
From lithium to emerging mono- and multivalent-cation-based rechargeable batteries: non-aqueous organic electrolyte and interphase perspectives
Heng
Zhang
 *a,
Lixin
Qiao
b,
Hannes
Kühnle
c,
Egbert
Figgemeier
*cd,
Michel
Armand
*b and
Gebrekidan Gebresilassie
Eshetu
*ce
*a,
Lixin
Qiao
b,
Hannes
Kühnle
c,
Egbert
Figgemeier
*cd,
Michel
Armand
*b and
Gebrekidan Gebresilassie
Eshetu
*ce
aKey Laboratory of Material Chemistry for Energy Conversion and Storage (Ministry of Education), School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Luoyu Road 1037, 430074, Wuhan, China. E-mail: hengzhang2020@hust.edu.cn
bElectrical energy storage department, CIC Energigune, Parque Tecnológico de Álava, Albert Einstein 48, 01510 Miñano, Álava, Spain. E-mail: marmand@cicenergigune.com
cInstitute for Power Electronics and Electrical Drives (ISEA), RWTH Aachen University, Campus-Boulevard 89, 52074 Aachen, Germany. E-mail: e.figgemeier@fz-juelich.de
dHelmholtz-Institute Münster (HI MS): Ionics in Energy Storage (IEK-12), Institute of Energy and Climate Research, Forschungszentrum Jülich GmbH, Corrensstrasse 46, 48149 Münster, Germany
eDepartment of Material Science and Engineering, Mekelle Institute of Technology—Mekelle University, Tigray, Ethiopia. E-mail: Gebrekidan.Eshetu@isea.rwth-aachen.de
First published on 9th November 2022
Abstract
Since the oil crisis in the 1970s, the importance of rechargeable batteries has been noted by both academia and industry. This has become more prominent with the increasing demand in e-mobility and integration of renewable sources in the energy ecosystem. However, despite the great success of lithium-ion batteries in portable consumer electronics and the above-mentioned domains, it is challenging to further expand their use to large-volume technical applications due to the limited resources of some key elements (lithium, cobalt, etc.). Accordingly, emerging mono-valent (e.g., sodium and potassium) and multi-valent (magnesium, calcium, zinc, aluminum, etc.) batteries are expected to overcome the resource limitation and related challenges. Herein, we present the historical development of non-aqueous organic electrolytes and electrode–electrolyte interphases and focus on the similarities and differences between lithium-based batteries and other complementary emerging battery technologies. Special attention is paid to some basic parameters related to solvents and salts, including donor numbers and Eigen values, to better understand the transport behavior in the bulk electrolyte. Moreover, key parameters impacting the features of the electrode–electrolyte interphase are critically analyzed for each battery configuration. Additionally, we discuss the possible strategies to enhance the physical (e.g., transport behavior and mechanical properties) and (electro)chemical properties of electrolytes and interphases, aiming at promoting the development of sustainable and high-performance mono- and multi-valent batteries for practical applications. Particularly, it is scrutinized whether the accumulated facts with respect to lithium can be smoothly transferred to other emerging battery systems or not.
 Heng Zhang | Prof. Dr. Heng Zhang received his PhD in Chemistry from Huazhong University of Science and Technology (HUST, China). Then, he joined CIC EnergiGUNE as a Post Doc Researcher (2016–2017) working with Prof. Dr. Michel Armand, and later he acted as the Research Line Manager of Polymer Electrolyte at CIC EnergiGUNE (January 2018–June 2020). Currently, he is a Full Professor of Organic Chemistry at HUST since August 2020. His research interests focus on electrolyte materials, particularly solid polymer electrolytes, for rechargeable batteries. |
 Lixin Qiao | Dr. Lixin Qiao obtained his PhD in Applied Chemistry and Polymeric Materials from CIC EnergiGUNE/University of the Basque Country (Vitoria-Gasteiz, Spain) in 2021. His research mainly focuses on innovative alkali metal salts and polymer electrolytes for metal batteries. |
 Hannes Kühnle | Hannes Kühnle received his Master's Degree in Technical Chemistry from Graz University of Technology (TU Graz, Austria) in 2017. In 2018, he joined the Chair for Ageing and Lifetime Prediction of Batteries of Prof. Dr. Egbert Figgemeier (ISEA, RWTH Aachen University, Germany) as a PhD student. His studies focus on lithium deposition on lithium-metal surfaces for rechargeable lithium-based batteries. His research interests further include designing tailored surfaces and interphases on metal-based rechargeable batteries, electrolytes and aging of batteries. |
 Egbert Figgemeier | Prof. Dr. Egbert Figgemeier received his PhD degree in chemistry from the University of Paderborn, Paderborn, Germany, in 1998. This was followed by academic research at University College Dublin, Dublin, Ireland, the University of Basel, Basel, Switzerland, and the University of Uppsala, Uppsala, Sweden. Since 2016, he has been the Group Leader with the Helmholtz Institute Münster, Forschungszentrum Jülich (Section Aachen), Münster, Germany. He also holds the Chair for “Ageing Processes and Lifetime Prediction of Batteries” at RWTH Aachen University, Aachen, Germany. His current research activities span from fundamental aging mechanisms in lithium-ion and lithium metal batteries to the development of designer electrolytes (salt anions and additives, prelithiation technologies and interphase/interface studies). |
 Michel Armand | Prof. Dr. Michel Armand received his PhD in Physics from Joseph Fourier University (1978). He has been the Directeur de Recherche at Centre National de la Recherche Scientifique (CNRS) since 1989 and Professor at University of Montreal (1995–2004). He has ushered theoretical concepts leading to practical applications in energy-related electrochemistry. He has been one of the pioneers in the use of intercalation electrodes (1972), the introduction of the polymer electrolytes for battery application (1978), new salts based on delocalized anions of the sulfonimide family (1986) and organic electrode materials. He joined CIC EnergiGUNE as a Group Leader in solid electrolyte in 2013, and his present activities include new highly conductive polymers and techniques to obtain cross-linked networks; new highly delocalized negative charges and new electrode materials. |
 Gebrekidan Gebresilassie Eshetu | Dr. Gebrekidan Gebresilassie Eshetu obtained his PhD at the end of 2013 in Materials Science and Chemistry from the LRCS (Centre National de la Recherche Scientifique - CNRS) Laboratory in Amiens, France, under the supervision of Prof. Dr. Michel Armand, Prof. Dr. Stephane Laruele and Dr. Sylvie Grugeon. Following this, he did his postdoctoral research at the Karlsruhe Institute of Technology (KIT), Helmholtz Institute Ulm (HIU) in the group of Prof. Dr. Stefano Passerini. Then, he worked in CIC EnergiGUNE (Spain, 2016–2018) as a Senior Postdoctoral Researcher on solid electrolytes for rechargeable batteries. Subsequently, he joined RWTH Aachen University in the group of Prof. Dr. Egbert Figgemeier. Currently, he is a Group Leader at RWTH Aachen University for surfaces, interface and interphases in rechargeable batteries. His research interest focuses on advanced electrolytes, modulating functional interfaces and interphases for rechargeable batteries. |
Broader contextThe global coalition in carbon neutrality (zero-carbon emissions) gives a strong impetus for the development of highly efficient electrochemical energy storage devices, particularly for e-mobility applications such as electric vehicles, integration of renewable energy sources and grid/utility storage. The new era of e-powered life has already been realized by the massive deployment of lithium-ion batteries (LIBs). In this case, it is of vital importance to diversify cell chemistry from classic LIBs stemming from the rocking-chair concept to other emerging mono-valent (e.g., sodium, potassium) and multi-valent (magnesium, calcium, zinc, and aluminum, etc.) cation-based rechargeable batteries with earth abundant and sustainable elements. In this review, the historical and present development of non-aqueous organic electrolytes and electrode–electrolyte interphases are outlined with focus on similarities and distinctions between lithium-based batteries and other transpiring rechargeable batteries. Furthermore, the possible directions and remedies in promoting the performance of emerging mono- and multi-valent batteries are discussed. The present work is anticipated to enable a better understanding and thus accelerate the practical deployment of emerging mono- and multi-valent batteries with improved energy and economic efficiencies. |
1. Introduction
Global efforts towards transitioning to a less (or totally independent) carbon-dependent energy economy and technology require the large-scale deployment of the electro-mobility sector and integration of more renewable sources in the energy ecosystem.1–3 To enable this ambitious paradigm shift, electrochemical energy storage systems with net-zero carbon emissions when operating have attracted significant attention, both from the research and industry sectors. Due to their high energy density and mature technological and industrial advancements, lithium (Li0)-based rechargeable batteries are the most prolific technologies in use currently, dominating the global market including portable electronic devices, electric vehicles (EVs) and grid/utility energy storage applications.4–6 However, due to the limited reserves (0.002 wt% of Earth's crust, Table 1), uneven geographical distribution and poor safety of Li, there is an urgent quest to find alternative sustainable chemistries.| Property | Li+ | Na+ | K+ | Mg2+ | Ca2+ | Zn2+ | Al3+ |
|---|---|---|---|---|---|---|---|
| Voltage (V vs. SHE) | −3.05 | −2.71 | −2.93 | −2.36 | −2.87 | −0.76 | −1.66 |
| Density (g cm−3) | 0.53 | 0.9 | 0.89 | 1.74 | 1.54 | 7.13 | 2.7 |
| Atomic radius (pm) | 148 | 180 | 220 | — | — | — | — |
| Shannon ionic radius (pm) | 76 | 102 | 138 | 72 | 100 | 75 | 53 |
| Stokes radius in PC (pm) | 480 | 460 | 360 | — | — | — | — |
| Polarization strength (pm−2) | 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
11100 |
47300 |
— | — | — | |
| Pauling electronegativity | 0.98 | 0.93 | 0.82 | 1.31 | 1.00 | 1.65 | 1.61 |
| Charge density (C mm−3) | 87 | 36 | 15 | 205 | 76 | 181 | 770 |
| Common coordination environments | Octahedral, tetrahedral | Octahedral, prismatic | Octahedral, prismatic | Octahedral | Octahedral | — | — |
| Abundance in earth's crust (wt%)/rank | 0.002/33rd | 2.36/6th | 2.09/7th | 2.60/8th | 4.86/5th | 0.004%/25th | 8.23/3rd |
| Specific capacity (mA h g−1) | 3861 | 1168 | 685 | 5505 | 1337 | 820 | 2980 |
| Volumetric capacity (mA h cm−3) | 2026 | 1128 | 591 | 3833 | 2073 | 5851 | 8040 |
| Melting point (°C) | 180.5 | 97.8 | 63.4 | 650.0 | 842.0 | 419.5 | 660.3 |
| Young's modulus (GPa) | 5 | 10 | 4 | 45 | 20 | 108 | 70 |
Consequently, non-lithium rechargeable batteries based on monovalent metal ions (sodium-Na+ and potassium-K+) and multivalent metal ions (magnesium-Mg2+, calcium-Ca2+, zinc-Zn2+, and aluminum-Al3+) have emerged as promising alternatives to lithium because of their strong capability in meeting the growing demand for future energy-storage applications.7–17 Rechargeable batteries based on these alternative metal elements can result in safer and higher power density and energy density systems built on abundant, low-cost, and environmentally friendly materials (Table 1), thus presenting high potential to achieve carbon neutrality and fulfill the ‘go green philosophy’. It needs to be emphasized that these emerging technologies will be potential complementary to the Li-ion, rather than being competitive or rival, and thus the notion of ‘post-Li batteries’ widely used in literature is somehow ambiguous and misleading.18 Moreover, no technology can satisfy all requirements of electrochemical energy storage applications, and thus energy storage systems need to be diversified. However, despite the increasing interest and extensive research efforts in developing the above-mentioned lithium-free metal–ion-based battery chemistries, their commercial prospects, and thus large-scale deployment are still hindered by fundamental scientific questions and engineering challenges.19 Moreover, their cost and performance parameters still need to be thoroughly evaluated and compared with lithium-based technologies.
In principle, a typical rechargeable battery consists of seven major components, namely, a negative electrode (anode), positive electrode (cathode), current collector, electrolyte, separator, electrode/electrolyte and electrode/current collector boundaries (interfaces and interphases) (Fig. 1).20 The electrolyte medium and electrode–electrolyte interphases, which are sandwiched between the highly reducing (negative) and highly oxidizing (positive) electrodes, may react and become significantly damaged. In fact, stable electrolytes and interphases are indispensable elements for the proper functioning of emerging mono-valent and multi-valent cation-based rechargeable batteries. Thus, the practical future deployment of these technologies will strongly depend on the accelerated discovery, optimization and development of customized electrolytes and full-scale understanding and design of their interphases. Compared to Li, the interphases formed on the above-mentioned Li-free mono- and multi-valent cations are more challenging due to their unique chemistries such as Eigen value, charge density, Lewis acidity, and size. Accordingly, owing to their importance, they have received increasing attention from the scientific research community.
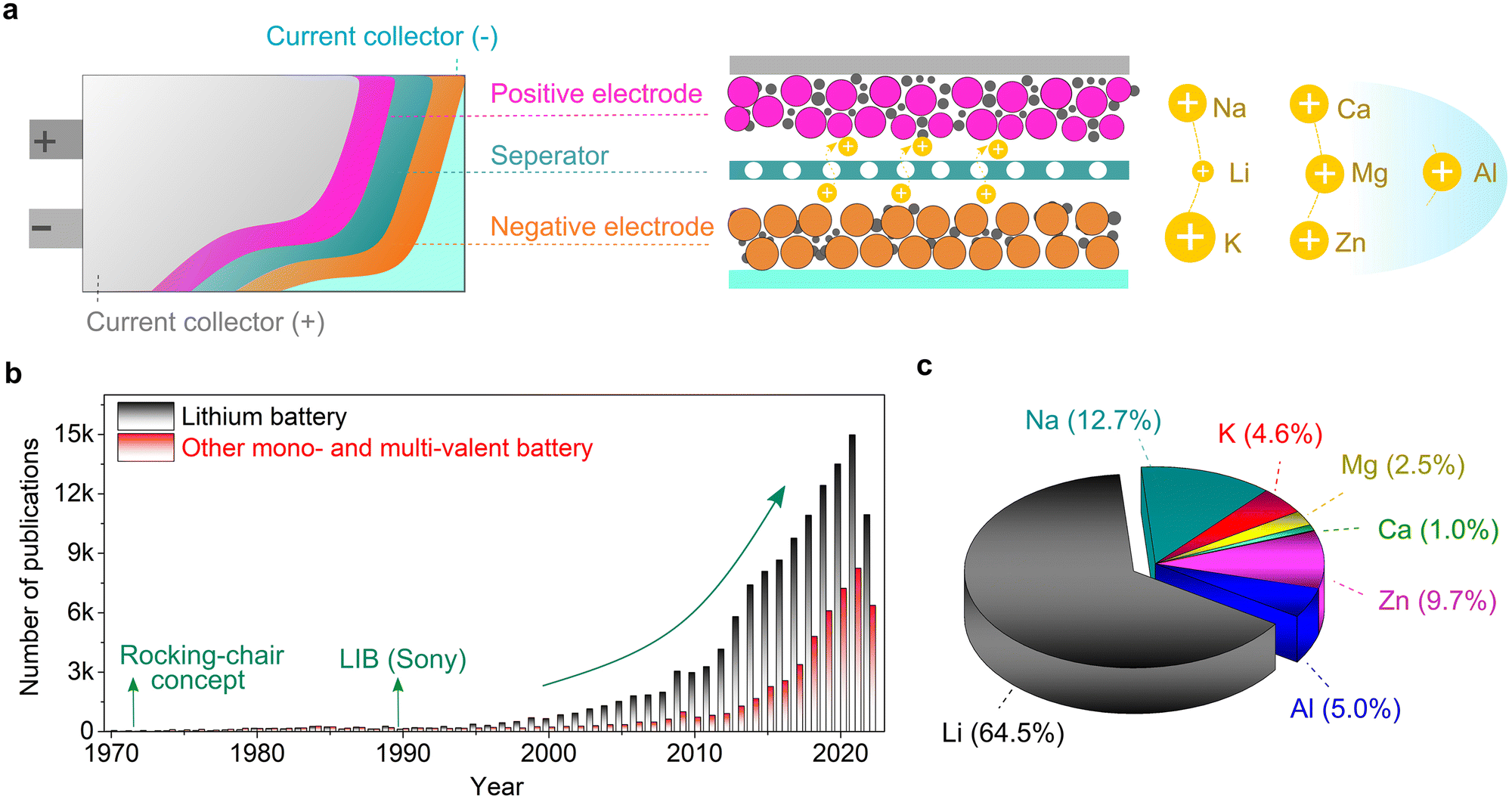 | ||
| Fig. 1 General background of mono- and multivalent cation-based rechargeable batteries. (a) Simplifed cell configurations for rechargeable batteries. Please note that the pouch cell structure has been simiplified to better present the inner structures of a single cell (i.e., negative and positive electrodes, separator, and current collector). The yellow circle with positive charge symbol represents the cations involved in rechargeable batteries, including mono- (Li+, Na+, and K+), di-valent (Mg2+, Ca2+, and Zn2+), and tri-valent cations (Al3+) examplified at the right side of the figure. (b) Research trend in lithium batteries and other mono- and multi-valent batteries vs. time (data from Scopus, 25th October 2022). (c) Distribution of publications in 2021 (total publications: 23218). Note that besides from lithium batteries, the sum of sodium, potassium, magnesium, calcium, zinc, and aluminum exceeds 35%. | ||
Although the rich experience and knowhow stockpiled from Li-based rechargeable batteries provide invaluable insights into the development of new electrolytes and understanding of interphases and interfaces, there is no guarantee that there will be a direct transition of facts from Li+- to emerging mono- (Na+ and K+) and multi- (Mg2+, Ca2+, Zn2+, and Al3+) metal cations. Thus, replacing Li+ with Na+, K+, Mg2+, Ca2+, Zn2+ and Al3+ requires deep revision and re-exploration of the electrolyte materials and interphase electrochemistry of the latter systems.
The aim of this review is to provide a comprehensive account of the progress, status, and prospects of various Mn+ (M = Metal, n = 1, 2, and 3) ion-conductive electrolytes (including solvents, salts, and additives) and interphases in the vicinity of the polarized electrodes. We anticipate that this critical review will stimulate new concepts, ideas, and research directions and open questions to further tailor and understand these electrolytes and interphases, eventually enabling the practical realization of the systems under consideration. Thus, to evaluate the viability and potential application of these new battery technologies, thinking outside the box and deploying a paradigm shift approach, including on the screening and designing of electrolytes and optimization of the electrolyte constituents and their interphases, are necessary.
For simplicity, this review is divided into six sections dealing with the fundamentals of rechargeable batteries, electrolytes, interphases, advanced characterization techniques, and aspects related to drop-in technology, i.e., assessment if these newly emerging technologies can be quickly inherited from the standard battery production and processes adopted for Li-based battery systems. These sections are further divided into different sub-sections.
2. Fundamentals of various rechargeable batteries
Rechargeable batteries are capable of storing and releasing electric power via reversible redox-reactions.1,2 For the developed non-aqueous organic electrolytes, the working principles of rechargeable batteries can be mainly classified into four types (Fig. 2), as follows: (1) rocking-chair battery comprised of two intercalation (IC)-type electrodes (e.g., lithium-ion batteries, sodium-ion batteries, and potassium-ion batteries, Fig. 2a),21,22 (2) metal-based rechargeable battery consisting of metallic anode and intercalation-type cathodes (e.g., lithium/sodium/potassium/magnesium/calcium/zinc metal batteries, Fig. 2b),23 (3) metal-based rechargeable batteries based on conversion-type cathodes (e.g., lithium/sodium/potassium/magnesium/calcium/zinc/aluminum sulfur batteries, Fig. 2c),15–17,24 and (4) metal-based rechargeable batteries with gaseous cathodes (lithium/sodium/potassium/magnesium/calcium/zinc/aluminum oxygen/air batteries, Fig. 2d).25–27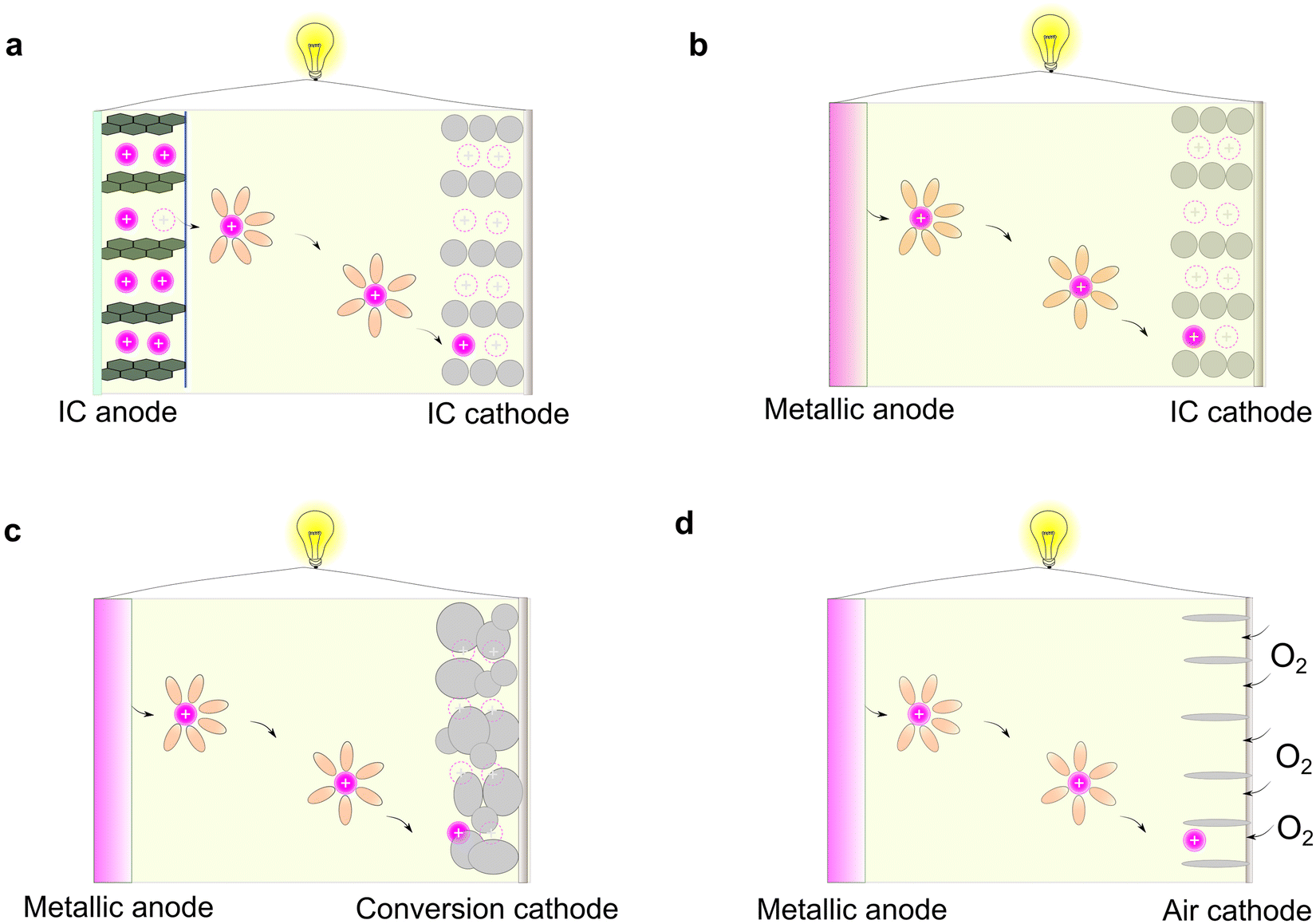 | ||
| Fig. 2 Working principles for various types of rechargeable batteries. (a) Rocking-chair batteries with two intercalation (IC)-type electrodes. (b) Metal-based rechargeable batteries with IC-type cathodes. (c) Metal-based rechargeable batteries with conversion-type cathodes (e.g., lithium–sulfur batteries, sodium–sulfur batteries, and magnesium–sulfur batteries). (d) Metal-based rechargeable batteries with gaseous cathodes (e.g., lithium–oxygen batteries, sodium–oxygen batteries, and aluminum–oxygen batteries). Please note that the red circle with positive charge symbol represents the cations of interest in this work (i.e., Li+, Na+, K+, Mg2+, Ca2+, Zn2+, and Al3+), and adjacent orange ellipsoid represent the solvent molecules. | ||
Among the different types of rechargeable batteries, those built on the “rocking-chair” concept have become the most prevailing battery technology, which greatly impact our daily life.28 This stems from the advantageous features of intercalation-type anodes vs. metallic anodes including (1) higher surface area, allowing the rapid transport of ions across the electrode–electrolyte interphases, (2) suppressed dendritic metal deposits during charging processes, thus avoiding internal short-circuit caused by the penetration of dendrites, and (3) lower sensitivity to moisture (air and humidity), enabling better processability of the batteries on a large scale.
In principle, a typical rocking-chair battery can also be treated as a concentration cell (i.e., two electrodes with different lithium concentrations), in which no metallic lithium is formed during continuous charge/discharge cycles.29 However, this requires extra and non-active hosting materials to decouple the ions and electrons, therefore resulting in a lower energy density compared to batteries paired with metallic anodes.4,30,31 Regarding the electrolyte, the most remarkable difference between the conversion electrode and intercalation electrode is their solvation and de-solvation processes of metal ions during the charge/discharge cycles. In the case of traditional intercalation-type electrodes, the inner solvent shell of the solvated metal ions has to be removed prior to their insertion in the host materials; however, this phenomenon is less prominent in the case of conversion-type electrode materials, given that the reduction/oxidation of metal ions occur concurrently with their de-solvation and solvation processes.32
3. Electrolytes
3.1. Basic concepts of non-aqueous electrolytes
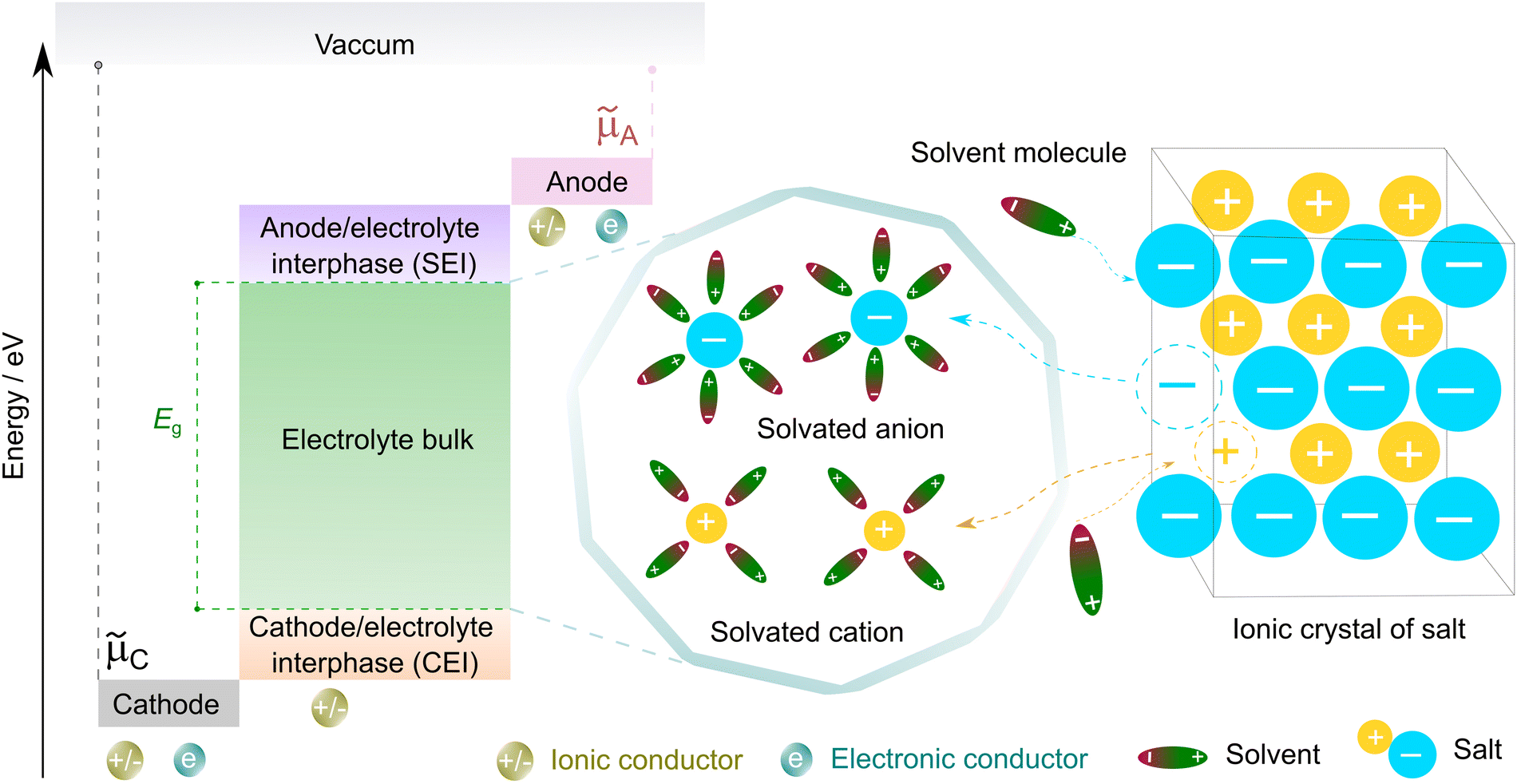 | ||
| Fig. 3 Working principle of non-aqueous electrolytes in rechargeable batteries. μC and μA represent the electrochemical potentials of the cathode and anode materials, respectively, and Eg denotes the thermodynamic stability window of the electrolyte. The open circuit energy diagram is adapted from ref. 33 with permission. Copyright 2010, the American Chemical Society. | ||
To form an ionic solution (i.e., liquid electrolyte) or a solid mixture (i.e., solid-state electrolyte), which can transport ionic species, alkali or alkaline metal salts with weakly coordinating anions (WCAs) together with highly polar solvents are generally employed. Most formulations are multi-component systems and single-solvent systems are rare. As seen in Fig. 3, the dissolution of solid ionic crystals through ion–solvent interactions (e.g., ion–dipole forces) yield solvated cations or anions, which are quite mobile under an electric field. Salts with WCAs [e.g., bis(trifluoromethanesulfonyl)imide (TFSI−)] have a low lattice energy, and thereby even doubly charged species can be easily separated via solvation processes. Dipolar aprotic solvents (e.g., carbonate, ester, and ether) with strong electron-donating/accepting ability and high relative permittivity can largely assist the breakdown of the crystal lattice of salt anions and transform ionic bonds to weaker interaction forces (i.e., solvation and dissolution). In most cases, the cation conductive electrolytes used in rechargeable batteries are achieved by the preferential solvation of cationic species utilizing solvents with strong enough electron-donating ability (i.e., high donor number). For example, Li+ cations in the commercial liquid electrolyte LP40 [i.e., 1.0 M LiPF6 in ethylene carbonate/ethyl methyl carbonate (EC/EMC; 4/6, v/v)] are tightly surrounded by carbonate solvents via Li+–O interactions, and the PF6− anions are mostly pushed outside the solvation shell with little interactions with the solvent molecules.
Different from the scenario of the aforementioned electrolytes based on polar solvents, highly ionic conductive electrolytes can also be formulated by combining a mixture of alkali halide and aluminum halide (e.g., LiCl–AlCl3, LiBr–AlBr3, and KBr–AlBr3) with low dielectric solvents [e.g., benzene, toluene (ε = 2.4 at 25 °C) and xylene].34–40 For example, the electrolyte of KBr–AlBr3 in benzene showed specific conductivities as high as ca. 4 × 10−3 S cm−1 at room temperature (RT; i.e., KBr:AlBr3:benzene = 1:3:12, by mole), enabling the efficient electro-plating of Al metal.41 AlBr3 exists as a dimer (e.g., Al2Br7−) in a solution of aromatic hydrocarbons, and the transport of ionic species in these electrolytes is effectively actualized by ions hopping from an ionic/neutral aggregate (e.g., K2[Al2Br7]+ and K[Al2Br7]) to the adjacent one instead of simple movement of ions in a viscous liquid, as indicated by Stokes law38 (Section 3.2). At the vicinity of the electrode, the Al2Br7− species breaks down with electron injection to deposit Al0, while the bromide ions form lower Al–Br complexes.
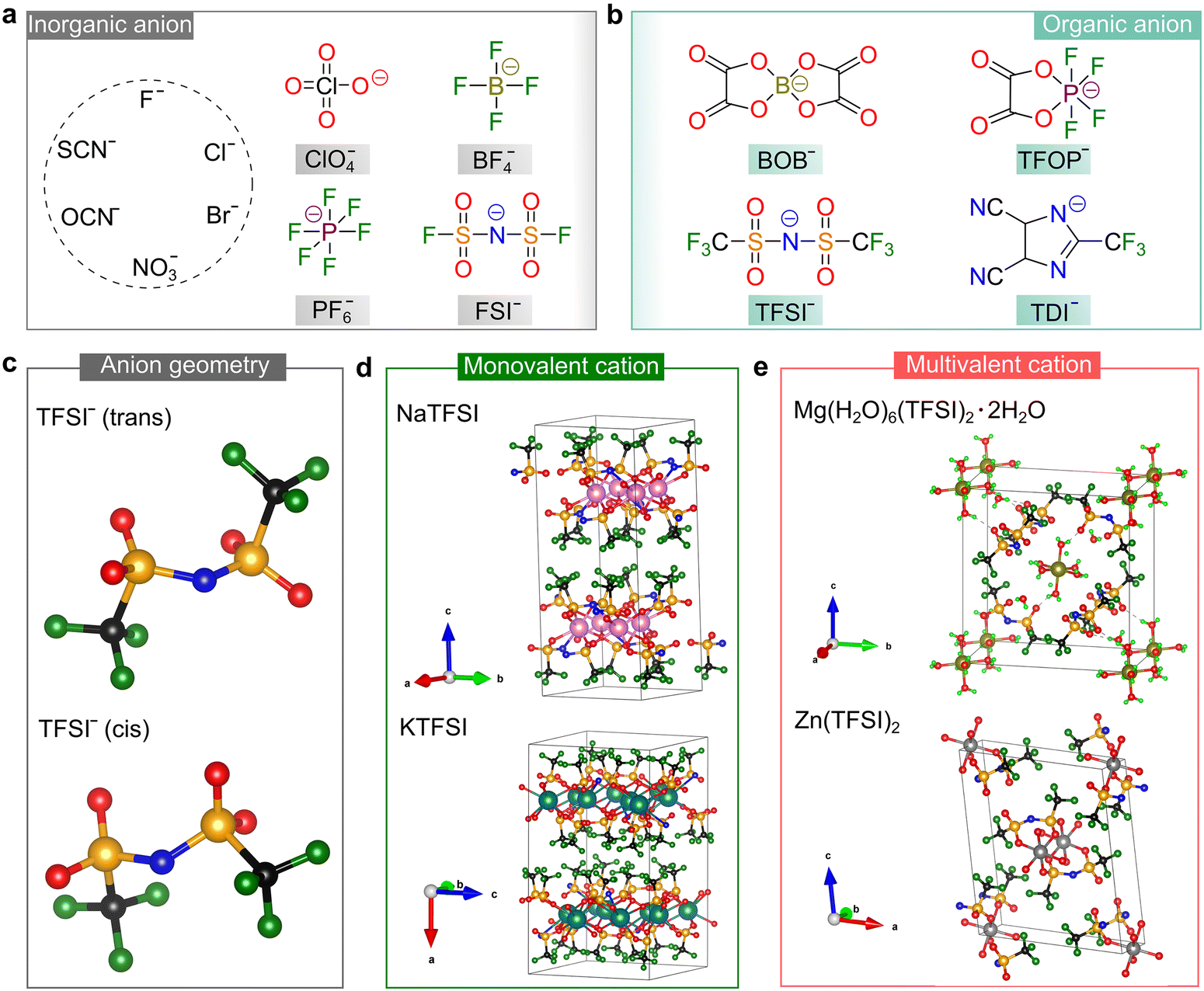 | ||
| Fig. 4 Salt anions developed for rechargeable batteries. (a and b) Chemical structures of some representative inorganic (a) and organic (b) anions designed for use in batteries. Abbreviations are as follows: perchlorate (ClO4−), tetrafluoroborate (BF4−), hexafluorophosphate (PF6−), bis(fluorosulfonyl)imide (FSI−), bis(oxalato)borate (BOB−), tetrafluorooxalatophosphate (TFOP−), bis(trifluoromethanesulfonyl)imide (TFSI−), and 4,5-dicyano-2-(trifluoromethyl)imidazole (TDI−). (c) Optimized geometries (gas phase) of the most popular sulfonimide anion, TFSI−, by DFT calculations, where geometries are taken from ref. 46. (d and e) Crystallographic structures of the TFSI-based conducting salts with monovalent (d) and multivalent cations (e). The pristine crystallographic data are taken from ref. 49–52 for NaTFSI, KTFSI, Mg(H2O)6(TFSI)2·2H2O, and Zn(TFSI)2, respectively. Lime green color (RGB: 0, 255, 0), black (RGB: 0, 0, 0), blue (RGB: 0, 0, 225), red (RGB: 255, 0, 0), dark green (RGB: 0, 128, 0), orange (RGB: 255, 165, 0), pink (RGB: 255, 192, 203), teal (RGB: 0, 128, 128), olive (RGB: 128, 128, 0), and gray (RGB: 128, 128, 128) spheres correspond to the H, C, N, O, F, S, Na, K, Mg, and Zn atoms, respectively. | ||
(1) The salt should dissolve and dissociate in aprotic solvents, thereby providing sufficient solubility (>0.1 M), which is required for battery operation.
(2) The solvated salts should be able to rapidly migrate under an electric field (i.e., high conductivity, >10−4 S cm−1 at the operating temperature), thus efficiently balancing the concentration of active species during the redox reaction of the battery electrodes.
(3) The salt itself should be chemically stable under trace amounts of moisture (e.g., water) and heat, thus ensuring the chemical, thermal, and electrochemical stability of the bulk electrolyte during the battery charge/discharge process.
(4) The salt should be readily accessible with abundant elements in efficient processes, and consequently be produced on a large-scale at an affordable cost.
(5) The salt should be non-toxic and environmentally benign, easily degradable when exposed to the environment, and preferably re-usable after the end of the battery cell life.
With respect to the family of inorganic anions, inorganic anions such as halide (F−, Cl−, and Br−), sulfate (SO42−), and phosphate (PO43−) are barely soluble in aprotic solvents, (e.g., 5.8 × 10−4 and 1.1 × 10−4 mol L−1 for LiF and NaF in EC/EMC, respectively42), and therefore can hardly afford electrolyte solutions/solid mixtures with sufficient ionic conductivities [e.g., at 25 °C, only ca. 10−6 S cm−1 for a saturated solution of LiF or NaF in EC/EMC,42 which is much lower than that required for battery use (i.e., ≥10−4 S cm−1)21]. This is mainly due to the strong binding energy (i.e., lattice energy) between mono- and multi-valent cations and these inorganic anions. Accordingly, WCAs with delocalized negative charges have become an increasingly important domain for searching suitable salts for rechargeable batteries.
It should be noted that the negative charges in relatively bulky inorganic anions (e.g., ClO4−, BF4−, PF6−, and AsF6−) are much more delocalized than inorganic halide (e.g., F− and Cl−) anions, which favors the dissociation of the corresponding metal salts in aprotic solvents. In alkyl carbonate solvent-based solution, the molar concentration of LiPF6 and NaPF6 can readily reach above 0.5 mol L−1 and the resulting electrolytes afford excellent ionic conductivities at room temperature [e.g., 9.3 × 10−3 S cm−1 for 1.0 M LiPF6-EC/EMC (1:1, v/v)43 and 6.5 × 10−3 S cm−1 for 1.0 M NaPF6-EC/DMC (1:1, v/v) at 25 °C44]. Effectively, the PF6− anion is an inorganic anion that fits most of the criteria of rechargeable batteries, i.e., endowed with well-balanced properties with concomitant trade-offs and restrictions, including ionic conductivity, anodic stability, and compatibility with current collector at different potentials.45 However, the PF6-based electrolytes are very sensitive toward ambient moisture, protic impurities, solvents and high temperature, showing inferior chemical and thermal stability.21,22
Alternatively, organic anions with strong electron-withdrawing groups [e.g., fluorinated alkyl chains (CmF2m+1) and cyano groups (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N)] and conjugated structures have been identified as important families for building highly conductive non-aqueous electrolytes. The chemical structures of some representative organic anions are presented in Fig. 4b. Among them, the TFSI− anion is the most popular anion employed for battery use due to its superior chemical and electrochemical stability and good solubility in aprotic solvents. In the gas phase, the TFSI− anions are likely to arrange in either a trans or cis conformation,46 as shown in Fig. 4c. The difference in total energies for both conformers is minimal (<5 kJ mol−1) and the –S–N–S– linkage of the sulfonimide center (–SO2N(−)SO2–) can easily rotate with a low energy barrier (<30 kJ mol−1),47 endowing the TFSI− anion with superior structural flexibility. These features are essential for building ionically conductive solid polymer electrolytes (SPEs), given that the transportation of ionic species in these electrolytes is offered by the segmental motion of the polymer backbone, and better flexibility of the salt anion means stronger plasticizing ability.48
N)] and conjugated structures have been identified as important families for building highly conductive non-aqueous electrolytes. The chemical structures of some representative organic anions are presented in Fig. 4b. Among them, the TFSI− anion is the most popular anion employed for battery use due to its superior chemical and electrochemical stability and good solubility in aprotic solvents. In the gas phase, the TFSI− anions are likely to arrange in either a trans or cis conformation,46 as shown in Fig. 4c. The difference in total energies for both conformers is minimal (<5 kJ mol−1) and the –S–N–S– linkage of the sulfonimide center (–SO2N(−)SO2–) can easily rotate with a low energy barrier (<30 kJ mol−1),47 endowing the TFSI− anion with superior structural flexibility. These features are essential for building ionically conductive solid polymer electrolytes (SPEs), given that the transportation of ionic species in these electrolytes is offered by the segmental motion of the polymer backbone, and better flexibility of the salt anion means stronger plasticizing ability.48
The unique properties of the TFSI− anion makes it a popular countercharge of emerging mono-valent and multi-valent cations, and the crystallographic structures determined by experimental approaches for some representative TFSI-based salts49–52 are shown in Fig. 4d and e. Generally, multivalent salts without any lattice solvents are more difficult to access compared to mono-valent salts, which is ascribed to the stronger coordinating ability of multivalent cations toward the water and polar organic solvents employed during the preparation processes. The crystallographic structures of neat TFSI-based mono-valent salts (including LiTFSI53,54) have been reported, while the anhydrous magnesium, calcium and aluminum salts of TFSI still need to be identified.
The dielectric constant represents the ability of a given solvent molecule to separate charges, as illustrated in Fig. 5a. Intuitively, solvents with a higher dielectric constant can more effectively suppress the formation of ion pairs or clusters, thus affording electrolyte solutions with higher ionic conductivity. Accordingly, the dielectric constant has been long recognized as the most important requirement for an ionizing solvent, and some textbooks written before the 1960s have highlighted that “a low dielectric constant results in a general decrease in the solubility of a salt”.58 However, as highlighted by Gutmann, using only the dielectric constant, it is difficult to explain why pyridine, having a much smaller dielectric constant (6.8) vs. anhydrous hydrogen cyanide (123), is an extremely useful solvents for a number of ionic compounds.55 Considering this, Gutmann suggested the use of empirical donor/acceptor numbers for describing the coordinating properties of a solvent. In this regard, the donor number (donicity, DN) is acknowledged to be a good metric to determine the electron-donating ability or Lewis basicity of a given solvent. As shown in Fig. 5b, the donor number (DN, in kcal mol−1) effectively stems from the negative enthalpy (ΔH) of the interactions between a donating solvent (D) and antimony (V) chloride (SbCl5) as a standard acceptor with the respective solvent in a 1:1 ratio in the inert solvent 1,2-dichloroethane, as defined by eqn (1):
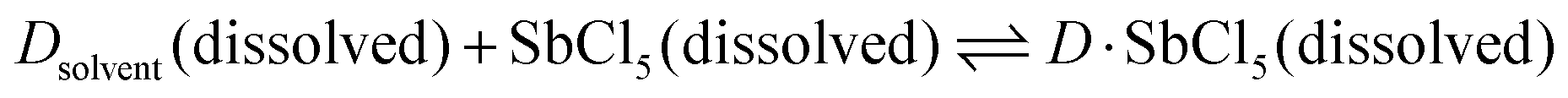
| DN = −ΔHD·SbCl5 | (1) |
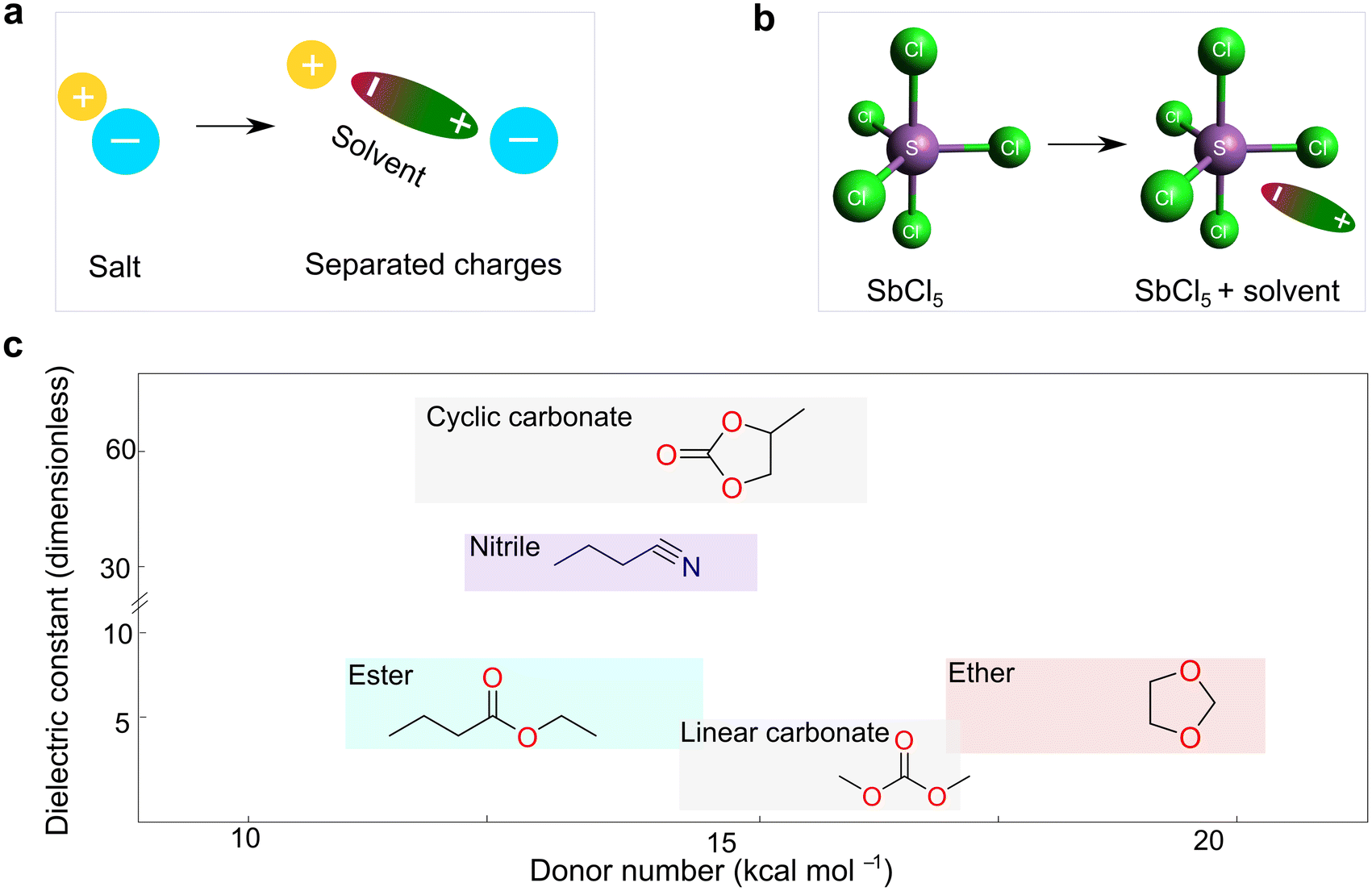 | ||
| Fig. 5 Basic properties of electrolyte solvents. (a) Sketch illustrating the dielectric properties of solvents. (b) Definition of donor number for solvents, as suggested by Gutmann.55 The geometry of SbCl5 is generated and optimized by the open-source molecular visualizer Avogadro. (c) Dual-entry plot showing donor number and dielectric constant for various families of organic solvents utilized for battery electrolytes. The donor number and dielectric constant values are taken from ref. 56 and 57, respectively. | ||
According to eqn (1), it can be observed that DN is a thermodynamic parameter, which describes the degree of the interactions between the donating solvent and SbCl5.
Fig. 5c depicts the correlation between the dielectric constant and donor number for a variety of electrolyte solvents presently used in rechargeable batteries. In practice, the liquid electrolytes that are being utilized for lithium batteries are composed of solvents with relatively high dielectric constants (≥7). In a typical example, linear carbonate solvents [e.g., dimethyl carbonate (DMC), diethyl carbonate (DEC), and EMC] are used as a diluent for cyclic carbonates [e.g., ethylene carbonate (EC) and propylene carbonate (PC)] due to the lower viscosity despite lower dielectric constants of the former [ε = 3 (DMC) vs. ε = 64 (PC) at 25 °C21], given that carbonate solvents have very similar donor numbers of around 16 kcal mol−1, nearly independent of their chemical structures. In comparison, the donicity of ethers is relatively higher than that of carbonates, but their relative permittivity is much lower, e.g., DN = 20 kcal mol−1 [1,2-dimethoxyethane (DME)] vs. DN = 16 (EC)56 and ε = 7 (DME) vs. ε = 90 (EC).21 However, it is interesting to note that ethers can chelate cations by virtue of the structural flexibility of their C–O linkage when there are multiple sites, endowing these solvents with strong solvating ability. For example, in liquid configurations, the solubility of some alkali metal fluorides is largely promoted in the presence of crown ether [1.3 M (acetone/18-crown-6-ether (9:1, by mole)) vs. 0.03 M (neat acetone) for KF59], while in solid configurations, poly(ethylene oxide) (PEO), a polymer version of DME, dissolves a large number of metal salts, which are less soluble in polycarbonates.60,61
Principally, the solvation structures are determined by the Mn+-solvent and Mn+-anion (from salt) interactions. In lean (dilute) electrolytes, the solvent–solvent and Mn+-additive interactions become considerable, both contributing to the electrostatic interactions, including ion–ion, ion–dipole, and dipole–dipole. In this regard, the dielectric constant of the solvent has a significant influence on the binding energy of these interactions, which can be described by the classical physical laws (eqn (2)–(4)).62
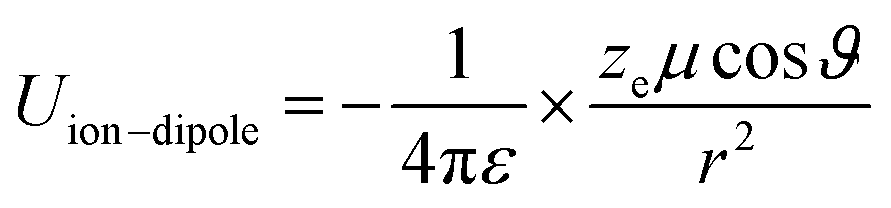 | (2) |
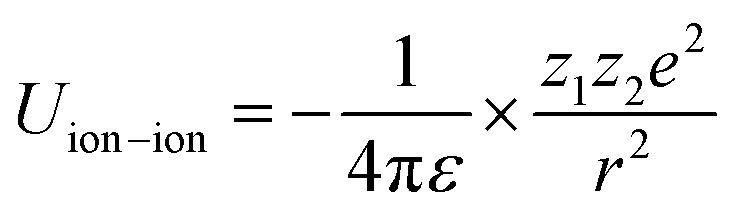 | (3) |
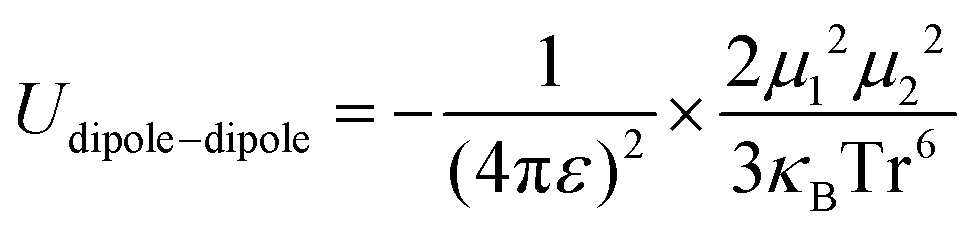 | (4) |
3.2. Transport of ions in non-aqueous electrolytes
The microscopic views of ion transportation in electrolyte solutions are a classic topic of electrochemistry, which have been well scrutinized in previous works, ref. 63–66. As summarized by Bockris et al.,67 the motion of ionic species can be generally categorized in three ways, as follows: (1) a flow of ions pushed by a concentration gradient, which is generally termed “diffusion”; (2) a flow of charges motivated by non-uniform electrostatic potential at various positions of the electrolyte, which is often as “migration or conduction”; and (3) a flow of the whole electrolyte produced by pressure and/or temperature difference, which is named “hydrodynamic” flow. In the battery field, the discussion is normally restricted to the first two cases, while the last one is a common physical phenomenon for all fluids.As early as the 1930s, Bernal and Flower68 proposed that the solvation sheath of ions in water consists of structure-enhanced (i.e., primary) and structure-broken (i.e., secondary) regions (Fig. 6a). Between these regions, water molecules remain unaffected with structures similar to bulk water (i.e., tetrahedrally bonded networks).68 This has been well translated to describe the microscopic images of ion–solvent interactions in non-aqueous solutions, in which the alkali or alkaline metal cations are solvated by aprotic dipolar solvents. In the steady-state, the diffusion and migration (conduction) of solvated ions follow Fick's first law (eqn (5)) and Ohm's law (eqn (6)), respectively, as follows:
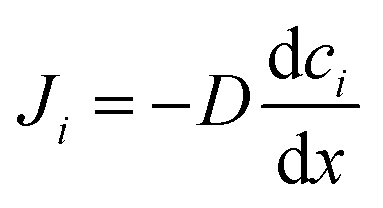 | (5) |
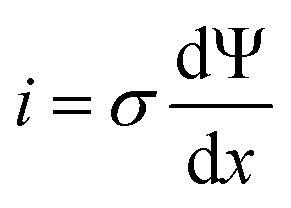 | (6) |
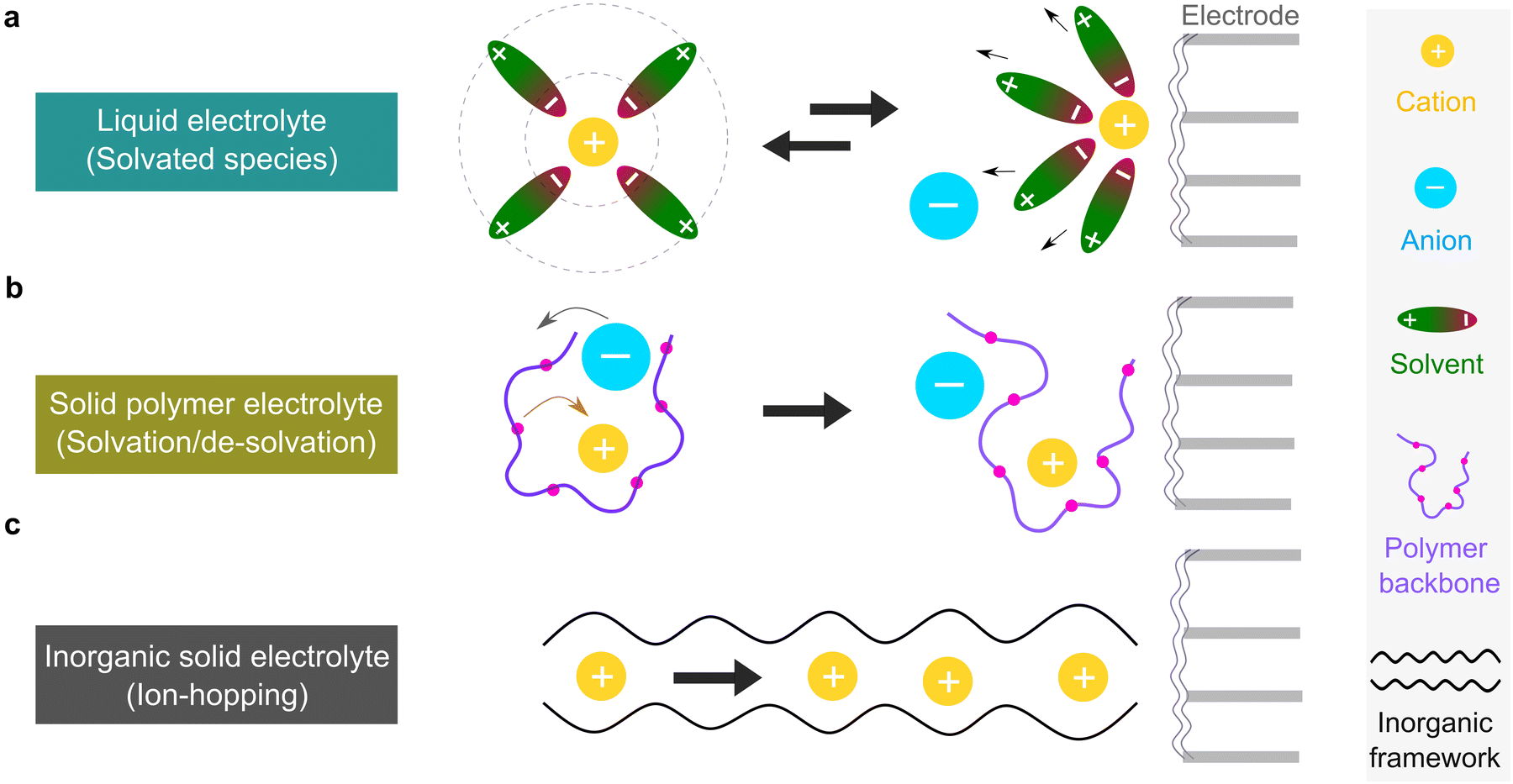 | ||
| Fig. 6 Transport mechanism of non-aqueous electrolytes. (a) Ion transport in liquid electrolyte by solvated species. (b) Ion transport in solid polymer electrolyte by solvation and de-solvation process. (c) Ion transport in inorganic solid electrolyte by ion-hopping process through vacancies. | ||
For liquid electrolyte, the flow of solvated ions is responsible for the transport of ions. The diffusion process can be related to the viscous flow of the electrolyte through the Stokes–Einstein relation (eqn (7)), as provided below:
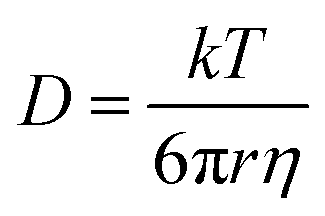 | (7) |
Furthermore, the ionic conductivity (Λ, S cm2 mol−1) is described by the Nernst-Einstein relation (eqn (8)):
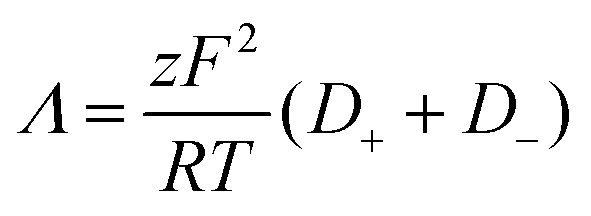 | (8) |
This scenario changes drastically in solvent-free polymer electrolyte due to the immobilization of the “solvent molecules”.60,63,64 In this case, the solvation and de-solvation processes between the ions and polymer chains are the major pathway for transporting the ionic species, as illustrated in Fig. 6b. The ionic conductivity (σ, S cm−1) can be mostly expressed by the free-volume equation (eqn (9)) based on the Vogel–Fulcher–Tammann (VFT) theory,69–71 as given below:
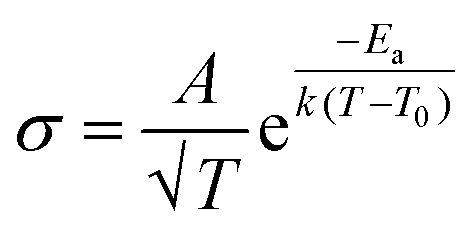 | (9) |
For inorganic solid electrolytes, as seen in Fig. 6c, the migration of ions is achieved via “ion-hopping” processes, which are enhanced when a fraction of the lattice sites contain vacancies or interstitials.72 It is possible to link the ionic conductivity with diffusion in an Arrhenius form (eqn (10)), as follows:
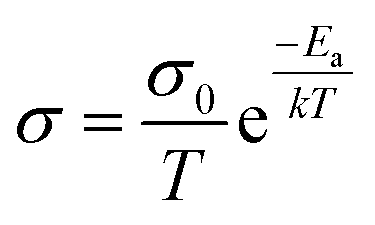 | (10) |
To understand the different transport behaviors between lithium and other monovalent and multivalent cations, it is significant to introduce another parameter, the Eigen number, which is a very important metric but unfortunately rarely discussed in the battery field. In the 1960s, Eigen and co-workers73 systematically studied the kinetics of inner sphere substitution processes of ligands in solution (Fig. 7a) and found that the exchange rate of water molecules is highly associated with the identity of the cations. These substitution (coordinating water) and hydrolysis (splitting water) processes are analogous to solvation and de-solvation processes, respectively, which are highlighted in the transportation of ions in electrolytes. With a wealth of ions, three types of behaviors are generally recognized, as detailed below:
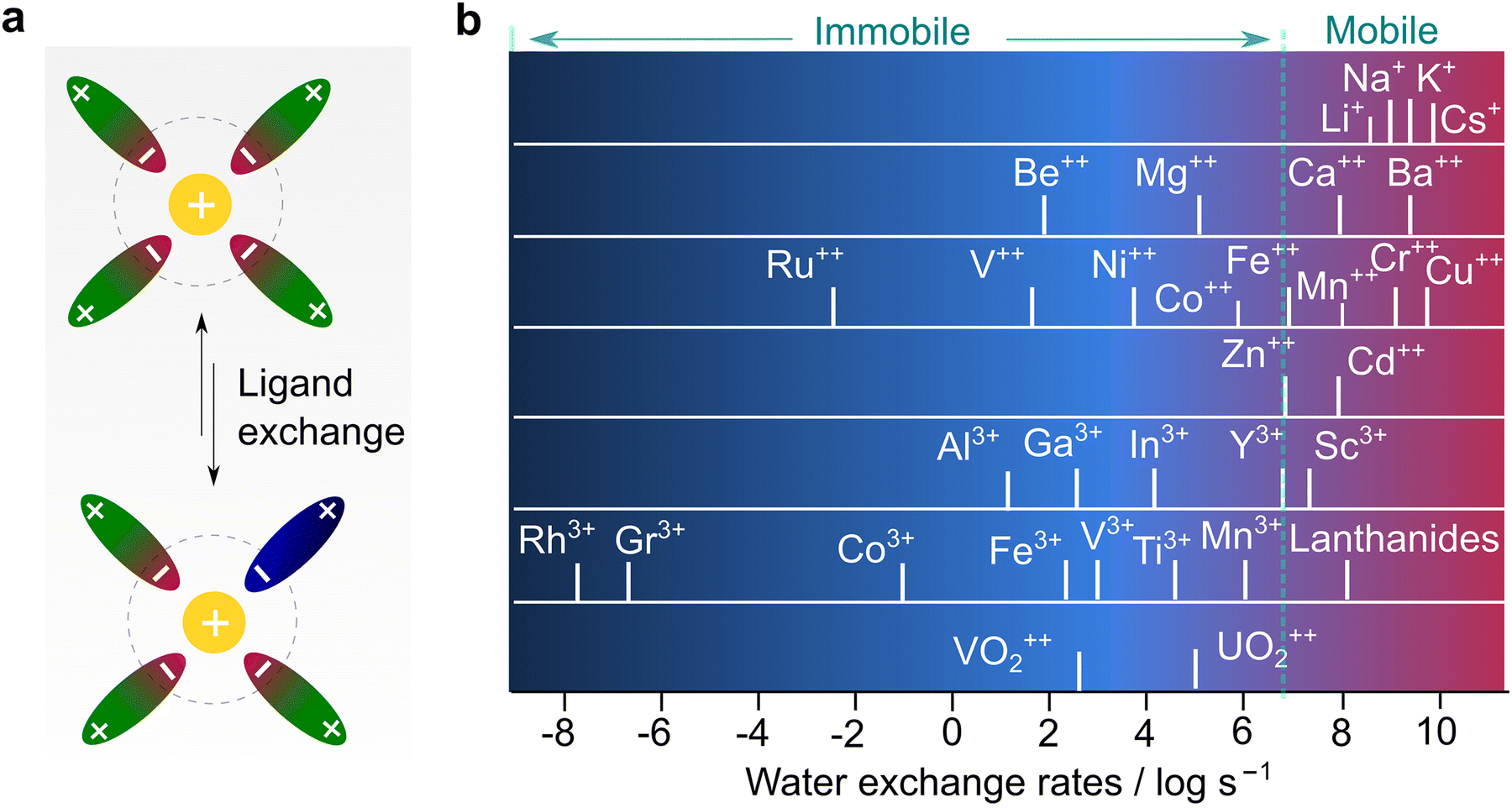 | ||
| Fig. 7 Correlation between ion transport and ligand exchange rate. (a) Ligand exchange process in electrolyte solution. (b) Exchange rates of water in the inner sphere of solvated metal ions, replotted with the data from Eigen's seminal work.73 | ||
(1) Ions with exchange rate constants higher than 107 s−1 (dash line in Fig. 7b), implying a relatively dynamic inner sphere of the solvation sheath (i.e., low kinetic energy in substituting the inner ligand). This includes all alkali (Li+, Na+, and K+) ions and alkaline earth ions beyond Ca2+, or some “d” or “s” shell cations (i.e., Cu2+, Sn2+, and Pb2+).
(2) Ions with exchange rate constants slightly lower than 107 s−1 (close to dash line in Fig. 7b), suggesting a ligand-dependent substitution process (i.e., H2O can replace certain types of ligands with poor interaction with metal ions). This covers most of the divalent transition metal ions, including those of interest for battery use (Mg2+, Zn2+, and Ca2+).14–17,19,24 In terms of kinetics, the metal ions follow the order of Ca2+ > Zn2+ > Mg2+, which reflects the ease at which the elements will be transported in a polymer electrolyte with hopping along the chains. The diffusion in candidate electrode materials also follows the same trend. Ca2+ intercalates in spinels, while Mg2+ diffusion at an appreciable rate is seen only in a sulfide environment (Chevrel phases). Zn2+ diffuses also slowly in γ-MnO2.
(3) Ions with extremely slow exchange rate constants (<104 s−1), indicating that the displacement of the inner ligand is very difficult, or hydrolysis is kinetically unfavorable. Trivalent metal ions and some divalent ones are part of this family. In particular, Al3+, which is an interesting element for making high-energy rechargeable batteries, falls in this catagory.13,24
3.3. Historical overview of non-aqueous electrolytes
Prior to discussing the emerging mono- and multi-valent cation-based electrolytes investigated in the last 10 years, it is important to give an overview of the development of non-aqueous electrolytes associated with rechargeable batteries. As early as the 13th century, metallic zinc was isolated and used. In the beginning of the 19th century, lithium was recognized as an element by Arfewdson74 and later isolated in metallic form by Davy75 through a voltaic pile containing fused lithium carbonate.76 In the same period, metallic sodium, potassium, magnesium, calcium, and aluminum were also obtained via electrolysis.75 Therefore, the alkali and alkaline metals of interest for the current rechargeable batteries were already available for electrochemists before the middle of the 19th century.At the end of the 19th century, non-aqueous solvents were employed by Plotnikov et al.77 to deposit Al metal from a solution of AlBr3-KBr in benzene, which was the time at which Al electroextraction from molten salt had just been industrialized (1886). Eleven years later, in 1897, Laszczynski and Gorski78 deposited metallic lithium from a non-aqueous solution of lithium chloride in pyridine. From the end of the 19th century to the first few decades of the 20th century, several metallic elements (e.g., lithium, magnesium, zinc, and aluminum) were plated from non-aqueous electrolytes. Patten et al.79 achieved the plating of zinc metal from ZnCl2/acetone and Evans et al.80 obtained metallic magnesium using an ether solution containing a Grignard reagent [ethyl magnesium bromide (EtMgBr)]. These early activities are a prelude to the prosperous development of electrochemistry in non-aqueous electrolytes for battery use,79–107 as mapped in Fig. 8.
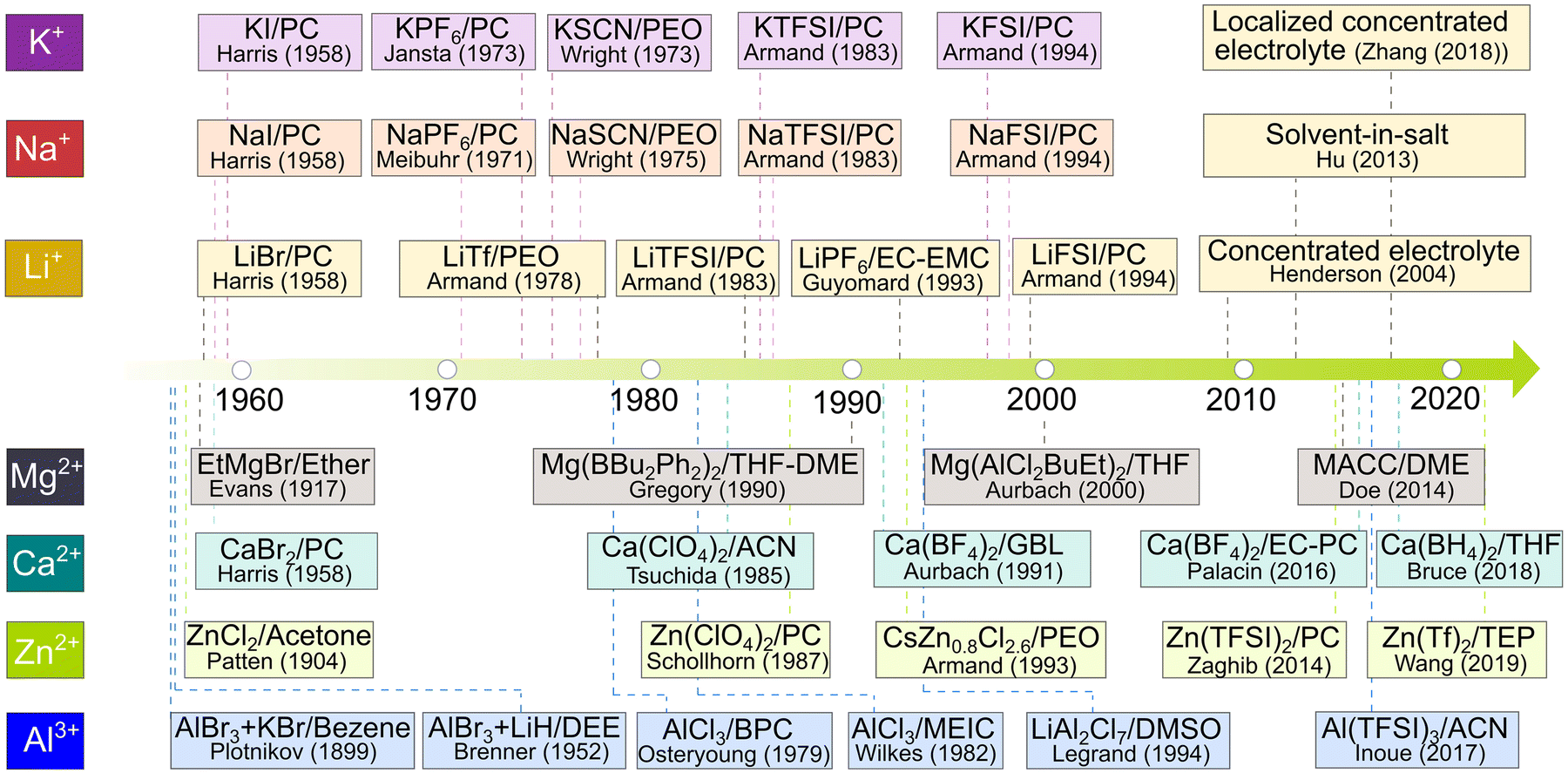 | ||
| Fig. 8 Historical evolution of non-aqueous electrolytes in relation to rechargeable batteries. For Li-based electrolytes, the respective source references for lithium bromide (LiBr)/propylene carbonate (PC), lithium triflate (LiTf)/poly(ethylene oxide) (PEO), lithium bis(trifluoromethanesulfonyl)imide (LiTFSI)/PC, lithium hexafluorophosphate (LiPF6)/ethylene carbonate (EC)-PC, lithium bis(fluorosulfonyl)imide (LiFSI)/PC, concentrated electrolyte, solvent-in-salt, and localized concentrated electrolytes are available from ref. 81, 83, 90, 95, 97, 99, 100 and 105, respectively. For Na-based electrolytes, the respective source references for sodium iodide (NaI)/PC, sodium hexafluorophosphate (NaPF6)/PC, sodium isocyanate (NaSCN)/PEO, sodium bis(trifluoromethanesulfonyl)imide (NaTFSI)/PC, and sodium bis(fluorosulfonyl)imide (NaFSI)/PC are available from ref. 83, 84, 87, 90 and 97, respectively. For K-based electrolytes, the respective source references for potassium iodide (KI)/PC, potassium hexafluorophosphate (KPF6)/PC, potassium isocyanate (KSCN)/PEO, potassium bis(trifluoromethanesulfonyl)imide (KTFSI)/PC, and potassium bis(fluorosulfonyl)imide (KFSI)/PC are available from ref. 83, 85, 86, 90 and 97, respectively. For Mg-based electrolytes, the respective source references for ethyl magnesium bromide (EtMgBr)/ether, magnesium dibutyldiphenylborate [Mg(Bu2Ph2)2]/tetrahydrofuran (THF)-1,2-dimethoxyethane (DME), magnesium butyldichloro(ethyl)aluminate [Mg(AlCl2BuEt)2]/THF, and magnesium aluminum chloride complex (MACC)/DME are available from ref. 80, 92, 98 and 101, respectively. For Ca-based electrolytes, the respective source references for calcium bromide (CaBr2)/PC, calcium perchlorate [Ca(ClO4)2]/PC, calcium tetrafluoroborate [Ca(BF4)2]/γ-butyrolactone (GBL), Ca(BF4)2/EC-PC, and calcium borohydride [Ca(BH4)2]/THF are available from ref. 83, 84, 93, 103 and 106, respectively. For Zn-based electrolytes, the respective source references for zinc chloride (ZnCl2)/acetone, zinc perchlorate (Zn(ClO4)2)/PC, cerium zinc chloride (CeZn0.8Cl2.6)/PEO, zinc bis(trifluoromethanesulfonyl)imide [Zn(TFSI)2]/PC, and zinc triflate (Zn(Tf)2)/triethyl phosphate (TEP) are available from ref. 79, 91, 94, 102 and 107, respectively. For Al-based electrolytes, the respective source references for aluminum bromide (AlBr3) and potassium bromide (KBr) in benzene, AlBr3 and lithium hydride (LiH) in diethyl ether (DEE), aluminum chloride (AlCl3) in n-butylpyridinium chloride (BPC), AlCl3 in 1-methyl-3-ethylimidazolium chloride (MEIC), and lithium aluminum chloride (LiAl2Cl7) in dimethyl sulfoxide (DMSO), and aluminum bis(trifluoromethanesulfonyl)imide (Al(TFSI)3)/acetonitrile (ACN) are available from ref. 79, 82, 88, 89, 96 and 104, respectively. | ||
In 1958, Harris and Tobias108 systematically investigated the fundamental properties of a series of non-aqueous electrolytes based on cyclic carbonates, including PC and EC, both of which are indispensable components in the current lithium-ion batteries (LIBs). This pioneer work is a milestone in the search for suitable electrolytes for LIBs and other emerging mono-valent cations - based batteries, such as sodium and potassium batteries. In the 1970s, the intercalation chemistry of layered compounds (e.g., TiS2) was thoroughly studied by several groups.109–112 In the early 1980s, Armand113 proposed the so-called rocking-chair batteries built with two intercalation compounds having different redox potentials, which is effectively the seminal concept for the current LIBs. This concept was later experimentally proven by Lazzari and Scrosati114 using LixWO2|LiClO4-PC|TiS2 cells. The discovery of the lithium cobalt oxide cathode by Goodenough et al.115 and carbonaceous anode by Yoshino et al.,116 together with the ingenious selection of LiPF6-based carbonate electrolyte, made the debut of LIBs in the commodity market possible in 1991.29,117–120
Following the same timeline of non-aqueous liquid electrolytes, solid electrolytes were significantly developed from the 1970s to 1990s. In 1973, Wright et al.85 discovered that a solid mixture of PEO and potassium thiocyanate (KSCN) affords decent ionic conductivities at a temperature above the melting transition of PEO (Tm = 65 °C). Subsequently, Armand et al.81 suggested that this new type of plastic solid electrolyte can be used for the fabrication of solid-state rechargeable batteries. This pioneering work brought polymer electrolyte into the center stage of solid electrolytes, which are supposed to be safer and more compatible with alkali metal anodes than carbonate - based liquid electrolytes.121–123
The success of LIBs in the 1990s generated great research interest in the battery community, and thus intensive efforts and financial funding were dedicated to their research. This effectively slowed the developments in the field of other types of rechargeable batteries based on other emerging mono- and di-valent cations. In the case of sodium- and potassium-based batteries, the attention paid to non-aqueous electrolytes was limited before the 2000s. In comparison, non-aqueous electrolytes for divalent cations and trivalent cations seemed to evolve gradually with the implementation of new salts and non-aqueous solvents. In the case of magnesium-based non-aqueous electrolytes, starting from the electrochemical deposition of metallic magnesium from Grignard reagent by Evans et al.80 in the 1900s, numerous magnesium salts have been developed for improving the Coulombic efficiencies (CEs) of magnesium batteries.10,12,17,124–126
3.4. Emerging mono- and multi-valent liquid electrolytes
In the case of Na-ion conducting electrolytes, the utilization of a binary mixture of EC and PC allows the facile dissociation of NaPF6, leading to relatively high ionic conductivities at room temperature (Fig. 9a). The resulting electrolyte allows stable cycling of the hard carbon-based anode with good capacity retention, which is recognized as the standard reference electrolyte for sodium-ion batteries.130–133 With the addition of small amounts of DMC, the ionic conductivities of the resulting electrolyte [i.e., NaPF6-EC/PC/DMC (45:45:10, by wt.), Fig. 9b] increased slightly and the hard carbon‖Na3V2(PO4)2F3 cells were cycled with a good rate-capability (Fig. 9c).134
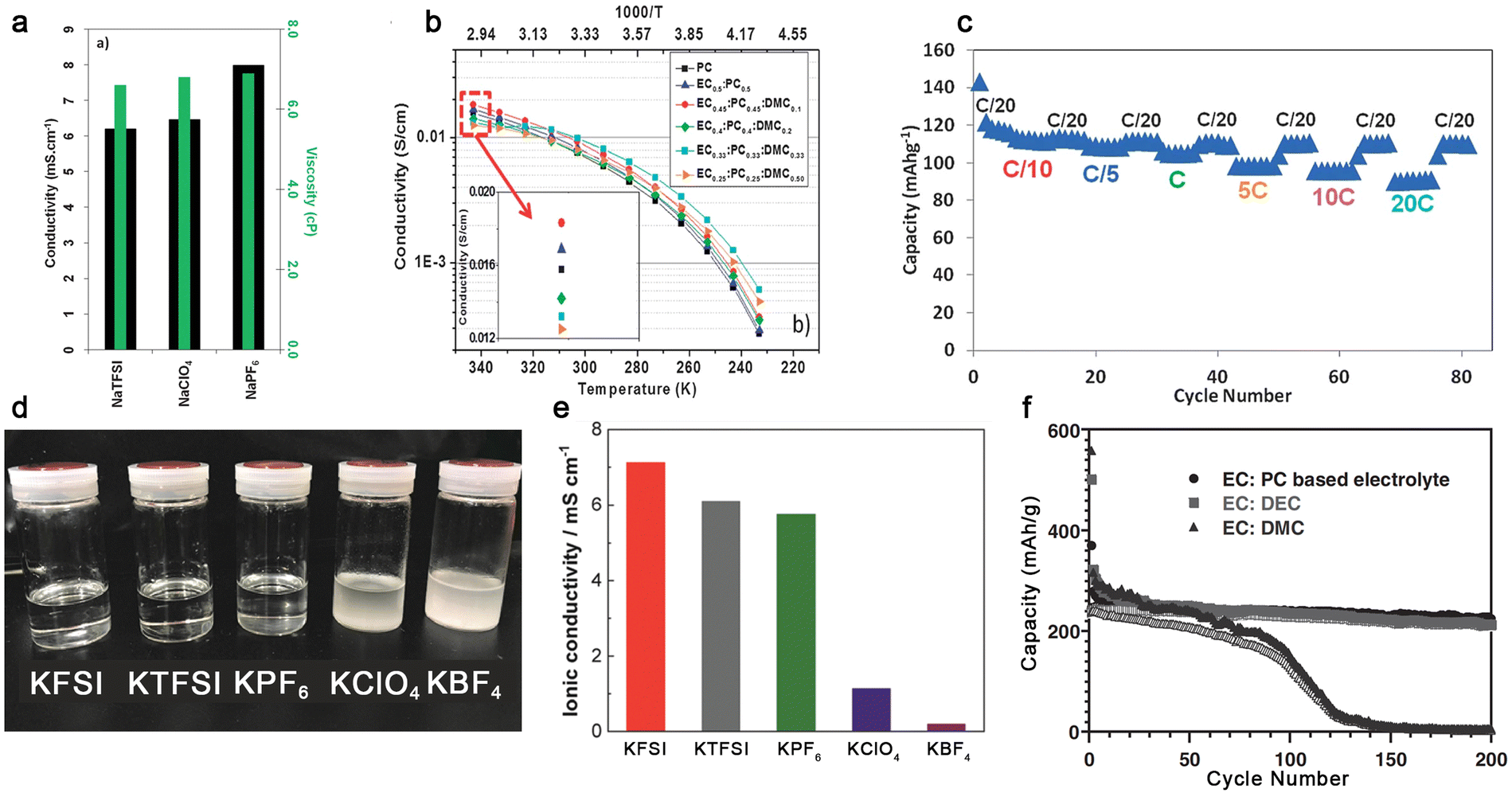 | ||
| Fig. 9 Na+-ion and K+-ion-conducting electrolytes based on organic carbonate solvents. (a) Ionic conductivities of various sodium salts in PC at 25 °C. Reproduced from ref. 130 with permission. Copyright 2012, The Royal Society of Chemistry. (b) Temperature dependence of ionic conductivities for NaTFSI-based electrolytes with different carbonate solvents. (c) Rate performance of the hard carbon‖Na3V2(PO4)2F3 cell utilizing 1.0 M NaPF6-EC/PC/DMC (45:45:10, by wt.). Reproduced from ref. 134 with permission. Copyright 2013, The Royal Society of Chemistry. (d) Solubility tests and € ionic conductivities of several types of potassium salts in PC. Reproduced from ref. 135 with permission. Copyright 2018, The Royal Society of Chemistry. (f) Cycling performance of K‖graphite cells with the electrolyte of 1.0 M KPF6 in EC/PC (1:1, by vol.), EC/DEC (1:1, by vol.), and EC/DMC (1:1, by vol.). Reproduced from ref. 136 with permission. Copyright 2016, Wiley-VCH. | ||
In the case of K-ion conducting electrolytes, the choice of salt anion seems to be vital. Some popular inorganic anions including ClO4− and BF4− are barely soluble in organic carbonate solvents (Fig. 9d) when paired with K+ cations due to the lower solvating power of K+ cations and the higher lattice energy of KClO4 and KBF4 compared to their Li- and Na-based analogues.135 Apparently, a saturated solution of KBF4 in PC is much less conductive than that with other salt anions [e.g., 8.0 × 10−3 (KFSI) vs. <1.0 × 10−3 S cm−1 (KBF4), Fig. 9e],135 which excludes the utilization of ClO4− and BF4− in K-based batteries. In addition to the selection of the salt anion, the nature of organic carbonates also has an impact on the cycling behavior of the graphite anode (Fig. 9f). It seems that linear carbonates including DEC and DMC are likely to decompose on the graphite anode during the intercalation and de-intercalation processes of K+ cations, causing a gradual decease in the attainable capacity.136
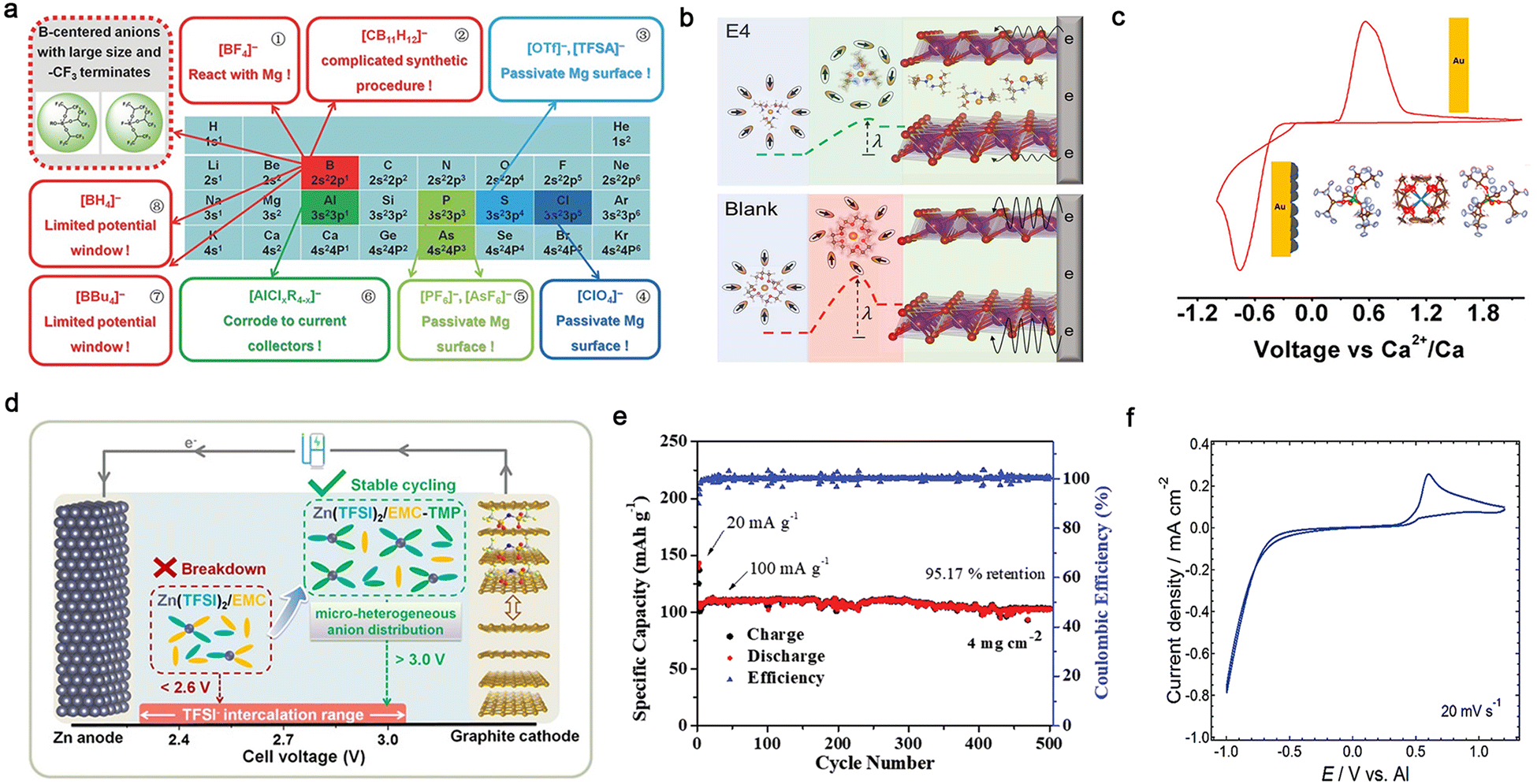 | ||
| Fig. 10 Multi-valent cation-based in-box organic liquid electrolytes. (a) Decorated periodic table for directing efficient Mg-ion electrolytes. Reproduced from ref. 153 with permission. Copyright 2017, Wiley-VCH. (b) Scheme of concerted ion and electron transfer in the cathode host limited by the solvation sheath reorganization. Reproduced from ref. 160 with permission. Copyright 2021, The American Association for the Advancement of Science. (c) Reversible Ca2+ plating and stripping at room temperature using calcium tetrakis(hexafluoroisopropyloxy)borate (Ca[B(hfip)4]2/DME) electrolyte. Reproduced from ref. 164 with permission. Copyright 2019, the American Chemical Society. (d) Design strategy of EMC-based electrolyte for high-voltage Zn/graphite cells. Reproduced from ref. 167 with permission. Copyright 2020, Wiley-VCH. (e) Cycling performance of VS2 cathode in trimethyl phosphate (TMP)–dimethyl carbonate (DMC) electrolyte. Reproduced from ref. 169 with permission. Copyright 2019, Wiley-VCH. (f) CV recorded on a Pt electrode in a solution of Al(Tf)3 in N-methylacetamide (NMA) and urea. Reproduced from ref. 173 with permission. Copyright 2015, The Royal Society of Chemistry. | ||
Hence, numerous types of electrolyte solutions have been developed for multi-valent batteries during the past decades to achieve reversible electrochemical behaviors and obtain high ionic conductivity, CEs, and anodic stabilities.14–17,19,24 Instead of employing the widely used carbonate solvents in LIBs, the solvents developed for multivalent batteries are normally ethereal solvents with higher donicity and cathodic stability. In the case of magnesium-based non-aqueous organic electrolytes, the first prototype rechargeable Mg-based cell was reported by Aurbach and co-workers in 2000,98 utilizing an electrolyte consisting of Mg organohaloaluminate salts. Since then, a variety of advanced Mg-ion conducting electrolytes with intriguing chemical/electrochemical features has been extensively explored, e.g., non-nucleophilic all-phenyl complex (APC) electrolytes,145,146 magnesium aluminum chloride complex (MACC) electrolytes,101,147,148 hexamethyldisilazide (HMDS)-based electrolytes,149 boron-centered electrolytes (Fig. 10a),150–157 aluminum-centered electrolytes,158,159 and Mg(TFSI)2-based electrolytes (Fig. 10b),160,161 which significantly boost the development and advancement of rechargeable magnesium batteries.
Similar to the Mg metal anode, the main obstacle of calcium-based non-aqueous batteries is that the SEI layers formed by the reactions between the Ca anode and conventional solvents [e.g., acetonitrile (ACN), PC, and tetrahydrofuran (THF)], and the reductive products from conventional calcium salts [e.g., Ca(ClO4)2 and Ca(BF4)2] generate passivating films, which impede the transport of Ca2+ through the surface and reversible electrochemical calcium deposition.14,93,162 A significant development in electrochemical calcium plating and stripping was achieved by Palacín et al. in 2016,103 who demonstrated that reversible redox processes occur in the electrolyte of Ca(BF4)2-EC/PC at 100 °C. However, poor reversibility of calcium redox reactions was detected in the Ca(ClO4)2- and Ca(TFSI)2-based electrolytes. The improved electrochemical performance of the Ca(BF4)2-based electrolyte is possibly ascribed to the surface layer formed on the electrode surface, which could enable easier migration of Ca2+ through the anode-electrolyte interphase (see Section 4). Reversible calcium plating and stripping at room temperature was demonstrated independently by the groups of Zhao-Karger163 and Nazar164 using an efficient electrolyte comprised of calcium tetrakis(hexafluoroisopropyloxy)borate (Ca[B(hfip)4]2) salt and DME solvent (Fig. 10c).165 The corresponding electrolyte displayed high ionic conductivity (>8.0 × 10−3 S cm−1), allowing the reversible deposition/dissolution of Ca2+ ions (CE: 80–90%), which opens a possible path towards room-temperature rechargeable calcium batteries. In addition, Bruce et al.106 demonstrated that with the Ca(BH4)2/THF electrolyte, calcium can be plated and stripped with quite high CEs (ca. 94–96%), which is possibly related to the presence of more ionically conductive species on the calcium surface accompanied by a small amount of CaH2 products.
Zinc, possessing the highest reduction potential [−0.76 V vs. standard hydrogen electrode (SHE)] among the mono-valent and multivalent metals, presents the highest tolerance to aprotic solvents and salt anions.24,166 Reversible electrochemical plating and stripping processes are realized in conventional solvents [e.g., PC,102 EMC,167 ACN,168 trimethyl phosphate (TMP),169 triethyl phosphate (TEP),107 and acetamide170,171], while several efficient non-aqueous organic electrolytes have been also explored for rechargeable zinc batteries. The ionic conductivity of the electrolyte is highly associated with the nature of the electrolyte solvent and salt.172 For example, with the same Zn(TFSI)2 salt, the ionic conductivity values were significantly improved when replacing acetamide with TMP solvent, e.g., 3.1 × 10−4 S cm−1 for Zn(TFSI)2/acetamide170vs. 1.5 × 10−2 S cm−1 for Zn(TFSI)2/TMP170 at room temperature. The addition of EMC to Zn(TFSI)2/TMP further increased the ionic conductivity up to 1.7 × 10−2 S cm−1, and the modified electrolyte could simultaneously ensure the reversible operation of 3-V-class Zn‖graphite cells (Fig. 10d). In addition to the solvent, salt anions also play a pivotal role in influencing the ionic conductivity of non-aqueous Zn-ion conducting electrolytes. With almost the same solvents, Zn(Tf)2-TMP/DEC electrolyte displays a lower ionic conductivity of 4.9 × 10−3 S cm−1,169 which can be ascribed to the higher dissociation of the TFSI− anion generated from its superior delocalized anionic structure. Nevertheless, the Zn‖VS2 cell with this electrolyte could be cycled for 500 cycles under a high loading (ca. 4 mg cm−2) with a high capacity retention of 95.2% (Fig. 10e).
In case of Al-based non-aqueous organic electrolytes, the choice of the solvent can significantly tune the physicochemical properties (e.g., ionic conductivity) owing to the highest charge density of Al3+ and its high Coulombic interactions with anions and solvents. It has been demonstrated that aluminum triflate (Al(Tf)3)-based electrolytes with higher dielectric constant solvents show higher ionic conductivities, e.g., 2.4 × 10−3 [Al(Tf)3/N-methylacetamide (NMA)] vs. 1.9 × 10−3 [Al(Tf)3/PC] vs. 1.6 × 10−4 S cm−1 [Al(Tf)3/ACN].173 The Al(Tf)3-based electrolyte containing a mixture of NMA/urea displayed electrochemically reversible behaviors (Fig. 10f) and a broader electrochemical window (ca. 3.5 V vs. Al/Al3+). Considering that the TFSI− anion is more resistant to electrochemical oxidation than the Tf− anion, the electrochemical window can be further extended by replacing Al(Tf)3 with Al(TFSI)3 in ACN solvent (Fig. 10f).104
In a seminal paper published in 1933, Onsager178 discussed the theoretic models for concentrated electrolytes, aiming to extend the Debye–Hückel model from the amply verified diluted solutions to much more complex concentrated solutions. The early trials on non-aqueous concentrated electrolyte developed for battery use can be traced back to the “polymer-in-salt” concept proposed by Angell et al.,179,180 in which an extremely high amount (>90 wt%) of low-melting lithium salt was employed to decouple the dynamics of ion transport from the segmental motion of polymer backbones.180
In 2013, Hu et al.100 proposed the use of a highly concentrated ether-based electrolyte consisting of 7 moles LiTFSI per liter of DME/1,3-dioxolane (DOL), which was named “solvent-in-salt (SIS)” electrolyte (Fig. 11a). As expected, the viscosity of the electrolyte gradually increased with an increase in the salt content, and the corresponding ionic conductivity became much lower for these SIS electrolytes [ca. 1.0 × 10−3 S cm−1 (7 moles salt per liter solvent) vs. ca. 1.5 × 10−2 S cm−1 (1 mole salt per liter solvent), Fig. 11b]. However, the transference numbers of the Li+ cations tended to be much higher for the electrolytes with a high salt content (>0.5), which is ascribed to the presence of ion-pairs in the electrolyte solution. Interestingly, a high concentration of LiTFSI in the ethereal solvent excluded the dissolution of polysulfide intermediates, as visualized in Fig. 11c, which could prevent the shuttling effect of soluble lithium polysulfides intermediates (Li2Sn, 4 ≤ n ≤ 8) observed in Li–S cells with the conventional salt content (<1.5 mol L−1). This trend can also be applied to the anodic dissolution processes of Al current collectors at high potentials. Henderson and co-workers174 demonstrated that Al corrosion in LiTFSI-carbonate electrolyte (a commonly known corrosive electrolyte) is remarkably suppressed by increasing the salt content (Fig. 11d) due to the absence of uncoordinated (i.e., free) solvent molecules in the concentrated electrolytes. These fascinating properties of salt-concentrated electrolytes have also been amply verified for a wide array of salt anions [e.g., PF6,181 FSI,182,183 (fluorosulfonyl)(trifluoromethanesulfonyl)imide (FTFSI),184 and (fluorosulfonyl)(n-nonafluorobutanesulfonyl)imide (FNFSI)184] and electrolyte solvent [EC,185 fluoroethylene carbonate (FEC),186 ACN,187etc.], as detailed in recent review articles.176,177,188
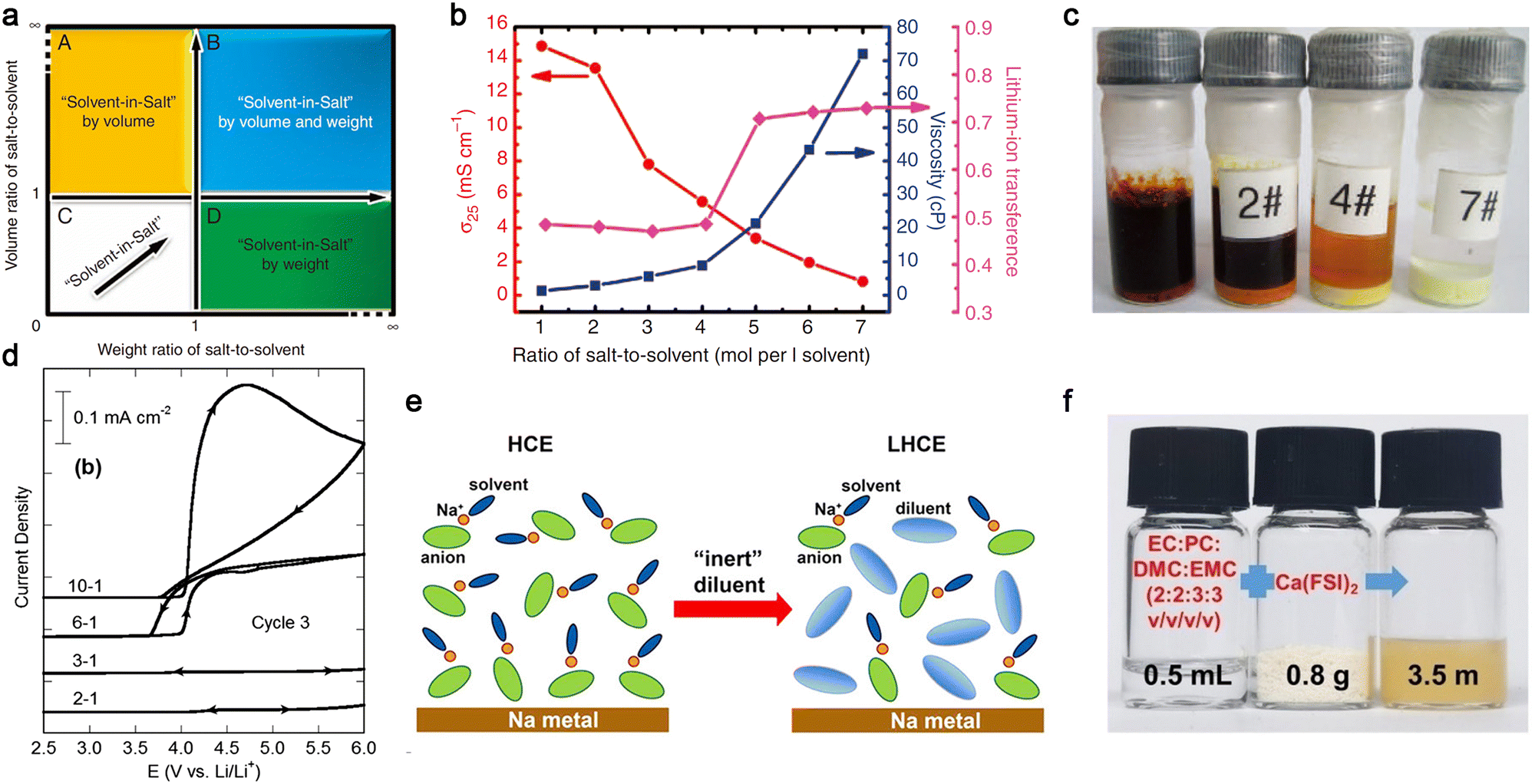 | ||
| Fig. 11 Salt-concentrated electrolyte developed for mono- and multi-valent batteries. (a) Sketch of the “solvent-in-salt” concept proposed by Hu et al.100 (b) Effect of salt concentration on viscosity and transport properties (i.e., ionic conductivity and lithium-ion transference number) of LiTFSI-DME/1,3-dioxolane (DOL) at room temperature. (c) Solubility of polysulfides in highly concentrated electrolyte after storage at room temperature for 18 days. (Code for electrolytes: 0 mol per liter solvent, 2#: 2 mol per liter solvent, 4#: 4 mol per liter solvent, and 7#: 7 mol per liter solvent). Reproduced from ref. 100 with permission. Copyright 2013, Macmillan Publishers Limited. (d) Anodic stability of Al foil in LiTFSI/EC electrolyte with various salt contents (EC:LiTFSI = 10, 6, 3, 2, and 1 by mole). Reproduced from ref. 174 with permission. Copyright 2014, The Royal Society of Chemistry. (e) Schematic illustration for the highly concentrated electrolyte (HCE) and localized concentrated electrolyte (LHCE). Reproduced from ref. 190 with permission. Copyright 2018, the American Chemical Society. (f) Digital photos of organic carbonate electrolyte, Ca(FSI)2 salt, and the resulting electrolyte. Reproduced from ref. 192 with permission. Copyright 2022, Wiley-VCH. | ||
Despite the interesting features of salt-concentrated electrolytes, the utilization of high amounts of metal salts, however, not only increases the density of the electrolyte solution, but also increases the cost, particularly for Li-based electrolytes, and longer wetting times before use due to their high viscosity. In this regard, Zhang et al.6,27,189 suggested the deployment of non-solvating solvents to make localized highly concentrated electrolyte (LHCE), in which the diluting solvent (e.g., fluoro-ether) itself possesses very low electron-donating ability but good electrochemical stabilities (Fig. 11e). This effectively allows a decrease in the concentration of metal salts with negligible effect on the electrochemical properties of the resulting electrolytes. Zhang et al.190 reported that the LHCE formed by dissolving 2.5 M NaFSI and DME-bis(2,2,2-trifluoroethyl) ether (BTFE) (1:2, by mole) affords relatively decent ionic conductivities (ca. 2.0 × 10−3 S cm−1 at room temperature) and impressive electrochemical stability, allowing the stable operation of Na‖Na3V2(PO4)3 cells (90.8% retention after 40000 cycles). Wang et al.191 suggested the utilization of a mixed Na-ion conducting electrolyte containing 2,2,2-trifluoro-N,N-dimethylacetamide (FDMA) as a solvating solvent and 1,1,2,2-tetrafluoroethyl methyl ether (MTFE) as an anti-solvent, significantly suppressing the dissolution of polysulfide species.
In addition to mono-valent electrolytes, the concept of salt-concentrated electrolyte has been also extended to divalent cation-based electrolytes. Recently, Tang et al.192 reported that a high concentration (ca. 3.5 moles salt per liter of solvent) of calcium bis(fluorosulfonyl)imide [Ca(FSI)2] can be achieved with organic carbonate solvents (Fig. 11f). Ca(FSI)2-based electrolytes are relatively conductive (ca. 5 × 10−4 S cm−1 at room temperature), enabling the reversible intercalation of Ca2+ ions in graphite cathode. The corresponding cell with 3,4,9,10-perylenetetracarboxylic dianhydride anode and graphite cathode showed low capacity decay after 300 cycles. It should be noted that the Eigen value of Ca2+ ions is much closer to that of mono-valent cations (e.g., Li+, Na+, and K+) compared to Mg2+ and Al3+ ions (Fig. 7); hence, the aforementioned interesting results effectively verify the applicability of Eigen values in developing designer electrolytes for multi-valent batteries.
The classic carbonate solvent, EC, has been applied in the battery field with tremendous efforts dedicated to the electrochemical analysis of the bulk electrolyte and EEI layers; however, the practical feasibility of salt-concentrated electrolytes has to be further evaluated by long-term cycling tests, particularly considering the interphase degradation of these new solvents at both negative and positive electrodes. For example, it remains unclear whether fluorinated anti-solvents (e.g., MTFE) can retain electrochemical compatibility with the electrode materials during aging tests, although these anti-solvents seem to be effective in decreasing the salt concentration and electrolyte viscosity. In relation to multi-valent cations, increasing the amount of salt will drastically increase the viscosity, and thus decrease the bulk conductivity and also result in longer wetting times. Moreover, the solvent/salt ratio is likely to increase for multi-valent cations due to their lower solvating ability compared to mono-valent cations, which may finally cause different solvating structures and chemical/electrochemical reactivities of the solvent molecules.
For battery applications, ILs with low viscosity and wide electrochemical windows are highly desirable. By replacing rigid anions (CF3SO3− and BF4−) with sulfonimide anions (TFSI− and FSI−), one may readily decrease the viscosity of ILs by virtue of the higher degree of the structural flexibility of the –SO2–N(−)–SO2– linkage, e.g., 90 cP [1-butyl-3-methylimidazolium triflate (BMITfO)200] vs. 52 cP [1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide (BMITFSI)200], and 42 cP [1-ethyl-3-methylimidazolium tetrafluoroborate (EMIBF4)201] vs. 34 cP [1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide (EMITFSI)202] at 25 °C. Note that TFSI-based ILs are quite conductive (ca. 8.0 × 10−3 S cm−1 for EMITFSI at room temperature) without any metal salts (Fig. 12a). Presently, TFSI-based ILs have been widely adopted to formulate IL-based electrolytes due to their low viscosity and high oxidative stability.193–197 Interestingly, one may enhance the anodic stability of TFSI-based ILs by replacing the oxygen atom with a stronger electron-withdrawing group (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NSO2CF3), which is beneficial for the operation of high-voltage cathode materials.
NSO2CF3), which is beneficial for the operation of high-voltage cathode materials.
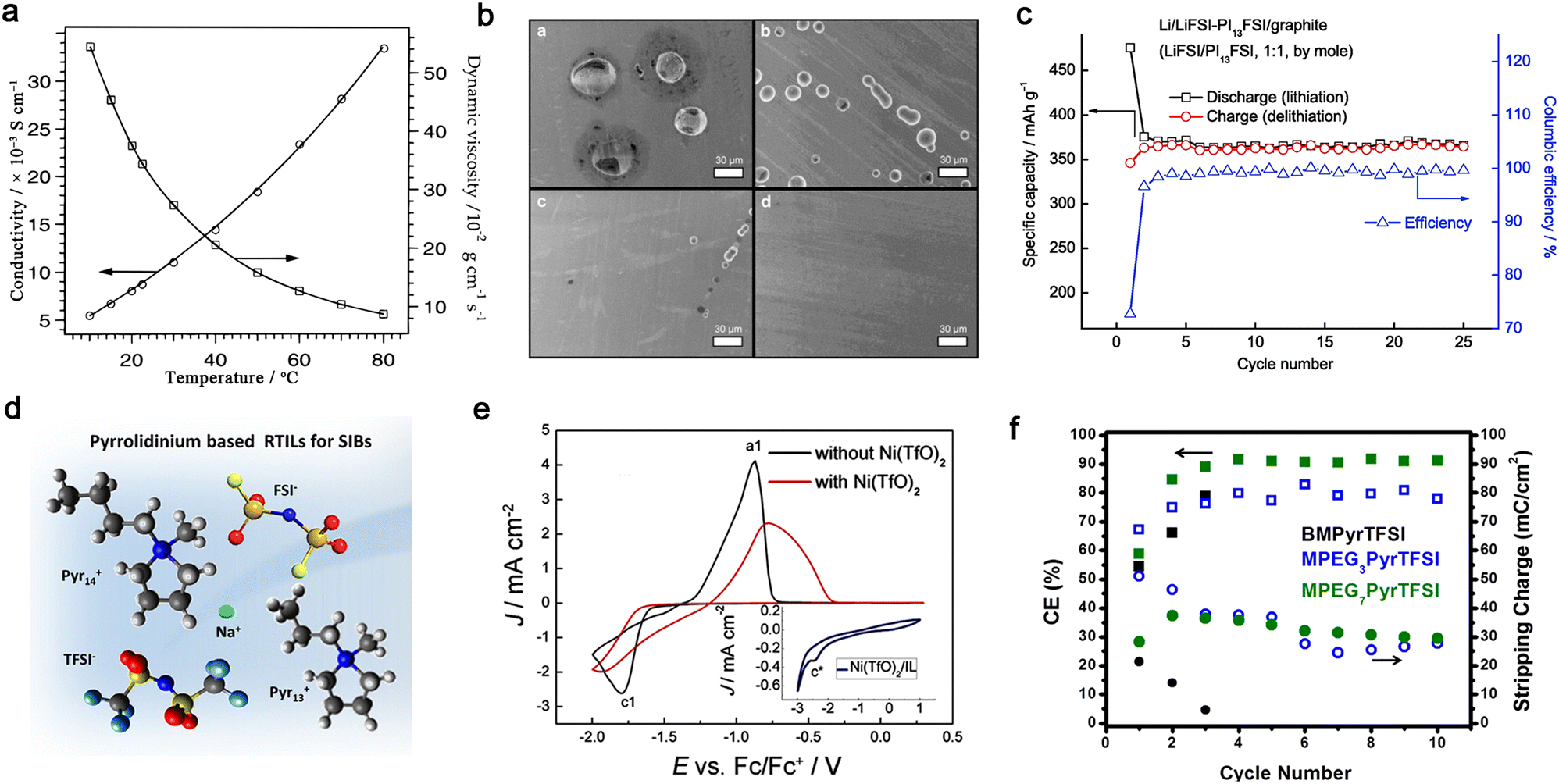 | ||
| Fig. 12 Ionic liquid-based non-aqueous organic liquid electrolytes for mono- and multivalent batteries. (a) Ionic conductivity and viscosity for 1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide (EMITFSI) at various temperatures. Reproduced from ref. 200 with permission. Copyright 1996, the American Chemical Society. (b) SEM images of Al electrodes obtained from polarization tests (4.6 V vs. Li/Li+ for 12 h). For clarity, the yellow circles are used to overlap figure labels in the source figure. Adapted from ref. 207 with permission. Copyright 2012, Elsevier. (c) Cycling performance of Li‖natural graphite cell with LiFSI/N-propylpiperidinium bis(fluorosulfonyl)imide (PI13FSI) (1:1, by mole) at 25 °C. Reproduced from ref. 209 with permission. Copyright 2014, Elsevier. (d) Pyrrolidinium-based IL electrolyte developed for sodium-ion batteries. Reproduced from ref. 211 with permission. Copyright 2019, the American Chemical Society. (e) CV profiles of the electrolytes of zinc triflate (Zn(TfO)2)/1-ethyl-3-methylimidazolium triflate (EMITfO) with or without Ni(TfO)2 (inset figure for the CV profile of 0.015 M Ni(TfO)2 in EMITfO). Reproduced from ref. 214 with permission. Copyright 2016, Wiley-VCH. (f) Coulombic efficiencies and stripping charges (circles) for each cycle in electrolytes with different ILs [i.e., 1-butyl-1-methylpyrrolidinium-bis(trifluoromethanesulfonyl)imide (BMPyrTFSI), 1-(2-(2-(2-methoxyethoxy)ethoxy)ethyl)-1-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide (MPEG3PyrTFSI), and methoxypolyethylene glycol bis(trifluoromethanesulfonyl)imide (MPEG7PyrTFSI)]. Reproduced from ref. 218 with permission. Copyright 2016, the American Chemical Society. | ||
With respect to the design of IL cations, the introduction of N-methyl-N-alkylpyrrolidinium by Forsyth et al.203–205 can be regarded as an important step forward in the case of imidazolium-based ILs. Pyrrolidinium-based ILs exhibit better electrochemical stability against oxidation/reduction compared to imidazolium-based ILs, rendering the corresponding electrolytes with better compatibility with electrode materials.204,205 Interestingly, one may obtain low-viscous ILs with multi-methoxyethyl substituted cations, e.g., 69 cP (N,N-diethyl-N-methyl-N-(2-methoxyethyl)ammonium bis(trifluoromethanesulfonyl)imide (N122,1O2TFSI)206) vs. 120 cP (N-(N-butyl)-N,N-diethyl-N-methylammonium bis(trifluoromethanesulfonyl)imide206) at 25 °C.
The Al current collector seems to behave differently in ILs compared to organic carbonate solvents. Balducci et al.207 demonstrated that the anodic dissolution of the Al current collector is greatly suppressed with TFSI-based ILs (Fig. 12b) due to the limited solubility of Al(TFSI)3 in N-butyl-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide (PYR14TFSI). Alternatively, the identity of the anion has an impact on the electrochemical compatibility between the graphite anode and ILs. The replacement of TFSI− with FSI− favors the formation of a stable SEI layer on the graphite anode, which can prevent the co-intercalation of EMI+ cations into graphene layers.208 An increase in salt content can further result in higher reversibility of the (de)intercalation processes for the graphite anode during cycling (Fig. 12c);209 however, the irreversible capacity loss in the first cycle seems to be quite pronounced for IL-based electrolytes, which needs to be addressed in future work.
The advantageous properties of ILs have been also inherited when designing Li-free mono- and multi-valent cation-based electrolytes.210–213 Chagas et al.211 observed that replacing the TFSI− anion with the FSI− anion led to lower viscosity in Na-based IL electrolytes (Fig. 12d), and thus improved the cycling stability of Na‖Na0.6Ni0.22Al0.11Mn0.66O2 cell. Endres et al.214 reported that the addition of nickel(II) triflate to a mixture of zinc triflate and 1-ethyl-3-methylimidazolium triflate (EMITfO) led to the uniform deposition of metallic zinc particles without the formation of any dendrites, and a high reversibility of Zn deposition/dissolution was achieved in the IL electrolytes (Fig. 12e). ILs containing dicyanamide [N(CN)2−] anions can plate Zn reversibly with high current density.215 Recently, to inhibit the uncontrollable growth of Zn dendrites, a polyzwitterion IL {i.e., poly([2-(methacryloyloxy)ethyl] dimethyl-(3-sulfopropyl)-co-tert-butyl acrylate)} was coated on a Zn anode, resulting in a long-term stable plating/stripping performance of over 2500 h and high average CE of 98.6% for 1500 cycles.216
Studies on ILs for rechargeable magnesium batteries are still in their infancy.217 Buttry et al.218 showed that TFSI-based ILs with longer ether chains in the pyrrolidinium cations substantially improved the reversibility of the electrodeposition/dissolution of Mg2+ ions (Fig. 12f). The chelating cation could possibly provide different cationic speciation for the transport of Mg2+ ions and suppress the reductive decomposition of the TFSI− anion. It has been reported that chloride species are of great significance in dictating the electrochemical performance of IL electrolyte -based magnesium batteries; however, these halogen species are corrosive to commonly used cell components (e.g., stainless steel).219 Recently, a chlorine-free electrolyte comprised of a non-nucleophilic magnesium bis(diisopropyl)amide and 1-butyl-1-methylpiperidinium bis(trifluoromethanesulfonyl)imide (PP14TFSI) showed a considerably reversible capability with a selenium cathode.220
Dai et al.221 proposed a cost-effective analog of conventional ILs (i.e., 1-ethyl-3-methylimidazolium chloroaluminate), i.e., a mixture of AlCl3 and urea (1.3:1, by mol). The AlCl4− anion and [AlCl2(urea)n]+ cation were found to be the active species responsible for the ionic transport, allowing Al‖graphite cell to perform for more than 150 cycles with a decent capacity of 74 mA h g−1. Recently, several new IL analogues with the same AlCl3 component were developed for high-performance rechargeable aluminum batteries.222–224 For example, improved cyclability and a higher capacity (e.g., 81 mA h g−1 after 1000 cycles) were obtained when replacing urea with 4-ethylpyridine. This enhanced cycling performance was ascribed to the better compatibility of 4-ethylpyridine/AlCl3 towards both Al and graphite electrodes.222
Notably, ILs are generally more viscous than conventional liquid electrolytes and may cause poor wettability toward porous separators and electrodes, despite their higher flash points and lower vapor pressure. Besides, the high viscosity of ILs may cause low bulk ionic conductivity and poor cell performance. In addition, the cations and anions of the IL generally undergo decomposition reactions at the negative and positive electrodes, respectively. Therefore, careful evaluation of the overall cell performances of IL-based electrolytes is urgently required to accelerate their practical deployment. Moreover, despite the numerous discoveries and studies, the deployment of ILs as a replacement for conventional solvents has not been realized to date.
3.5. Emerging mono- and multi-valent organic solid electrolytes
Among the various polymer matrices, the classic polyether PEO shows several beneficial features such as superior solvation ability, low glass transition temperature (ca. −60 °C), facile processability and low cost, and thus are widely utilized in PE-based batteries.48,226 It has been well accepted that lithium salts not only play a crucial role in determining the glass transition temperature, ionic conductivity, lithium-ion transference number, and wettability of PEs, but also dictates the qualities (e.g., mechanical stability and solubility) of the solid electrolyte interphase (SEI) and cathode–electrolyte interphase (CEI) films formed on anode and cathode active materials, respectively.123,226 As described in Section 3.1, LiTFSI with inherently excellent solubility and plasticizing capability generated from its superior delocalized anionic structure has become the most widely used conductive salt in PE-based lithium batteries since its first employment by Armand in 1989.28,235 The prevailing LiTFSI-based SPEs show high ionic conductivities (ca. 6.8 × 10−4 S cm−1 at 70 °C); however, their low transference numbers (ca. 0.2) and poor compatibility with high-energy electrode materials (LiCoO2, NMC, etc.) significantly limit their further utilization.235,238
Hence, recently, several lithium novel anions have been innovatively proposed to alleviate the dilemma of LiTFSI-based PEs, including (fluorosulfonyl)(trifluoromethanesulfonyl)imide (FTFSI−),239 (difluoromethanesulfonyl)(trifluoromethanesulfonyl)imide (DFTFSI−),235,240,241 ether-functionalized anion (EFA−),242 tricyanomethanide (TCM−),235 bis(difluoromethanesulfonyl)imide (DFSI−),243 (benzenesulfonyl)(trifluoromethanesulfonyl)imide (BTFSI−)227 and (trifluoromethanesulfonyl)(N-ethyl-N-methylsulfamoyl)imide (TFEMSI−).244 Among them, the LiDFTFSI/PEO electrolyte displays higher lithium-ion conductivity than that of the LiTFSI/PEO electrolyte [e.g., 2.0 × 10−4 (LiDFTFSI/PEO) vs. 1.5 × 10−4 S cm−1 (LiTFSI/PEO) at 70 °C], owing to the decreased anion mobility caused by the hydrogen bonding interactions formed between the H atoms of the anion and O atoms of PEO. More importantly, the electrochemically labile –CF2H moiety in the anion can be facilely decomposed to form an excellent SEI film, comprising both ionically conductive lithium hydride (LiH) and mechanically stable lithium fluoride (LiF) species, resulting in superior compatibility toward the lithium metal anode (Fig. 13a).240
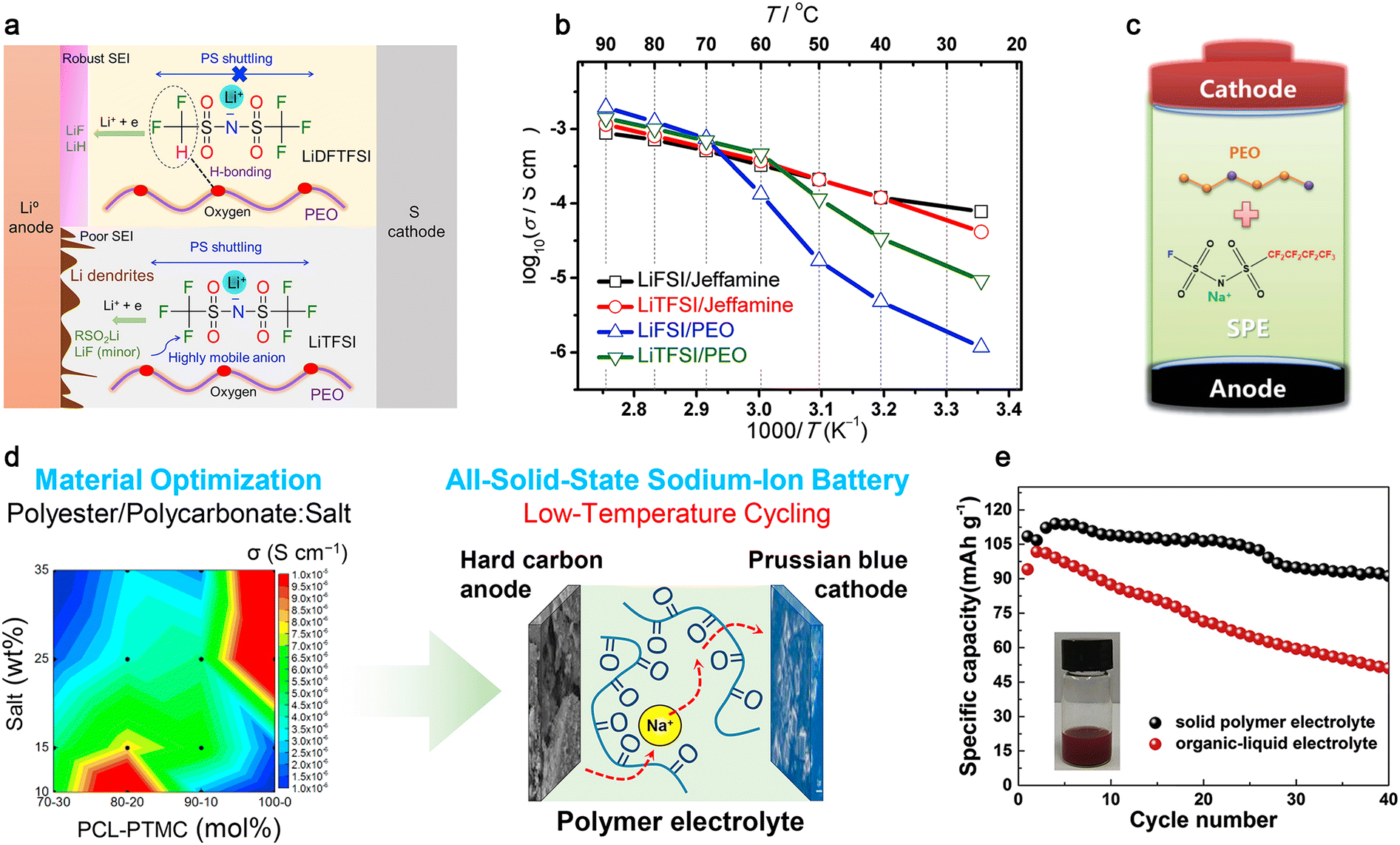 | ||
| Fig. 13 Classic polymer electrolytes for mono-valent batteries. (a) Role of the two salts in SPE-based Li-S batteries. Reproduced from ref. 240 with permission. Copyright 2019, Elsevier. (b) Arrhenius plots for different SPEs. Reproduced from ref. 250 with permission. Copyright 2018, Elsevier (c) Structure of NaFNFSI and schematic illustration of the formed interfacial films on the electrodes. Reproduced from ref. 262 with permission. Copyright 2017, The Royal Society of Chemistry. (d) Scheme of the polycarbonate-based cell operating at low temperature. Reproduced from ref. 265 with permission. Copyright 2019, Elsevier. (e) Cycling performance of SPE or liquid electrolyte-based cells. Reproduced from ref. 269 with permission. Copyright 2018, Elsevier. | ||
In addition to tuning the lithium anionic structures, replacing PEO with other alternative polymer matrices, such as polycarbonates,245–247 polyesters247,248 and polyetheramines,233,249–254 is another feasible method to enhance the ionic conductivity, TLi+, and anodic stability of PEs. For example, poly(CL-co-TMC) copolymers combining ε-caprolactone (CL) and trimethylene carbonate (TMC) units presented a high ionic conductivity of 4.0 × 10−5 S cm−1 at 25 °C and high TLi+ of 0.6 at 40 °C when coupled with LiTFSI salt.248,255 Recently, we proposed the use of a series of polyetheramine-based polymer matrices consisting of commercial polyetheramines (Jeffamine®) and poly(ethylene-alt-maleimide) as both side and main chains,233,249–253 respectively, which displayed more than one order of magnitude higher ionic conductivity (Fig. 13b) and better cycling performance compared with the conventional LiTFSI/PEO electrolyte at room temperature. This is attributed to the amorphous nature of the Jeffamine-based PEs. However, the application of SPEs is still hindered by their intrinsic low ionic conductivities and inferior contact toward high-voltage electrodes. Thus, incorporating plasticizing inorganic active/inactive fillers, conventional organic solvents or ILs in SPEs to obtain composite polymer electrolytes (CPEs) or gel polymer electrolytes (GPEs) is considered an elegant approach to overcome these issues, thereby gaining much attention during the past few decades.231,234,236,237
The study of PEs for sodium batteries started in almost the same period as lithium batteries.87 Conventional SPEs comprised of sodium sulfonimide salts (e.g., NaTFSI and NaFSI) and PEO matrices show comparable ionic conductivities to the analogous Li-based SPEs, which are normally around 10−8–10−7 S cm−1 at RT and approaching above 10−4 S cm−1 when the temperature is higher than 60 °C.232,256–260 However, all these TFSI- and FSI-based SPEs display low cation transference numbers (below 0.5) and potential corrosion toward the aluminum current collector.261 Replacing the –CF3 moiety of LiTFSI with longer perfluorinated alkyl chains has been demonstrated to be an efficient methodology to alleviate the corrosion of the aluminum current collector and simultaneously enhance the selectivity of cationic transport. Inspired by this concept, PEO-based SPEs containing sulfonimide anions with –C4F9 group were investigated (Fig. 13c), showing good cycling performances in Na|SPE|NaCu1/9Ni2/9Fe1/3Mn1/3O2 cell.262 Besides to the commonly used PEO matrices, polycarbonate-based SPEs, e.g., poly(trimethylene carbonate) (PTMC), were also prepared for sodium batteries. The ionic conductivities of NaClO4/PTMC and NaTFSI/PTMC electrolytes are similar to their lithium salt counterparts, despite their higher glass transition temperatures.263,264 In this regard, polycarbonate-based SPEs seem to be suitable electrolytes, which allow the operation of hard carbon‖Prussian blue cells even at an unprecedently low temperature of 22 °C (Fig. 13d).265
Compared with Li- and Na-based batteries, the research of PEs for potassium is still in its infancy, although its research started before Li- and Na-based batteries.85 There are only a few studies on SPEs for potassium batteries, despite the fact that they share a similar ionic transportation mechanism as that of lithium-based electrolytes229.9,266–268 For example, an interesting SPE comprised of poly(propylene carbonate) (PPC) and KFSI salt showed an ionic conductivity of 1.4 × 10−5 S cm−1 at 20 °C, resulting in a stable cycling performance (Fig. 13e).269 Similarly, GPEs for K-based batteries also show high ionic conductivities of around 10−3–10−4 S cm−1, which are comparable to that of liquid electrolytes.268,270
 | ||
| Fig. 14 Classic polymer electrolytes for multi-valent batteries (a). Cycling stability of the composite polymer electrolyte-based Mg cell. Reproduced from ref. 272 with permission. Copyright 2015, Elsevier. (b) Schematic illustration of in situ preparation of polytetrahydrofuran (PTHF)-borate-based GPE and the cell assembly procedure. Reproduced from ref. 273 with permission. Copyright 2019, Wiley-VCH (c) Composition of formulation and schematic structure of the ionic-liquid-based gel electrolyte. Reproduced from ref. 275 with permission. Copyright 2020, the American Chemical Society. (d) Arrhenius plots of the ionic conductivity of ionic liquid-based gel electrolyte containing 0.2 M concentration of calcium salt. Reproduced from ref. 275 with permission. Copyright 2020, the American Chemical Society. (e) Ionic conductivity and glass transition temperature of the electrolytes with different concentrations of Zn(BF4)2. Reproduced from ref. 281 with permission. Copyright 2020, Wiley-VCH. (f) Scheme of the preparation process of GPEs. Reproduced from ref. 284 with permission. Copyright 2019, Wiley-VCH. | ||
In the case of rechargeable calcium batteries, there are much less studies on Ca-ion conducting PEs.228 Recently, a highly conductive and IL-based GPE was obtained via the single-step photo-cross-linking of poly(ethylene glycol) diacrylate for a prototype calcium battery (Fig. 14c).275 Interestingly, the Ca(ClO4)2-based electrolytes showed the highest ionic conductivities compared to Ca(BF4)2- and Ca(TFSI)2-based electrolytes (Fig. 14d). This seems to be contradictory to the tendency observed in Li- or Na-based based PEs (i.e., TFSI > ClO4 > BF4), suggesting a deviation in the transport mechanism of Ca-ion conducting electrolytes. Nevertheless, the ionic conductivities of all these Ca-ion conducting electrolytes are lower than that of their lithium analogue, LiTFSI, e.g., 1.7 × 10−4 S cm−1 for Ca(ClO4)2 electrolyte vs. ca. 8 × 10−3 S cm−1 for LiTFSI electrolyte276 under a similar concentration range at room temperature. This can be attributed to the greater dissociation of LiTFSI in the electrolyte compared to the calcium salts.275
Differing from calcium batteries, PEs for rechargeable zinc batteries have gained increasing attention. Thus, there is plenty of revealing research into PE-based rechargeable zinc batteries; however, most of the studies focus on water-containing hydrogel polymer electrolytes, which have been previously reviewed in the literature148,236,277 and not within the scope of this perspective. Effectively, similar to other mono-valent and multi-valent cations - based batteries, the ionic conductivities of SPEs for rechargeable zinc batteries are undesirable at room temperature, being in the range of 10−6 S cm−1 irrespective of the identity of the zinc salt, e.g., ZnBr2, ZnCl2, and Zn(Tf)2.278,279 Similar to mono-valent cation-based batteries, introducing inorganic fillers, organic solvents, and/or ILs in SPEs can significantly enhance their ionic conductivity; however, the selection of the type of desired plasticizer requires careful consideration. Recently, amorphous PEs based on PEO280 or poly(1,3-dioxolane)281 were prepared, showing a maximum ionic conductivity of 1.96 × 10−2 S cm−1 at room temperature with Zn(BF4)2 as a conducting salt (Fig. 14e).281
The development and application of PEs for rechargeable aluminum batteries are still at their inception, where only a few reports have been published13,282.283 Here, again, the knowledge accumulated for mono-valent batteries in unlikely to be directly transferred to rechargeable aluminum batteries, given that most of the commonly used polymer matrices (e.g., PEO, PVDF, and polyacrylonitrile (PAN)) can form strong interactions with the electrochemically active cations.13 Inspired by the inherent safety of ILs, a free-standing GPE was constructed by the polymerization of acrylamide monomers in the presence of an IL of AlCl3/1-ethyl-3-methylimidazolium chloride (EMICl) (Fig. 14f), showing an ionic conductivity of 5.8 × 10−4 S cm−1 at 25 °C and a reversible charge/discharge capacity around 120 mA h g−1 under a current density of 60 mA g−1.284 In another work, a stable quasi-solid-state electrolyte was prepared by encapsulating the same IL of AlCl3/[EMIm]Cl in a metal–organic framework (MOF), which not only protected the IL from moisture, but also created sufficient ionic transport network in the electrolyte, resulting in a long-term cycling of more than 2000 cycles of Al‖graphite cells.285
In short, the development of multivalent conducting organic electrolytes tends to fall behind their mono-valent conducting counterparts, which is directly associated with the drastic difference between these two families of cations, including different solvation/de-solvation behaviors, Eigen values, and charge density (Section 2). This further suggests that thinking outside the box will be necessary for the design of solid polymer electrolytes with high conductivities and electrochemical stability for multi-valent cations - based rechargeable batteries.
4. Interphases in mono-and-multivalent cation-based rechargeable batteries
In principle, the reduction and oxidation of electrolyte constituents in the vicinity of the electrochemically reactive electrodes (i.e., solid-liquid interface) are undesirable given that they lead to cell failure due to various processes, including the continuous consumption of the metal cations (Mn+, n = +1, +2 and +3, e.g., Li+, Na+, K+, Mg2+, Ca2+, Zn2+, and Al3+), electrolyte dry-out, thus increase in impedance and cell polarization, ultimately resulting in accelerated cell failure. The continuous consumption of electrolytes during battery operation results in the accumulation of decomposition products, and thus SEI thickening (swelling). However, in reality, this electrolyte decomposition is almost unavoidable.The state-of-the-art and emerging electrolytes, i.e., non-aqueous carbonate, IL-based liquid, and solid electrolytes, are (electro)chemically unstable against polarized negative and positive electrode active materials when exposed beyond their safe electrochemical stability window. Outside their stability domain, electrolytes undergo single or multi-electron reduction and oxidation reactions at the negative and positive electrodes, respectively, and the sacrificial electrochemically and/or chemically induced decomposition products of the electrolyte components (solvents, salt anions and additives) result in the formation of a passivating film of different solid products, including hardly soluble inorganic metal (e.g., Li, Na, K, Mg, Ca, Zn, and Al) salts in the bulk electrolyte and various metal-free and metal-containing organic components, being precipitated on the electrode surfaces.
Ideally, the passivating film should kinetically stabilize the electrolyte constituents at potentials far beyond their thermodynamic stability limits, enabling the cell reactions to function reversibly. In the early studies, the passivation layer on alkali metals, graphite and other anode materials was found to inherit the properties of the solid electrolyte, i.e., permeable to Li+ ions, but blocking anions, solvent molecules and electrons, to prevent further unwanted electrolyte decomposition reactions. Accordingly, this layer earned the name ‘solid electrolyte interphase (SEI)’ by Peled in 1979,286 referring to the passivation layer formed on the Li metal surface in the vicinity of the electrolyte solution.287 In the current context, these layers are often referred to as ‘electrode electrolyte interphases (EEI)’, with particular reference to the ‘solid electrolyte interphase (SEI)’ and ‘cathode electrolyte interphase (CEI)’ for that formed on the negative and positive polarized electrode active materials, respectively. However, it is noteworthy to mention that passivation films need to inherit the properties of the solid electrolyte to take over the name ‘solid electrolyte interphase’.
It is of paramount importance to distinguish ‘interPHASES’ from ‘interFACES’, given that their roles are unique in the case of electrochemical systems.288–290 In the existing scientific literature, ‘interPHASE’ and ‘interFACE’ are somewhat ill-defined with no clarity and consistency, which are more often interchangeably used to describe the electrified boundary between the electrode and electrolyte systems (Fig. 15). In scientific publications (Fig. 15), authors and research groups largely refer to the well-known SEI as the “solid electrolyte interFACE”, while the appropriate terminology is the “solid electrolyte interPHASE”. The term ‘interFACE’ can be defined as a 2D (<1 nm thickness) point of contact of the solid surface of the electrode at which ionic double layers are formed during polarization and the immediate layer of counter ions of the electrolyte. It can also refer to the boundary among/between the EEI components (e.g., Li2CO3 and LiF). Alternatively, the term ‘interPHASE’ describes a 3D layer of multi-mosaic structure, which possesses volume, with ion diffusivity, microscopic structure and chemical constitution distinctively different from the electrode, electrolyte and electrode–electrolyte interface. In short, ‘interFACE’ refers to the physical boundary (2D object, Fig. 15a) between the electrode and electrolyte elements, whereas the chemical decomposition appearing at the interface is considered the interphase (3D object, Fig. 15a), i.e., interphase refers to an interfacial region with a defined volume. The SEI film on the anode side for LIBs, for instance, is viewed as a multi-layered structure with an inorganic inner layer near the electrode/SEI (e.g., Li2CO3, LiF, Li2O, Li2S, and Li3N), which allows Li+ transport, and an organic (ca. dilithium ethylene glycol dicarbonate (Li2EDC, [CH2OCO2Li]2), ROCO2Li (R = CH3, C2H5) ROLi, etc., where R depends on the solvent) outer layer, which is heterogeneous, porous, and permeable to both Mn+ (e.g., Li+) and electrolyte solvent molecules, near the SEI/electrolyte boundary. In an electrochemical device, interphases between electrolyte and electrodes constitute the only “legitimate” sites where redox reactions occur and function as a double-edged sword.22
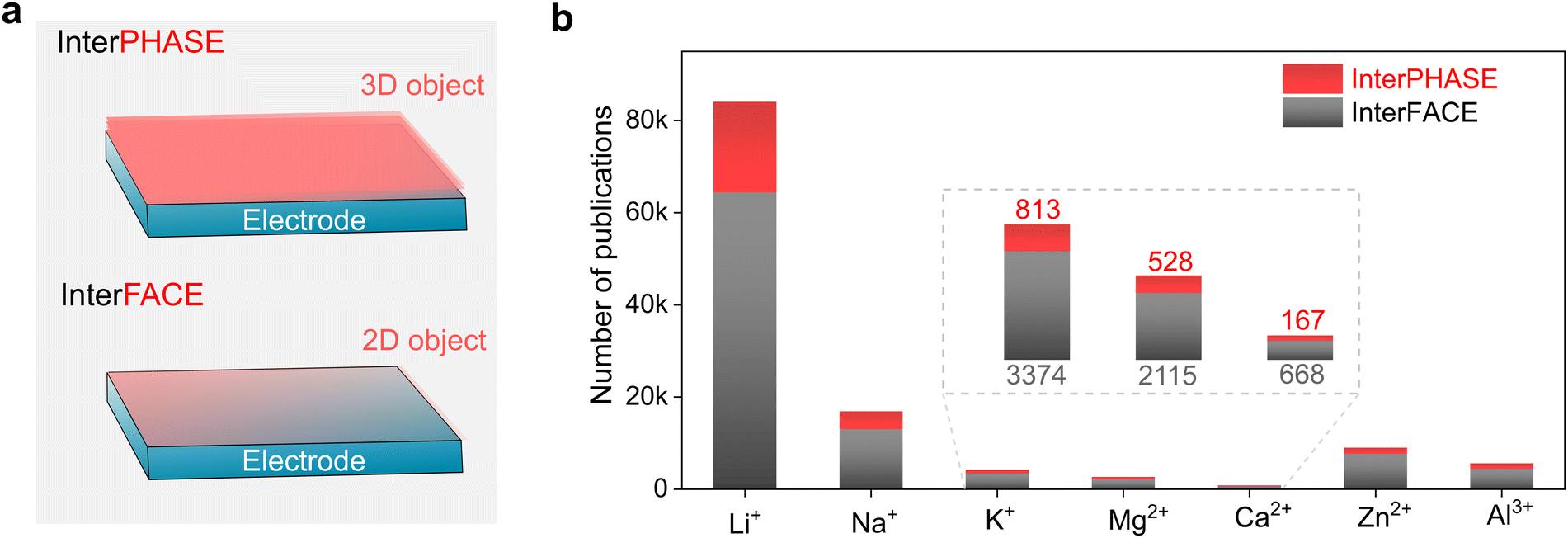 | ||
| Fig. 15 Difference between ‘interPHASE’ and ‘interFACE’. (a) Schematic illustration of the definition of these two terminologies. (b) Volume of publications on the use of ‘interPHASE’ and ‘interFACE’ in different types of rechargeable batteries. The values are obtained from a Scopus search using the keywords (M battery + interPHASE or M battery + interFACE; M = Li+, Na+, K+, Mg2+, Ca2+, Zn2+, and Al3+), as accessed on the 25th of October 2022. | ||
Electrode–electrolyte interphases are the most critical elements given that they strongly impact the various battery performance metrics, including efficiency (Coulombic, voltage, and energy), cycle life, rate capability, self-discharge behavior, safety, and cost. It is noteworthy to recount that the wetting (conditioning) of electrodes is an important factor in reducing the SEI formation time and manufacturing process, which directly impact the battery-pack cost. In absolute terms, the formation of an SEI in LIBs is reported to last for ca. 0.5–2 weeks for the entire process,287 with an estimated cost (involving wetting and formation) of up to $32 to $33 per kW h of usable energy for the battery-pack cost (out of a total cost of ∼$500 per kW h).291 The formation time, and thus corresponding cost can be much higher for emerging Li-free mono- and multi-valent cation-based batteries, which is linked to their less stable passivating films and other linked complexities. Consequently, the formation of interphases is dictated by the cation (Mn+)-solvation/de-solvation process including ionic migration through the interface and interphase, the nature of the ion pairs, the composition, and the crystalline properties of the interphase, and structure.133,292,293 The de-solvation process is also well known to alter the electrochemical stability of the electrolyte. The solvation and de-solvation processes are notably dictated by the magnitude of the interactions between the ion–ion and ion–solvent molecules, accumulation of ions at the electrode surface and other related variables.294,295 The reversible reduction and oxidation reactions in the vicinity of the polarized electrodes are closely related to the nature and dose (concentration) of the electrolyte constitutes (e.g., solvents, salt anions, and additives), nature of the active materials, electrode conductive additives, and operating parameters including temperature, and reduction/oxidation current rate. Besides, the effectiveness of the protection capability of the EEI is predominantly contributed by a combination of various factors including its overall chemical composition, thickness, ion transport, morphology, homogeneity, mechanical strength, chemical, electrochemical and thermal stabilities, and compatibility with other components of the battery system.
4.1. Thermodynamic consideration of interphase formation and misconceptions
Fig. 16 demonstrates the generic formation mechanism of the anode (solid–electrolyte interphase, SEI) and cathode–electrolyte interphase (CEI) according to the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) energy levels. The HOMO and LUMO values represent the abilities of molecules to donate and accept electrons, respectively, and the gap between the HOMO and LUMO determines the chemical reactivity, tendencies of interphase formation and chemical hardness-softness of the molecule296,297 under consideration. | ||
| Fig. 16 Energy diagrams of a rechargeable battery with metallic anode and semiconductor cathode. (a) Energy levels of the system in thermodynamic equilibrium at open-circuit voltage. (b) Shifts of the Fermi energies after the solvation process. (c) Shifts of the Fermi energies upon discharge. (d) Final energy level rearrangement after the formation of the SEI. Both electrodes have a chemical potential that can be approximated to the Fermi energy of the anode (EF−) and the cathode (EF+). The latter having valence and conduction bands with energies EV+ and EC+, respectively. The figure is recreated from ref. 298 with permission. Copyright 2022, Wiley-VCH. | ||
The Fermi energies of the negative (anode) and positive (cathode) electrodes are designated by their electrochemical potentials, μA and μC, respectively (Fig. 16a).298 Traditionally, to ensure the electrochemical stability of electrolytes, two conditions must meet, as follows: (1) the energy gap (Eg) of the electrolyte between the HOMO and LUMO should be larger than the voltage difference, VOC, between the electrodes (i.e., Eg > VOC), and (2) the HOMO and LUMO levels should be positioned “outside” the electrochemical potentials of the anode and cathode electrodes (i.e., ELUMO > μA and EHOMO < μC), respectively. Ideally, a molecule with a small or no (zero) HOMO–LUMO gap is assumed to be chemically reactive.299 However, the reductive and oxidative decomposition of the electrolyte constituents in batteries is inevitable, given that the working potentials of the negative and positive electrodes lie below and above the electrochemical stability window of the electrolyte, respectively. Subsequently, electrolyte decomposition can only be minimized by the formation of a robust passivation layer on both electrodes, which will block or suppress the electron transfer after restructuring the energy levels of the system (Fig. 16c and d).
However, it has to be carefully noted that the HOMO and LUMO are thermodynamic concepts defined within the electronic structure theory involving the electronic properties of isolated molecules (Fig. 16a). Specifically, the energy levels of these isolated entities do not indicate the actual pattern of the species participating in redox reactions.300 On the contrary, the redox potentials are directly related to the Gibbs free energy differences of the reactants and products, and the nature of the cation–solvent or cation–anion interaction has a strong impact on the reduction stabilities of the solvent/anion system given that it significantly affects the solvation structure and the solvent coordination number around the cation center. Thus, the HOMO–LUMO gap has been proven in several cases to be a few eV broader than the actual electrolyte reduction–oxidation gap (Fig. 16b), clearly indicating that the HOMO–LUMO concept is not a definite indicator of the stability window of electrolytes.
It has been demonstrated that the complexation of solvents in the presence of cations lowers both the LUMO and HOMO energies with respect to their pristine versions (Fig. 16b).301 It has been reported that ion (Li+, Na+, K+, Mg2+, and Ca2+)-ester (EC, DEC, PC, and FEC) complexes present an excellent linear relationship between their LUMO energy level change and the binding energies as well as the ratio of carbon atomic orbital contribution in the LUMO. Ion-ether complexes exhibit significant LUMO energy level changes given that their LUMOs are mainly composed of metal atomic orbitals.301 All ion–solvent complexes exhibit much lower LUMO energy levels compared with their individual components (pure solvents), showing that the solvents decompose more easily on metal anodes once they are complexed with the metal cations in the electrolyte (Fig. 17). This observation agrees with bond length analyses that the C–O bond length of the carbonyl group in an ester and –C–O–C– group in ether increases after binding with a cation.
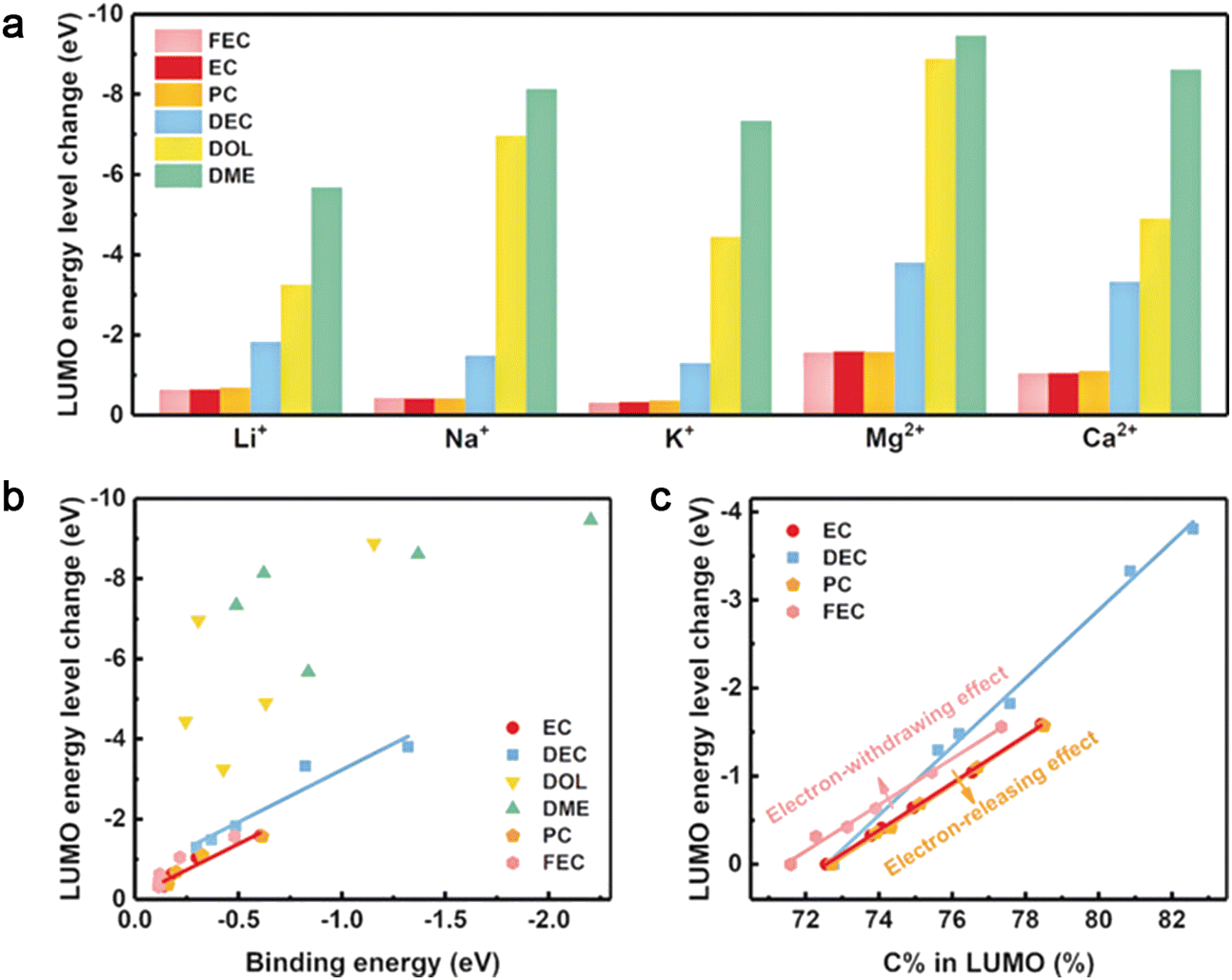 | ||
| Fig. 17 Correlation between lowest unoccupied molecular orbital (LUMO) energies and electrolyte stability on the anode. (a) Summary of LUMO energy level changes of the ion–solvent complexes compared with pure solvents. (b) Correlation between the LUMO energy level change and binding energy. (c) Correlation between the LUMO energy level change and the ratio of C atomic orbital contribution in LUMOs. Reproduced from ref. 301 with permission. Copyright 2018, Wiley-VCH. | ||
Consistent with the trend of the electron-withdrawing effect of mono-metal cations (Li+ > Na+ > K+), the ΔLUMO and ΔHOMO (Δ = difference in LUMO or HOMO between pure solvents and ion–solvent complexes) of ion–solvent complexes are found to follow the order of Li+ > Na+ > K+ in ester-based solvents. This shows that K+-ester complexes lead to higher reduction and lower oxidative stabilities. As a rule of thumb, coulombic interactions of stronger Lewis acids with a solvent or negatively charged species are noteworthy.302 The strength of the Mn+-solvent interaction heavily depends on the nature of the cation and solvent. For instance, despite the similar physico-chemical properties of mono-valent cations such as Li+, Na+ and K+, their reactivity and solvation structure differ owing to their differences in charge density, Lewis acidity and other governing parameters. The comparison between the analogous divalent cations, Ca2+ and Zn2+ reveals a significant cation size effect, which is evidenced in the considerably reduced cation–solvent bond lengths, and thus stronger solvent bonding for Zn2+ compared to Ca2+.
Besides the size and Lewis acidity, the charge density of the metal cation greatly affects its (de-) solvation strength, and thus the nature and effectiveness of the passivation layer. Compared with monovalent alkali metal cations, divalent ions such as Mg2+, Ca2+ and Zn2+ tend to form more stable ion pairs in a given solution. Probably, Mg2+ ions have stronger solvation than Li+ ions, and therefore their charge transfer resistance is expected to be considerably much higher.
De-solvation of a metal cation at the electrode/electrolyte interphase/interface has been reported to be rate-determining step of the interphasial reactions in rechargeable batteries, and thus its evaluation is of paramount significance. Comparison of the de-solvation energies of Li+, Na+, and Mg2+ ions in organic electrolyte solvents reveals that Na+ presents a lower de-solvation energy compared to Li+ despite the fact that they have similar solvation structures, which is attributed to the lower Lewis acidity of Na (Fig. 18).303,304 Alternatively, Mg-ion complexes show extraordinarily larger de-solvation energies due to their double positive charge and higher coordination number, which are associated with the change in the solvation sphere.
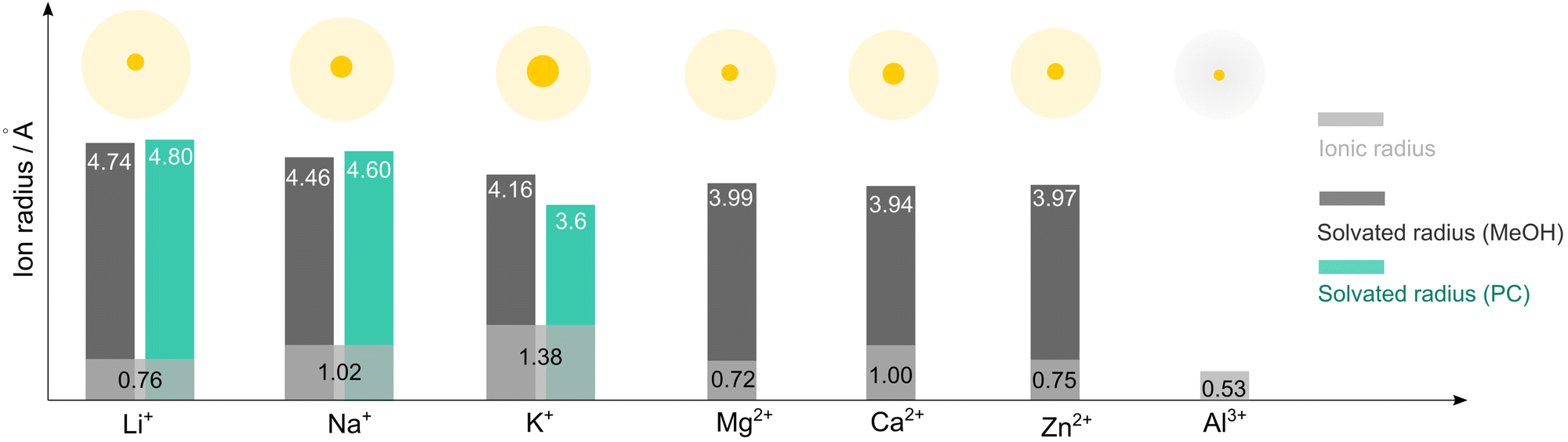 | ||
| Fig. 18 Importance of solvation and de-solvation of cations on electrode–electrolyte interphases. Comparison of Stokes radii of various cations in non-aqueous solvents, where the values are extracted from ref. 304 and 340. | ||
Investigation of the solvation structures, coordination numbers, and de-solvation energy of Li+, Na+, K+ and Mg2+ in the presence of 27 organic solvents showed that K+ complexes exhibit a lower de-solvation energy than the other tested cations.305 The solvation behavior is also found to be affected by the salt, solvent, additive and their concentrations, all cumulatively affecting the nature and efficacy of the EEI layers.
In the design of an ideal EEI, both the kinetic and thermodynamic parameters should be considered. The functional properties for an ideal and robust EEI layer are as follows:
(1) High cation selectivity and permeability, and negligible solvent and/or solvated species penetration.
(2) Electronic insulation to inhibit electron tunneling (te− ≈ 0), and thus electron transfer for catalyzing the reduction/oxidation of the electrolyte.
(3) Low solubility and inertness in the chemical/electrochemical environment (solvent solution and intermediate decomposition products).
(4) High chemical and thermal stabilities over a wide range of operating temperatures, and mechanical toughness (combination of strength and ductility) to maintain stability against cascading chemical reactions/corrosion, thermal oscillations and electrode volume change during battery operation, respectively.
(5) High compatibility with electrolyte constituents, electrodes, conductive additives, binders, etc.
(6) Wider electrochemical stability to low and high voltages to avoid additional side reactions.
(7) High ionic conductivity (t+ ≈ 1). The conductivity of the EEI should be homogeneous, which requires the uniform chemical distribution and thickness of the EEI layer.
(8) Good adhesion to the electrode active materials.
4.2. Evidence of the presence of interphases in rechargeable batteries
Depending on the electrolyte composition, voltage window, etc., the EEI layer in Na- and K-based systems presents similar species to their Li counterparts. For instance, for MPF6-based electrolytes in non-aqueous solvents (ca. EC, DEC, and DMC), the passivating species are noted to be inorganic (M2CO3, MF, MOH, M2O, M2S, M2C2O4, MxPFy, MxPF3−xO, etc.) and organic mainly composed of metal alkyl carbonates (ROCO2M), metal alkoxides (ROM), oligo(ethylene oxides) (OEO), (CH2OCO2M)2 ester linkages, etc., where M = Li, Na and K. However, due caution should be taken given that the proportion of organic and inorganic species and their distribution can vary depending on the nature of the mono-valent cation. Although they belong to the same group in the periodic table (Group IA), because of their different electronic structures, i.e., Li (1s2, 2s1), Na (1s2, 2s2 2p6, 3s1) and K (1s2, 2s2 2p6, 3s2 3p6, 4s1), they have different reactivity with their environment, including towards the electrolyte.306Although the transport of metal cations is generally sluggish, reports on Ca2+, Zn2+ and Al3+ cation-based battery cells prove the presence of ion-conductive interphases. However, despite the well-established evidence on the existence of solid electrolyte interphases in Li-, Na-, K-, Ca-, Zn-, and Al-based rechargeable batteries, there is still a debate in the scientific community regarding Mg-based electrodes. Similar to Li, due to its low redox potential, Mg is susceptible to spontaneous reaction with traces of atmospheric components (O2, H2O, and CO2), reactions (R1)–(R3), respectively, salt anions (ClO4−, BF4−, CF3SO3−, etc.) and archetypal aprotic solvents, resulting in the formation of solid passivating films, which are insoluble organic and inorganic Mg salts.307 However, the resulting film is both electronically and ionically insulating in the solid phase, hindering reversible cell chemistry. Hence, it is still under debate whether the film formed is a simple passivation layer or is endowed with the governing basic properties of the SEI because the SEI layer earns its name for being conductive to metal cations while blocking electrons. The impedance of Mg electrodes was found to be several orders of magnitude higher than that of Li electrodes in the same solution, evidencing the insulating behavior of the precipitated compounds in Mg-based systems.307 Early reports also evidence that the electrochemical deposition of Mg-ion-containing species on Mg electrodes covered by passivating surface films is impossible.
| Mg + 1/2O2 → MgO | (R1) |
| MgO + CO2 → MgCO3 | (R2) |
| Mg + 2H2O → Mg(OH)2 + H2 | (R3) |
If Peled's model of the SEI structure, in which the SEI layer is considered to be an inorganic/organic mosaic structure, is applied, the ion transport through the bulk inorganic (MgO, MgF2, MgCO3, Mg (OH)2, MgS, etc.) part is the rate-determining step. The insulating nature of these compounds to Mg2+ defies the deriving factor behind the SEI nomenclature.
In contrary to this long-held account, Gao et al.308 argued that the surface film formed by the electrolyte decomposition on Mg electrodes can function as an SEI layer. Their claim is based on the limited number of reports highlighting the reversible Mg deposition/stripping in Mg(TFSI)2-glyme electrolyte309–311 and successful operation of Mg‖S batteries with small hysteresis at a discharge voltage of 1.5–1.6 V close to the theoretical value of 1.8 V.153,312,313
4.3. Origin of degradation processes in rechargeable batteries
According to a collection of seminal studies over the past four decades, the thermodynamic instability of electrolytes is proven to play a critical role in almost all pathways through which rechargeable batteries lose their intended performances. The major contributing phenomena are presented as below.Contrary to the intuitive belief that an interphase is formed during the first few cycles and parasitic decomposition of the electrolyte constituents is prevented thereafter, the rate of growth (dx/dt) of the interphase demonstrates a parabolic relation (eqn (11)), proving that it can only slow down (reducing k), but never be completed during the entire life span of the battery cell.
Actually, although EEI formation takes place mainly in the first few charge/discharge cycles, it continues to grow, and change composition and thickness throughout the entire life of the battery. It only slows down at a rate that follows the general mathematical relationship314 given below:
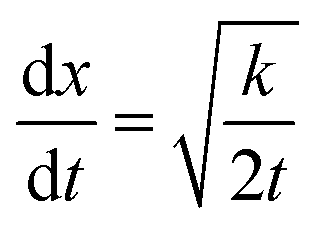 | (11) |
The thickness of the SEI layer, for instance on Li, can be roughly estimated using the following equation (eqn (12)).315
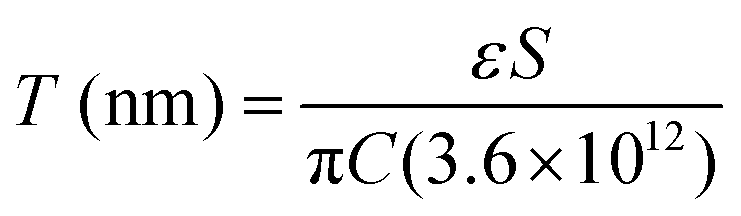 | (12) |
| MPF6 ↔ MF + PF5 | (R4) |
| MPF6 ↔ M+ + PF6− | (R5) |
| PF5 + H2O → POF3 + HF | (R6) |
The degree of decomposition and hydrolysis of MPF6 highly depends on the nature of the cation (M = Li+, Na+, and K+). Considering their Madelung energies, a parameter linked to the electrostatic energy in ionic crystals, their chemical and thermal stability follow the order of KPF6 > NaPF6 > LiPF6.317,318 Thus, KPF6 is expected to undergo a lesser extent (if it exists) hydrolysis reaction compared to its Li and Na counterparts (i.e., LiPF6 and NaPF6), making it beneficial to potassium-ion battery (KIB) chemistry. This effect becomes more pronounced with divalent cations, M(PF6)2 (M = Ca2+, Mg2+, Zn2+, etc.).
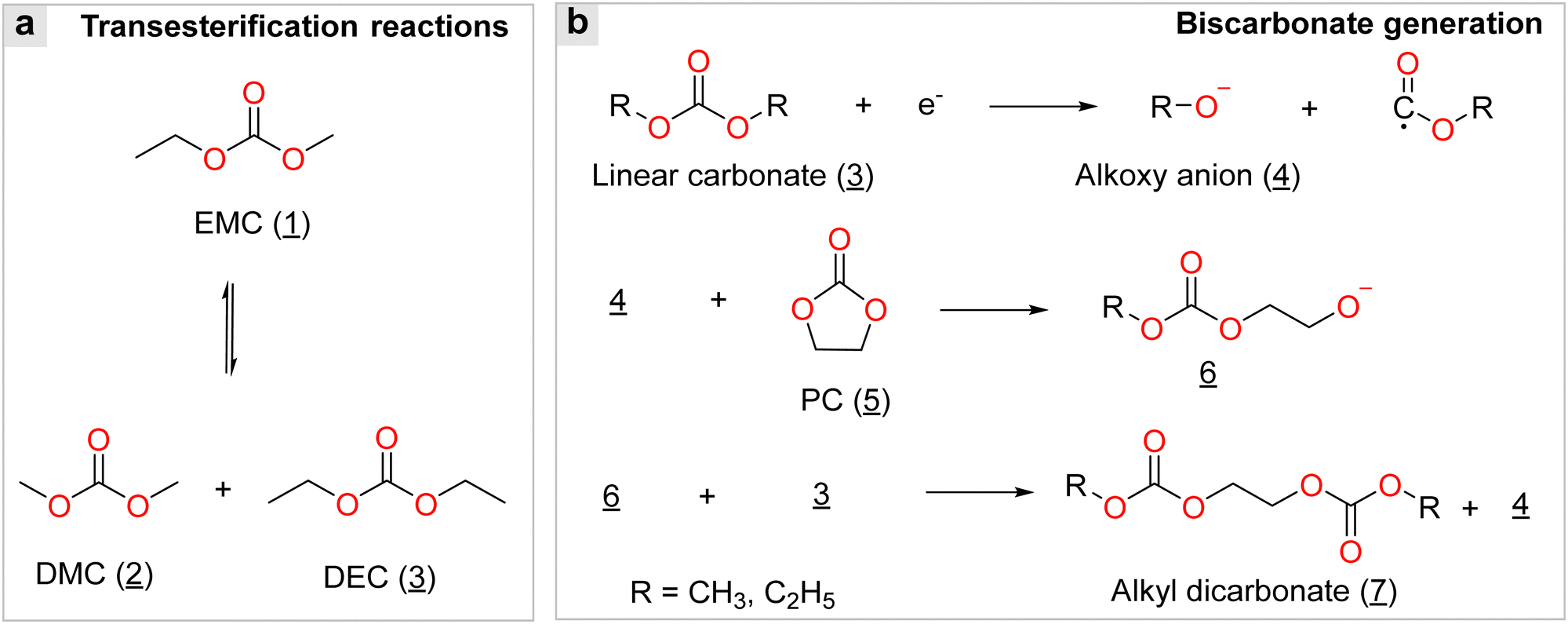
4.4. Quality indicators for electrode–electrolyte interphases
| ΔG = ΔH − TΔS | (13) |
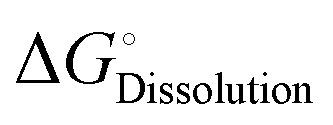 , eqn (14)):
, eqn (14)):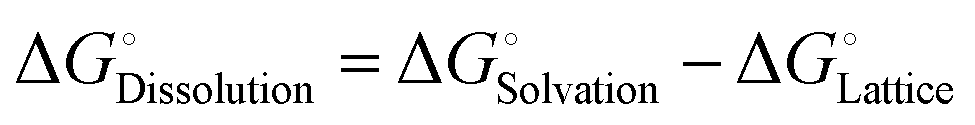 | (14) |
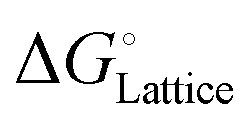 and
and 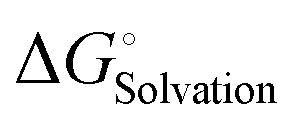 are the lattice and solvation Gibbs energies of the salt, respectively.323 Among the mono-valent cations, K+ ions have the weakest Lewis acidity, and thus weakest interactions with Lewis bases such as anions and solvents than their analogous lighter alkali metal ions of Li+ and Na+ ions. Thus, K+ ions show a lower solvation energy than Li+ and Na+ ions for aqueous and non-aqueous systems, resulting in their lower solubility (Fig. 19b).
are the lattice and solvation Gibbs energies of the salt, respectively.323 Among the mono-valent cations, K+ ions have the weakest Lewis acidity, and thus weakest interactions with Lewis bases such as anions and solvents than their analogous lighter alkali metal ions of Li+ and Na+ ions. Thus, K+ ions show a lower solvation energy than Li+ and Na+ ions for aqueous and non-aqueous systems, resulting in their lower solubility (Fig. 19b).
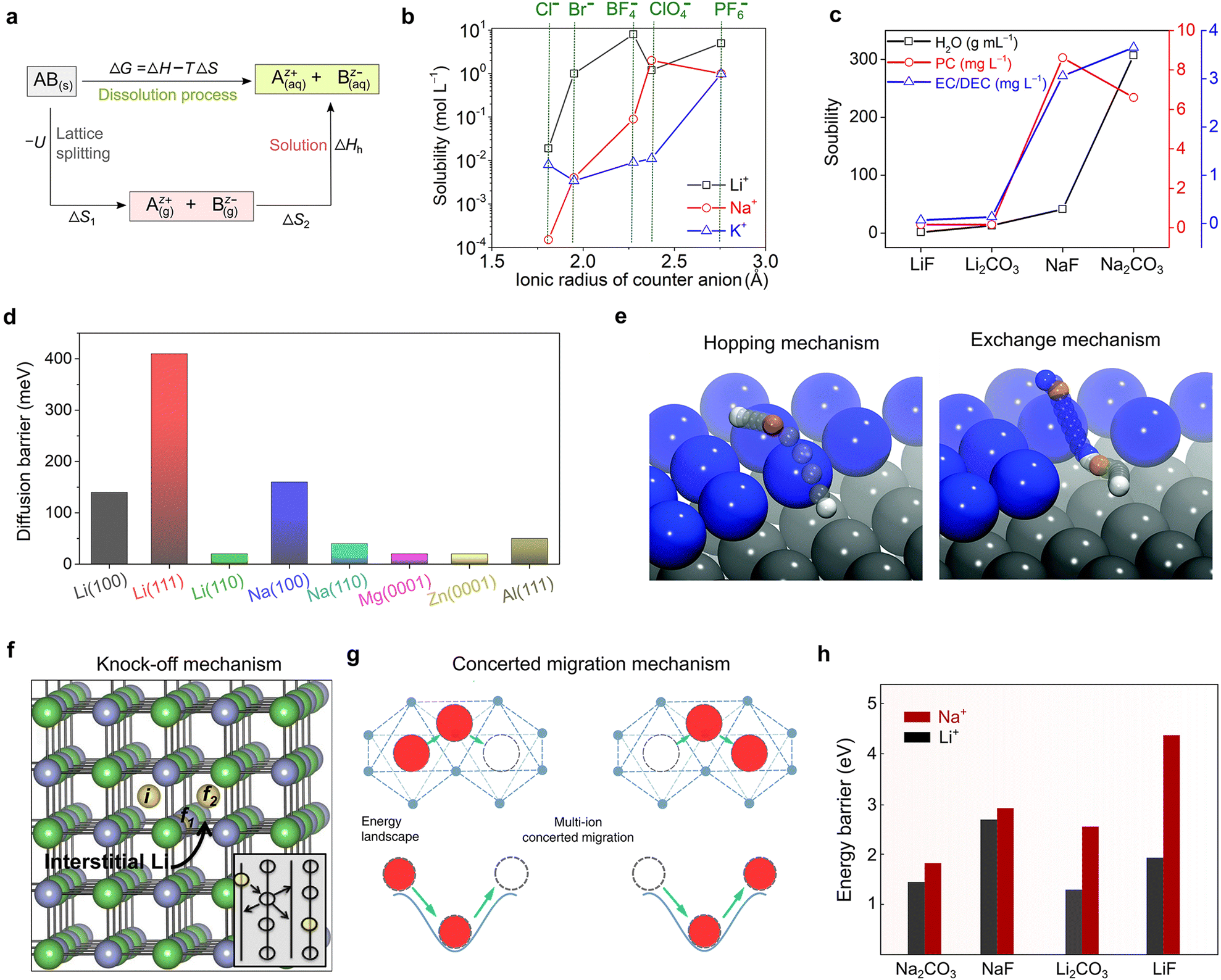 | ||
| Fig. 19 Quality indicators for electrode–electrolyte interphases. (a) Energy parameters necessary during the dissolution process of EEI building compounds, adapted from ref. 322 with permission, Copyright 2022, The Authors. (b) Solubility of several Li, Na, and K salts in PC as a function of the ionic radius of the counter anion, adapted from ref. 323 with permission, Copyright 2022, The Chemical Society of Japan. (c) Solubilities of LiF, Li2CO3, NaF, and Na2CO3 in H2O, PC and EC:DEC.321 (d) Self-diffusion barrier for various types of metal surfaces. Value are taken from ref. 332. (e) Hopping and exchange mechanisms on the Mg(0001) surface. Orange balls represent the transition state. Adapted from ref. 332 with permission. Copyright 2018, The Royal Society of Chemistry. (f and g) Knock-off and concerted migration mechanisms. Adapted from ref. 334 and 335 with permission, respectively. Copyright 2015, the American Chemical Society. Copyright 2017, Macmillan Publishers Limited. (h) Diffusion energy barriers of various types of species in certain EEI building species. Adapted from ref. 337 with permission. Copyright 2017, Wiley-VCH. | ||
Although there are some studies on the solubility of the EEI components for LIBs and limited accounts for sodium ion batteries (NIBs), studies related to KIB, divalent and trivalent cation-based rechargeable batteries are rare. Although the available data and comparisons with the solubility of EEI compounds in water can provide invaluable insight; unfortunately, this approach may not be valid for all alkali salts in all solvents. Ma et al. investigated the solubility of MF and M2CO3 (M = Li and Na) in different solvents, namely PC, and EC:DEC.321 The solubility of the same compounds was also evaluated in H2O.324 Their experiments demonstrated that Li salts (e.g., LiF and Li2CO3) have remarkably lower solubility in all the employed solvents compared to their Na counterparts (NaF, Na2CO3, etc.; Fig. 19c).
It is generally supposed that an EEI layer with a high Young's modulus (E) provides better mechanical stability to the passivation layer, thus enhancing the cycling performance of battery cells. According to atomistic modelling of the principal SEI building materials, the modulus is found to be between 2.4 and 58.1 GPa, with organic components ranging from 2.5 to 18.9 GPa. Among the expected phases, the magnitudes of the moduli follow the order of PEO < LiEC < LiMC < Li2EDC < Li2CO3 < LiF, with polymeric < organic < amorphous inorganic components < inorganic crystalline components.325 However, it is noteworthy to mention that an EEI presenting a large elastic strain limit (εY) may favor highly reversible batteries despite its low E. Thus, it is of vital interest to find a trade-off between these two parameters to regulate the mechanical property of the SEI or EEI in general. The maximum elastic deformation energy (U) is proposed to bridge both parameters (U ∝ (E × εY), eqn (15)), thus robustly predicting the stability of the EEI.326
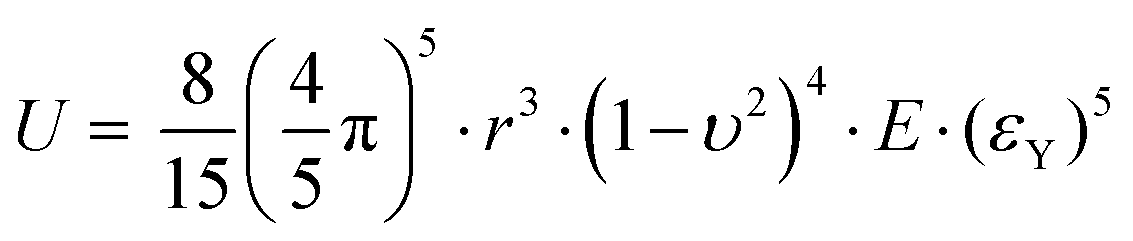 | (15) |
The thermal and chemical stabilities of the EEI layers are of supreme importance given that they dictate the cycle life, rate capability and safety of battery systems. Specifically, the acid-base reaction between the salt/salt degradation products (HF, PF5, POF3, etc.) and EEI building species, and transformation of one EEI component to another are among the critical reactions affecting the chemical stability of the interphases. The basicity of the principal EEI compounds follows the order of M2O > MOR > M2CO3 > RCO2M, ROCO2M > M2C2O4, where M = Li, Na, and K. The acid-base reactions, transformations, and decompositions of the EEI building materials result in the generation of new compounds, which are possibly either insoluble, soluble/partially soluble, or gaseous compounds in the electrolyte solution (reactions (R7), (R8) and (R9)).128,328–330
| MPF6 + M2CO3 → 3MF + POF3 + CO2 | (R7) |
| M2CO3 + 2HF → 2MF + CO2 + H2O | (R8) |
| ROCO2M+ 2HF → MF + CO2 + H2O + CH3F | (R9) |
The behavior of the transport of M (metal) ions through the EEI medium depends strongly on the solvation and de-solvation process, and thus the Eigen values (i.e., ionic migration through the interface/interphase), the nature of ion pairs, composition, and crystalline properties of the interphase.338 The magnitude of the interactions between ion–ion and ion–molecules, and the accumulation of ions at the electrode surface significantly influence the solvation/de-solvation process.
The ionic conductivities of the generated EEI building compounds heavily dictate the nature and pattern of the metal ion transport. The fraction of inorganic to organic compounds, the crystallinity of the inorganic compounds, diffusion pathway through the EEI species, etc. contribute significantly to the kinetics of the metal cation transport in and through the interphase and interface regions. In general, the inorganic species of the EEI layer are expected to be inferior to the organic species in terms of ionic conductivity, and thus their proportion has a significant impact in determining the transport properties of ionic species in the EEI layer. It has been reported that while Li+ prefers a knock-off or direct hopping diffusion mechanism, the Na+ ion is supported by vacancy diffusion or knock-off mechanisms, evidencing the differences in their diffusion mechanism and kinetic process.339 These variations can be ascribed to the differences in the intrinsic properties between Li and Na, including Stokes radius, reduction potential, and charge density (Table 1). In general, multivalent ionic conduction in inorganic solids and across the EEI is lower with respect to its monovalent analogues because of the challenges posed by the high charge density, in which ions of high charge density induce stronger Coulombic interactions and slower ligand exchange rates.
The charge densities (ρ, C mm−3, Table 1) of cations and anions can be calculated according to the following formula (eqn (16)):137
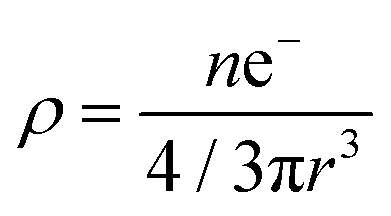 | (16) |
Considering a coordination number (CN) of six, the ionic radii of Mg2+ and Zn2+ are 0.72 Å and 0.74 Å, respectively, which are nearly similar to that of Li+ (0.76 Å).341 Similarly, the ionic radii of Ca2+ and Na+ are the same, i.e., 1.0 Å and 1.02 Å, respectively. In contrast, the radius of Al3+ is significantly smaller (0.54 Å). For divalent cations, the ionic conduction of the more polarizable (softer) Ca2+ (76 C mm−3) may be easier than Mg2+ (205 C mm−3) and Zn2+ (181 C mm−3), and the radii used for charge density calculations are taken for six-coordinate ions. Due to the large charge density of Mg2+, it is expected that the ionic conductivity of MgO, MgF2, and MgS is orders of magnitude lower than that of their Li(Na) counterparts.342 Achieving high Al3+ mobility is even more challenging because of its higher charge density (ca. 770 C mm−3 for radius taken for four-coordinated ions, Table 1), further amplifying the complications of conduction seen in multi-valent ions.343,344
| 2Li2CO3 → 4Li+ + 4e− + 2CO2 + O2 | (R10) |
In general, the organic components of the EEI are reported to be electrochemically less stable than the inorganic phases;348 however, this needs further work given there are scarcity of reports on the electrochemical stability of the passivating species. It should be noted that there is a lack of research on this topic despite its detrimental impact on the long-term operation and aging of batteries, and thus deserves further dedicated studies.
4.5. Translation of interphases from half cells to full cells
Most of the current interphase-related characterizations are carried-out in half-cell configurations and/or samples extracted from cycled half-cells. In these set-ups, the metal ions (Mn+) used as counter electrodes provide an unlimited metal (M) source. However, despite their simplicity and serving as vessels for the initial evaluation of interphases in new materials and experimental set-ups, investigations involving half-cells, i.e., largely battery characteristics after completion of cycling in the same environment, mask the complex interactions, compatibility issues, and cross-contamination at the SEI/CEI resulting from the dissolutive products in practical full cell configurations (Fig. 20a). The interphasial processes occurring at the cathode and anode surfaces are sometimes considered as isolated from each other, but increasing evidence reveals that clear forms of ‘conversation or dialogue’ exist between the cathode and anode, and their respective interphases, given that the interphasial species generated on one electrode often appear on the other with unforeseen detrimental impacts. Thus, the information obtained from interfaces and interphases in half-cells may not accurately account and predict the interface and interphase processes, and thus the performance deterioration in full cells.22 Moreover, in contrast to the half-cell set-up, full-cell is operated by control of the cell voltage, implying that the individual electrode potentials are subject to change over operation, which depends on the used materials and electrochemical operating conditions (Fig. 20b).349 In summary, converting the results obtained from half-cells to full cells is not straightforward, and thus tests involving a combination of half-cell and full cell set-ups are of supreme importance to bridge the discrepancy between half-cell laboratory and practical full cells and cell formats (e.g., multilayer pouch cells or cylindrical cells).350 | ||
| Fig. 20 Interphases from half-cells to full-cells. (a) Schematic illustration on the cross-talk between SEI and CEI species. (b) Characterization set-up between typical Li cells and versatile set-ups for other emerging mono- and multi-valent batteries. Reproduced from ref. 351 with permission. Copyright 2019, the American Chemical Society. | ||
Most importantly, compared to Li-based rechargeable batteries, scientific research on interphases involving emerging mono (Na+ and K+)- and multivalent-based batteries is still in its infancy, and thus serious consideration of both half-cell and full-cell set-ups is needed to fully understand the governing phenomena of the systems, and thus accelerate their large-scale commercialization.351
4.6. Critical role of electrolyte solvents and additives as enablers and precursors of robust interphases
Precise engineering of the EEI layers can be realized in many ways, including by modifying the electrolyte constituents through additives, metal salt anions, and solvents. In addition to the importance of metal salts and their concentration (Section 3.4), the nature and chemistry of the electrolyte solvents, and molecular and salt-type additives are hailed as one of the most enabling remedies to guide the interface and interphase reactions, and thus result in the formation of highly regulated interphases in Li-based batteries. They also greatly impact the properties of the EEI layers of emerging mono- and multi-valent cation-based batteries. Electrolyte solvents and additives engineer the formation pathway, chemical make-up, physiochemistry, growth, structure, morphology, overall nature and physical properties of interphases including (electro)chemical, mechanical, and thermal stabilities, and ionic transport.352–354For mono-valent cation-based batteries, the formation of inorganic-rich species on the anode side is desirable to suppress the dissolution of the SEI components and, thus increase the mechanical stability of the SEI layer. This is rationalized by the enhanced performances of the highly salt-concentrated electrolytes (see Fig. 11). However, for divalent cation (e.g., Ca2+ and Mg2+)-based batteries, organic-rich species on the anode side are preferred for facilitating the rapid transport of these divalent cations through the interphases. For instance, by utilizing electrolyte solvents with higher donicity (e.g., dimethylacetamide (DMAc), 1-methylimidazole (MeIm); Gutmann DN >25 kcal mol−1) vs. conventional carbonates (e.g., EC/PC; Gutmann DN <18 kcal mol−1), the solvating structure of calcium cations changes drastically, forming a solvent-dominated solvent sheath.355 Consequently, an organic-rich SEI layer is formed on the surface of calcium metal, thereby leading to an enhanced cycling performance of calcium metal batteries. However, the benefits of organic-rich interphases in multi-valent cation-based rechargeable batteries may be a trade-off with the mechanical stability, and thus accompanied by volume changes during long-term charging and discharging processes.
Electrolyte additives with high donicity can modulate the solvating structures of cationic species, thus tuning the electrochemical properties of the corresponding electrolytes. However, it should be noted that additive properties in emerging mono- and multi-valent cation batteries are quite different in comparison to Li-based batteries due to the difference in their inherent chemistries. For instance, among the various screened additives, i.e., fluoroethylene (FEC), difluoroethylene carbonates (DFEC), vinylene carbonate (VC), and ethylene sulfite (ES), Komaba et al.356 found that only FEC improved the electrochemical performance of hard carbon or NaNi1/2Mn1/2O2 half-cells. Alternatively, Yang et al.357 reported that FEC is less effective when applied to K+ ion-based batteries. An extensive literature survey indicated that studies dealing with electrolyte additives for K+ and multi-valent cation-based rechargeable batteries are rarely conducted, and only a handful of accounts are available. However, multifunctional additives with a synergistic effect can be a qualifying avenue to overcome the interphase-related limitations in multi-valent cation-based batteries.
5. Advanced characterization techniques
Detailed understanding of the chemical make-up, morphologies, mechanical and chemical properties, and ion transport and migration kinetics is of paramount importance to direct the further design of robust electrolytes, interphases and interfaces in rechargeable batteries.358–361 Given the elusive formation mechanism, highly dynamical nature, nano-scale dimension, susceptibility to various uncontrollable variables, complicated microstructures and delicate chemical compositions, heterogeneity, unknown structure-property relationships and lack of reliable in situ quantitative characterization tools, the in-depth understanding, and thus mastering of interphases remain the most challenging issue in electrochemical energy storage systems.In situ, in operando, and ex situ techniques (Fig. 21) are among the most widely used measurements to study the interphases in lithium and non-lithium rechargeable batteries.358–362 Although in situ measurement refers to analysing interphases within the cell and set up, ex situ measurement involves the analysis of the interphase, where cycling is separated from the characterization setup and a transfer holder is needed to move the battery cell.
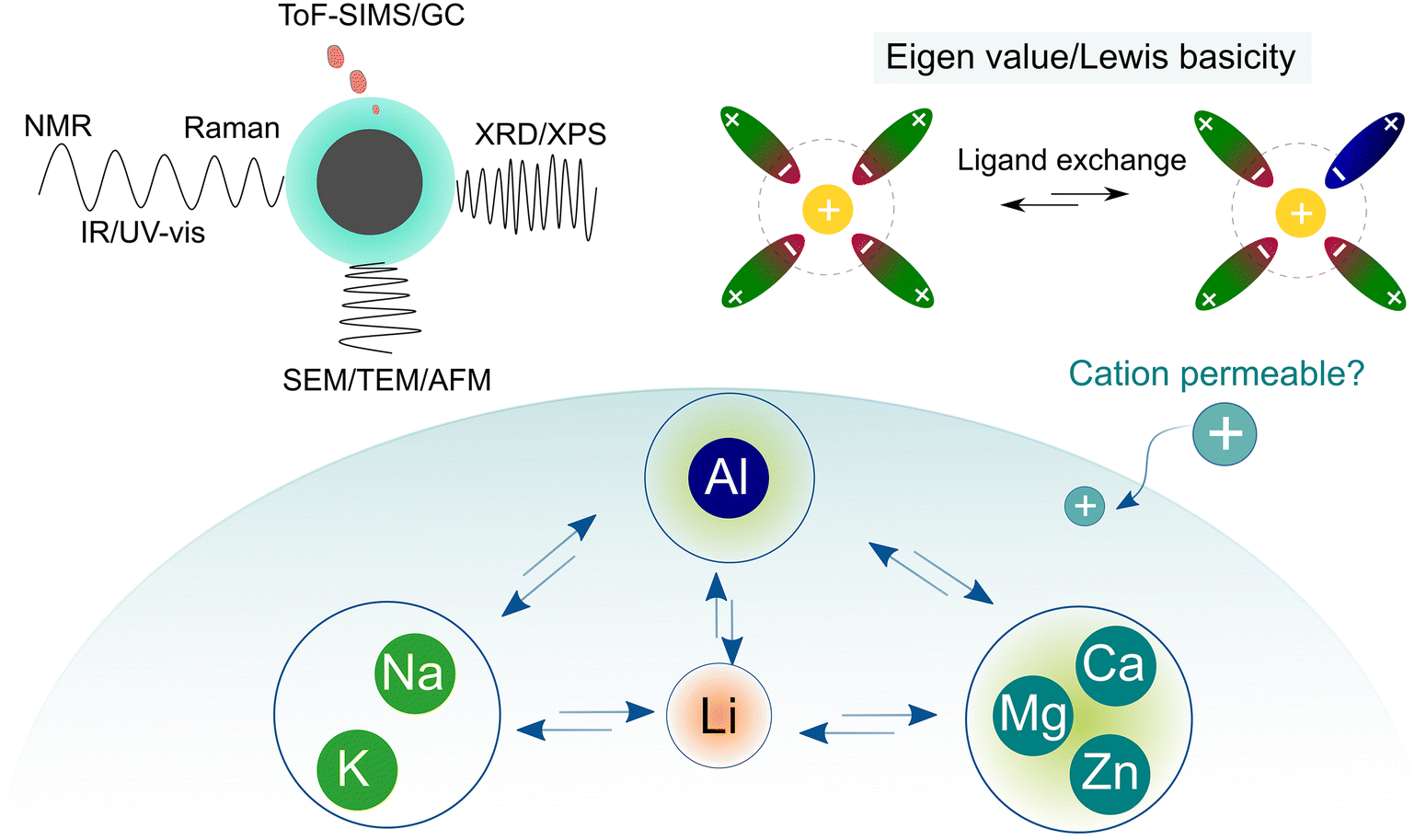 | ||
| Fig. 21 Possible directions for further improving the performance of electrolyte and electrode–electrolyte interphases for emerging mono and multivalent batteries. Abbreviations are given as follows: nuclear magnetic resonance (NMR) spectroscopy, infrared (IR) spectroscopy, ultraviolet and visible (UV-Vis) spectroscopy, time-of-flight secondary ion mass spectrometry (ToF-SIMS), gas chromatography (GC), X-Ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), scanning (SEM) and transmission electron microscope (TEM), and atomic force microscopy (AFM). | ||
Interchangeably used with in situ analysis, the operando technique refers to conducting the interphase analysis during battery cycling without pausing the cycling to conduct the analysis. Ex situ investigation of the interphases/interfaces in batteries is problematic given that it involves the disassembly of the cell to expose the material of interest, which can lead to the modification of the real time behaviour of the intended sample and accompanying information. This can be linked to water and oxygen contamination, and evaporation of the electrolyte solvent, which lead to drastic changes in the chemical composition of the EEI layers. Also, although they are more expensive, less accessible, and complicated, in situ and operando techniques are among the most suitable tools to investigate the formation, structure, and evolution of the interphases in rechargeable batteries. Of significant importance, the spatial resolution, sensitivity and selectivity should be considered as the central elements, while considering the investigation technique of choice.
Comprehensive understanding of the interphases in emerging mono- and multi-valent cation-based rechargeable batteries is of supreme significance, and thus inevitably resorts to the use of multi-technique approaches, offering complementary information that becomes mandatory.
With respect to the electrolyte, in situ and/or in operando characterization techniques are also urgently needed, particularly with the burgeoning interest in implementing solid electrolytes in building high-energy and safe rechargeable batteries.358–361,363 Interestingly, the analysis of the bulk solid electrolyte shares some common features as that of electrode–electrolyte interphases, given that both components are solid materials and are difficult to be isolated.364 In recent years, a wide array of emerging analytic techniques has been specifically developed for batteries, including solid-state nuclear magnetic resonance (NMR),358,361 nano(micro)scale X-ray tomography,365 and synchrotron X-ray techniques,366 which are out of the scope of the present work and can be readily accessed in recent review articles.358–361,363,366
6. Drop-in concept: from Li to emerging rechargeable battery technologies
In bulk electrolyte, it seems that the knowledge accumulated in the design of Li-based salts and solvents can be partially utilized in other monovalent cation-based electrolytes, including Na- and K-based electrolytes. For example, the delocalization of the negative charges can facilitate the dissociation of cations, irrespective of the nature of the cationic species. However, for multi-valent cation-based electrolytes, the high dissociation of cations does not necessarily mean that a higher concentration of charge carriers will be achieved, given that the Lewis acidity of these cations tends to be stronger than that of mono-valent ones.The detailed research accounts presented in this review undoubtedly evidence the fundamental distinctions between the chemistries of the interphases in Li and Li-free emerging mono- (Na+ and K+) and multi (Mg2+, Ca2+, Zn2+, and Al3+) cation-based rechargeable batteries. Peculiar dissimilarities in the charge density, degree of Lewis acidity, ligand exchange rate, equivalent volume, reduction potentials, de-solvation energy of Mn+ or M, solubility and kinetics of M-based EEI compounds, etc. certainly dictate the functional properties of the EEI layer including its nature, composition, and properties. Therefore, though the experiences and knowhow stockpiled from LIBs provide baseline information and insights into the development of electrolytes and understanding of the interphases, there is no guarantee that the direct and smooth transition of behaviors from Li- to emerging mono- and multi-valent cation-based battery chemistries is valid. Thus, the interphases in the latter may not follow the known rules for Li-based batteries in many cases, thus requesting new perspectives and thinking outside the box, while considering new battery chemistries and technologies.367
Moreover, limited data is available on interphases formed in multi-valent cation-based batteries, and thus deep understanding employing advanced characterization tools capable of shedding light on this few-nanometer region is urgently needed for its comprehension.
7. Conclusions and outlooks
In this current perspective, we comparatively outlined the fundamental physical (e.g., transport properties) and (electro)chemical aspects of the electrolyte and interphases in lithium batteries and other mono- and multi-valent batteries. Based on the meticulous examinations of the historical and contemporary developments in these intriguing domains, the authors concluded the following (Fig. 21):(1) The electron-donicity and dielectric properties of electrolyte solvents govern the transport properties of non-aqueous organic electrolytes. The rapid transport of solvated ions is readily seen for electrolytes based on mono-valent ions; however, this becomes quite difficult for multivalent ones due to their bulky solvation sheath. This implies that the search for highly conductive multi-valent cations based electrolytes requires fundamental innovation in the case of conducting salt and electrolyte solvents, which can finally replace the classic coupled-decoupled transport behavior of the current organic electrolytes developed for battery use.
(2) The Eigen values proposed in the 1960s are suitable not only for screening highly conductive electrolytes but also for high-performance electrode materials; however, their application in the battery domain still needs to be further explored. Also, it has to be highlighted that the sluggish exchange rate of the solvent molecules surrounding the Mg2+ and Al3+ ions effectively puts forward the towering challenges in building high-performing batteries based on these two elements (Fig. 21).
(3) The development of solid-state batteries based on other emerging mono- and multi-valent batteries is certainly of supreme importance for the utilization of electrochemical energy systems; however, considerable efforts are required for building highly conductive electrolyte and stable electrode–electrolyte interphases.
(4) A detailed literature survey clearly evidences the fundamental dissimilarities between the chemistries of the passivation layers formed on Li-based and emerging mono- and valent cation-based batteries. This is linked to the differences in the size, charge density, Lewis acidity, solubility of the EEI building compounds, equivalent volume of the metals, reduction potential of metal ions, desolvation energy, etc. These differences undoubtedly affect the chemical make-up (composition), physiochemistry, nature, ion transport, and overall quality of the passivation layers and a detailed and comprehensive investigation considering all the underlying differences is urgently needed.
(5) Despite the current research efforts and developments in interphase design and engineering, various challenges remain to be addressed in relation to the interphases formed in emerging mono- and multi-valent cation-based rechargeable batteries, including understanding the solvation chemistry and dynamics of cation (Mn+) migration at the interphasial/interfacial regions, the identification and prediction of the EEI composition, and the mitigation of dendritic growth and related issues.
(6) Advanced characterization techniques related to the electrolyte and electrode–electrolyte interphases are highly desirable, including both in situ and ex situ methods (Fig. 21). The characterizations can be undertaken in combination with multi-scale modelling approaches368–371 to provide atomistic insight into the design of conductive mono and multivalent electrolytes, as well as regulating the interphase/interface properties.
(7) Advanced multi-scale simulation calculations afford a powerful service to understand and master the chemical make-up, structure and function of the interphase layers, essentially bridging the gap between existing experimental characterization methods in examining the accuracy of the interphasial/interfacial reactions. To actualize the full-range potential of theory-guided experimental design for investigating electrolytes and interphases, it requires theorists and experimentalists to collaborate from the outset.
(8) Despite the fact that the majority of knowledge and concepts accumulated in lithium-based batteries can be transferrable to other emerging mono-, and partly multi-valent batteries, serious precautions have to be taken. The development of electrolytes and understanding of the interphases for lithium-free mono and multi-valent rechargeable batteries call for new brand and out-of-the-box thinking.
(9) Although half cells provide important data to understand battery systems, they do not completely get transformed into full cells, and there are no effective predictive methods to correlate a single interphase performance in half cells with the actual performance in full cells. This calls for the simultaneous consideration and evaluation of both the SEI and CEI in full cell set-ups to assess the major phenomena including microscopic reaction mechanism of the electrolyte, accurate identification of the components, and structure, ion transport and migration, and mechanical properties of the interphase layers.
Accordingly, we anticipate that besides to the popular lithium-based batteries, other emerging mono- and multi-valent cations - based reharegeable batteries can certainly gain deeper penetration in the commodity market with ingenious design of the electrolyte and electrode–electrolyte interphases, and thus becoming important building blocks for future energy networks.
Glossary
| ACN | Acetonitrile |
| AlBr3 | Aluminum bromide |
| AlCl3 | Aluminum chloride |
| Al(TFSI)3 | Aluminum bis(trifluoromethanesulfonyl)imide |
| BF4− | Tetrafluoroborate anion |
| BMITfO | 1-Butyl-3-methylimidazolium triflate |
| BMITFSI | 1-Butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide |
| BOB− | Bis(oxalato)borate anion |
| BPC | n-Butylpyridinium chloride |
| BTFE | Bis(2,2,2-trifluoroethyl) ether |
| BTFSI− | (Benzenesulfonyl)(trifluoromethanesulfonyl)imide anion |
| Ca(BF4)2 | Calcium tetrafluoroborate |
| Ca(BH4)2 | Calcium borohydride |
| Ca[B(hfip)4]2 | Calcium tetrakis(hexafluoroisopropyloxy)borate |
| CaBr2 | Calcium bromide |
| Ca(ClO4)2 | Calcium perchlorate |
| CEI | Cathode electrolyte interphase |
| CEs | Coulombic efficiencies |
| CeZn0.8Cl2.6 | Cerium zinc chloride |
| ClO4− | Perchlorate anion |
| CPEs | Composite polymer electrolytes |
| N(CN)2− | Dicyanamide |
| DEC | Diethyl carbonate |
| DEE | Diethyl ether |
| DFSI− | Bis(difluoromethanesulfonyl)imide anion |
| DFTFSI− | (Difluoromethanesulfonyl)(trifluoromethanesulfonyl)imide anion |
| DFEC | Difluoroethylene carbonate |
| DMAc | Dimethylacetamide |
| DMC | Dimethyl carbonate |
| DME | 1,2-Dimethoxyethane |
| DMSO | Dimethyl sulfoxide |
| DN | Donor number |
| DOL | 1,3-Dioxolane |
| EC | Ethylene carbonate |
| EEI | Electrode electrolyte interphase |
| EFA− | Ether-functionalized anion |
| EMC | Ethyl methyl carbonate |
| EMIBF4 | 1-Ethyl-3-methylimidazolium tetrafluoroborate |
| EMICl | 1-Ethyl-3-methylimidazolium chloride |
| EMITfO | 1-ethyl-3-methylimidazolium triflate |
| EMITFSI | 1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide |
| ES | Ethylene sulfite |
| EtMgBr | Ethyl magnesium bromide |
| EVs | Electric vehicles |
| FDMA | N,N-Dimethylacetamide |
| FEC | Fluoroethylene carbonate |
| FNFSI− | (Fluorosulfonyl)(n-nonafluorobutanesulfonyl)imide anion |
| FSI− | Bis(fluorosulfonyl)imide anion |
| FTFSI− | (Fluorosulfonyl)(trifluoromethanesulfonyl)imide anion |
| GBL | γ-Butyrolactone |
| GPEs | Gel polymer electrolytes |
| HOMO | Highest occupied molecular orbital |
| ILs | Ionic liquids |
| KBr | Potassium bromide |
| KFSI | Potassium bis(fluorosulfonyl)imide |
| KI | Potassium iodide |
| KPF6 | Potassium hexafluorophosphate |
| KSCN | Potassium isocyanate |
| KTFSI | Potassium bis(trifluoromethanesulfonyl)imide |
| LHCE | Localized highly concentrated electrolyte |
| LiAl2Cl7 | Lithium aluminum chloride |
| LiBr | Lithium bromide |
| LIBs | Lithium-ion batteries |
| Li2EDC | Dilithium ethylene glycol dicarbonate |
| LiFSI | Lithium bis(fluorosulfonyl)imide |
| LiH | Lithium hydride |
| LiPF6 | Lithium hexafluorophosphate |
| LiTf | Lithium triflate |
| LiTFSI | Lithium bis(trifluoromethanesulfonyl)imide |
| LUMO | Lowest unoccupied molecular orbital |
| MACC | Magnesium aluminum chloride complex |
| MEIC | 1-Methyl-3-ethylimidazolium chloride |
| MeIm | 1-Methylimidazole |
| MOF | Metal–organic framework |
| MTFE | 1,1,2,2-Tetrafluoroethyl methyl ether |
| NaFSI | Sodium bis(fluorosulfonyl)imide |
| NaI | Sodium iodide |
| NaPF6 | Sodium hexafluorophosphate |
| NaSCN | Sodium isocyanate |
| NaTFSI | Sodium bis(trifluoromethanesulfonyl)imide |
| NIBs | Sodium ion batteries |
| N122,1O2TFSI | N,N-diethyl-N-methyl-N-(2-methoxyethyl)ammonium bis(trifluoromethanesulfonyl)imide |
| OEO | Oligo(ethylene oxides) |
| PC | Propylene carbonate |
| PEO | Poly(ethylene oxide) |
| PEs | Polymer electrolytes |
| PF6− | Hexafluorophosphate anion |
| PPC | Poly(propylene carbonate) |
| PP14TFSI | 1-Butyl-1-methylpiperidinium bis(trifluoromethanesulfonyl)imide |
| PTHF | Polytetrahydrofuran |
| PTMC | Poly(trimethylene carbonate) |
| PYR14TFSI | N-Butyl-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide |
| RT | Room temperature |
| SEI | Solid electrolyte interphase |
| SHE | Standard hydrogen electrode |
| SIS | Solvent-in-salt |
| SPEs | Solid polymer electrolytes |
| TCM− | Tricyanomethanide anion |
| THF | Tetrahydrofuran |
| TDI− | 4,5-Dicyano-2-(trifluoromethyl)imidazole anion |
| TEP | Triethyl phosphate |
| TMP | Trimethyl phosphate |
| TFEMSI− | (Trifluoromethanesulfonyl)(N-ethyl-N-methylsulfamoyl)imide anion |
| TFOP− | Tetrafluorooxalatophosphate anion |
| TFSI− | Bis(trifluoromethanesulfonyl)imide anion |
| VC | Vinylene carbonate |
| VFT | Vogel–Fulcher–Tammann |
| WCAs | Weakly coordinating anions |
| ZnCl2 | Zinc chloride |
| Zn(ClO4)2 | Zinc perchlorate |
| Zn(TFSI)2 | Zinc bis(trifluoromethanesulfonyl)imide |
| Zn(Tf)2 | Zinc triflate |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Fundamental Research Funds for the Central Universities, HUST (2020kfyXJJS095). H. K., G. G. E., and E. F. acknowledge the funding received from the European Commission via the H2020 IMAGE project (Grant Agreement Number: 769929—IMAGE—H2020-GV-2016-2017/H2020-GV-2017), and G. G. E. and E. F. for HELENA (under grant agreement No. 101069681).Notes and references
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- S. Ferrari, M. Falco, A. B. Muñoz-García, M. Bonomo, S. Brutti, M. Pavone and C. Gerbaldi, Adv. Energy Mater., 2021, 11, 2100785 CrossRef CAS.
- J.-G. Zhang, W. Xu, J. Xiao, X. Cao and J. Liu, Chem. Rev., 2020, 120, 13312–13348 CrossRef CAS.
- S. Dhir, S. Wheeler, I. Capone and M. Pasta, Chem, 2020, 6, 2442–2460 CAS.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 CrossRef CAS.
- M. Zhou, P. Bai, X. Ji, J. Yang, C. Wang and Y. Xu, Adv. Mater., 2021, 33, e2003741 CrossRef PubMed.
- R. Mohtadi, O. Tutusaus, T. S. Arthur, Z. Zhao-Karger and M. Fichtner, Joule, 2021, 5, 581–617 CrossRef CAS.
- Y. Lu, L. Li, Q. Zhang, Z. Niu and J. Chen, Joule, 2018, 2, 1747–1770 CrossRef CAS.
- P. Saha, M. K. Datta, O. I. Velikokhatnyi, A. Manivannan, D. Alman and P. N. Kumta, Prog. Mater. Sci., 2014, 66, 1–86 CrossRef CAS.
- O. M. Leung, T. Schoetz, T. Prodromakis and C. Ponce de Leon, J. Electrochem. Soc., 2021, 168, 056509 CrossRef CAS.
- M. Armand, P. Axmann, D. Bresser, M. Copley, K. Edström, C. Ekberg, D. Guyomard, B. Lestriez, P. Novák, M. Petranikova, W. Porcher, S. Trabesinger, M. Wohlfahrt-Mehrens and H. Zhang, J. Power Sources, 2020, 479, 228708 CrossRef CAS.
- M. Li, J. Lu, X. Ji, Y. Li, Y. Shao, Z. Chen, C. Zhong and K. Amine, Nat. Rev. Mater., 2020, 5, 276–294 CrossRef CAS.
- P. Canepa, G. Sai Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson and G. Ceder, Chem. Rev., 2017, 117, 4287–4341 CrossRef CAS PubMed.
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS PubMed.
- C. Heubner, T. Lein, M. Schneider and A. Michaelis, J. Mater. Chem. A, 2020, 8, 16854–16883 RSC.
- M. A. Schroeder, L. Ma, G. Pastel and K. Xu, Curr. Opin. Electrochem., 2021, 29, 100819 CrossRef CAS.
- R. Borah, F. R. Hughson, J. Johnston and T. Nann, Mater. Today Adv., 2020, 6, 100046 CrossRef.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS.
- X. Judez, G. G. Eshetu, C. Li, L. M. Rodriguez-Martinez, H. Zhang and M. Armand, Joule, 2018, 2, 2208–2224 CrossRef CAS.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 CrossRef CAS.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- M. Li, X. Bi, R. Wang, Y. Li, G. Jiang, L. Li, C. Zhong, Z. Chen and J. Lu, Matter, 2020, 2, 32–49 CrossRef.
- S. Chen, J. Zheng, L. Yu, X. Ren, M. H. Engelhard, C. Niu, H. Lee, W. Xu, J. Xiao, J. Liu and J.-G. Zhang, Joule, 2018, 2, 1548–1558 CrossRef CAS.
- A. Mauger, C. M. Julien, J. B. Goodenough and K. Zaghib, J. Electrochem. Soc., 2020, 167, 070507 CrossRef CAS.
- H. Zhang, C. Li, G. G. Eshetu, S. Laruelle, S. Grugeon, K. Zaghib, C. Julien, A. Mauger, D. Guyomard, T. Rojo, N. Gisbert-Trejo, S. Passerini, X. Huang, Z. Zhou, P. Johansson and M. Forsyth, Angew. Chem., Int. Ed., 2020, 59, 534–538 CrossRef CAS.
- G. Cui, Matter, 2020, 2, 805–815 CrossRef.
- X. Shen, H. Liu, X.-B. Cheng, C. Yan and J.-Q. Huang, Energy Storage Mater., 2018, 12, 161–175 CrossRef.
- H. Wang, S. Chen, C. Fu, Y. Ding, G. Liu, Y. Cao and Z. Chen, ACS Mater. Lett., 2021, 3, 956–977 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- W. A. Plotnikow and S. Jakubson, Zeitschrift für Physikalische Chemie, 1928, 138A, 251–259 CrossRef.
- W. A. Plotnikow and S. Jakubson, Zeitschrift für Physikalische Chemie, 1930, 147A, 227–230 CrossRef.
- G. A. Capuano and W. G. Davenport, J. Electrochem. Soc., 1971, 118, 1688–1695 CrossRef CAS.
- E. Peled, A. Mitavski, A. Reger and E. Gileadi, J. Electroanal. Chem. Interfacial Electrochem., 1977, 75, 677–695 CrossRef CAS.
- A. Reger, E. Peled and E. Gileadi, J. Phys. Chem., 1979, 83, 869–873 CrossRef CAS.
- A. Reger, E. Peled and E. Gileadi, J. Phys. Chem., 1979, 83, 873–879 CrossRef CAS.
- M. Gálová, Surf. Technol., 1980, 11, 357–369 CrossRef.
- E. Peled and E. Gileadi, J. Electrochem. Soc., 1976, 123, 15–19 CrossRef CAS.
- D. Shanmukaraj, S. Grugeon, G. Gachot, S. Laruelle, D. Mathiron, J.-M. Tarascon and M. Armand, J. Am. Chem. Soc., 2010, 132, 3055–3062 CrossRef CAS.
- H.-B. Han, S.-S. Zhou, D.-J. Zhang, S.-W. Feng, L.-F. Li, K. Liu, W.-F. Feng, J. Nie, H. Li, X.-J. Huang, M. Armand and Z.-B. Zhou, J. Power Sources, 2011, 196, 3623–3632 CrossRef CAS.
- A. Bhide, J. Hofmann, A. Katharina Dürr, J. Janek and P. Adelhelm, Phys. Chem. Chem. Phys., 2014, 16, 1987–1998 RSC.
- R. Younesi, G. M. Veith, P. Johansson, K. Edström and T. Vegge, Energy Environ. Sci., 2015, 8, 1905–1922 RSC.
- H. Zhang, O. Arcelus and J. Carrasco, Electrochim. Acta, 2018, 280, 290–299 CrossRef CAS.
- J. N. Canongia Lopes, K. Shimizu, A. A. H. Pádua, Y. Umebayashi, S. Fukuda, K. Fujii and S.-I. Ishiguro, J. Phys. Chem. B, 2008, 112, 1465–1472 CrossRef CAS PubMed.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- K. Matsumoto, T. Matsui, T. Nohira and R. Hagiwara, J. Fluorine Chem., 2015, 174, 42–48 CrossRef CAS.
- L. Xue, C. W. Padgett, D. D. DesMarteau and W. T. Pennington, Solid State Sci., 2002, 4, 1535–1545 CrossRef CAS.
- L. Xue, D. D. DesMarteau and W. T. Pennington, Solid State Sci., 2005, 7, 311–318 CrossRef CAS.
- M. J. Earle, U. Hakala, B. J. McAuley, M. Nieuwenhuyzen, A. Ramani and K. R. Seddon, Chem. Commun., 2004, 1368–1369 RSC.
- J. L. Nowinski, P. Lightfoot and P. G. Bruce, J. Mater. Chem. A, 1994, 4, 1579–1580 RSC.
- J. Kotwiński, M. Marzantowicz, M. Leszczynska, A. Gągor, I. Abrahams and F. Krok, Solid State Ionics, 2019, 330, 9–16 CrossRef.
- V. Gutmann, Coordination Chemistry in Non-Aqueous Solutions, Springer-Verlag, New-York, 1968 Search PubMed.
- F. Cataldo, Eur. Chem. Bull., 2015, 4, 92–97 Search PubMed.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, third edn, VCH, New York, 2003 Search PubMed.
- M. C. Day and F. Selbin, Theoretical Inorganic Chemistry, Reinhold Publ. Corp., New York, 1962 Search PubMed.
- D. A. Wynn, M. M. Roth and B. D. Pollard, Talanta, 1984, 31, 1036–1040 CrossRef CAS.
- M. Armand, Annu. Rev. Mater. Res., 1986, 16, 245–261 CrossRef CAS.
- L. Meabe, S. Rodriguez Peña, M. Martinez-Ibañez, Y. Zhang, E. Lobato, H. Manzano, M. Armand, J. Carrasco and H. Zhang, J. Phys. Chem. C, 2020, 124, 17981–17991 CrossRef CAS.
- X. Chen, X. Q. Zhang, H. R. Li and Q. Zhang, Batteries Supercaps, 2019, 2, 128–131 CrossRef CAS.
- M. A. Ratner and D. F. Shriver, Chem. Rev., 1988, 88, 109–124 CrossRef CAS.
- M. A. Ratner, P. Johansson and D. F. Shriver, MRS Bull., 2000, 25, 31–37 CrossRef CAS.
- O. Borodin, J. Self, K. A. Persson, C. Wang and K. Xu, Joule, 2020, 4, 69–100 CrossRef CAS.
- J. C. Bachman, S. Muy, A. Grimaud, H.-H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed.
- J. O. M. Bockris and A. K. N. Reddy, Modern Electrochemistry 1 Ionics, Second Edn, 2002 Search PubMed.
- J. D. Bernal and R. H. Fowler, J. Chem. Phys., 1933, 1, 515–548 CrossRef CAS.
- H. Vogel, Phys. Z., 1921, 22, 645–646 CAS.
- G. S. Fulcher, J. Am. Ceram. Soc., 1925, 8, 339–355 CrossRef CAS.
- G. Tamman and W. Hesse, Z. Anorg. Allg. Chem., 1926, 156, 245–257 CrossRef.
- K. Funke, Sci. Technol. Adv. Mater., 2013, 14, 043502 CrossRef CAS.
- M. Eigen, Pure Appl. Chem., 1963, 6, 97–115 CAS.
- J. J. Berzelius, Z. Phys. Chem., 1818, 21, 44–48 Search PubMed.
- M. E. Weeks and M. E. Larson, J. Chem. Educ., 1937, 14, 403 CrossRef CAS.
- M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS.
- M. I. Konovalov and V. A. Plotnikov, Russ. J. Phys. Chem., 1899, 31, 1020 Search PubMed.
- S. V. Laszczynski and S. V. Gorski, Z. Elektrochem., 1897, 4, 290–293 Search PubMed.
- H. E. Patten, J. Phys. Chem., 1904, 8, 483–487 CrossRef CAS.
- J. M. Nelson and W. V. Evans, J. Am. Chem. Soc., 1917, 39, 82–83 CrossRef CAS.
- M. B. Armand, J. M. Chabagno and M. Duclot, 1979, Poly-ethers as solid electrolytes, ed. P. Vashitshta, Mundy J. N., G. K. Shenoy, in Fast ion Transport in Solids. Electrodes and Electrolytes, North Holland Publishers, Amsterdam Search PubMed.
- D. E. Couch and A. Brenner, J. Electrochem. Soc., 1952, 99, 234–244 CrossRef CAS.
- W. S. Harris, Electrochemical Studies in Cyclic Esters, Dissertation submitted to University of California, 1958 DOI:10.2172/4305596.
- S. G. Meibuhr, J. Electrochem. Soc., 1971, 118, 709–711 CrossRef CAS.
- D. E. Fenton, J. M. Parker and P. V. Wright, Polymer, 1973, 14, 589 CrossRef CAS.
- J. Jansta, F. P. Dousek and J. Říha, J. Electroanal. Chem. Interfacial Electrochem., 1973, 44, 263–273 CrossRef CAS.
- P. V. Wright, Br. Polym. J., 1975, 7, 319–327 CrossRef CAS.
- J. Robinson and R. A. Osteryoung, J. Am. Chem. Soc., 1979, 101, 323–327 CrossRef CAS.
- J. S. Wilkes, J. A. Levisky, R. A. Wilson and C. L. Hussey, Inorg. Chem., 1982, 21, 1263–1264 CrossRef CAS.
- M. Armand and F. E. K. C. Moursli, Bisperhaloacyl or bisperhalosulfonyl imides of alkali metals, their solid solutions with polymers, and their use as battery electrolytes, FR2527602A1, 1983 Search PubMed.
- E. Gocke, W. Schramm, P. Dolscheid and R. Schöllhorn, J. Solid State Chem., 1987, 70, 71–81 CrossRef CAS.
- T. D. Gregory, R. J. Hoffman and R. C. Winterton, J. Electrochem. Soc., 1990, 137, 775–780 CrossRef CAS.
- D. Aurbach, R. Skaletsky and Y. Gofer, J. Electrochem. Soc., 1991, 138, 3536–3545 CrossRef CAS.
- D. Baril, Y. Chabre and M. B. Armand, J. Electrochem. Soc., 1993, 140, 2687–2690 CrossRef CAS.
- D. Guyomard and J. M. Tarascon, J. Electrochem. Soc., 1993, 140, 3071–3081 CrossRef CAS.
- L. Legrand, A. Tranchant and R. Messina, Electrochim. Acta, 1994, 39, 1427–1431 CrossRef CAS.
- C. Michot, M. Armand, J. Y. Sanchez, Y. Choquette and M. Gauthier, Ionic conducting material having good anticorrosive properties, US6254797B1, 1995 Search PubMed.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- J. Grondin, D. Talaga, J.-C. Lassègues and W. A. Henderson, Phys. Chem. Chem. Phys., 2004, 6, 938–944 RSC.
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- R. E. Doe, R. Han, J. Hwang, A. J. Gmitter, I. Shterenberg, H. D. Yoo, N. Pour and D. Aurbach, Chem. Commun., 2014, 50, 243–245 RSC.
- A. Guerfi, J. Trottier, I. Boyano, I. De Meatza, J. A. Blazquez, S. Brewer, K. S. Ryder, A. Vijh and K. Zaghib, J. Power Sources, 2014, 248, 1099–1104 CrossRef CAS.
- A. Ponrouch, C. Frontera, F. Bardé and M. R. Palacín, Nat. Mater., 2016, 15, 169–172 CrossRef CAS.
- M. Chiku, S. Matsumura, H. Takeda, E. Higuchi and H. Inoue, J. Electrochem. Soc., 2017, 164, A1841–A1844 CrossRef CAS.
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J.-G. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef PubMed.
- D. Wang, X. Gao, Y. Chen, L. Jin, C. Kuss and P. G. Bruce, Nat. Mater., 2018, 17, 16–20 CrossRef CAS.
- A. Naveed, H. Yang, J. Yang, Y. Nuli and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 2760–2764 CrossRef CAS PubMed.
- W. S. Harris, Electrochemical Studies in Cyclic Esters, Dissertation submitted to University of California, Berkeley, CA, 1958 Search PubMed.
- J. Rouxel, M. Danot and J. Bichon, Bull. Soc. Chim. Fr., 1971, 3930–3935 CAS.
- A. Leblanc-Soreau, M. Danot, L. Trichet and J. Rouxel, Mater. Res. Bull., 1974, 9, 191–197 CrossRef.
- M. S. Whittingham, Science, 1976, 192, 1126–1127 CrossRef CAS PubMed.
- D. A. Winn and B. C. H. Steele, Mater. Res. Bull., 1976, 11, 551–557 CrossRef CAS.
- M. B. Armand, in Materials for Advanced Batteries, ed. D. W. Murphy, J. Broadhead and B. C. H. Steele, Springer US, Boston, MA, 1980, pp. 145–161 Search PubMed.
- M. Lazzari and B. Scrosati, J. Electrochem. Soc., 1980, 127, 773–774 CrossRef CAS.
- K. Mizushima, P. C. Jones, P. J. Wiseman and J. B. Goodenough, Mater. Res. Bull., 1980, 15, 783–789 CrossRef CAS.
- K. Sanechika and A. Yoshino, JPS60253157, 1983.
- Y. Nishi, J. Power Sources, 2001, 100, 101–106 CrossRef CAS.
- A. Yoshino, Angew. Chem., Int. Ed., 2012, 51, 5798–5800 CrossRef CAS PubMed.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- J. Xie and Y. C. Lu, Nat. Commun., 2020, 11, 2499 CrossRef CAS PubMed.
- M. Armand, Solid State Ionics, 1994, 69, 309–319 CrossRef CAS.
- M. Armand, Adv. Mater., 1990, 2, 278–286 CrossRef CAS.
- H. Zhang, Y. Chen, C. Li and M. Armand, SusMat, 2021, 1, 24–37 CrossRef.
- J. Muldoon, C. B. Bucur, A. G. Oliver, T. Sugimoto, M. Matsui, H. S. Kim, G. D. Allred, J. Zajicek and Y. Kotani, Energy Environ. Sci., 2012, 5, 5941–5950 RSC.
- Z. Ma, D. R. MacFarlane and M. Kar, Batteries Supercaps, 2019, 2, 115–127 CrossRef.
- Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson and G. Ceder, Chem. Rev., 2021, 121, 1623–1669 CrossRef CAS PubMed.
- W. Linert, A. Camard, M. Armand and C. Michot, Coord. Chem. Rev., 2002, 226, 137–141 CrossRef CAS.
- Z. Song, X. Wang, H. Wu, W. Feng, J. Nie, H. Yu, X. Huang, M. Armand, H. Zhang and Z. Zhou, J. Power Sources Adv., 2022, 14, 100088 CrossRef CAS.
- T. Thomberg, R. Väli, J. Eskusson, T. Romann and A. Jänes, J. Electrochem. Soc., 2018, 165, A3862–A3870 CrossRef CAS.
- A. Ponrouch, E. Marchante, M. Courty, J.-M. Tarascon and M. Rosa Palacin, Energy Environ. Sci., 2012, 5, 8572–8583 RSC.
- A. Ponrouch, A. R. Goni and M. Rosa Palacin, Electrochem. Commun., 2013, 27, 85–88 CrossRef CAS.
- A. Ponrouch, D. Monti, A. Boschin, B. Steen, P. Johansson and M. R. Palacin, J. Mater. Chem. A, 2015, 3, 22–42 RSC.
- D. Monti, E. Jonsson, A. Boschin, M. R. Palacin, A. Ponrouch and P. Johansson, Phys. Chem. Chem. Phys., 2020, 22, 22768–22777 RSC.
- A. Ponrouch, R. Dedryvere, D. Monti, A. E. Demet, J. M. A. Mba, L. Croguennec, C. Masquelier, P. Johansson and M. R. Palacin, Energy Environ. Sci., 2013, 6, 2361–2369 RSC.
- T. Hosaka, K. Kubota, H. Kojima and S. Komaba, Chem. Commun., 2018, 54, 8387–8390 RSC.
- J. Zhao, X. Zou, Y. Zhu, Y. Xu and C. Wang, Adv. Funct. Mater., 2016, 26, 8103–8110 CrossRef CAS.
- S. Huang, P. Du, C. Min, Y. Liao, H. Sun and Y. Jiang, J. Fluoresc., 2013, 23, 621–627 CrossRef CAS PubMed.
- R. Attias, M. Salama, B. Hirsch, Y. Goffer and D. Aurbach, Joule, 2019, 3, 27–52 CrossRef CAS.
- Z. Liang and C. Ban, Angew. Chem., Int. Ed., 2020, 60, 11036–11047 CrossRef PubMed.
- J. Muldoon, C. B. Bucur and T. Gregory, Angew. Chem., Int. Ed., 2017, 56, 12064–12084 CrossRef CAS PubMed.
- S.-Y. Ha, Y.-W. Lee, S. W. Woo, B. Koo, J.-S. Kim, J. Cho, K. T. Lee and N.-S. Choi, ACS Appl. Mater. Interfaces, 2014, 6, 4063–4073 CrossRef CAS PubMed.
- L. Qiao, U. Oteo, M. Martinez-Ibañez, A. Santiago, R. Cid, E. Sanchez-Diez, E. Lobato, L. Meabe, M. Armand and H. Zhang, Nat. Mater., 2022, 21, 455–462 CrossRef CAS PubMed.
- M. Mao, T. Gao, S. Hou and C. Wang, Chem. Soc. Rev., 2018, 47, 8804–8841 RSC.
- Z. Zhang, S. Dong, Z. Cui, A. Du, G. Li and G. Cui, Small Methods, 2018, 2, 1800020 CrossRef.
- O. Mizrahi, N. Amir, E. Pollak, O. Chusid, V. Marks, H. Gottlieb, L. Larush, E. Zinigrad and D. Aurbach, J. Electrochem. Soc., 2008, 155, A103–A109 CrossRef CAS.
- N. Pour, Y. Gofer, D. T. Major and D. Aurbach, J. Am. Chem. Soc., 2011, 133, 6270–6278 CrossRef CAS PubMed.
- T. Liu, Y. Shao, G. Li, M. Gu, J. Hu, S. Xu, Z. Nie, X. Chen, C. Wang and J. Liu, J. Mater. Chem. A, 2014, 2, 3430–3438 RSC.
- H. Dong, J. Li, J. Guo, F. Lai, F. Zhao, Y. Jiao, D. J. L. Brett, T. Liu, G. He and I. P. Parkin, Adv. Mater., 2021, 33, 2007548 CrossRef CAS PubMed.
- H. S. Kim, T. S. Arthur, G. D. Allred, J. Zajicek, J. G. Newman, A. E. Rodnyansky, A. G. Oliver, W. C. Boggess and J. Muldoon, Nat. Commun., 2011, 2, 427 CrossRef PubMed.
- W. Ren, D. Wu, Y. NuLi, D. Zhang, Y. Yang, Y. Wang, J. Yang and J. Wang, ACS Energy Lett., 2021, 6, 3212–3220 CrossRef CAS.
- A. Du, Z. Zhang, H. Qu, Z. Cui, L. Qiao, L. Wang, J. Chai, T. Lu, S. Dong, T. Dong, H. Xu, X. Zhou and G. Cui, Energy Environ. Sci., 2017, 10, 2616–2625 RSC.
- J. Luo, Y. Bi, L. Zhang, X. Zhang and T. L. Liu, Angew. Chem., Int. Ed., 2019, 58, 6967–6971 CrossRef CAS PubMed.
- Z. Zhang, Z. Cui, L. Qiao, J. Guan, H. Xu, X. Wang, P. Hu, H. Du, S. Li, X. Zhou, S. Dong, Z. Liu, G. Cui and L. Chen, Adv. Energy Mater., 2017, 7, 1602055 CrossRef.
- Z. Zhao-Karger, M. E. Gil Bardaji, O. Fuhr and M. Fichtner, J. Mater. Chem. A, 2017, 5, 10815–10820 RSC.
- T. J. Carter, R. Mohtadi, T. S. Arthur, F. Mizuno, R. Zhang, S. Shirai and J. W. Kampf, Angew. Chem., Int. Ed., 2014, 53, 3173–3177 CrossRef CAS PubMed.
- H. Zhao, J. Zhou and P. Jena, Angew. Chem., Int. Ed., 2016, 55, 3704–3708 CrossRef CAS PubMed.
- R. Mohtadi, M. Matsui, T. S. Arthur and S.-J. Hwang, Angew. Chem., Int. Ed., 2012, 51, 9780–9783 CrossRef CAS PubMed.
- K. Tang, A. Du, X. Du, S. Dong, C. Lu, Z. Cui, L. Li, G. Ding, F. Chen, X. Zhou and G. Cui, Small, 2020, 16, 2005424 CrossRef CAS PubMed.
- K.-C. Lau, T. J. Seguin, E. V. Carino, N. T. Hahn, J. G. Connell, B. J. Ingram, K. A. Persson, K. R. Zavadil and C. Liao, J. Electrochem. Soc., 2019, 166, A1510–A1519 CrossRef CAS.
- S. Hou, X. Ji, K. Gaskell, P.-F. Wang, L. Wang, J. Xu, R. Sun, O. Borodin and C. Wang, Science, 2021, 374, 172–178 CrossRef CAS PubMed.
- Y. Zhao, A. Du, S. Dong, F. Jiang, Z. Guo, X. Ge, X. Qu, X. Zhou and G. Cui, ACS Energy Lett., 2021, 6, 2594–2601 CrossRef CAS.
- L. Stievano, I. de Meatza, J. Bitenc, C. Cavallo, S. Brutti and M. A. Navarra, J. Power Sources, 2021, 482, 228875 CrossRef CAS.
- Z. Li, O. Fuhr, M. Fichtner and Z. Zhao-Karger, Energy Environ. Sci., 2019, 12, 3496–3501 RSC.
- A. Shyamsunder, L. E. Blanc, A. Assoud and L. F. Nazar, ACS Energy Lett., 2019, 4, 2271–2276 CrossRef CAS.
- K. V. Nielson, J. Luo and T. L. Liu, Batteries Supercaps, 2020, 3, 766–772 CrossRef CAS.
- T. Zhang, Y. Tang, S. Guo, X. Cao, A. Pan, G. Fang, J. Zhou and S. Liang, Energy Environ. Sci., 2020, 13, 4625–4665 RSC.
- Z. Chen, Y. Tang, X. Du, B. Chen, G. Lu, X. Han, Y. Zhang, W. Yang, P. Han and J. Zhao, Angew. Chem., Int. Ed., 2020, 59, 21769–21777 CrossRef CAS PubMed.
- N. Zhang, Y. Dong, Y. Wang, Y. Wang, J. Li, J. Xu, Y. Liu, L. Jiao and F. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 32978–32986 CrossRef CAS PubMed.
- A. Naveed, H. Yang, Y. Shao, J. Yang, N. Yanna, J. Liu, S. Shi, L. Zhang, A. Ye and B. He, Adv. Mater., 2019, 31, 1900668 CrossRef PubMed.
- H. Qiu, X. Du, J. Zhao, Y. Wang, J. Ju, Z. Chen, Z. Hu, D. Yan, X. Zhou and G. Cui, Nat. Commun., 2019, 10, 5374 CrossRef PubMed.
- H. Qiu, R. Hu, X. Du, Z. Chen, J. Zhao, G. Lu, M. Jiang, Q. Kong, Y. Yan, J. Du, X. Zhou and G. Cui, Angew. Chem., Int. Ed., 2022, 61, e202113086 CAS.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- T. Mandai and P. Johansson, J. Mater. Chem. A, 2015, 3, 12230–12239 RSC.
- D. W. McOwen, D. M. Seo, O. Borodin, J. Vatamanu, P. D. Boyle and W. A. Henderson, Energy Environ. Sci., 2014, 7, 416–426 RSC.
- Y. Yamada and A. Yamada, J. Electrochem. Soc., 2015, 162, A2406–A2423 CrossRef CAS.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- S. Sayah, A. Ghosh, M. Baazizi, R. Amine, M. Dahbi, Y. Amine, F. Ghamouss and K. Amine, Nano Energy, 2022, 98, 107336 CrossRef CAS.
- L. Onsager, Chem. Rev., 1933, 13, 73–89 CrossRef CAS.
- C. A. Angell, C. Liu and E. Sanchez, Nature, 1993, 362, 137–139 CrossRef CAS.
- C. A. Angell, Electrochim. Acta, 2017, 250, 368–375 CrossRef CAS.
- C. Geng, D. Buchholz, G.-T. Kim, D. V. Carvalho, H. Zhang, L. G. Chagas and S. Passerini, Small Methods, 2019, 3, 1800208 CrossRef CAS.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- P. Cheng, H. Zhang, Q. Ma, W. Feng, H. Yu, X. Huang, M. Armand and Z. Zhou, Electrochim. Acta, 2020, 363, 137198 CrossRef CAS.
- Z. Fang, Q. Ma, P. Liu, J. Ma, Y.-S. Hu, Z. Zhou, H. Li, X. Huang and L. Chen, ACS Appl. Mater. Interfaces, 2016, 9, 4282–4289 CrossRef PubMed.
- B. Aktekin, G. Hernández, R. Younesi, D. Brandell and K. Edström, ACS Appl. Energy Mater., 2022, 5, 585–595 CrossRef CAS PubMed.
- L. Suo, W. Xue, M. Gobet, S. G. Greenbaum, C. Wang, Y. Chen, W. Yang, Y. Li and J. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1156–1161 CrossRef CAS PubMed.
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, J. Am. Chem. Soc., 2014, 136, 5039–5046 CrossRef CAS PubMed.
- X. Cao, H. Jia, W. Xu and J.-G. Zhang, J. Electrochem. Soc., 2021, 168, 010522 CrossRef CAS.
- X. Ren, S. Chen, H. Lee, D. Mei, M. H. Engelhard, S. D. Burton, W. Zhao, J. Zheng, Q. Li, M. S. Ding, M. Schroeder, J. Alvarado, K. Xu, Y. S. Meng, J. Liu, J.-G. Zhang and W. Xu, Chem, 2018, 4, 1877–1892 CAS.
- J. Zheng, S. Chen, W. Zhao, J. Song, M. H. Engelhard and J.-G. Zhang, ACS Energy Lett., 2018, 3, 315–321 CrossRef CAS.
- J. Wu, Y. Tian, Y. Gao, Z. Gao, Y. Meng, Y. Wang, X. Wang, D. Zhou, F. Kang, B. Li and G. Wang, Angew. Chem., Int. Ed., 2022, 61, e202205416 CAS.
- J. Li, C. Han, X. Ou and Y. Tang, Angew. Chem., Int. Ed., 2022, 61, e202116668 CAS.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- G. Gebresilassie Eshetu, M. Armand, B. Scrosati and S. Passerini, Angew. Chem., Int. Ed., 2014, 53, 13342–13359 CrossRef CAS PubMed.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan, W. Zheng, S. Zhang and J. Zhang, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS.
- G. G. Eshetu, M. Armand, H. Ohno, B. Scrosati and S. Passerini, Energy Environ. Sci., 2016, 9, 49–61 RSC.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC.
- H. Ohno, Electrochemical Aspects of Ionic Liquids, 2005, pp. i–xiv Search PubMed.
- M. K. Douglas, R. MacFarlane and J. M. Pringle, Fundamentals of Ionic Liquids, 2017, pp. i–ix Search PubMed.
- P. Bonhote, A.-P. Dias, M. Armand, N. Papageorgiou, K. Kalyanasundaram and M. Graetzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef CAS PubMed.
- Z. B. Zhou, H. Matsumoto and K. Tatsumi, Chem. – Eur. J., 2004, 10, 6581–6591 CrossRef CAS PubMed.
- K. Liu, Y.-X. Zhou, H.-B. Han, S.-S. Zhou, W.-F. Feng, J. Nie, H. Li, X.-J. Huang, M. Armand and Z.-B. Zhou, Electrochim. Acta, 2010, 55, 7145–7151 CrossRef CAS.
- S. Forsyth, J. Golding, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2001, 46, 1753–1757 CrossRef CAS.
- P. C. Howlett, D. R. MacFarlane and A. F. Hollenkamp, Electrochem. Solid-State Lett., 2004, 7, A97–A101 CrossRef CAS.
- P. C. Howlett, N. Brack, A. F. Hollenkamp, M. Forsyth and D. R. MacFarlane, J. Electrochem. Soc., 2006, 153, A595–A606 CrossRef CAS.
- Z. B. Zhou, H. Matsumoto and K. Tatsumi, Chem. – Eur. J., 2005, 11, 752–766 CrossRef CAS PubMed.
- R.-S. Kühnel, M. Lübke, M. Winter, S. Passerini and A. Balducci, J. Power Sources, 2012, 214, 178–184 CrossRef.
- M. Ishikawa, T. Sugimoto, M. Kikuta, E. Ishiko and M. Kono, J. Power Sources, 2006, 162, 658–662 CrossRef CAS.
- C. Liu, X. Ma, F. Xu, L. Zheng, H. Zhang, W. Feng, X. Huang, M. Armand, J. Nie, H. Chen and Z. Zhou, Electrochim. Acta, 2014, 149, 370–385 CrossRef CAS.
- L. Wu, A. Moretti, D. Buchholz, S. Passerini and D. Bresser, Electrochim. Acta, 2016, 203, 109–116 CrossRef CAS.
- L. G. Chagas, S. Jeong, I. Hasa and S. Passerini, ACS Appl. Mater. Interfaces, 2019, 11, 22278–22289 CrossRef CAS PubMed.
- S. Brutti, M. A. Navarra, G. Maresca, S. Panero, J. Manzi, E. Simonetti and G. B. Appetecchi, Electrochim. Acta, 2019, 306, 317–326 CrossRef CAS.
- A. Ponrouch and M. R. Palacin, Curr. Opin. Electrochem., 2018, 9, 1–7 CrossRef CAS.
- Z. Liu, T. Cui, G. Pulletikurthi, A. Lahiri, T. Carstens, M. Olschewski and F. Endres, Angew. Chem., Int. Ed., 2016, 55, 2889–2893 CrossRef CAS PubMed.
- T. J. Simons, D. R. MacFarlane, M. Forsyth and P. C. Howlett, ChemElectroChem, 2014, 1, 1688–1697 CrossRef CAS.
- Y. Lv, Y. Xiao, S. Xu, F. Huo, Y. Chen, M. Zhao, L. Liu, C. Su, Y. Zhu and S. Chen, J. Mater. Chem. A, 2022, 10, 16952–16961 RSC.
- N. Zhu, K. Zhang, F. Wu, Y. Bai and C. Wu, Energy Mater. Adv., 2021, 2021, 9204217 Search PubMed.
- T. Watkins, A. Kumar and D. A. Buttry, J. Am. Chem. Soc., 2016, 138, 641–650 CrossRef CAS PubMed.
- H. Zhang, L. Qiao and M. Armand, Angew. Chem., Int. Ed., 2022, e202214054 Search PubMed.
- W. Ren, D. Wu, Y. NuLi, X. Zhang, J. Yang and J. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 32957–32967 CrossRef CAS PubMed.
- M. Angell, C.-J. Pan, Y. Rong, C. Yuan, M.-C. Lin, B.-J. Hwang and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 834–839 CrossRef CAS PubMed.
- C. Li, J. Patra, J. Li, P. C. Rath, M.-H. Lin and J.-K. Chang, Adv. Funct. Mater., 2020, 30, 1909565 CrossRef CAS.
- A. J. Lucio, I. Efimov, O. N. Efimov, C. J. Zaleski, S. Viles, B. B. Ignatiuk, A. P. Abbott, A. R. Hillman and K. S. Ryder, Chem. Commun., 2021, 57, 9834–9837 RSC.
- L. Zhang, Q. Ma, G. Wang, Z. Liu and L. Zhang, J. Electroanal. Chem., 2021, 888, 115176 CrossRef CAS.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- J. Castillo, L. Qiao, A. Santiago, X. Judez, A. Sáenz de Buruaga, G. Jiménez-Martín, M. Armand, H. Zhang and C. Li, Energy Mater., 2022, 2, 200003 Search PubMed.
- L. Qiao, S. Rodriguez Peña, M. Martínez-Ibañez, A. Santiago, I. Aldalur, E. Lobato, E. Sanchez-Diez, Y. Zhang, H. Manzano, H. Zhu, M. Forsyth, M. Armand, J. Carrasco and H. Zhang, J. Am. Chem. Soc., 2022, 144, 9806–9816 CrossRef CAS PubMed.
- X. Qu, Y. Tang, A. Du, S. Dong and G. Cui, Chem. – Asian J., 2021, 16, 3272–3280 CrossRef CAS PubMed.
- H. Yin, C. Han, Q. Liu, F. Wu, F. Zhang and Y. Tang, Small, 2021, 17, 2006627 CrossRef CAS PubMed.
- H. Zhang and M. Armand, Isr. J. Chem., 2021, 61, 94–100 CrossRef CAS.
- N. Boaretto, L. Meabe, M. Martinez-Ibañez, M. Armand and H. Zhang, J. Electrochem. Soc., 2020, 167, 070524 CrossRef CAS.
- L. Qiao, X. Judez, T. Rojo, M. Armand and H. Zhang, J. Electrochem. Soc., 2020, 167, 070534 CrossRef CAS.
- I. Aldalur, M. Armand and H. Zhang, Batteries Supercaps, 2020, 3, 30–46 CrossRef CAS.
- D. Zhou, D. Shanmukaraj, A. Tkacheva, M. Armand and G. Wang, Chem, 2019, 5, 2326–2352 CAS.
- H. Zhang, U. Oteo, H. Zhu, X. Judez, M. Martinez-Ibañez, I. Aldalur, E. Sanchez-Diez, C. Li, J. Carrasco and M. Forsyth, Angew. Chem., Int. Ed., 2019, 58, 7829–7834 CrossRef CAS PubMed.
- K. Wu, J. Huang, J. Yi, X. Liu, Y. Liu, Y. Wang, J. Zhang and Y. Xia, Adv. Energy Mater., 2020, 10, 1903977 CrossRef CAS.
- X. Yu and A. Manthiram, Energy Storage Mater., 2020, 34, 282–300 CrossRef.
- https://www.bollore.com/en/activites-et-participations-2/stockage-delectricite-et-systemes/ .
- G. G. Eshetu, X. Judez, C. Li, M. Martinez-Ibañez, I. Gracia, O. Bondarchuk, J. Carrasco, L. M. Rodriguez-Martinez, H. Zhang and M. Armand, J. Am. Chem. Soc., 2018, 140, 9921–9933 CrossRef CAS PubMed.
- H. Zhang, U. Oteo, X. Judez, G. G. Eshetu, M. Martinez-Ibañez, J. Carrasco, C. Li and M. Armand, Joule, 2019, 3, 1689–1702 CrossRef CAS.
- U. Oteo, M. Martinez-Ibañez, I. Aldalur, E. Sanchez-Diez, J. Carrasco, M. Armand and H. Zhang, ChemElectroChem, 2019, 6, 1019–1022 CrossRef CAS.
- H. Zhang, F. Chen, O. Lakuntza, U. Oteo, L. Qiao, M. Martinez-Ibañez, H. Zhu, J. Carrasco, M. Forsyth and M. Armand, Angew. Chem., Int. Ed., 2019, 58, 12070–12075 CrossRef CAS PubMed.
- L. Qiao, U. Oteo, Y. Zhang, S. R. Peña, M. Martínez-Ibañez, A. Santiago, R. Cid, L. Meabe, H. Manzano, J. Carrasco, H. Zhang and M. Armand, Energy Storage Mater., 2020, 32, 225–233 CrossRef.
- M. Martinez-Ibañez, E. Sanchez-Diez, U. Oteo, I. Gracia, I. Aldalur, H. B. Eitouni, M. Joost, M. Armand and H. Zhang, Chem. Mater., 2022, 34, 3451–3460 CrossRef.
- J. Zhang, J. Zhao, L. Yue, Q. Wang, J. Chai, Z. Liu, X. Zhou, H. Li, Y. Guo, G. Cui and L. Chen, Adv. Energy Mater., 2015, 5, 1501082 CrossRef.
- K. Kimura, J. Motomatsu and Y. Tominaga, J. Phys. Chem. C, 2016, 120, 12385–12391 CrossRef CAS.
- B. Sun, J. Mindemark, K. Edström and D. Brandell, Solid State Ionics, 2014, 262, 738–742 CrossRef CAS.
- J. Mindemark, B. Sun, E. Törmä and D. Brandell, J. Power Sources, 2015, 298, 166–170 CrossRef CAS.
- I. Aldalur, H. Zhang, M. Piszcz, U. Oteo, L. M. Rodriguez-Martinez, D. Shanmukaraj, T. Rojo and M. Armand, J. Power Sources, 2017, 347, 37–46 CrossRef CAS.
- I. Aldalur, M. Martinez-Ibañez, M. Piszcz, L. M. Rodriguez-Martinez, H. Zhang and M. Armand, J. Power Sources, 2018, 383, 144–149 CrossRef CAS.
- I. Aldalur, M. Martinez-Ibañez, M. Piszcz, H. Zhang and M. Armand, Batteries Supercaps, 2018, 1, 149–159 CrossRef CAS.
- I. Aldalur, M. Martinez-Ibañez, A. Krztoń-Maziopa, M. Piszcz, M. Armand and H. Zhang, J. Power Sources, 2019, 423, 218–226 CrossRef CAS.
- I. Aldalur, X. Wang, A. Santiago, N. Goujon, M. Echeverría, M. Martínez-Ibáñez, M. Piszcz, P. C. Howlett, M. Forsyth, M. Armand and H. Zhang, J. Power Sources, 2019, 448, 227424 CrossRef.
- T. Dong, J. Zhang, G. Xu, J. Chai, H. Du, L. Wang, H. Wen, X. Zang, A. Du, Q. Jia, X. Zhou and G. Cui, Energy Environ. Sci., 2018, 11, 1197–1203 RSC.
- J. Mindemark, E. Törmä, B. Sun and D. Brandell, Polymer, 2015, 63, 91–98 CrossRef CAS.
- J. Serra Moreno, M. Armand, M. B. Berman, S. G. Greenbaum, B. Scrosati and S. Panero, J. Power Sources, 2014, 248, 695–702 CrossRef CAS.
- A. Boschin and P. Johansson, Electrochim. Acta, 2015, 175, 124–133 CrossRef CAS.
- X. Judez, H. Zhang, C. Li, J. A. González-Marcos, Z. Zhou, M. Armand and L. M. Rodriguez-Martinez, J. Phys. Chem. Lett., 2017, 8, 1956–1960 CrossRef CAS PubMed.
- C. Zhao, L. Liu, Y. Lu, M. Wagemaker, L. Chen and Y.-S. Hu, Angew. Chem., Int. Ed., 2019, 58, 17026–17032 CrossRef CAS PubMed.
- L. Liu, X. Qi, S. Yin, Q. Zhang, X. Liu, L. Suo, H. Li, L. Chen and Y.-S. Hu, ACS Energy Lett., 2019, 4, 1650–1657 CrossRef CAS.
- X. Qi, Q. Ma, L. Liu, Y.-S. Hu, H. Li, Z. Zhou, X. Huang and L. Chen, ChemElectroChem, 2016, 3, 1741–1745 CrossRef CAS.
- Q. Ma, J. Liu, X. Qi, X. Rong, Y. Shao, W. Feng, J. Nie, Y.-S. Hu, H. Li, X. Huang, L. Chen and Z. Zhou, J. Mater. Chem. A, 2017, 5, 7738–7743 RSC.
- C. Sångeland, R. Mogensen, D. Brandell and J. Mindemark, ACS Appl. Polym. Mater., 2019, 1, 825–832 CrossRef.
- J. Mindemark, R. Mogensen, M. J. Smith, M. M. Silva and D. Brandell, Electrochem. Commun., 2017, 77, 58–61 CrossRef CAS.
- C. Sångeland, R. Younesi, J. Mindemark and D. Brandell, Energy Storage Mater., 2019, 19, 31–38 CrossRef.
- J. Zheng, L. Schkeryantz, G. Gourdin, L. Qin and Y. Wu, ACS Appl. Energy Mater., 2021, 4, 4156–4164 CrossRef CAS.
- S. Trano, F. Corsini, G. Pascuzzi, E. Giove, L. Fagiolari, J. Amici, C. Francia, S. Turri, S. Bodoardo, G. Griffini and F. Bella, ChemSusChem, 2022, 15, e202200294 CrossRef CAS PubMed.
- M. Hamada, R. Tatara, K. Kubota, S. Kumakura and S. Komaba, ACS Energy Lett., 2022, 7, 2244–2246 CrossRef CAS.
- H. Fei, Y. Liu, Y. An, X. Xu, G. Zeng, Y. Tian, L. Ci, B. Xi, S. Xiong and J. Feng, J. Power Sources, 2018, 399, 294–298 CrossRef CAS.
- H. Gao, L. Xue, S. Xin and J. B. Goodenough, Angew. Chem., Int. Ed., 2018, 57, 5449–5453 CrossRef CAS PubMed.
- B. Park and J. L. Schaefer, J. Electrochem. Soc., 2020, 167, 070545 CrossRef.
- Y. Shao, N. N. Rajput, J. Hu, M. Hu, T. Liu, Z. Wei, M. Gu, X. Deng, S. Xu, K. S. Han, J. Wang, Z. Nie, G. Li, K. R. Zavadil, J. Xiao, C. Wang, W. A. Henderson, J.-G. Zhang, Y. Wang, K. T. Mueller, K. Persson and J. Liu, Nano Energy, 2015, 12, 750–759 CrossRef CAS.
- A. Du, H. Zhang, Z. Zhang, J. Zhao, Z. Cui, Y. Zhao, S. Dong, L. Wang, X. Zhou and G. Cui, Adv. Mater., 2019, 31, 1805930 CrossRef PubMed.
- L. Wang, Z. Li, Z. Meng, Y. Xiu, B. Dasari, Z. Zhao-Karger and M. Fichtner, Energy Storage Mater., 2022, 48, 155–163 CrossRef.
- S. Biria, S. Pathreeker, F. S. Genier and I. D. Hosein, ACS Appl. Polym. Mater., 2020, 2, 2111–2118 CrossRef CAS.
- J. Asenbauer, N. Ben Hassen, B. D. McCloskey and J. M. Prausnitz, Electrochim. Acta, 2017, 247, 1038–1043 CrossRef CAS.
- H. Zhao, N. Deng, W. Kang, Z. Li, G. Wang and B. Cheng, Energy Storage Mater., 2020, 26, 334–348 CrossRef.
- W. P. Hagan, R. J. Latham, R. G. Linford and S. L. Vickers, Solid State Ionics, 1994, 70–71, 666–669 CrossRef CAS.
- M. M. Silva, M. J. Smith and P. Lightfoot, Port. Electrochim. Acta, 1999, 17, 3–10 CrossRef CAS.
- Z. Zhao, J. Wang, Z. Lv, Q. Wang, Y. Zhang, G. Lu, J. Zhao and G. Cui, Chem. Eng. J., 2021, 417, 128096 CrossRef CAS.
- L. Ma, S. Chen, X. Li, A. Chen, B. Dong and C. Zhi, Angew. Chem., Int. Ed., 2020, 59, 23836–23844 CrossRef CAS PubMed.
- C. Li, C.-C. Hou, L. Chen, S. Kaskel and Q. Xu, Energy Chem., 2021, 3, 100049 CrossRef CAS.
- Z. Liu, H. Du, Y. Cui, L. Du, Z. Zhao, X. Wang, Z. Lv, M. Sun, Z. Liu, K. Li, G. Zhang, M.-C. Lin and G. Cui, J. Power Sources, 2021, 497, 229839 CrossRef CAS.
- Z. Yu, S. Jiao, S. Li, X. Chen, W.-L. Song, T. Teng, J. Tu, H.-S. Chen, G. Zhang and D.-N. Fang, Adv. Funct. Mater., 2019, 29, 1806799 CrossRef.
- Z. Huang, W.-L. Song, Y. Liu, W. Wang, M. Wang, J. Ge, H. Jiao and S. Jiao, Adv. Mater., 2021, 34, 2104557 CrossRef PubMed.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047–2051 CrossRef CAS.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- C. Villevieille, Adv. Mater. Interfaces, 2022, 9, 2101865 CrossRef CAS.
- S. Trasatti and R. Parsons, Pure Appl. Chem., 1986, 58, 437–454 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, 2nd edn, Wiley-VCH, 2008 Search PubMed.
- D. L. Wood, J. Li and C. Daniel, J. Power Sources, 2015, 275, 234–242 CrossRef CAS.
- D. J. Siegel, L. Nazar, Y.-M. Chiang, C. Fang and N. P. Balsara, Trends Chem., 2021, 3, 807–818 CrossRef CAS.
- S. K. Vineeth, C. B. Soni, Y. Sun, V. Kumar and Z. W. Seh, Trends Chem., 2022, 4, 48–59 CrossRef CAS.
- X. Chen, X. Shen, T.-Z. Hou, R. Zhang, H.-J. Peng and Q. Zhang, Chem, 2020, 6, 2242–2256 CAS.
- P. Kulkarni, D. Ghosh and R. G. Balakrishna, Sustainable Energy Fuels, 2021, 5, 1619–1654 RSC.
- A. Khanmohammadi and F. Ravari, Phys. Chem. Res., 2017, 5, 57–68 CAS.
- N. Kumar and D. J. Siegel, J. Phys. Chem. Lett., 2016, 7, 874–881 CrossRef CAS PubMed.
- M. Angel Munoz-Marquez, M. Zarrabeitia, S. Passerini and T. Rojo, Adv. Mater. Interfaces, 2022, 9, 2101773 CrossRef.
- J.-i Aihara, Phys. Chem. Chem. Phys., 2000, 2, 3121–3125 RSC.
- P. Peljo and H. H. Girault, Energy Environ. Sci., 2018, 11, 2306–2309 RSC.
- X. Chen, H.-R. Li, X. Shen and Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 16643–16647 CrossRef CAS PubMed.
- H. Wang, D. Zhai and F. Kang, Energy Environ. Sci., 2020, 13, 4583–4608 RSC.
- M. Okoshi, Y. Yamada, A. Yamada and H. Nakai, J. Electrochem. Soc., 2013, 160, A2160–A2165 CrossRef CAS.
- Y. Matsuda, H. Nakashima, M. Morita and Y. Takasu, J. Electrochem. Soc., 1981, 128, 2552–2556 CrossRef CAS.
- M. Okoshi, Y. Yamada, S. Komaba, A. Yamada and H. Nakai, J. Electrochem. Soc., 2016, 164, A54–A60 CrossRef.
- W. Liu, P. Liu and D. Mitlin, Adv. Energy Mater., 2020, 10, 2002297 CrossRef CAS.
- Z. Lu, A. Schechter, M. Moshkovich and D. Aurbach, J. Electroanal. Chem., 1999, 466, 203–217 CrossRef CAS.
- T. Gao, S. Hou, K. Huynh, F. Wang, N. Eidson, X. Fan, F. Han, C. Luo, M. Mao, X. Li and C. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 14767–14776 CrossRef CAS PubMed.
- I. Shterenberg, M. Salama, H. D. Yoo, Y. Gofer, J.-B. Park, Y.-K. Sun and D. Aurbach, J. Electrochem. Soc., 2015, 162, A7118–A7182 CrossRef CAS.
- J. G. Connell, B. Genorio, P. P. Lopes, D. Strmcnik, V. R. Stamenkovic and N. M. Markovic, Chem. Mater., 2016, 28, 8268–8277 CrossRef CAS.
- N. N. Rajput, X. Qu, N. Sa, A. K. Burrell and K. A. Persson, J. Am. Chem. Soc., 2015, 137, 3411–3420 CrossRef CAS PubMed.
- Z. Zhao-Karger, X. Zhao, D. Wang, T. Diemant, R. J. Behm and M. Fichtner, Adv. Energy Mater., 2015, 5, 1401155 CrossRef.
- X. Yu and A. Manthiram, Chem. Mater., 2016, 28, 896–905 CrossRef.
- A. J. Smith, J. C. Burns, X. Zhao, D. Xiong and J. R. Dahn, J. Electrochem. Soc., 2011, 158, A447–A452 CrossRef CAS.
- Z. Wang, Z. Sun, J. Li, Y. Shi, C. Sun, B. An, H.-M. Cheng and F. Li, Chem. Soc. Rev., 2021, 50, 3178–3210 RSC.
- E. N. Keyzer, H. F. J. Glass, Z. Liu, P. M. Bayley, S. E. Dutton, C. P. Grey and D. S. Wright, J. Am. Chem. Soc., 2016, 138, 8682–8685 CrossRef CAS PubMed.
- G. G. Eshetu, S. Grugeon, H. Kim, S. Jeong, L. Wu, G. Gachot, S. Laruelle, M. Armand and S. Passerini, ChemSusChem, 2016, 9, 462–471 CrossRef CAS PubMed.
- A. W. Ells, R. May and L. E. Marbella, ACS Appl. Mater. Interfaces, 2021, 13, 53841–53849 CrossRef CAS PubMed.
- H. Yoshida, T. Fukunaga, T. Hazama, M. Terasaki, M. Mizutani and M. Yamachi, J. Power Sources, 1997, 68, 311–315 CrossRef CAS.
- K. Tasaki and S. J. Harris, J. Phys. Chem. C, 2010, 114, 8076–8083 CrossRef CAS.
- L. A. Ma, A. J. Naylor, L. Nyholm and R. Younesi, Angew. Chem., Int. Ed., 2021, 60, 4855–4863 CrossRef CAS PubMed.
- Z. Tian, Y. Zou, G. Liu, Y. Wang, J. Yin, J. Ming and H. N. Alshareef, Adv. Sci., 2022, 9, 2201207 CrossRef CAS PubMed.
- T. Hosaka and S. Komaba, Bull. Chem. Soc. Jpn., 2022, 95, 569–581 CrossRef CAS.
- D. I. Iermakova, R. Dugas, M. R. Palacín and A. Ponrouch, J. Electrochem. Soc., 2015, 162, A7060–A7066 CrossRef CAS.
- J. D. McBrayer, C. A. Apblett, K. L. Harrison, K. R. Fenton and S. D. Minteer, Nanotechnology, 2021, 32, 502005 CrossRef CAS PubMed.
- Y. Gao, X. Du, Z. Hou, X. Shen, Y.-W. Mai, J.-M. Tarascon and B. Zhang, Joule, 2021, 5, 1860–1872 CrossRef CAS.
- Y. Xu, K. Dong, Y. Jie, P. Adelhelm, Y. Chen, L. Xu, P. Yu, J. Kim, Z. Kochovski, Z. Yu, W. Li, J. LeBeau, Y. Shao-Horn, R. Cao, S. Jiao, T. Cheng, I. Manke and Y. Lu, Adv. Energy Mater., 2022, 12, 2200398 CrossRef.
- G. G. Eshetu, S. Grugeon, G. Gachot, D. Mathiron, M. Armand and S. Laruelle, Electrochim. Acta, 2013, 102, 133–141 CrossRef CAS.
- L. Zheng, H. Zhang, P. Cheng, Q. Ma, J. Liu, J. Nie, W. Feng and Z. Zhou, Electrochim. Acta, 2016, 196, 169–188 CrossRef CAS.
- Z. Song, L. Zheng, P. Cheng, X. Wang, H. Wu, Q. Ma, J. Liu, W. Feng, J. Nie, H. Yu, X. Huang, M. Armand, H. Zhang and Z. Zhou, J. Power Sources, 2022, 526, 231105 CrossRef CAS.
- M. Jäckle and A. Groß, J. Chem. Phys., 2014, 141, 174710 CrossRef PubMed.
- M. Jäckle, K. Helmbrecht, M. Smits, D. Stottmeister and A. Groß, Energy Environ. Sci., 2018, 11, 3400–3407 RSC.
- M. Jäckle and A. Groß, J. Chem. Phys., 2019, 151, 234707 CrossRef PubMed.
- H. Yildirim, A. Kinaci, M. K. Chan and J. P. Greeley, ACS Appl. Mater. Interfaces, 2015, 7, 18985–18996 CrossRef CAS PubMed.
- X. He, Y. Zhu and Y. Mo, Nat. Commun., 2017, 8, 15893 CrossRef CAS PubMed.
- B. Lee, E. Paek, D. Mitlin and S. W. Lee, Chem. Rev., 2019, 119, 5416–5460 CrossRef CAS PubMed.
- F. A. Soto, P. Yan, M. H. Engelhard, A. Marzouk, C. Wang, G. Xu, Z. Chen, K. Amine, J. Liu, V. L. Sprenkle, F. El-Mellouhi, P. B. Balbuena and X. Li, Adv. Mater., 2017, 29, 1606860 CrossRef PubMed.
- Q. Zhao, S. Stalin and L. A. Archer, Joule, 2021, 5, 1119–1142 CrossRef CAS.
- F. A. Soto, A. Marzouk, F. El-Mellouhi and P. B. Balbuena, Chem. Mater., 2018, 30, 3315–3322 CrossRef CAS.
- R. Shannon, Acta Crystallogr., Sect. A: Found. Crystallogr., 1976, 32, 751–767 CrossRef.
- Z. W. B. Iton and K. A. See, Chem. Mater., 2022, 34, 881–898 CrossRef CAS.
- J. Popovic, Energy Technol., 2021, 9, 2001056 CrossRef CAS.
- Z. Rong, R. Malik, P. Canepa, G. Sai Gautam, M. Liu, A. Jain, K. Persson and G. Ceder, Chem. Mater., 2015, 27, 6016–6021 CrossRef CAS.
- T. Nestler, F. Meutzner, A. A. Kabanov, M. Zschornak, T. Leisegang and D. C. Meyer, Chem. Mater., 2019, 31, 737–747 CrossRef CAS.
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Chem. Mater., 2016, 28, 266–273 CrossRef CAS.
- N. Mahne, S. E. Renfrew, B. D. McCloskey and S. A. Freunberger, Angew. Chem., Int. Ed., 2018, 57, 5529–5533 CrossRef CAS PubMed.
- A. T. S. Freiberg, J. Sicklinger, S. Solchenbach and H. A. Gasteiger, Electrochim. Acta, 2020, 346, 136271 CrossRef CAS.
- R. L. Sacci, J. M. Black, N. Balke, N. J. Dudney, K. L. More and R. R. Unocic, Nano Lett., 2015, 15, 2011–2018 CrossRef CAS PubMed.
- T. Placke, G. G. Eshetu, M. Winter and E. Figgemeier, Lithium-ion Batteries Enabled by Silicon Anodes, Institution of Engineering and Technology, 2021, pp 349–404 Search PubMed.
- G. G. Eshetu, H. Zhang, X. Judez, H. Adenusi, M. Armand, S. Passerini and E. Figgemeier, Nat. Commun., 2021, 12, 5459 CrossRef CAS PubMed.
- R. Dugas, J. D. Forero-Saboya and A. Ponrouch, Chem. Mater., 2019, 31, 8613–8628 CrossRef CAS PubMed.
- J. Forero-Saboya, C. Bodin and A. Ponrouch, Electrochem. Commun., 2021, 124, 106936 CrossRef CAS.
- X. Deng, L. Li, G. Zhang, X. Zhao, J. Hao, C. Han and B. Li, Energy Storage Mater., 2022, 53, 467–481 CrossRef.
- Q. Wei, L. Zhang, X. Sun and T. L. Liu, Chem. Sci., 2022, 13, 5797–5812 RSC.
- Z. Hou, R. Zhou, Y. Yao, Z. Min, Z. Lu, Y. Zhu, J.-M. Tarascon and B. Zhang, Angew. Chem., Int. Ed., 2022, e202214796 Search PubMed.
- S. Komaba, T. Ishikawa, N. Yabuuchi, W. Murata, A. Ito and Y. Ohsawa, ACS Appl. Mater. Interfaces, 2011, 3, 4165–4168 CrossRef CAS PubMed.
- H. Yang, C.-Y. Chen, J. Hwang, K. Kubota, K. Matsumoto and R. Hagiwara, ACS Appl. Mater. Interfaces, 2020, 12, 36168–36176 CrossRef CAS PubMed.
- R. Bhattacharyya, B. Key, H. Chen, A. S. Best, A. F. Hollenkamp and C. P. Grey, Nat. Mater., 2010, 9, 504–510 CrossRef CAS PubMed.
- J. Z. Lee, T. A. Wynn, M. A. Schroeder, J. Alvarado, X. Wang, K. Xu and Y. S. Meng, ACS Energy Lett., 2019, 4, 489–493 CrossRef CAS.
- D. H. S. Tan, A. Banerjee, Z. Chen and Y. S. Meng, Nat. Nanotechnol., 2020, 15, 170–180 CrossRef CAS PubMed.
- X. Liu, Z. Liang, Y. Xiang, M. Lin, Q. Li, Z. Liu, G. Zhong, R. Fu and Y. Yang, Adv. Mater., 2021, 33, 2005878 CrossRef CAS PubMed.
- Y. Zhou, M. Su, X. Yu, Y. Zhang, J.-G. Wang, X. Ren, R. Cao, W. Xu, D. R. Baer, Y. Du, O. Borodin, Y. Wang, X.-L. Wang, K. Xu, Z. Xu, C. Wang and Z. Zhu, Nat. Nanotechnol., 2020, 15, 224–230 CrossRef CAS PubMed.
- Y. Chu, Y. Shen, F. Guo, X. Zhao, Q. Dong, Q. Zhang, W. Li, H. Chen, Z. Luo and L. Chen, Electrochem. Energy Rev., 2020, 3, 187–219 CrossRef.
- Y. Xiang, X. Li, Y. Cheng, X. Sun and Y. Yang, Mater. Today, 2020, 36, 139–157 CrossRef CAS.
- J. Scharf, M. Chouchane, D. P. Finegan, B. Lu, C. Redquest, M.-C. Kim, W. Yao, A. A. Franco, D. Gostovic, Z. Liu, M. Riccio, F. Zelenka, J.-M. Doux and Y. S. Meng, Nat. Nanotechnol., 2022, 17, 446–459 CrossRef CAS PubMed.
- Z. Gong and Y. Yang, J. Energy Chem., 2018, 27, 1566–1583 CrossRef.
- Z. Zhang, X. Yang, P. Li, Y. Wang, X. Zhao, J. Safaei, H. Tian, D. Zhou, B. Li, F. Kang and G. Wang, Adv. Mater., 2022 DOI:10.1002/adma.202206970.
- A. A. Franco, A. Rucci, D. Brandell, C. Frayret, M. Gaberscek, P. Jankowski and P. Johansson, Chem. Rev., 2019, 119, 4569–4627 CrossRef CAS PubMed.
- O. Borodin, Curr. Opin. Electrochem., 2019, 13, 86–93 CrossRef CAS.
- H. Zhang, F. Chen and J. Carrasco, Energy Storage Mater., 2021, 36, 77–90 CrossRef.
- F. Chen, Curr. Opin. Electrochem., 2022, 35, 101086 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |