
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Destruction of per/poly-fluorinated alkyl substances by magnetite nanoparticle-catalyzed UV-Fenton reaction†
Danielle R.
Schlesinger
 ,
Collin
McDermott
,
Nam Q.
Le
,
Jesse S.
Ko
,
James K.
Johnson
,
Plamen A.
Demirev
and
Zhiyong
Xia
*
,
Collin
McDermott
,
Nam Q.
Le
,
Jesse S.
Ko
,
James K.
Johnson
,
Plamen A.
Demirev
and
Zhiyong
Xia
*
The Johns Hopkins University Applied Physics Laboratory, Laurel, MD 20723, USA. E-mail: zhiyong.xia@jhuapl.edu
First published on 21st September 2022
Abstract
Novel economically-sustainable and environmentally-friendly technologies for per- and poly-fluoroalkyl substance (PFAS) destruction are becoming increasingly important as PFAS contamination has increased in drinking water throughout the globe. UV-Fenton chemistry catalyzed by nanosize magnetite (Fe3O4) particles in aqueous environments is a promising method for production of reactive oxygen species (ROS), capable of disrupting PFAS carbon-fluorine bonds. Here we demonstrate greater than 90% degradation efficiency for a wide variety of PFAS compounds by ROS generated in a UV-Fenton reaction, involving naturally-occurring Fe3O4 nanoparticles. In order to demonstrate that ROS are indeed formed under our experimental conditions, we utilized a fluorescent probe to confirm the presence of ROS in the reaction solution. PFAS destruction efficiency is increased at elevated pH levels; however, by varying both the Fe3O4 and hydrogen peroxide (H2O2) concentrations in solution, high efficiency can be achieved at neutral pH conditions, like those found in drinking water. PFAS destruction occurred with UV exposure times on the order of minutes. Nano Fe3O4 retained its oxidation state and catalytic efficiency after multiple cycles of PFAS destruction, indicating that the material is reusable. High resolution mass spectrometry was utilized to demonstrate destruction efficiency and to identify the degradation products. ROS generation utilizing naturally-occurring Fe3O4 nanoparticles has been shown to be an efficient method for PFAS destruction with potential scalability for drinking water decontamination.
Water impactPFAS contamination in water is a critical challenge the world is facing as PFAS are extremely difficult to degrade with cost-effective, non-toxic, and efficient methods. UV-Fenton chemistry with reusable magnetite nanoparticles destroys recalcitrant PFAS in contaminated waters, and this work provides a first step in the development of new water treatment technologies to ensure clean and sustainable water resources. |
1. Introduction
Per- and poly-fluoroalkyl substances (PFAS) are a family of synthetic chemicals that are known to cause a variety of health-related issues, including endocrine disruption, high cholesterol levels, birth defects, and cancers.1,2 PFAS originate from a wide range of sources, including aqueous film-forming foams, non-stick cookware, and stain-resistant carpets.3 As such, PFAS have been recently identified as major emerging and persistent contaminants in drinking water and throughout environmental ecosystems.4,5 Once released to the environment, PFAS are extremely hard to degrade due to the high bond dissociation energy of the carbon–fluorine (C–F) bond (536 kJ mol−1), which increases with increasing fluorination on molecules;6,7 as a result, PFAS are also referred to as ‘forever chemicals’. Currently, the U.S. Environmental Protection Agency (EPA) advises that the combined concentration of two common PFAS compounds, perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS), to be less than 70 ng L−1 (parts per trillion [ppt]) in drinking water.8 However, at least 20 million people in the U.S. are regularly exposed, particularly through their tap water, to levels beyond the recommended limits, and for this reason, efficient, cost-effective, and potentially environmentally benign methods for PFAS removal and degradation must be developed.9Thus far, the dominant PFAS remediation strategy is PFAS capture.10–14 The major drawback of these technologies is the generation of PFAS contaminated media, which requires further treatment for decontamination or long term storage. These ancillary waste streams could further be released into the environment and cause secondary PFAS contamination. An ideal PFAS decontamination strategy is to develop and utilize techniques that completely destroy the PFAS compounds by breaking C–F bonds, completely ridding the current and any downstream systems of species. While some destruction technologies have been developed recently, these methods are often costly, require high energy input, are difficult to install, non-eco-friendly (e.g. generating other toxic byproducts or utilizing hazardous materials), inefficient (require a many step process or take a long time), and difficult to scale up for waste water treatment.15–21 In this study, we present results for a novel highly efficient, environmentally friendly method for the degradation of PFAS species.
This work leverages UV-Fenton chemistry to induce oxidative defluorination in PFAS. Conventional Fenton chemistry involves a redox reaction, either oxidation of ferrous iron (Fe2+) or reduction of ferric Fe3+ (often referred to as a Fenton-like reaction) by hydrogen peroxide (H2O2) in the presence of UV radiation in order to produce reactive oxygen species (ROS), such as hydroxyl (HO˙) and peroxyl (HOO˙) free radicals (eqn (1)–(3)).22
| Fe2+ + H2O2 → Fe3+ + HO˙ + OH− | (1) |
| Fe3+ + H2O2 → Fe2+ + HOO˙ + H+ | (2) |
| 2H2O2 → HO˙ + HOO˙ + H2O | (3) |
To address these challenges, here we propose nanosize magnetite (Fe3O4) as the catalyst for the UV-Fenton reaction, substituting for typically used aqueous Fe species. Magnetite nanoparticles provide a high surface area structure to maximize interaction of PFAS with the material while supporting efficient generation of ROS. Recently, Fe3O4 and other Fe bearing minerals have been identified as capable of inducing heterogeneous Fenton processes in a variety of sytems.28–30 Compared to other Fe bearing minerals, Fe3O4 is a naturally occurring ferrimagnetic mineral. Fe3O4 contains iron in its two common oxidation states, ferrous Fe(II) and ferric Fe(III), indicating that the material may be reusable in the long term, as it can undergo continuous redox reactions. Finally, Fe3O4 is an environmentally benign and cost-effective material that can be removed or filtered from water readily, following the remediation process. Our results indicate that utilization of Fe3O4 nanoparticles as the Fenton catalyst is a promising solution for PFAS destruction, achieving high destruction efficiency under typical drinking water conditions.
2. Materials and methods
2.1 Materials
PFOA (100 μg mL−1 in MeOH) and PFOS (100 μg mL−1 in MeOH) were purchased from AccuStandard (New Haven, CT, USA) and diluted to 1 mg L−1 (1 ppm) stock solutions in deionized (DI) water. PFAS challenge water containing 18 PFAS species was prepared by spiking DI water with the 18 PFAS compounds as listed in the EPA 537.1 method. Specifically, the 18 PFAS challenge contains 1500 ng L−1 of each of the following species: perfluorohexanoic acid (PFHxA), perfluoroheptanoic acid (PFHpA), perfluorooctanoic acid (PFOA), perfluorononanoic acid (PFNA), perfluorodecanoic acid (PFDA), perfluorotridecanoic acid (PFTrDA), perfluorotetradecanoic acid (PFTeDA), perfluorobutanesulfonic acid (PFBS), perfluorohexanesulfonic acid (PFHxS), perfluorooctanesulfonic acid (PFOS), N-ethylperfluorooctanesulfonamidoacetic acid (NEtFOSAA), N-methylperfluorooctanesulfonamido acetic acid (NMeFOSAA), perfluorododecanoic acid (PFDoDA), hexafluoropropylene oxide dimer acid (HFPODA), 9-chlorohexadecafluoro-3-oxanone-1-sulfonic acid (9Cl-PF3ONS), 11-chloroeicosafluoro-3-oxaundecane-1-sulfonic acid (11Cl-PF3OUdS), 4,8-dioxa-3H-perfluorononanoic acid (DONA), and perfluoroundecanoic acid (PFUnDA).Hydrogen peroxide (H2O2, 50 wt% in H2O, stabilized) was purchased from Sigma-Aldrich (Burlington, MA). Iron oxide (magnetite) Fe3O4 nanopowder/nanoparticles (>98%, 20–30 nm nominal particle size) was purchased from US Research Nanomaterials Inc. (Houston, TX, USA). A stock solution of 3000 ppm Fe3O4 was prepared by suspending the nanoparticles in DI water in a volumetric flask.
2.2 Fenton reaction experimental set-up
Batch reactions of UV-Fenton were conducted in 250 mL Pyrex beakers. Prior to each reaction, all beakers were washed with HNO3 to reduce any trace level contamination. Solutions of PFAS, Fe3O4, and H2O2 were added to DI water at varied volumes to achieve desired final concentrations. Each solution was mixed and pH was monitored with a Mettler Toledo Five Easy F20 pH mV−1 meter. Solution pH was adjusted to the desired pH utilizing solutions of 1 M HNO3 and 1 M NaOH. Samples were then placed into a Spectrolinker XL-1500 UV Crosslinker oven containing six 15 watts UV-C bulbs (wavelength 254 nm). The oven was turned on to optimal crosslink settings of 120 μJ cm−2, and solutions were exposed to UV-C radiation between 5 min to 1 h (most commonly 30 min). Following removal from the UV oven, solutions were readily reacting and left alone for up to 24 h. Solutions were then transferred to 250 mL, ultra-clean plastic bottles stored in a fridge at 20 °C until analysis.2.3 PFAS testing
Quantitative analysis of PFAS concentration was conducted by solid phase extraction and LC/MS/MS following EPA method 537.1 (2018).14,31 Briefly, C13-labeled analogs of the target compounds were added to each sample. Samples were passed through a solid phase extraction cartridge with a weak anion exchange sorbent. The final solvent solution was then injected into the LC/MS/MS system with electrospray ionization in negative ion mode for analysis. The PFAS percent destruction was determined by comparing the final concentration of PFAS detected in solution to the initial PFAS concentration of the prepared sample.2.4 ROS detection with fluorescence spectroscopy
Samples with lower concentrations of magnetite (0, 50, 250, and 500 ppb) and at 1 μM H2O2 concentration were prepared as previously described. After reaction in the UV-C oven, 25 μL of 20 mM 2′-7′-dichlorofluorescin diacetate (DCFH-DA), a widely-used probe for detecting ROS, was added.32,33 The solution was then analyzed for fluorescence intensity using a Horiba Scientific Fluoromax+ spectrometer. Each sample was placed into a 10 mL crystal cuvette and analyzed at an excitation wavelength (λex) of 495 nm (slit 5 nm) and emission wavelengths (λem) from 500–600 nm (slit 2 nm).2.5 High resolution mass spectrometry (HRMS) analysis
Qualitative analysis of PFAS degradation products under varying reaction conditions was performed using a HRMS Orbitrap Exploris 240 (Thermo Fisher Scientific, San Jose, CA). Samples for analysis were prepared by filtration through 0.2 μm PTFE syringe filters followed by dilution in methanol in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio of sample to methanol. Elemental composition assignments from accurate mass measurements were performed using the Web-based software ChemCalc.34 Additional details, including list with tentatively assigned elemental compositions of degradation products, can be found in the online ESI,† respectively.
:10 ratio of sample to methanol. Elemental composition assignments from accurate mass measurements were performed using the Web-based software ChemCalc.34 Additional details, including list with tentatively assigned elemental compositions of degradation products, can be found in the online ESI,† respectively.
2.6 X-ray absorption near-edge structures (XANES)
Fe3O4 nanoparticles were analyzed for Fe speciation on the Beamline for Materials Measurement (6-BM) at the National Synchrotron Light Source II at Brookhaven National Laboratory with details included in the ESI† S2. All results were analyzed using Athena software.35,36 Spectra were normalized by fitting a first-order polynomial to the pre-edge region and by fitting a second order polynomial to normalize the post-edge region to 1.0.3. Results and discussion
3.1 Nano-magnetite catalyzed UV-Fenton chemistry efficiency and optimization
Batch reactions of UV-Fenton reagents in the presence of PFAS species were conducted in order to determine the efficiency of the reaction and optimal conditions for destructing PFAS species. PFOA and PFOS were initially tested, as these are two of the most commonly encountered PFAS in contaminated waters.37 PFOA and PFOS are also recommended for monitoring by the US EPA.8 Challenge water samples were prepared with 1500 ppt of PFOA and 1500 ppt of PFOS in DI water.Generally, four major parameters influence the effectiveness of the Fenton reaction: UV-C exposure time, H2O2 concentration, Fe3O4 nanoparticle concentration, and pH values. The first three variables were initially tested to determine optimal reaction conditions (Table ST1). These initial reactions were all slightly acidic with a pH of 6 at the start of the reaction. To fully evaluate these parameters, a factorial design experiment was implemented with the data analyzed using JMP® 15.1.0 software (Fig. 1).38 Among the three variables, H2O2 concentration was shown to have the greatest impact on PFOS and PFOA reduction with the peak reduction appearing near 3.5 M for reaction time groups (5, 32.5 and 60 min). In comparison, the effects of UV-C exposure time and Fe3O4 concentration both showed weaker impacts on the PFOS and PFOA reduction rates. UV-C exposure time seems to reach peak efficiency in reduction for both PFOS and PFOA at and after 30 min, indicating that 30 min is a sufficient to catalyze the Fenton reaction. For both PFOS and PFOA, there is an increase in destruction efficiency at the highest concentration of Fe3O4 used, however, the overall change in efficiency is small. For further investigation, 1000 ppm of Fe3O4 was selected as it provided ample PFAS destruction when coupled with higher concentrations of H2O2.
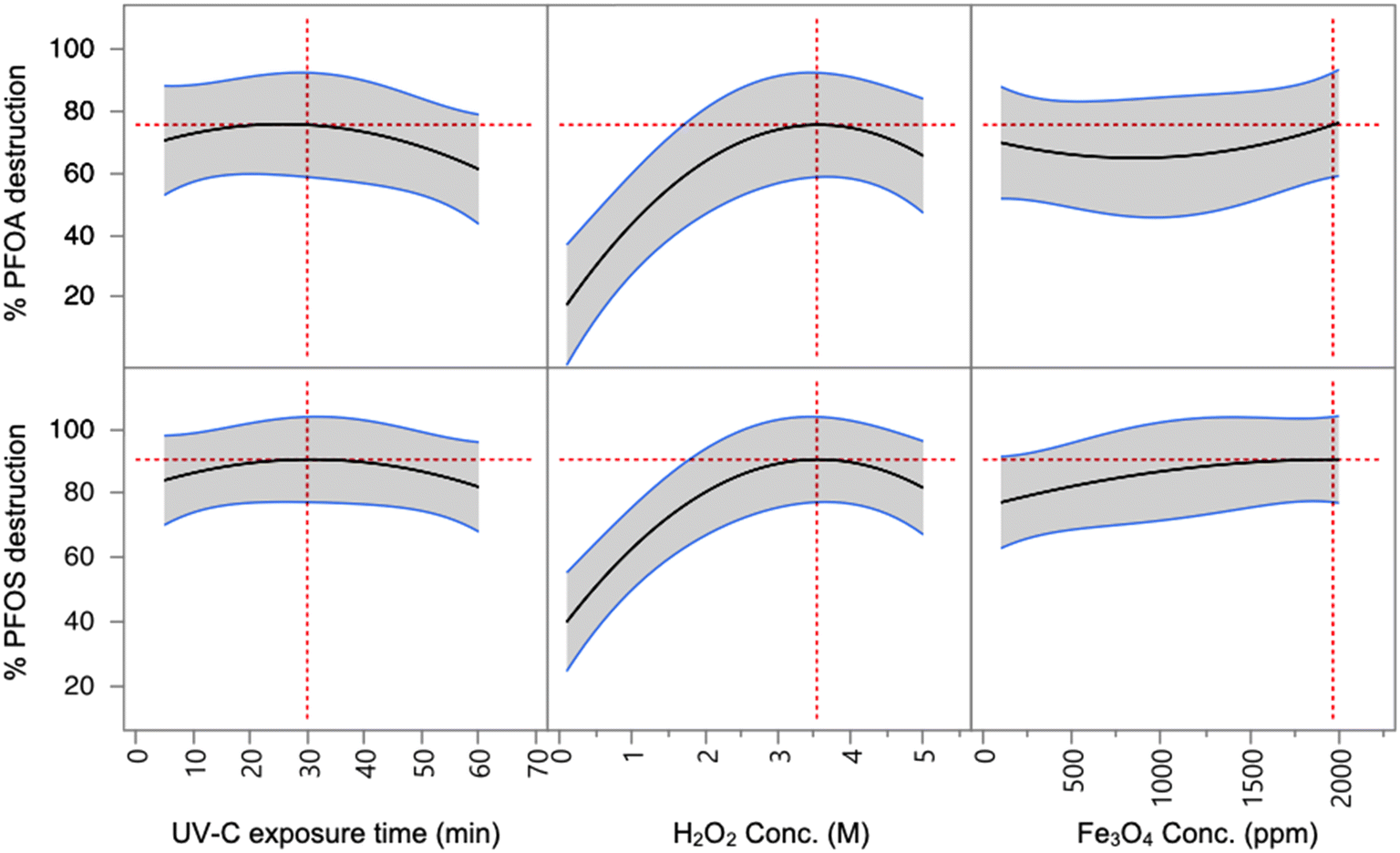 | ||
| Fig. 1 Effects of UV-C exposure time, H2O2 concentration, and Fe3O4 concentration on PFOA and PFOS destruction efficiency. The grey shaded region indicates the 95% confidence interval, and the red dashed lines indicate the determined optimal reaction condition for each variable. | ||
Based on the above analysis, a controlled experiment was carried out under a UV-C exposure time of 30 min in order to address the effect of pH value on the PFAS destruction. All tests were carried out at pH 5, 7 and 9. Challenge water solutions were again prepared with 1500 ppt each of PFOA and PFOS in DI water. At each pH value, the solutions were tested at 0.1, 2.55, and 5 M H2O2 and at 100 and 1000 ppm Fe3O4 (Fig. 2). These tests further confirmed the highly efficient PFAS destruction at higher concentrations of H2O2 and showed that pH had a significant impact on overall PFAS destruction efficiency. Reactions at low pH (5) showed consistently low PFAS destruction efficiency (10–30%) at all concentrations of H2O2 and Fe3O4 tested with the effects on PFOS being less pronounced than that of PFOA. The low overall PFAS reduction efficiency at low pH may be a combination of increased radical scavenging by H+ and dissolution of Fe3O4 to aqueous Fe species, which are less efficient for the UV-Fenton process.39 Similarly, the tests conducted with only 0.1 M H2O2 did not show high efficiency for either PFOA or PFOS destruction. For PFOA, destruction efficiency at 0.1 M H2O2 was less than 20% at all three pH conditions and at all concentrations of Fe3O4, whereas PFOS destruction went up to as high as 70% at higher pH values. At mid-range concentration of H2O2 (2.55 M), destruction efficiency for both PFOA and PFOS was impacted by both pH and Fe3O4 concentration. Experiments at basic pH showed consistently higher PFAS destruction efficiency at a H2O2 concentration of 2.55 M. The influence on PFAS destruction at elevated pH values may be explained by the fact that ˙OH redox potential is reduced at high pH, which may also impact the extent of reaction with organic species in solution.40,41 Additionally, higher pH may also assist in the reaction, as Fe3O4 is more stable at higher pH.42,43 At lower pH, Fe3O4 may undergo some extent of dissolution to aqueous Fe ions, which have been shown to be only partially efficient for UV-Fenton processes for PFAS destruction.26,27 Changes in pH can potentially facilitate adsorption of PFAS species to the magnetite, which would appear as reduction of PFAS concentration in subsequent analyses and can thus be erroneously interpreted as PFAS destruction. To clarify effects of pH change, we performed experiments at 50×-reduced H2O2 concentrations of 0.1 M. We also changed 10-fold the magnetite concentration keeping all other conditions the same. At reduced peroxide concentration and for 100 ppm magnetite in solution, the difference in PFAS destruction percentage at three different pH is within the experimental error (Fig. 2). For example, the percent removal (destruction) of PFOA in these samples is nearly equal (within 5% of one another). This result indicates that under our experimental conditions pH changes are not significantly influencing adsorption of PFOA to the magnetite surface.
 | ||
| Fig. 2 Percent destruction of PFOA and PFOS at variable pH, H2O2 concentration, and Fe3O4 concentration conditions. All samples underwent 30 minutes of UV-C exposure. (A) PFOA with 100 ppm Fe3O4, (B) PFOS with 100 ppm Fe3O4, (C) PFOA with 1000 ppm Fe3O4, (D) PFOS with 1000 ppm Fe3O4. Percent error for samples was calculated to be ±7.0% for PFOA and ±4.9% for PFOS, based on replicate data points. | ||
Despite high efficiency achieved under most conditions at basic pH, comparable PFAS destruction can be achieved at neutral pH by increased concentrations of Fe3O4 and H2O2 (Fig. 2). At high concentration of Fe3O4 (1000 ppm), the destruction efficiency at neutral pH was increased at 2.55 M H2O2 with the PFOA destruction efficiency being 75.7% (±4.8%) and PFOS destruction efficiency being 96.3% (±2.0%). This showed that Fe3O4 nanoparticles are critical to the efficiency of the UV-Fenton process, and that water treatment processes for PFAS could be conducted at neutral pH (i.e., drinking water pH values) by adjusting the concentration of Fe3O4 and H2O2. At high H2O2 concentrations (5 M), PFOA and PFOS destruction efficiency was found to be greater than 90% with the efficiency not affected by either pH or Fe3O4 concentration (Fig. 2). By utilizing high concentrations of H2O2, it is possible to achieve maximum PFAS destruction efficiency at neutral pH and low concentration of Fe3O4. Ultimately, a balance of UV-Fenton reagents and pH conditions could be established for waste water treatment that would maximize PFAS destruction efficiency while lowering the amount of Fenton reagents needed.
In order to demonstrate that ROS are generated, a fluorescent probe, DCFH-DA, was utilized to confirm the presence of ROS in the reaction solution. Samples with varied concentrations of Fe3O4 nanoparticles and a single H2O2 concentration were reacted in the UV-C oven. In order to prevent saturation of the fluorescence detector, selected concentrations were much lower than those used for PFAS destruction. With increase in magnetite concentration, there was a clear increase in the ROS-probe fluorescence signal (Fig. SF3). The control sample, without any added Fe3O4, shows negligible fluorescence. This result confirms both the presence of ROS after UV-Fenton reaction, and shows that Fe3O4 nanoparticles play a critical role in catalyzing the UV-Fenton reaction. This observation, combined with the fact that very little PFAS destruction is observed at low concentrations of Fe3O4 and H2O2 (Fig. 2), demonstrates that ROS likely play a role in PFAS destruction. Our observations are further corroborated by previous studies that have shown a link between ROS production and depletion of PFAS species through a variety of advanced oxidation processes.44–47 For example, electron paramagnetic resonance spectroscopy has demonstrated the rapid and efficient oxidation of PFAS by hydroxyl radicals in the presence on an iron-decorated clay-cyclodextrin polymer composites.48
3.2 High resolution mass spectrometry (HRMS) analysis
HRMS has been demonstrated as one of the most useful techniques for untargeted analysis of PFAS and their degradation products. For example, HRMS has been recently applied for unambiguous identification of novel PFAS compounds and their grouping into classes based on CF2 Kendrick mass defect analysis.49 HRMS has been also used for elucidating the degradation products of PFAS in water after controlled γ-irradiation.50 To confirm PFAS degradation in our experiments, high resolution negative ion mass spectra of a control PFOA sample and PFOA samples after reactions under varying conditions are plotted in SF1 and SF2 with observed masses and tentative elemental composition assignments in Tables 1 and 2. For the control, the most intense ion peaks correspond to intact PFOA anion [C8F15O2]−˙ at m/z 412.9664 (molecular ion) and its major fragment [C7F15]−˙ at m/z 368.9766. The absolute intensity of the intact PFOA peak in spectra from reacted samples is decreasing considerably – a qualitative indication for efficient PFOA degradation. At 5 M H2O2 concentration and pH 9, the initial PFOA has been completely degraded and no molecular ion is observed (Fig. SF1.C). A number of additional peaks are observed in spectra from reacted PFOA and are interpreted as PFOA degradation products, when compared to unreacted controls. Some of these products (ions at m/z 163, 235, 363) have been reported in prior studies of ROS-induced degradation of polyfluorinated species by mass spectrometry, helping to elucidate the degradation pathways of PFAS under diverse reaction conditions.51,52 Such conditions include photochemical and microwave discharge-induced53 degradation of PFOA in water.| Measured mass [Da] | Composition | Calculated mass [Da] |
|---|---|---|
| 412.96585 | C 8 F 15 O 2 | 412.96582 |
| 368.97605 | C 7 F 15 | 368.97600 |
| 362.95926 | C 7 F 13 O 2 | 362.96907 |
| 296.93994 | ||
| 266.93172 | ||
| 260.89190 | ||
| 240.95259 | ||
| 234.98052 | C 4 F 9 O | 234.98054 |
| 189.09113 | ||
| 176.97526 | ||
| 162.9596 | C3F5O2 | 162.98184 |
| Measured mass [Da] | Composition | Calculated mass [Da] |
|---|---|---|
| 498.92961 | C 8 F 17 O 3 S | 498.92965 |
| 448.93296 | C 7 F 15 O 3 S | 448.93286 |
| 398.93609 | C 6 F 13 O 3 S | 398.93604 |
| 189.09119 | ||
| 176.97532 |
3.3 UV-Fenton chemistry for destruction of 18 PFAS species
With the promising results seen for PFOS and PFOA, we further expanded our work to treating 18 PFAS compounds, as defined in EPA 537.1. Challenge water containing 18 PFAS compounds with a starting concentration of 750 ppt for each contaminant, and the solutions were prepared with the following test conditions: 2.55 M H2O2 at pH 7 and 9, and 5 M H2O2 at pH 7 and 9. Each reaction condition contained 1000 ppm Fe3O4 and was left in the UV-C oven to react for 30 min. Detection of the 18 PFAS species was done with LC/MS/MS and PFAS remaining in solution was quantified (Fig. 3). | ||
| Fig. 3 Percent destruction of 18 PFAS species at (A) pH 7 and (B) pH 9 and two concentrations of H2O2. All samples were prepared with 1000 ppm Fe3O4 and underwent 30 minutes of UV-C exposure. Instrumental analysis measurement uncertainty for detected concentration (in units of ppt) are as follows: PFBS – 23.2%; HFPODA* – 28.3%; PFHxA – 21.8%; PFHxS – 21.6%; PFHpA – 23.2%; DONA* – 22.9%; PFOA – 22.5%; PFFOS – 25.2%; 9Cl-PF3ONS – 22.3%; PFNA – 21.5%; PFDA – 23.2%; 11Cl-PF3OUdS* – 25.9%; PFUnDA – 22.8%; NMeFOSAA – 27.1%; PFDoDA – 24.4%; NEtFOSAA* – 25.8%; PFTrDA – 26.5%; PFTeDA – 21.6%. *These PFAS species are branched, which may impact their reactivity when compared with the linear compounds. | ||
At a pH of 9 (Fig. 3B) greater than 90% PFAS destruction can be achieved for 14 out of the 18 PFAS compounds. At pH 7, greater than 90% destruction was observed for 9 out of the 18 compounds. This is consistent with prior results indicating that higher pH conditions were more suitable for achieving greater destruction efficiency. At both pH 7 and 9, there was a decrease in destruction efficiency at 5 M H2O2, when compared with 2.55 M H2O2 for some PFAS tested. This drop was especially pronounced in the pH 7 samples. Previous studies observed a similar phenomenon in testing the utilization efficiency of H2O2 with Fe3O4 and found that at higher concentrations of H2O2, there was an observed decrease in this efficiency most likely due to scavenging of ROS by excessive H2O2 present in the solution.28 This potential scavenging may not have impacted destruction efficiency in tests with just PFOA and PFOS, where the total PFAS concentration was 3000 ppt, and ROS were expected to be in significant excess for PFAS oxidation. The experiments conducted with the 18 PFAS challenge water were done with higher total concentration of PFAS (13500 ppt total), and as such, ROS scavenging could have had a greater impact on overall reaction efficiency when less in excess of total PFAS. This would be consistent with concentrations of ROS reported from Fenton reactions in previous literature, which were measured in the range of tens54 to hundreds55 of parts per billion (ppb) after 30 min.
This hypothesis was tested by determining PFOA destruction efficiency in a range of highly concentrated samples. Samples were prepared with PFOA at five different starting concentrations with increasing magnitude: 1.5, 50, 150, 500, and 1000 ppb. UV-Fenton was conducted with 1000 ppm Fe3O4 and 5 M H2O2, and 30 min of UV-C exposure for all samples. Solutions were run at pH 7 and repeated at pH 9 for comparison. The detected percent destruction of PFOA is shown in Fig. 4. At both pH 7 and pH 9, destruction efficiency remained consistent for all PFOA starting concentrations except the highest, 1000 ppb. At this high concentration, destruction efficiency was reduced by an average of 16% (±4.8%) at pH 7 and an average of 36% (±4.8%) at pH 9. This reduction in efficiency was at a concentration two orders of magnitude higher than those tested with the 18 PFAS challenge water experiments, and this indicated that UV-Fenton exhibited high efficiency in any of the PFAS concentrations that were expected to be in contaminated samples in waste water streams. The drop in efficiency with the 5 M H2O2 samples, particularly at pH 7, therefore, may be due to a combination of effects; the first was increased ˙OH scavenging at the lower pH condition, which had been shown to play a dominant role in the UV-Fenton process, particularly when catalyzed by Fe3O4.28 Additionally, if increased radical scavenging occurred at excess H2O2, this would be expected to be more pronounced at the lower pH, as the reaction rate of PFAS destruction may be slower, as has been indicated by similar studies that observed this change in reaction rate of organic contaminant oxidation as a function of pH.28 The exact mechanistic reason for the observed change in destruction efficiency at high H2O2 concentration remains an interesting area for further study, requiring additional experimentation to elucidate the exact mechanism associated with Fe3O4 catalyzed UV-Fenton for PFAS destruction.
 | ||
| Fig. 4 Percent destruction with increasing PFOA concentration at pH 7 and at pH 9. All samples were prepared with 1000 ppm Fe3O4 and 5 M H2O2, and underwent 30 min of UV-C exposure. | ||
Another variable that appeared to play a significant role in destruction efficiency was the molecular structure of the individual PFAS. In all reaction conditions for the 18 PFAS challenge water experiments, PFAS destruction was the least efficient for shorter chain PFAS compounds. In Fig. 3, compounds are listed in order of increasing number of carbons in the molecule backbone. For example, the three PFAS with consistently low removal efficiency, even at pH 9, were the three shortest chain compounds: PFBS (4 carbons), HFPODA (6 carbons), and PFHA (6 carbons). This pattern was observed previously when studying the simple defluorination pattern of PFAS, and it was determined that defluorination efficiency decreased with shorter chain molecules due their reduced hydrophobic sorption capacity to a catalytic reaction site.56–58 This further elucidates some of the mechanisms at play in Fe3O4 mediated Fenton chemistry, as previous studies found that ˙OH and superoxide radicals, such as O2˙− were the species directly involved in the oxidation process.28 It is therefore possible that in solution PFAS are hydrophobically partitioned closer to the Fe3O4 surface, thereby bringing the species close to the site of the catalytic reaction and allowing for sterically improved interaction of PFAS with ROS. Fe3O4 has been previously shown to adsorb a variety of organic acids and other organic contaminants, and this adsorption is enhanced by the hydrophobicity of the adsorbing compound.59,60 The longer chain PFAS, which are more hydrophobic with increasing chain length and fluorination, may more readily adsorb to the Fe3O4 nanoparticles, making them more available for efficient oxidation and destruction. This hypothesis is consistent with prior theoretical work that showed longer chain PFAS adsorb more strongly to silicate surfaces in aqueous solution than short chain PFAS due to lower solubility of the long chains in water.61 Furthermore, theoretical calculations using density functional theory have shown that the C–F bond dissociation energies tend to decrease with increasing chain length of PFAS,62 which could also contribute to the greater destruction efficiency of longer chain molecules. Our control experiments of PFAS in solution with only Fe3O4 or at very low concentrations of Fe3O4 and H2O2 did not indicate adsorption of PFAS to an extent that would impact concentration detection (Fig. SF4). It is still likely, however, that longer-chain PFAS can become more closely associated with the nanoparticles thus contributing to the increase in the rate of the catalytic reaction.
3.4 Reusability of Fe3O4 nanoparticles in repeat cycles of PFAS destruction
Fe3O4 is a multivalent mineral species, containing iron in both Fe(II) and Fe(III) oxidation states in its structural composition. While both Fe(II) and Fe(III) can participate in Fenton and Fenton-like reactions, respectively, numerous studies have shown that Fe(II) is kinetically favorable in catalyzing the production of ROS.22,63–65 Over time, it is possible that magnetite nanoparticles would become more oxidized, and the Fenton catalysis process (2) may rely more on Fe(III), reducing the efficiency of ROS production and therefore destruction of PFAS species. Fe3O4 nanoparticles were thus evaluated for their reusability after multiple cycles of use. Samples were prepared as described for previous UV-Fenton batch reactions. However, after 24 h following each reaction period, new PFOA and PFOS containing solution was added to the remaining Fe3O4 and H2O2 in solution and UV-Fenton reaction was rerun with another 30 min of UV-C exposure. Fe3O4 was collected for Fe K-edge XANES analysis, and the remaining solution was collected for PFOA and PFOS detection analysis.Fe XANES analysis (Fig. SF5) was utilized to determine the speciation of the Fe3O4 and if the Fe3O4 mineral composition was altered over the course of multiple cycles of UV-Fenton. Four UV-Fenton reaction conditions were tested (the same reaction conditions tested against the 18 PFAS challenge water): 2.55 M H2O2 at pH 7 and 9, and 5 M H2O2 at pH 7 and 9. For each solution condition, the UV-Fenton reaction was run for 3 cycles using the same starting concentration of PFOA/PFOS each time and reusing the Fe3O4 from the previous cycle. Fe XANES spectra did not indicate any significant change in the Fe3O4 speciation or local bonding in all conditions tested (see on-line ESI†).
The results from the Fe3O4 XANES analysis were further corroborated by detection of PFOA and PFOS remaining in solution following multiple cycles of UV-Fenton with the same Fe3O4 nanoparticles. Fig. 5 shows that in nearly all four aforementioned solution conditions, Fe3O4 UV-Fenton catalysis efficiency was retained. At pH 9 conditions, for both 2.55 M H2O2 (Fig. SF5B) and 5 M H2O2 (Fig. SF5D), PFOA and PFOS destruction was retained above 90% efficiency for 1, 2, and 3 cycles of UV-Fenton. For pH 7 conditions, for both 2.55 M H2O2 (Fig. SF5A) and 5 M H2O2 (Fig. SF5C), PFOS destruction remained consistent at greater than 90% efficiency after 1, 2, and 3 cycles of UV-Fenton. PFOA, however, did show a drop in destruction efficiency over the course of the three cycles at the lower pH conditions. After the second cycle, PFOA destruction efficiency dropped by 3%, followed by another 6–8% after the third cycle. This drop was consistent regardless of H2O2 concentration, and further suggested that pH had a critical effect on Fe3O4 stability and thereby catalysis efficiency for UV-Fenton. This additionally emphasizes that the molecular structure of the specific target PFAS can impact its destruction efficiency. For example, PFOA efficiency was impacted by pH change, while PFOS was not, as observed with the 18 PFAS challenge water study. Overall, the results of the Fe XANES study combined with the detection of PFOA and PFOS after multiple cycles of UV-Fenton suggested that Fe3O4 nanoparticles were highly stable, especially at higher pH, and can be reused over time. Fe3O4 nanoparticles were therefore promising catalyst materials for PFAS destruction by UV-Fenton both for efficiency in destroying multiple types of PFAS species, and in Fe3O4 recyclability.
 | ||
| Fig. 5 Percent destruction of PFOA and PFOS at variable pH and H2O2 concentration. All samples contained 1000 ppm of Fe3O4 and underwent 30 minutes of UV-C exposure. (A) 2.55 M H2O2, pH 7, (B) 2.55 M H2O2, pH 9, (C) 5 M H2O2, pH 7, (D) 5 M H2O2, pH 9. Percent error for samples was calculated to be ±7.0% for PFOA and ±4.9% for PFOS. The 3 cycles data for pH 9 and 5 M H2O2 is not included in this report, as that data point was a significant outlier, and it was determined that there was an issue with sample preparation that resulted in an inaccurate measurement. | ||
4. Conclusion
UV-Fenton catalyzed by Fe3O4 nanoparticles showed greater than 90% rates of PFAS destruction for a wide variety of PFAS. UV-Fenton is a PFAS destruction technique, moving away from previous technologies relying on separation or adsorption, which typically produce downstream waste.66,67 The work presented in this paper indicates that Fe3O4 is an additionally useful catalyst, as degradation requires a very short UV-C exposure period when compared to aqueous Fe species;26 does not require additional species added to solution and further water treatment or filtering;27 can be performed under neutral pH conditions (appropriate for drinking water); and can be optimized by varying Fe3O4 and/or H2O2 concentrations. Finally, Fe3O4 is a naturally occurring and potentially environmentally-benign material, which can be reused. If required, it can be easily filtered or removed from solution with a magnet during the waste-water treatment process. Our results suggest that nano-Fe3O4 induced UV-Fenton can be considered and potentially implemented as a novel and efficient technique for destruction of the more than 300 PFAS that may be found in contaminated waste streams and drinking water. Future work will be done to improve the utilization efficiency of H2O2 in order to assess the economic feasibility of scale-up for this process. Further experiments need to be performed in more complex matrices. For example, water from a previously contaminated waste stream, including dissolved organic matter, could potentially inhibit or enhance the reaction. We plan to conduct further investigations of nano-Fe3O4 catalyzed UV-Fenton, to continue to improve reaction efficiency and economic feasibility and probe degradation products for a larger class of PFAS, in order to evaluate its full potential as an alternative for PFAS destruction.Funding sources
The authors acknowledge funding support from the Independent Research and Development (IRAD) Fund from the Research and Exploratory Development Mission Area of the Johns Hopkins University Applied Physics Laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr. Claresta Joe-Wong for assisting in sample preparation. The authors acknowledge the Beamline for Materials Measurement (6-BM) of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704.References
- J. M. Jian, Y. Guo, L. Zeng, L. Liang-Ying, X. Lu, F. Wang and E. Y. Zeng, Global Distribution of Perfluorochemicals (PFCs) in Potential Human Exposure Source–A Review, Environ. Int., 2017, 108, 51–62, DOI:10.1016/j.envint.2017.07.024
.
- S. Gaballah, A. Swank, J. R. Sobus, X. M. Howey, J. Schmid, T. Catron, J. McCord, E. Hines, M. Strynar and T. Tal, Evaluation of Developmental Toxicity, Developmental Neurotoxicity, and Tissue Dose in Zebrafish Exposed to GenX and Other PFAS, Environ. Health Perspect., 2020, 128(4), 1–22, DOI:10.1289/EHP5843
- S. Garg, P. Kumar, V. Mishra, R. Guijt, P. Singh, L. F. Dumée and R. S. Sharma, A Review on the Sources, Occurrence and Health Risks of per-/Poly-Fluoroalkyl Substances (PFAS) Arising from the Manufacture and Disposal of Electric and Electronic Products, J. Water Process Eng., 2020, 38 DOI:10.1016/j.jwpe.2020.101683
- D. Cui, X. Li and N. Quinete, Occurrence, Fate, Sources and Toxicity of PFAS: What We Know so Far in Florida and Major Gaps, TrAC, Trends Anal. Chem., 2020, 130, 115976, DOI:10.1016/j.trac.2020.115976
- J. L. Guelfo and D. T. Adamson, Evaluation of a National Data Set for Insights into Sources, Composition, and Concentrations of per- and Polyfluoroalkyl Substances (PFASs) in U.S. Drinking Water, Environ. Pollut., 2018, 236, 505–513, DOI:10.1016/j.envpol.2018.01.066
- Y. J. Yu, F. L. Zhang, T. Y. Peng, C. L. Wang, J. Cheng, C. Chen, K. N. Houk and Y. F. Wang, Sequential C-F Bond Functionalizations of Trifluoroacetamides and Acetates via Spin-Center Shifts, Science, 2021, 371(6535), 1232–1240, DOI:10.1126/science.abg0781
- U. Mazurek and H. Schwarz, Carbon—Fluorine Bond Activation — Looking at and Learning from Unsolvated Systems, ChemInform, 2003, 34(35), 1321–1326, DOI:10.1002/chin.200335229
- Epa, U. of Water, O. FACT SHEET PFOA & PFOS Drinking Water Health Advisories. 2016, 1–4.
- A. Reade, T. Quinn and J. S. Schreiber, PFAS in Drinking Water 2019: Scientific and Policy Assessment for Addressing per- and Polyfluoroalkyl Substances (PFAS) in Drinking Water. PFAS Drink., Water 2019 Sci., 2019, 1–102 Search PubMed
- D. Barpaga, J. Zheng, K. S. Han, J. A. Soltis, V. Shutthanandan, S. Basuray, B. P. McGrail, S. Chatterjee and R. K. Motkuri, Probing the Sorption of Perfluorooctanesulfonate Using Mesoporous Metal-Organic Frameworks from Aqueous Solutions, Inorg. Chem., 2019, 58(13), 8339–8346, DOI:10.1021/acs.inorgchem.9b00380
- S. Garg, J. Wang, P. Kumar, V. Mishra, H. Arafat, R. S. Sharma and L. F. Dumée, Remediation of Water from Per-/Poly-Fluoroalkyl Substances (PFAS) - Challenges and Perspectives, J. Environ. Chem. Eng., 2021, 9(4) DOI:10.1016/j.jece.2021.105784
- F. Dixit, R. Dutta, B. Barbeau, P. Berube and M. Mohseni, PFAS Removal by Ion Exchange Resins: A Review, Chemosphere, 2021, 272, 129777, DOI:10.1016/j.chemosphere.2021.129777
- H. Guo, J. Zhang, L. E. Peng, X. Li, Y. Chen, Z. Yao, Y. Fan, K. Shih and C. Y. Tang, High-Efficiency Capture and Recovery of Anionic Perfluoroalkyl Substances from Water Using PVA/PDDA Nanofibrous Membranes with Near-Zero Energy Consumption, Environ. Sci. Technol. Lett., 2021, 8(4), 350–355, DOI:10.1021/acs.estlett.1c00128
- J. K. Johnson, C. M. Hoffman, D. A. Smith and Z. Xia, Advanced Filtration Membranes for the Removal of Perfluoroalkyl Species from Water, ACS Omega, 2019, 4(5), 8001–8006, DOI:10.1021/acsomega.9b00314
- A. J. Lewis, T. Joyce, M. Hadaya, F. Ebrahimi, I. Dragiev, N. Giardetti, J. Yang, G. Fridman, A. Rabinovich, A. A. Fridman, E. R. McKenzie and C. M. Sales, Rapid Degradation of PFAS in Aqueous Solutions by Reverse Vortex Flow Gliding Arc Plasma, Environ. Sci.: Water Res. Technol., 2020, 6(4), 1044–1057, 10.1039/c9ew01050e
- B. R. Pinkard, S. Shetty, D. Stritzinger, C. Bellona and I. V. Novosselov, Destruction of Perfluorooctanesulfonate (PFOS) in a Batch Supercritical Water Oxidation Reactor, Chemosphere, 2021, 279, 130834, DOI:10.1016/j.chemosphere.2021.130834
- A. L. Duchesne, J. K. Brown, D. J. Patch, D. Major, K. P. Weber and J. I. Gerhard, Remediation of PFAS-Contaminated Soil and Granular Activated Carbon by Smoldering Combustion, Environ. Sci. Technol., 2020, 54(19), 12631–12640, DOI:10.1021/acs.est.0c03058
- S. Witt, N. Rancis, M. Ensch and V. Maldonado, Electrochemical Destruction of “Forever Chemicals”: The Right Solution at the Right Time, Electrochem. Soc. Interface, 2020, 29(2), 73–76, DOI:10.1149/2.F11202IF
- B. Wu, S. Hao, Y. Choi, C. P. Higgins, R. Deeb and T. J. Strathmann, Rapid Destruction and Defluorination of Perfluorooctanesulfonate by Alkaline Hydrothermal Reaction, Environ. Sci. Technol. Lett., 2019, 6(10), 630–636, DOI:10.1021/acs.estlett.9b00506
- S. M. Con, Z. Chen, C. Li, J. Gao, H. Dong, Y. Chen, B. Wu and C. Gu, E Ffi Cient Reductive Destruction of Per Fl Uoroalkyl Substances under Self-Assembled Micelle Con Fi Nement. 2020.
- J. Cui, P. Gao and Y. Deng, Destruction of Per- A Nd Polyfluoroalkyl Substances (PFAS) with Advanced Reduction Processes (ARPs): A Critical Review, Environ. Sci. Technol., 2020, 54(7), 3752–3766, DOI:10.1021/acs.est.9b05565
- S. Goldstein, D. Meyerstein and G. Czapski, The Fenton Reagents, Free Radical Biol. Med., 1993, 15(4), 435–445, DOI:10.1016/0891-5849(93)90043-T
- E. E. Ebrahiem, M. N. Al-Maghrabi and A. R. Mobarki, Removal of Organic Pollutants from Industrial Wastewater by Applying Photo-Fenton Oxidation Technology, Arabian J. Chem., 2017, 10, S1674–S1679, DOI:10.1016/j.arabjc.2013.06.012
- B. Jain, A. K. Singh, H. Kim, E. Lichtfouse and V. K. Sharma, Treatment of Organic Pollutants by Homogeneous and Heterogeneous Fenton Reaction Processes, Environ. Chem. Lett., 2018, 16(3), 947–967, DOI:10.1007/s10311-018-0738-3
- S. M. Kim and A. Vogelpohl, Degradation of Organic Pollutants by the Photo-Fenton-Process, Chem. Eng. Technol., 1998, 21(2), 187–191, DOI:10.1002/(SICI)1521-4125(199802)21:2<187::AID-CEAT187>3.0.CO;2-H
- H. Tang, Q. Xiang, M. Lei, J. Yan, L. Zhu and J. Zou, Efficient Degradation of Perfluorooctanoic Acid by UV-Fenton Process, Chem. Eng. J., 2012, 184, 156–162, DOI:10.1016/j.cej.2012.01.020
- A. Santos, S. Rodríguez, F. Pardo and A. Romero, Use of Fenton Reagent Combined with Humic Acids for the Removal of PFOA from Contaminated Water, Sci. Total Environ., 2016, 563–564, 657–663, DOI:10.1016/j.scitotenv.2015.09.044
- W. Li, Y. Wang and A. Irini, Effect of PH and H2O2 Dosage on Catechol Oxidation in Nano-Fe3O4 Catalyzing UV-Fenton and Identification of Reactive Oxygen Species, Chem. Eng. J., 2014, 244, 1–8, DOI:10.1016/j.cej.2014.01.011
- Y. Chen, C. J. Miller and T. D. Waite, Heterogeneous Fenton Chemistry Revisited: Mechanistic Insights from Ferrihydrite-Mediated Oxidation of Formate and Oxalate, Environ. Sci. Technol., 2021, 55, 14414–14425, DOI:10.1021/acs.est.1c00284
- S. Zhang, M. Sun, T. Hedtke, A. Deshmukh, X. Zhou, S. Weon, M. Elimelech and J. H. Kim, Mechanism of Heterogeneous Fenton Reaction Kinetics Enhancement under Nanoscale Spatial Confinement, Environ. Sci. Technol., 2020, 54(17), 10868–10875, DOI:10.1021/acs.est.0c02192
- EPA, EPA Method 537.1, 2018, vol. 1, (2009), pp. 1–50 Search PubMed.
- O. Myhre, J. M. Andersen, H. Aarnes and F. Fonnum, Evaluation of the Probes 2′,7′-Dichlorofluorescin Diacetate, Luminol, and Lucigenin as Indicators of Reactive Species Formation, Biochem. Pharmacol., 2003, 65(10), 1575–1582, DOI:10.1016/S0006-2952(03)00083-2
- B. Kalyanaraman, V. Darley-Usmar, K. J. A. Davies, P. A. Dennery, H. J. Forman, M. B. Grisham, G. E. Mann, K. Moore, L. J. I. Roberts and H. Ischiropoulos, Measuring Reactive Oxygen and Nitrogen Species with Fluorescent Probes: Challenges and Limitations, Free Radical Biol. Med., 2012, 52(1), 1–6, DOI:10.1016/j.freeradbiomed.2011.09.030.Measuring
- L. Patiny and A. Borel, ChemCalc: A Building Block for Tomorrow's Chemical Infrastructure, J. Chem. Inf. Model., 2013, 53(5), 1223–1228, DOI:10.1021/ci300563h
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: Data Analysis for X-Ray Absorption Spectroscopy Using IFEFFIT, J. Synchrotron Radiat., 2005, 12(4), 537–541, DOI:10.1107/S0909049505012719
- B. Ravel and M. Newville, ATHENA and ARTEMIS: Interactive Graphical Data Analysis Using IFEFFIT, Phys. Scr., T, 2005, 115, 1007–1010, DOI:10.1238/Physica.Topical.115a01007
- National Institute of Environmental Health Sciences. Perfluoroalkyl and Polyfluoroalkyl Substances (PFAS). 2019, No. March, 2.
- SAS Institute Inc., JMP, SAS Institute Inc., Cary, NC, 2021 Search PubMed.
- X. R. Xu, X. Y. Li, X. Z. Li and H. B. Li, Degradation of Melatonin by UV, UV/H2O2, Fe2+/H2O2 and UV/Fe2+/H2O2 Processes, Sep. Purif. Technol., 2009, 68(2), 261–266, DOI:10.1016/j.seppur.2009.05.013
- C. Fang, M. Megharaj and R. Naidu, Electrochemical Advanced Oxidation Processes (EAOP) to Degrade per- and Polyfluoroalkyl Substances (PFASs), J. Adv. Oxid. Technol., 2017, 20(2), 20170014, DOI:10.1515/jaots-2017-0014
-
K. B. Krauskopf and D. K. Bird, Introduction to Geochemistry, McGraw-Hill, Inc., 3rd edn, 1995 Search PubMed
-
G. Faure, Principles and Applications of Inorganic Geochemistry, Macmillan Publishing Company, 1991 Search PubMed
- B. Beverskog, Revised Diagrams for Iron At 25-300 ° C, Science, 1996, 38(12), 2121–2135 CAS
- Y. Qu, C. Zhang, F. Li, J. Chen and Q. Zhou, Photo-Reductive Defluorination of Perfluorooctanoic Acid in Water, Water Res., 2010, 44(9), 2939–2947, DOI:10.1016/j.watres.2010.02.019
- X. J. Lyu, W. W. Li, P. K. S. Lam and H. Q. Yu, Insights into Perfluorooctane Sulfonate Photodegradation in a Catalyst-Free Aqueous Solution, Sci. Rep., 2015, 5, 1–6, DOI:10.1038/srep09353
- Y. Bao, S. Deng, X. Jiang, Y. Qu, Y. He, L. Liu, Q. Chai, M. Mumtaz, J. Huang, G. Cagnetta and G. Yu, Degradation of PFOA Substitute: GenX (HFPO-DA Ammonium Salt): Oxidation with UV/Persulfate or Reduction with UV/Sulfite?, Environ. Sci. Technol., 2018, 52(20), 11728–11734, DOI:10.1021/acs.est.8b02172
- J. S. Ko and J. K. Johnson, Novel Niobium-Doped Titanium Oxide Towards Electrochemical Destruction of Forever Chemicals. 0–25.
- S. Kundu and A. Radian, Surface Confinement of Per-Fluoroalkyl Substances on an Iron-Decorated Clay-Cyclodextrin Composite Enables Rapid Oxidation by Hydroxyl Radicals, Chem. Eng. J., 2022, 431(P2), 134187, DOI:10.1016/j.cej.2021.134187
- R. B. Young, N. E. Pica, H. Sharifan, H. Chen, H. K. Roth, G. T. Blakney, T. Borch, C. P. Higgins, J. J. Kornuc, A. M. McKenna and J. Blotevogel, PFAS Analysis with Ultrahigh Resolution 21T FT-ICR MS: Suspect and Nontargeted Screening with Unrivaled Mass Resolving Power and Accuracy, Environ. Sci. Technol., 2022, 56(4), 2455–2465, DOI:10.1021/acs.est.1c08143
- D. Patch, N. O'Connor, I. Koch, T. Cresswell, C. Hughes, J. B. Davies, J. Scott, D. O'Carroll and K. Weber, Elucidating Degradation Mechanisms for a Range of Per- and Polyfluoroalkyl Substances (PFAS) via Controlled Irradiation Studies, Sci. Total Environ., 2022, 832, 154941, DOI:10.1016/j.scitotenv.2022.154941
- M. Trojanowicz, K. Bobrowski, B. Szostek, A. Bojanowska-Czajka, T. Szreder, I. Bartoszewicz and K. Kulisa, A Survey of Analytical Methods Employed for Monitoring of Advanced Oxidation/Reduction Processes for Decomposition of Selected Perfluorinated Environmental Pollutants, Talanta, 2018, 177, 122–141, DOI:10.1016/j.talanta.2017.09.002
- T. Yamamoto, Y. Noma, S. I. Sakai and Y. Shibata, Photodegradation of Perfluorooctane Sulfonate by UV Irradiation in Water and Alkaline 2-Propanol, Environ. Sci. Technol., 2007, 41(16), 5660–5665, DOI:10.1021/es0706504
- S. Horikoshi, A. Tsuchida, H. Sakai, M. Abe and N. Serpone, Microwave Discharge Electrodeless Lamps (MDELs). VI. Performance Evaluation of a Novel Microwave Discharge Granulated Electrodeless Lamp (MDGEL) - Photoassisted Defluorination of Perfluoroalkoxy Acids in Aqueous Media, J. Photochem. Photobiol., A, 2011, 222(1), 97–104, DOI:10.1016/j.jphotochem.2011.05.007
- S. Qiu, D. He, J. Ma, T. Liu and T. D. Waite, Kinetic Modeling of the Electro-Fenton Process: Quantification of Reactive Oxygen Species Generation, Electrochim. Acta, 2015, 176, 51–58, DOI:10.1016/j.electacta.2015.06.103
- T. Zhou, X. Wu, J. Mao, Y. Zhang and T. T. Lim, Rapid Degradation of Sulfonamides in a Novel Heterogeneous Sonophotochemical Magnetite-Catalyzed Fenton-like (US/UV/Fe3O4/Oxalate) System, Appl. Catal., B, 2014, 160–161(1), 325–334, DOI:10.1016/j.apcatb.2014.05.036
- M. C. Hansen, M. H. Børresen, M. Schlabach and G. Cornelissen, Sorption of Perfluorinated Compounds from Contaminated Water to Activated Carbon, J. Soils Sediments, 2010, 10(2), 179–185, DOI:10.1007/s11368-009-0172-z
- H. Park, C. D. Vecitis, J. Cheng, W. Choi, B. T. Mader and M. R. Hoffmann, Reductive Defluorination of Aqueous Perfluorinated Alkyl Surfactants: Effects of Ionic Headgroup and Chain Length, J. Phys. Chem. A, 2009, 113(4), 690–696, DOI:10.1021/jp807116q
- U. Rao, Y. Su, C. M. Khor, B. Jung, S. Ma, D. M. Cwiertny, B. M. Wong and D. Jassby, Structural Dependence of Reductive Defluorination of Linear PFAS Compounds in a UV/Electrochemical System, Environ. Sci. Technol., 2020, 54(17), 10668–10677, DOI:10.1021/acs.est.0c02773
- W. Cheng, R. Marsac and K. Hanna, Influence of Magnetite Stoichiometry on the Binding of Emerging Organic Contaminants, Environ. Sci. Technol., 2018, 52(2), 467–473, DOI:10.1021/acs.est.7b04849
- E. Tombácz, I. Y. Tóth, D. Nesztor, E. Illés, A. Hajdú, M. Szekeres and L. Vékás, Adsorption of Organic Acids on Magnetite Nanoparticles, PH-Dependent Colloidal Stability and Salt Tolerance, Colloids Surf., A, 2013, 435, 91–96, DOI:10.1016/j.colsurfa.2013.01.023
- J. A. R. Willemsen and I. C. Bourg, Molecular Dynamics Simulation of the Adsorption of Per- and Polyfluoroalkyl Substances (PFASs) on Smectite Clay, J. Colloid Interface Sci., 2021, 585, 337–346, DOI:10.1016/j.jcis.2020.11.071
- M. J. Bentel, Y. Yu, L. Xu, Z. Li, B. M. Wong, Y. Men and J. Liu, Defluorination of Per- and Polyfluoroalkyl Substances (PFASs) with Hydrated Electrons: Structural Dependence and Implications to PFAS Remediation and Management, Environ. Sci. Technol., 2019, 53(7), 3718–3728, DOI:10.1021/acs.est.8b06648
- M. L. Kremer, Oxidation Reduction Step in Catalytic Decomposition of Hydrogen Peroxide by Ferric Ions, Trans. Faraday Soc., 1963, 59, 2535–2542, 10.1039/tf9635902535
- C. Jiang, S. Pang, F. Ouyang, J. Ma and J. Jiang, A New Insight into Fenton and Fenton-like Processes for Water Treatment, J. Hazard. Mater., 2010, 174(1–3), 813–817, DOI:10.1016/j.jhazmat.2009.09.125
- S. Wang, A Comparative Study of Fenton and Fenton-like Reaction Kinetics in Decolourisation of Wastewater, Dyes Pigm., 2008, 76(3), 714–720, DOI:10.1016/j.dyepig.2007.01.012
- J. Horst, J. McDonough, I. Ross, M. Dickson, J. Miles, J. Hurst and P. Storch, Water Treatment Technologies for PFAS: The Next Generation, Groundwater Monit. Rem., 2018, 38(2), 13–23, DOI:10.1111/gwmr.12281
- B. N. Nzeribe, M. Crimi, S. Mededovic Thagard and T. M. Holsen, Physico-Chemical Processes for the Treatment of Per- And Polyfluoroalkyl Substances (PFAS): A Review, Crit. Rev. Environ. Sci. Technol., 2019, 49(10), 866–915, DOI:10.1080/10643389.2018.1542916
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ew00058j |
| This journal is © The Royal Society of Chemistry 2022 |