
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnisotropic oxidative growth of goethite-coated sand particles in column reactors during 4-chloronitrobenzene reduction by Fe(II)/goethite†
Adel
Soroush
a,
R. Lee
Penn
 b and
William A.
Arnold
*a
b and
William A.
Arnold
*a
aDepartment of Civil, Environmental, and Geo- Engineering, University of Minnesota – Twin Cities, 500 Pillsbury Drive SE, Minneapolis, Minnesota 55455, USA. E-mail: arnol032@umn.edu; Fax: +1 612 626 7750; Tel: +1 612 625 8582
bDepartment of Chemistry, University of Minnesota – Twin Cities, Smith Hall, Pleasant St SE, Minneapolis, MN 55455, USA
First published on 30th November 2021
Abstract
Reduction of nitroaromatic compounds (NACs), an important class of groundwater pollutants, by Fe(II) associated with iron oxides, a highly reactive reductant in anoxic aquifers, has been studied widely, but there are significant differences between the well-controlled, batch reactor conditions of the laboratory and the complicated conditions encountered in the field. Continuous flow column reactors containing goethite-coated sand and aqueous carbonate buffer were continuously exposed to 0.05 mM 4-chloronitrobenzene (4-ClNB) and 0.5 mM Fe(II) to emulate more realistic scenarios and to allow study of the oxidative growth of goethite particles using both saturated and unsaturated flow conditions. The experiments were designed to test how attachment to a surface affected particle growth and how particle growth affected the extent of reaction over time. After reaction, particles from different sections of each column were collected, and the goethite was detached from the sand grains for characterization using transmission electron microscopy. The amount of oxidative growth varied as a function of distance from the column inlet, with the most growth observed at the inlet end (bottom) of the column. Similar to previous work using batch reactors, newly oxidized Fe(III) was mostly added to the goethite particle tips, resulting in up to an 81% increase in length under saturated flow and a 50% increase in length under unsaturated flow after 220 pore volumes. With saturated flow, reactant concentrations and the extent of the reaction are important factors determining the extent of mineral growth. For unsaturated column conditions, however, flow path substantially impacts mineral growth in the column. Reactors sacrificed after 220 pore volumes under saturated flow conditions resulted in an overall 70% increase in goethite mass while the unsaturated flow column resulted in a 40% increase in goethite mass, more variable mineral growth as a function of distance from the inlet, and overall, 50% less 4-ClNB conversion. The results demonstrate that quantitative characterization of oxidative mineral growth of goethite nanoparticles attached to an underlying mineral is practical and elucidates the major variables impacting the reactivity of mineral nanoparticles in contaminated groundwater systems.
Environmental significanceAdvancing remediation techniques for contaminated groundwater is vital to ensure equitable access to clean water. Reactions occurring at natural mineral nanoparticle surfaces, such as iron oxides, lead to contaminant transformations, but the reactions have primarily been studied in batch reactors. Column reactors containing sand coated with goethite nanoparticles and carbonate buffer that are continuously exposed to a flow of contaminant-dosed water provide a more realistic model of field conditions. Analyzing nanoparticles after reaction in column reactors elucidates factors that control nanoparticle reactivity, including any effect of attachment to the underlying sand particles. Results advance our understanding of mineral-mediated contaminant degradation in groundwater under iron reducing conditions and improve the ability to quantify the reactivity of iron oxide mineral nanoparticles. |
Introduction
Iron oxides, oxyhydroxides, and hydroxides (hereafter referred to as iron oxides), such as goethite (α-FeOOH) and hematite (Fe2O3), occur as mineral nanoparticles, coatings of mineral nanoparticles on other minerals, or components of rocks.1–5 The iron oxides play important roles in the environment due to their roles in surface-mediated redox reactions, which affect the fate and transport of trace elements, nutrients, and groundwater contaminants.6–15 The reduction of many common groundwater contaminants (e.g., nitroaromatic compounds, NACs), which are commonly used in munitions, insecticides, herbicides, and pharmaceuticals, as well as feedstocks for industrial chemicals16–22 are well studied.23,24 The release of nitro munitions, such as 2,4,6-trinitrotoluene (TNT), and hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) and their associated by- or/and degradation products into the aquatic environment through load, assembly, and packing processes has put many munition plants under the National Priorities List for Superfund cleanup.25 The continuous regeneration of surface-bound Fe(II) with low standard reduction potential (EH0) via microbial iron-reduction11 and generation of reducing capacity by in situ redox manipulation26,27 produce conditions suitable for reducing oxidized contaminants, such as NACs, in soils, sediments and aquifers.28Reduction of NACs results in the formation of the corresponding anilines19 with concurrent oxidative mineral growth as a result of oxidation of Fe(II) to Fe(III) (eqn (1) and (2)).
| Ar–NO2 + 6H+ + 6e− → Ar–NH2 + 2H2O | (1) |
| 6Fe(aq)2+ + 12H2O → 6α-FeOOH + 18H+ + 6e− | (2) |
Studies on the reductive degradation of NACs by Fe(II) associated with iron oxide minerals in batch reactors provide valuable information that is useful for assessing contaminant reactivity and design of remediation treatment. Past results indicate the importance of the evolving mineralogy in the subsurface environment including 1) the production/consumption of reactive sites and 2) changes in the total surface area of the solid phase.29,30 Such experiments, however, fall short in terms of simulating real environmental systems, which are more complicated due to the heterogeneity of the solid materials, dissolved species, and flow conditions. Iron oxides in aquifers typically occur as components of heterogeneous materials, such as coatings of nanoscale particles on bulk materials like sand.1,4 Additionally, continuous or cyclical exposure to aqueous Fe(II) and other dissolved species is expected. Column studies enable continuous or cyclical dosing of reactants and other dissolved species and concurrent removal of the dissolved reaction products, which more closely simulate natural groundwater systems. Although a number of studies have examined the reduction of model pollutant molecules by Fe(II) associated with mineral surfaces in columns,35–37 these studies did not include detailed comparisons of the solid state materials before and after reaction. Reversible attachment of iron oxide particles on other sediment or soil constituents, like quartz sand grains, provides the opportunity to study how mineral particles change in detail. Furthermore, iron oxide coated sands have featured prominently in recent works focused on the removal or remediation of a wide range of toxic species (e.g., dissolved arsenic and heavy metals, organic pollutants),38–41 but how the solid materials change from a solid-state perspective remains underexplored.
Here, we report results from experiments in which the reductive degradation of 4-ClNB by Fe(II) on goethite coated quartz sand in continuous flow column experiments was monitored. While the experiments are still idealized as compared to real groundwater systems, the goals of this work are to develop a methodology to study particle growth under saturated and unsaturated flow conditions during reaction with a contaminant and assess how these changes affect the extent of contaminant transformation. Attachment of goethite particles onto the quartz sand was accomplished by exploiting the reversible electrostatic attraction between the goethite and quartz sand grains, which enabled collection of goethite particles for characterization before and after reactions. 4-ClNB was used as a model organic pollutant and goethite as the iron oxide mineral to facilitate comparisons to previous works.29–31 4-ClNB reductively transforms to a known and easily detected product, 4-chloroaniline (4-ClAn), allowing calculation of mass and electron balances.13,19 Goethite nanoparticles are abundant in subsurface soils and sediments and can be synthesized using routine laboratory techniques.42 Effects of different hydrological parameters, including degree of saturation of the column materials and reaction time, on the rate of reductive degradation of 4-ClNB and concurrent oxidative goethite growth were characterized, and results were compared to data obtained using repetitively dosed (multi-spike) batch reactors. Post-reaction materials were collected from different regions (zones) as a function of distance from the column inlet and detached goethite particles characterized using transmission electron microscopy (TEM). Particle size distributions for both pre- and post-reaction goethite nanoparticles were determined and compared, and nonparametric statistics were applied. Results offer a new insight into the reactivity of Fe(II) associated with goethite coated sands with NACs under conditions more comparable to groundwater systems.
Materials and methods
Ultrapure water (18.2 MΩ cm) obtained from a MilliPore Milli-Q Advantage A10 system was used for the preparation of all the solutions used in synthesis, goethite-coated sand preparation, column and batch reactors, and sample preparation for subsequent characterization. When deoxygenated solutions were required (batch reactors, column experiments, and ferrozine assay), the water was sparged with ultrapure N2 (5 h L−1) prior to adding other reagents. Solutions prepared using HCl (BDH Aristar, 12.1 M, 36–38%) and NaOH (Fisher Scientific, 19 M, 50% w/w) were used to adjust pH. NaHCO3 (Fisher Chemical, ACS reagent), Fe(NO3)3 (Fisher Chemical, ACS reagent), FeCl2·4H2O (Fisher Chemical, ACS reagent), Ottawa sand (50–60 mesh, US Silica, Berkeley Springs, West Virginia), ferrozine [3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine-p,p′-disulfonic acid monosodium salt hydrate, Sigma-Aldrich 97%], NaCl (Mallinckrodt, ACS reagent), NaBr (Fisher Scientific, ACS reagent), 4-ClNB (Acros, 99%), 4-ClAn (Acros, 98%), ammonium acetate (Sigma-Aldrich 97%), HPLC grade acetonitrile (Sigma-Aldrich), and ammonium hydroxide (Sigma-Aldrich 28%) were all used as received.Goethite synthesis
Goethite nanoparticles were synthesized using the method described by Anschutz and Penn.42 First, a suspension of six-line ferrihydrite was prepared by drop-wise addition of 0.48 M NaHCO3 into 0.4 M Fe(NO3)3 followed by microwave annealing for three 40 second intervals with shaking in between, until boiling followed by immediate plunging into an ice bath to cool to approximately 20 °C. The resulting suspension was dialyzed (Spectra/Por 7, 2 kD, 38 mm × 24 mm) for four days with three water exchanges per day. The pH of the aqueous suspension of ferrihydrite was adjusted to 12 using a 5 M NaOH solution and then aged at 90 °C for 3 days, which resulted in the formation of goethite (α-FeOOH) nanorods. The mass loading of the final aqueous suspension was measured (23.3 mg mL−1), and the suspension was stored at pH 4 and at 4 °C.Particle characterization
An aliquot of the goethite suspension was allowed to air dry, and the resulting material ground using a mortar and pestle to produce a powder, which was analyzed using a PANalytical X'Pert PRO X-ray diffractometer equipped with a cobalt source (1.7909 Å) and an X'Celerator detector. The measurement was conducted from 10–90° 2θ with an effective dwell time of 100 s per step and step size of 0.0167° 2θ.Particle sizes (length and width) were quantified from calibrated images collected using an FEI Tecnai T12 TEM operated at 120 kV. To prepare the sample, a drop of diluted particle suspension was placed onto a Cu 200 mesh holey carbon coated grid (SPI supplies) and allowed to air-dry. ImageJ software was used to manually measure the lengths and the widths of approximately 500 particles from calibrated micrographs.43 The averages and standard deviations were determined. The surface area of the particles was measured by Brunauer–Emmett–Teller (BET) analysis from N2 sorption data collected using a Quantachrome Autosorb iQ2-MP at 77 K with outgassing at 90 °C for 12 h.
Preparation of goethite-coated sand
Ottawa sand was sieved, and particles between mesh 50 and mesh 60 were collected and soaked in 0.1 M HCl solution for 24 h. The particles were then washed several times using ultrapure water to remove any impurities from the surface of the sand grains. A 3 mL aliquot of the goethite suspension (70 mg of goethite) was added to a 100 mL suspension of 50 g of the acid-washed sand in water, and the pH was adjusted to 6.8 using 1 M HCl and 1 M NaOH solutions. The mixture was rotated end-over-end (Glas-Col; 40 rpm) for 24 h in screw-cap polyethylene bottles. The mixture was allowed to settle, and the clear and colorless supernatant was discarded. The resulting yellow goethite-coated sand was washed using ultrapure water three times and allowed to air-dry. A small amount of the dried goethite-coated sand was placed on conductive carbon tape applied to an aluminum stub (Structure Probe, Inc.), and the resulting sample was sputter-coated with 5 nm of iridium (Leica EM, ACE 600) and then examined using scanning electron microscopy (SEM; Hitachi SU8230).Fe(II) sorption isotherms
Fe(II) sorption experiments were conducted in an anaerobic atmosphere using a NexGen system glovebox (Vacuum Atmosphere Company) with 100% N2 gas. The oxygen level of the environment in the glovebox was maintained below 1 ppm by reaction with copper catalysts and moisture was removed by molecular sieves. Sorption of Fe(II) to the sand and goethite-coated sand was quantified by equilibrating materials in 35 mL serum bottles. The bottles were filled with 9.0 g of either bare or goethite-coated sand and 10 mM deoxygenated sodium bicarbonate buffer solution. A range of Fe(II) concentrations, from 0.0 mM to 1.0 mM, was achieved in the bottles by spiking specific volumes of an acidified, deoxygenated 175 mM or 4.37 mM FeCl2 stock solution. The pH of the solution was then adjusted to 7.0 using 1 M HCl and 1 M NaOH. After 20 hours of equilibration via rotating end-over-end (Glas-Col; 40 rpm), samples were collected and filtered (Acrodisc 13 mm syringe filters with 0.2 μm nylon membranes) and the aqueous concentration of Fe(II) in the supernatant determined using the ferrozine assay.44 The mass sorbed was determined by difference between the initial and final aqueous concentrations. The Langmuir sorption model was used,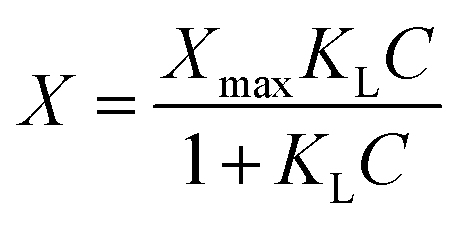 | (3) |
Batch control experiments
To test whether dissolved species produced by dissolution of the quartz sand grains could impact the kinetics of the reductive degradation of 4-ClNB by Fe(II) on goethite, and therefore comparisons between batch and column reactors, multi-spike batch experiments were performed. HCl-washed Ottawa sand (100 g) was soaked in 1 L of deoxygenated 10 mM NaHCO3 solution at pH 7 for one month. The supernatant is hereafter referred to as sand-conditioned water. Four batch reactors were prepared using 10 mM NaHCO3 buffer prepared using either the sand-conditioned or ultra-pure water. In each 35 mL serum bottle reactor, an aliquot of the acidified Fe(II) stock solution (175 mM FeCl2) was added so that the concentration of Fe(II) was 1 mM, and the reactors were allowed to equilibrate for 21 h. Then, 5 cycles of the reaction were performed, with each cycle initiated by injection of 4-ClNB stock solution so that the initial concentration was 0.1 mM. Between each cycle, the Fe(II) concentration in the aqueous phase was quantified, the Fe(II) concentration returned to 1 mM by adding the appropriate volume of Fe(II) stock solution, the pH adjusted to 7.0 using 1 M HCl and 1 M NaOH solutions, and the reactors allowed to re-equilibrate for 21 h. At the end of each cycle, 1 mL of solution was removed and filtered (Acrodisc 13 mm syringe filters with 0.2 μm nylon membranes), and the concentration of 4-ClNB was determined by high pressure liquid chromatography (HPLC) and Fe(II) was quantified using the ferrozine assay. The next cycle was started once no detectable 4-ClNB (<0.001 mM) remained. After the fifth spike, particles were collected. In the case of reactors containing only goethite, the particles were simply washed. In the case of reactors containing goethite-coated sand, the goethite particles were separated from the sand and air dried for TEM measurements using the procedure described below.Column experiments
Borosilicate glass columns (Kimble FLEX-COLUMNS®) of various lengths were fitted with polypropylene end caps and packed with bare sand or goethite-coated sand. Two flow adapters (Kimble, Rockwood, TN) were secured to both ends of the column to prevent sediment migration and leaching of the liquid phase as well as allow for adjustment of the column length. The column internal diameter and column length were optimized according to three experimental parameters. First, the column internal diameter was selected so as to avoid wall-effects45 by satisfying the condition that DC/dP > 40, where DC is the internal column diameter and dP is the sand particle diameter (here ∼250 μm). Second, the minimum column length was selected so it was possible to slice the column into three equally sized portions. To achieve these conditions, columns with different heights and diameters were assembled and the concentration of Fe(II) and 4-ClNB were measured in the effluent in preliminary experiments. The third criterion was to have reaction occur throughout the column and having both reagents present in the effluent is necessary for this to occur. A column assembled using a length of 3 cm and internal diameter of 2.5 cm satisfied the criteria. Flow rates of 0.5 or 1 mL min−1 were used. These flow rates are not realistic in terms of flow rates observed in the field, but these flow rates provide a balance between what was experimentally feasible, both in terms of experimental time frames and ensuring that reaction occurred throughout the length of the column, with simulating groundwater velocities of real environmental systems. For this initial study, we chose to emphasize experimental practicality. Although the chosen flow rate leads to velocities larger than typical groundwater velocity, a lower flow rate would have led to unreasonably small column lengths. Faster flow rates would allow use of a longer column, but those faster rates would be even more dissimilar from real groundwater flow rates. Further optimization of flow rate and column height could be performed by changing the goethite content on the sand, and that is a priority for future studies.The schematic of the column set up is presented in the ESI† (Fig. S1). High-pressure piston pumps (Chrom Tech, P-LSP10S) were used to pump solutions through the column. Because CO2 is more soluble in water than air, assembled columns were initially flushed with CO2 for 5 minutes to replace air trapped in columns during the packing46 and then saturated with a 10 mM NaCl (Mallinckrodt, ACS reagent) background solution at a flow rate of 0.5 mL min−1 outside of the glovebox.
All tracer (NaBr) breakthrough curves were fit to the solution of the one-dimensional advection–dispersion equation for a step input initial condition and semi-finite boundary conditions:47,48
 | (4) |
T is equal to the number of pore volumes (T = ντ/L), and ν is the linear velocity of fluid and is equal to Q/Aε, where A is the cross-sectional area of the column, ε is porosity, and τ is elapsed time. The Peclet number (Pe) is defined as νL/DPe where L is the column length and DPe is the hydrodynamic dispersion coefficient. R is the retardation coefficient that accounts for linear and reversible equilibrium sorption, which is equal to one for a conservative tracer. The tracer analysis was conducted before and after the reaction to determine if any changes to the hydrodynamic flow in the column occurred. A 10 mM NaCl solution was used as the electrolyte background and was run through the column for three pore volumes. Subsequently, a 10 mM NaBr (Fisher Scientific, ACS reagent) solution was pumped through the column as the conservative chemical tracer. The effluent concentration of NaBr was measured by bromide ion selective electrode (Cole-Parmer) and was plotted against the elapsed time. The pore volume was identified as the point at which C/C0 = 0.5. Finally, the hydrodynamic dispersion coefficient and porosity of each column were calculated through non-linear curve fitting by OriginLab (2016).
After tracer analysis, columns containing either sand or goethite-coated sand were conditioned using 10 mM NaHCO3 (pH 7.0) as influent for three pore volumes, followed by 1 mM Fe(II) in 10 mM NaHCO3 (pH 7.0) solution for another three pore volumes (0.5 mL min−1) leading to an effluent Fe(II) concentration of ∼70% of influent concentration. The introduction of the Fe(II) solution alone served to promote formation of reactive Fe(II)-bound species, which initiate the reaction when 0.1 mM 4-ClNB in 10 mM NaHCO3 (pH 7.0) was subsequently fed at a rate of 0.5 mL min−1 by a different pump along with the 1 mM Fe(II) solution for a total flow rate of 1 mL min−1. Samples were collected at the end of the column every 9 min using fraction collectors (BIORAD 2110), and the reaction was quenched and stabilized by filtering samples (Acrodisc 13 mm syringe filters with 0.2 μm nylon membranes) and adding 50 μL 1 M of HCl solution to each sample to lower the pH < 4 to prevent any precipitation of iron materials.49 The aqueous Fe(II) and 4-ClNB concentrations in the reservoir and at the column effluent were measured using the ferrozine assay and HPLC, respectively. Initial experiments were run for 18 pore volumes (PV) and later experiments for 220 PV. Control experiments (no Fe(II)) were also performed for both sand and goethite-coated sand packed columns.
For the unsaturated column, a column pre-saturated with a 10 mM NaCl solution was subjected to three cycles of evacuation (4.1 CFM (7.0 m3 h−1) vacuum pump) in the exchange chamber of the glove box. Because both ends of the column were open to the atmosphere, some water was lost from the column, leaving some parts of the column dry, resulting in the unsaturated condition. For saturated columns, the columns were not subjected to the cycles of evacuation, thus maintaining the initial saturated condition. Information about all of the column experiments performed is summarized in Table S1.†
Analytical methods
The 4-ClNB and 4-ClAn concentrations were measured via HPLC using an Agilent 1200 Infinity equipped with a Zorbax SB-C18 (4.6 × 300 mm, 5 μm particle size column). The mobile phase was composed of a 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 mixture of A: 12 mM ammonium acetate (Sigma-Aldrich 97%) in 90 vol% water and 10 vol% HPLC grade acetonitrile (Sigma-Aldrich) adjusted to pH 7.0 with ammonium hydroxide (Sigma-Aldrich 28%), and B: acetonitrile. The flow rate was 0.7 mL min−1, and the detection wavelength was set to 254 nm. A six-point calibration curve with concentrations of 4-ClNB and 4-ClAn ranging from 0 to 0.1 mM were used.
:70 mixture of A: 12 mM ammonium acetate (Sigma-Aldrich 97%) in 90 vol% water and 10 vol% HPLC grade acetonitrile (Sigma-Aldrich) adjusted to pH 7.0 with ammonium hydroxide (Sigma-Aldrich 28%), and B: acetonitrile. The flow rate was 0.7 mL min−1, and the detection wavelength was set to 254 nm. A six-point calibration curve with concentrations of 4-ClNB and 4-ClAn ranging from 0 to 0.1 mM were used.
To quantify Fe(II) sorption, 0.15 mL of filtered sample was added to 0.20 mL of 10 mM Ferrozine reagent and 2.65 mL of ultrapure water and seven standards ranging from 0 mM to 0.05 mM were created. The absorbance at 562 nm (UV-1601PC, Shimadzu) was quantified for each standard and sample, and the standard calibration curve composed of seven points was used for determination of Fe(II) concentrations.
Particle characterization after reaction
To evaluate whether the detachment process fundamentally altered the particle size distribution of goethite particles harvested from goethite-coated sand particles as compared to the as-synthesized goethite particles, the detachment process was conducted using unreacted goethite-coated sand. Dry goethite-coated sand was suspended in ultrapure water, the pH adjusted to 9.0 (using 1 M NaOH), and aged for 24 h. At pH 9, the quartz sand grains and goethite nanoparticles have negative charges and are expected to electrostatically repel each other. Finally, the samples were sonicated using an Aquasonic (150HT) bath sonicator for 5 min and the supernatant collected. A drop of this sample was placed onto a Cu 200 mesh holey carbon coated grid and allowed to air-dry and then examined using TEM. Average particle size and distribution of unreacted, detached particles were used as a reference for comparison to particles after exposure to Fe(II) and/or 4-ClNB.To collect samples of solid materials from columns, the solid matrix was removed from the column as an intact cylinder by opening one end and pushing the flow adaptor from the other end. The material was sliced using a thin plastic ruler (0.5 mm thickness) into three 1 cm zones (0–1, 1–2, and 2–3 cm from the influent end denoted as Z-1, Z-2, and Z-3, see ESI†). For columns packed with goethite-coated sand, each zone of material was then subjected to the detachment process as described above.
Pairwise Kolmogorov–Smirnov (K–S) tests were used to statistically compare particle size distributions of pre-reaction and post-reaction particles using MATLAB (MathWorks, 2019). This test has previously been used to make comparisons of particles collected from Fe(II)/goethite batch reactors under different conditions.30 This test describes differences in both location and shape of the empirical cumulative distribution functions of the two samples. In the K–S test, D values describe the extent of difference between two distributions, and the P value indicates the probability that two data sets are statistically similar, respectively. The null hypothesis for this test is that two data sets are statistically indistinguishable and for the P < 0.05 (significance level), the null hypothesis is rejected. Details for particle size calculations and determination of electron balances are presented in the ESI† (section S2).
Results and discussion
Materials characterization and Fe(II) sorption to goethite
The XRD pattern of the synthesized goethite matches the goethite reference PDF 029-0713 (Fig. S2a†), and no other crystalline phases, like ferrihydrite or hematite, were detected. TEM characterization (Fig. S2b†) revealed that only goethite particles, with acicular morphology, an average length of 74 ± 25 nm, and average width of 8 ± 4 nm (n = 500 particles), were present. Smaller particles indicative of residual ferrihydrite were not observed. A specific surface area of 119 m2 g−1 was measured by N2 sorption BET. SEM images of goethite-coated sand grains are consistent with uniform coverage of goethite particles on the sand grain surfaces (representative image shown in Fig. 1a).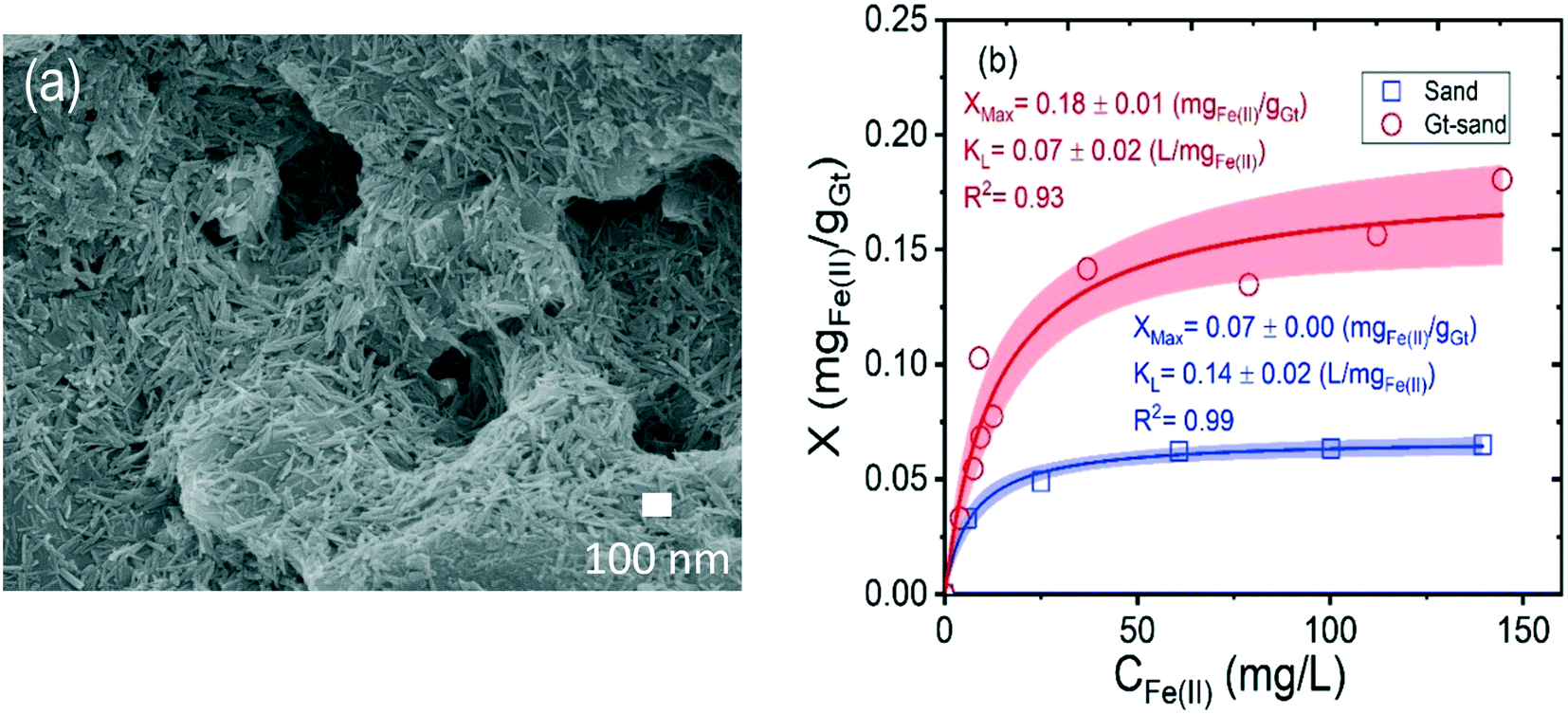 | ||
| Fig. 1 SEM image of goethite-coated sand grain (a) and sorption isotherms of Fe(II) (b) on bare and goethite-coated sand (GT-sand) at pH 7 in 10 mM NaHCO3 buffer. The solid lines in (b) represent fits using the Langmuir adsorption model. Errors reported with the parameters are 95% confidence intervals, and 95% confidence bands are shown in red and blue shades. | ||
The sorption isotherms of Fe(II) onto the sand and goethite-coated sand at pH 7 are shown in Fig. 1b. The latter material has higher Fe(II) sorption capacity, and this difference is attributed to the difference in the structure of iron oxide and quartz and the higher surface area of the heterogeneous goethite-coated sand grains as compared to bare sand grains. For the sand, a sorption plateau is clearly reached, but it is only approached for goethite coated sand, consistent with past work indicating increasing sorption with increasing aqueous Fe(II).50 Thus, Freundlich isotherms for the data in Fig. 1b are given in Fig. S3.† Overall, the extent of sorption on the goethite-coated sand is ∼2-fold higher, on a surface area basis (Fig. S3†) to that of Dixit et al.,50 which could be due to differences in abundances of specific sorption sites. Surface complexation modeling to evaluate this is beyond the scope of this study. As previous work has shown,32,33,51 the semi-conducting nature of iron oxides means that sorption of Fe(II) is different from sorption onto other mineral surfaces, with the possibility of electron transfer between aqueous Fe(II) and structural Fe(III) followed by bulk electron conduction and Fe(II)–Fe(III) atom exchange. Implications of this in terms of recrystallization in the column studies is discussed below.
Particle detachment
No statistically significant difference in the particle length distributions of the initial particles (74 ± 25 nm) and those attached and subsequently detached from sand (74 ± 27 nm; D = 0.048, P = 0.605, Fig. S4†) was observed. While the average widths are also identical (8 ± 4 nm and 8 ± 3 nm), the distribution of widths is statistically different (D = 0.115, P = 0.002; Fig. S4†). The histograms plotting the lengths and widths for original and detached particles (Fig. S4†) have similar shapes, and the D values are small. Thus, we conclude that the attachment/detachment processes do not significantly affect the distribution of particle sizes (e.g., no new particle fragments have been formed as a consequence of the attachment and detachment processes). Thus, results tracking the particle sizes of post-reaction particles after using the detachment procedure will enable meaningful comparisons between experiments.Batch reactors
The presence of trace dissolved silica did not significantly impact the reactivity of the goethite particles. Average dimensions (Table 1) and histograms depicting the lengths and widths (Fig. S5†) of particles collected from batch reactors after five cycles of reaction with 4-ClNB using carbonate buffer prepared in ultrapure water and carbonate buffer equilibrated with quartz sand demonstrated that, in both cases, oxidative mineral growth occurred mostly on the particle tips, as reported previously,29,30,52 and the average lengths increased from 74 nm to 106 nm (Table 1). Results indicate that the presence of the underlying sand grains in columns prepared using goethite-coated sand will not alter solution chemistry sufficiently so as to change the relative reactivity of the tip versus side crystal surfaces. Thus, mineral growth observed in different column experiments below can be attributed to the reactivity of Fe(II) with 4-ClNB and changes in the column conditions and not to a matrix effect caused by the presence of sand.| Sample | Reactor | Length (nm) | Width (nm) | D (length) | D (width) | N |
|---|---|---|---|---|---|---|
| a Number of particles measured. b D values are for comparison of 1 + 2 without sand to 1 + 2 with sand after reaction. | ||||||
| Sand-conditioned carbonate buffer | Reactor 1 | 111 ± 38 | 9 ± 4 | 0.410 | 0.124 | 504 |
| Reactor 2 | 101 ± 34 | 9 ± 4 | 0.366 | 0.268 | 506 | |
| Average | 106 ± 36 | 9 ± 4 | 0.045b | 0.087b | 1010 | |
| Carbonate buffer | Reactor 1 | 104 ± 32 | 9 ± 3 | 0.252 | 0.084 | 502 |
| Reactor 2 | 106 ± 37 | 9 ± 4 | 0.510 | 0.260 | 502 | |
| Average | 105 ± 35 | 9 ± 4 | 0.045b | 0.087b | 1004 | |
Control column reactors
The breakthrough curves obtained from control experiments, which used columns packed with bare sand and goethite-coated sand grains and fed with either Fe(II) solution or 4-ClNB solution, precluding the reaction from occurring, are shown in Fig. S6.† The 4-ClNB and Fe(II) breakthrough curves show deviations from those for the NaBr tracer, in which the effluent concentration reached one-half the influent concentration after one pore volume with a fit retardation coefficient (R) equal to 1. The reproducibility of the column packing/tracer data is shown in Fig. S7.† These results indicate that Fe(II) sorbs to the mineral surface (as expected based on Fig. 1), and the sorption of 4-ClNB is minimal. Because recrystallization (without particle growth) can occur under high Fe(II) conditions over tens of days32,53,54 we evaluated whether any particle growth occurred over the time period of our column reactors (tens of hours). A column filled with goethite-coated sand was fed a 0.5 mM Fe(II) solution in 10 mM sodium bicarbonate at pH 7, similar to the condition used in reactive column experiments (see below), after which the columns were sacrificed for characterization. The breakthrough curve of Fe(II) shows that after ∼10 pore volumes the concentration of Fe(II) reaches the initial value (C/C0 ≈ 1) and indicates sorption equilibrium and no Fe(II) consumption. After 110 pore volumes, no change in the mass loading of total iron per unit mass of goethite coated sand was observed (Fig. S8 and Table S2†).While we cannot preclude that electron transfer and atom exchange of Fe(II) leading to recrystallization are occurring based on these data, little to no electron transfer and atom exchange is expected over the time frame of the column experiment. Strehlau et al. measured the goethite particle size distribution after 18 days of stirring in 10 mM carbonate buffer with 1 mM Fe(II) and compared the average particle dimensions, and their distribution, with those of the initial particles.30 They observed no significant change in the average particle size and size distribution under these conditions. Additionally, results from reactive columns shown below indicate that the goethite tip surface area comprises the reactive surface area, indicating the mass addition is driven by oxidative growth due to reaction with 4-ClNB.
Reactive column reactors
The rate and extent of 4-ClNB reduction by Fe(II) on the column packed with bare sand were drastically less as compared to the column packed with the goethite-coated sand (Fig. S9†). This drop is attributed to reductive degradation and concurrent oxidative mineral growth. For the column packed with bare sand grains, the Fe(II) drops to an average of 95% of the influent concentration after just five pore volumes and remains at that level throughout the experiment. The 4-ClNB concentration in the effluent, however, shows a delayed breakthrough, at approximately 2–2.5 pore volumes as compared to the breakthrough observed with the conservative tracer. The effluent 4-ClNB concentration eventually equals that of the influent (at ∼15 pore volumes), demonstrating minimal reaction with sand. In addition, a maximum in concentration (0.5 μM) of 4-ClAn, the product of 4-ClNB reduction, was observed after five PV, and after that, 4-ClAn was no longer detectable. Thus, for the bare sand column, we conclude that there is initially some reaction of 4-ClNB but that a small number of reactive sites are exhausted early in the experiment, which, combined with the limited adsorption of Fe(II) onto the quartz sand grains, leads to this short-lived and minimal reactivity. In contrast, the concentrations of Fe(II) and 4-ClNB in the effluent from the goethite-sand column of Fig. S9† reached steady concentrations of 60% of the influent Fe(II) and 40% of the influent 4-ClNB concentrations, indicating continuous degradation of the 4-ClNB with a concurrent drop in the dissolved Fe(II) concentration.The concentrations of 4-ClNB and 4-ClAn in the effluent from two goethite-sand columns were then monitored over the course of 220 PV (Fig. 2 and S10†). The 4-ClNB concentration in the effluent increased and the concentration of the 4-ClAn decreased, approaching a steady state condition at ∼100 pore volumes (Fig. 2). The initial higher extent of conversion seen in Fig. 2 and S10† (the replicate experiment) is likely due to the fact that columns were equilibrated with the solution of Fe(II) prior to introduction of 4-ClNB. This means that there was initially increased reducing capacity as compared to the later condition when both were introduced into the column simultaneously and continuously.
 | ||
| Fig. 2 Concentrations of 4-ClNB in both influent (feed) and effluent, 4-ClAn in effluent, and mass balance (4-ClNB + 4-ClAn) for 220 PV (a) and tracer analysis before and after 4-ClNB reduction for 220 PV (b) 95% confidence bands are shown in blue and red shades. Prior to initiating flow of the 4-ClNB solution, 10 mM NaHCO3 for 3 PV and then 1 mM Fe(II) in 10 mM NaHCO3 for 3 PV were fed into each column. For all experiments, the flow rates of each Fe(II) and 4-ClNB were 0.5 mL min−1 and columns with inner diameters of 2.5 cm and heights of 3 cm were used. | ||
Results examining the breakthrough of the NaBr (Fig. 2b) before and after reaction for 220 PV, however, show substantial differences. The porosity decreased slightly, from 53% to 47% while the hydrodynamic dispersion coefficient increased by more than fourfold, from 1.07 × 10−4 cm2 s−1 to 4.58 × 10−4 cm2 s−1. This increase in the hydrodynamic dispersion coefficient likely results from oxidative mineral growth and is consistent with the same flow rate but altered flow distribution or pathways through the columns. As the oxidative mineral growth occurs, hydrodynamic properties of the column evolve and the flow path of the solution changes. New hydrodynamic conditions lead to changes in access to reactive sites, and therefore, less reactivity as a result of reduced access of reactants to reactive sites and higher 4-ClNB effluent concentrations. The overall fluctuations in concentrations in Fig. 2a are likely due to these changes. While the overall trend is lower conversion (which is consistent with increased hydrodynamic dispersion coefficient and lower porosity), the fluctuations in concentration could be due to altered (longer or shorter) flow paths as the minerals grow.
The mass balance (4-ClNB + 4-ClAn) is 85–115% of the feed concentration (Fig. 2 and S10†). Deficits in mass balance could be attributed to the formation of the hydroxylamine, an unstable intermediate, which was not measured in this study.21 Overall, the variability is considered as within experimental error and/or mixing of different parcels of water traveling over different flow paths. The quantitative analysis of oxidative mineral growth resulting from the reduction of 4-ClNB to 4-ClAn is further discussed below with the measurements of particle dimensions.
Unsaturated flow conditions
The extent and conversion rate of 4-ClNB to 4-ClAn, as well as the change in porosity and hydrodynamic dispersion, in an unsaturated column filled with goethite-coated sand (Fig. 3) were substantially different than observed for experiments using saturated conditions (Fig. 2 and S10†). The concentrations of 4-ClNB initially increased with time, reached the feed concentration after 50 PV, and then decreased. Similar to what was observed for the columns reacted under the saturated flow condition, the concentration of 4-ClAn decreased when 4-ClNB increased and increased when 4-ClNB concentrations fell. At ∼75 PV, minimal 4-ClNB conversion to 4-ClAn was observed, which we attribute to the channeling of flow in the column, resulting in a drastic reduction of contact of fresh feed solution with the goethite particles. We also see two breaks in both 4-ClNB and 4-ClAn concentrations in the effluent at around 100 PV and 120 PV (Fig. 3a). We attribute these observations to changes in the flow path, possibly due to pore blockage resulting from mineral oxidative growth or shifting of the packed grains. As with the results from the column used in collecting the data for Fig. 2, the initial high conversion of 4-ClAn (from 0 to 1.5 pore volumes) of this unsaturated column is likely due to pre-loading the column with Fe(II). Non-uniform distribution of oxidative growth under advective flow was observed by Hansel et al., where the distribution of secondary mineralization of iron oxides within the column was highly dictated by the flow-induced Fe(II) profile under dissimilatory iron reduction conditions.55 Thus, we hypothesize that in our unsaturated flow system in which the distribution of reactants (Fe(II) and 4-ClNB) were not uniform, non-uniform mineral oxidative growth was likely and would result in flow path changes. The tracer data support this interpretation, showing substantial changes in the hydrodynamic properties for the unsaturated columns with a dramatic increase in porosity and decrease in hydrodynamic dispersion coefficient (Fig. 3b) over the course of the experiment. The breakthrough curves show characteristics of an unsaturated flow condition both before and after reaction.56 The increase in porosity and decrease in the hydrodynamic dispersion coefficient after reaction (Fig. 3b) indicate that the column was more saturated at the conclusion of the experiment. The (water filled) porosity of the column increased from 25% to 40%, and the hydrodynamic dispersion coefficient dropped two-fold, from 2.85 × 10−3 cm2 s−1 to 1.14 × 10−3 cm2 s−1, which is attributed to changes of flow paths in an already unsaturated column. | ||
| Fig. 3 Concentration of 4-ClNB in both influent (feed) and effluent, 4-ClAn, and mass balance (a) and tracer analysis before and after 4-ClNB reduction for unsaturated flow (b) 95% confidence bands are shown in blue and red shades. All the columns were conditioned with 10 mM NaHCO3 and then 1 mM Fe(II) for 3 pore volumes each before adding 4-ClNB to the feed solution. For all experiments, the flow rates of each Fe(II) and 4-ClNB were 0.5 mL min−1 and columns with ID: 2.5 cm and height: 3 cm were used. | ||
Particle growth
The extent of oxidative growth of goethite particles and how it varies as a function of position in the columns were studied by measuring the size of goethite particles collected from each of three 1 cm zones (Z-1, Z-2, Z-3, Fig. S1†) within the column. The particle lengths and widths, and the fraction of new goethite mass added to the column in each zone (section S2, ESI†) are presented in Table 2, from both the column run for a short period (Fig. S7,† red circles) and those of long duration (Fig. 2 and 3). The change in particle length is greatest at the bottom (Z-1) of the columns where the concentrations of reagents are the largest (Fig. 4).| Sample | Column zone (Fig. S1†) | Length (nm) | Width (nm) | Mass addedb (%) | D (length) | D (width) | N |
|---|---|---|---|---|---|---|---|
| a Errors represent one standard deviation. b Fraction of goethite mass added to each zone based on the particle dimensions determined by TEM (section S2†). c D: K–S test parameter for the comparison of any set with detached, unreacted particles. The higher the D values are, the greater the difference in the data distributions. d N = number of particles measured for each data set. | |||||||
| Unreacted, detached particles | — | 74 ± 27a | 8 ± 3 | — | — | — | 500 |
| Saturated flow (18 PV, Fig. S7†) | Z-1 | 87 ± 28 | 10 ± 5 | 98.5 | 0.213 | 0.203 | 446 |
| Z-2 | 74 ± 27 | 8 ± 4 | 0 | 0.073 | 0.111 | 446 | |
| Z-3 | 75 ± 25 | 8 ± 3 | 1.5 | 0.066 | 0.285 | 369 | |
| Saturated flow (220 PV, Fig. 2) | Z-1 | 134 ± 68 | 9 ± 3 | 63 | 0.480 | 0.188 | 505 |
| Z-2 | 109 ± 51 | 8 ± 3 | 23 | 0.340 | 0.096 | 507 | |
| Z-3 | 96 ± 39 | 8 ± 2 | 14 | 0.266 | 0.053 | 510 | |
| Unsaturated flow (220 PV, Fig. 3) | Z-1 | 111 ± 33 | 10 ± 4 | 75.5 | 0.456 | 0.290 | 502 |
| Z-2 | 84 ± 39 | 9 ± 5 | 25.5 | 0.133 | 0.112 | 487 | |
| Z-3 | 77 ± 30 | 6 ± 3 | 0 | 0.053 | 0.330 | 502 | |
 | ||
| Fig. 4 The distribution of goethite particle length and width after reaction with 4-ClNB in column reactors for 220 PV under saturated (a and b) and unsaturated (c and d) conditions. The measurements from three zones (Z-1), middle (Z-2), and top (Z-3) are compared to the detached, unreacted particles. All the reactors were conditioned by 10 mM NaHCO3 and fed with 1 mM Fe(II) and 0.1 mM 4-ClNB. P values show if data sets are statistically different (for P < 0.05) and D values show how histograms are different from the one for the detached particles. Larger D values indicate the more difference from the detached particles. For all experiments, the flow rates of Fe(II) and 4-ClNB were each 0.5 mL min−1 and columns with ID: 2.5 cm and height: 3 cm were used. See Table 2 for average length and width data. | ||
Comparing the particle size distributions for pre-reaction and post-reaction particles after 18 PV (Fig. S11†) under saturated conditions shows that the reaction primarily occurred in zone 1, with no significant size increase observed in zones 2 and 3, due to the short exposure to the 4-ClNB and Fe(II). After 220 PV under saturated conditions (Fig. 4a and b), an increase in average particle length was observed in all three zones (P < 0.05), with the most growth observed in the zone (Z1) closest to the influent, where reagent concentrations, and thus reaction rate, are largest. While the widths do significantly increase (Fig. 4b), the data are consistent with oxidative growth primarily occurring at the tips, thus increasing the lengths of the goethite particles. For the unsaturated flow conditions (Fig. 4c and d), particles at the bottom (Z-1) and middle (Z-2) of the column are longer than the unreacted, detached goethite particles, and no significant increase in length of goethite particles at the top of the column was observed. As with the saturated flow conditions, the data are consistent with oxidative growth mainly occurring on the tips of the particles, with the increase in length substantially greater than the increase in width on a per mass basis. The unsaturated condition results in a more dynamic system in which the oxidative growth is not only a function of the reactant concentration profile throughout the column (here different column zones) but also a function of flow path.
In both Fig. 2 and 3, it is apparent that both 4-ClNB and Fe(II) are present in zone 3 (both reactants are detected the end/top of the column), but the extent of particle growth that occurred in this zone (Table 2) is smaller than zones 1 and 2. Decreased extent of particle growth could be due to slower reaction rate (both the concentrations of the reactants are lower than those in the influent) or slowing of the reaction by changes in solution chemistry (change in pH, elevated 4-ClAn concentration).
The extent of transformation, however, is affected by the mineral oxidative growth. Fig. 2 and 3 clearly indicate the change in the extent of transformation of 4-ClNB to 4-ClAn. For the saturated column the extent of the transformation decreases over time. It can be attributed to the decreasing porosity of the column, which reduces the contact time between reactant and reactive sites. In addition, the reactivity and the number of reactive sites of the newly formed solids could be different, which could lead to the decrease in the extent of transformation. While the increase in goethite mass does result in more surface area, particle size characterization demonstrates growth primarily on the goethite crystal tips, which means that the total “tip” surface area would remain relatively constant, even with drastic increases in goethite crystal length. Furthermore, as the goethite crystals elongate, new overlaps between goethite could effectively reduce accessible, reactive surface area. For the unsaturated column on the other hand, the extent of 4-ClNB transformation to 4-ClAn increases over time. Fig. 3 shows that the (water filled) porosity of the column increases in the course of reaction, which means reactants have more contact time with goethite particles, resulting in the higher extent of transformation. As the unsaturated column becomes more saturated over time, the more uniform flow would be expected to lead to increased access of reactants to the goethite surface, which would be expected to increase the extent of transformation.
Phenomenological approach to the kinetics of 4-ClNB reduction
To compare the reactivity of different column systems towards NACs and assess potential changes in surface area/Fe(II) on the observed extent of transformation, we used the approach of Heijman et al.,57 in which the apparent zero-order rate constant, kobs, was defined as: | (5) |
Connecting 4-ClNB reduction to mineral growth
By quantifying the total 4-ClNB consumed and 4-ClAn produced combined with the 1:6 stoichiometric ratio for the reduction of 4-ClNB by Fe(II) (eqn (1) and (2)),30 the theoretical maximum goethite produced during the reaction was calculated. Then, the mass of goethite produced was calculated using the observed average particle dimensions (see section S2†), assuming no new particles form, which is supported by the TEM observation. These two values were compared, and the results are shown in Fig. 5. Results show a good agreement between new goethite mass added to the column based on TEM observation and the predicted values for the goethite mass added to the column based on stoichiometry. For the column sacrificed after 18 PV, however, a disagreement between two values is observed. This observation is attributed to the duration of the reaction not being of long enough to have sufficient particle growth to obtain robust results.
 | ||
| Fig. 5 Goethite mass (normalized to initial goethite mass in the columns) formed over the course of reactions as calculated by conversion of 4-ClNB, production of 4-ClAn, or increase in goethite size observed by TEM. Black symbols represent values calculated based on 4-ClAn production (method 1, ESI†) and filled symbols with represent values calculated based on 4-ClNB consumption (method 2, ESI†). Symbols are for the 18 pore volume (PV) reactor (upward triangle), 220 PV reactor (hexagons), 220 PV unsaturated reactor (right triangle), and batch experiments with (squares) and without sand-conditioned water (stars). Empty and filled symbols for hexagons and stars overlapped. | ||
Another observation in Fig. 5 is the large difference between the total 4-ClNB converted over the 220 pore volumes under the saturated and unsaturated conditions. Although both columns were filled with the same mass of goethite-coated sand grains and fed with solutions of the same concentrations and using the same flow rate, the amount of goethite formation in the saturated column was almost twice that formed in the unsaturated column. This is consistent with more reactive surface area being exposed to the Fe(II) and 4-ClNB over the course of the experiment, most likely because of the changing flow pathway in the column. Fig. 3 also shows that in the middle of the experiment (∼75 pore volumes) the concentration of 4-ClNB in the effluent reaches almost the initial value, which likely means channeling through the column, with decreased oxidative goethite growth during this period of the experiment.
Implications
In this study, the quantitative investigation of oxidative goethite mineral growth using conditions that are more relevant to field settings, i.e. continuous exposure to an oxidizing organic contaminant, 4-ClNB, and Fe(II), in the presence of other mineral surfaces is presented. The approach enables the recovery of goethite particles from columns packed with goethite-coated sand grains by exploiting electrostatic attraction and repulsion. Results demonstrate no significant change in average particle size nor particle size distribution because of the procedure used to prepare the goethite-sand and to harvest goethite nanoparticles from the goethite-sand. The results demonstrate that the presence of dissolved silica, which is present due to the contact between solution and exposed quartz sand grain surfaces,58,59 did not detectably affect the oxidative goethite growth. We also found that under saturated flow columns, the mineral growth is mainly a function of the extent of reaction while for the column with unsaturated flow, the mineral growth behavior is more complicated and appears to depend on the flow path. Finally, results indicate that our facile attachment/detachment of goethite particles to/from the quartz sand grains enables quantitative characterization of oxidative growth, with growth primarily observed on the tips versus the sides of the acicular goethite nanocrystals. Results demonstrate the goethite particles primarily grow longer when attached to the bare sand grain surfaces, which is consistent with results obtained in batch reactors using aqueous suspensions of goethite nanoparticles. Results from the unsaturated flow condition, which is more relevant to environmental conditions in the vadose zone after precipitation and runoff, indicate the importance of the evolution of flow path and particle growth. Sampling from different zones of the column is beneficial in the study of the kinetics of 4-ClNB reduction throughout the column, and results will facilitate the development of comprehensive mass transport models for the evolving reactive system under advective flow.Although the column dimensions and fluid flow rate in this study are substantially higher than typical groundwater conditions, this approach enables the study of more complicated scenarios that may influence iron oxide nanoparticle reactivity, including the presence of NOM and other competing trace elements as well as model development to assess how evolving mineralogy and transport might influence the success of remediation scenarios. Moreover, other experimental conditions like pH, mixing conditions (batch, flow), and water chemistry (oxide structure and composition, Fe(II) concentration, and carbonate and phosphate concentration) will affect the end product and the extent of contaminant transformation. Studies of the dynamics of iron oxide reduction by bacteria and formation of new material upon contaminant exposure are also possible.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was funded by the U.S. National Science Foundation grants ECS-1507496 and ECS-1904858. Parts of this work were carried out in the Characterization Facility, University of Minnesota, which receives partial support from NSF through the MRSEC program. Thanks to Emily Hughes and Alanna Hildebrandt for their assistance in the laboratory, and the reviewers for their helpful comments.References
- R. L. Penn, C. Zhu, H. Xu and D. R. Veblen, Iron oxide coatings on sand grains from the Atlantic coastal plain: High-resolution transmission electron microscopy characterization, Geology, 2002, 29, 843–846 CrossRef.
- M. C. Goldberg, E. R. Weiner and P. M. Boymel, Adsorption of goethite onto quartz and kaolinite, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 1491–1498 RSC.
- A. Scheidegger, M. Borkovec and H. Sticher, Coating of silica sand with goethite: preparation and analytical identification, Geoderma, 1993, 58, 43–65 CrossRef CAS.
- C. van der Zee, D. R. Roberts, D. G. Rancourt and C. P. Slomp, Nanogoethite is the dominant reactive oxyhydroxide phase in lake and marine sediments, Geology, 2003, 31, 993–996 CrossRef CAS.
- U. Schwertmann, Transformation, in Encyclopedia of Earth Sciences Series, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2008, ch. 14, pp. 363–369 Search PubMed.
- W. Stumm and B. Sulzberger, The cycling of iron in natural environments: Considerations based on laboratory studies of heterogeneous redox processes, Geochim. Cosmochim. Acta, 1992, 56, 3233–3257 CrossRef CAS.
- B. R. Coughlin and A. T. Stone, Nonreversible Adsorption of Divalent Metal Ions (MnII, CoII, NiII, CuII, and PbII) onto Goethite: Effects of Acidification, Fe‖ Addition, and Picolinic Acid Addition, Environ. Sci. Technol., 1995, 29, 2445–2455 CrossRef CAS PubMed.
- M. Elsner, R. P. Schwarzenbach and S. B. Haderlein, Reactivity of Fe(II)-Bearing Minerals toward Reductive Transformation of Organic Contaminants, Environ. Sci. Technol., 2004, 38, 799–807 CrossRef CAS PubMed.
- G. A. Waychunas, C. S. Kim and J. F. Banfield, Nanoparticulate Iron Oxide Minerals in Soils and Sediments: Unique Properties and Contaminant Scavenging Mechanisms, J. Nanopart. Res., 2005, 7, 409–433 CrossRef CAS.
- C. Colombo, G. Palumbo, J. Z. He, R. Pinton and S. Cesco, Review on iron availability in soil: interaction of Fe minerals, plants, and microbes, J. Soils Sediments, 2014, 14, 538–548 CrossRef CAS.
- C. Schmidt, S. Behrens and A. Kappler, Ecosystem functioning from a geomicrobiological perspective - a conceptual framework for biogeochemical iron cycling, Environ. Chem., 2010, 7, 399–405 CrossRef CAS.
- R. J. Bowell, Sorption of arsenic by iron oxides and oxyhydroxides in soils, Appl. Geochem., 1994, 9, 279–286 CrossRef CAS.
- T. B. Hofstetter, C. G. Heijman, S. B. Haderlein, C. Holliger and R. P. Schwarzenbach, Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions, Environ. Sci. Technol., 1999, 33, 1479–1487 CrossRef CAS.
- I. Hwang and B. Batchelor, Reductive dechlorination of tetrachloroethylene in soils by Fe(II)-based degradative solidification/stabilization, Environ. Sci. Technol., 2001, 35, 3792–3797 CrossRef CAS PubMed.
- K. Pecher, S. B. Haderlein and R. P. Schwarzenbach, Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides, Environ. Sci. Technol., 2002, 36, 1734–1741 CrossRef CAS PubMed.
- V. R. Vermeul, M. D. Williams, J. E. Szecsody, J. S. Fruchter, C. R. Cole and J. E. Amonette, Creation of a Subsurface Permeable Reactive Barrier Using in Situ Redox Manipulation, in Handbook of Groundwater Remediation using Permeable Reactive Barriers, Academic Press, 2003, ch. 6, pp. 163–192 Search PubMed.
- P. N. Dave and L. V. Chopda, Application of Iron Oxide Nanomaterials for the Removal of Heavy Metals, J. Nanotechnol., 2014, 398569 Search PubMed.
- A. Agrawal and P. Tratnyek, Reduction of Nitro Aromatic Compounds by Zero-Valent Iron Metal, Environ. Sci. Technol., 1995, 30, 153–160 CrossRef.
- J. Klausen, S. P. Tröber, S. B. Haderlein and R. P. Schwarzenbach, Reduction of Substituted Nitrobenzenes by Fe(II) in Aqueous Mineral Suspensions, Environ. Sci. Technol., 1995, 29, 2396–2404 CrossRef CAS PubMed.
- S. B. Haderlein, K. W. Weissmahr and R. P. Schwarzenbach, Specific adsorption of nitroaromatic explosives and pesticides to clay minerals, Environ. Sci. Technol., 1996, 30, 612–622 CrossRef CAS.
- J. Pichtel, Distribution and Fate of Military Explosives and Propellants in Soil: A Review, Appl. Environ. Soil Sci., 2012, 2012, 1–33 CrossRef.
- P. Kovacic and R. Somanathan, Nitroaromatic compounds: Environmental toxicity, carcinogenicity, mutagenicity, therapy and mechanism, J. Appl. Toxicol., 2014, 34, 810–824 CrossRef CAS PubMed.
- Y. Tong, M. J. Berens, B. A. Ulrich, J. Bolotin, J. H. Strehlau, T. B. Hofstetter and W. A. Arnold, Exploring the Utility of Compound-Specific Isotope Analysis for Assessing Ferrous Iron-Mediated Reduction of RDX in the Subsurface, Environ. Sci. Technol., 2021, 55, 6752–6763 CrossRef CAS PubMed.
- T. A. Douglas, M. E. Walsh, C. J. McGrath, C. A. Weiss, A. M. Jaramillo and T. P. Trainor, The fate of nitroaromatic (TNT) and nitramine (RDX and HMX) explosive residues in the presence of pure metal oxides, ACS Symp. Ser., 2011, 1069, 197–215 CrossRef CAS.
- S. S. Talmage, D. M. Opresko, C. J. Maxwell, C. J. E. Welsh, F. M. Cretella, P. H. Reno and F. B. Daniel, Nitroaromatic munition compounds: Environmental effects and screening values, Rev. Environ. Contam. Toxicol., 1999, 161, 1–156 CrossRef CAS PubMed.
- J. D. Istok, J. E. Amonette, C. R. Cole, J. S. Fruchter, M. D. Humphrey, J. E. Szecsody, S. S. Teel, V. R. Vermeul, M. D. Williams and S. B. Yabusaki, In Situ Redox Manipulation by Dithionite Injection: Intermediate-Scale Laboratory Experiments, Ground Water, 1999, 37, 884–889 CrossRef CAS.
- H. K. Boparai, S. D. Comfort, P. J. Shea and J. E. Szecsody, Remediating explosive-contaminated groundwater by in situ redox manipulation (ISRM) of aquifer sediments, Chemosphere, 2008, 71, 933–941 CrossRef CAS PubMed.
- T. J. Strathmann, Redox Reactivity of Organically Complexed Iron(II) Species with Aquatic Contaminants in Aquatic Redox Chemistry, ACS Symp. Ser., 2011, 1071, 283–313 CrossRef CAS.
- C. L. Chun, R. L. Penn and W. A. Arnold, Kinetic and Microscopic Studies of Reductive Transformations of Organic Contaminants on Goethite, Environ. Sci. Technol., 2006, 40, 3299–3304 CrossRef CAS PubMed.
- J. H. Strehlau, M. S. Stemig, R. L. Penn and W. A. Arnold, Facet-Dependent Oxidative Goethite Growth As a Function of Aqueous Solution Conditions, Environ. Sci. Technol., 2016, 50, 10406–10412 CrossRef CAS PubMed.
- J. H. Strehlau, J. D. Schultz, A. M. Vindedahl, W. A. Arnold and R. L. Penn, Effect of nonreactive kaolinite on 4-chloronitrobenzene reduction by Fe(ii) in goethite-kaolinite heterogeneous suspensions, Environ. Sci.: Nano, 2017, 4, 325–334 RSC.
- S. C. Southall, S. Micklethwaite, S. A. Wilson and A. J. Frierdich, Changes in Crystallinity and Tracer-Isotope Distribution of Goethite during Fe(II)-Accelerated Recrystallization, ACS Earth Space Chem., 2018, 2, 1271–1282 CrossRef CAS.
- S. D. Taylor, J. Liu, X. Zhang, B. W. Arey, L. Kovarik, D. K. Schreiber, D. E. Perea and K. M. Rosso, Visualizing the iron atom exchange front in the Fe(II)-catalyzed recrystallization of goethite by atom probe tomography, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2866–2874 CrossRef CAS PubMed.
- E. D. Burton, R. T. Bush, L. A. Sullivan and D. R. G. Mitchell, Schwertmannite transformation to goethite via the Fe(II) pathway: Reaction rates and implications for iron–sulfide formation, Geochim. Cosmochim. Acta, 2008, 72, 4551–4564 CrossRef CAS.
- J. Greskowiak, H. Prommer, G. Massmann, C. D. Johnston, G. Nützmann and A. Pekdeger, The impact of variably saturated conditions on hydrogeochemical changes during artificial recharge of groundwater, Appl. Geochem., 2005, 20, 1409–1426 CrossRef CAS.
- J. Šimůnek and M. T. van Genuchten, Contaminant Transport in the Unsaturated ZoneTheory and Modeling, in The Handbook of Groundwater Engineering, Routledge Handbooks Online, 2016, ch. 8, pp. 203–229 Search PubMed.
- R. Simon, D. Colón, C. L. Tebes-Stevens and E. J. Weber, Effect of redox zonation on the reductive transformation of p-cyanonitrobenzene in a laboratory sediment column, Environ. Sci. Technol., 2000, 34, 3617–3622 CrossRef CAS.
- J. N. Ryan, M. Elimelech, R. A. Ard, R. W. Harvey and P. R. Johnson, Bacteriophage PRD1 and silica colloid transport and recovery in an iron oxide-coated sand aquifer, Environ. Sci. Technol., 1999, 33, 63–73 CrossRef CAS.
- N. Boujelben, J. Bouzid and Z. Elouear, Adsorption of nickel and copper onto natural iron oxide-coated sand from aqueous solutions: Study in single and binary systems, J. Hazard. Mater., 2009, 163, 376–382 CrossRef CAS PubMed.
- V. K. Gupta, V. K. Saini and N. Jain, Adsorption of As(III) from aqueous solutions by iron oxide-coated sand, J. Colloid Interface Sci., 2005, 288, 55–60 CrossRef CAS PubMed.
- M. M. Benjamin, R. S. Sletten, R. P. Bailey and T. Bennett, Sorption and filtration of metals using iron-oxide-coated sand, Water Res., 1996, 30, 2609–2620 CrossRef CAS.
- A. J. Anschutz and R. L. Penn, Reduction of crystalline iron(III) oxyhydroxides using hydroquinone: Influence of phase and particle size, Geochem. Trans., 2005, 6, 60–66 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, NIH Image to ImageJ: 25 years of image analysis, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- L. L. Stookey, Ferrozine-A New Spectrophotometric Reagent for Iron, Anal. Chem., 1970, 42, 779–781 CrossRef CAS.
- R. M. Fand and R. Thinakaran, The Influence of the Wall on Flow Through Pipes Packed With Spheres, J. Fluids Eng., 2008, 112, 84 CrossRef.
- Y. H. Aly, C. Liu, D. P. McInnis, B. A. Lyon, J. Hatton, M. McCarty, W. A. Arnold, K. D. Pennell and M. F. Simcik, In Situ Remediation Method for Enhanced Sorption of Perfluoro-Alkyl Substances onto Ottawa Sand, J. Environ. Eng., 2018, 144, 04018086 CrossRef.
- A. Kreft and A. Zuber, On the physical meaning of the dispersion equation and its solutions for different initial and boundary conditions, Chem. Eng. Sci., 1978, 33, 1471–1480 CrossRef CAS.
- M. T. van Genuchten and J. C. Parker, Boundary Conditions for Displacement Experiments through Short Laboratory Soil Columns, Soil Sci. Soc. Am. J., 1984, 48, 703–708 CrossRef.
- M. J. Berens, T. B. Hofstetter, J. Bolotin and W. A. Arnold, Assessment of 2,4-Dinitroanisole Transformation Using Compound-Specific Isotope Analysis after In Situ Chemical Reduction of Iron Oxides, Environ. Sci. Technol., 2020, 54, 5520–5531 CrossRef CAS PubMed.
- S. Dixit and J. G. Hering, Sorption of Fe(II) and As(III) on goethite in single- and dual-sorbate systems, Chem. Geol., 2006, 228, 6–15 CrossRef CAS.
- C. A. Gorski and M. M. Scherer, Fe2+ Sorption at the Fe Oxide-Water Interface: A Revised Conceptual Framework Aquatic Redox Chemistry, ACS Symp. Ser., 2011, 1071, 315–343 CrossRef CAS.
- A. M. Vindedahl, M. S. Stemig, W. A. Arnold and R. L. Penn, Character of Humic Substances as a Predictor for Goethite Nanoparticle Reactivity and Aggregation, Environ. Sci. Technol., 2016, 50, 1200–1208 CrossRef CAS PubMed.
- C. Mikutta, J. G. Wiederhold, O. A. Cirpka, T. B. Hofstetter, B. Bourdon and U. Von Gunten, Iron isotope fractionation and atom exchange during sorption of ferrous iron to mineral surfaces, Geochim. Cosmochim. Acta, 2009, 73, 1795–1812 CrossRef CAS.
- R. M. Handler, A. J. Frierdich, C. M. Johnson, K. M. Rosso, B. L. Beard, C. Wang, D. E. Latta, A. Neumann, T. Pasakarnis, W. A. P. J. Premaratne and M. M. Scherer, Fe(II)-catalyzed recrystallization of goethite revisited, Environ. Sci. Technol., 2014, 48, 11302–11311 CrossRef CAS PubMed.
- C. M. Hansel, S. G. Benner, J. Neiss, A. Dohnalkova, R. K. Kukkadapu and S. Fendorf, Secondary mineralization pathways induced by dissimilatory iron reduction of ferrihydrite under advective flow, Geochim. Cosmochim. Acta, 2003, 67, 2977–2992 CrossRef CAS.
- M. B. Kirkham, Pore volume, in Principles of Soil and Plant Water Relations, Academic Press, 2014, ch. 14, pp. 229–241 Search PubMed.
- C. G. Heijman, E. Grieder, C. Holliger and R. P. Schwarzenbach, Reduction of Nitroaromatic Compounds Coupled to Microbial Iron Reduction in Laboratory Aquifer Columns, Environ. Sci. Technol., 1995, 29, 775–783 CrossRef CAS PubMed.
- P. V. Brady and J. V. Walther, Kinetics of quartz dissolution at low temperatures, Chem. Geol., 1990, 82, 253–264 CrossRef CAS.
- F. K. Crundwell, On the Mechanism of the Dissolution of Quartz and Silica in Aqueous Solutions, ACS Omega, 2017, 2, 1116–1127 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1en00788b |
| This journal is © The Royal Society of Chemistry 2022 |