (Δt < 10 ns) decay channels that become important at higher DEHP concentrations. The decay dynamics shows a concentration dependence, with faster signal decays at higher DEHP concentrations, evidenced in Fig. 11 by the decreasing spread of the yield traces on the logarithmic scale. Thus, the ratio of the yields at the latest and earliest delay times increases from 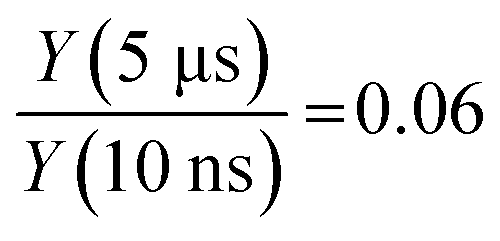 at a concentration of 1.5 M and to a value of 0.23 at a concentration of 0.0025 M. While the long-lived states invariably survive in significant amounts for at least 5 μs, their decay accelerates in more concentrated DEHP solutions.
at a concentration of 1.5 M and to a value of 0.23 at a concentration of 0.0025 M. While the long-lived states invariably survive in significant amounts for at least 5 μs, their decay accelerates in more concentrated DEHP solutions.
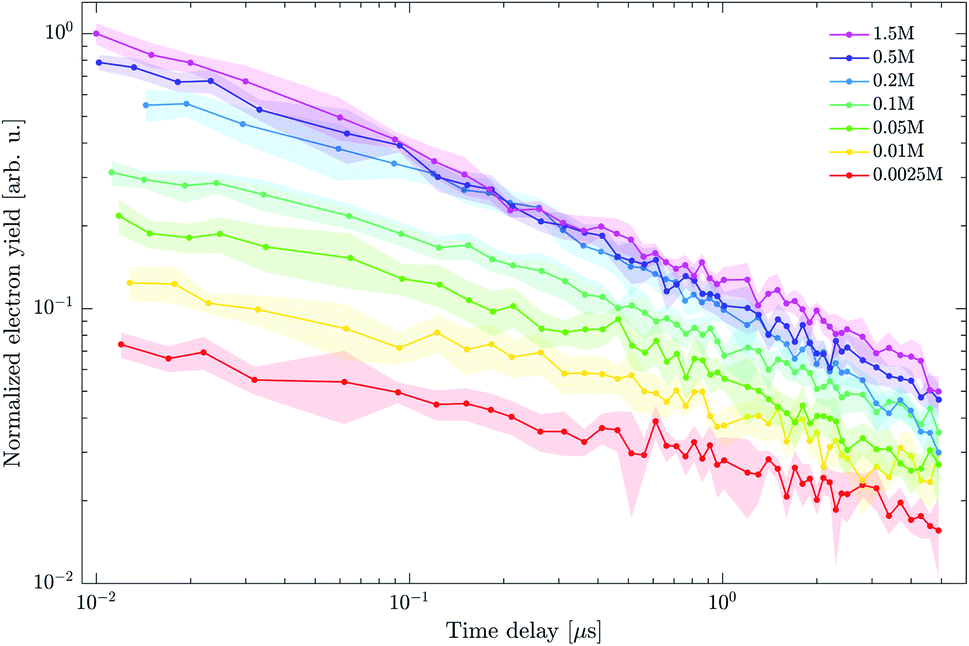 | ||
| Fig. 11 Electron yield decay dynamics as a function of DEHP concentration extracted from the pump–probe eTOF traces. Shaded regions illustrate the estimated signal uncertainty and lines are connecting the measured data points to guide the eye. Data is normalized to the signal at 10 ns from the measurement with highest DEHP concentration (1.5 M). | ||
Discussion of the long-lived states assignment
All the evidence so far speaks for the existence of long-lived states, but what are their nature? In the following we discuss the assignment of the long-lived states based on the presented experimental data, in particular the IE and decay dynamics. For the latter, we analyzed the data from Fig. 11 with two different phenomenological models (first order decay kinetics and diffusion-limited kinetics).A commonly encountered long-lived state in the literature on similar aromatic systems is the triplet (T1) state. For example, efficient and fast formation of the triplet T1 state following excitation to the Sn manifold was reported for the benzoic acid monomer in the gas phase43–45 and in acetonitrile.38 In the latter, photoexcitation at 267 nm resulted in the formation of T1 (∼2.5 ps) with a large quantum yield (∼0.65). No photofragments were observed. A T1 lifetime of 9 μs was measured in N2 saturated solutions and 0.1 μs in air saturated solutions. The photo-physics of DEHP was reported in ref. 46 following excitation with 20 ns, 266 nm pulses in dilute 1,1,2-trichlorotrifluoroethane solutions. The authors report that long-lived triplet states decay via a first-order decay process (∼80 ns) in O2 saturated solutions, while second-order triplet–triplet annihilation kinetics was reported in Ar-saturated solutions with half-lives of a few microseconds, leading to deactivation into the singlet manifold and to the formation of biradicals on a longer timescale (∼20 μs). Similar results were found in smaller analogues of DEHP, such as dibutyl phthalate,39 where triplet states were also suggested as primary photoproducts.
From the above, it seems reasonable to assume that a likely candidate for the long-lived states is the triplet (T1) state. This assignment is supported by the calculated IE (5.4 eV for VIE and 4.6 eV for AIE, see ESI, Section 2)† which is in reasonable agreement with the experiment (Fig. 5 and 10). Contributions from other candidates, such as photo-fragments or solvated electrons do not appear plausible for the same reasons already given for the band assignment in the ns single pulse PES (see above).
Let us now consider the decay dynamics (Fig. 11) as a further test of the triplet state assignment. The photophysics of the triplet (T1) state47 includes various unimolecular processes (e.g. phosphorescence and intersystem crossing) and efficient quenching reactions by contaminants (most often O2). The intrinsic T1 lifetimes (in the absence of quenchers) are typically on the order of microseconds, so that the <5 μs decay (Fig. 11) must have another explanation. Provided quenchers are present in excess (or if the process is unimolecular), the quenching follows pseudo-first order kinetics, described by a single exponential decay with a rate constant equal to the sum of rate constants for all quenching reactions starting from the same initial state.
Therefore, in a first phenomenological approach we use an exponential decay model to analyze the data. A minimum of three independent exponential decay terms were required to obtain a reasonable fit to the data in Fig. 11
Yfit(t) = a1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−k1t) + a2exp(−k2t) + a3exp(−k3t) exp(−k1t) + a2exp(−k2t) + a3exp(−k3t) |
The need for three exponential decays suggests that the electron yield dynamics cannot be explained by triplet quenching processes only (e.g., oxygen quenching). Either additional decay channels (e.g., bimolecular processes) or additional states seem to contribute to the signal (e.g., intermediate states or initial states other than the triplet). It is currently not possible to assign these three timescales to a physical reaction mechanism. Further experiments examining the effect of quenchers (i.e., oxygen) on the decay kinetics are necessary since in our present experiment the oxygen content could not be determined or controlled. As a caveat, we note that a sufficient number of exponential decays can eventually describe any decay dynamics, even when physical mechanisms other than first-order reactions are at play.48
As an alternative picture let us consider a situation where the above-mentioned quenching reactions are absent and only bimolecular processes (self-quenching and triplet–triplet annihilation46,47) are observed on the time scale of the experiment. We found that the following ansatz leads to a reasonable fit to the data in Fig. 11
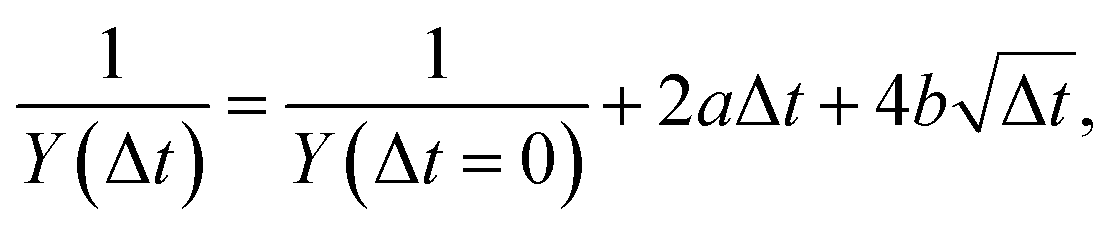
Finally, we would like to note that our electron yield measurements cannot directly distinguish triplet state contributions from other intermediate states with a similar IE. In both approaches (exponential and diffusion-limited) the involvement of intermediate states would entail more complicated kinetic expressions to reproduce the data with a physically meaningful parameterization. For example, excimer (excited DEHP dimer) formation is one of the outcomes of the triplet–triplet annihilation process.47 Since their IE is similar to the triplet state, the electron yield could well contain contributions from both species.
In conclusion, we cannot at this moment assign the electron yield decay dynamics to a precise physical mechanism. We analyzed the data considering two limiting cases, composite first order (exponential) kinetics and diffusion-limited kinetics. Neither model affords a conclusive analysis of the data in terms of physically interpretable parameters, possibly indicating that intermediate levels are involved in the observed “long-lived states” dynamics. Further kinetics experiments are necessary to obtain a clear picture, e.g., by quantifying the influence of experimental parameters such as the oxygen concentration, the viscosity of the liquid or the temperature.
Results and discussion – size effects
In this section, we focus on the effect of droplet size on the electron yield decay dynamics. Fig. 12 shows results obtained from the ns-pump (4.7 eV) and fs-probe (3.1 eV) experiment on neutral droplets with a DEHP concentration of 0.2 M. The droplet diameter was varied from 500 nm to 1200 nm by adjusting the settings of the AAC. We also show additional measurements for even larger particles, for which we can only roughly estimate a size range. These droplets formed in the ADL at higher operating pressures, presumably in a coagulation process from a 500 nm droplet sample. Based on the observation of a strong light scattering signal, we qualitatively assign these particles to a size range of a few micrometers. The traces in Fig. 12 are normalized to the respective signal at ∼10 ns in order to account for different number concentrations of droplets in the spectrometer as a consequence of different droplet generation conditions.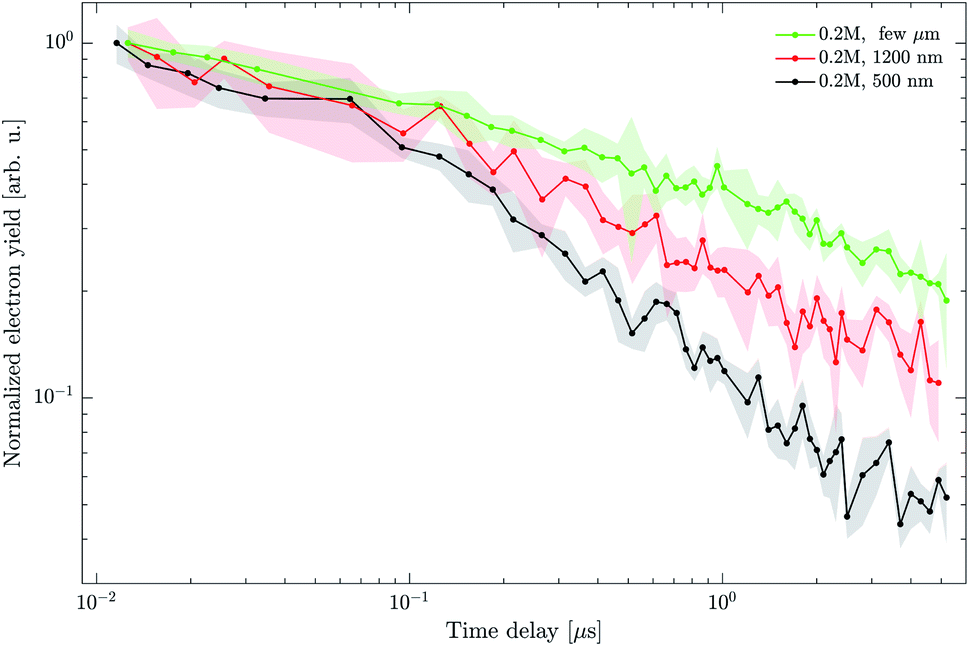 | ||
| Fig. 12 Electron yield decay dynamics for different diameters of neutral droplets containing 0.2 M DEHP. Shaded regions illustrate the estimated signal uncertainty and lines are connecting the measured data points to guide the eye. Each individual trace is normalized to its signal at 10 ns. | ||
The electron yield shows a decay rate which decreases with increasing droplet diameter from 500 nm (black) to 1200 nm (red) to a few micrometers (green).
How can the observed size-dependence be explained? First, we need to consider the effects of inelastic electron scattering which determines the experimental probing depth by limiting the distance from the surface from which photoelectrons can escape to vacuum and be detected. For water, the probing depth of <10 nm at an eKE of a few eV results from a value of ∼3 nm for the inelastic mean-free path (IMFP).14,31 Taking data on solid benzene49,50 as a rough estimate for the IMFP in DEHP/DEHS mixtures we arrive at a range of ∼1–10 nm. The probing depth is likely slightly higher than in water (a lower density of scatterers leads to a longer IMFP), but presumably does not exceed ∼10 nm. Consequently, we can assume that our measurements (Fig. 12) are only sensitive to long-lived states that are present within a thin layer less than ∼10 nm away from the surface. The range of diffusion in DEHP/DEHS droplets on a timescale of ∼200 ns to ∼2 μs lies between ∼2 nm and ∼6 nm (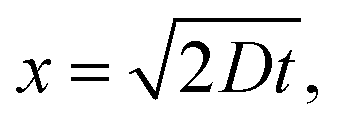 assuming D = 10−2 nm2 ns−1).51 These distances are smaller than the probing depth and indicate that the long-lived states do not diffuse out of the layer defined by the probing depth within our pump–probe time-window.
assuming D = 10−2 nm2 ns−1).51 These distances are smaller than the probing depth and indicate that the long-lived states do not diffuse out of the layer defined by the probing depth within our pump–probe time-window.
In addition to the probing depth, the light intensity distribution inside the droplet must be taken into account. For droplets with diameters in the submicrometer range, interacting with visible or UV light, the internal light intensity distribution is not homogeneous,1,5,13 leading to spatially dependent photoexcitation and photoionization probabilities. Therefore, it is important to examine whether the observed size-dependence could arise from the internal electric field magnitude or size-dependent spatial overlap of pump and probe pulses. The probability of forming long-lived states (we assume a triplet) in an infinitesimal volume dV is proportional to
| [T1(dV)] ∝ ΦTσS[S0]Ipump(dV) |
| Y(dV) ∝ σT[T1(dV)]Iprobe2(dV) |
| Y(dV) ∝ Ipump(dV)Iprobe2(dV) |
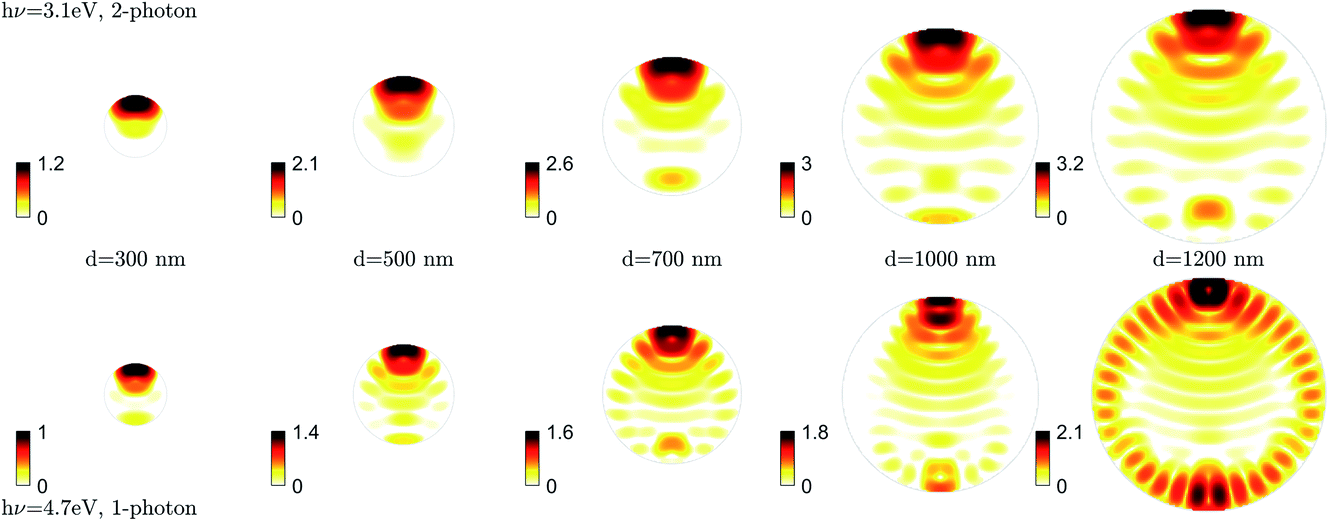 | ||
| Fig. 13 ADDA simulations top row Iprobe2, bottom row Ipump. Light propagation direction is upwards and the polarization vector lies horizontally. The color axis is given as the internal intensity (Iprobe2, Ipump) relative to the incident intensity outside the droplet. The color scale is given on a logarithmic axis and signal below the input intensity (<1) is shown as white. | ||
We used the discrete-dipole approximation52 to calculate Ipump(dV) and Iprobe(dV)2 assuming refractive indices of N = 1.52 + i0.001 (4.7 eV) and N = 1.49 + i0.0001 (3.1 eV). For the real part of the refractive index we used values for DEHP13 (DEHS is assumed to have the same value53). For the imaginary part, we scaled the DEHP values at the corresponding wavelength by 0.1 to account for a concentration of 0.2 M (DEHS does not significantly absorb at these wavelengths).
Fig. 13 shows the resulting internal light intensity distributions in the plane spanned by the laser propagation and polarization directions. The color scale shows the logarithm of the internal light intensity relative to the incident light intensity. The effect of nano-focusing is clearly visible in the asymmetry of the distribution and the intensity enhancement which increases for larger particles (left to right). Iprobe(dV)2 (top row) shows a large light intensity enhancement with particle size, with maximum values increasing by almost 1 order of magnitude going from a particle diameter of 500 nm to 1000 nm (see color bars). By comparison, Ipump(dV) increases by a factor of ∼2 in the same size range. The larger enhancement in the probe process (Iprobe(dV)2) reflects the dependence of the 2-photon ionization probability on the square of the intensity.
The most obvious effect of droplet size is the different magnitude of light intensity enhancement. Data in Fig. 12 are normalized to signal at ∼10 ns so that total relative signal at longer times should reflect the survival probability of the long-lived states since
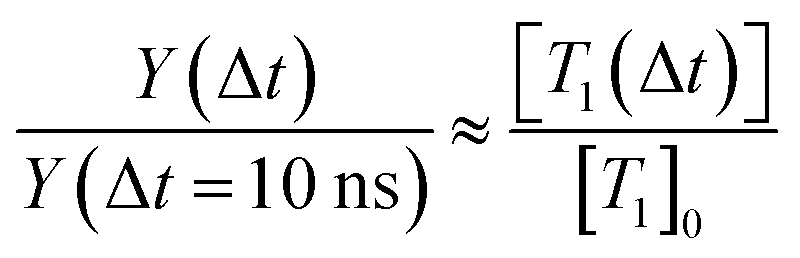
In addition, the spatial distribution of the light intensity needs to be considered. From Fig. 13, the spatial distribution of both pump and probe pulses seems qualitatively similar. To investigate the spatial distribution of the light intensity in more detail, we integrated the 3-dimensional internal light distributions over the whole solid angle (Ω)
| I(r) = ∫∫I(r,Ω)r2dΩ |
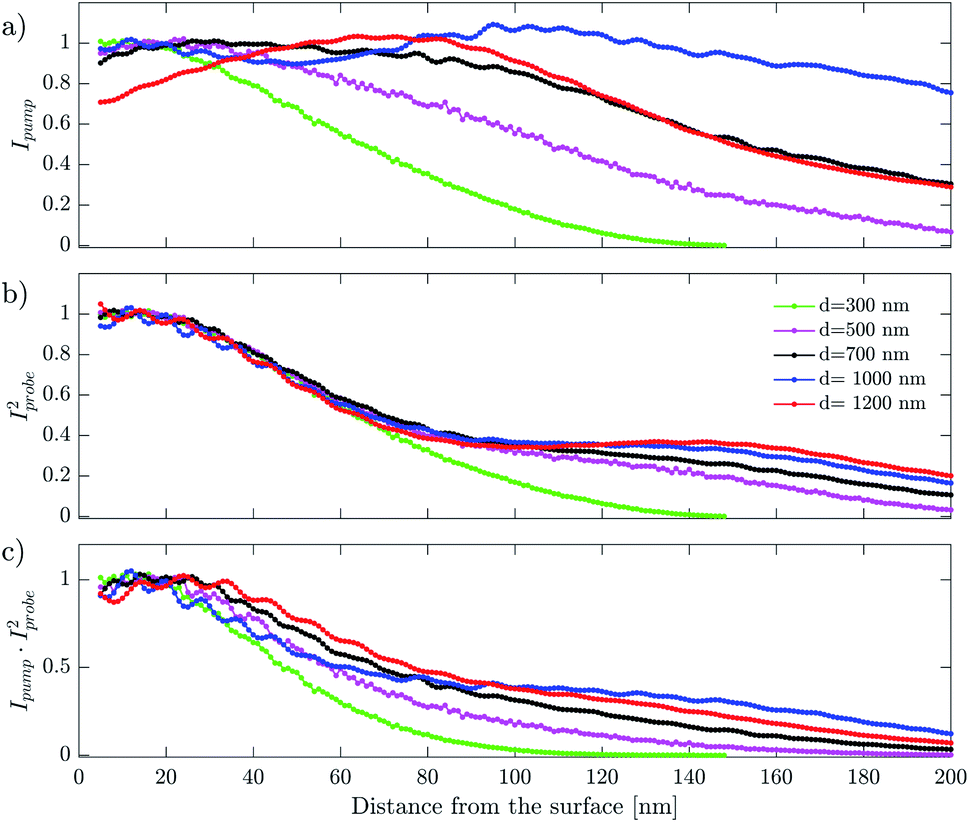 | ||
| Fig. 14 Internal light intensity distribution integrated over the whole solid angle for droplets of different diameter (d). Data for d = 1000 nm and 1200 nm were smoothed to average out the oscillations coming from the Mie resonances. Panel (a) is a plot of the pump intensity (Ipump), panel (b) is a plot of the probe intensity squared (Iprobe2) and panel (c) is their product. | ||
The internal light intensity distribution of pump pulses Ipump (Fig. 14a) is maximal close to the surface and extends further into the droplet interior for larger droplets. In the probe case, Iprobe2 (Fig. 14b), the distribution shows a pronounced maximum close to the surface and a weaker tail extending to the droplet interior for larger sizes. The product IpumpIprobe2, which determines the spatial overlap of our pump–probe experiment is shown in Fig. 14c. It shows one dominant feature close to the droplet surface. For analyzed droplets diameters this feature is essentially size-independent up to ∼30 nm away from the surface. This distance is larger than the probing depth (<10 nm) and we can therefore conclude that the size-dependent distribution of light inside the droplets plays no role on the observed decay dynamics.
From the above, it is clear that the observed increase in the electron survival probability with droplet size cannot be explained by diffusion out of the finite probing depth nor by variations in the internal light distribution. This leaves the influence of the droplet surface on the reactions taking place within a thin layer <10 nm from the surface as a possible cause for the observed size-dependence of the dynamics.
Accelerated reaction rates have been previously reported in micrometer sized droplets, but their physical origin is still being discussed.54 A possible explanation can be proposed in the case of diffusion-limited reaction, where the reaction time is lowered by the decreased dimensionality at the droplet surface.55 This constraint is relaxed as the droplet size increases and the reaction slows down, in qualitative agreement with our data. This hypothesis needs further experimental testing and must be interpreted with care. As highlighted in ref. 54, detailed and critical analysis of different effects is necessary before assigning the altered kinetics to a particular surface effect.
Results and discussion – charge effects
For the final section of this paper, we continue our discussion by presenting experiments on charged droplets. Surface charge has been proposed as a mechanism to explain accelerated reactivity observed in nanometer and micrometer sized droplets.54,56In Fig. 15, we show the electron yield decay dynamics recorded on the droplets with a diameter of 500 nm with varying degrees of charging. The electron yield is normalized to its signal at ∼10 ns. The charge states correspond to the measurements shown in Fig. 3. The droplet charge is assumed to be homogeneously distributed on the droplet surface. This assumption is supported by computational studies on electrospray ionization.54,57,58 In this picture, the electric field magnitude E on the droplet surface of diameter d can be calculated from the droplet charge state (q = ne) by the simple model of a charged conductive sphere (see also ref. 54)
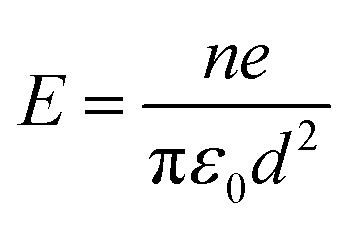
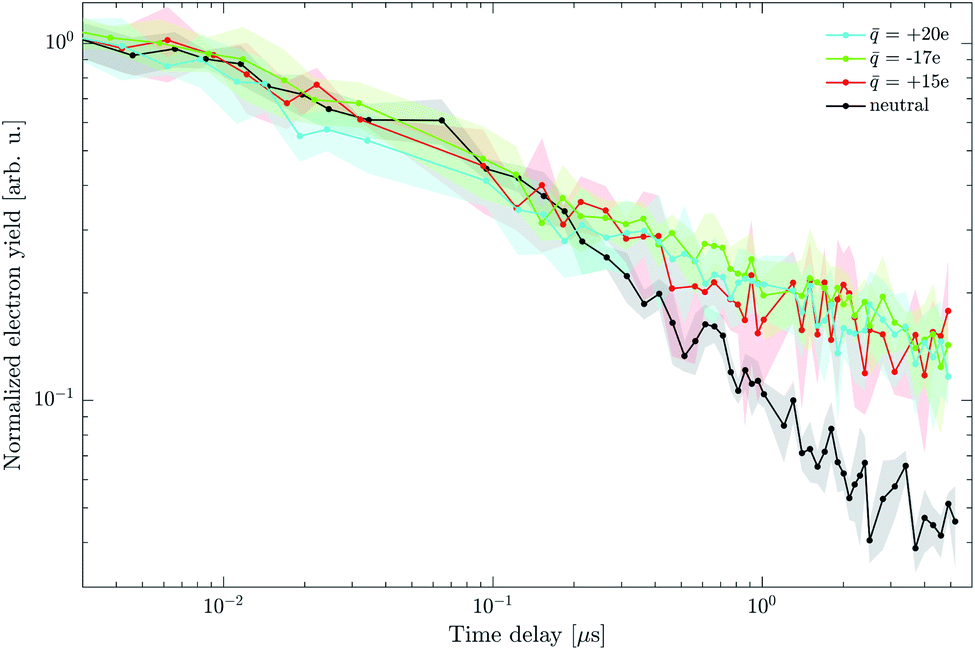 | ||
| Fig. 15 Effect of charge on electron yield dynamics. Droplets of 500 nm in diameter with charge states of +15e, +20e and −17e (colored traces) show a significantly slower electron decay rate than neutral droplets of the same size (black). Shaded regions illustrate the estimated signal uncertainty and lines are connecting the measured data points to guide the eye. Each individual trace is normalized to its signal at 10 ns. | ||
The number density of the charged droplet sample was significantly lower than in the neutral case, which is reflected in the larger signal uncertainty. The temporal evolution of the electron yield of charged droplets clearly deviates from the neutral sample and leads to higher electron yield at times above ∼200 ns. The electron yield is enhanced for both charge polarities and no clear dependence on the electric field magnitude was observed within the relatively large uncertainty.
First, it is important to discuss the nature of the droplet charging process since we are investigating excited state photophysics where quenching by impurities could have a strong effect. The droplets are charged by collisions with ions generated by the discharge in the gas flow. The species responsible for charging therefore depends on the polarity of the discharge, specific charge settings as well as the gas composition. In air, positive corona discharge primarily consists of small, protonated water clusters.59 The situation is more complicated in the negative case,60 where the generation of ozone leads to the presence of hydrated cluster anions of OH−and NO3−. The number of absorbed ions directly determines the droplet charge, which at the charge states used in our experiment corresponds to a negligible concentration of these ions. The effects of ions on the photo-physics can therefore be safely neglected and cannot explain the difference from the neutral case. This is further confirmed by the fact that both polarities (different ions) show the same effect even though different ions would be formed.
But how then can the presence of charge influence measured photo-electron yields? Let us consider two general mechanisms by which an electric field can affect the electron yield. The first one applies to ionization processes that proceed via charge-pair states. In this case, the electric field can enhance the electron signal by facilitating charge separation and thus increasing the ionization probability. Examples of this mechanism are found in benzene excimers excited to charge-transfer states just below the ionization threshold.33 Electric fields of about 10−3 V nm−1 produced a measurable increase in free charge generation.
The second mechanism works through the influence of the electric field on the diffusion of a particle. This has been widely investigated in the context of the recombination of charged particle pairs61 (e.g. solvated electron) and of neutral-charged pairs.62 For ion-pair geminate recombination,61 electric fields of ∼10−2 V nm−1 resulted in an increase in the escape probability. However, in the case of neutral reactants, the effects of the electric field are not obvious.
The electric field effect on the electron yield could bring additional insight into the decay mechanism. Within the assumptions of our kinetic model, we can exclude the effect of charged particle diffusion since only neutral species are considered. We therefore propose that surface charge facilitates the charge-pair separation that takes place during the ionization process.
The electric charge effects could be a hint that excimer states, in addition to the triplet state, could contribute to the signal since their ionization often proceeds via a charge-separation mechanism.33 This could be rationalized by a triplet–triplet annihilation mechanism which proceeds via an excimer intermediate (T + T ⇆ E →). Future studies should therefore extend the kinetic analysis to account for the possible effect of diffusion-limited monomer-excimer reactivity.63,64 An intriguing observation is that our exponential kinetic model yields a time constant of ∼240 ns, commensurate with the time delay after which the charged droplet electron yield departs from the neutral case in Fig. 15.
Conclusions
We performed a photoionization study of submicrometer droplets consisting of solutions of DEHP in DEHS and investigated the effect of pulse duration on the resonant photoionization process. When comparing single-pulse photoelectron spectra obtained upon ionization with 4.7 eV nanosecond and femtosecond pulses, we observed a pronounced difference. The presence of an intense higher kinetic energy band in the spectra of the nanosecond case was attributed to contributions from long-lived states populated within the duration of the nanosecond pulse. This assignment was confirmed by pump–probe photoelectron spectra, in which only the long-lived states contributions were observed. The relative contribution of the long-lived states decreased at higher DEHP concentrations, suggesting a more efficient quenching process in the presence of more DEHP molecules. The decay dynamics of the electron yield was measured by a pump–probe experiment using 4.7 eV nanosecond pump and 3.1 eV femtosecond probe pulses. The electron signal survived for at least 5 μs and showed an accelerated decay rate at higher DEHP concentrations, consistent with the single-pulse nanosecond spectra. We tentatively assign the long-lived states to a DEHP triplet, which is supported by the experimental photoelectron spectrum and DFT calculations. The decay dynamics were analyzed in terms of two limiting cases (1st order multi-exponential and diffusion-limited), but neither of these phenomenological approaches affords a conclusive interpretation of the fit parameters. The results might hint at the involvement of an additional intermediate state in the observed “long-lived states” dynamics.We further investigated the influence of particle size and charge state on the decay dynamics of the electron yield. We observed a decrease of the decay rate with increasing droplet size. Because of the relatively low probing depth (<10 nm), the experiment preferentially probes the kinetics near the surface. Therefore, we tentatively attribute the decrease of the rate observed for larger droplets to an altered diffusion of the reactants near the surface. Further experiments are necessary before a detailed mechanism can be proposed. The decay rate of the electron yield also decreases in charged compared with neutral droplets. This effect does not depend on the charge polarity. We propose that the electric field on the surface of the droplets increases the ionization probability by facilitating charge-separation in the ionization process. This result hints that excimer states could be important intermediates in the long-lived states decay kinetics.
Although many questions remain open, the present study highlights the potential of droplet photoelectron imaging to gain insight into the photophysics and photochemistry in confined systems. The combination of pump–probe photoelectron spectroscopy with droplet beams of controlled size (submicrometer regime) and charge enables the study of kinetics at a surface with high time resolution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Dr Martin Fierz for his help and lending us devices for aerosol charging experiments. We thank David Stapfer, Dr Egor Chasovskikh and Dr Thomas E. Gartmann for technical contributions to the experimental setup, in particular our droplet photoelectron spectrometer. We also thank Daniel Zindel, Markus Steger and Dr Erich C. Meister for technical support regarding chemical and electronic aspects of the project. This project has received funding from the European Unions Horizon 2020 research and innovation program from the European Research Council under the Grant Agreement No. 786636, and the research was supported by the NCCR MUST, funded by the Swiss National Science Foundation (SNSF), through ETH-FAST, and through SNSF project No. 200020_200306.Notes and references
- L. Ban, B. L. Yoder and R. Signorell, Annu. Rev. Phys. Chem., 2020, 71, 315–334 CrossRef CAS PubMed.
- J. Shu, K. R. Wilson, M. Ahmed and S. R. Leone, Rev. Sci. Instrum., 2006, 77, 043106 CrossRef.
- K. R. Wilson, H. Bluhm and M. Ahmed, in Fundamentals and Applications in Aerosol Spectroscopy, ed. R. Signorell and J. P. Reid, CRC Press, 2010, pp. 367–399 Search PubMed.
- E. Antonsson, H. Bresch, R. Lewinski, B. Wassermann, T. Leisner, C. Graf, B. Langer and E. Rühl, Chem. Phys. Lett., 2013, 559, 1–11 CrossRef CAS.
- K. R. Wilson, Z. Shengli, S. Jinian, E. Rühl, S. R. Leone, G. C. Schatz and M. Ahmed, Nano Lett., 2007, 7, 2014–2019 CrossRef CAS.
- C. C. Su, Y. Yu, P. C. Chang, Y. W. Chen, I. Y. Chen, Y. Y. Lee and C. C. Wang, J. Phys. Chem. Lett., 2015, 6, 817–823 CrossRef CAS PubMed.
- B. R. Bzdek and J. P. Reid, J. Chem. Phys., 2017, 147, 220901 CrossRef PubMed.
- B. R. Bzdek, J. P. Reid and M. I. Cotterell, Commun. Chem., 2020, 3, 10–13 CrossRef.
- M. I. Jacobs, B. Xu, O. Kostko, N. Heine, M. Ahmed and K. R. Wilson, J. Phys. Chem. A, 2016, 120, 8645–8656 CrossRef CAS PubMed.
- E. Antonsson, M. Patanen, C. Nicolas, J. J. Neville, S. Benkoula, A. Goel and C. Miron, Phys. Rev. X, 2015, 5, 1–7 Search PubMed.
- O. Sublemontier, C. Nicolas, D. Aureau, M. Patanen, H. Kintz, X. Liu, M. A. Gaveau, J. L. Le Garrec, E. Robert, F. A. Barreda, A. Etcheberry, C. Reynaud, J. B. Mitchell and C. Miron, J. Phys. Chem. Lett., 2014, 5, 3399–3403 CrossRef CAS PubMed.
- M. I. Jacobs, B. Xu, O. Kostko, A. A. Wiegel, F. A. Houle, M. Ahmed and K. R. Wilson, J. Phys. Chem. A, 2019, 123, 6034–6044 CrossRef CAS PubMed.
- R. Signorell, M. Goldmann, B. L. Yoder, A. Bodi, E. Chasovskikh, L. Lang and D. Luckhaus, Chem. Phys. Lett., 2016, 658, 1–6 CrossRef CAS.
- R. Signorell, Phys. Rev. Lett., 2020, 124, 205501 CrossRef CAS PubMed.
- O. Kostko, M. I. Jacobs, B. Xu, K. R. Wilson and M. Ahmed, J. Chem. Phys., 2019, 151, 2–5 CrossRef PubMed.
- S. Amanatidis, B. L. Yoder and R. Signorell, J. Chem. Phys., 2017, 146, 224204 CrossRef PubMed.
- M. I. Jacobs, O. Kostko, M. Ahmed and K. R. Wilson, Phys. Chem. Chem. Phys., 2017, 19, 13372–13378 RSC.
- L. Ban, T. E. Gartmann, B. L. Yoder and R. Signorell, Phys. Rev. Lett., 2020, 124, 13402 CrossRef CAS PubMed.
- K. R. Wilson, M. Jimenez-Cruz, C. Nicolas, L. Belau, S. R. Leone and M. Ahmed, J. Phys. Chem. A, 2006, 110, 2106–2113 CrossRef CAS PubMed.
- K. R. Wilson, D. S. Peterka, M. Jimenez-Cruz, S. R. Leone and M. Ahmed, Phys. Chem. Chem. Phys., 2006, 8, 1884 RSC.
- S. Tigrine, N. Carrasco, D. K. Bozanic, G. A. Garcia and L. Nahon, Astrophys. J., 2018, 867, 164 CrossRef CAS.
- S. Hartweg, G. A. Garcia, D. K. Božanić and L. Nahon, J. Phys. Chem. Lett., 2021, 12, 2385–2393 CrossRef CAS PubMed.
- F. Tavakoli and J. S. Olfert, Aerosol Sci. Technol., 2013, 47, 916–926 CrossRef CAS.
- X. Wang, F. E. Kruis and P. H. McMurry, Aerosol Sci. Technol., 2005, 39, 611–623 CrossRef CAS.
- M. Goldmann, J. Miguel-Sánchez, A. H. C. West, B. L. Yoder and R. Signorell, J. Chem. Phys., 2015, 142, 224304 CrossRef PubMed.
- B. Dick, Phys. Chem. Chem. Phys., 2014, 16, 570–580 RSC.
- J.-B. Du, Y.-L. Tang, Z.-W. Long, S.-H. Hu and T. Li, Russ. J. Phys. Chem. A, 2014, 88, 819–822 CrossRef CAS.
- R. Signorell, M. Goldmann, B. L. Yoder, A. Bodi, E. Chasovskikh, L. Lang and D. Luckhaus, Chem. Phys. Lett., 2016, 658, 1–6 CrossRef CAS.
- A. Roy, R. Seidel, G. Kumar and S. E. Bradforth, J. Phys. Chem. B, 2018, 122, 3723–3733 CrossRef CAS PubMed.
- F. Buchner, H. H. Ritze, J. Lahl and A. Lübcke, Phys. Chem. Chem. Phys., 2013, 15, 11402–11408 RSC.
- D. Luckhaus, Y. Yamamoto, T. Suzuki and R. Signorell, Sci. Adv., 2017, 3, e1603224 CrossRef PubMed.
- Y. I. Yamamoto, S. Karashima, S. Adachi and T. Suzuki, J. Phys. Chem. A, 2016, 120, 1153–1159 CrossRef CAS PubMed.
- T. W. Scott, C. L. Braun and A. C. Albrecht, J. Chem. Phys., 1982, 76, 5195–5202 CrossRef CAS.
- C. Wohlfahrt, in Static Dielectric Constants of Pure Liquids and Binary Liquid Mixtures, ed. O. Madelung, Springer-Verlag, Berlin/Heidelberg, pp. 5–228 Search PubMed.
- R. Weinkauf, P. Aicher, G. Wesley, J. Grotemeyer and E. W. Schlag, J. Phys. Chem., 1994, 98, 8381–8391 CrossRef CAS.
- A. L. Thompson and T. J. Martínez, Faraday Discuss., 2011, 150, 293–311 RSC.
- T. Suzuki, Molecules, 2014, 19, 2410–2433 CrossRef PubMed.
- C. Yang, H. Su, X. Sun and M. W. George, J. Chem. Phys., 2012, 136, 204507 CrossRef PubMed.
- N. Liu, M.-H. Wu, G. Xu, Q. Yuan, L. Tang and J. Zhou, in 2009 International Conference on Energy and Environment Technology, IEEE, 2009, vol. 3, pp. 455–458 Search PubMed.
- Y. A. Jeilani, B. H. Cardelino and V. M. Ibeanusi, J. Am. Soc. Mass Spectrom., 2011, 22, 1999–2010 CrossRef CAS PubMed.
- M. Lacko, P. Papp and Š. Matejčík, J. Chem. Phys., 2018, 148, 214305 CrossRef PubMed.
- T. K. Lau, W. Chu and N. Graham, Chemosphere, 2005, 60, 1045–1053 CrossRef CAS PubMed.
- J. Li, F. Zhang and W.-H. Fang, J. Phys. Chem. A, 2005, 109, 7718–7724 CrossRef CAS PubMed.
- G. Meijer, M. S. De Vries, H. E. Hunziker and H. R. Wendt, J. Phys.Chem., 1990, 94, 4394–4396 CrossRef CAS.
- S. I. Kamei, H. Abe, N. Mikami and M. Ito, J. Phys. Chem., 1985, 89, 3636–3641 CrossRef CAS.
- G. Hizal, Q. Q. Zhu, C. H. Fischer, T. Majima and W. Schnabel, J. Photochem. Photobiol., A, 1992, 69, 33–39 CrossRef CAS.
- J. B. Birks, Photophysics of aromatic molecules, Wiley-Interscience, London, New York, 1970 Search PubMed.
- D. J. S. Birch, A. D. Dutch, R. E. Imhof, B. Nadolski and I. Soutar, J. Photochem., 1987, 38, 239–254 CrossRef CAS.
- T. Goulet and J. P. Jay-Gerin, Solid State Commun., 1985, 55, 619–623 CrossRef CAS.
- T. Goulet and J.-P. Jay-Gerin, Int. J. Radiat. Appl. Instrum., Part C, 1986, 27, 229–239 CAS.
- K. R. Harris, B. Ganbold and W. S. Price, J. Chem. Eng. Data, 2015, 60, 3506–3517 CrossRef CAS.
- M. A. Yurkin and A. G. Hoekstra, J. Quant. Spectrosc. Radiat. Transfer, 2011, 112, 2234–2247 CrossRef CAS.
- L. De Lorenzi, M. Fermeglia and G. Torriano, J. Chem. Eng. Data, 1997, 42, 919–923 CrossRef CAS.
- G. Rovelli, M. I. Jacobs, M. D. Willis, R. J. Rapf, A. M. Prophet and K. R. Wilson, Chem. Sci., 2020, 11, 13026–13043 RSC.
- S. Mondal, S. Acharya, R. Biswas, B. Bagchi and R. N. Zare, J. Chem. Phys., 2018, 148, 244704 CrossRef PubMed.
- S. Banerjee, E. Gnanamani, X. Yan and R. N. Zare, Analyst, 2017, 142, 1399–1402 RSC.
- L. Konermann, E. Ahadi, A. D. Rodriguez and S. Vahidi, Anal. Chem., 2013, 85, 2–9 CrossRef CAS PubMed.
- C. F. Chamberlayne and R. N. Zare, J. Chem. Phys., 2020, 152, 184702 CrossRef CAS PubMed.
- D. Y. H. Pui, S. Fruin and P. H. McMurry, Aerosol Sci. Technol., 1988, 8, 173–187 CrossRef CAS.
- K. Sekimoto and M. Takayama, Int. J. Mass Spectrom., 2007, 261, 38–44 CrossRef CAS.
- A. Mozumder, J. Chem. Phys., 1974, 60, 4300–4304 CrossRef CAS.
- S. Y. Reigh, K. J. Shin and M. Tachiya, J. Chem. Phys., 2008, 129, 1–12 CrossRef PubMed.
- M. N. Berberan-Santos and J. M. G. Martinho, J. Chem. Phys., 1991, 95, 1817–1824 CrossRef CAS.
- K. Sienicki and M. A. Winnik, J. Chem. Phys., 1987, 87, 2766–2772 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information on the experimental and theoretical methods. See DOI: 10.1039/d1fd00108f |
| This journal is © The Royal Society of Chemistry 2022 |