 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Studying the impact of the pre-exponential factor on templated nucleation
Vivek
Verma
 *a,
Hamish
Mitchell
a,
Mingxia
Guo
a,
Benjamin K.
Hodnett
b and
Jerry Y. Y.
Heng
ac
*a,
Hamish
Mitchell
a,
Mingxia
Guo
a,
Benjamin K.
Hodnett
b and
Jerry Y. Y.
Heng
ac
aDepartment of Chemical Engineering, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK. E-mail: v.verma@imperial.ac.uk
bSynthesis and Solid State Pharmaceutical Centre, Department of Chemical Sciences, Bernal Institute, University of Limerick, Limerick, V94 T9PX, Ireland
cInstitute of Molecular Science and Engineering, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
First published on 13th December 2021
Abstract
Traditionally, the enhancement of nucleation rates in the presence of heterogeneous surfaces in crystallisation processes has been attributed to the modification of the interfacial energy of the system according to the classical nucleation theory. However, recent developments have shown that heterogeneous surfaces instead alter the pre-exponential factor of nucleation. In this work, the nucleation kinetics of glycine and diglycine in aqueous solutions have been explored in the presence and absence of a heterogeneous surface. Results from induction time experiments show that the presence of a heterogeneous surface increases the pre-exponential factor by 2-fold or more for both glycine and diglycine, while the interfacial energy remains unchanged for both species. This study suggests that the heterogeneous surface enhances the nucleation rate via hydrogen bond formation with both glycine and diglycine. This is verified by hydrogen bond propensity calculations, molecular functionality analysis, and calculation of the time taken for a solute molecule to attach to the growing nucleus, which is an order of magnitude shorter than the estimated lifetime of the hydrogen bond. The effect of the heterosurface is of greater magnitude for diglycine than for glycine, which may be due to the heightened molecular complementarity between the hydrogen bond donor and acceptor sites on diglycine and the heterosurface.
1. Introduction
The universal nature of crystallization has been explored in every aspect of science, from organics to inorganics to bio-organics, and from atmospherics to minerals. Crystallisation is a two-step process, with nucleation being the initial step of forming a stable new phase from a supersaturated medium. Nucleation is subsequently followed by growth as well as ripening, agglomeration, aggregation, and phase transformation. In both steps, supersaturation is the driving force, caused by the difference between the actual concentration of the crystallizing solute and the saturation concentration (solubility). There are a variety of methods for generating supersaturation, such as cooling, anti-solvent addition, evaporation, and the addition of precipitants, amongst others. Since the physical properties of the crystals, such as shape, structure, defects, and polymorphism are determined at the nucleation stage, this step is usually rate limiting for the crystallisation process.1,2The nucleation process occurring at interfaces is both profoundly important and widely encountered in different areas of science, such as material synthesis,3–5 biomineralisation,6 battery operation,7,8 geochemistry,9,10 geologic CO2 sequestration,11 cement hardening,12 and drug production.13,14 It is essential to analyse, predict, control, and fine-tune these systems by comprehending nucleation and quantifying the relevant thermodynamic and kinetic parameters. Heterogeneous nucleation occurs in ‘impure’ liquids, for example, where surface walls or nucleating agents (not crystal seeds) are present, rather than in an idealized supersaturated liquid, where internal fluctuations in density trigger the crossing of the kinetic barrier to nucleation, resulting in homogeneous nucleation. Ward and colleagues laid a modern scientific foundation for the study of heterogeneous nucleation in organic molecules.15–17 Their work on benzoic acid nucleation on the freshly cleaved surface of β-succinic acid or L-valine delivered by sublimation led to the concept of ledge-directed epitaxy (LDE). It was postulated that benzoic acid nucleation at a pair of intersecting cleavage planes on the heterosurface was a result of the favourable lattice matching.16
Over the past few decades, the ‘state-of-the-art’ approaches responsible for studying heterogeneous nucleation have been used for the non-specific adsorption of molecules on the heterosurface,18 specific interactions such as functional group matching,19 or selective interactions such as lattice matching.20 The materials that have successfully served the function of the heterosurface for heterogeneous nucleation are pharmaceutical excipients,14,21–23 silanised glass substrates,13,24,25 amino acids,26 self-assembled monolayers,27 bio-compatible28 and synthetic polymers,29,30 and porous substrates.31 Protein crystallization has also been achieved with chemically modified surfaces.32 These surfaces were used either to induce nucleation and/or study the nucleation kinetics of the drug molecules. Parambil et al.33 have reviewed the existing literature discussing both experimental and simulation studies directed towards deeper understanding of the underpinning mechanisms. Despite the significant number of publications in this area, it remains unclear how heterogeneous nucleation occurs and how the thermodynamic and kinetic factors are influenced by heterogeneous nucleation.
In recent years, functional group matching between the nucleating solute and the heterosurface has emerged directly or indirectly as the leading responsible factor for the occurrence of heterogeneous nucleation. Diao et al.34 showed that the nucleation density of aspirin increased dramatically in the presence of two polymers, in particularly poly(4-acryloylmorpholine) and poly(2-carboxyethyl acrylate). Aspirin’s hydrogen bond donor (HBD) sites can interact most efficiently within these polymers since they have hydrogen bond acceptor (HBA) sites. Later, Diao et al.35 also showed that nanopores of different shapes (spherical, square and hexagonal), specifically angular nanopores (15–120 nm), promoted the nucleation rate of aspirin. Although the porosity in polymers with specific wall angles enhanced the nucleation rate, pointing towards lattice matching as the potential reason, this was only evident for the polymers with HBA properties. Polymers lacking HBA properties did not influence the nucleation rate of aspirin.
The concept of hydrogen bonding complementarity in the template induced nucleation of pharmaceutical ingredients was further explored by Chadwick et al.19,36 for the heterogeneous nucleation of acetaminophen (AAP) in the presence of graphite, δ-mannitol, α-lactose monohydrate, L-histidine, poly(methylmethacrylate), and poly-n-butylmethacrylate. Interestingly, graphite was among the poorest in accelerating the heterogeneous nucleation of AAP. Later, Bueno et al.22 studied the influence the range of different heterosurfaces such as δ-mannitol, silica, microcrystalline cellulose (MCC), carboxymethyl cellulose, α/β-lactose, and polycaprolactone (PCL) on the heterogeneous nucleation of fenofibrate (FF). They found that FF nucleated slowest in the presence of the PCL heterosurface. This is because except for PCL, all the heterosurfaces studied possessed HBDs which could easily form hydrogen bonds with the HBAs of fenofibrate. Therefore, this study shows the influence of functional group complementarity in template induced nucleation systems.
Recently, Ouyang et al.37 were able to successfully nucleate and stabilise the carbamazepine (CBMZ) metastable form (FII) at lower supersaturations, at which normally only the stable form of CBMZ (FIII) crystallises, when crystallised in the presence of a phenyl functionalised silica heterosurface. The possible mechanism for this observation was the stabilisation of CBMZ molecules on the heterosurface due to the aromatic–aromatic interaction between the surface of phenyl-functionalized silica and the CBMZ molecule. Later, Ouyang et al.38 expanded the work of using functionalized silica (SiO2, SiO2–NH2, SiO2–COOH) as a heterosurface in the nucleation of vanillin. The functionalised silica was able to significantly enhance the nucleation rate of vanillin, which is evident by the reduction in the induction time from 4 hours to 20 minutes. The heterosurface also increased the growth rate of the nucleated vanillin crystal. These increases in the nucleation and growth rates were potentially due to the formation of hydrogen bonds between the vanillin molecules and the SiO2–NH2 and SiO2–COOH heterosurfaces. Recently Li et al.39 discovered that the use of silica particles as a heterosurface improved the crystallisation rate of lysozyme by reducing the induction time even in the presence of an impurity protein, thaumatin.
A recent study by Verma et al.14 studied the influence of a heterosurface (microcrystalline cellulose (MCC)) on the nucleation kinetic parameters of seven different active pharmaceutical ingredients (APIs), namely carbamazepine (CBMZ), acetaminophen (AAP), caffeine (CAF), clozapine (CPB), risperidone (RIS), phenylbutazone (PBZ), and FF. Crystallisation of the latter five APIs was accelerated in the presence of MCC compared to the former two, CBMZ and AAP. The maximum acceleration was observed for FF, which was 16 times faster than what was observed for homogeneous nucleation. This acceleration was possibly because of the formation of a stable hydrogen bond between the HBAs of the latter five APIs with the HBD sites on MCC. This study confirmed that the functional group complementarity between the API and heterosurface is an important factor in improving the API nucleation rate. This confirmation was supported by the hypothesis that the lifetime of the single adsorbed molecule on the surface of a hetero substance is sufficiently long to allow for the adsorption of other API molecules or clusters to attach to the adsorbed molecule, thereby leading to the formation of stable nuclei that remain attached to the heterosurface. This hypothesis was recently proved by Cazade et al.40 using Monte Carlo (MC) and molecular dynamics (MD) simulations. The MD simulation results for the adsorption of RIS onto MCC surfaces concluded that the hydrogen-bonded lifetime of 32 ns is comparatively longer than that of the RIS–RIS attachment lifetime in the solution. It is also an order of magnitude longer than the time to add a single molecule of RIS to a growing crystal at a moderate supersaturation.
Verma et al.14 also reported that the heterogeneous nucleation of APIs in the presence of MCC had minimal influence on the interfacial energy of APIs in the nucleating solution. This is contradictory to the reported literature claiming that heterosurfaces decrease the interfacial energy, thereby reducing the nucleation energy barrier for nucleation, resulting in an increased nucleation rate.2,41 Instead, they claimed that the increase in the nucleation rate in the presence of MCC was due to the increase in the pre-exponential factor (A) in the presence of MCC. The value of A increased more than 2-fold for the API, exhibiting an increase in the nucleation rate in the presence of MCC. This signifies that the rate of subcritical size cluster formation is at least 2-fold greater in the presence of MCC for most of the APIs, thereby promoting heterogeneous nucleation. This is consistent with their previous findings on the nucleation of clozapine–methanol solvate in the presence of MCC, confirming that the pre-exponential factor is the major contributing factor to heterogeneous nucleation.42
Glycine and diglycine were selected as molecules of interest for this work. The reason for this is that glycine is the simplest and most studied amino acid, consisting of a non-heavy hydrogen side chain, which eliminates the need to consider the effect of the side chain of the amino acid when analysing the effect of the heterosurface. Glycine exhibits a total of six polymorphs (α, β, γ, δ, ε, and ζ),43 but the latter three only form under extreme pressures.43 At atmospheric conditions, only α, β, and γ will form.43 The γ polymorph is the thermodynamically stable form of glycine, while α- and β-glycine exist as metastable and unstable polymorphs, respectively.44 However, the nucleation of γ-glycine is kinetically hindered when compared to that of α-glycine, meaning that under agitation at atmospheric conditions, α-glycine is the initially forming polymorph. The solution-mediated phase transformation of glycine occurs slowly, taking around 20 hours to begin.45 There have been numerous studies into the effects of process conditions on the polymorphic outcome of glycine crystallisation, which have identified factors such as humidity,46 pH,47 the usage of microdroplets,48 and the addition of precipitants such as sodium chloride.45 It is possible to distinguish between the α and γ forms visually – α-glycine forms as rod-like monoclinic crystals belonging to the space group P21/n,49 whereas γ-glycine forms trigonal pyramidal crystals belonging to P31 or P32.50
Recently, Vesga et al.51 showed that for the cooling crystallisation of aqueous solutions of glycine, agitation was a critical factor in determining the polymorphic outcome – the presence of agitation led to the preferential formation of α-glycine, while γ-glycine was able to nucleate under quiescent conditions.51 They also showed that the degree of supersaturation influenced the polymorphic outcome, with higher concentrations leading to the co-existence of both α-glycine and γ-glycine.
Diglycine was selected for a similar reason as it is the simplest dipeptide, consisting of two glycine residues. There have been comparatively fewer studies on the crystallisation of diglycine, but it has been shown to exhibit three polymorphs, α, β, and γ.52 However, the β and γ polymorphs are notoriously elusive, requiring many recrystallisations to form.52 As well as this, the α-polymorph is the thermodynamically stable form, meaning that diglycine does not suffer from the same polymorphic issues as glycine.53 As a result, the α-polymorph is the most extensively studied polymorph as it forms most readily. The α-polymorph forms plate-like crystals belonging to the space group P21/c.54
This work further explores the hypothesis that the heterosurface (glass beads) will enhance the nucleation rates of the amino acid (glycine) and dipeptide (diglycine) through heterogeneous nucleation. Previous work from the authors revealed that functional group complementarity between the heterosurface and the nucleating solute encouraged hydrogen bond formation between them, resulting in the heterogeneous nucleation of the solute. However, their work was only restricted to small organic drug molecules. The aim of this work is to advance the hydrogen bond complementarity and hydrogen bond lifetime hypotheses to small biomolecules, such as amino acids and dipeptides. The goal is to crystallise glycine and diglycine in the absence and presence of silica at different supersaturations to calculate heterogeneous nucleation parameters and to use the classical nucleation theory (CNT) to deduce the kinetic parameters of nucleation for each. The possible knowledge of hydrogen bonding behaviour between heterosurfaces and the crystallising solute, alongside appropriate crystallisation conditions, will result in the generation of crystals with the desired size, morphology, and polymorphic outcome.
2. Experimental section
2.1. Materials
Glycine (Gly, >99%, suitable for electrophoresis) and diglycine (Digly, >99% by titration) were supplied by Sigma-Aldrich and used without further purification. Deionized water was generated by a PURELAB Chorus 1 water purification system (ELGA LabWater (High Wycombe, UK)). Glass beads with an average diameter of 50 μm were supplied by Sigmund Linder GmbH (Warmensteinach, Germany). The molecular structures of the heterosurface and the two organic compounds are presented in Fig. 1.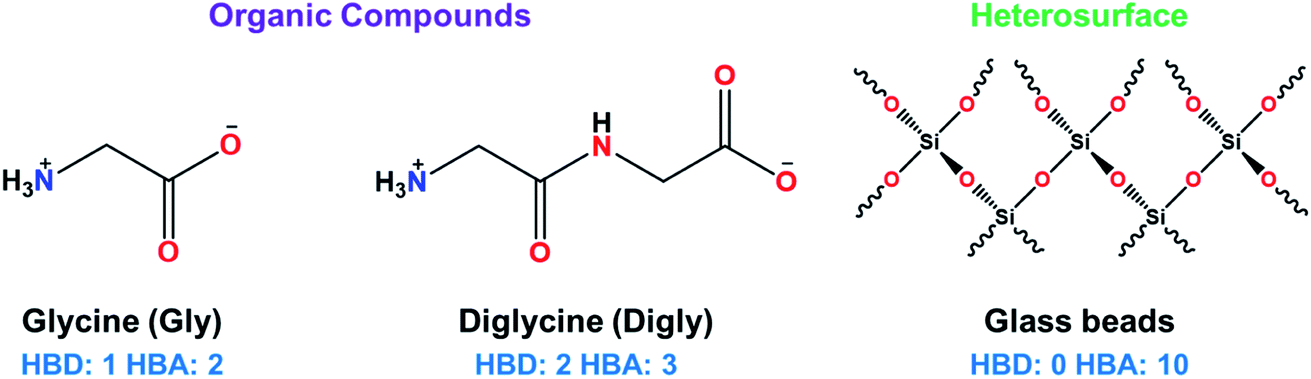 | ||
| Fig. 1 Chemical structures of the heterosurface (glass beads) and the two organic compounds (glycine and diglycine) used in this study with the number of hydrogen bond donor (HBD) and hydrogen bond acceptor (HBA) groups in each structure shown. | ||
2.2. Determination of the metastable zone width (MSZW) for the glycine and diglycine solutions in water in the absence of silica
The MSZW for both glycine and diglycine were determined using the following procedure. A known amount of glycine/diglycine was added to a lidded bottle containing 200 mL of deionised water. The solution was placed in a water bath controlled by a Grant GP200 circulator (Royston, UK) at 45 °C for an hour to allow the solids to dissolve. After this, the solutions were checked visually to ensure the solids had dissolved and the solution was filtered via a Sartorius 0.22 μm pore size PTFE syringe filter (Göttingen, Germany) and transferred to a 250 mL glass bottle submerged in a water bath connected to a JULABO F32-GB thermoregulator (Seelbach, Germany). The thermoregulator was programmed to follow a set heating profile. Firstly, the system was held at 30 °C (50 °C for diglycine) for one hour to allow the system to equilibrate and to ensure solid dissolution. The temperature was then decreased to 25 °C (45 °C for diglycine), and the system was held for a further 30 minutes to ensure thermal equilibrium. From here, a cooling ramp of 0.1 °C min−1 was imposed until the system reached 5 °C. At this point, the system was maintained at 5 °C for 1 hour. The onset of nucleation was determined via a Nikon D90 camera fitted with an AF-S 18–105 mm lens, which took regularly timed images of the solution, and the metastable zone widths were calculated from these images.2.3. Determination of the induction time of glycine and diglycine in the absence of glass beads at different supersaturations
All induction time experiments in the absence of glass bead particles were carried out via the methodology outlined below (see Fig. 2). A known amount of glycine/diglycine was added to a lidded bottle containing 100 mL of deionised water. The solution was placed in a water bath controlled by a Grant GP200 circulator (Royston, UK) at 45 °C for an hour to allow the solids to dissolve. After this, the solutions were checked visually to ensure that the solids had dissolved and 50 mL of solution was then filtered via a syringe filter and transferred to a lidded 100 mL bottle. The bottle was then placed back in the water bath at 45 °C for an hour to further ensure that no crystallisation or precipitation had occurred. The concentration of the filtrate was verified via a Thermo Scientific NanoDrop OneC UV-Vis spectrophotometer (Waltham, Massachusetts). The bottle was then placed in a second water bath controlled by a JULABO F32-GB thermoregulator, which was set at a lower temperature to generate supersaturation. The onset of nucleation was determined via a Nikon D90 camera fitted with an AF-S 18–105 mm lens, which took regularly timed images of the solution. The images were then analysed to determine the onset of nucleation. After nucleation had occurred, the solution was removed from the bath and the entire procedure was repeated at least three times to obtain an average value for the induction time, alongside an indication of the standard deviation. This was then repeated for two more different crystallisation temperatures, allowing for induction time measurements for three different supersaturations for both glycine and diglycine. The conditions for each experiment are given in Table 1.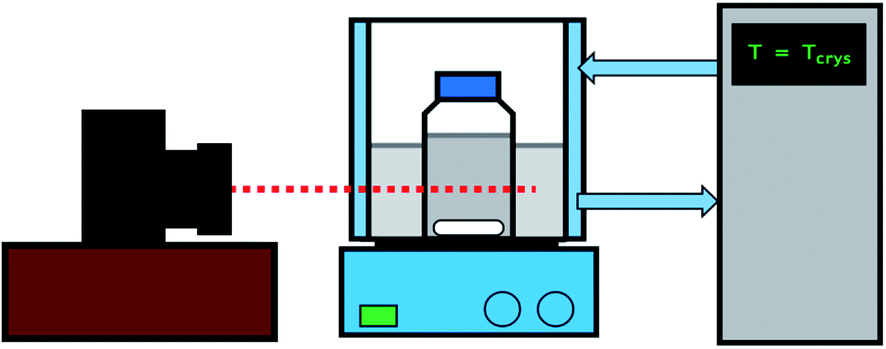 | ||
| Fig. 2 Schematic representation of the experimental set-up. | ||
| Compound | Supersaturation (S) | Saturation temperature, Tsat/°C (solubility (mg mL−1)) | Supersaturation temperature, Tcry/°C (solubility (mg mL−1)) | Solubility data | MSZW (°C) |
|---|---|---|---|---|---|
| Glycine (Gly) | 1.17 | 26 (234.35) | 17.5 (201.10) | 56 | 9.0 (ref. 55) |
| 1.22 | 15.0 (192.25) | ||||
| 1.33 | 10.0 (175.70) | ||||
| Diglycine (Digly) | 1.30 | 43 (298.24) | 30.0 (228.76) | 56 | 17.5 (this work) |
| 1.40 | 26.5 (213.00) | ||||
| 1.50 | 23.0 (198.32) |
2.4. Crystallisation of glycine and diglycine in the presence of glass beads at different supersaturations
The procedure of determining the induction time for the presence of glass beads is similar to that outlined in Section 2.3 (see Fig. 2). However, there are two key differences. Firstly, once the solutions were filtered, a known mass of glass beads corresponding to 50% of the maximum theoretical yield of glycine and diglycine (calculated as the difference in concentration between the initial supersaturation and the solubility) was added to the bottles containing filtered solution. Secondly, it was not possible to monitor crystallisation via visual observation methodologies, as the presence of glass beads rendered the solutions cloudy from the offset. Instead, 150 μL of the solution was collected via a syringe into an Eppendorf tube, and then filtered using 200 nm PTFE membrane syringe filters. From the supernatant, 100 μL was then diluted in 500 μL of deionised water in a second Eppendorf tube, to prevent any further crystallisation from occurring and to bring the concentration to within the limits of calibration. The concentration of the diluted sample was then characterised via a Thermo Scientific NanoDrop OneC UV-Vis spectrophotometer (Waltham, Massachusetts) by collecting absorbance data at 220 nm (glycine) and 230 nm (diglycine), and the concentration of the original sample was back-calculated via a linear correlation between concentration and absorbance. This correlation was calculated by dissolving known quantities of glycine and diglycine at concentrations well below the solubility limit and measuring their absorbance at 220 nm (glycine) and 230 nm (diglycine) to form a linear calibration curve. From this, it was possible to plot desupersaturation profiles for each experiment, from which the induction time could be estimated. These experiments were again repeated a minimum of three times for each of the conditions described in Table 1.2.5. Solid state characterisation of the glycine/diglycine–glass bead composites
2.6. Hydrogen bond propensity calculations
To find the most probable HBAs and HBDs in the glycine and diglycine, hydrogen bond propensities were calculated57 using the program Mercury 3.10.1 as follows. The training dataset for the statistical models was composed of molecules extracted from the Cambridge Structural Database (CSD) that contain all the functional groups present in the target APIs. A logistic regression was then applied to the training dataset that allowed the predictions in the form of H-bond propensities upon consideration of the environmental variables for the functional groups (e.g., aromaticity, steric density) of the target API. The H-bond propensity for a donor–acceptor pair can take a value between 0 and 1, where 0 indicates no likelihood of H-bond formation and 1 indicates that a hydrogen bond will always be found.3. Results and discussion
3.1. Determination of the MSZW
Table 1 summarizes the key process parameters used during the various crystallizations of glycine and diglycine from their respective saturated aqueous solutions in the presence of the heterosurface, glass beads. The MSZW determined during this study for Digly is included in Table 1, along with the previously reported MSZW for Gly.55 Bonnin-Paris et al.55 showed that the MSZW of glycine aqueous solution is approximately 9 °C at a cooling rate of 0.1 °C min−1. Recently, Ramakers et al.58 studied the influence of the addition of antisolvents such as ethanol, methanol, and dimethyl formamide to the aqueous glycine solution on the MSZW of glycine. The MSZW of glycine is narrower in aqueous solution but wider when the initial glycine solution has a greater antisolvent mass fraction. The MSZW of Digly (Tsat = 43 °C) was determined to be 17.5 °C at a cooling rate of 0.1 °C min−1. Table 1 further indicates that most of the crystallization temperatures used to generate the desired supersaturations reside inside or within 0.5 °C of the MSZW based on the data presented, thereby favouring heterogeneous nucleation in the presence of glass beads at the applied supersaturations.3.2. Induction time of glycine and diglycine in the absence and presence of glass beads
Table 2 summarises the induction time of glycine and diglycine in both the presence and absence of glass beads. The extent of the change in induction in the absence and the presence of the heterosurface between both compounds varied considerably. As such, the glass beads display a positive yet discriminating influence over the nucleation of Gly and Digly. Table 2 also summarises the nucleation rate for both Gly and Digly in the absence and presence of glass beads (J* and J, respectively), calculated according to eqn (1), previously reported by Mealey et al.,59 along with the corresponding nucleation rate ratios (J/J*).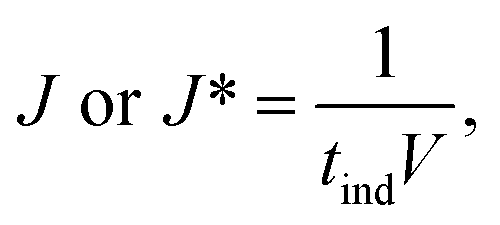 | (1) |
| Compounds | Supersaturation (S) |
RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnS (J mol−1) lnS (J mol−1) |
Homogeneous | Heterogeneous | Nucleation rate ratio (J/J*) | ||
|---|---|---|---|---|---|---|---|
| T ind (min) ± SD (min) | Nucleation rate (J*, nuclei per m3 per s) | T ind (min) ± SD (min) | Nucleation rate (J, nuclei per m3 per s) | ||||
| Glycine (Gly) | 1.17 | 379 | 211 ± 34 | 1.58 | 120 ± 30 | 2.78 | 1.76 |
| 1.22 | 346 | 50 ± 11 | 6.67 | 45 ± 15 | 7.41 | 1.11 | |
| 1.33 | 671 | 17 ± 2 | 19.61 | 10 ± 5 | 33.33 | 1.70 | |
| Diglycine (Digly) | 1.30 | 661 | 188 ± 44 | 1.77 | 60 ± 15 | 5.56 | 3.13 |
| 1.40 | 838 | 80 ± 26 | 4.17 | 30 ± 15 | 11.11 | 2.67 | |
| 1.50 | 998 | 44 ± 9 | 7.58 | 15 ± 5 | 22.22 | 2.93 | |
When comparing the nucleation behaviour of Gly and Digly, the nucleation rate ratio clearly indicates that glass beads have a pronounced effect on the nucleation of Digly compare to Gly. One of the possible reasons for this change in the nucleation rate in presence of glass beads is the abundant presence of hydrogen bond accepting oxygen atoms on the silibead molecules, resulting in hydrogen bond formation between the heterosurface and the crystallising solutes. Both Gly and Digly are rich in hydrogen bond donors (HBDs), which present complementarity to the HBA of glass beads, thereby favouring the formation of hydrogen bonds. These hydrogen bonds facilitate the formation of clusters of Gly and Digly on the surface of the glass beads, resulting in the faster nucleation. Similar results were observed by Verma et al.40 for the nucleation of various APIs in the absence and presence of microcrystalline cellulose (MCC). It was observed that the APIs with only HBAs could easily form hydrogen bonds with the HBD sites of MCC, which resulted in the stable cluster of API molecules on the surface of MCC, therefore enhancing the nucleation rate of the APIs possessing only HBAs. Fig. 3 presents the desupersaturation profile of Gly and Digly in the presence of glass beads. Irrespective of the supersaturation, the rates of the desupersaturation of Gly and Digly aqueous solutions in the presence of the heterosurface are similar, as observed by the slope of the desupersaturation profiles. The standard deviations are slightly bigger due to the stochastic nature of the solution, as the volume is only 50 mL.
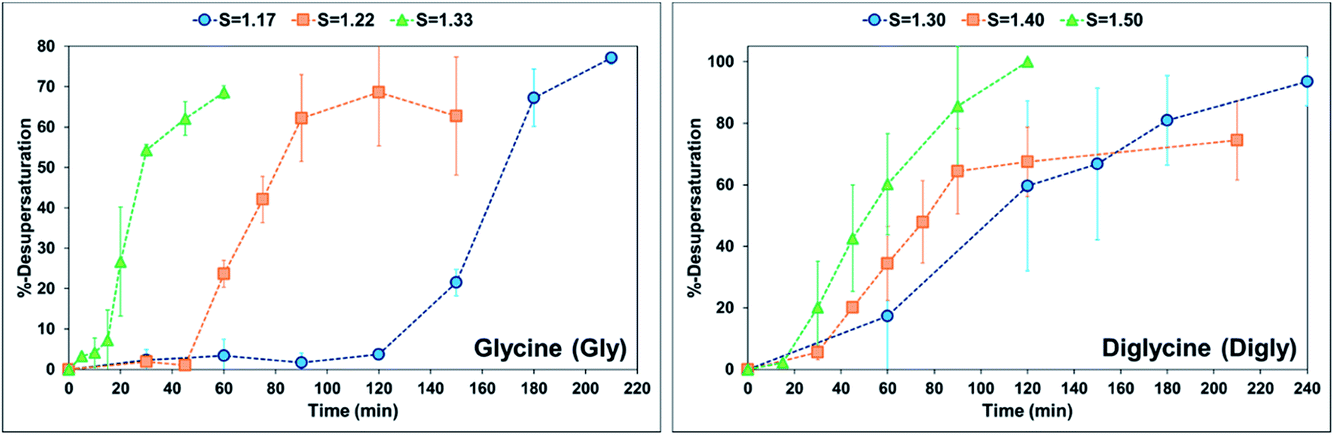 | ||
| Fig. 3 %-Desupersaturation of glycine (left) and diglycine (right) in the presence of glass beads at the supersaturations indicated; volume = 50 mL. | ||
Despite similar molecular functionalities on Gly and Digly – amide and carboxylic groups – the extent of nucleation is different in each case. For example, the nucleation rate of Gly increased by more than 1.5 times at the lowest S of 1.17 in the presence of glass beads, whereas the corresponding increase in the case of Digly at its lowest S of 1.30 is more than 3-fold. This change in the nucleation rate of crystallising solute in the presence of the heterosurface, as shown in Table 2, is a clear indication that heterogeneous nucleation can be influenced by the interplay of supersaturation and the presence or absence of a heterosurface.
3.3. Solid state characterisation of isolated solids
Fig. 4 presents the powder X-ray diffraction (PXRD) patterns of the isolated solids after the full desupersaturation of Gly and Digly aqueous solutions in the presence of glass beads at the desired supersaturations. The PXRD patterns confirm the presence of the metastable α-form for Gly and the stable α-form for Digly in the presence of glass beads.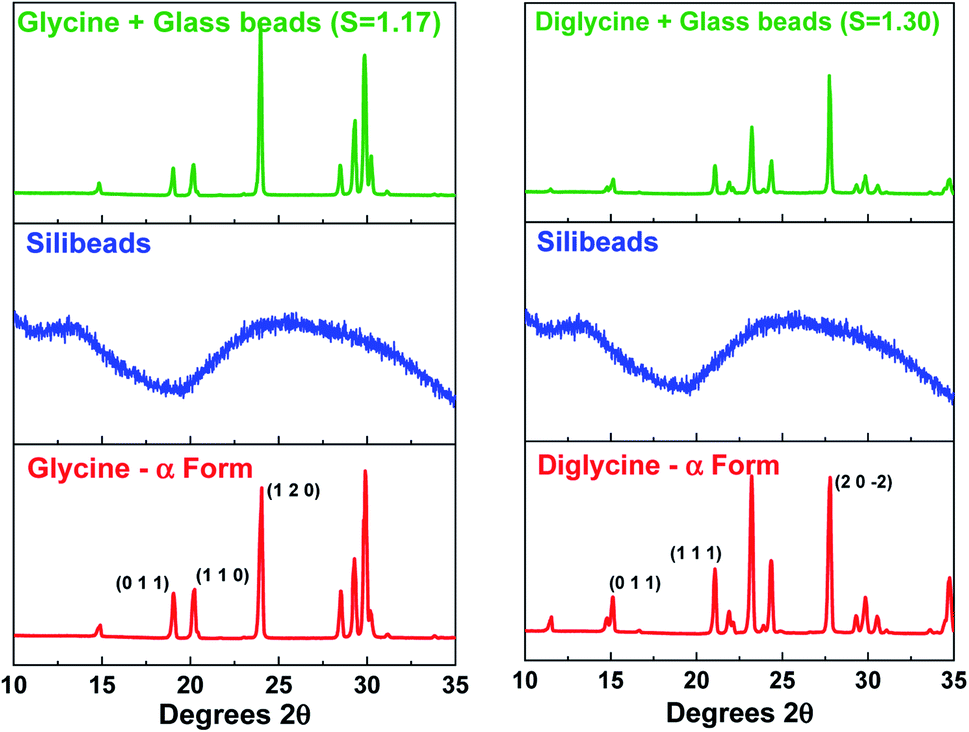 | ||
| Fig. 4 Powder X-ray diffraction patterns of the composite solids isolated after the complete desupersaturation of both glycine (left) and diglycine (right) in the presence of glass beads at the indicated supersaturations, along with the glass beads and glycine and diglycine patterns. | ||
The solids isolated after the homogeneous and heterogeneous nucleation of Gly and Digly were observed under an inverted light microscope. Crystals (100 μm) of the metastable α-form of Gly were observed in the presence and absence of glass beads, while crystals (100 μm) of the stable α-form of Digly were crystallised in the absence and presence of the heterosurface, as seen in Fig. 5. Most of the Gly and Digly crystals seem to be associated with the silibead micro particles, supporting the arguments made in Section 3.2. Similar finding of drug crystals attached to the heterosurface have previously been reported extensively in the literature.14,21,22,42
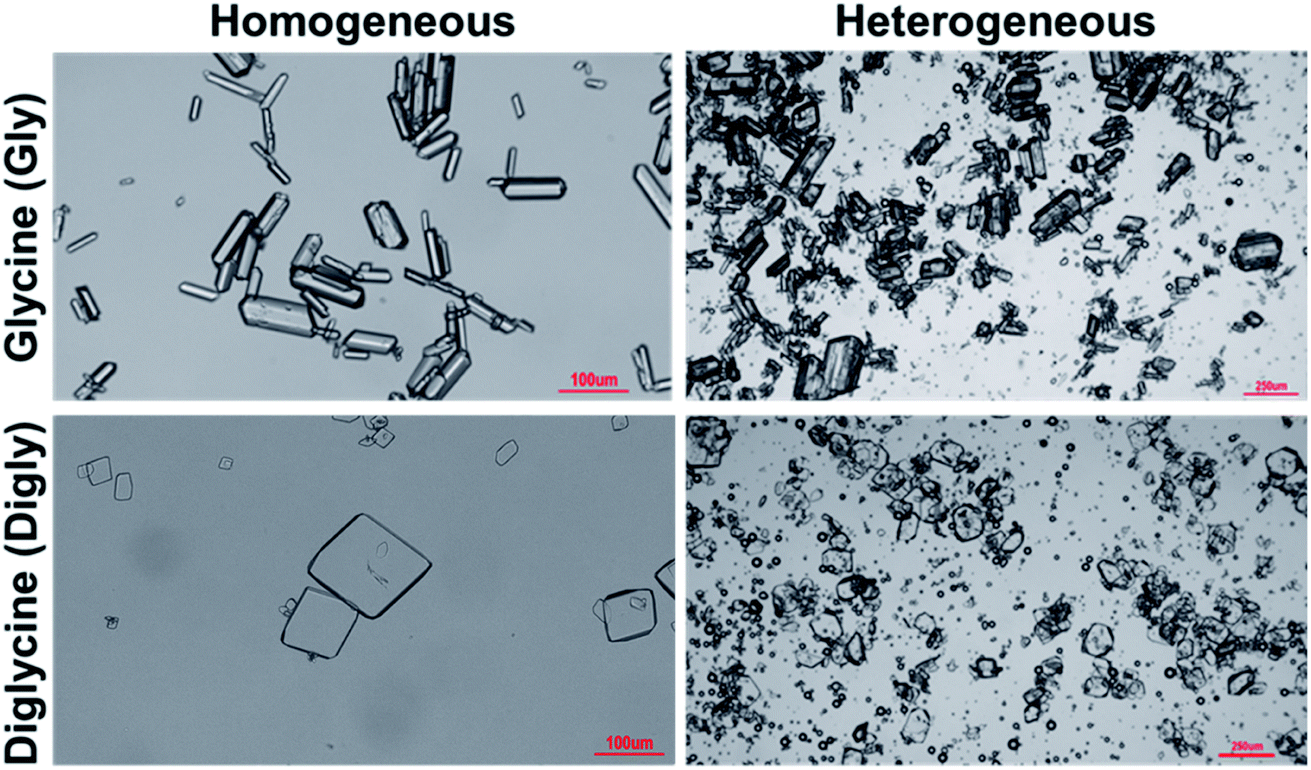 | ||
| Fig. 5 Microscope images of the solids isolated after the homogeneous and heterogeneous nucleation of glycine and diglycine in the absence and presence of glass beads. | ||
3.4. Effect of glass beads on the kinetic parameters of glycine and diglycine
The energy barrier associated with this desupersaturation is called the free energy barrier to nucleation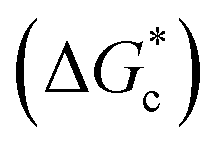 , which is mainly dependent on the temperature, supersaturation, and the interfacial energy at the crystal–solution interface. An equation for
, which is mainly dependent on the temperature, supersaturation, and the interfacial energy at the crystal–solution interface. An equation for 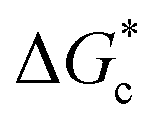 , which is based upon the classical nucleation theory, is shown in eqn (2).
, which is based upon the classical nucleation theory, is shown in eqn (2).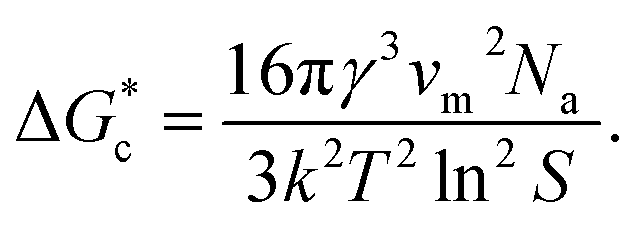 | (2) |
According to eqn (2), 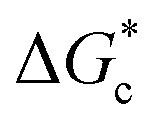 is inversely proportional to the supersaturation (S), therefore at low S, the
is inversely proportional to the supersaturation (S), therefore at low S, the 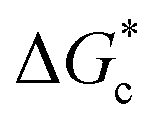 is relatively large, and thus the possibility of the conversion of the solution to crystals is a rare event. A sufficiently large S is required to cross this energy barrier for nucleation to occur. The formation of nuclei has been postulated to occur either through a single step process of exchanging monomer units with other growing clusters in a structured way, or via a two-step process, whereby an unstructured aggregation of solute molecules is followed by the defined arrangement to form a crystal.60
is relatively large, and thus the possibility of the conversion of the solution to crystals is a rare event. A sufficiently large S is required to cross this energy barrier for nucleation to occur. The formation of nuclei has been postulated to occur either through a single step process of exchanging monomer units with other growing clusters in a structured way, or via a two-step process, whereby an unstructured aggregation of solute molecules is followed by the defined arrangement to form a crystal.60
The knowledge of supersaturation and the induction time of Gly and Digly in the absence and presence of a heterosurface were used to calculate the interfacial energy (γ* and γ) and the pre-exponential factor (A* and A) in the absence and presence of glass beads, respectively, for each compound. The kinetic and the thermodynamic parameters were derived using eqn (3) published by Kashchiev et al.61 and used by the authors in the past.14,42
| J(S) = ASexp(−B/ln2S) | (3) |
| B = 16πv02γef3/3(kT)3, | (4) |
Eqn (3) can be re-written as follows:
| ln(J/S) = lnA − (B/ln2S) | (5) |
Best fit linear equations, as presented in Fig. 6(A) and (B), were used to calculate γ* and γ, and A* and A for Gly and Digly crystallization systems in the absence and presence of glass beads, respectively, plotted using eqn (5). According to previously published literature, an interface “promotes nucleation by lowering the interfacial energy of aggregates”,32,63 but interestingly the data presented in Fig. 6(C) suggests that the presence of a heterosurface does not appreciably influence the interfacial energy of either the Gly or Digly crystallisation systems. Surprisingly, the major contributing factor to the heterogeneous nucleation of Gly and Digly is the pre-exponential factor (A*). As reported earlier, we interpret A as the number of clusters of a size less than the critical radius which form per cubic metre of supersaturated solution each second.14Fig. 6(C) suggests that the presence of glass beads coincides with an increase in the pre-exponential (A*) factor of ca. 2-fold for the crystallization of Gly and Digly. These results are in support of the previous findings from the group, where the presence of MCC in the crystallisation system resulted in a minimal change to the interfacial energy of the crystallising solute but instead resulted in an increase of the pre-exponential factor by 2-fold or more.14,42 Similar results were obtained by Heffernan et al.,64 where the presence of structurally similar impurities (demethoxycurcumin (DMC) and bisdemethoxycurcumin (BDMC)) in the curcumin solution did not influence the interfacial energy of the crystallising solute, but instead changed the pre-exponential factor.64 Another report by Bodnár et al.65 also presents similar findings, where the presence of docusate sodium (DOSS) in a mefenamic acid solution in dimethylacetamide (DMA)–water mixtures resulted in an approximately 50% increase in the pre-exponential factor, while the interfacial energy remained uninfluenced. Hence, the nucleation of organics in the presence of heterosurfaces in either a dissolved or undissolved state is influenced by the change in the pre-exponential factor, while the interfacial energy remains uninfluential.
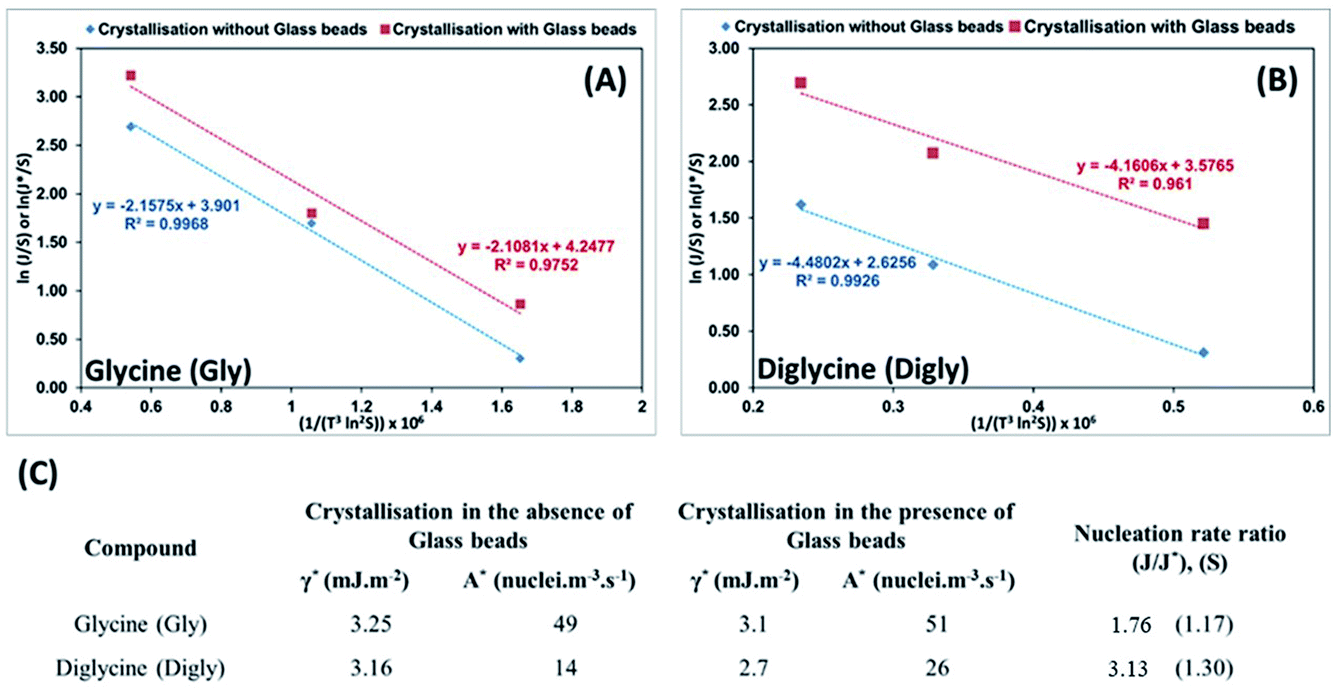 | ||
| Fig. 6 (A and B) Plots of ln(J/S) or ln(J*/S) against (1/(T3ln2S)) × 106, which illustrate the dependence of the nucleation rate on supersaturation for the crystallization of glycine (A) and diglycine (B) in the absence (blue diamond) and presence (red square) of glass beads at different supersaturations; the lines shown are the best linear fits and include the respective line equations and R2 values. (C) Interfacial energies and pre-exponential factors for the crystallization of glycine and diglycine from aqueous solutions in the absence and presence of glass beads, and the related nucleation rate ratios at the lowest supersaturations. | ||
3.5. Analysis of the intermolecular interactions
The presence of hydrogen bond donor (HBD) or acceptor (HBA) sites on the organic molecules allow them to form inter- or intra-molecular hydrogen bonds. This hydrogen bonding network has been proven to influence the crystallisation kinetics,14,22 as well as providing thermodynamic stability to the growing crystals.66 The crystallising molecules in this study, glycine and diglycine, possess 1 and 2 HBDs, respectively, and 2 and 3 HBAs, respectively, as shown in Fig. 1 and Table 3. To quantify the contribution of HBAs and HBDs to the formation of the crystal structures of Gly and Digly, the logit hydrogen-bonding propensity (LHP) model was used, based on the statistical analysis of hydrogen bonds in the Cambridge Structural Database (CSD).57 The hydrogen bond propensity (π) can be defined as a probability measure of an intermolecular hydrogen bond forming between two molecules within a crystal structure, with the assumption that the strongest possible donor–acceptor pairs will form this bond.57 The values of π for Gly (0.98) and Digly (0.87) are reported in Table 3, suggesting that Gly has a slightly higher affinity to form hydrogen bonds with itself compared to Digly. This result is consistent with other work on glycine polymorphism in aqueous solutions, which has shown that α-glycine is constructed from zwitterionic dimers,47 and that the presence of glycine dimers in solution (as a result of hydrogen bonding) is conducive to the preferential formation of α-glycine over γ-glycine.67 Comparing the values of π with the nucleation rate ratio (J/J*), suggests that the solute with a larger value of π resulted in a lower nucleation rate ratio, and vice versa. Both Gly and Digly can form hydrogen bonds with the glass beads due to hydrogen bonding complementarity between the crystallising solute and the heterosurface. Considering the cumulative number of HBDs and HBAs on Gly and Digly, and the value of π, suggests that the amide (–NH2) and carboxylic groups (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of Digly have a higher propensity to form hydrogen bonds with the oxygen molecules (–O–) of the glass beads compared to Gly. This, therefore, results in the comparatively faster nucleation rate of Digly to that of Gly in the presence of glass beads.
O) of Digly have a higher propensity to form hydrogen bonds with the oxygen molecules (–O–) of the glass beads compared to Gly. This, therefore, results in the comparatively faster nucleation rate of Digly to that of Gly in the presence of glass beads.
| Compound | Number of HBDs | Number of HBAs | Hydrogen bond propensity, π | Time required to add one molecule (ps) | Nucleation rate ratio (J/J*), (S) |
|---|---|---|---|---|---|
| Glycine (Gly) | 1 | 2 | 0.98 | 0.66 | 1.76 (1.17) |
| Diglycine (Digly) | 2 | 3 | 0.87 | 2.01 | 3.13 (1.30) |
Heterogeneous nucleation is governed heavily by the lifetimes of the hydrogen bond interactions, as reported in past literature.14,22,40,42 Previous literature has reported that the average lifetime of single molecules attached to a surface via hydrogen bonding ranges from <10 ns to <70 ns. A previous paper from the authors suggests that the lifetime of the hydrogen bond between a risperidone molecule and the surface of microcrystalline cellulose is about 30 ns, as computed by molecular dynamics (MD) simulations.40Table 3 also presents the time required (tm) to add a single molecule to growing crystals of either Gly or Digly. These values were calculated from the growth phases of Fig. 3 for each of the APIs using eqn (6).14
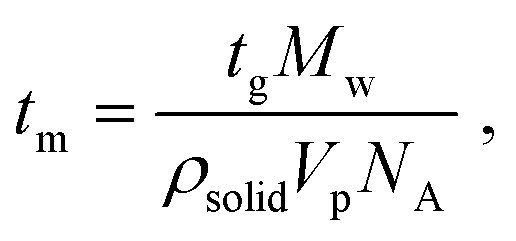 | (6) |
The times required to add a single molecule to a growing crystal of Gly and Digly are 0.66 ps and 2.01 ps, respectively, which are much smaller than the average lifetime of the hydrogen bond (30 ns) between a molecule of the crystallising solute and the heterosurface. This also confirms that Gly is a fast-nucleating molecule compared to Digly. Alternatively, the adsorbed solute molecule can exist attached to a surface for a time scale which will allow the attachment of multiple solute molecules from the solution phase and facilitate the formation of stable nuclei and eventually fully grown crystals. However, in the solution phase, solute–solute interactions are much shorter-lived, increasing the difficulty of nucleus formation, as reported previously.22 Therefore, the modest enhancement in the nucleation rate of Gly could be explained using the “lifetime effect”, where once a Gly molecule adheres to the silibead surface via hydrogen bond formation, it remains adhered for a long enough period for other Gly molecules or clusters to attach to this silibead-bound Gly molecule. This would result in stable nuclei which subsequently grow into a well-formed crystal. Therefore, the heterogeneous nucleation observed for Gly in the presence of glass beads is postulated to be due to the “lifetime effect”, whereas for Digly it is the combination of the “lifetime effect” and molecular complementarity.
4. Conclusions
The homogeneous and heterogeneous nucleation of glycine and diglycine in the absence and presence of glass beads were performed from aqueous solutions. It was observed that there was a modest increase in the nucleation rate of Gly compared to Digly. This confirms that glass beads displayed a positive yet discriminating effect on the heterogeneous nucleation of Gly and Digly. PXRD and microscope images confirmed the presence of the α-forms of both Gly and Digly after both the homogeneous and heterogeneous crystallisation processes. Experimental data suggests that the presence of glass beads did not dramatically influence the interfacial energies of Gly and Digly, but instead increased the pre-exponential factor by a factor of at least 2. Furthermore, molecular functionality analysis, hydrogen bond propensity calculations, and the time required to add a single molecule to the growing crystal of Gly and Digly all suggest that the heterogeneous nucleation of Gly is the consequence of the hydrogen bond “lifetime effect”, whereas for Digly it is a combination of this effect as well as molecular complementarity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The results incorporated in this paper have received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement no. 101026339. KH thanks the Science Foundation Ireland (SFI) for support (award grant numbers 12/RC/2275_P2). JH and MG thank the EPSRC for funding SCoBiC (EP/N015916/1). HM is grateful for the financial support from the Engineering and Physical Science Research Council (EP/T518207/1 and EP/T005556/1).References
- Q. Li and Y.-S. Jun, The apparent activation energy and pre-exponential kinetic factor for heterogeneous calcium carbonate nucleation on quartz, Commun. Chem., 2018, 1(1), 56 CrossRef.
- S. Karthika, T. K. Radhakrishnan and P. Kalaichelvi, A Review of Classical and Nonclassical Nucleation Theories, Cryst. Growth Des., 2016, 16(11), 6663 CrossRef CAS.
- C. Lindberg, A. Whiticar, K. A. Dick, N. Sköld, J. Nygård and J. Bolinsson, Silver as Seed-Particle Material for GaAs Nanowires—Dictating Crystal Phase and Growth Direction by Substrate Orientation, Nano Lett., 2016, 16(4), 2181 CrossRef CAS PubMed.
- Y. Xia, X. Xia and H.-C. Peng, Shape-Controlled Synthesis of Colloidal Metal Nanocrystals: Thermodynamic versus Kinetic Products, J. Am. Chem. Soc., 2015, 137(25), 7947 CrossRef CAS PubMed.
- J. Zeng, J. Huang, C. Liu, C. H. Wu, Y. Lin, X. Wang, S. Zhang, J. Hou and Y. Xia, Gold-Based Hybrid Nanocrystals Through Heterogeneous Nucleation and Growth, Adv. Mater., 2010, 22(17), 1936 CrossRef CAS PubMed.
- P. J. M. Smeets, K. R. Cho, R. G. E. Kempen, N. A. J. M. Sommerdijk and J. J. De Yoreo, Calcium carbonate nucleation driven by ion binding in a biomimetic matrix revealed by in situ electron microscopy, Nat. Mater., 2015, 14(4), 394 CrossRef CAS PubMed.
- A. Pei, G. Zheng, F. Shi, Y. Li and Y. Cui, Nanoscale Nucleation and Growth of Electrodeposited Lithium Metal, Nano Lett., 2017, 17(2), 1132 CrossRef CAS PubMed.
- S. Lau and L. A. Archer, Nucleation and Growth of Lithium Peroxide in the Li–O2 Battery, Nano Lett., 2015, 15(9), 5995 CrossRef CAS PubMed.
- Y. Hu, B. Lee, C. Bell and Y.-S. Jun, Environmentally Abundant Anions Influence the Nucleation, Growth, Ostwald Ripening, and Aggregation of Hydrous Fe(III) Oxides, Langmuir, 2012, 28(20), 7737 CrossRef CAS PubMed.
- Q. Li, C. I. Steefel and Y.-S. Jun, Incorporating Nanoscale Effects into a Continuum-Scale Reactive Transport Model for CO2-Deteriorated Cement, Environ. Sci. Technol., 2017, 51(18), 10861 CrossRef CAS PubMed.
- Y.-S. Jun, L. Zhang, Y. Min and Q. Li, Nanoscale Chemical Processes Affecting Storage Capacities and Seals during Geologic CO2 Sequestration, Acc. Chem. Res., 2017, 50(7), 1521 CrossRef CAS PubMed.
- J. J. A. Thomas, New Approach to Modeling the Nucleation and Growth Kinetics of Tricalcium Silicate Hydration, J. Am. Ceram. Soc., 2007, 90(10), 3282 CrossRef CAS.
- J. V. Parambil, S. K. Poornachary, R. B. H. Tan and J. Y. Y. Heng, Template-induced polymorphic selectivity: the effects of surface chemistry and solute concentration on carbamazepine crystallisation, CrystEngComm, 2014, 16(23), 4927 RSC.
- V. Verma, J. Zeglinski, S. Hudson, P. Davern and B. K. Hodnett, Dependence of Heterogeneous Nucleation on Hydrogen Bonding Lifetime and Complementarity, Cryst. Growth Des., 2018, 18(11), 7158 CrossRef CAS.
- P. W. Carter, A. C. Hillier and M. D. Ward, Nanoscale Surface Topography and Growth of Molecular Crystals: The Role of Anisotropic Intermolecular Bonding, J. Am. Chem. Soc., 1994, 116(3), 944 CrossRef CAS.
- P. W. Carter and M. D. Ward, Topographically directed nucleation of organic crystals on molecular single-crystal substrates, J. Am. Chem. Soc., 1993, 115(24), 11521 CrossRef CAS.
- P. W. Carter and M. D. Ward, Directing Polymorph Selectivity During Nucleation of Anthranilic Acid on Molecular Substrates, J. Am. Chem. Soc., 1994, 116(2), 769 CrossRef CAS.
- I. Weissbuch, L. Addadi, M. Lahav and L. Leiserowitz, Molecular Recognition at Crystal Interfaces, Science, 1991, 253(5020), 637 CrossRef CAS PubMed.
- K. Chadwick, J. Chen, A. S. Myerson and B. L. Trout, Toward the Rational Design of Crystalline Surfaces for Heteroepitaxy: Role of Molecular Functionality, Cryst. Growth Des., 2012, 12(3), 1159 CrossRef CAS.
- V. López-Mejías, A. S. Myerson and B. L. Trout, Geometric Design of Heterogeneous Nucleation Sites on Biocompatible Surfaces, Cryst. Growth Des., 2013, 13(8), 3835 CrossRef.
- V. Verma, R. V. G. Peddapatla, C. M. Crowley, A. M. Crean, P. Davern, S. Hudson and B. K. Hodnett, Experimental Study on the Influence of Excipients on the Heterogeneous Crystallization and Dissolution Properties of an Active Pharmaceutical Ingredient, Cryst. Growth Des., 2018, 18(1), 338 CrossRef CAS.
- R. Arribas Bueno, C. M. Crowley, P. Davern, B. K. Hodnett and S. Hudson, Heterogeneous Crystallization of Fenofibrate onto Pharmaceutical Excipients, Cryst. Growth Des., 2018, 18(4), 2151 CrossRef CAS.
- R. Arribas Bueno, C. M. Crowley, B. K. Hodnett, S. Hudson and P. Davern, Influence of Process Parameters on the Heterogeneous Nucleation of Active Pharmaceutical Ingredients onto Excipients, Org. Process Res. Dev., 2017, 21(4), 559 CrossRef CAS.
- J. V. Parambil, S. K. Poornachary, S. J. Hinder, R. B. H. Tan and J. Y. Y. Heng, Establishing template-induced polymorphic domains for API crystallisation: the case of carbamazepine, CrystEngComm, 2015, 17(33), 6384 RSC.
- H. Yang, C. L. Song, Y. X. S. Lim, W. Chen and J. Y. Y. Heng, Selective crystallisation of carbamazepine polymorphs on surfaces with differing properties, CrystEngComm, 2017, 19(44), 6573 RSC.
- F. J. Link and J. Y. Y. Heng, Enhancing the crystallisation of insulin using amino acids as soft-templates to control nucleation, CrystEngComm, 2021, 23(22), 3951 RSC.
- J. Aizenberg, A. J. Black and G. M. Whitesides, Control of crystal nucleation by patterned self-assembled monolayers, Nature, 1999, 398(6727), 495 CrossRef CAS.
- H. B. Eral, V. López-Mejías, M. O’Mahony, B. L. Trout, A. S. Myerson and P. S. Doyle, Biocompatible Alginate Microgel Particles as Heteronucleants and Encapsulating Vehicles for Hydrophilic and Hydrophobic Drugs, Cryst. Growth Des., 2014, 14(4), 2073 CrossRef CAS.
- V. López-Mejías, J. W. Kampf and A. J. Matzger, Polymer-Induced Heteronucleation of Tolfenamic Acid: Structural Investigation of a Pentamorph, J. Am. Chem. Soc., 2009, 131(13), 4554 CrossRef PubMed.
- L. Y. Pfund, C. P. Price, J. J. Frick and A. J. Matzger, Controlling Pharmaceutical Crystallization with Designed Polymeric Heteronuclei, J. Am. Chem. Soc., 2015, 137(2), 871 CrossRef CAS PubMed.
- J. Ling and K. Chadwick, Heterogeneous Crystallization Inside Microporous Polymer Particles as a Process Intensification Technology for the Manufacture of Drug Formulations, Org. Process Res. Dev., 2017, 21(6), 827 CrossRef CAS.
- Y.-X. Liu, X.-J. Wang, J. Lu and C.-B. Ching, Influence of the Roughness, Topography, and Physicochemical Properties of Chemically Modified Surfaces on the Heterogeneous Nucleation of Protein Crystals, J. Phys. Chem. B, 2007, 111(50), 13971 CrossRef CAS PubMed.
- J. V. Parambil, S. K. Poornachary, J. Y. Y. Heng and R. B. H. Tan, Template-induced nucleation for controlling crystal polymorphism: from molecular mechanisms to applications in pharmaceutical processing, CrystEngComm, 2019, 21(28), 4122 RSC.
- Y. Diao, A. S. Myerson, T. A. Hatton and B. L. Trout, Surface Design for Controlled Crystallization: The Role of Surface Chemistry and Nanoscale Pores in Heterogeneous Nucleation, Langmuir, 2011, 27(9), 5324 CrossRef CAS PubMed.
- Y. Diao, T. Harada, A. S. Myerson, T. Alan Hatton and B. L. Trout, The role of nanopore shape in surface-induced crystallization, Nat. Mater., 2011, 10(11), 867 CrossRef CAS PubMed.
- K. Chadwick, A. Myerson and B. Trout, Polymorphic control by heterogeneous nucleation - A new method for selecting crystalline substrates, CrystEngComm, 2011, 13(22), 6625 RSC.
- J. Ouyang, J. Chen, W. Chen, I. Rosbottom, M. Guo and J. Y. Y. Heng, Application of Phenyl-Functionalized Porous Silica for the Selective Crystallization of Carbamazepine Metastable Form II, Ind. Eng. Chem. Res., 2021, 60(2), 939 CrossRef CAS.
- J. Ouyang, X. Xing, J. Chen, L. Zhou, Z. Liu and J. Y. Y. Heng, Effects of solvent, supersaturation ratio and silica template on morphology and polymorph evolution of vanillin during swift cooling crystallization, Particuology, 2022, 65, 93 CrossRef CAS.
- X. Li and J. Y. Y. Heng, Protein crystallisation facilitated by silica particles to compensate for the adverse impact from protein impurities, CrystEngComm, 2021, 23(47), 8386 RSC.
- P.-A. Cazade, V. Verma, B. K. Hodnett and D. Thompson, Extended Lifetime of Molecules Adsorbed onto Excipients Drives Nucleation in Heterogeneous Crystallization, Cryst. Growth Des., 2021, 21(4), 2101 CrossRef CAS PubMed.
- R. J. Davey, S. L. M. Schroeder and J. H. terHorst, Nucleation of Organic Crystals—A Molecular Perspective, Angew. Chem., Int. Ed., 2013, 52(8), 2166 CrossRef CAS PubMed.
- V. Verma, P. Bannigan, M. Lusi, C. M. Crowley, S. Hudson, B. K. Hodnett and P. Davern, The heterogeneous crystallization of a novel solvate of clozapine base in the presence of excipients, CrystEngComm, 2018, 20(31), 4370 RSC.
- C. L. Bull, G. Flowitt-Hill, S. de Gironcoli, E. Kucukbenli, S. Parsons, C. H. Pham, H. Y. Playford and M. G. Tucker, [zeta]-Glycine: insight into the mechanism of a polymorphic phase transition, IUCrJ, 2017, 4(5), 569 CrossRef CAS PubMed.
- G. L. Perlovich, L. K. Hansen and A. Bauer-Brandl, The Polymorphism of Glycine. Thermochemical and structural aspects, J. Therm. Anal. Calorim., 2001, 66(3), 699 CrossRef CAS.
- X. Yang, J. Lu, X.-J. Wang and C.-B. Ching, Effect of sodium chloride on the nucleation and polymorphic transformation of glycine, J. Cryst. Growth, 2008, 310(3), 604 CrossRef CAS.
- K. Srinivasan, Crystal growth of α and γ glycine polymorphs and their polymorphic phase transformations, J. Cryst. Growth, 2008, 311(1), 156 CrossRef CAS.
- C. S. Towler, R. J. Davey, R. W. Lancaster and C. J. Price, Impact of Molecular Speciation on Crystal Nucleation in Polymorphic Systems: The Conundrum of γ Glycine and Molecular ‘Self Poisoning’, J. Am. Chem. Soc., 2004, 126(41), 13347 CrossRef CAS PubMed.
- S. K. Poornachary, J. V. Parambil, P. S. Chow, R. B. H. Tan and J. Y. Y. Heng, Nucleation of Elusive Crystal Polymorphs at the Solution–Substrate Contact Line, Cryst. Growth Des., 2013, 13(3), 1180 CrossRef CAS.
- G. Albrecht and R. B. Corey, The Crystal Structure of Glycine, J. Am. Chem. Soc., 1939, 61(5), 1087 CrossRef CAS.
- Y. Iitaka, The crystal structure of γ-glycine, Acta Crystallogr., 1961, 14(1), 1 CrossRef CAS.
- M. J. Vesga, D. McKechnie, P. A. Mulheran, K. Johnston and J. Sefcik, Conundrum of γ glycine nucleation revisited: to stir or not to stir?, CrystEngComm, 2019, 21(13), 2234 RSC.
- S. A. Moggach, D. R. Allan, S. Parsons and L. Sawyer, Effect of pressure on the crystal structure of [alpha]-glycylglycine to 4.7 GPa; application of Hirshfeld surfaces to analyse contacts on increasing pressure, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62(2), 310 CrossRef PubMed.
- S. M. Clarke, B. A. Steele, M. P. Kroonblawd, D. Zhang, I. F. W. Kuo and E. Stavrou, An Isosymmetric High-Pressure Phase Transition in α-Glycylglycine: A Combined Experimental and Theoretical Study, J. Phys. Chem. B, 2020, 124(1), 1 CrossRef CAS PubMed.
- E. W. Hughes and W. J. Moore, The Crystal Structure of β-Glycylglycine, J. Am. Chem. Soc., 1949, 71(8), 2618 CrossRef CAS.
- J. Bonnin-Paris, S. Bostyn, J.-L. Havet and H. Fauduet, Determination of the Metastable Zone Width of Glycine Aqueous Solutions for Batch Crystallisations, Chem. Eng. Commun., 2011, 198(8), 1004 CrossRef CAS.
- M. Guo, H. Z. Chang, E. Liang, H. Mitchell, L. Zhou, Q. Yin, J. E. Guinn and Y. Y. J. Heng, The effect of chain length and side chains on the solubility of peptides in water from 278.15K to 313.15K: A case study in glycine homopeptides and dipeptides, J. Mol. Liq., 2021, 352, 118681 CrossRef.
- P. T. A. Galek, L. Fabian, W. D. S. Motherwell, F. H. Allen and N. Feeder, Knowledge-based model of hydrogen-bonding propensity in organic crystals, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63(5), 768 CrossRef CAS PubMed.
- L. A. I. Ramakers, J. McGinty, W. Beckmann, G. Levilain, M. Lee, H. Wheatcroft, I. Houson and J. Sefcik, Investigation of Metastable Zones and Induction Times in Glycine Crystallization across Three Different Antisolvents, Cryst. Growth Des., 2020, 20(8), 4935 CrossRef CAS.
- D. Mealey, J. Zeglinski, D. Khamar and Å. C. Rasmuson, Influence of solvent on crystal nucleation of risperidone, Faraday Discuss., 2015, 179, 309 RSC.
- J. De Yoreo James, U. P. A. Gilbert Pupa, A. J. M. Sommerdijk Nico, R. L. Penn, S. Whitelam, D. Joester, H. Zhang, D. Rimer Jeffrey, A. Navrotsky and F. Banfield Jillian, et al., Crystallization by particle attachment in synthetic, biogenic, and geologic environments, Science, 2015, 349(6247), aaa6760 CrossRef CAS PubMed.
- D. Kashchiev and G. M. van Rosmalen, Review: Nucleation in solutions revisited, Cryst. Res. Technol., 2003, 38(7–8), 555 CrossRef CAS.
- http://www.chemspider.com/, 2021.
- V. López-Mejías, J. L. Knight, C. L. Brooks and A. J. Matzger, On the Mechanism of Crystalline Polymorph Selection by Polymer Heteronuclei, Langmuir, 2011, 27(12), 7575 CrossRef PubMed.
- C. Heffernan, M. Ukrainczyk, J. Zeglinski, B. K. Hodnett and Å. C. Rasmuson, Influence of Structurally Related Impurities on the Crystal Nucleation of Curcumin, Cryst. Growth Des., 2018, 18(8), 4715 CrossRef CAS.
- K. Bodnár, S. P. Hudson and Å. C. Rasmuson, Promotion of Mefenamic Acid Nucleation by a Surfactant Additive, Docusate Sodium, Cryst. Growth Des., 2019, 19(2), 591–603 CrossRef.
- G. R. Desiraju, Crystal Engineering: From Molecule to Crystal, J. Am. Chem. Soc., 2013, 135(27), 9952 CrossRef CAS PubMed.
- Y. Yani, P. S. Chow and R. B. H. Tan, Glycine Open Dimers in Solution: New Insights into α-Glycine Nucleation and Growth, Cryst. Growth Des., 2012, 12(10), 4771 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |