Characteristics and mechanism of electrochemical peroxymonosulfate activation by a Co–N@CF anode for pollutant removal†
Received
19th September 2021
, Accepted 7th November 2021
First published on 8th November 2021
Abstract
Herein, carbon felt (CF) modified with N and Co (Co–N@CF) was prepared as an anode to effectively activate peroxymonosulfate (PMS) for tetracycline (TC) removal. The influencing factors, kinetic characteristics and the transformation of generated active radicals were further investigated. The results show that N and Co modification of CF via a galvanostatic electrodeposition method results in a significant decrease in the charge transfer resistance (Rct) of the CF. The fabricated Co–N@CF anode exhibits high catalytic performance and stability towards PMS activation, wherein the TC removal efficiency reached 92.2% within 1 h under optimal conditions. Radical quenching tests and electron paramagnetic resonance spectroscopy experiments demonstrated that SO4˙−, OH˙ and O2˙− are the main contributors towards TC removal, while 1O2 generation is inhibited significantly. The PMS decomposition process driven by the activator enhances the generation of OH˙. The electricity energy demand per order (EE/O) in the reaction system (Co–N@CF anode + PMS) is only 0.072 kWh m−3, which makes it an energy-saving approach for pollutant removal. The newly developed fabrication of the Co–N@CF anode for PMS activation could provide insights into the SO4˙−-based electrochemical advanced oxidation process (EAOP) applied in wastewater treatment.
Water impact
Antibiotic wastewater pollution is a big threat to water environments. Herein, we report an electrochemical peroxymonosulfate (PMS) activation method to enhance tetracycline degradation. The fabricated Co–N@CF anode for PMS activation shows promise for practical application due to its low energy consumption, good reusability and neutral reaction conditions. This work makes an important breakthrough in electrochemical PMS activation for wastewater treatment.
|
1. Introduction
The increasing quantity of pollutants discharged into the environment has become a complex challenge and has thus attracted increasing research attention.1–3 Among those pollutants, tetracycline (TC) is one of the typical antibiotics mainly expelled from pharmaceutical factories, the livestock industry and hospitals, which is not readily decomposed in the natural environment.4–7 Furthermore, as well as damaging the environment, TC can be accumulated in the human body through the food chain and result in severe damage to human health.8 Adsorption methods, biodegradation processes, coagulation and chemical processes have been applied to remove antibiotics from the environment.9–13 However, antibiotics are difficult to remove from the environment using traditional technologies due to their complex chemical structures and poor biodegradability.14 Advanced oxidation processes (AOPs) are feasible methods by which to degrade antibiotics via the generation of active free radicals.15–17 Compared to conventional Fenton-like processes, AOP-based sulfate radicals (SO4˙−) generated via persulfate (PS) activation have drawn an increasing amount of focus in pollutant removal considering the long life of the radicals generated in this process, as well as the wide pH range and specific selectivity.18 To achieve a high yield of sulfate radicals, the application of PS activator with enhanced performance has become an indispensable issue.19
There are numerous ways to active persulfate to produce sulfate radicals.13,20–24 Recently, transition-metal activation has received more research attention owing to its energy-saving potential, low cost and high efficiency.25 Transition metals exhibit favorable performance in activating persulfate because they readily give up their electrons to break the O–O bond of PS such as peroxodisulfate (PDS) and peroxymonosulfate (PMS). Generally, PMS with its asymmetric structure can be activated more easily compared to PDS13. It has been observed that PMS exhibits a higher catalytic performance than PDS.26 Among the transition metals, cobalt is considered to be the most effective metal to activate PS.22,27 Therefore, various cobalt-based heterogeneous catalysts have been fabricated that exhibit high efficiency in the removal of organic pollutants via the PS activation process.13,28–32 However, secondary pollution caused by heavy-metal ion leakage has adverse effects on human health.33 Thus, an environmentally-friendly and economical activator with high performance should be synthesized. It has been reported that cobalt oxide loaded on activated carbon shows excellent performance in PS activation, exhibiting higher activity than an unsupported heterogeneous Co3O4 catalyst and homogeneous Co2+ ions.34 However, most methods of activation have restricted implementation due to their high energy demand, high cost and complex equipment management.
Due to its high performance, easy operation and environmental safety, electrochemical PS activation is a promising way to produce radicals and can be applied in AOPs.35,36 Compared with cathodic activation, anodic activation is a more effective method to use to activate PMS to generate radicals,37 as PMS is negatively charged and there are electrostatic attractions between PMS molecules and the anode surface.38 Carbon-based materials are widely used as substrates for metal nanoparticles due to their stability, good electrical conductivity and low cost.39 The dispersion of cobalt on carbon felt (CF) via chemical binding reduces metal leaching, promotes electron transport and enhances catalytic activity.40 Nitrogen with its suitable atomic size and high valence electrons can tightly bind to carbon atoms.41 Many studies have reported the use of nitrogen-doped carbon materials in electrochemical applications.42–44 Thus, nitrogen-doped carbon materials could be used as suitable substrates in the electrochemical activation of PMS. Furthermore, reports have shown that single oxygen (1O2) is the main active species during the process of cathodic PMS activation,45 while in the case of anode oxidation, SO4˙− is the dominant reactive radical.13 Nevertheless, few studies have focused on the degrading of pollutants by cobalt loaded on CF via the assistance of electricity, therefore, the mechanism of PS activation and the transformation of various generated radicals also need further investigation.
Herein, an N- and Co-doped CF (Co–N@CF) was fabricated via a simple electrodeposition and calcination method and applied as an anode for the electrochemical activation of PMS. Dropcasting, hydrothermal and electrodeposition methods are commonly used to fabricate electrodes. The dropcasting method requires a binder to combine catalysts, thus reducing the conductivity of the electrode,46 and the hydrothermal method consumes a large quantity of energy.47 Thus, electrodeposition was selected to prepare a Co–N@CF anode. The results show that the Co–N@CF anode exhibits good performance towards pollutant removal under optimal conditions. Furthermore, the transformation of the generated radicals during PMS activation was studied via EPR tests and quenching experiments, and subsequently, the main oxidative radicals that contribute towards TC removal were also verified. Our results provide a feasible method to fabricate a CF-based anode to effectively activate PMS and could be applied in wastewater treatment.
2. Experimental section
2.1 Materials
All of the chemical reagents were purchased and used without any further purification. Sodium sulfate (Na2SO4), sodium hydroxide (NaOH), cobalt nitrate hexahydrate (Co(NO3)2·6H2O), and sodium thiosulfate (Na2S2O3) were purchased from Tianjin Kemiou Chemical Reagent Co., Ltd. Potassium PMS (KHSO5), 2-methylimidazole (C4H6N2), p-benzoquinone (C6H4O2) and TC (C22H24N2O8) were obtained from Shanghai Macklin Biochemical Co., Ltd. Sulfuric acid (H2SO4) and ethanol (CH3CH2OH) were purchased from Chengdu Chemical Reagent Co., Ltd. tert-Butyl alcohol ((CH3)3COH) and L-histidine (C6H9N3O2) were provided by Fuchen Chemical Reagent Co., Ltd.
2.2 Preparation of the different anodes by cyclic voltammetry and galvanostatic electrodeposition methods
The CF was cut into several pieces with dimensions of 2 cm × 2 cm. Then, these CF pieces were ultrasonically cleaned in 0.1 M HCl, ethanol and deionized water, sequentially, and dried in an oven at 60 °C overnight. A schematic diagram of the preparation process of the different anodes is shown in Fig. S1.† The preparations of the different anodes by cyclic voltammetry or galvanostatic electrodeposition methods were conducted in a typical three-electrode system with CF, Pt plate and Ag/AgCl as the working, counter and reference electrodes, respectively. A Co@CF anode was prepared via a galvanostatic electrodeposition method with CF poised on a potential of −1.0 V for 1200 s using an electrochemical workstation (CHI-600E, Chenghua, China) in 0.1 M Co(NO3)2·6H2O solution. As a comparison, a Co@CF-cv anode was also fabricated via a cyclic voltammetry electrodeposition method using CF poised on a potential ranging from −1.1 V to 0.1 V at a scan rate of 10 mV s−1 for 10 cycles. After electro-deposition, the Co@CF or Co@CF-cv was taken out and washed with deionized water several times, followed by drying in an oven at 60 °C for 3 h.
To obtain Co- and N-modified CF, the as-prepared Co@CF or Co@CF-cv were immersed in 0.25 M 2-methylimidazole ethanol solution for 24 h. Subsequently, the Co@CF and Co@CF-cv were dried in an oven and then put into a tube furnace and annealed at 600 °C for 2 h at a heating rate of 5 °C min−1 under a N2 atmosphere to obtain Co–N@CF or Co–N@CF-cv, respectively.
2.3 Characterization
The charge transfer resistance (Rct) values of the fabricated anodes (bare CF, Co@CF, Co@CF-cv, Co–N@CF and Co–N@CF-cv) were determined by electrochemical impedance spectroscopy (EIS) testing and simulated using the ZSimWin software. EIS tests were performed in a typical three-electrode system containing 0.1 M Na2SO4 and the frequency was ranged from 0.01 to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz. A scan electron microscope (SEM, JEOL JSM-6510LV) was used to determine the microstructures of the prepared anodes. X-ray electron spectroscopy (XPS) was carried out using Al Kα radiation. The crystal structures were measured using X-ray diffractometry (XRD, Rigaku UltimaIV) with Cu Kα radiation. EPR spectroscopy (Bruker ZMXmicro-6/1) was applied to identify the generation of radical species using 5,5-dimethy1-1-pyrroline (DMPO) and 2,2,6,6-tetramethyl-4-piperidinol (TMP) as the spin-trapping regents. All EPR tests were conducted at room temperature. The experimental parameters of the EPR spectrometer were as follows: (1) center field: 3500.0 G; (2) sweep width 100.0 G; (3) sweep time: 60 s; (4) sample g-factor: 2.00; (5) receiver gain: 30 dB; (6) modulation amplitude: 1.0 G.
000 Hz. A scan electron microscope (SEM, JEOL JSM-6510LV) was used to determine the microstructures of the prepared anodes. X-ray electron spectroscopy (XPS) was carried out using Al Kα radiation. The crystal structures were measured using X-ray diffractometry (XRD, Rigaku UltimaIV) with Cu Kα radiation. EPR spectroscopy (Bruker ZMXmicro-6/1) was applied to identify the generation of radical species using 5,5-dimethy1-1-pyrroline (DMPO) and 2,2,6,6-tetramethyl-4-piperidinol (TMP) as the spin-trapping regents. All EPR tests were conducted at room temperature. The experimental parameters of the EPR spectrometer were as follows: (1) center field: 3500.0 G; (2) sweep width 100.0 G; (3) sweep time: 60 s; (4) sample g-factor: 2.00; (5) receiver gain: 30 dB; (6) modulation amplitude: 1.0 G.
2.4 Degradation experiments
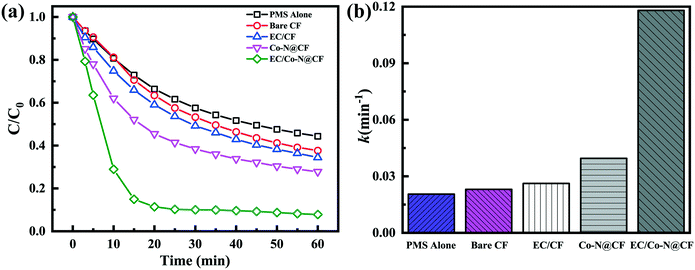
All of the degradation experiments were conducted in a 150 mL reactor containing 0.1 mM of TC. The distance between the anode and counter electrode (Pt plate) was 2.0 cm face-to-face in the reactor, with 50 mM Na2SO4 as the electrolyte. At selected reaction intervals, the sample was taken out and the TC concentration was determined using a UV-vis spectrophotometer (Shimadzu, UV-1780) at an absorbance wavelength of 357 nm. The concentration of TC was determined according to a concentration–absorbance standard curve (Fig. S2†). The reaction rate constant (k) was calculated according to the following equation:| | 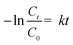 | (1) |
where C0, t, Ct and k are initial concentration of TC, the reaction time, the concentration at the selected reaction time and the reaction rate constant, respectively.
3. Results and discussion
3.1 Characterization of the Co- and N-doped CF
Fig. 1b shows the charge transfer resistance (Rct) values of bare CF, Co@CF, Co@CF-cv, Co–N@CF and Co–N@CF-cv obtained from the EIS tests shown in Fig. 1a. The results show that the Rct of Co–N@CF is the lowest among the different fabricated anodes and the Co modification of CF significantly reduces the Rct value of bare CF (Fig. 1b). The Rct value of Co@CF is lower than that of Co@CF-cv, which indicates that the galvanostatic electrodeposition method is more effective for the electrodeposition of Co on CF than the cyclic voltammetry electrodeposition method. Owing to the doping of N, the electronic arrangement of the original carbon structure is improved, which results in N-doped anodes exhibiting higher electrochemical performance than non-N-doped materials.47,48 Thus, Co- and N-doping could further significantly reduce the Rct value of bare CF (Fig. 1b), resulting in the high electrochemical activity of the Co–N@CF anode. Therefore, Co–N@CF was chosen as the anode for PMS activation in the following experiments.
 |
| | Fig. 1 (a) EIS tests and fitting curves and (b) the Rct values of different fabricated anodes. | |
The surface of bare CF can be observed to be clean and smooth (Fig. 2(a–c)), while the Co–N@CF is covered by a thin film (Fig. 2(d–f)), indicating that Co and N were deposited on the surface of the CF. Furthermore, the morphology of the Co–N@CF remained almost unchanged after four consecutive usages of the anode (Fig. 2(g–i)), indicating its favorable stability.
 |
| | Fig. 2 SEM images of (a–c) bare CF, (d–f) Co–N@CF and (g–i) Co–N@CF after four consecutive uses. | |
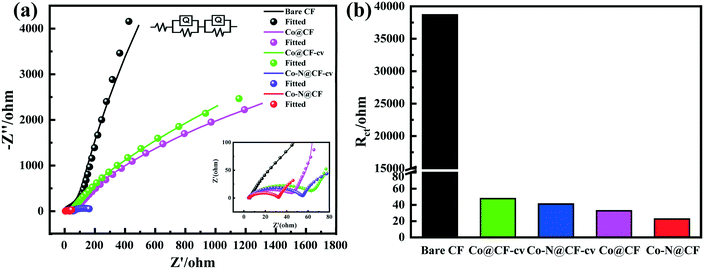
The high-resolution XPS C 1s and O 1s spectra of bare CF are shown in Fig. S3,† where it can be seen that the C 1s spectrum can be well fitted to three peaks at 284.80 eV, 285.8 eV, 288.33 eV, corresponding to C–C, C–O and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively (Fig. S3a†) and in the high-resolution O 1s spectrum peaks are located at 531.19 eV and 532.60 eV, which correspond to C–O and CO (Fig. S3b†). Fig. 3a shows the XPS survey spectrum of the Co- and N-doped material compared with that of the bare CF (Fig. 3a), demonstrating that Co and N were doped on the CF surface successfully, and the elemental contents of Co and N were 7.33% and 15.76% in Co–N@CF, respectively (Table S1†). The coexistence of Co and N makes the activation of PMS more efficient via the promotion of electron transfer.49Fig. 3b shows the Co 2p high-resolution spectrum, where the main peaks of Co 2p are located at 780.90 and 796.10 eV, corresponding to Co 2p3/2 and Co 2p1/2.50 The fitting peaks of Co 2p3/2 were deconvoluted at 778.31, 780.12 and 783.17 eV, which can be attributed to Co, Co3+ and Co2+, respectively. In addition, there is a satellite peak located at 786.03 eV in high-resolution Co 2p spectrum, which can be ascribed to the shakeup excitation of high-spin Co2+. The high-resolution C 1s spectrum could be well fitted well into three different peaks at 284.80, 285.64, and 287.85 eV, corresponding to C–C, C–O–C and O–CO, respectively (Fig. 3c). Fig. 3d shows the high-resolution N 1s spectrum, which is split into four peaks of nitrogen at 398.40, 399.07, 401.10 and 404.81 eV, corresponding to pyridinic N, pyrrolic N, graphite N and oxidized N, respectively.51 Generally, pyridinic N (accounting for 7.94% of nitrogen) and pyrrolic N (accounting for 25.37% of nitrogen) with lone-pair electrons for metal coordination combine with cobalt.52 In the high-resolution spectrum of O 1s, the three peaks located at 529.87, 531.04 and 532.98 eV correspond to Co–O, C–O, CO, respectively (Fig. 3e). The XRD analysis shows a clear diffraction peak at 26.4° in the diffraction patterns of CF and Co–N@CF, which is a typical peak of graphitic carbon.53 The XRD peaks of Co–N@CF located at 26.4°, 36.5°,42.4°,61.5°,73.6° and 77.5° can be ascribed to the (002) planes of carbon, and the (111), (200), (220), (311) and (222) planes of CoO, respectively (Fig. 3f). These results indicate that CoO was electrodeposited on N-doped CF successfully, consistent with the XPS analysis results.
O, respectively (Fig. S3a†) and in the high-resolution O 1s spectrum peaks are located at 531.19 eV and 532.60 eV, which correspond to C–O and CO (Fig. S3b†). Fig. 3a shows the XPS survey spectrum of the Co- and N-doped material compared with that of the bare CF (Fig. 3a), demonstrating that Co and N were doped on the CF surface successfully, and the elemental contents of Co and N were 7.33% and 15.76% in Co–N@CF, respectively (Table S1†). The coexistence of Co and N makes the activation of PMS more efficient via the promotion of electron transfer.49Fig. 3b shows the Co 2p high-resolution spectrum, where the main peaks of Co 2p are located at 780.90 and 796.10 eV, corresponding to Co 2p3/2 and Co 2p1/2.50 The fitting peaks of Co 2p3/2 were deconvoluted at 778.31, 780.12 and 783.17 eV, which can be attributed to Co, Co3+ and Co2+, respectively. In addition, there is a satellite peak located at 786.03 eV in high-resolution Co 2p spectrum, which can be ascribed to the shakeup excitation of high-spin Co2+. The high-resolution C 1s spectrum could be well fitted well into three different peaks at 284.80, 285.64, and 287.85 eV, corresponding to C–C, C–O–C and O–CO, respectively (Fig. 3c). Fig. 3d shows the high-resolution N 1s spectrum, which is split into four peaks of nitrogen at 398.40, 399.07, 401.10 and 404.81 eV, corresponding to pyridinic N, pyrrolic N, graphite N and oxidized N, respectively.51 Generally, pyridinic N (accounting for 7.94% of nitrogen) and pyrrolic N (accounting for 25.37% of nitrogen) with lone-pair electrons for metal coordination combine with cobalt.52 In the high-resolution spectrum of O 1s, the three peaks located at 529.87, 531.04 and 532.98 eV correspond to Co–O, C–O, CO, respectively (Fig. 3e). The XRD analysis shows a clear diffraction peak at 26.4° in the diffraction patterns of CF and Co–N@CF, which is a typical peak of graphitic carbon.53 The XRD peaks of Co–N@CF located at 26.4°, 36.5°,42.4°,61.5°,73.6° and 77.5° can be ascribed to the (002) planes of carbon, and the (111), (200), (220), (311) and (222) planes of CoO, respectively (Fig. 3f). These results indicate that CoO was electrodeposited on N-doped CF successfully, consistent with the XPS analysis results.
 |
| | Fig. 3 XPS spectra of bare CF and Co–N@CF. (a) Survey spectra of bare CF and Co–N@CF; (b)–(e) XPS spectra of Co–N@CF, for (b) Co 2p, (c) C 1s, (d) N 1s, and (e) O 1s; (f) XRD patterns of bare CF and Co–N@CF. | |
3.2 Anode performance in the TC degradation process
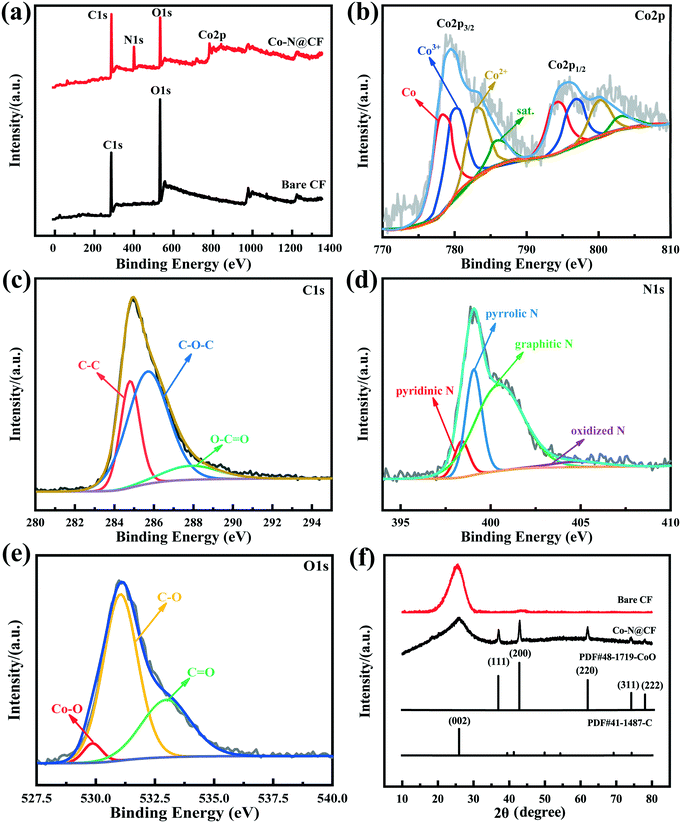
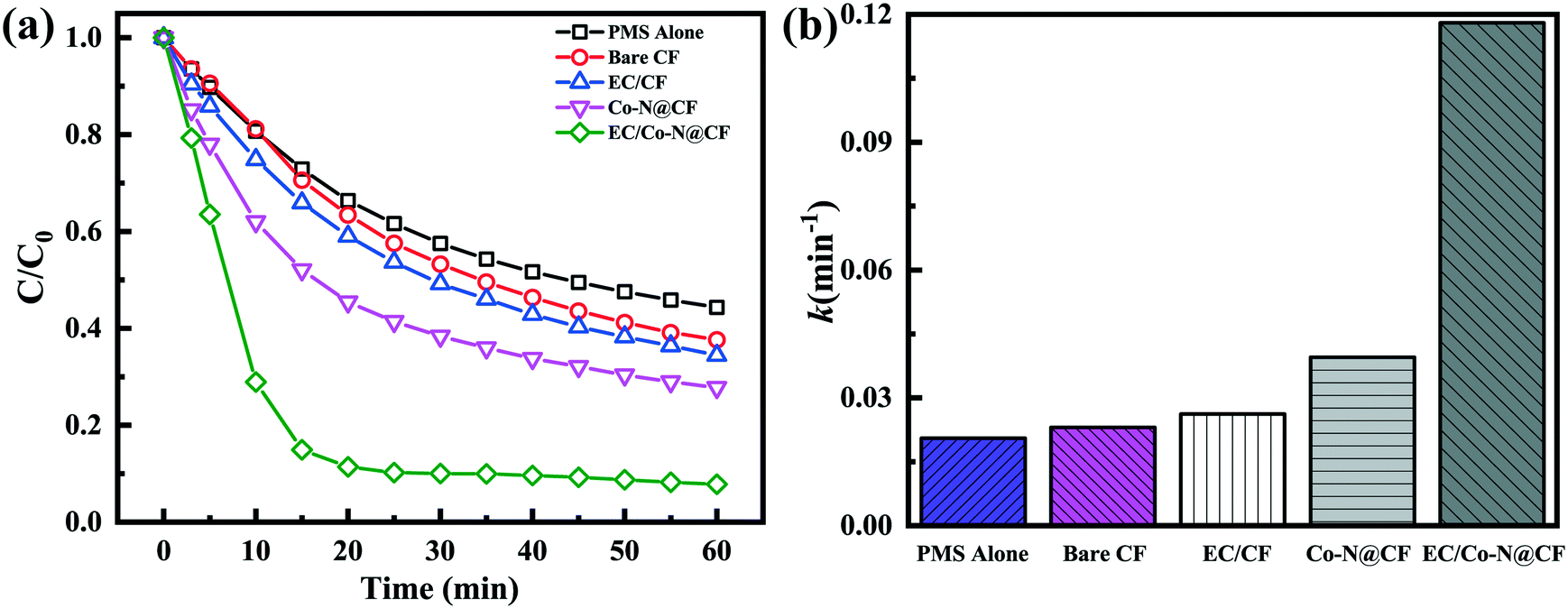
As shown in Fig. 4a, the degradation efficiency of TC reached 55.6% within 1 h owing to the self-decomposition of PMS and generated strong oxidized radicals to decompose TC. In the presence of CF, the TC removal efficiency increased to 62.3% which was attributed to the additional adsorption of CF. While, in the presence of PMS activator of Co–N@CF, the TC removal efficiency was elevated to 72.2%, indicating the enhanced ability toward PMS activation of CF modified by N and Co.
 |
| | Fig. 4 (a) Degradation efficiency of TC and (b) the corresponding reaction rate constants of the different reaction systems. | |
To further investigate the characteristics of the electrochemical activation of PMS, the CF and Co–N@CF anodes were introduced. Using the Co–N@CF anode as the PMS activator (EC/Co–N@CF), the TC removal efficiency was enhanced significantly to 92.2% and the reaction kinetics rate constant (k) was 4.5 times higher than that of the CF anode (EC/CF) (Fig. 4b), which showed that the modification of CF by N and Co contributed towards improving the activation performance of the CF anode. In the EC/Co–N@CF process, radicals such as SO4˙− and active oxygen radicals (OH˙ and 1O2) were generated (Fig. S4†).
3.3 Influencing factors of the degradation of TC and the recyclability of the Co–N@CF anode
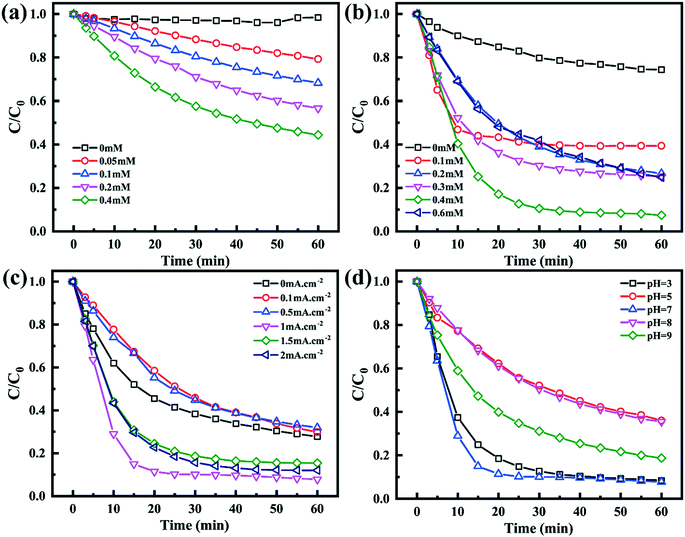
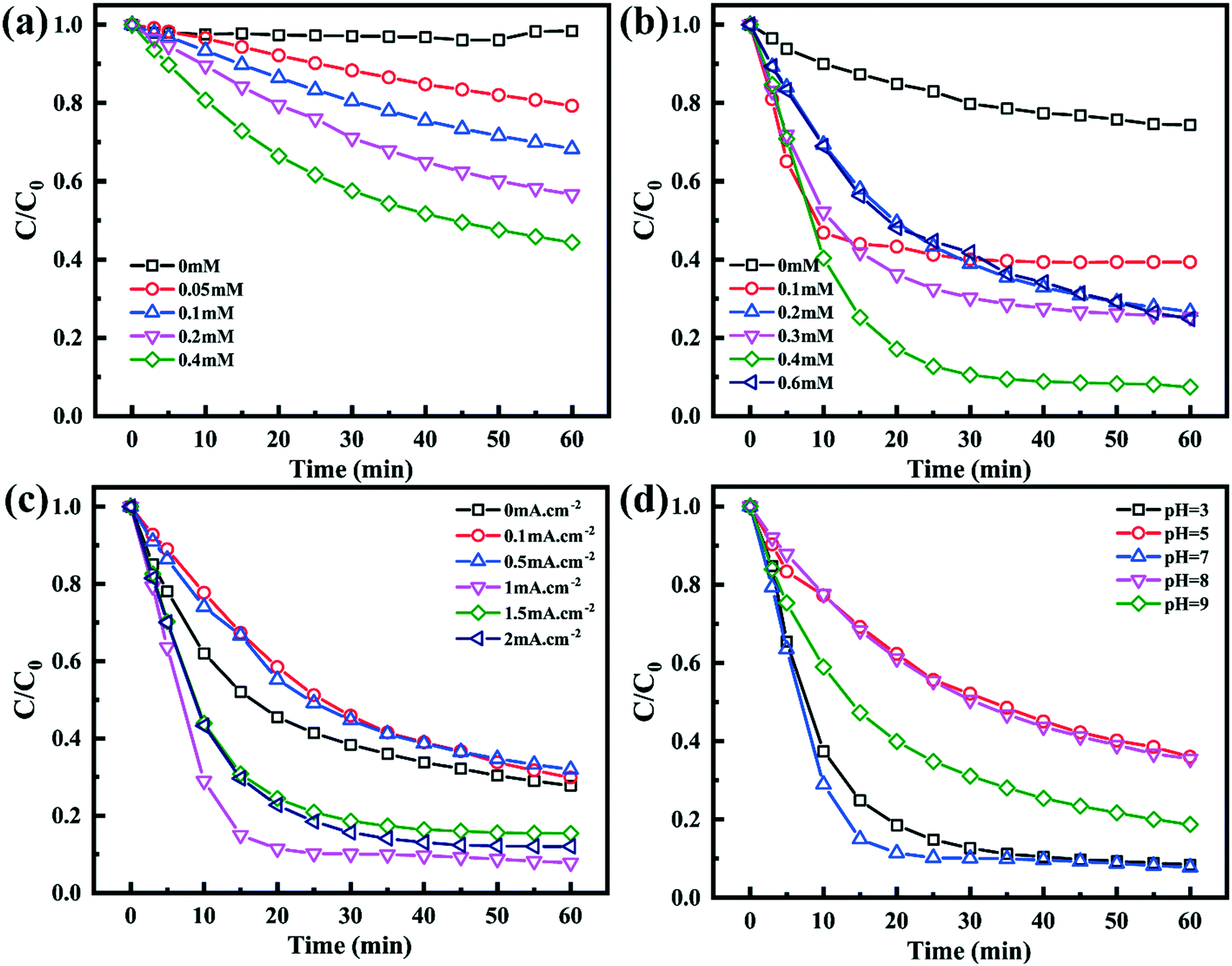
As shown in Fig. 5a, the TC degradation efficiencies increased with an increase in the content of PMS and were further enhanced when PMS was activated by the Co–N@CF anode. For example, the degradation efficiency of TC reached 92.2% in a reaction system containing 0.4 mM of PMS activated by the Co–N@CF anode (Fig. 5b), which was 36.6% higher than that in the absence of the Co–N@CF anode. Excess PMS resulted in poor TC removal efficiency, which was also noted (Fig. 5b). This was attributed to the too high amount of PMS consuming SO4˙− to generate lower activity radicals (SO5˙−).54 The reaction stoichiometric efficiency (RSE) of EC/Co–N@CF was 32% according to eqn (2):| | 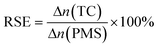 | (2) |
where Δn(TC) and Δn(PMS) represent the change in the number of moles of TC and PMS, respectively.55,56
 |
| | Fig. 5 Degradation efficiency of TC in a reaction system containing different concentrations of PMS in the (a) absence or (b) presence of the Co–N@CF anode and the degradation efficiency of TC influenced by (c) different anode current densities and (d) pH. | |
The maximum degradation efficiency of TC was observed when the anode current density was 1 mA cm−2, but a further increase in the anode current density resulted in a decrease in the degradation efficiency (Fig. 5c). There are two possible reasons that might explain this phenomenon, one being that a high current density may cause hydrolysis, and the generated H2 bubbles attach to the surface of the Co–N@CF anode, inhibiting the reactant mass transfer between the anode surface and bulk solution. The other is that it is likely that the produced radicals such as SO4˙− or OH˙ are quenched by the anode upon an increase in the applied current density.57
Aside from the PMS concentration and current density, pH also plays an important role in TC removal. The results showed that the appropriate pH value for the TC degradation was 7.0, implying that there was no need to add additional reagents to adjust the pH (Fig. 5d), which also indicates the practical application prospects of this system for use in real wastewater treatment. Under acidic conditions at low pH, excess H+ consumes SO4˙− and OH˙ according to eqn (3) and (4).58 While, under alkaline conditions, TC is negatively charged, resulting in electrostatic repulsion and worse adsorption toward radicals.46 The influence of the initial concentration of TC on the pollutant removal efficiency was also investigated, and the results showed that too high a TC concentration may generate more intermediates to consume free radicals, resulting in poor removal efficiency (Fig. S5†).
| | | SO4˙− + H+ + e− → HSO4˙− | (4) |
The TC removal efficiency remained almost constant after four consecutive usages (Fig. S6a†) and the kinetic reaction constant also remained at around 0.05 min−1 (Fig. S6b†), indicating that the Co–N@CF anode exhibits desirable stability and recyclability. The electricity energy demand per order (EE/O) was calculated using eqn (5) (an illustration of this equation is shown in Text S1†). The EE/O value in the reaction system containing PMS activated by the Co–N@CF anode was only 0.072 kWh m−3 (Table S2†), which was 9.56 and 3.36 times lower than those of the reaction system equipped with the Co–N@CF anode in the absence of PMS and reaction system containing PMS activated by the CF anode, respectively. While, in other research, such as that on an EC/ACF/PMS process, the EE/O was 1.89 kWh m−3,45 which was 25.25 times higher than the value in our current work (EC/Co–N@CF/PMS). This result shows that the Co–N@CF anode is an energy-saving catalyst, which is beneficial for its practical application.
| | 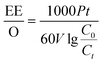 | (5) |
3.4 Kinetic characteristics of the calculation of the activation energy
Compared with those in the reaction systems containing PMS activated by Co–N@CF, the TC removal efficiency and reaction rate constant (k) were significantly improved at different temperatures (20, 30 and 40 °C) in the reaction systems containing PMS activated by the Co–N@CF anode (Fig. S7† and 6a and b), indicating that PMS can be effectively activated by the Co–N@CF anode and that consequently the pollutant removal efficiency was enhanced.
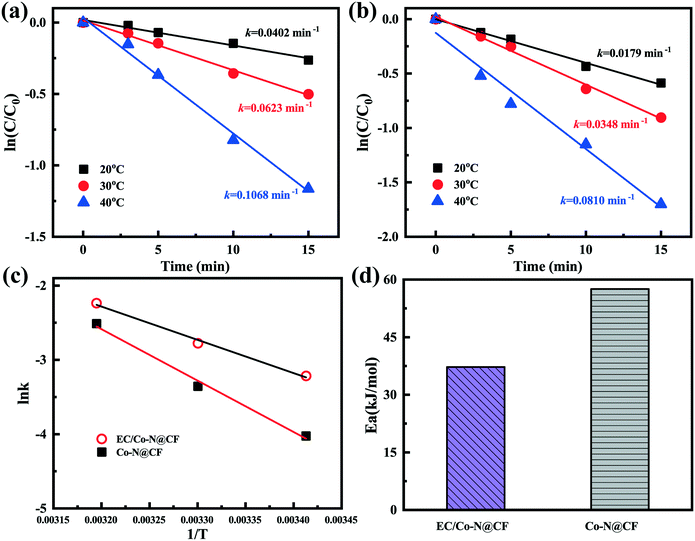 |
| | Fig. 6 Linear fitted curve of the degradation results at various temperatures for the reaction systems containing PMS activated by the (a) Co–N@CF anode and (b) Co–N@CF, (c) the relationship between lnk and 1/T, and (d) the calculated energy demanded during PMS activation by the Co–N@CF anode and Co–N@CF. | |
The activation energies calculated using eqn (6) (detailed information about eqn (6) is shown in Text S2†) for the reaction systems containing PMS activated by the Co–N@CF anode and Co–N@CF were 37.23 kJ mol−1 and 57.53 kJ mol−1 (Fig. 6d), respectively. This indicates the energy demanded during the PMS activation process by the Co–N@CF anode was 20.3 kJ mol−1 lower than that compared with in the presence of Co–N@CF, resulting in an increased reaction rate. The activation energy of the Co–N@CF anode is much lower than those of other cobalt-based carbon-support catalysts.59,60 With the synergistic effect of Co and N modification and being electricity driven, the PMS activation becomes more kinetically favorable.
| | 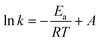 | (6) |
3.5 The proposed mechanism of TC degradation
To verify the main reactive radicals generated during the process of PMS activation by the Co–N@CF anode, TBA, p-BQ and L-histidine were selected as quenchers for OH˙, O2˙− and 1O2,47,51 respectively, while Na2S2O3 was chosen as a scavenger for the radicals OH˙ and SO4˙−. The reaction rate constants in the presence of the radical quenchers are shown in Table S3.†Fig. 7a shows that the TC removal efficiency was inhibited and decreased to 40.3% in the presence of 100 mM of TBA, and that k was also significantly decreased to 0.01533 min−1 (Table S4†), which indicated the existence of OH˙. Since L-histidine quenches both OH˙ and 1O2,61 the results showed a decreased removal efficiency of TC with increasing L-histidine concentration (Fig. 7b), indicating that both OH˙ and 1O2 were the main generated radicals. As shown in Fig. 7c, the TC removal efficiency decreased significantly from 92.2% to 33.5% in the reaction system spiked with 1 mM of p-BQ (O2˙− quencher), suggesting that O2˙− may play an important role in TC degradation. The degradation efficiency of TC was inhibited significantly and decreased to only 15.25% in the reaction system spiked with 1 mM of Na2S2O3 (Fig. 7d), which was caused by the presence of Na2S2O3, a scavenger for OH˙ and SO4˙−, showing that these are the active radicals (Fig. 7d). Based on the aforementioned information, radicals including OH˙ and SO4˙− as well as O2˙− were generated in PMS activation and contributed towards the degradation of TC.
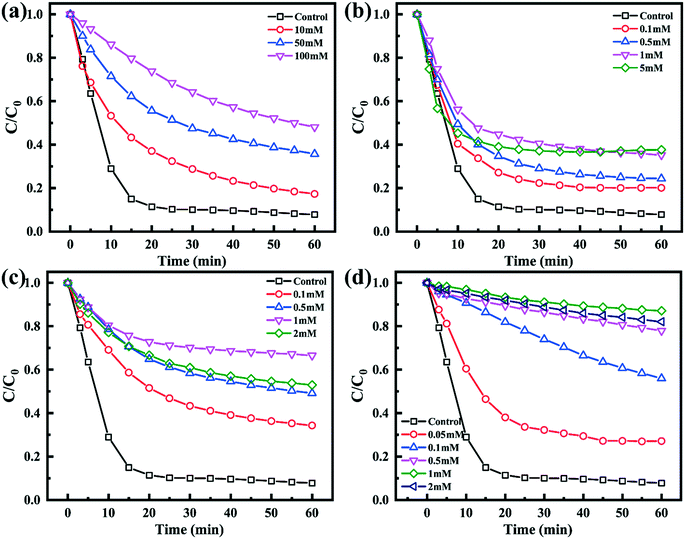 |
| | Fig. 7 (a) Effects of TBA, (b) L-histidine, (c) p-BQ and (d) Na2S2O3 as the different quenching reagents on the pollutant removal efficiency. | |
EPR tests were further conducted to investigate the generated radicals and their transformation under different conditions. In Fig. 8a, a characteristic 4-line spectrum with an intensity ratio of 1:2:2:1 can be observed, which is typical of a DMPO–OH˙ adduct (hyperfine splitting constants of αH = αN = 14.7 G),62 indicating the presence of OH˙ in the reaction system.63 Besides this, a typical DMPO–SO4˙− (hyperfine splitting constants of αN = 13.2 G, αH = 9.6, 1.48, 0.78 G) signal was also detected,64 which is adjacent to the EPR peaks for OH˙. Meantime, a signal for TMP–1O2 with a typical 3-line spectrum in an intensity ratio of 1:1:1 can also be observed in Fig. 8b. This demonstrates that OH˙, SO4˙− and 1O2 were generated via the PMS activation process. While, the DMPO–O2˙− adduct was not observed in the EPR tests, which could be ascribed to its kinetic instability and thus ready transformation to a DMPO–OH˙ adduct.65 Furthermore, the time course of the EPR tests showed that the peak intensities of the generated radicals (OH˙, SO4˙− and 1O2) decreased with increasing reaction time (Fig. 8).
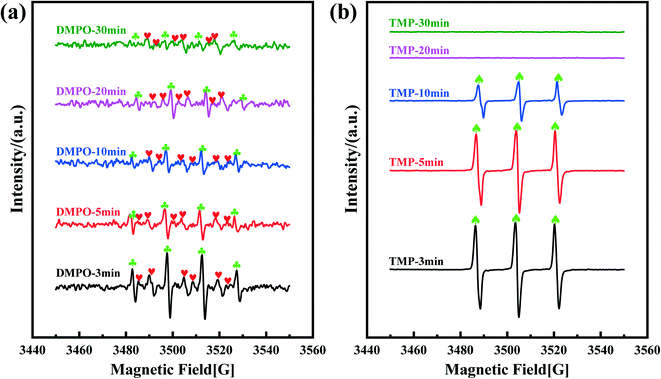 |
| | Fig. 8 EPR spectra of DMPO–OH˙ ( ), DMPO–SO4˙− ( ), DMPO–SO4˙− ( ) (a) and TMP–1O2 (♠) (b) obtained from the reaction system containing PMS activated by the Co–N@CF anode at different reaction times. Reaction conditions: T = 298 K; PMS = 0.4 mM; current density = 1 mA cm−2; pH = 7; Na2SO4 = 50 mM. ) (a) and TMP–1O2 (♠) (b) obtained from the reaction system containing PMS activated by the Co–N@CF anode at different reaction times. Reaction conditions: T = 298 K; PMS = 0.4 mM; current density = 1 mA cm−2; pH = 7; Na2SO4 = 50 mM. | |
As previously reported, PMS can self-decompose to produce SO4˙−, OH˙, O2˙− and 1O2 according to eqn (7)–(10).66 As shown in Fig. 9a and g, when only PMS was spiked in the reaction system, signals for DMPO–SO4˙−, DMPO–OH˙ and TMP–1O2 were observed in the EPR spectrum, confirming that the radicals (SO4˙−, OH˙, 1O2) were generated during the PMS self-decomposition process. While, in the presence of Co–N@CF, the EPR peak intensity of DMPO–OH˙ (Fig. 9b) and TMP–1O2 (Fig. 9h) were enhanced, indicating that Co–N@CF efficiently activates PMS (eqn (11)). When PMS was activated by the Co–N@CF anode, the EPR signal intensity of DMPO–OH˙ and DMPO–SO4˙− remained almost constant (Fig. 9c), while the signal intensity of the TMP–1O2 adduct decreased significantly (Fig. 9i) compared with that activated by Co–N@CF (Fig. 9b and h). Thus, it was proposed that 1O2 generation was inhibited during the process of PMS activation by the Co–N@CF anode (Fig. 10). However, the degradation efficiency and rate were significantly improved in the reaction system containing the Co–N@CF anode, thus, we speculated that SO4˙− and OH˙ but not 1O2 were the predominant radicals that contribute towards pollutant degradation.
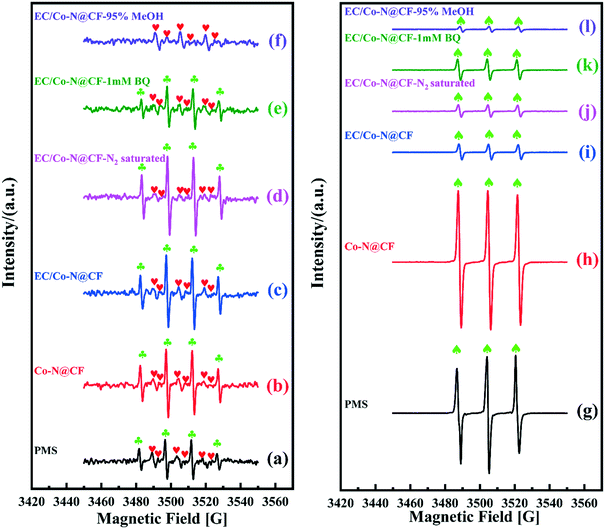 |
| | Fig. 9 EPR spectra of DMPO–OH˙ ( ), DMPO–SO4˙− ( ), DMPO–SO4˙− ( ) (a–f) and TMP–1O2 (♠) (g–l) obtained from the reaction system containing PMS with self-decomposition or activated by the Co–N@CF anode and Co–N@CF under different reaction conditions. ) (a–f) and TMP–1O2 (♠) (g–l) obtained from the reaction system containing PMS with self-decomposition or activated by the Co–N@CF anode and Co–N@CF under different reaction conditions. | |
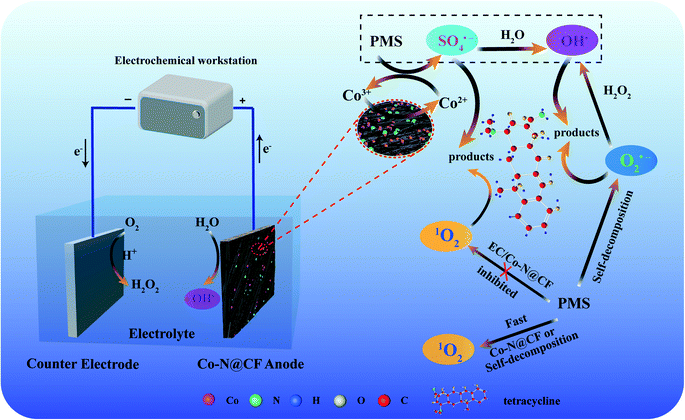 |
| | Fig. 10 Schematic diagram of PMS activation by the Co–N@CF anode for pollutant removal. | |
To determine the transformation pathways of O2˙− and OH˙, the following EPR experiments were conducted. In the reaction system of EC/Co–N@CF with the addition of 1 mM of BQ (O2˙− quencher), the EPR peak intensity of the DMPO–OH˙ adduct was decreased (Fig. 9e), while the TMP–1O2 peak intensity was almost unchanged (Fig. 9k) compared with those in the absence of BQ (Fig. 9c and i). These results illustrate that the transformation of O2˙− to OH˙ (eqn (12)) was inhibited because of the shortage of O2˙− in the presence of BQ (O2˙− quencher) and that the presence of O2˙− makes no contribution towards 1O2 generation (eqn (13)). Furthermore, the self- or activator-driven decomposition of PMS could be considered as the main pathways to produce OH˙ (eqn (7) and (14)), which was further proved by the EPR tests in reaction systems spiked with MeOH, and the results showed that the EPR peak of the DMPO–OH˙ adduct was almost not detectable (Fig. 9f). This is as a result of the shortage of SO4˙− in the presence of MeOH (the SO4˙− quencher),67 confirming that OH˙ should be derived from the hydrolysis of SO4˙− in aqueous solution (eqn (14)). Besides this, a negligible amount of OH˙ and H2O2 may be generated on the surfaces of the Co–N@CF anode and counter electrode according to eqn (15) and (16), respectively.45,47 As previous reported, O2˙− is mainly generated by PMS self-decomposition (eqn (8)–(10)) or O2 reduction (eqn (17)).68
Meanwhile, in the reaction system of EC/Co–N@CF saturated by N2, the EPR peak intensity of the DMPO–OH˙ (Fig. 9d) and TMP–1O2 adduct (Fig. 9j) remained almost unchanged, considering the absence of O2 in the reaction system saturated by N2, with the results showing that the transformation of the oxygen-containing radicals through the pathways for O2 reduction to generate O2˙− and 1O2 (eqn (17) and (18)) could be neglectable (Fig. 10). Therefore, we can conclude that O2˙− was mainly generated by PMS self-decomposition.
| | | HSO5− + SO52− → HSO4− + SO4˙− + 1O2 | (7) |
| | | HSO5− + H2O → HSO4− + H2O2 | (8) |
| | | H2O2 + OH˙ → HO2˙ + H2O | (9) |
| |  | (11) |
| | | O2˙− + H2O2 → OH˙ + OH− + O2 | (12) |
| | | SO4˙− + H2O → OH˙ + SO42− + H+ | (14) |
| | | O2 + 2H+ + 2e− → H2O2 | (15) |
| | 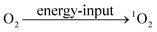 | (18) |
4. Conclusion
Herein, a Co–N@CF anode was fabricated via a simple electrodeposition method and further applied to efficiently activate PMS to degrade TC. Under optimal conditions, the TC degradation efficiency reached 92.2% within 1 h using the Co–N@CF anode as the PMS activator. N and Co modification of the CF via the galvanostatic electrodeposition method significantly decreased the Rct value of CF, which contributed towards the high PMS activation performance of the fabricated Co–N@CF anode. Quenching experiments and EPR tests demonstrated that OH˙, O2˙− and SO4˙− were the main contributors for pollutant degradation. More importantly, we found that the generation of 1O2 was significantly inhibited during the PMS activation process by the Co–N@CF anode. The facile prepared Co–N@CF anode with good PMS activation performance exhibits practical application prospects due to low cost and low energy consumption, good reusability and no need for pH adjustment (neutral reaction conditions). Our current study offers a strategy for the electroactivation of PMS, which could be applied in SO4˙−-based advanced oxidation processes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the Strategic Research and Development Program of Shaanxi Province (2019ZDLSF06-02) and the Special Project for Serving Local S&T Development of Education Department, Shaanxi Province (21JC022).
References
- B. Chen, M. Wang, M. Duan, X. Ma, J. Hong, F. Xie, R. Zhang and X. Li, In search of key: Protecting human health and the ecosystem from water pollution in China, J. Cleaner Prod., 2019, 228, 101–111 CrossRef CAS.
- L. Chen, F. Caro, C. J. Corbett and X. Ding, Estimating the environmental and economic impacts of widespread adoption of potential technology solutions to reduce water use and pollution: Application to China's textile industry, Environ. Impact Assess. Rev., 2019, 79, 106293 CrossRef.
- R. Hu, Pollution control and remediation of rural water resource based on urbanization perspective, Environ. Technol. Innovation, 2020, 20, 101136 CrossRef CAS.
- G. Chen, Y. Yu, L. Liang, X. Duan, R. Li, X. Lu, B. Yan, N. Li and S. Wang, Remediation of antibiotic wastewater by coupled photocatalytic and persulfate oxidation system: A critical review, J. Hazard. Mater., 2021, 408, 124461 CrossRef CAS.
- A. A. Elezz, A. Easa, F. Atia and T. Ahmed, The potential impact data of Tylosin and Enrofloxacin veterinary antibiotics on germination and accumulation in barley seed as a forage crop and good dietary sources using LC/MS-MS, Data Brief, 2019, 25, 104326 CrossRef.
- R. Vadakke Pariyarath, Y. Inagaki and Y. Sakakibara, Phycoremediation of tetracycline via bio-Fenton process using diatoms, J. Water Process. Eng., 2021, 40, 101851 CrossRef.
- C. Wang, C. Lin and G. Liao, Degradation of antibiotic tetracycline by ultrafine-bubble ozonation process, J. Water Process. Eng., 2020, 37, 101463 CrossRef.
- J. J. López-Peñalver, M. Sánchez-Polo, C. V. Gómez-Pacheco and J. Rivera-Utrilla, Photodegradation of tetracyclines in aqueous solution by using UV and UV/H2O2 oxidation processes, J. Chem. Technol. Biotechnol., 2010, 85, 1325–1333 CrossRef.
- Y. Wang, H. Zhang, J. Zhang, C. Lu, Q. Huang, J. Wu and F. Liu, Degradation of tetracycline in aqueous media by ozonation in an internal loop-lift reactor, J. Hazard. Mater., 2011, 192, 35–43 CAS.
- T. Saitoh, K. Shibata, K. Fujimori and Y. Ohtani, Rapid removal of tetracycline antibiotics from water by coagulation-flotation of sodium dodecyl sulfate and poly(allylamine hydrochloride) in the presence of Al(III) ions, Sep. Purif. Technol., 2017, 187, 76–83 CrossRef CAS.
- X. Zhang, Y. Li, M. Wu, Y. Pang, Z. Hao, M. Hu, R. Qiu and Z. Chen, Enhanced adsorption of tetracycline by an iron and manganese oxides loaded biochar: Kinetics, mechanism and column adsorption, Bioresour. Technol., 2021, 320, 124264 CrossRef CAS.
- J. Huang, Z. Li, J. Zhang, Y. Zhang, Y. Ge and X. Cui, In-situ synchronous carbonation and self-activation of biochar/geopolymer composite membrane: Enhanced catalyst for oxidative degradation of tetracycline in water, Chem. Eng. J., 2020, 397, 125528 CrossRef CAS.
- H. Song, L. Yan, Y. Wang, J. Jiang, J. Ma, C. Li, G. Wang, J. Gu and P. Liu, Electrochemically activated PMS and PDS: Radical oxidation versus nonradical oxidation, Chem. Eng. J., 2020, 391, 123560 CrossRef CAS.
- N. A. Al-Dhabi, G. A. Esmail and M. Valan Arasu, Effective degradation of tetracycline by manganese peroxidase producing Bacillus velezensis strain Al-Dhabi 140 from Saudi Arabia using fibrous-bed reactor, Chemosphere, 2021, 268, 128726 CrossRef CAS PubMed.
- X. Zheng, W. Fu, F. Kang, H. Peng and J. Wen, Enhanced photo-Fenton degradation of tetracycline using TiO2-coated α-Fe2O3 core–shell heterojunction, J. Ind. Eng. Chem., 2018, 68, 14–23 CrossRef CAS.
- J. Wang, D. Zhi, H. Zhou, X. He and D. Zhang, Evaluating tetracycline degradation pathway and intermediate toxicity during the electrochemical oxidation over a Ti/Ti4O7 anode, Water Res., 2018, 137, 324–334 CrossRef CAS.
- N. Nasseh, B. Barikbin and L. Taghavi, Photocatalytic degradation of tetracycline hydrochloride by FeNi3/SiO2/CuS magnetic nanocomposite under simulated solar irradiation: Efficiency, stability, kinetic and pathway study, Environ. Technol. Innovation, 2020, 20, 101035 CrossRef CAS.
- J. Li, M. Xu, G. Yao and B. Lai, Enhancement of the degradation of atrazine through CoFe2O4 activated peroxymonosulfate (PMS) process: Kinetic, degradation intermediates, and toxicity evaluation, Chem. Eng. J., 2018, 348, 1012–1024 CrossRef CAS.
- X. Pei, H. Ren, D. Xing, G. Xie, G. Cao, J. Meng, N. Ren and B. Liu, Electron transfer mechanism of peroxydisulfate activation by sewage sludge-derived biochar for enhanced degradation of sulfamethoxazole, Environ. Sci.: Water Res. Technol., 2021, 7, 1563–1575 RSC.
- Y. Qi, R. Qu, J. Liu, J. Chen, G. Al-Basher, N. Alsultan, Z. Wang and Z. Huo, Oxidation of flumequine in aqueous solution by UV-activated peroxymonosulfate: Kinetics, water matrix effects, degradation products and reaction pathways, Chemosphere, 2019, 237, 124484 CrossRef CAS PubMed.
- Y. Ji, C. Dong, D. Kong, J. Lu and Q. Zhou, Heat-activated persulfate oxidation of atrazine: Implications for remediation of groundwater contaminated by herbicides, Chem. Eng. J., 2015, 263, 45–54 CrossRef CAS.
- C. Cai, H. Zhang, X. Zhong and L. Hou, Ultrasound enhanced heterogeneous activation of peroxymonosulfate by a bimetallic Fe–Co/SBA-15 catalyst for the degradation of Orange II in water, J. Hazard. Mater., 2015, 283, 70–79 CrossRef CAS.
- A. Fernandes, P. Makoś and G. Boczkaj, Treatment of bitumen post oxidative effluents by sulfate radicals based advanced oxidation processes (S-AOPs) under alkaline pH conditions, J. Cleaner Prod., 2018, 195, 374–384 CrossRef CAS.
- P. Chen, Y. Gou, J. Ni, Y. Liang, B. Yang, F. Jia and S. Song, Efficient Ofloxacin degradation with Co(II)-doped MoS2 nano-flowers as PMS activator under visible-light irradiation, Chem. Eng. J., 2020, 401, 125978 CrossRef CAS.
- L. Niu, G. Zhang, G. Xian, Z. Ren, T. Wei, Q. Li, Y. Zhang and Z. Zou, Tetracycline degradation by persulfate activated with magnetic γ-Fe2O3/CeO2 catalyst: Performance, activation mechanism and degradation pathway, Sep. Purif. Technol., 2021, 259, 118156 CrossRef CAS.
- J. Lee, U. von Gunten and J.-H. Kim, Persulfate-Based Advanced Oxidation: Critical Assessment of Opportunities and Roadblocks, Environ. Sci. Technol., 2020, 54, 3064–3081 CrossRef CAS PubMed.
- Q. Yang, H. Choi, S. R. Al-Abed and D. D. Dionysiou, Iron–cobalt mixed oxide nanocatalysts: Heterogeneous peroxymonosulfate activation, cobalt leaching, and ferromagnetic properties for environmental applications, Appl. Catal., B, 2009, 88, 462–469 CrossRef CAS.
- F. Ren, W. Zhu, J. Zhao, H. Liu, X. Zhang, H. Zhang, H. Zhu, Y. Peng and B. Wang, Nitrogen-doped graphene oxide aerogel anchored with spinel CoFe2O4 nanoparticles for rapid degradation of tetracycline, Sep. Purif. Technol., 2020, 241, 116690 CrossRef CAS.
- L. Lai, J. Yan, J. Li and B. Lai, Co/Al2O3-EPM as peroxymonosulfate activator for sulfamethoxazole removal: Performance, biotoxicity, degradation pathways and mechanism, Chem. Eng. J., 2018, 343, 676–688 CrossRef CAS.
- B. Ramasamy, N. Pratihary, K. Sekar and T. Das, Cobalt promoted bifunctional graphene composite (Co@pGSC) for heterogeneous peroxymonosulfate activation, Chem. Eng. J., 2020, 399, 125752 CrossRef CAS.
- J. Cao, S. Sun, X. Li, Z. Yang, W. Xiong, Y. Wu, M. Jia, Y. Zhou, C. Zhou and Y. Zhang, Efficient charge transfer in aluminum-cobalt layered double hydroxide derived from Co-ZIF for enhanced catalytic
degradation of tetracycline through peroxymonosulfate activation, Chem. Eng. J., 2020, 382, 122802 CrossRef CAS.
- Q. Gao, G. Wang, Y. Chen, B. Han, K. Xia and C. Zhou, Utilizing cobalt-doped materials as heterogeneous catalysts to activate peroxymonosulfate for organic pollutant degradation: a critical review, Environ. Sci.: Water Res. Technol., 2021, 7, 1197–1211 RSC.
- Y. Guan, J. Ma, X. Li, J. Fang and L. Chen, Influence of pH on the Formation of Sulfate and Hydroxyl Radicals in the UV/Peroxymonosulfate System, Environ. Sci. Technol., 2011, 45, 9308–9314 CrossRef CAS PubMed.
- J. C. Espinosa, P. Manickam-Periyaraman, F. Bernat-Quesada, S. Sivanesan, M. Álvaro, H. García and S. Navalón, Engineering of activated carbon surface to enhance the catalytic activity of supported cobalt oxide nanoparticles in peroxymonosulfate activation, Appl. Catal., B, 2019, 249, 42–53 CrossRef CAS.
- K. Govindan, M. Raja, M. Noel and E. J. James, Degradation of pentachlorophenol by hydroxyl radicals and sulfate radicals using electrochemical activation of peroxomonosulfate, peroxodisulfate and hydrogen peroxide, J. Hazard. Mater., 2014, 272, 42–51 CrossRef CAS PubMed.
- C. A. Martínez-Huitle and M. Panizza, Electrochemical oxidation of organic pollutants for wastewater treatment, Curr. Opin. Electrochem., 2018, 11, 62–71 CrossRef.
- L. Bu, S. Zhou, Z. Shi, L. Deng and N. Gao, Removal of 2-MIB and geosmin by electrogenerated persulfate: Performance, mechanism and pathways, Chemosphere, 2017, 168, 1309–1316 CrossRef CAS.
- W. Wang, M. Chen, D. Wang, M. Yan and Z. Liu, Different activation methods in sulfate radical-based oxidation for organic pollutants degradation: Catalytic mechanism and toxicity assessment of degradation intermediates, Sci. Total Environ., 2021, 772, 145522 CrossRef CAS.
- A. Shahzeydi, M. Ghiaci, L. Jameie and M. Panjepour, Immobilization of N-doped carbon porous networks containing copper nanoparticles on carbon felt fibers for catalytic applications, Appl. Surf. Sci., 2019, 485, 194–203 CrossRef CAS.
- X. Zhao, Q. An, Z. Xiao, S. Zhai and Z. Shi, Seaweed-derived multifunctional nitrogen/cobalt-codoped carbonaceous beads for relatively high-efficient peroxymonosulfate activation for organic pollutants degradation, Chem. Eng. J., 2018, 353, 746–759 CrossRef CAS.
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, Nitrogen-Doped Graphene and Its Application in Electrochemical Biosensing, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS PubMed.
- X. Li, Y. Fang, X. Lin, M. Tian, X. An, Y. Fu, R. Li, J. Jin and J. Ma, MOF derived Co3O4 nanoparticles embedded in N-doped mesoporous carbon layer/MWCNT hybrids: extraordinary bi-functional electrocatalysts for OER and ORR, J. Mater. Chem. A, 2015, 3, 17392–17402 RSC.
- M. Park, Y.-J. Jung, J. Kim, H. I. Lee and J. Cho, Synergistic Effect of Carbon Nanofiber/Nanotube Composite Catalyst on Carbon Felt Electrode for High-Performance All-Vanadium Redox Flow Battery, Nano Lett., 2013, 13, 4833–4839 CrossRef CAS.
- S. Wang, X. Zhao, T. Cochell and A. Manthiram, Nitrogen-Doped Carbon Nanotube/Graphite Felts as Advanced Electrode Materials for Vanadium Redox Flow Batteries, J. Phys. Chem. Lett., 2012, 3, 2164–2167 CrossRef CAS PubMed.
- Z. Liu, H. Ding, C. Zhao, T. Wang, P. Wang and D. D. Dionysiou, Electrochemical activation of peroxymonosulfate with ACF cathode: Kinetics, influencing factors, mechanism, and application potential, Water Res., 2019, 159, 111–121 CrossRef CAS PubMed.
- W. Xie, Y. Shi, Y. Wang, Y. Zheng, H. Liu, Q. Hu, S. Wei, H. Gu and Z. Guo, Electrospun iron/cobalt alloy nanoparticles on carbon nanofibers towards exhaustive electrocatalytic degradation of tetracycline in wastewater, Chem. Eng. J., 2021, 405, 126585 CrossRef CAS.
- J. Di, M. Zhu, R. Jamakanga, X. Gai, Y. Li and R. Yang, Electrochemical activation combined with advanced oxidation on NiCo2O4 nanoarray electrode for decomposition of Rhodamine B, J. Water Process. Eng., 2020, 37, 101386 CrossRef.
- D. Shao, C. Wang, L. Wang, X. Guo, J. Guo, S. Zhang and Y. Lu, A N-doped Cobalt@graphitized carbon material derived from ZIF-67 assisted polyvinylidene fluoride hollow fiber membrane for supercapacitors, J. Alloys Compd., 2021, 863, 158682 CrossRef CAS.
- J. Ananpattarachai, P. Kajitvichyanukul and S. Seraphin, Visible light absorption ability and photocatalytic oxidation activity of various interstitial N-doped TiO2 prepared from different nitrogen dopants, J. Hazard. Mater., 2009, 168, 253–261 CrossRef CAS PubMed.
- H. Shao, X. Zhao, Y. Wang, R. Mao, Y. Wang, M. Qiao, S. Zhao and Y. Zhu, Synergetic activation of peroxymonosulfate by Co3O4 modified g-C3N4 for enhanced degradation of diclofenac sodium under visible light irradiation, Appl. Catal., B, 2017, 218, 810–818 CrossRef CAS.
- J. Miao, W. Geng, P. J. J. Alvarez and M. Long, 2D N-Doped Porous Carbon Derived from Polydopamine-Coated Graphitic Carbon Nitride for Efficient Nonradical Activation of Peroxymonosulfate, Environ. Sci. Technol., 2020, 54, 8473–8481 CrossRef CAS.
- Q. Jia, Y. Gao, Y. Li, X. Fan, F. Zhang, G. Zhang, W. Peng and S. Wang, Cobalt nanoparticles embedded in N-doped carbon on carbon cloth as free-standing electrodes for electrochemically-assisted catalytic oxidation of phenol and overall water splitting, Carbon, 2019, 155, 287–297 CrossRef CAS.
- H. Dai, W. Zhou and W. Wang, Co/N co-doped carbonaceous polyhedron as efficient peroxymonosulfate activator for degradation of organic pollutants: Role of cobalt, Chem. Eng. J., 2021, 417, 127921 CrossRef CAS.
- Y. Tang, J. Kang, M. Wang, C. Jin, J. Liu, M. Li, S. Li and Z. Li, Catalytic degradation of oxytetracycline via FeVO4 nanorods activating PMS and the insights into the performance and mechanism, J. Environ. Chem. Eng., 2021, 9, 105864 CrossRef CAS.
- M. Amasha, A. Baalbaki and A. Ghauch, A comparative study of the common persulfate activation techniques for the complete degradation of an NSAID: The case of ketoprofen, Chem. Eng. J., 2018, 350, 395–410 CrossRef CAS.
- Y. Qi, J. Wei, R. Qu, G. Al-Basher, X. Pan, A. A. Dar, A. Shad, D. Zhou and Z. Wang, Mixed oxidation of aqueous nonylphenol and triclosan by thermally activated persulfate: Reaction kinetics and formation of co-oligomerization products, Chem. Eng. J., 2021, 403, 126396 CrossRef CAS.
- C. Wang, Y. Yu, L. Yin, J. Niu and L. Hou, Insights of ibuprofen electro-oxidation on metal-oxide-coated Ti anodes: Kinetics, energy consumption and reaction mechanisms, Chemosphere, 2016, 163, 584–591 CrossRef CAS PubMed.
- Y. Huang, Y. Huang, C. Huang and C. Chen, Efficient decolorization of azo dye Reactive Black B involving aromatic fragment degradation in buffered Co2+/PMS oxidative processes with a ppb level dosage of Co2+-catalyst, J. Hazard. Mater., 2009, 170, 1110–1118 CrossRef CAS PubMed.
- T. Huang, J. Chen, Z. Wang, X. Guo and J. C. Crittenden, Excellent performance of cobalt-impregnated activated carbon in peroxymonosulfate activation for acid orange 7 oxidation, Environ. Sci. Pollut. Res., 2017, 24, 9651–9661 CrossRef CAS PubMed.
- K.-Y. A. Lin, Y.-C. Chen and C.-F. Huang, Magnetic carbon-supported cobalt prepared from one-step carbonization of hexacyanocobaltate as an efficient and recyclable catalyst for activating Oxone, Sep. Purif. Technol., 2016, 170, 173–182 CrossRef CAS.
- S. Cheng, C. Shen, H. Zheng, F. Liu and A. Li, OCNTs encapsulating Fe-Co PBA as efficient chainmail-like electrocatalyst for enhanced heterogeneous electro-Fenton reaction, Appl. Catal., B, 2020, 269, 118785 CrossRef CAS.
- D. Wang, D. Huang, S. Wu, G. Fang, F. Zhu, N. Chen, S. Liu, C. Zhu and D. Zhou, Pyrogenic Carbon Initiated the Generation of Hydroxyl Radicals from the Oxidation of Sulfide, Environ. Sci. Technol., 2021, 55, 6001–6011 CrossRef CAS PubMed.
- K. Wang, D. Huang, W. Wang, Y. Ji and J. Niu, Enhanced perfluorooctanoic acid degradation by electrochemical activation of peroxymonosulfate in aqueous solution, Environ. Int., 2020, 137, 105562 CrossRef CAS PubMed.
- X. Pan, J. Chen, N. Wu, Y. Qi, X. Xu, J. Ge, X. Wang, C. Li, R. Qu, V. K. Sharma and Z. Wang, Degradation of aqueous 2,4,4′-Trihydroxybenzophenone by persulfate activated with nitrogen doped carbonaceous materials and the formation of dimer products, Water Res., 2018, 143, 176–187 CrossRef CAS PubMed.
- D. Zhong, Y. Jiang, Z. Zhao, L. Wang, J. Chen, S. Ren, Z. Liu, Y. Zhang, D. C. W. Tsang and J. C. Crittenden, pH Dependence of Arsenic Oxidation by Rice-Husk-Derived Biochar: Roles of Redox-Active Moieties, Environ. Sci. Technol., 2019, 53, 9034–9044 CrossRef CAS PubMed.
- H. Song, L. Yan, J. Jiang, J. Ma, Z. Zhang, J. Zhang, P. Liu and T. Yang, Electrochemical activation of persulfates at BDD anode: Radical or nonradical oxidation?, Water Res., 2018, 128, 393–401 CrossRef CAS PubMed.
- J. Yao, Y. Zhang and Z. Dong, Enhanced degradation of contaminants of emerging concern by electrochemically activated peroxymonosulfate: Performance, mechanism, and influencing factors, Chem. Eng. J., 2021, 415, 128938 CrossRef CAS.
- Y. Liu, X. Gan, B. Zhou, B. Xiong, J. Li, C. Dong, J. Bai and W. Cai, Photoelectrocatalytic degradation of tetracycline by highly effective TiO2 nanopore arrays electrode, J. Hazard. Mater., 2009, 171, 678–683 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ew00676b |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Ying
Gao
ab,
Junli
Li
ab,
Jiawu
Liu
ab,
Jianfeng
Zhang
*ab,
Ying
Gao
ab,
Junli
Li
ab,
Jiawu
Liu
ab,
Jianfeng
Zhang
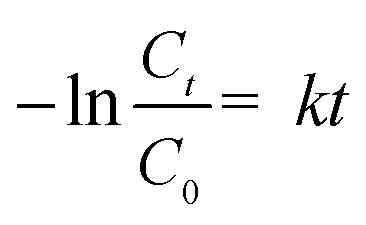



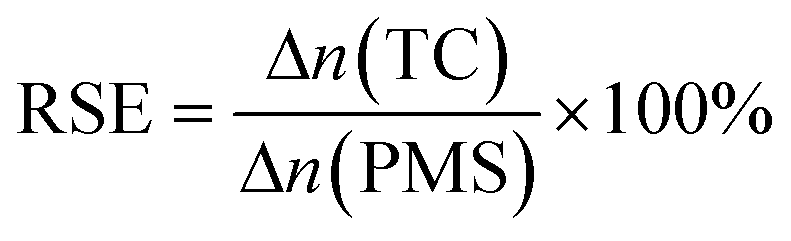





 ), DMPO–SO4˙− (
), DMPO–SO4˙− ( ) (a) and TMP–1O2 (♠) (b) obtained from the reaction system containing PMS activated by the Co–N@CF anode at different reaction times. Reaction conditions: T = 298 K; PMS = 0.4 mM; current density = 1 mA cm−2; pH = 7; Na2SO4 = 50 mM.
) (a) and TMP–1O2 (♠) (b) obtained from the reaction system containing PMS activated by the Co–N@CF anode at different reaction times. Reaction conditions: T = 298 K; PMS = 0.4 mM; current density = 1 mA cm−2; pH = 7; Na2SO4 = 50 mM.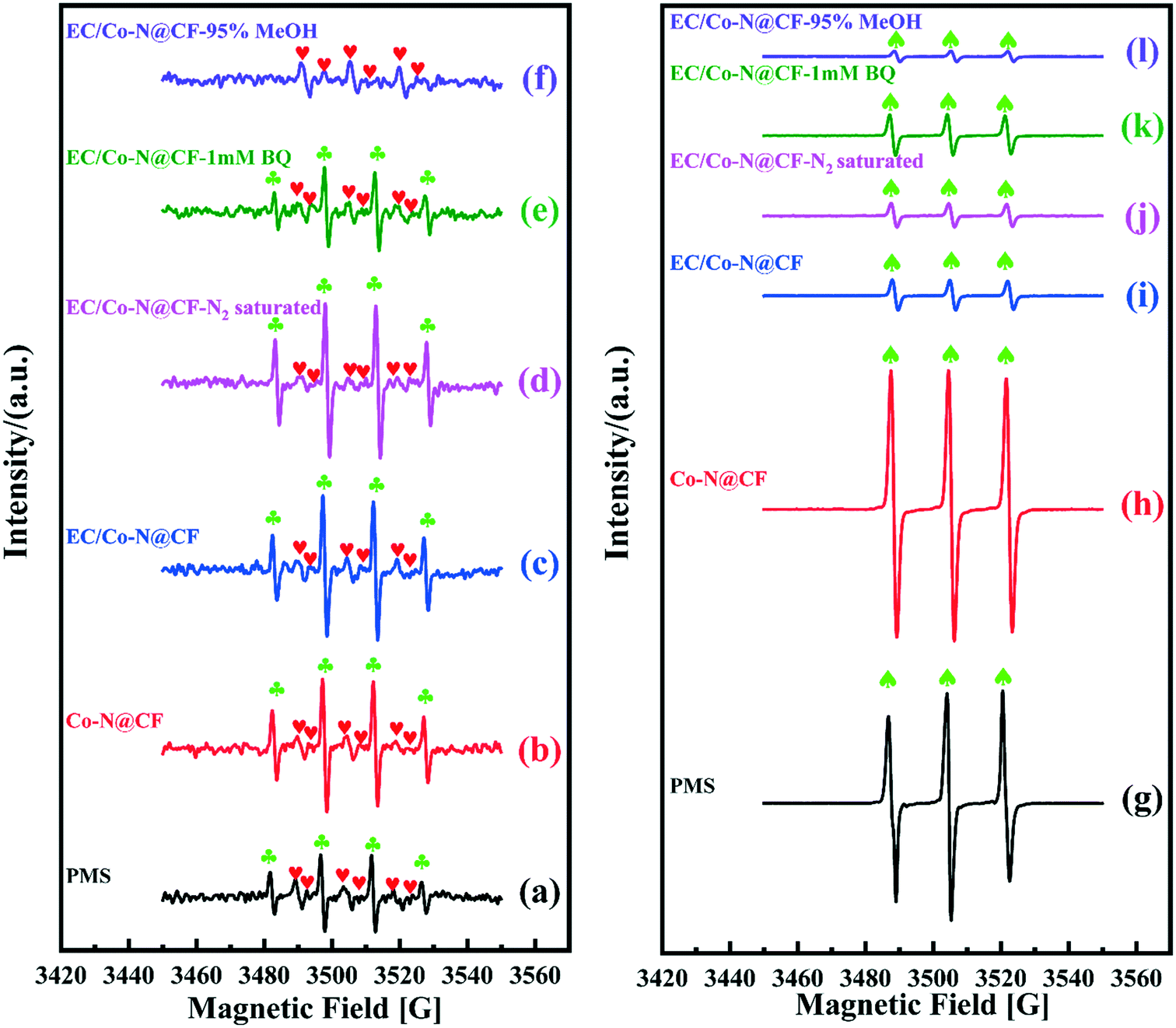
 ), DMPO–SO4˙− (
), DMPO–SO4˙− ( ) (a–f) and TMP–1O2 (♠) (g–l) obtained from the reaction system containing PMS with self-decomposition or activated by the Co–N@CF anode and Co–N@CF under different reaction conditions.
) (a–f) and TMP–1O2 (♠) (g–l) obtained from the reaction system containing PMS with self-decomposition or activated by the Co–N@CF anode and Co–N@CF under different reaction conditions.

