
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePinus nigra bark from a mercury mining district studied with high resolution XANES spectroscopy†
Fabrizio
Bardelli
 *ab,
Valentina
Rimondi
c,
Pierfranco
Lattanzi
d,
Mauro
Rovezzi
e,
Marie-Pierre
Isaure
f,
Andrea
Giaccherini
c and
Pilario
Costagliola
c
*ab,
Valentina
Rimondi
c,
Pierfranco
Lattanzi
d,
Mauro
Rovezzi
e,
Marie-Pierre
Isaure
f,
Andrea
Giaccherini
c and
Pilario
Costagliola
c
aUniversità di Roma La Sapienza, Roma, Italy. E-mail: fabrizio.bardelli@gmail.com
bCNR-Nanotec, Italy
cUniversità di Firenze, Italy. E-mail: pilario.costagliola@unifi.it; andrea.giaccherini@gmail.com; valentina.rimondi@unifi.it
dCNR, IGG, Italy. E-mail: pierfrancolattanzi@gmail.com
eUniv. Grenoble Alpes, CNRS, IRD, Irstea, OSUG, FAME, Météo France, Grenoble, France. E-mail: mauro.rovezzi@esrf.eu
fUniversité de Pau et des Pays de l'Adour, E2S UPPA, CNRS, IPREM, Pau, France. E-mail: marie-pierre.isaure@univ-pau.fr
First published on 5th August 2022
Abstract
Tree bark near former mercury (Hg) mines and roasting plants is known to have exceptionally high (up to several mg kg−1) Hg concentrations. This study explores the change of Hg speciation with depth (down to 25–30 mm from the outermost surface) in black pine (Pinus nigra) bark by means of high-resolution X-ray absorption near edge structure (HR-XANES) spectroscopy at the Hg LIII-edge. Principal component analysis and linear combination fitting applied to the HR-XANES spectra suggested that in the outermost layer (∼0–2 mm from the surface), roughly 50% of Hg is in the form of nanoparticulate metacinnabar (nano-β-HgS). A progressive increase in Hg-organic species (Hg bound to thiol groups) is found in deeper bark layers, while nano-β-HgS may decrease below the detection limit in the deepest layers. Notably, bark layers did not contain cinnabar (α-HgS), which was found in the nearby soils along with β-HgS (bulk), nor Hg0, which is the main Hg species in the atmosphere surrounding the sampled trees. These observations suggested that nano-β-HgS, at least in part, does not originate from mechanically trapped wind-blown particulates from the surrounding soil, but may be the product of biochemical reactions between gaseous elemental Hg and the bark tissue.
Environmental significanceA detailed knowledge of Hg speciation in environmental samples is critical to correctly predict the potential impacts. However, performing X-ray near-edge absorption spectroscopy (XANES) in samples with Hg amounts <100 mg kg−1 is challenging, because of the low signal-to-noise ratio of the resulting spectra. Moreover, the large core-hole lifetime broadening at the Hg LIII-edge results in poor spectral features, preventing a robust analysis of the data. The use of high-resolution XANES (HR-XANES) allows overcoming both limitations. In this work, HR-XANES was exploited for the first time to study Hg-contaminated tree bark samples. The results suggest that the bark, which was considered inert, may instead actively control the Hg biogeochemistry by transforming gaseous Hg into an insoluble, and thus scarcely bioavailable, Hg(II)-sulfide. |
1 Introduction
Mercury (Hg) is one of the pollutants of most concern due to its high toxicity, persistence in the atmosphere, and bioaccumulation ability.1 It is usually released to the atmosphere by anthropogenic sources, such as Hg and gold (Au) artisanal mining, coal-fired power stations and steel plants, or from waste management processes, such as landfills and incinerators.2–4 It can then circulate in the atmosphere and deposit in aquatic and terrestrial ecosystems, becoming part of the global Hg cycle.5,6 There are, consequently, increasing efforts to quantify and precisely define the processes controlling the Hg global budget.7,8The largest part of Hg present on or in the vegetation originates from gaseous (atmospheric), elemental Hg.9,10 Forests, in particular, are essential to regulate the Hg biogeochemistry cycle, due to the strong affinity of Hg to organic matter in soils and vegetation. For this reason, vegetation has been proposed as a biomonitor to evaluate the extent of Hg deposition in terrestrial ecosystems.11–14 On the other hand, forests may recycle Hg back to the atmosphere, because Hg uptake may involve both reversible and irreversible mechanisms.15 Wildfires are another variable to consider; indeed, in the 1997–2006-decade, Hg release due to biomass combustion accounted for 8% of the global (natural + anthropogenic) Hg emissions.16,17 While this contribution is minor on a global scale, local or regional impacts could be much more pronounced,16 and biomass burning is believed to be a major driver of the inter-annual Hg variations in the troposphere.18
Forest vegetation removes atmospheric gaseous Hg0 through stomatal and non-stomatal uptake.19–21 Similarly, tree bark may concentrate significant amounts of total Hg, and is proposed to be efficient monitors of airborne Hg.12,14,22–26 In particular, a study investigating the potential of Pinus pinaster as a biosorbent of Hg2+ suggested that the metal is adsorbed from aqueous solution by an ion exchange mechanism.27 However, the mechanism of Hg uptake at the bark/atmosphere interface, where Hg is essentially present as Hg0,28 is only partially known.29 Hanson et al. (1997)30 indicated that Hg0 is also re-emitted from bark, although at rates probably minor compared to those of soil or foliage.
After plant tissue senescence, the plant detritus (i.e., litterfall), composed also of tree bark, brings Hg back to the soil reservoir.31–33 Here, Hg may eventually be sequestered by forming insoluble Hg-phases,34 evade back to the atmosphere, or even methylate,35 possibly increasing its geo/bio availability.36 It is therefore important to know the exact speciation of Hg contained in tree bark, which, in turn, would contribute to control the Hg exchange rates with the environment.
Black pine (Pinus nigra) bark from Monte Amiata (Italy) is known to contain exceptionally high (up to several mg kg−1) Hg content, because of the proximity to former Hg mines and roasting plants of the third largest Hg district in the world.12,37 Over a century of mining and roasting caused Hg dispersion in the atmosphere and in the drainage network. Moreover, the area hosts a geothermal field, which is exploited to produce energy. These geothermal power plants are an additional source of Hg in the environment.38
In our previous study, conventional X-ray absorption near edge structure spectroscopy (XANES) data suggested that Hg speciation in superficial bark (<16 mm) included metacinnabar (β-HgS), possibly Hg0, and organic Hg-species (namely, Hg bound to cysteine or tannic acid).29 However, the comparatively low energy resolution of the data and the limited set of Hg references prevented a reliable determination of the trend of Hg speciation with depth. The study of the Hg content and speciation with depth is important to evaluate if bark can be used as a biomonitor for atmospheric Hg pollution. For example, Arnold et al., 201839 claimed that Pinus bark is not a good passive sampler for Hg air concentrations, because they found no correlation between Hg bark and total gaseous Hg concentrations. However, it should be noted that the total gaseous Hg concentration that the authors used to expose saplings was much lower than that detected in the area surrounding the trees sampled in this work. Moreover, these authors did not differentiate between inner and outer bark.
In this study, the change of Hg speciation with depth (down to ∼30 mm from the outermost surface) in Pinus nigra outer bark from Monte Amiata is explored by exploiting, for the first time for this subject, high resolution XANES (HR-XANES). The Hg speciation in bark is compared with that in the corresponding soils collected nearby the sampled trees, and the implications for the Hg biogeochemical cycle are discussed.
2 Materials and methods
2.1 Sampling
Outer bark layers of black pine (Pinus nigra J. F. Arnold) trees were sampled in 2016 in and around the Abbadia San Salvatore town in the Monte Amiata region (Fig. 1 and Table 1), where mining activity for Hg was conducted up to the 1980s. As local soil dust is expected to be the most likely contribution to particulates,12,29 bark layers (0–50 mm portions) were handpicked at about 150 cm above ground (Fig. S1†), following the procedure described in Rimondi et al. (2020)37 in order to minimize the amount of particles deposited on bark layers which originated from soil resuspension. Tree bark samples were collected at the four cardinal directions to take into account possible changes in Hg concentrations with preferential wind directions. However, as no systematic changes in the Hg concentrations were observed with the cardinal direction, for each tree, the sample with the highest Hg concentration was used. Immediately after sampling, bark samples were sealed in plastic bags and then stored at −20 °C to prevent the loss of volatile Hg species and limit possible changes in Hg speciation.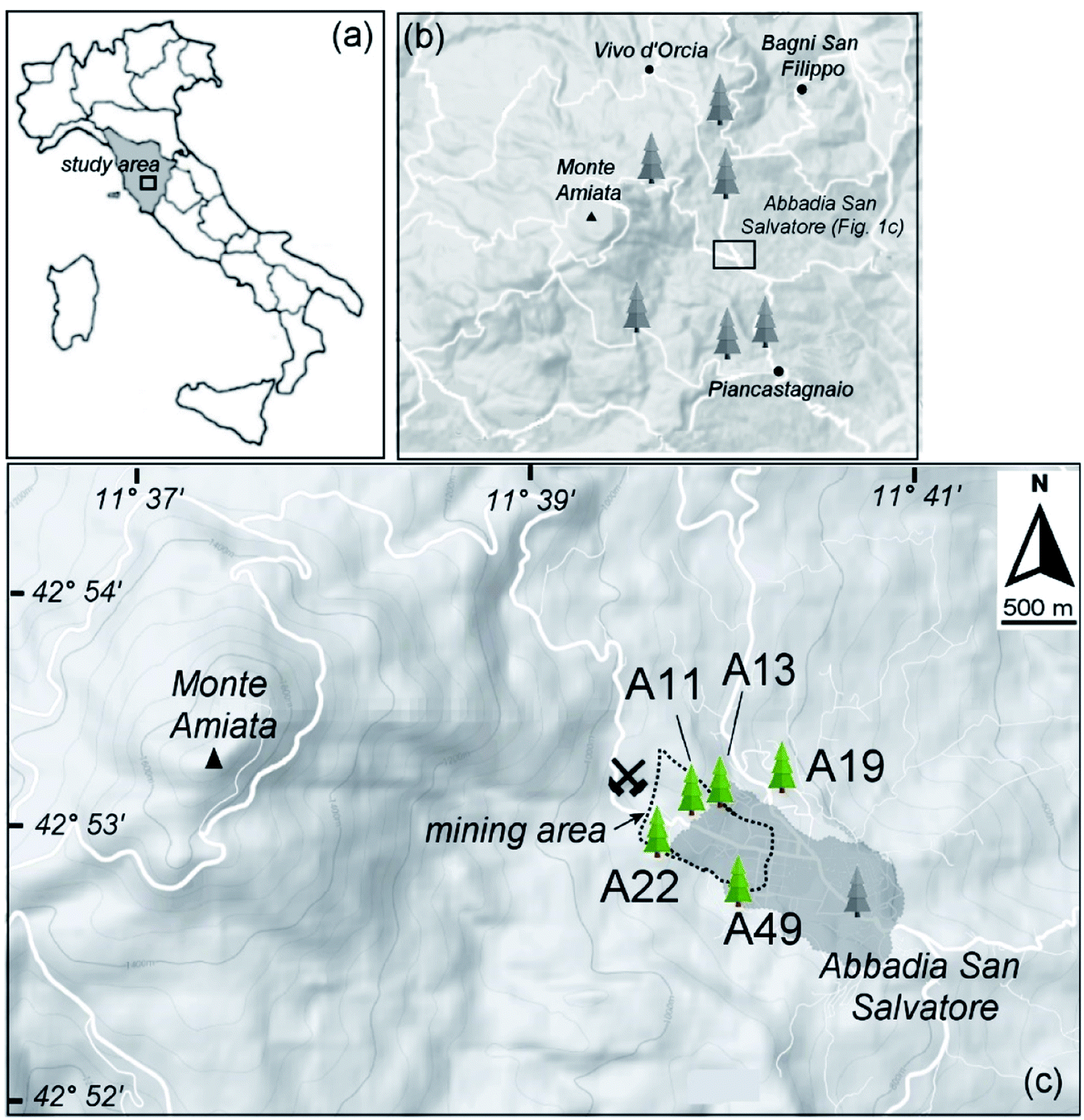 | ||
| Fig. 1 (a) Study area and sampling sites considered in the present work; (b) sampling sites considered in Rimondi et al. (2020).37 (c) Zoomed view of the investigated area. The mining and smelting area near to the Abbadia San Salvatore town (darker grey area) is indicated by the dotted line contour. The trees selected for the HR-XANES study are reported (green tree symbols). | ||
| Sample ID | Tree age (years) | Depth (mm) | Hg in bark (mg kg−1) | Hg in soil37 (mg kg−1) | Hg0 in air40 (ng m−3) |
|---|---|---|---|---|---|
| A11 | 100 | 0–2 | 8.6 ± 1.9 | 480 | 9.86–15.7 |
| 2–4 | 20.4 ± 6.4 | ||||
| 4–8 | 5.3 ± 0.7 | ||||
| A13 | 150 | 0–2 | 16.5 ± 2.4 | 186 | 16.0–17.9 |
| 2–8 | 9.8 ± 0.3 | ||||
| 8–15 | 8.9 ± 1.0 | ||||
| 15–25 | 1.4 ± 0.08 | ||||
| A19 | 100 | 0–5 | 13.7 ± 1.3 | 66 | 3.82–4.24 |
| 5–10 | 11.3 ± 0.3 | ||||
| 10–19 | 7.6 ± 0.3 | ||||
| 19–29 | 2.6 ± 0.03 | ||||
| A22 | 150 | 0–2 | 8.3 ± 0.05 | 66 | 7.48–14.8 |
| 2–9 | 4.3 ± 1.3 | ||||
| 9–15 | 3.8 ± 0.01 | ||||
| 15–25 | 3.0 ± 0.1 | ||||
| A49 | 100 | 0–2 | 9.0 ± 2.3 | 163 | 25–116 |
| 2–6 | 7.7 ± 0.02 | ||||
| 6–13 | 5.9 ± 0.03 | ||||
| 13–27 | 1.5 ± 0.01 |
The Pinus nigra bark typically consists of easily split layers, having a thickness of some millimeters. Bark blocks of about 30 mm from the bark surface were sampled. The phloem (living tissue) was not reached, so that the sampled bark layers were composed of tissue with no active translocation of soluble organic compounds. More details about the sampling procedure can be found in Rimondi et al., 2020.37 Soils were collected as composite samples: three splits from the first 10 cm ground layer from random locations around the tree, outside the tree canopy, and after removal of the tree litter.
2.2 Analytical methods
In a previous study,37 tree bark layers were analyzed for the Hg concentration in bulk 15 mm-thick slices. In the present study, we investigated the Hg concentration and speciation as a function of the depth from the bark surface. Each bark was prepared on a cutting board, where slices of approximately 2–10 mm were separated with the help of a ceramic knife, following the inherent structure of the Pinus nigra bark. For this reason, no attempt was made to achieve pre-determined layer thicknesses, which varied from one sample to another. Each subsample (i.e., layer) was finely crumbled (<500 μm) on the cutting board and transferred to polypropylene flasks for further analysis. This procedure was performed under a chemical hood to limit possible Hg contamination from external sources. The concentration of gaseous Hg inside the hood was determined with a Lumex instrument and was close to the instrument detection limit, i.e. orders of magnitude lower than in the natural environment where the bark layers were sampled. Accurate cleaning of materials was performed between the samples to avoid cross contamination. The aliquot used for total Hg analysis was analyzed by means of a Direct Hg Analyzer (DMA-80 Evo, Milestone) following the US EPA 7473 method, under wet conditions to avoid possible Hg volatilization. Another aliquot was heated to 110 °C until constant weight to calculate the water loss and correct the total Hg values obtained on the wet aliquot. To estimate the analytical precision, three replicate analyses of the same slice were routinely performed; the results were generally reproduced within 15–20% of the average value. Accuracy was tested with international standards (NIST 1573a, 1575, and 2711a) and was within 10%.To achieve the best possible signal-to-noise ratio in absorption spectra, nineteen bark samples from five trees with the highest Hg total content (>1 mg kg−1) were chosen. The Hg content of the sample portions (about 0.5 g) brought to the synchrotron facility are reported in Table 1. Despite the relatively high amount of Hg in the samples, HR-XANES measurements remained challenging. In particular, the acquisition of spectra of the samples containing less than 10 mg kg−1 of Hg with statistics comparable to those of the higher Hg-concentration samples would have taken too long compared to the available beamtime. Therefore, the deepest bark layer spectra, which correspond to the lowest Hg-concentrations, have a lower signal to noise ratio.
Soils were air dried under a chemical hood, sieved at 2 mm and ground for geochemical investigations. Total Hg contents, determined with a Milestone DMA-80 instrument using the same protocol described for bark samples to prevent the loss of volatile Hg species, exhibited high analytical variability. Therefore, a larger mass of sample (1 g) was digested in a sand bath with aqua regia (HCl/HNO3 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and analyzed by inductively coupled plasma optical emission spectrometry (ICP-OES, PerkinElmer Optima 8000 instrument) equipped with a hydride generator (PerkinElmer B0507957). Two of the samples were processed in duplicate to evaluate method precision, which was within 15%. The analytical accuracy, checked against CCRMP STSD-1 (ICP-OES) international standards, was within 10%. The mercury concentration in soil samples ranged between 66 and 480 mg kg−1 (Table 1).
:1) and analyzed by inductively coupled plasma optical emission spectrometry (ICP-OES, PerkinElmer Optima 8000 instrument) equipped with a hydride generator (PerkinElmer B0507957). Two of the samples were processed in duplicate to evaluate method precision, which was within 15%. The analytical accuracy, checked against CCRMP STSD-1 (ICP-OES) international standards, was within 10%. The mercury concentration in soil samples ranged between 66 and 480 mg kg−1 (Table 1).
2.3 X-ray spectroscopy
High resolution X-ray absorption near-edge structure spectroscopy measurements were performed at the French CRG FAME-UHD beamline,41 BM16 port, at the European Synchrotron Radiation Facility (ESRF, France). The monochromator was equipped with a couple of cryogenically cooled Si 220 mirrors with a dynamical sagittal focusing of the second crystal. The beam spot size on the sample was 200 μm (H) × 100 μm (V) FWHM. All spectra were measured at the Hg LIII-edge (12284 eV) in fluorescence yield detection mode with a crystal analyzer spectrometer. The Hg Lα1 (3d5/2 → 2p3/2) fluorescence line was selected using the 555 reflection of twelve spherically bent (radius 1 m) Si analyzer crystals (diameter 100 mm), aligned at an 81.8° Bragg angle in a vertical Rowland circle geometry. The total energy bandwidth of the twelve crystal analyzers was 1.38 eV, measured with individual elastic peak scans. Spectra were collected at a temperature of ∼10 K, using a He flow cryostat, to avoid possible photon-induced redox reactions. The position of the sample with respect to the X-ray beam was changed at each scan to minimize beam damage and to account for the inherent inhomogeneity of the samples. Calibration of the monochromator was performed with an elemental Hg reference (see below for preparation details). Compared to our previous study,29 the experimental setup with the crystal analyzer spectrometer offered significantly increased energy resolution. This resulted in more distinctive spectral features (see Fig. S2†), which were essential to effectively reveal the Hg speciation.
Portions of the same samples used for the chemical characterization were newly ground for synchrotron analysis in an agate mortar, then mixed and homogenized with cellulose and pressed into a pellet. This procedure should have ensured a more homogeneous distribution of Hg in the analyzed samples. Extreme care was taken to avoid cross contamination among the samples having different concentrations. Depending on the concentration of Hg, five to twenty consecutive scans were acquired and then averaged to improve statistics.
Reference Hg species measured by HR-XANES included: (i) inorganic Hg compounds, in particular, sulphides (α-HgS, β-HgS), which may derive from entrapment of windblown particles from the surrounding soil, and (ii) organic-bound Hg species, which may derive from the binding of Hg with organic matter in soil and/or in the bark. The Hg references HgCl2, HgO, CH3HgCl, and Hg-acetate (Hg(CH3COO)2) were purchased from Merck or Sigma Aldrich (purity > 95%). α-HgS (cinnabar) and β-HgS (metacinnabar) were natural specimens,42 which were evaluated for purity by X-ray powder diffraction. The elemental Hg reference (Hg0) was prepared by thoroughly mixing liquid Hg with epoxy resin, which was then cut with a microtome to make a film as thin as possible, suitable for acquisition in the transmission mode. The Hg-tannic acid reference (Hg-phenol) was prepared by dissolving 500 mg of tannic acid in Milli-Q® water, adding 2.5 mL of Hg(NO3)2 1000 mg L−1 solution, adjusting the pH to 5, and then freeze drying the solution. Liquid references, Hg-cysteine ([Hg] = 18 mM, M/L = 1/2, pH = 2.2, and Hg(Cys)2), CH3HgOH ([Hg] = 43 mM; pH = 9.4), CH3Hg-cysteine ([Hg] = 21 mM, M/L = 1/2, pH = 7.2, and CH3Hg-(Cys)2), and CH3Hg-glycine ([Hg] = 10 mM, M/L = 1/1, and pH = 7.1), were synthetized as described in Isaure et al. (2020), and measured as frozen liquids at 20 K. In Hg(SR)2 and Hg(SR)3, Hg is bonded with two or three thiolate groups, respectively (SR = cyclohexanethiolate).44,45 Hg(SR)2 has a polymeric structure consisting of bent S–Hg–S bonds (160.4°) linked together by weaker and farther Hg–S bonds. The Hg atoms have distorted tetrahedral coordination, and each thiolate acts as an asymmetric bridge between two Hg atoms. Hg(SR)3 contains Hg atoms in a distorted trigonal-planar coordination environment, with one angle larger than and one lower than 120°.46 The details of the synthesis of the above references are reported in Isaure et al. (2020),43 except for the Hg(SR)2 and Hg(SR)3 references, whose preparations are reported in Alsina et al. (1992).46
Further references are available in the ESI† of some published papers.45,47,48 These included nanoparticulate β-HgS (nano-β-HgS) and Hg bound to cysteine groups or to L-glutathione groups, both synthetized at pH 7.5 (Hg(Cys)2 and Hg(GSH)2, with GSH = γ-Glu–Cys–Gly),47 bulk and nanoparticulate HgSe,48 and Hg bound to thiolate and NO3 groups (Hg(SR)2 + (NO3)2).45 The HR-XANES spectra of all reference compounds are reported in Fig. S3† in the ESI.†
Principal component analysis (PCA) and linear combination fitting (LCF) were applied to the nineteen HR-XANES spectra to obtain quantitative information on the sample speciation. PCA allows determining the number of abstract components responsible for the main spectral features, which, in an actual system, can be related to the number of Hg species. It can also suggest the best candidates among the set of available references through a procedure named target transformation (TT49–51).
LCF consists of a weighted linear combination of the reference standards able to best fit a given experimental spectrum. PCA and LCF were performed using the SIXPACK and IFEFFIT packages, respectively.52,53
2.4 Electron microscopy microanalysis
All external layers of the five bark samples, one intermediate layer, A13_2–6 mm, and two soil samples, A13 and A11, were also analyzed using scanning electron microscopy coupled with energy dispersive spectroscopy (SEM-EDS), with an EVO-MA15 Zeiss model, equipped with the Oxford INCA 250 microanalysis software, at the Interdepartmental Center for Electron Microscopy and Microanalysis Services (MEMA) of the University of Florence. All the samples were metallized with a few nm of graphite and analyzed at 20 kV acceleration voltage, 700 pA electron current, and 8–10 mm working distance. Due to the discrete distribution of Hg, the surface of soil and bark was investigated favoring the natural atomic number contrast of any probable Hg particle with respect to the bark substrate, that is, increasing the contrast and lowering the brightness of the acquisition mode (brightness ∼5% and 80–90% of contrast). The surface was then observed at 2000×.3 Results
3.1 Soil samples
Principal component analysis performed on the five samples of soils showed that all the experimental soil spectra (Fig. S4†) can be satisfactorily described by two principal components (see Fig. S7†). The target transformation procedure suggested bulk β-HgS and α-HgS as the best candidates among the set of reference compounds (see Table S3† for a list of the most relevant statistical indicators of the TT). SEM observations confirmed the presence of micron-sized HgS particles (Fig. S5†), although their morphology (crystal habit) did not allow discrimination between α-HgS and β-HgS. Initially, the nano-β-HgS reference was included in the LCF along with bulk β-HgS and α-HgS. However, the three-component LCF did not significantly improve the match with the experimental data, and the bulk β-HgS reference resulted always as the major or only component along with α-HgS. Accordingly, LCF analysis suggested that the main Hg speciation in the soil samples is composed of an admixture of variable relative fractions of β-HgS and α-HgS (∼60 and ∼40% on average, respectively). The spectra and LCF curves of soil samples (n = 5) are reported in Fig. S4† and the corresponding numerical results in Table S2.†Notably, thanks to the higher energy resolution of the data, the presence of elemental Hg or organic-bound Hg (at least in amounts larger than ∼5–10%, which was estimated to be the maximum sensitivity of the HR-XANES data), which was a possibility in our previous study,42 can be ruled out. The different spectral features arising from the contribution of different relative amounts of α-HgS and β-HgS are clear, as highlighted in Fig. 2, where the spectra of the two representative soil samples, displaying the highest (lowest) and lowest (highest) β-HgS (α-HgS) contents (i.e. A19 and A49, respectively) and the corresponding LCF, are shown.
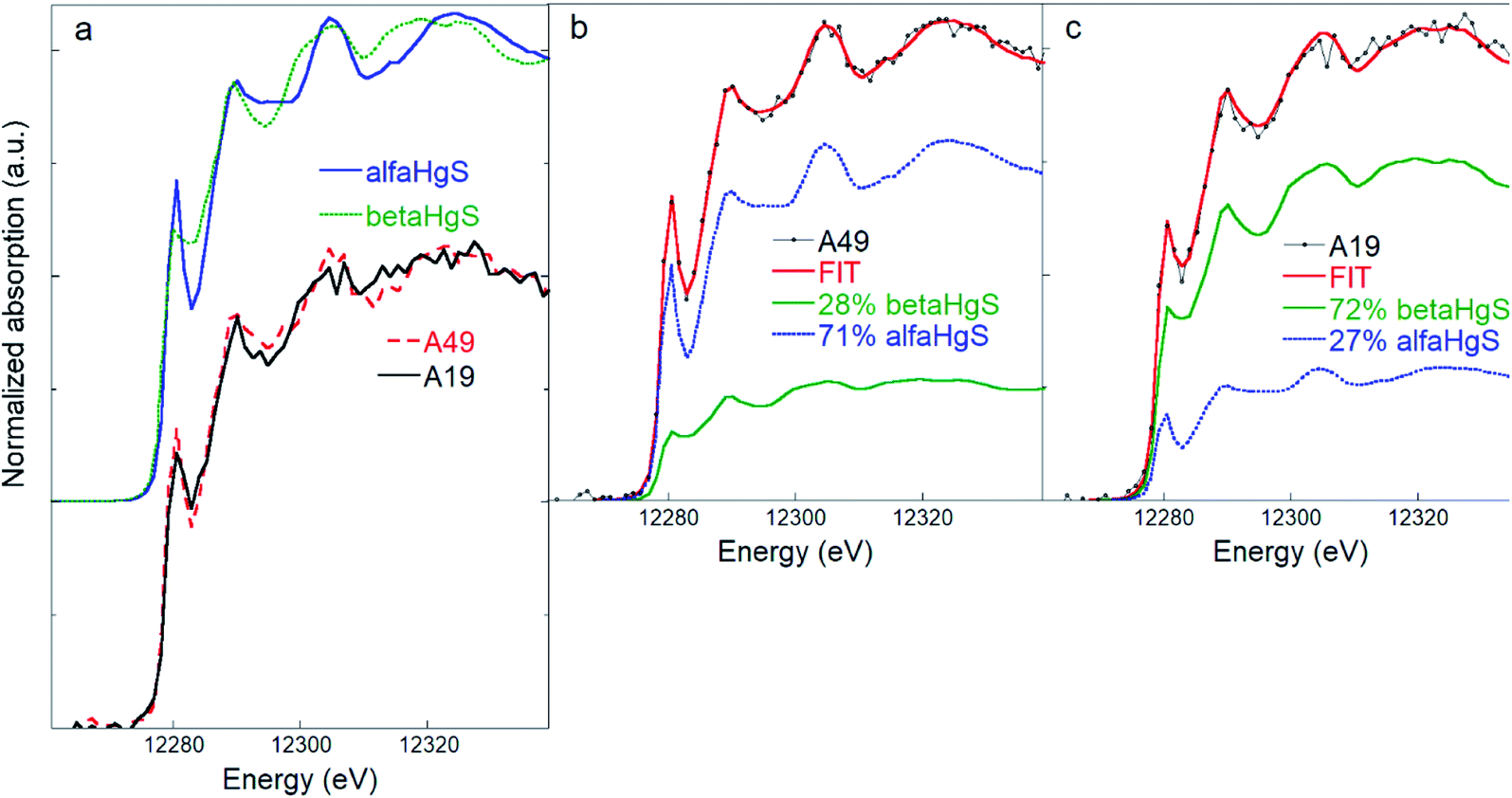 | ||
| Fig. 2 (a) Comparison of β-HgS (metacinnabar) and α-HgS (cinnabar) and of two representative soil spectra (A19 and A49) highlighting the different spectral features. (b) and (c) Linear combination fitting (LCF) of the soil samples A49 (b) and A19 (c) performed using the β-HgS and α-HgS reference spectra, showing how different relative amounts of β-HgS and α-HgS reflect in different spectral features in the mixed phase. The numerical values of the LCF are reported in Table S2.† | ||
3.2 Bark samples
All samples' spectra, including those with the lowest Hg concentrations, show a well-defined Hg LIII absorption edge step and a workable XANES in the region 12260–12340 eV (Fig. S6†). In general, for a given sample, the single spectrum was not much different from one from another, even if they were acquired at different points of the sample to prevent beam damage (see the Materials and methods); therefore, the sum of all spectra usually had the only effect of improving the counting statistics. However, in some cases, the differences were large enough, reflecting the intrinsic inhomogeneity of the samples, which was reduced, but clearly not completely removed, by grinding and thoroughly mixing the samples. To take into account this inhomogeneity, outlier spectra were included in the final averages. The majority of the spectra shows intermediate spectral features between bulk or nano-β-HgS and some organic Hg references, in particular, Hg(Cys)2, Hg(SR)2, Hg(GSH)2, and CH3HgCl (Fig. S3†). A visual screening of the bark spectra showed that the samples collected from surface layers have a closer match with the β-HgS references, while those from the deepest layers have a more pronounced peak in the pre-edge region, and a good match with the Hg(Cys)2, Hg(SR)2, Hg(GSH)2, or CH3HgCl references. The latter have a distinctive peak at 12280 eV, which is less intense in the tetragonal β-HgS reference (Fig. S3†), and which has been attributed to the degree of the linearity of the S–Hg–S bonds in Hg thiol compounds.21 For the Hg(Cys)2 reference, a second absorption peak is observed at 12288.5 eV (Fig. S3†). This feature shifts toward 12290 eV in the Hg(SR)2 and CH3HgCl references, as found in the spectra of the deeper bark layers (Fig. S6†).
Principal component analysis, performed on the nineteen bark samples' spectra, suggested that all the spectra can be satisfactorily described with three principal components (see Fig. S7†). The target transformation procedure indicated β-HgS, nano-β-HgS, Hg(SR)2, Hg(GSH)2, CH3Hg(Cys)2, Hg(Cys)2, CH3HgCl, and Hg(SR)3 as the acceptable candidates among the set of reference compounds (see Table S3† for a list of the most relevant statistical indicators for the TT). Despite the information from PCA, all combinations of three (or more)-component LCF were attempted. However, in the final refinements, fits with more than two-components were discarded, because of the negligible changes in the χν2 and R2 statistical indicators with respect to two-component fits. More details on the LCF analyses are reported in a dedicated section at the end of the ESI† section.
Quantitative two-component LCF of the surface layer samples resulted in good matches using a combination of the nano-β-HgS and Hg(Cys)2 references (Fig. 3). The nano-β-HgS spectrum has a lower amplitude structure compared with its bulk counterpart, as is expected from a nanoparticulate material, and it was found to significantly improve the fits in the region ∼25 eV above the edge with respect to the bulk β-HgS one. When the nano-β-HgS fraction diminished below the sensitivity of the LCF (i.e. 5–10%, as estimated considering negligible changes in the χν2 and R2 statistical indicators), which usually occurred in the case of the deepest layers (Table S1†), the best fits were obtained using a combination of the Hg(Cys)2 and Hg(SR)2 references. This can be explained by the fact that the spectral features of Hg(Cys)2 and Hg(SR)2 are similar, while those of nano-β-HgS are significantly different. Therefore, when nano-β-HgS is present, the sensitivity required to discern between Hg(Cys)2 or Hg(SR)2 is highly reduced, and only one among these Hg organic references suffices to obtain good matches with the experimental data.
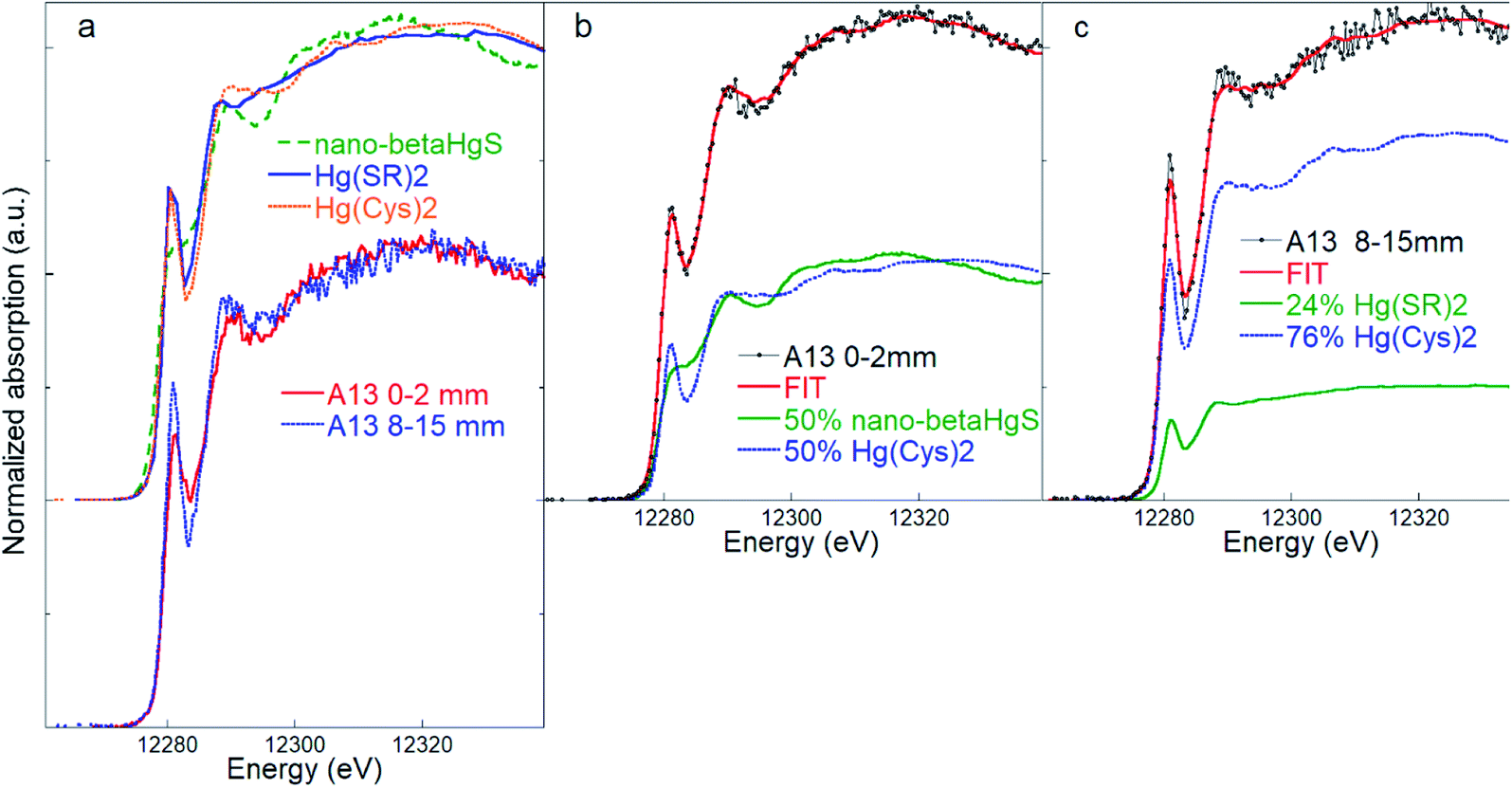 | ||
| Fig. 3 (a) Comparison of the inorganic nano-β-HgS (nanoparticulate metacinnabar) and Hg(SR)2 and Hg(Cys)2 organic references, and of bark spectra representative of the outermost (0–2 mm) and inner (8–15 mm) layers of sample A13. (b) Linear combination fitting (LCF) of the bark samples A13 (layer 0–2 mm) and A13 (layer 8–15 mm) (c) using references Hg(SR)2 and Hg(Cys)2. The numerical values of the LCF are reported in Table S2.† | ||
It has to be noted that, for deeper layer samples, good results were also obtained with two-component fits performed using Hg(SR)2 or Hg(Cys)2 and the CH3HgCl reference.
With only one exception out of nineteen spectra, the distribution of the Hg species with depth showed the same general trend: the inorganic β-HgS fraction decreases from the outermost to the innermost layer, with a corresponding increase of the organic-bound Hg species. This trend is highlighted in a bar plot in Fig. 4, where the average total (bulk) Hg content is also reported. The inorganic Hg fraction is dominant in the outermost layer (46 ± 10% on average) and decreases down to 16 ± 15% on average in the innermost layer, while the organic Hg fraction increases accordingly. Likewise, the total Hg content decreases monotonically from the outermost to the innermost layer. In agreement with the above results, SEM-EDS observations detected the rare presence of discernible HgS particles (about one micrometer size) in the outermost bark layers (Fig. S5†), while no such particles could be observed in the inner portions.
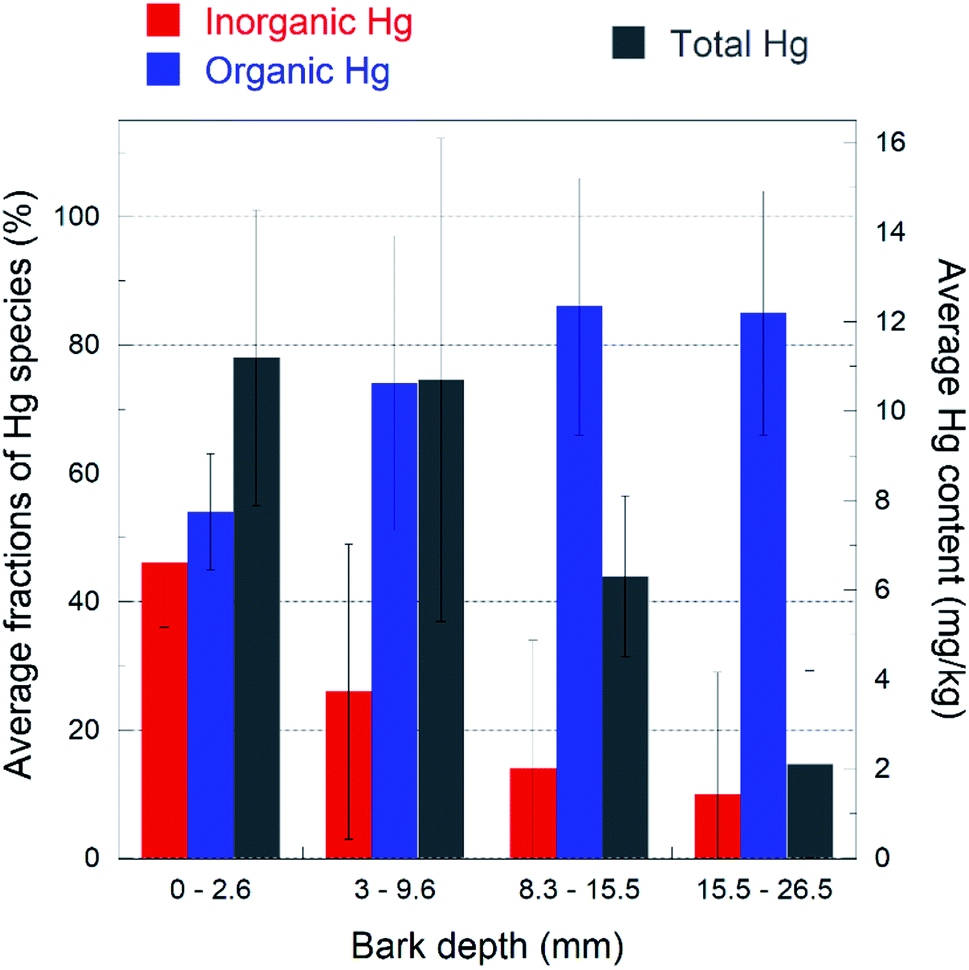 | ||
| Fig. 4 Bar plot highlighting the trend of the inorganic (nano-βHgS, red color) and Hg-organic (blue color) fractions (%) as a function of the depth of the bark layer. Total Hg content (mg kg−1) is also reported (gray color). The fractions shown are the averages of the values reported in Table S1,† for each layer. The bark layer thicknesses have been averaged, because they were slightly different for each bark sample (see the Materials and methods). The data show that the organic Hg fraction increases with depth at the expense of the inorganic one, and the total Hg content decreases with depth. The error bars represent the standard deviations for each layer (n = 5). | ||
4 Discussion
The decrease of the total Hg content from the bark surface toward the deeper layers suggests that most of this pollutant originates from an external source (i.e. wind-blown particulate or gaseous atmospheric Hg0) and then diffuses to the inner part of the bark. In fact, many authors believe that systemic Hg uptake in tree tissues from soils is negligible,15,39,54–56 particularly from roots, where Hg is in poorly soluble phases, such as in the Monte Amiata district.42 Accordingly, in this work, no organic bonded, and thus more bioavailable, Hg was detected in soil samples. Manceau et al.21 detected α-HgS and nanoparticulate β-HgS in foliage from two Hg mining districts in China.In a previous study,29 it was suggested that β-HgS in the bark originated from mechanically trapped soil particles, likely as resuspension of the surrounding soil, where Hg sulphides were detected. This is supported by SEM-EDS observations, which revealed the presence of micrometer-size HgS particles at the bark surface. However, in some samples (Table S1†), β-HgS also occurs in significant amounts (>30%) in the deepest layers (down to 29 mm), where micron-sized HgS particles were not detected by SEM-EDS, suggesting that in deeper layers, the origin of β-HgS may be different. In addition, bulk β-HgS was detected in the soil surrounding the samples, while nanoparticulate β-HgS was detected in bark samples, suggesting a different origin for the Hg-sulphide species in soils and bark. Indeed, β-HgS may occur as a biogenic nanoparticulate, in agreement with recent studies,21,44,57 which show that β-HgS nanoparticulate clusters may be produced in low temperature systems, through formation of complexes with thiolates or cysteine. Specifically, Manceau et al.21 reported the formation of nanoparticulate β-HgS at the surface of leaves exposed to a Hg-bearing atmosphere by the reaction of gaseous Hg species via intermediate compounds, such as Hg(SR)2, which was detected in the deeper bark layers along with Hg-cysteine. At present, we cannot discriminate beyond any doubt between β-HgS of inorganic (metallurgical waste via soil) or biochemical origin. However, the results of the present work indirectly favor the second option because α-HgS was identified in the soil in almost the same proportion as that of β-HgS, but was notably absent from the bark matrix, suggesting that the origin of the two Hg-sulphides may be different.
Manceau and coauthors21,58 proposed two possible mechanisms for the formation of the Hg sulphide species in plant leaves or organic matter, both based on a biomineralization process: enzymatic sulphidization or chemical dealkylation. The authors, however, note that both mechanisms require Hg atoms to face each other, a possibility that increases with the total Hg content. In agreement with this scenario, in this work, higher nanoparticulate β-HgS amounts correlate with the total Hg content at the bark surface (Fig. 4).
The present HR-XANES data confirm that Hg is bonded with two S atoms, but the match with the Hg(Cys)2 or Hg(SR)2 references, or with a linear combination of them, is not exact. The actual local Hg coordination in the organic fraction is probably a more complex one, with the two primary thiol groups at an intermediate angle (>160° and <175°) and possibly, with one or more secondary bonds with amine, carboxyl, or hydroxyl groups, as proposed by Manceau et al.21
The possible formation of methylated species would be obviously of paramount importance for its ecotoxicological consequences. Indeed, LCF of some samples gives permissive evidence of the possible presence of methylated species, such as CH3HgCl. However, methylated Hg species in general are expected to be present at very low levels in the environment compared to other Hg species, μg kg−1vs. the mg kg−1 levels measured in the bark samples,59i.e. at much lower levels than it is required to introduce a significant contribution in the HR-XANES spectra (>5–10% of the total Hg content). Therefore, it is likely that the improvement of LCF observed when including the CH3HgCl reference is an artefact, probably arising from the poor ability of HR-XANES to distinguish adjacent elements, such as S and Cl. Substantial evidence against the presence of CH3HgCl comes from SEM-EDS observations, which did not show Hg bound to Cl.
The absence of Hg0 in bark layers, present as the dominant airborne Hg form in the surrounding atmosphere of the Monte Amiata mining district60 and which may have been present in the liquid form in the bark samples (detectable as solid (frozen) Hg0 in this work), is noteworthy. The results of this study suggest that gaseous elemental Hg is transformed at the bark surface by the reaction with thiol-containing proteins; although we cannot totally exclude a loss back to the atmosphere, despite the great care taken to prevent it (see the Materials and methods section). Possible transformations into comparatively scarcely mobile forms in the bark layers represent a sink that may slow down the diffusion of airborne Hg in the environment. On the other hand, bark may contribute to the occurrence of Hg in the litterfall through bark foliage and ultimately to soils. It has to be noted that no organic Hg compounds have been observed in soils surrounding trees, possibly because their concentration is lower than the detection limit of the HR-XANES technique (5–10% of the total Hg). This indicates that Hg sulphidation in bark may provide Hg to litterfall mainly as a sulphide, a theoretically poorly geo-available form of Hg, as found to occur in tree leaves.21 This conclusion supports the concept that the bark, which was considered inert, may actively contribute to the Hg biogeochemistry and eventually to the Hg cycle in the environment. However, it should be noted that the reactivity of nanoparticulate Hg species is not known at present, and more research on this subject would be important.
5 Conclusions
The results of this study confirm that Pinus nigra bark can be an efficient trap for airborne Hg. Furthermore, the observed trend of the Hg speciation with depth in the bark suggests that it behaves not only as a passive Hg sink, e.g. physically blocking or absorbing wind-blown Hg-bearing particulate or aerodispersed gaseous Hg, but can also support reactions that transform Hg into organic-bound compounds. An important step in this process could be the formation of insoluble, and thus scarcely bioavailable, biogenic nano-β-HgS. Among aspects worthy of further investigation are the possible presence of low concentrations of methylated species, which is of high concern because of their toxicity, and the determination of the reactivity, and effective bioavailability, of nanoparticulate Hg sulphide phases compared to their bulk counterparts.Conflicts of interest
There are no conflicts of interest to declare.Author contributions
Experiment planning: VR, PC, and PL. Preparation of Hg reference compounds: MPI. Beamline setup: MR and FB. Data acquisition: MR, FB, AG, VR, and PL. Spectra elaboration and fitting: FB. Preliminary draft: VR, FB, and PL. Final draft: all authors.Acknowledgements
We thank Laura Chiarantini and the electron microscopy facility (MEMA) of Università di Firenze for assistance with the SEM-EDS observations. We acknowledge the European Synchrotron Radiation Facility (ESRF), Grenoble, for granting beamtime for the HR-XANES experiment (EV-294) which was performed at beamline BM16. The study was funded by ordinary funds of a Ministero dell'Università e Ricerca (MIUR) grant to PC and VR. We also want to thank the three anonymous reviewers, whose perceptive comments greatly improved the manuscript.References
- R. A. Bernhoft, Mercury Toxicity and Treatment: A Review of the Literature, J. Environ. Public Health, 2012, 2012, 460508 CrossRef PubMed.
- H. Agarwalla, R. N. Senapati and T. B. Das, Mercury emissions and partitioning from Indian coal-fired power plants, J. Environ. Sci., 2021, 100, 28–33 CrossRef CAS PubMed.
- D. van Velzen, H. Langenkamp and G. Herb, Review: mercury in waste incineration, Waste Manage. Res., 2002, 20, 556–568 CrossRef CAS PubMed.
- S. Pervez, A. Koshle and Y. Pervez, Study of spatiotemporal variation of atmospheric mercury and its human exposure around an integrated steel plant, India, Atmos. Chem. Phys., 2010, 10, 5535–5549 CrossRef CAS.
- W. H. Schroeder and J. Munthe, Atmospheric mercury—An overview, Atmos. Environ., 1998, 32, 809–822 CrossRef CAS.
- Dynamics of mercury pollution on regional and global scales: Atmospheric processes and human exposures around the world, ed. N. Pirrone and K. R. Mahaffey, Springer, 2005, pp. 1–734 Search PubMed.
- D. O'Connor, D. Hou, Y. S. Ok, J. Mulder, L. Duan, Q. Wu, S. Wang, F. M. G. Tack and J. Rinklebe, Mercury speciation, transformation, and transportation in soils, atmospheric flux, and implications for risk management: A critical review, Environ. Int., 2019, 126, 747–761 CrossRef PubMed.
- P. Panagos, M. Jiskra, P. Borrelli, L. Liakos and C. Ballabio, Mercury in European topsoils: Anthropogenic sources, stocks and fluxes, Environ. Res., 2021, 201, 111556 CrossRef CAS PubMed.
- J. A. Ericksen, M. S. Gustin, D. E. Schorran, D. W. Johnson, S. E. Lindberg and J. S. Coleman, Accumulation of atmospheric mercury in forest foliage, Atmos. Environ., 2003, 37, 1613–1622 CrossRef CAS.
- D. F. Grigal, Mercury Sequestration in Forests and Peatlands, J. Environ. Qual., 2003, 32, 393–405 CAS.
- F. Baroudi, J. Al-Alam, O. Delhomme, S. Chimjarn, Z. Fajloun and M. Millet, The use of Pinus nigra as a biomonitor of pesticides and polycyclic aromatic hydrocarbons in Lebanon, Environ. Sci. Pollut. Res., 2021, 28(8), 10283–10291 CrossRef CAS PubMed.
- L. Chiarantini, V. Rimondi, M. Benvenuti, M. W. Beutel, P. Costagliola, C. Gonnelli, P. Lattanzi and M. Paolieri, Black pine (Pinus nigra) barks as biomonitors of airborne mercury pollution, Sci. Total Environ., 2016, 569–570, 105–113 CrossRef CAS PubMed.
- R. J. Hutnik, J. R. McClenahen, R. P. Long and D. D. Davis, Mercury accumulation in Pinus nigra (Austrian Pine), Northeast. Nat., 2014, 21, 529–540 CrossRef.
- S. Viso, S. Rivera, A. Martinez-Coronado, J. M. Esbrí, M. M. Moreno and P. Higueras, Biomonitoring of Hg0, Hg2+ and particulate hg in a mining context using tree barks, Int. J. Environ. Res. Public Health, 2021, 18, 5191 CrossRef CAS PubMed.
- R. Naharro, J. M. Esbrí, J. Á. Amorós, F. J. García-Navarro and P. Higueras, Assessment of mercury uptake routes at the soil-plant-atmosphere interface, Geochem.: Explor., Environ., Anal., 2018, 19, 146–154 CrossRef.
- H. R. Friedli, A. F. Arellano, S. Cinnirella and N. Pirrone, Initial estimates of mercury emissions to the atmosphere from global biomass burning, Environ. Sci. Technol., 2009, 43, 3507–3513 CrossRef CAS PubMed.
- B. Gworek, W. Dmuchowski and A. H. Baczewska-Dąbrowska, Mercury in the terrestrial environment: a review, Environ. Sci. Eur., 2020, 32, 128 CrossRef CAS.
- F. Slemr, C. A. Brenninkmeijer, A. Rauthe-Schöch, A. Weigelt, R. Ebinghaus, E. G. Brunke, L. Martin, T. G. Spain and S. O'Doherty, El Niño-Southern Oscillation influence on tropospheric mercury concentrations, Geophys. Res. Lett., 2016, 43, 1766–1771 CrossRef CAS.
- A. Laacouri, E. A. Nater and R. K. Kolka, Distribution and uptake dynamics of mercury in leaves of common deciduous tree species in Minnesota, U.S.A, Environ. Sci. Technol., 2013, 47, 10462–10470 CrossRef CAS PubMed.
- J. Stamenkovic and M. S. Gustin, Nonstomatal versus stomatal uptake of atmospheric mercury, Environ. Sci. Technol., 2009, 43, 1367–1372 CrossRef CAS PubMed.
- A. Manceau, J. Wang, M. Rovezzi, P. Glatzel and X. Feng, Biogenesis of Mercury-Sulfur Nanoparticles in Plant Leaves from Atmospheric Gaseous Mercury, Environ. Sci. Technol., 2018, 52, 3935–3948 CrossRef CAS PubMed.
- M. Birke, U. Rauch and F. Hofmann, Tree bark as a bioindicator of air pollution in the city of Stassfurt, Saxony-Anhalt, Germany, J. Geochem. Explor., 2018, 187, 97–117 CrossRef CAS.
- J. A. Rodríguez Martín, N. Nanos, J. C. Miranda, G. Carbonell and L. Gil, Volcanic mercury in Pinus canariensis, Naturwissenschaften, 2013, 100, 739–747 CrossRef PubMed.
- M. Chrabąszcz and L. Mróz, Pol. J. Environ. Stud., 2017, 26, 453–466 CrossRef.
- M. Rajfur, Assessment of the possibility of using deciduous tree bark as a biomonitor of heavy metal pollution of atmospheric aerosol, Environ. Sci. Pollut. Res., 2019, 26, 35945–35956 CrossRef CAS PubMed.
- M. Sut-Lohmann, J. Jonczak, A. Parzych, V. Šimanský, N. Polláková and T. Raab, Accumulation of airborne potentially toxic elements in Pinus sylvestris L. bark collected in three Central European medium-sized cities, Ecotoxicol. Environ. Saf., 2020, 200, 110758 CrossRef CAS PubMed.
- G. Vázquez, J. González-Álvarez, S. Freire, M. López-Lorenzo and G. Antorrena, Removal of cadmium and mercury ions from aqueous solution by sorption on treated Pinus pinaster bark: Kinetics and isotherms, Bioresour. Technol., 2002, 82, 247–251 CrossRef.
- W. F. Fitzgerald and C. H. Lamborg, Geochemistry of Mercury in the Environment, Treatise Geochem., 2007, 9–9, 1–47 Search PubMed.
- L. Chiarantini, V. Rimondi, F. Bardelli, M. Benvenuti, C. Cosio, P. Costagliola, F. Di Benedetto, P. Lattanzi, G. Eraldine Sarret, F. Bardelli, C. Cosio, P. Lattanzi, G. Sarret, M. Benvenuti, C. Cosio, P. Costagliola, F. Di and P. Lattanzi, Mercury speciation in Pinus nigra barks from Monte Amiata (Italy): An X-ray absorption spectroscopy study, Environ. Pollut., 2017, 227, 83–88 CrossRef CAS PubMed.
- P. J. Hanson, T. A. Tabberer and S. E. Lindberg, Emissions of mercury vapor from tree bark, Atmos. Environ., 1997, 31, 777–780 CrossRef CAS.
- Y. Yang, R. D. Yanai, C. T. Driscoll, M. Montesdeoca and K. T. Smith, Concentrations and content of mercury in bark, wood, and leaves in hardwoods and conifers in four forested sites in the northeastern USA, PLoS One, 2018, 13, e0196293 CrossRef PubMed.
- X. Wang, Z. Bao, C. J. Lin, W. Yuan and X. Feng, Assessment of Global Mercury Deposition through Litterfall, Environ. Sci. Technol., 2016, 50, 8548–8557 CrossRef CAS PubMed.
- A. Gómez-Armesto, M. Méndez-López, P. Pérez-Rodríguez, D. Fernández-Calviño, M. Arias-Estévez and J. C. Nóvoa-Muñoz, Litterfall Hg deposition to an oak forest soil from southwestern Europe, J. Environ. Manage., 2020, 269, 110858 CrossRef PubMed.
- J. Zhou, Z. Wang and X. Zhang, Deposition and Fate of Mercury in Litterfall, Litter, and Soil in Coniferous and Broad-Leaved Forests, J. Geophys. Res.: Biogeosci., 2018, 123, 2590–2603 CrossRef CAS.
- B. D. Hall and V. L. St. Louis, Methylmercury and total mercury in plant litter decomposing in upland forests and flooded landscapes, Environ. Sci. Technol., 2004, 38, 5010–5021 CrossRef CAS PubMed.
- K. Schlüter, Review: Evaporation of mercury from soils. An integration and synthesis of current knowledge, Environ. Geol., 2000, 39, 249–271 CrossRef.
- V. Rimondi, P. Costagliola, R. Benesperi, M. Benvenuti, M. W. Beutel, A. Buccianti, L. Chiarantini, P. Lattanzi, D. Medas and P. Parrini, Black pine (Pinus nigra) barks: A critical evaluation of some sampling and analysis parameters for mercury biomonitoring purposes, Ecol. Indic., 2020, 112, 106110 CrossRef CAS.
- M. L. Parisi, N. Ferrara, L. Torsello and R. Basosi, Life cycle assessment of atmospheric emission profiles of the Italian geothermal power plants, J. Cleaner Prod., 2019, 234, 881–894 CrossRef.
- J. Arnold, M. S. Gustin and P. J. Weisberg, Evidence for Nonstomatal Uptake of Hg by Aspen and Translocation of Hg from Foliage to Tree Rings in Austrian Pine, Environ. Sci. Technol., 2018, 52, 1174–1182 CrossRef CAS PubMed.
- D. S. McLagan, F. Monaci, H. Huang, Y. D. Lei, C. P. J. Mitchell, F. Wania, D. S. McLagan, F. Monaci, H. Huang, Y. D. Lei, C. P. J. Mitchell and F. Wania, Characterization and Quantification of Atmospheric Mercury Sources Using Passive Air Samplers, J. Geophys. Res.: Atmos., 2019, 124, 2351–2362 CrossRef CAS.
- O. Proux, E. Lahera, W. Del Net, I. Kieffer, M. Rovezzi, D. Testemale, M. Irar, S. Thomas, A. Aguilar-Tapia, E. F. Bazarkina, A. Prat, M. Tella, M. Auffan, J. Rose and J.-L. Hazemann, High-Energy Resolution Fluorescence Detected X-Ray Absorption Spectroscopy: A Powerful New Structural Tool in Environmental Biogeochemistry Sciences, J. Environ. Qual., 2017, 46, 1146–1157 CrossRef CAS PubMed.
- V. Rimondi, F. Bardelli, M. Benvenuti, P. Costagliola, J. E. J. E. Gray and P. Lattanzi, Mercury speciation in the Mt. Amiata mining district (Italy): Interplay between urban activities and mercury contamination, Chem. Geol., 2014, 380, 110–118 CrossRef CAS.
- M. P. Isaure, M. Albertelli, I. Kieffer, R. Tucoulou, M. Petrel, E. Gontier, E. Tessier, M. Monperrus and M. Goñi-Urriza, Relationship Between Hg Speciation and Hg Methylation/Demethylation Processes in the Sulfate-Reducing Bacterium Pseudodesulfovibrio hydrargyri: Evidences From HERFD-XANES and Nano-XRF, Front. Microbiol., 2020, 11, 1–11 CrossRef PubMed.
- A. Manceau, C. Lemouchi, M. Enescu, A.-C. Gaillot, M. Lanson, V. Magnin, P. Glatzel, B. A. Poulin, J. N. Ryan, G. R. Aiken, I. Gautier-Luneau and K. L. Nagy, Formation of Mercury Sulfide from Hg(II)–Thiolate Complexes in Natural Organic Matter, Environ. Sci. Technol., 2015, 49, 9787–9796 CrossRef CAS PubMed.
- A. Manceau, C. Lemouchi, M. Rovezzi, M. Lanson, P. Glatzel, K. L. Nagy, I. Gautier-Luneau, Y. Joly and M. Enescu, Structure, Bonding, and Stability of Mercury Complexes with Thiolate and Thioether Ligands from High-Resolution XANES Spectroscopy and First-Principles Calculations, Inorg. Chem., 2015, 54, 11776–11791 CrossRef CAS PubMed.
- T. Alsina, W. Clegg, K. A. Fraser and J. Sola, Homoleptic cyclohexanethiolato complexes of mercury(II), J. Chem. Soc., Dalton Trans., 1992, 1393–1399 RSC.
- J. P. Bourdineaud, M. Gonzalez-Rey, M. Rovezzi, P. Glatzel, K. L. Nagy and A. Manceau, Divalent Mercury in Dissolved Organic Matter Is Bioavailable to Fish and Accumulates as Dithiolate and Tetrathiolate Complexes, Environ. Sci. Technol., 2019, 53, 4880–4891 CrossRef CAS PubMed.
- A. Manceau, J. P. Bourdineaud, R. B. Oliveira, S. L. F. Sarrazin, D. P. Krabbenhoft, C. A. Eagles-Smith, J. T. Ackerman, A. R. Stewart, C. Ward-Deitrich, M. E. Del Castillo Busto, H. Goenaga-Infante, A. Wack, M. Retegan, B. Detlefs, P. Glatzel, P. Bustamante, K. L. Nagy and B. A. Poulin, Demethylation of Methylmercury in Bird, Fish, and Earthworm, Environ. Sci. Technol., 2021, 55, 1527–1534 CrossRef CAS PubMed.
- E. R. Malinowski, Theory of the distribution of error eigenvalues resulting from principal component analysis with applications to spectroscopic data, J. Chemom., 1987, 1, 33–40 CrossRef CAS.
- E. R. Malinowski, Theory of Error in Factor Analysis, Anal. Chem., 1977, 49, 606–612 CrossRef CAS.
- T. Ressler, J. Wong, J. Roos and I. L. Smith, Quantitative speciation of mn-bearing particulates emitted from autos burning (methylcyclopentadienyl) manganese tricarbonyl-added gasolines using XANES spectroscopy, Environ. Sci. Technol., 2000, 34, 950–958 CrossRef CAS.
- S. M. Webb, SIXpack: A graphical user interface for XAS analysis using IFEFFIT, Phys. Scr., T, 2005, T115, 1011–1014 CrossRef CAS.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. Zhou, D. Obrist, A. Dastoor, M. Jiskra and A. Ryjkov, Vegetation uptake of mercury and impacts on global cycling, Nat. Rev. Earth Environ., 2021, 2, 269–284 CrossRef.
- E. I. H. Siwik, L. M. Campbell and G. Mierle, Distribution and trends of mercury in deciduous tree cores, Environ. Pollut., 2010, 158, 2067–2073 CrossRef CAS PubMed.
- J. A. Fleck, D. F. Grigal and E. A. Nater, Mercury uptake by trees: An observational experiment, Water, Air, Soil Pollut., 1999, 115, 513–523 CrossRef CAS.
- S. A. Thomas and J. F. Gaillard, Cysteine Addition Promotes Sulfide Production and 4-Fold Hg(II)-S Coordination in Actively Metabolizing Escherichia coli, Environ. Sci. Technol., 2017, 51, 4642–4651 CrossRef CAS PubMed.
- M. Enescu, K. L. Nagy and A. Manceau, Nucleation of mercury sulfide by dealkylation, Sci. Rep., 2016, 6(1), 1–6 CrossRef PubMed.
- S. M. Ullrich, T. W. Tanton and S. A. Abdrashitova, Mercury in the aquatic environment: A review of factors affecting methylation, Crit. Rev. Environ. Sci. Technol., 2001, 31, 241–293 CrossRef CAS.
- O. Vaselli, P. Higueras, B. Nisi, J. María Esbrí, J. Cabassi, A. Martínez-Coronado, F. Tassi and D. Rappuoli, Distribution of gaseous Hg in the Mercury mining district of Mt. Amiata (Central Italy): A geochemical survey prior the reclamation project, Environ. Res., 2013, 125, 179–187 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2em00239f |
| This journal is © The Royal Society of Chemistry 2022 |