
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow membrane characteristics influence the performance of CO2 and CO electrolysis†
Sahil
Garg
a,
Carlos A.
Giron Rodriguez
a,
Thomas E.
Rufford
 b,
John R.
Varcoe
c and
Brian
Seger
*a
b,
John R.
Varcoe
c and
Brian
Seger
*a
aSurface Physics and Catalysis (Surf Cat) Section, Department of Physics, Technical University of Denmark, 2800 Kgs. Lyngby, Denmark. E-mail: brse@fysik.dtu.dk
bSchool of Chemical Engineering, The University of Queensland, St Lucia, 4072, Brisbane, Queensland, Australia
cDepartment of Chemistry, University of Surrey, Guildford, UK
First published on 15th September 2022
Abstract
Due to the ability to produce sustainably carbon-based chemicals and fuels, CO2 electrolysis and the closely related CO electrolysis are advancing rapidly from fundamental studies toward industrial applications. Many near-room temperature CO2 and CO electrolysis (CO(2)E) technologies adopt features from proton exchange membrane fuel cells and H2 electrolyzers. However, CO(2)E's selectivity and overall performance are highly sensitive to a multitude of parameters, adding an extra degree of complexity. One often-overlooked parameter in optimizing these devices is the ion exchange membranes (IEM). Here we critically review the IEM performance variables of most relevance to CO(2)E, which leads to identifying several parameters in need of substantial more scientific understanding. We begin with a summary of the working principles of the three main IEM types for CO(2)E, then focus on anion exchange membranes (AEM) since AEMs provide the most favorable local alkaline environment for CO(2)E at the cathode. Critical issues for AEMs in CO2E include (i) ion and water transport in the membrane, (ii) ionic conductivity, and (iii) chemical stability. We conclude with an overview of the state-of-the-art IEM reported in high current density (j ≥ 100 mA cm−2) CO2 and CO electrolysis devices.
Broader contextCO2 and CO electrolysis has progressed beyond fundamental catalytic studies towards developing industrially scalable devices able to produce high-value chemicals sustainably. While much work has focused on the catalysis of this process, this work reviews the membrane influence on device performance, which is an important, but often unheralded part of CO2 and CO electrolysis. Membranes contribute substantially to energy efficiency losses within the device and can create environments that affect catalytic selectivity. Thus, only by understanding all parts of a CO2 electrolysis device can one fully understand and thus optimize device performance. As this work demonstrates, CO2 electrolysis is substantially more complex than water electrolysis, yet the insight gained from the review and analysis in this work additionally does provide an analysis approach towards even more complex electrosynthesis reactions that are aiming for upscaling towards industrial applications. |
Introduction
CO2 electrolysis (CO2E) is poised to play a pivotal role in advancing and decarbonizing the energy and chemical manufacturing sectors by transforming renewable energy sources such as solar or wind into fuels and chemicals.1 This technology has the added benefit of recovering the cost from energy-intensive CO2 capture processes by valorizing the CO2-concentrated industrial waste streams into useful products (e.g., CO or hydrocarbons).2 Compared to high-temperature CO2 utilization processes, using low-temperature (i.e., < 100 °C) and pressure, CO2E allows for reduced infrastructure costs compared to competing high-temperature (700–900 °C) solid-oxide-based CO2 electrolysis.3 Furthermore, CO being the first stable intermediate in CO2E, CO electrolysis (COE) allows for either the flexibility of a tandem CO2/CO electrolysis device or simply procuring CO derived from biomass.4While low-temperature water electrolysis to green H2 is rapidly becoming commercialized and implemented in society, there are parameters specific to CO2E and COE that are considerably more challenging. The gaseous reactant stream, non-selective catalysts, and formation of carbonate species represent additional challenges on top of developing active catalysts and designing devices that can achieve high current densities, high energy efficiencies, and durable operation over thousands of hours.5–7 These have led to different device designs as well as the need for new understandings in terms of gas diffusion electrodes (GDE) and ion-exchange membranes (IEM).
This review focuses on an analysis of membranes in relation to the application in CO2E and COE (together denoted as CO(2)E), with a primary emphasis on anion exchange membranes (AEM). However, device designs and GDEs will be introduced to provide the necessary context.
The most mature CO(2)E technologies adapt materials and engineering features from other electrochemical devices such as polymer electrolyte membrane (PEM) fuel cells8 and chlor-alkali electrolyzers for chlorine production.9 CO(2)E devices that follows most closely to the aforementioned industrial processes use a membrane electrode assembly (MEA) sandwiched between a cathode and anode, as shown in Fig. 1a.10 The MEA configuration is commonly referred to as a zero-gap electrolyzer and the actual membrane can either be an AEM, cation exchange (CEM), or bipolar membrane (BPM). The compressed nature of an MEA means the local environment imparted by the membrane can have a substantial effect on catalytic activity and device performance.
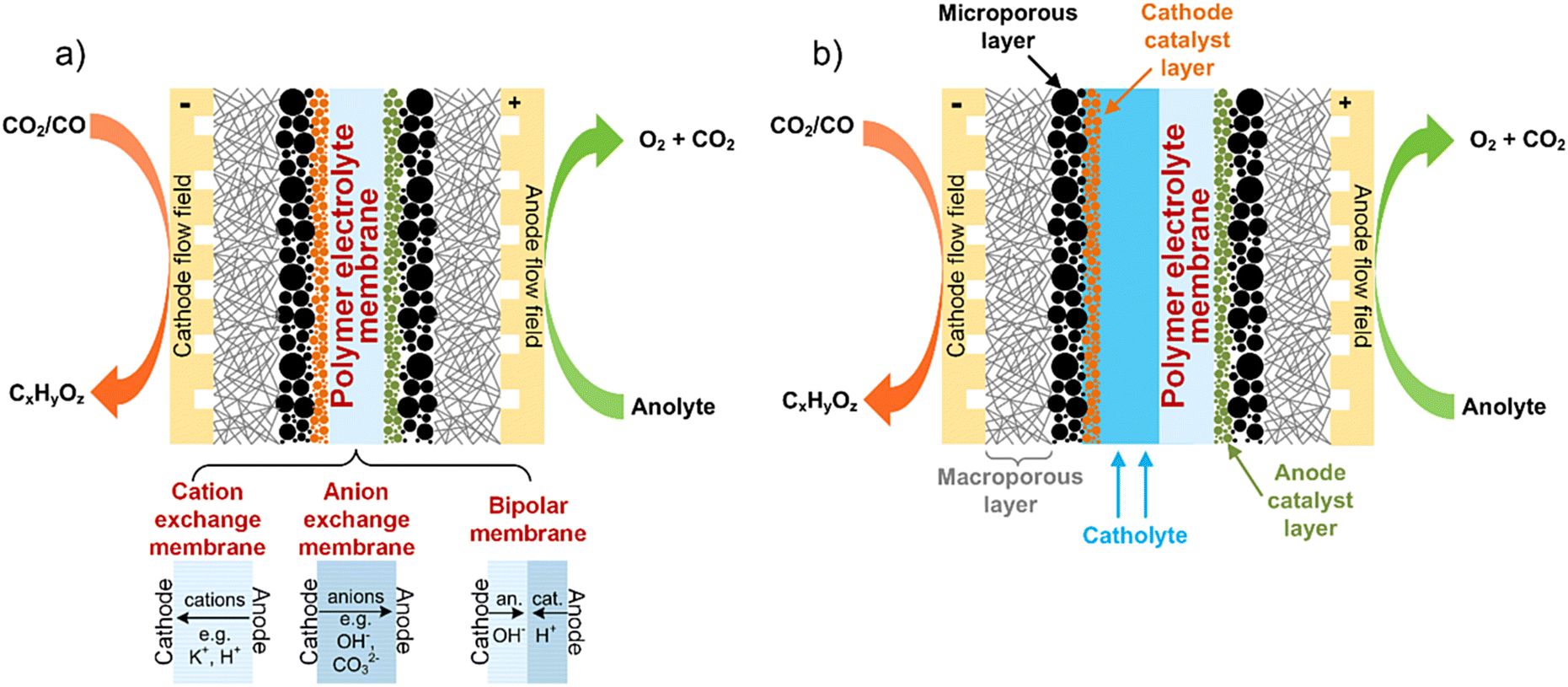 | ||
| Fig. 1 Schematic of a typical (a) zero-gap and (b) catholyte-based electrolyzer for CO2 and CO electrolysis. The three membrane types shown in (a) could also be used in (b) electrolyzer design. | ||
However with CO(2)E producing liquid products, alternative design variations (cf. an MEA type) have also been studied that entails circulating a catholyte in a chamber between the cathode and membrane Fig. 1b).3 The commercial viability of the catholyte approach is restricted by the large ohmic drop across the catholyte (and thus lower energy efficiency); however, it does have the advantage of providing a controllable environment around the membrane that helps when focusing on GDE optimizations. Additionally, the catalyst does not feel the local environment of the membrane, thus this dilutes the impact of what type of membrane is used.
To maximize the efficiency of the membrane there are a substantial number of characteristics that need to be optimized for CO(2)E devices, with the major parameters being:
1. Maximizing ionic conductivity (with whatever conductive ions are predominant).
2. Minimizing electrical conductivity.
3. Optimizing water transport (amount and diffusion kinetics).
4. Inhibiting crossover of undesired species across the membrane.
5. Maximizing mechanical and chemical robustness.
6. Providing a local pH environment at the cathode optimal CO(2)E catalysis (i.e. basic environment).
Only by optimizing all these parameters, can membranes for CO(2)E devices have the same success that membranes have had in the fuel cell, water electrolysis, and chlor-alkali fields. However, the final point is quite difficult for CEMs to achieve; hence this review will focus extensively on AEMs.
Recently, numerous excellent reviews have been published, focusing mainly on catalysts for CO(2)E,1,11–22 electrolyzer design including membranes,23,24 GDEs,25–27 electrolytes,3,28,29 and engineering issues.30,31 This review, on the other hand, aims to demonstrate how physical properties (e.g., water uptake, membrane hydration, ionic conductivity, ion selectivity), chemical stabilities, and structures (including polymer backbone and cationic head-group functionalities) can affect the ion and water transport across an AEM, and ultimately, the CO(2)E performance (e.g., energy efficiency, selectivity, and operando durability and performance stability). Herein, we provide the background to the fundamental challenges associated with the current state-of-the-art AEM-based CO(2)E electrolyzers and the general design approaches that have been employed to address these challenges.
Figures of merit to describe membrane properties
To aid our discussion of membrane functions, properties, and performance, we first define key figures of merit.The ion-exchange capacity (IEC) is the number of ion equivalents per unit mass of the dry polymer (eq. g−1), which is equal to the mol g−1 anions if the anions are monovalent (e.g., Cl− or OH−).32 It is most often directly measured using various titration techniques, but it can also be estimated using spectroscopic data. The IEC will vary as a function of the anion, as the mass of the dry AEM is a function of the mass of the anion(s) present – it is vital to transparently state the anion form of the AEM when it is weighed (it is bad practice to weigh the AEM directly after titration where the anion-form present is not controlled and where other contaminants may be present, e.g., AgCl particles if an AgNO3 titrant was used). Equivalent weight (EW) is simply the property that is inversely proportional to the IEC. Generally, the goal is to have a high IEC, but this is not necessarily a direct indicator of conductivity as other issues such as insufficiently developed conductive channels between ionic domains or excessive water swelling may lower conductivity.32,33
The ionic conductivity (σ) is an intrinsic property that is directly proportional to both the intrinsic mobility of a given ion (μion) and the concentration within the conductive channels (cion). It relates to the through-plane membrane resistance (Rmem), the active contact area (A) of the membrane sandwiched between the electrodes, and the thickness of the membrane (Lmem) using eqn (1):
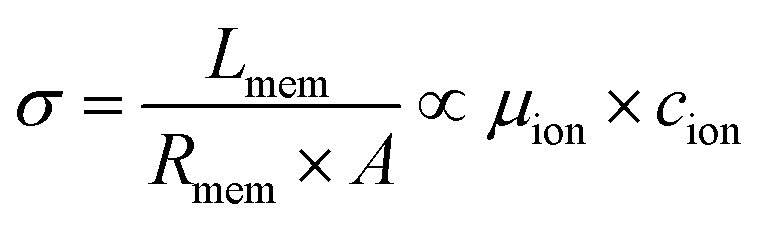 | (1) |
It is important to note that Lmem and μion, and cion (hence σ) are all functions of the AEM's hydration level (and IEC), thus complicating the analysis.
Accurate characterization of the water content in an AEM is essential since this facilitates anion transport in an AEM. To quantify this, the hydration number or water uptake (λ) is often used, which is defined as the average number of H2O molecules per ion-exchange site (per anion for monovalent anions),34 which is calculated using eqn (2). For each type of AEM, λ is a function of the environment that an AEM is exposed to, the thermo-mechanical history of the AEM, as well as the anion present. In addition, the water content of the AEM is often expressed as a gravimetric water uptake, which is the mass of water absorbed by the hydrated AEM (masswater) divided by the mass of the dehydrated AEM sample. Note that both λ and WU are average bulk properties and do not give you information on the nano/micro distribution or the state/nature of the water: bulk, free, and bound (sometimes misleadingly described as “freezing” and “non-freezing” water).35,36 New THz-based techniques are now being developed to allow the states of water in IEMs to be more fully characterized.37
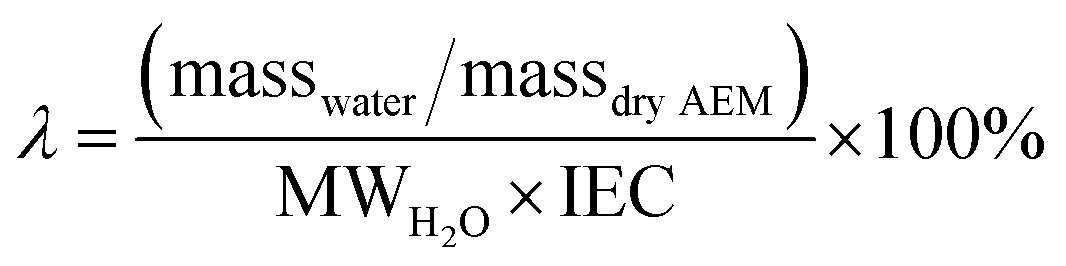 | (2) |
The degree of swelling is the degree to which the AEM (area, thickness, or volume) changes with hydration. Although high water content can increase ionic conductivity and lead to higher chemical stabilities in alkali solutions (OH− anions with full hydration shells are less nucleophilic compared to OH−s that are less hydrated), excessive water-uptake can lead to excessive swelling, which leads to mechanically weaker AEMs and/or lower conductivities (diluted concentration of charge carriers – lower cion).38 The degree of swelling (S) in terms of thickness (through-plane) can be estimated (providing hydrated (th) and dehydrated (td) thickness are available) as follows:
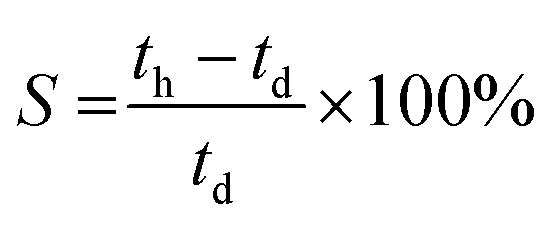 | (3) |
Role of ion-exchange membrane types
Among different membranes used in CO(2)E electrolyzers, AEM typically provides an alkaline environment if they are transferring hydroxide (OH−), carbonate (CO32−), and bicarbonate (HCO3−) to a lesser extent, whereas, the CEMs typically provide either an acidic environment with H+, or a more neutral environment with K+, Cs+, etc. As long as the catalyst is in close proximity to the membrane (or ionomer), this will influence the localized pH at the catalyst surface. Conversely, if the CO2E catalyst is spatially distant from the polymer electrolyte/membrane, the catalyst will only experience the bulk aqueous electrolyte (that has penetrated the catalyst layer). Thus catalytic activity will be independent of membrane type.39 However an increasing distance between catalyst and membrane significantly increases ohmic resistance (Fig. 1b).40 Thus it is essential to have the catalyst as close to the membrane as possible, entailing that catalysts will generally need to cope with the local environment that is affected by the presence of a membrane.From a catalysis standpoint, the rate-limiting step for CO2E is well established to be related to a non-proton coupled electron transfer (n-PCET) step,1 whereas the concomitant O2 evolution half-reaction is limited by a PCET step.41 Because of the n-PCET step, CO2E favors a more alkaline pH to reduce overpotentials (supplement to the equilibrium potentials, see Table 1) at the cathode. Furthermore, aqueous-based CO2E always competes with H2 evolution (HER) at the cathode, which has a PCET rate-limiting step, thus operating in alkaline conditions has the added advantage of shifting selectivity to CO2E versus HER. Thus, the local alkaline environments that AEMs provide greatly help in promoting activity and selectivity for CO2E.
| CO2E reaction | Equilibrium standard potential (E°, V vs. RHE) |
|---|---|
| a pKa formic acid: 3.75. b pKa acetic acid: 4.76. | |
| CO2 + H2O + 2e− → HCOO− + OH− | −0.20 (for pH < 4), −0.20 + 0.059*[pH-4] (for pH > 4)a |
| CO2 + H2O + 2e− → CO + 2OH− | −0.11 |
| CO2 + 6H2O + 8e− → CH4 + 8OH− | 0.17 |
| 2CO2 + 5H2O + 8e− → CH3COO− + 7OH− | 0.07 (for pH < 5), 0.07 + 0.059*[pH-5] (for pH > 5)b |
| 2CO2 + 8H2O + 12e− → C2H4 + 12OH− | 0.08 |
| 2CO2 + 9H2O + 12e− → C2H5OH + 12OH− | 0.09 |
| HER | |
| 2H2O + 2e− → H2 + 2OH− | 0 |
| OER | |
| 2H2O → O2(g) + 4H+ + 4e− | 1.23 |
| CO electrolysis (COE) reactions | |
| 2CO + 3H2O + 4e− → CH3COO− + 3OH− | 0.45 |
| CO + 5H2O + 6e− → CH4 + 6OH− | 0.26 |
| 2CO + 6H2O + 8e− → C2H4(g) + 8OH− | 0.17 |
| 2CO + 7H2O + 8e− → C2H5OH(aq) + 8OH− | 0.19 |
Despite the promise of AEM-based CO2E electrolyzers, many substantial technical challenges persist with one of the biggest issues relating to the CO2 reduction half-reaction itself. Reduction reactions (either CO2E, COE, or the competing HER) stoichiometrically produce OH− ions at the cathode interface, as shown in Table 1. These OH− ions are energetically favored to further convert incoming CO2 into HCO3−/CO32− as described in eqn (4) and (5). Unfortunately, these reactions occur almost immediately with rate constants for eqn (4) and (5): 2.23 × 103 M−1 s−1 and 6 × 109 M−1 s−1, respectively.42
| CO2 + OH− ⇌ HCO3− (pKa = 7.8*) | (4) |
| OH− + HCO3− ⇌ CO32− (pKa = 10.3) | (5) |
Furthermore, the transport of CO2-derived HCO3−/CO32− and CO2E products (e.g., as formate, and ethanol) through the AEM from the cathode to the anode reduces the overall conversion of CO2E electrolyzers (Fig. 2). The issue of having (bi)carbonate passing through the AEM is compounded by the fact that the anodic half-reaction is typically water oxidation producing O2 and H+. The acidic nature of protons shifts the carbonate-CO2 equilibrium back to gaseous CO2, thus the anode emits CO2 along with the O2.43–46 Stoichiometrically, every O2 evolved produces four protons, and thus this allows two carbonates to neutralize into CO2 per every O2 produced (or four bicarbonates). Having an electrochemical process with an anode emitting a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CO2:O2 ratio gaseous effluent is counter to the rationale for using CO2E systems and is highly wasteful of the CO2 reactant (lowers conversion efficiencies).47–49 Resolving this issue is a daunting task and is noted as the most significant barrier to commercializing CO2E.50
:1 CO2:O2 ratio gaseous effluent is counter to the rationale for using CO2E systems and is highly wasteful of the CO2 reactant (lowers conversion efficiencies).47–49 Resolving this issue is a daunting task and is noted as the most significant barrier to commercializing CO2E.50
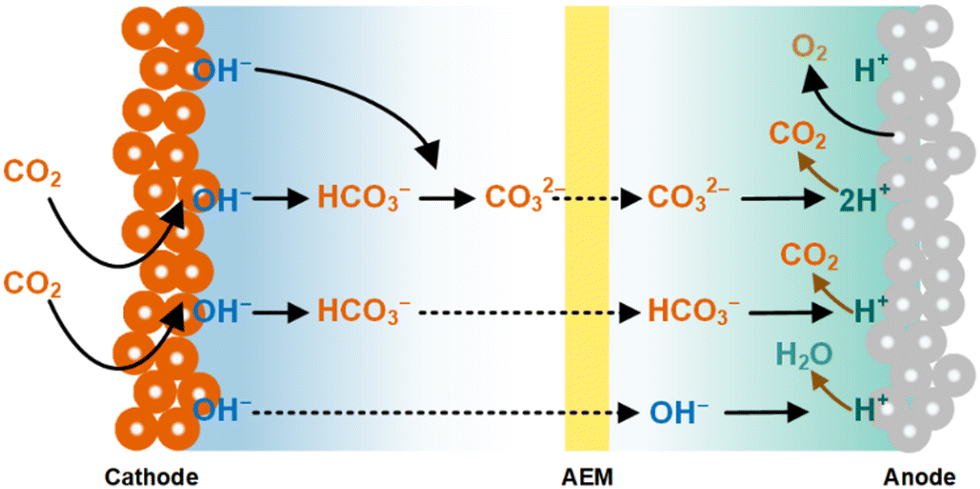 | ||
| Fig. 2 Transport of CO2-derived HCO3−/CO32− species through the AEM and subsequent evolution of CO2 (from HCO3−/CO32−) with O2 in a typical CO2 electrolyzer. Adapted from Ma et al.52 | ||
An undesirable solution to the co-evolution of O2 and CO2 at the anode would be to use a downstream separation unit to recover the CO2 and recycle it back at the cathode; however, this would obviously make the overall balance of plant more expensive both in terms of capital costs and energy use.51
In addition to CO2 degassing at the anode, the reaction between the H+ (from OER) and CO32− (continuously supplied from the cathodic CO2E reactions) creates a steady-state condition in the anolyte with a pH of ∼7.9 or potentially lower dependent on the depletion of the buffer capacity.52,53 While a 1 M aqueous K2CO3 has a pH of 11.6, modeling has shown the pH at the cathode is mostly likely to be near 14 (from the continuous generation of OH−) at the high current densities needed for commercial electrolysis.40 Thus there is at least 6 pH units between the anode and cathode providing a very strong driving force for CO32− to transfer across the membrane. However, the pH variation also results in a shift in the equilibrium potential of the O2 evolving anode. With the anode operating more acidic than the cathode, a pH variation of 6 entails a Nernstian thermodynamic loss of 354 mV (59 mV per pH unit) at room temperature. A depleted anode buffer capacity of bicarbonate could substantially increase this Nernstian loss.
While the anion management challenges need to be overcome, cation management also needs to be monitored. It has recently been shown that non-H+ cations are essential for CO2E to proceed.54–56 With the rate-limiting step being an n-PCET step (adsorbed CO2 intermediate [*CO2] formation for everything except Cu, where it is the *OCCO), the interfacial electric field across the Helmholtz layer plays a dominant role in determining the kinetics. Increasing the electric field enhances the kinetics, and this favors the cations that can most densely pack charge within the Helmholtz layer. Thus the smaller the hydrated radii (Cs+ < K+ < Na+ < Li+), the higher the catalytic activity.54 However, having high concentrations of metal cations close to the cathode interface (in conjunction with high local pH) will lead to oversaturation and a salting-out effect. For example, Table 2 shows that Group 1 alkali metal carbonates/bicarbonates have lower solubility than their hydroxide counterparts, thus enhancing precipitation issues. Given that group 2 and 3 alkali metal cations have even lower solubility than group 1 cations, their availability close to the reaction interface would worsen the situation. Even in the absence of an aqueous catholyte, the cations from the anolyte can crossover the AEM to the cathode via diffusion and migration and react with HCO3−/CO32− to make solid precipitates of alkali metal (bi)carbonates.57 These solid precipitates fill the pores of the cathode GDE and the flow-field thus increasing the water accumulation at the cathode,58 and obstructing both the CO2 flow to the catalyst as well as the effluent gas from the catalyst.59,60 The use of higher solubility cationic salts (e.g. CsOH/CsHCO3 having 3–4 times higher solubility than K+ and Na+ analogues, see Table 2) could potentially delay or avoid salt precipitation in the GDE.
| Compound | Solubility (g/100 g H2O) | Solubility (M) |
|---|---|---|
| LiHCO3 | — | — |
| Li2CO3 | 1.30 | 0.18 |
| KHCO3 | 36.20 | 3.62 |
| K2CO3 | 111 | 8.03 |
| KOH | 121 | 21.57 |
| CsHCO3 | 209 (at 15 °C) | 10.78 |
| Cs2CO3 | 261 (at 15 °C) | 8.01 |
| CsOH | 300 (at 30 °C) | 20.01 |
| NaHCO3 | 10.30 | 1.23 |
| Na2CO3 | 30.74 | 2.90 |
| NaOH | 100 | 25 |
| BaCO3 | 0.0014 (at 20 °C) | 7.10 × 10−5 |
| Ba(OH)2 | 4.91 | 0.40 |
| La(OH)3 | 0.000020 (at 20 °C) | 1.03 × 10−6 |
If we take a brief analysis of cation exchange membranes (CEMs), there is a common misconception that CEMs such as Nafion are intrinsically acidic and the acidic environment favors HER over CO(2)E selectivity, which is not correct. Nafion is only acidic when protons are being transferred, whereas if an anolyte such as KHCO3 is used, it is the positive cations (i.e., K+ in this case) that transfer through the membrane, entailing the membrane is of a relatively neutral pH.61
A bigger issue with CEMs relates to what happens to the transferring cations. As CO2/CO reduction produces OH−s (that further get converted to carbonates in the presence of CO2) and cations such as K+, Cs+, etc. will form either hydroxyl species or carbonates which then will build up and eventually precipitate. A CEM that transports purely H+ ions would allow water formation, which could then easily back diffuse into the anode or evaporate into the outlet cathodic gas. Conversely, a device completely absent of non-protonic cations entails there will be an insufficient electric field for CO(2)E.63
An alternative approach to using AEMs and CEMs, as shown by Vermaas and Smith,64 was to employ a BPM (for CO2E), which allows simultaneous transport of cations and anions at each electrode. In a BPM, ion transport depends on membrane orientation. With the cation exchange layer (CEL) next to the anode and the anion exchange layer (AEL) next to the cathode (i.e., forward bias as shown in Fig. 1a), this will consume anions from the cathode and cations from the anode. If hydroxyl groups are produced and maintained at the cathode as in CO electrolysis, the BPM then combines a cathodic OH− with an anodic H+ to form H2O. However, if carbonates are formed cathodically, this leads to the evolution of H2O and CO2 at the BPM interface, which is undesired as gas bubble evolution could lead to BPM delamination (especially if the BPM is made from the lamination of an AEM and CEM).
To resolve the carbonate/CO2 issue a BPM can have the relative location on the AEL and CEL inverted (i.e., reverse bias). This entails water will be dissociated to H+ and OH+ at the AEL/CEL interface, with H+ migrating to the cathode. As the concentration of H+ and OH− within the CEL and AEL layer respectively are on the order of ∼1 M,61,65 this entails a pH = 0 in the CEL and a pH = 14 on the AEL. From a simple Nernstian analysis, a thermodynamic driving force for a pH shift of 14 corresponds to a loss of 820 mV per mol water dissociated, and this does not even include any kinetic overpotential or mass transfer induced losses due to water dissociation. Moreover, with the H+ emitting CEL of the BPM towards the cathode side, any formed CO32− or HCO3− will acidify and release the CO2 at the cathode. While it is highly beneficial that the CO2 stays on the cathode side, the acidic environment needed for this also favors HER. However, work with BPMs typically involves using a catholyte66,67 that buffers the pH to more alkaline conditions albeit at an increased ohmic loss related to the catholyte thickness.
Thus, in summary, AEMs have issues due to CO2 crossover and mixing with O2, CEMs have selectivity issues and/or salting out of carbonates on the cathode, and BPMs need excessive voltage, thus there is currently not a clear winner in terms of preferred membrane type.
CO electrolysis
One workaround to prevent CO2 crossover in AEMs during CO2E is to shift towards COE, where only cathodically generated OH− transports across the AEM (the absence of CO2 prevents HCO3− and CO32− formation). The switch from mixed OH−/HCO3−/CO32− conduction to pure OH− conduction will lead to higher AEM conductivities (Section 5), but this is offset by the potential higher rate of chemical degradation of the AEM (Section 6). Given that metal hydroxides are more soluble than metal carbonates (see Table 2), COE also significantly mitigates the precipitation issue. One potential downside of COE is that CO has a relatively low solubility of 1 mM compared to 33 mM for CO2 (25 °C, 1 atm) in water. However, CO has a solubility 26% higher than H2,62 and as H2 fuel cells have minimal mass transport limitations in optimized devices, CO mass transport issues are unlikely to prevent the commercialization of this approach.One issue that will still be encountered in both CO2E and COE is liquid products crossing over the membrane to the anode and oxidizing into CO2, which is exacerbated with the use of AEMs.44,46,50 For example, formate and acetate, being the negatively charged anions, can crossover primarily through electromigration;44,46 while neutral products (such as ethanol or propanol) diffuse through the membrane due to concentration gradient and electroosmotic drag.60,68 Li et al.44 reported the permeation rates of 0.017 and 0.025 mmol h−1 for ethanol and methanol respectively, for an AEM (Neosepta with 170 μm thickness) at 50 mA current. Additionally, they reported a flux ratio of 0.009 for ethanol to ions with an AEM, which was reduced to 0.006 for a BPM (Fumasep FBM) at 50 mA. Recently, Miao et al.69 reported that almost 75% of the ethanol formed at the cathode diffuses through the AEM at higher current densities (≥100 mA cm−2). Liquid product crossover can happen even at open circuit potential via diffusion, but the crossover issue is exacerbated at high current densities because of an increase in both the concentration gradient and ion transport across the AEM. Zhang et al.46 inserted a catholyte that already contained standard liquid CO2E products and observed a relatively similar diffusion rate through the AEM for all products at equal concentrations under open-circuit conditions (Fig. 3a). However, by applying a potential and increasing the current density, crossover increased for all liquid products (an increase in formate crossover from ca. 7% at 50 mA cm−2 to ca. 20% at 300 mA cm−2). This suggests that liquid product crossover through electromigration is much more significant than diffusion or permeation. Among the different AEMs studied by Zhang et al.,46 Selemion AMV showed the highest formate crossover (Fig. 3b). As Selemion AMV showed the highest crossover despite it having a higher thickness (between 110 and 150 μm) than both Sustainion and Fumasep (both having ∼50 μm thickness) entails there can be quite differing diffusion coefficients between the different chemistry membranes.
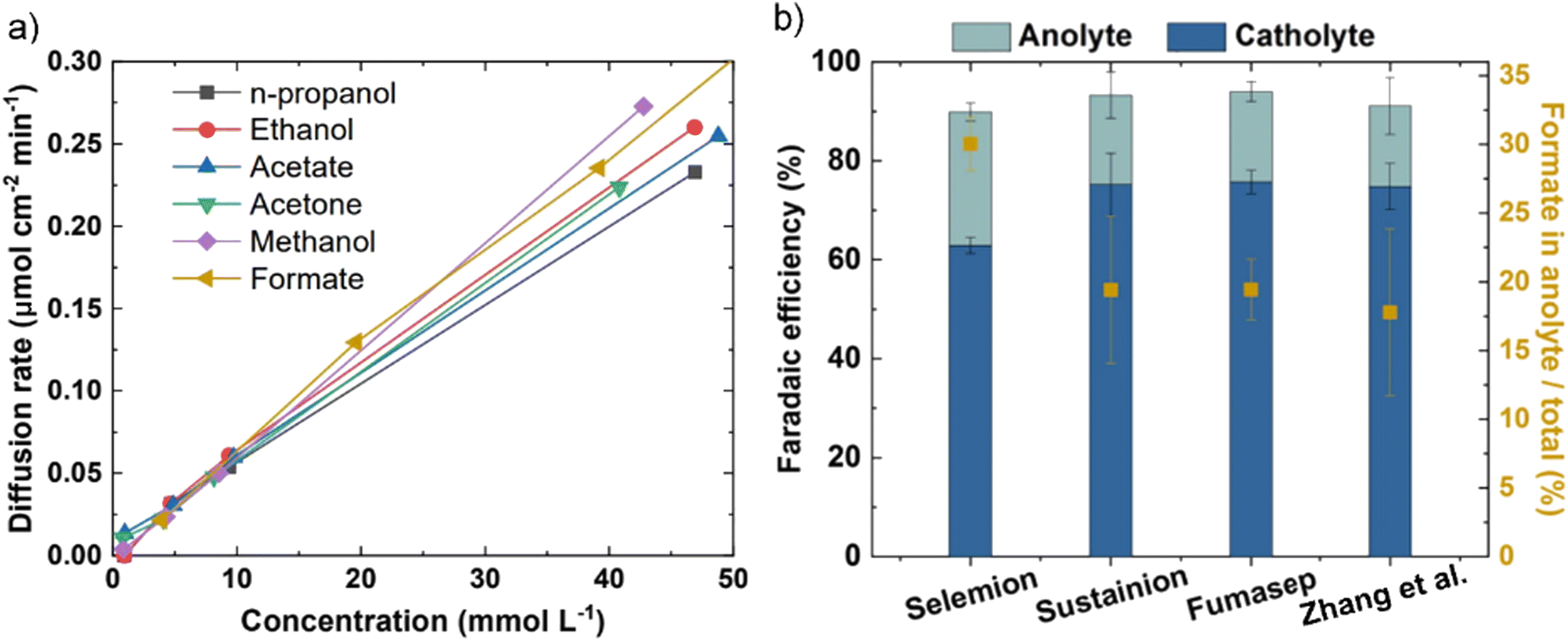 | ||
| Fig. 3 (a) The liquid product crossover (diffusion) rates through the AEM (Sustainion X37-5) as a function of their concentration in the solution at open-circuit conditions. (b) Comparison of faradaic efficiency of formate and its crossover using different AEMs over an Sn-based GDE with 1.0 M KOH as electrolyte and 50 sccm CO2 flowrate at 200 mA cm−2, Figures adapted with permission from Zhang et al.46 | ||
Ion transport in AEMs for CO(2)E
Anion transport mechanisms in AEMs
AEMs are generally made of hydrophobic polymer (e.g., polysulfone, polyphenylene or polystyrene) backbones containing hydrophilic cationic functionalities (e.g., quaternary ammonium, alkylphosponium, pyridinium, or imidazolium), that are either part of the polymer main chain (PiperIon, Aemion) or anchored as side chains. The availability of both a hydrophobic backbone and hydrophilic cations in the AEM usually contributes to a hydrophobic/hydrophilic phase segregated structure, where hydrophilic cationic species form ion and water-containing channels that assist in transporting the anions across the AEM. For a further discussion on AEM structures and chemistries, we refer the readers to reviews by Varcoe et al.,32 Merle et al.,47 Gorgieva et al.,70 and Maurya et al.71 The hydration of any particular AEM is a function of the anion present, the humidity of the immediate environment, and thermo-hydration history, and is critical for efficient conduction of anions through the hydrophilic ionic channels (as long as water contents are not excessive where the cationic sites become widely separated). It should be kept in mind that most anions, such as Cl− and CO32−, transport through hydrated AEMs via vehicular and exchange site-based hopping mechanisms, while OH− ions (like H+ in PEMs) can additionally utilize structure diffusion (e.g., Grotthus) mechanisms (hence the higher intrinsic mobilities of OH− anions, discussed below); the relative contributions of each mechanism being a function of hydration levels. Here, we discuss the transport mechanisms in AEMs from a CO2E and COE perspective.From fuel cell studies,72–76 it is known that the OH− (the major charge carrier in COE) can migrate through a Grotthus-type transport mechanism. In other words, OH− migration happens due to the interconversion of OH− between ‘hypercoordinated’ OH−(H2O)4 and H9O5− by the formation and cleavage of hydrogen bonds between neighboring hydration shells in a hydrogen-bonded network of water molecules (Fig. 4).77 In addition, OH− can also be transported by non-structural diffusion mechanisms, where the transfer of OH− anions can occur directly between a (non-static) cationic group on a polymeric chain (of the AEM) and neighboring cationic groups (with or without the help of water vehicles), either in the same chain or a proximate adjacent chain (Fig. 4). The dominant transport mechanism that operates is a function of water content. A highly hydrated AEM entails an expansion of the hydrophilic channels that weakens the OH− binding to the cationic sites, resulting in a higher relative contribution of structure diffusion. In contrast, AEMs with low water content have reduced ionic conductivity because of smaller hydrophilic channels, leading to an increased level of a surface-site hopping.76
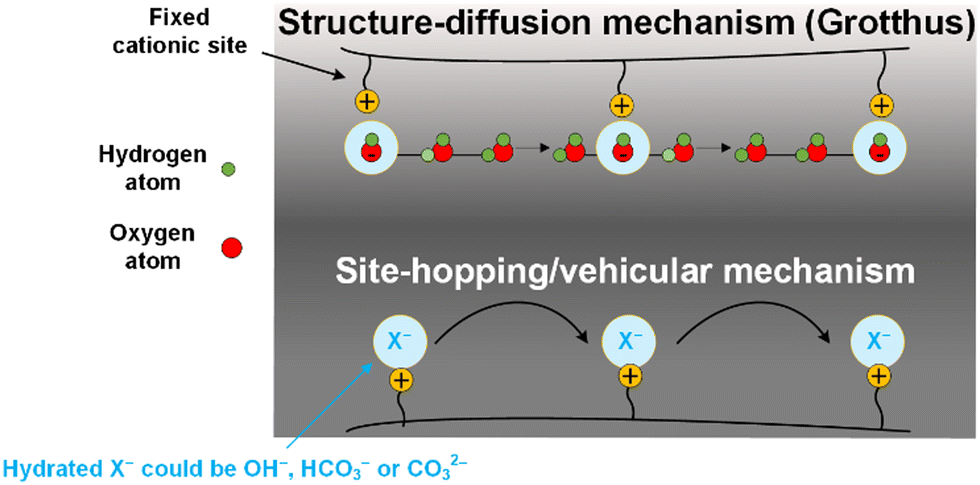 | ||
| Fig. 4 Typical ion and water transport mechanisms in CO(2) electrolyzer. | ||
In the case of CO2E, HCO3−/CO32− are the major charge carriers across the AEM, which cannot conduct via structure diffusion (Fig. 4).78 Because of this difference in conduction mechanism, and that both HCO3−/CO32− have larger hydrated radii and masses than OH−, the intrinsic mobility of HCO3−/CO32− anions is ∼5 times lower than for OH− (see Table 6),79 resulting in a 4–5 times reduction in the AEM's conductivity80 (when assuming no significant differences in water content and nano-/micro-phase segregation/channel structure on the change of anion). For example, Jouny et al.81 showed an increase in cell voltage (Fig. 5) when switching from COE to CO2E. Initially, when COE started, the cell potential remained stable because only OH− was conducting through the AEM. Immediately after the authors switched to CO2E, OH− conduction remained dominant, but the cell potential rose slightly (∼100 mV) due to the higher activation overpotential for CO2 reduction compared to CO (Fig. 5). After this switch from COE to CO2E, the feed CO2 then started to react with KOH to form CO32−, which gradually became the dominating conduction species leading to a slow rise in cell potential (see Fig. 5 after 30 min) owing to the lower conductivity of CO32− through the AEM. Interestingly, when the cathode was switched back to COE, the cell potential reduced slightly to a fairly stable plateau, but this was ∼200 mV higher than the initial start-up COE reaction. This directly reflects the presence of CO32− in the polymer electrolyte membrane. Additionally, the decrease in the local pH as the electrolyte shifts from pure OH− to increasing levels of CO32− can also lead to an increase in cathodic overpotential for COE.
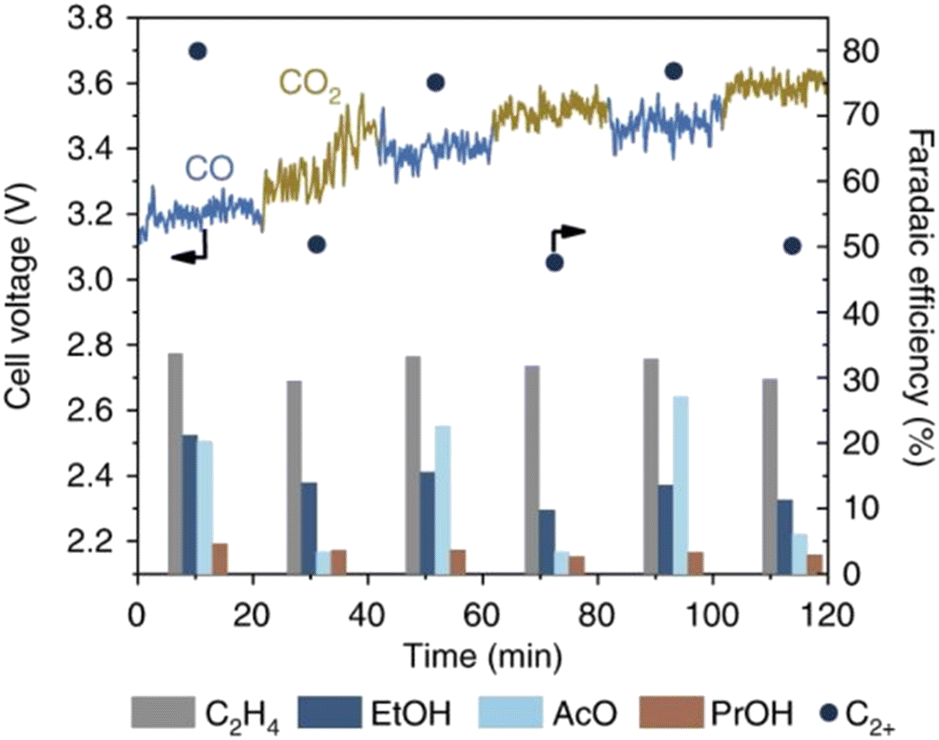 | ||
| Fig. 5 Comparison of C2+ product selectivities for CO2E and COE over micrometer copper in 1 M KOH at 300 mA cm−2. Reproduced with permission.81 Copyright 2018, Springer Nature. | ||
Cation transport in AEMs
An ideal AEM should be permselective to anions and impermeable to cations (such as K+ or Na+). In AEM-based zero-gap electrolyzers, cation crossover from the anode to the cathode is in fact necessary (assuming there are no cations or cationic ionomer present at the cathode interface before electrolysis) to create the electric field at the electrode interface necessary for CO(2)E to occur.56,82 However, cation crossover can also lead to the precipitation of solid (bi)carbonates at the cathode GDE, which clogs up the reactor and limit the mass transport of CO2 to the catalyst surface. Janáky and co-workers57 confirmed the presence of potassium bicarbonate (KHCO3) and related carbonate species (K4H2(CO3)3)·1.5H2O at the cathode GDE (including GDE pores and the cathode flow field) in a zero-gap reactor using X-ray diffraction-based spectroscopy techniques as shown in Fig. 6. However, it is important to point out that the SEM-EDX and micro-CT view of the GDE in Fig. 6 was performed postmortem and ex situ, meaning that there could be a potential change in the salt precipitation (found in GDE) after exposure to the feed gas (i.e., CO2). For example, after electrolysis, the CO2 passing through the cathode flow-field (including the catalyst layer) could potentially react with the alkaline cathodic environment and subsequently affect the salt precipitation. In addition, while doing ex situ analysis of the AEM (post electrolysis), CO2 present in the air can react with the OH− in the AEM to form (bi)carbonates and thus could affect the physical properties of the AEM, especially in the case of COE where AEM transports OH−s. Despite the negative effect of carbonation on the AEM conductivity (when handled in the air), the effect is found to be reversible.83,84 For instance, depending upon how long the AEM is kept in contact with air while assembling the cell for COE, it will take some time for the AEM to fully exchange all the HCO3−/CO32− species with OH−s produced from the cathodic reactions during the start of COE and then a stable cell potential (at least no significant change arising from membrane loss) should be expected. One way to completely ensure that the charge carriers through the AEM during COE are OH−s is by initially performing HER through N2/Ar purging and once the cell potential becomes stable, then switch to CO. Unfortunately, to the best of our knowledge, experiments have yet to be done to determine the time needed to purge carbonates, but we would expect this to be on the order of 5–10 minutes based off previous ion exchange experiences.61 | ||
| Fig. 6 Cross-sectional (a) SEM-EDX and (b) micro-CT view of the GDE after CO2E in an MEA-based electrolyzer with 1 M KOH as anolyte at 50 °C and 3 V cell potential. The red color represents Ag atoms while the green represents K atoms in both (a) and (b). Reproduced with permission.57 Copyright 2021, Springer Nature. | ||
Cation migration in an AEM occurs through two different modes: (a) diffusion-driven by a difference in the concentration gradient of ions across the AEM; and (b) migration-driven due to the electrical potential gradient between the cathode and anode.57 Although it is impossible to completely stop the transport of cations through an AEM (none are 100% permselective, see Table 3), the cation crossover can be slowed or minimized by modifying the operating conditions of the electrolyzer and the chemistry of the AEM. For example, the transference number of Cs+ (≈1 × 10−5 for 0.01 M CsOH with PiperIon AEM) is ca. an order of magnitude lower than that of K+ (≈1 × 10−4 for 0.01 KOH with Sustainion® AEM), meaning that using Cs+ based anolyte could greatly help decelerate the precipitation phenomenon. However, given that PiperIon and Sustainion® have different AEM chemistry (piperidinium in main chains vs. imidazolium in side chains), a direct comparison of cation's transference numbers is difficult, and the lack of literature on cation transference numbers through AEMs is something that needs to be addressed. In addition, AEMs that have a high concentration of cationic species in the polymeric backbone could inhibit the migration of cations via electrostatic repulsion. While increasing the cationic group concentration in the AEM (e.g., quaternary ammonium content) is synthetically easy to achieve, a higher cationic content commonly leads to high water uptake, resulting in reduced electrostatic repulsion (lower permselectivities), excessive swelling, and weakened mechanical integrity.85
| AEM | Permselectivity | |
|---|---|---|
| Experimental dataa | Commercial datab | |
| a Estimated by keeping the membrane between 0.5 M and 1 M NaCl solutions. b Estimated by electrophoresis, 2 mA cm−2. | ||
| Fumasep FAD | 86.0 | >91 |
| Neosepta AM-1 | 91.8 | >96b |
| Neosepta AFN | 88.9 | >96b |
| Neosepta AMX | 90.7 | >96b |
| Ralex AMH-PES | 89.3 | >90 |
| Selemion DSV | 89.9 | Not available |
| Selemion APS | 88.4 | Not available |
Another way to stop cation crossover from the anolyte is to use pure water at the anode as is done in the water electrolysis field. However, with pure water, it is essential that a large triple-phase boundary is produced. While this can be easily achieved with CEM materials such as Nafion via coating catalysts with ionomers and hot-pressing catalysts to membranes, anion-exchange ionomers are less developed and thus their stability and durability currently make this more challenging. Pure water at the anode also leads to insufficient cations at the cathode interface, as this is needed for CO2E catalysis.56 For instance, the Janáky group57 showed a one-third drop in current density when using pure water as an anolyte as compared to 0.1 M KOH (Fig. 7a). With water as an anolyte, the current density decreased from 200 mA cm−2 to <130 mA cm−2 within one hour of CO2E (Fig. 7a). After initially providing a sufficient electric field at the cathode (AEM were kept in 1 M KOH before use, so cations were available), this drop was attributed to the reduced availability of cations as they slowly diffused away into the pure water at the anolyte.57 In addition, Fig. 7b shows that the decrease in current density with water as an anolyte could be attributed to the change in overpotentials of cathodic and anodic reactions (as seen from the increased charge transfer resistance), however, it is difficult to deconvolute which overpotential (anodic or cathodic) dominates. Similarly, Yin et al.87 used water as an anolyte and found that the charge transfer resistance could be reduced by ∼3 Ω cm2 by increasing the temperature from 30 to 80 °C. The authors spray-coated an Au/C (mixed with quaternary ammonia poly(N-methylpiperidine-co-p-terphenyl), QAPPT, ionomer) cathode onto a QAPPT membrane, where the Au/C catalyst was prepared by mixing carbon black in glycerol solution and subsequently reduced by HAuCl4 and NaBH4. This entailed that there could be Na+ cations present in the catalyst layer necessary for CO2E.
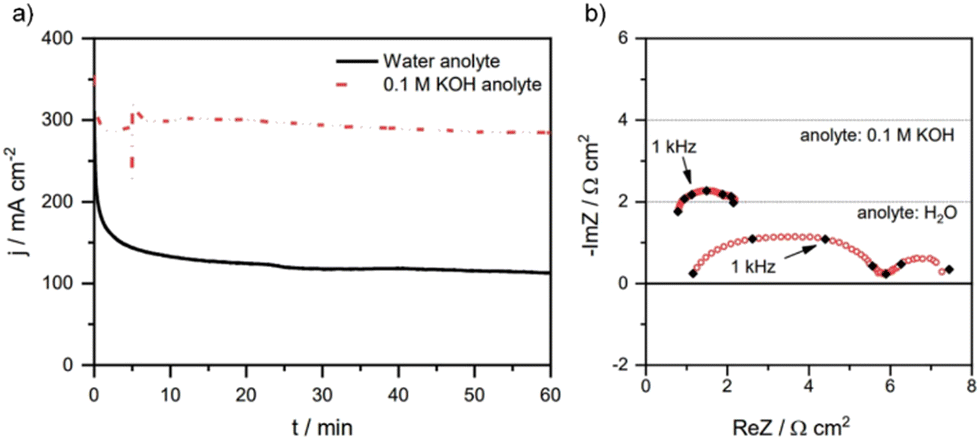 | ||
| Fig. 7 (a) Chronoamperometric tests and (b) impedance spectra recorded during CO2E with 0.1 M KOH or pure water as an anolyte. Conditions: Sustainion X37-50 AEM, Tcathode = 60 °C, 12.5 sccm CO2 feed rate, and constant cell potential of 3.1 V. Note: the impedance spectra recorded in (b) shows overall charge transfer resistance of the process (including both cathodic and anodic reactions). Reproduced with permission.57 Copyright 2021, Springer Nature. | ||
Water transport in AEMs for CO(2)E
Water transport mechanisms in AEMs
CO(2)E is unique when comparing to gaseous fuel cells or aqueous water electrolyzers in that the anode side uses liquid water, whereas the cathode side uses water vapor. Given that sufficient hydration is necessary for facile ion conduction, understanding water transport in AEMs is essential. While some CO(2)E device designs use a catholyte between the gas diffusion electrode and membrane, this review/analysis will focus primarily on a zero-gap MEA type device since a lack of a catholyte entails lower ohmic losses. However, the MEA approach of direct interaction of a vapor side at the cathode interacting with membranes complicates and provides challenges for water management.Thermodynamically, a 100% relative humidity (RH) vapor phase has the same chemical potential as liquid water, however, the water uptake by an ion-exchange membrane (both AEM and CEM) differs between the vapor and liquid phase.88 This seemingly contradiction of thermodynamics is well known as Schroeder's Paradox but its origin is still not fully understood.89 Because of this, the gaseous cathodic side and the aqueous anodic side of the membrane will have a difference in water uptake. Fig. 8 shows Schroeder Paradox in conventional AEMs (FAA3 and PAP-TP-85) with higher water uptake when in contact with liquid water than with saturated vapor (this paradox is most evident with the AEMs in OH− form).90 There are two possible explanations to justify the observed paradox:91
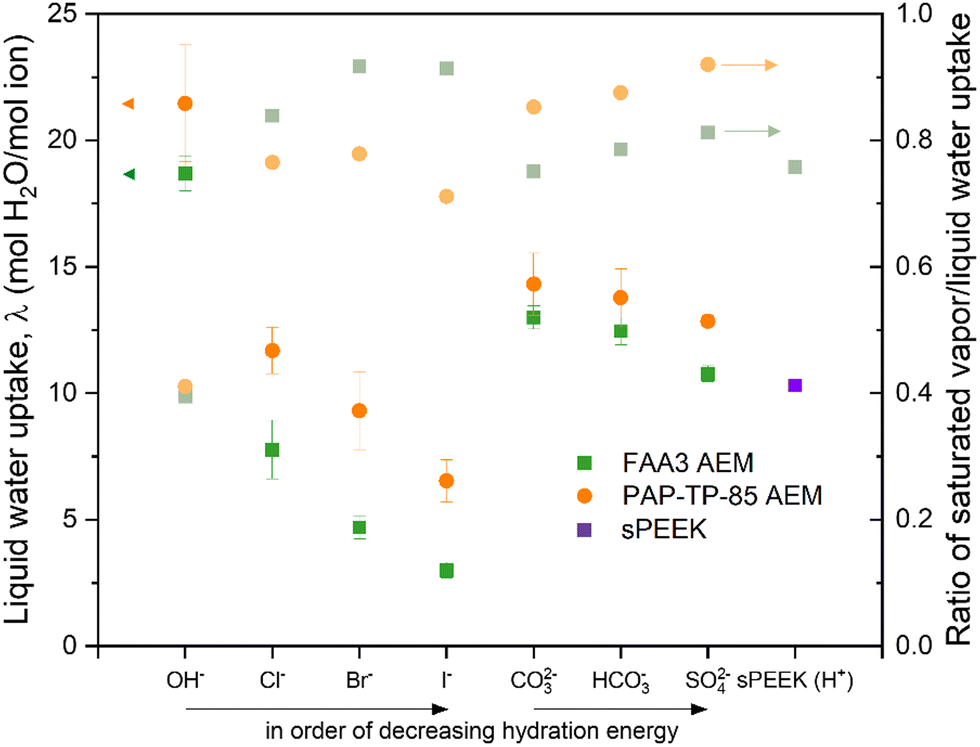 | ||
| Fig. 8 Liquid water uptake (right y-axis) and the ratio of saturated vapor over liquid water uptake (left y-axis, Schroeder's Paradox) for two different AEMs in their different anions forms and compared with sulfonated poly ether ether ketone (SPEEK) at room temperature. Reproduced with permission.90 Copyright 2020, Elsevier. | ||
(1) Membrane materials are not at equilibrium when exposed to different conditions during water uptake measurements. Typically, it takes a longer time for membranes to reach the steady state in saturated vapor than when in contact with liquid water because of the slow kinetics of membrane hydration in vapor-equilibrated samples.92 This is attributed to a lower concentration of water molecules at the membrane surface when in contact with saturated vapor. Even in liquid water, the membranes can take up to 200 h to reach equilibrium (membranes such as Nafion require boiling treatments to obtain the fully expanded hydrated forms in realistic timeframes).93 Therefore, this non-equilibrium behavior under different conditions could give rise to Schroeder's Paradox.
(2) The surface morphology of the membrane could vary under disparate humidified/liquid water conditions because the availability of feed gases (e.g., CO2 or CO) in saturated conditions (with water) could increase the concentration of hydrophobic functionalities at the interface.91 This phenomenon may be worse for thinner membranes where interfacial effects could affect the free energy of the membranes.91,94–96
The above discussion considered water uptakes under more stagnant conditions. For a CO(2) electrolyzer we need to consider water transport under more practical operating conditions. The overall water transport/flux (JAEM,w) across the membrane is a function of diffusion (Jdiff), electro-osmotic drag (JEOD), and hydraulic permeation (JHP, also known as back convection), as shown in Fig. 9 and expressed by the following equation:
| JAEM,w = Jdiff − JEOD ± JHP | (6) |
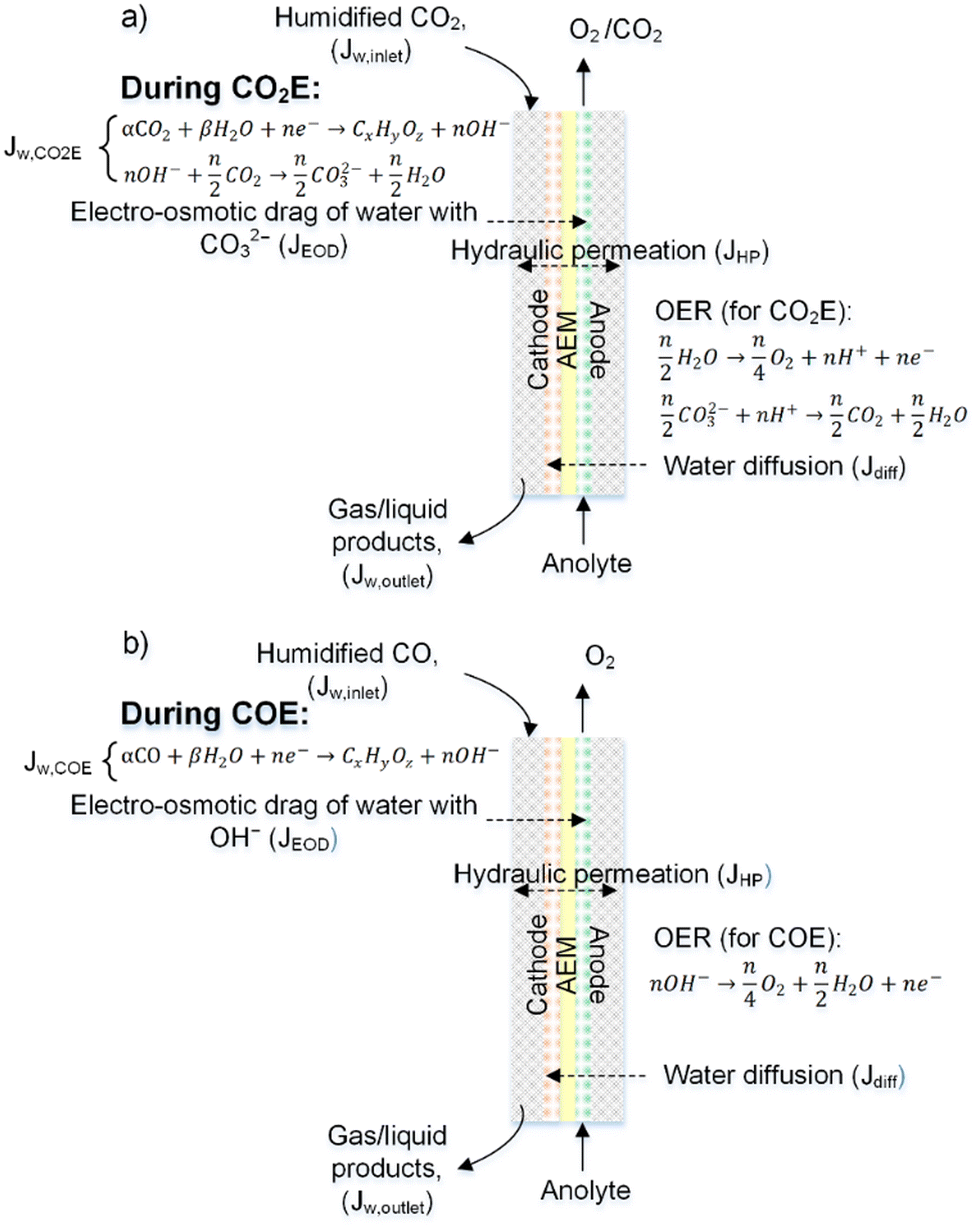 | ||
| Fig. 9 Typical water transport during (a) CO2 electrolysis and (b) CO electrolysis. This diagram shows water fluxes due to: reaction stoichiometry (JW,CO(2)E) electroosmotic drag (JEOD), diffusion (JDiff), hydraulic permeation (JHP), gas inlet (JInlet) and gas outlet (Joutlet). | ||
Beyond the flux across the membrane, there is also the stoichiometric amount of water used at the cathode (JW,CO2E or JW,COE) and the water flux in (JW,inlet) and out (JW,outlet) of the reactor all of which are notable contributors to the overall water balance that needs to be optimized for CO2 reduction. All of the aforementioned water fluxes are shown in Fig. 9 to visually demonstrate where they occur in a CO(2)E device.
In a typical MEA-based CO(2) electrolyzer, water is supplied to the cathode: (a) via humidified CO(2) feed and (b) from anolyte crossover through the AEM. The diffusion rate of the latter case is affected by (1) water consumption at the cathode due to CO(2)E and HER stoichiometry; (2) hydration of the produced OH− (HCO3−/CO32− from CO(2)E) species; and (3) humidity in the cathodic gas stream, all of which creates a change in chemical potential of water between the anode and the cathode. This can be modeled according to Fick's law, however, with a liquid anolyte and a gaseous cathode, chemical potentials need to be used rather than the typical concentration terms. Thus, by applying Fick's law, the water flux from the anode to the cathode due to diffusion (Jdiff) through the AEM per unit electrode area (A) can be written as:
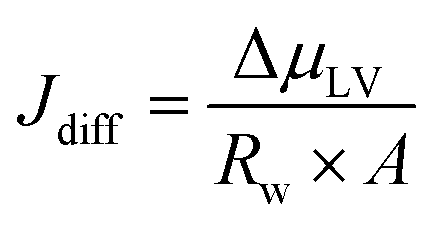 | (7) |
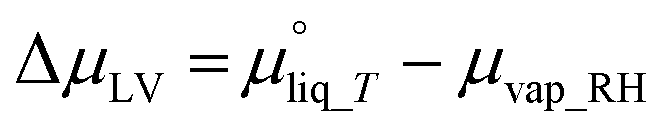 | (8) |
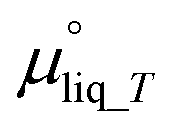 is the chemical potential of liquid water at temperature T and μvap_RH is the chemical potential of humidified gas (or water vapor) at a specific RH, calculated using:
is the chemical potential of liquid water at temperature T and μvap_RH is the chemical potential of humidified gas (or water vapor) at a specific RH, calculated using: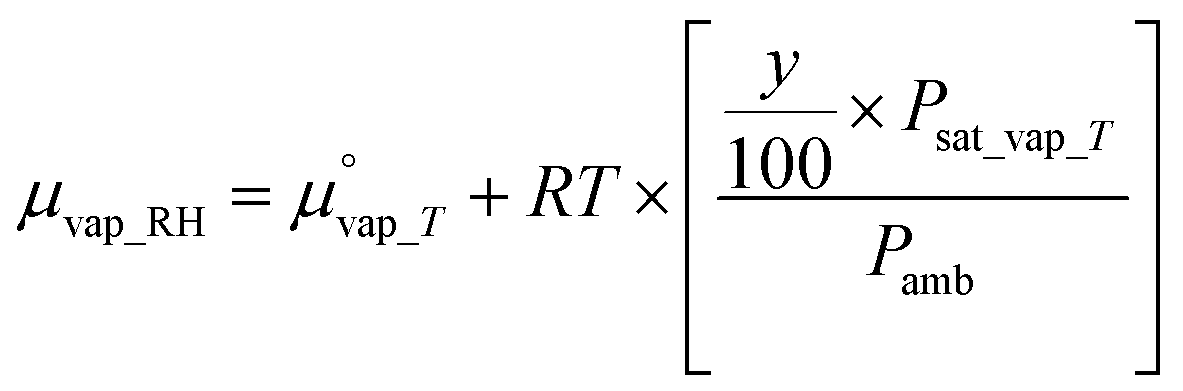 | (9) |
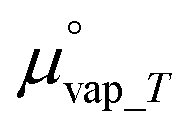 is the chemical potential of water vapor at T, R is the universal gas constant, y is the relative humidity (%), Psat_vap_T is the saturated vapor pressure at T, and Pamb is the ambient pressure.
is the chemical potential of water vapor at T, R is the universal gas constant, y is the relative humidity (%), Psat_vap_T is the saturated vapor pressure at T, and Pamb is the ambient pressure.
We can deconvolute the effective water permeation resistance (Rw) into the internal water permeation resistance of the membrane (Rw,internal) and the interfacial water permeation resistance at the membrane–vapor interface (Rw,interface) at the gaseous side (eqn (10)). Rw,internal and Rw,interface can easily be derived in relation to the diffusion coefficient D′ and an effective mass transfer coefficient (k′) using eqn (11) and (12), respectively as:
| Rw = Rw,internal + Rw,interface | (10) |
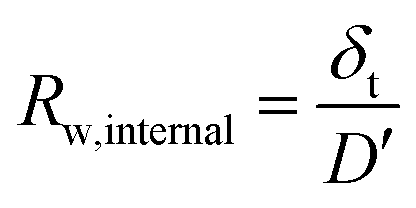 | (11) |
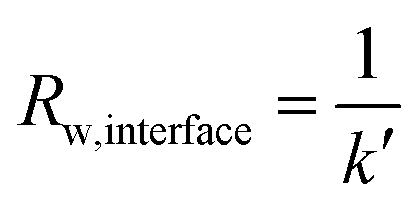 | (12) |
Experimentally Rw can be estimated by simply exposing the membrane to liquid water on one side and setting the relative humidity gas on the other side, and then measuring Jdiff at different ΔμLV using eqn (7). By measuring Rw as a function of δt, D′ can then be estimated from the slope, and k′ can be found from the y-axis intercept. However, the meaningfulness of D′ is significantly complicated by the fact that D′ is known to be a function of λ.98,99 This means that cathodes with relative humidity less than 100% will have the D′ vary across the thickness (δt) of the membranes. Effectively, D′ will be smaller near the gas phase side compared to the liquid side. To the best of our knowledge, no one has investigated varying D′ values across a membrane, but work has been done on determining overall Rw for liquid–vapor AEM systems.
Luo et al.100 showed that the overall Rw for AEMs with a liquid–vapor system is almost an order of magnitude higher than liquid–liquid water on both sides. By comparing these two systems, they could isolate the Rw,interface for the liquid–vapor system (or more precisely the membrane–vapor interface), showing that the ratio Rw,interface/Rw (for the membrane–vapor interface) is more than 80%. This demonstrates that having humidified gas on one side of the membrane results in the Rw,interface playing the dominant role in the overall water permeability resistance (R). The reason for this substantial interfacial resistance has been attributed to the anisotropic dehydration of the membrane surface at relative humidities less than 100%.101 Since Rw,interface across the membrane vanishes when exposed to liquid on both sides, λ shows a flat profile within the membrane. However, with liquid water on one side and saturated vapor on another, a non-zero Rw,interface will be observed on the saturated vapor side (Fig. 10a). After longer equilibration times (membrane exposed to a saturated vapor stream), the difference between the λliq and λsat_vap will decrease allowing the membrane to behave in a similar way as having liquid on both sides (Fig. 10a). After the saturated vapor was switched with a dry cathode gas, a more dramatic λ variation across a membrane was seen (Fig. 10b). A first-order diffusion model suggests λ follows an exponential decay from the liquid side to the vapor side. However, such a simple model is unlikely to be fully representative due to the complexities of polymers. A lack of literature data prevents further meaningful analysis.
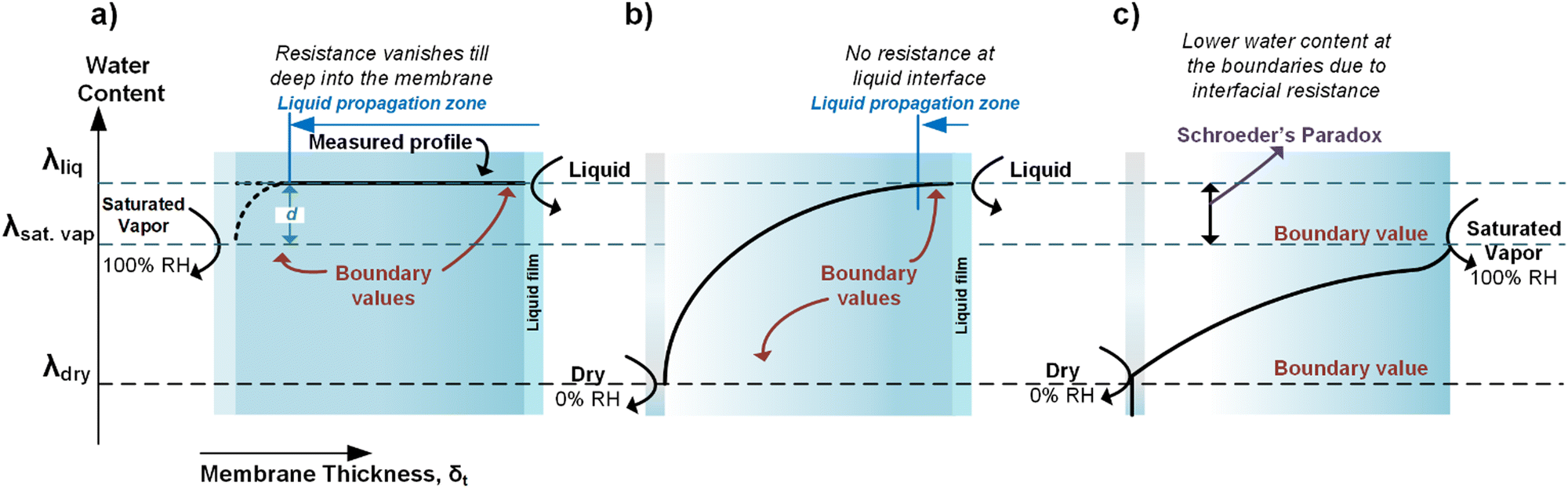 | ||
| Fig. 10 Schematic of how water profile would look inside the membrane when exposed to different conditions (meaning varied interfacial resistance) at the cathode and anode. This figure was adapted from Kusoglu and Weber.102 | ||
Fig. 10c demonstrates a slightly different and complex evolution in the membrane's λ profile when both the anode and cathode are gaseous streams with varying humidity. Although to the best of our knowledge, no experimental studies exist using this approach, it would have the advantage of controlling and locking the total cation concentration (e.g. Cs+) in the device and thus will be analyzed. Given that the water activity at the membrane surface is unity for both liquid water and saturated vapor, the presence of Rw,interface in the vapor case can cause a drop in λ (and thus showing the so-called Schroeder's Paradox).
The above analysis has yet to consider how CO(2)E influences the water balance. When considering CO(2)E and eqn (7) and (8), 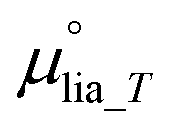 is not expected to change (aqueous anolyte), but μvap_RH (see eqn (9)) will vary as water reacts stoichiometrically in most CO(2)E reactions (see Table 1).
is not expected to change (aqueous anolyte), but μvap_RH (see eqn (9)) will vary as water reacts stoichiometrically in most CO(2)E reactions (see Table 1).
For the case where CO2 is reduced to C2H4 and with the assumption that all electrogenerated OH− are converted to CO32−, 2 mol of H2O is consumed at the cathode for every 1 mole of C2H4 produced. However, in the case of COE, the H2O/C2H4 ratio is 6 with all OH− diffusing to the anode. Table 4 shows the water consumption/generation at the cathode (nc) and anode (na) for other prominent products as calculated via the following general equation:
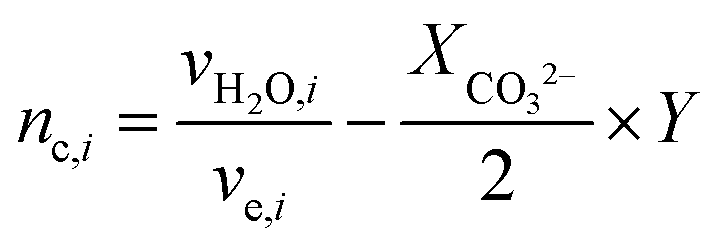 | (13) |
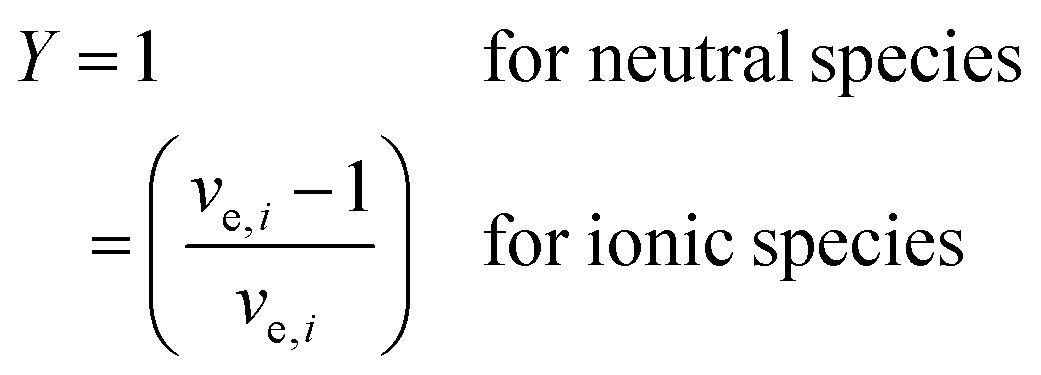 | (14) |
| Product, i | @Cathode | @Anode | |||
|---|---|---|---|---|---|
|
|
|
n c,i |
|
|
|
| a n c,i assumes XCO32− = 1 for CO2E and XCO32− = 0 for COE. | |||||
| CO2E | |||||
| HCOO− | −0.50 | −0.50 | 0.25 | −0.50 | −0.25 |
| CO | 0 | 0 | 0 | 0 | 0 |
| CH4 | −2.00 | −2.00 | 0.25 | 0 | 0 |
| CH3COO− | −0.75 | −1.50 | 0.19 | −0.50 | −0.06 |
| C2H4 | −1.00 | −2.00 | 0.17 | 0 | 0 |
| C2H5OH | −1.50 | −3.00 | 0.25 | 0 | 0 |
| H2 | 0 | −2.00 | 0.50 | 0 | 0 |
| COE | |||||
| CH3COO | −1.50 | −3.00 | 0.75 | +1.00 | +0.25 |
| CH4 | −5.00 | −5.00 | 0.83 | +3.00 | +0.50 |
| C2H4 | −3.00 | −6.00 | 0.75 | +4.00 | +0.50 |
| C2H5OH | −3.50 | −7.00 | 0.88 | +4.00 | +0.50 |
| H2 | 0 | −2.00 | 1.00 | 0 | 0 |
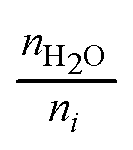
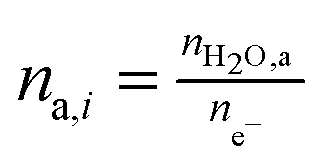
During CO2E the OH− produced will typically react with CO2 to form either HCO3−, or more likely at high current densities, CO32− and H2O. To account for the extra water produced when carbonates are formed, eqn (13) introduces the term XCO32−, relating to the fraction of OH− converted into CO32−. Since the focus of this work is primarily on high current density CO2 electrolysis, all values listed in Table 4 assume a purely carbonate electrolyte (i.e. XCO32− = 1). The value Y is a parameter to take into consideration that ionic species such as formate or acetate have a slightly different water transfer due to the stoichiometry for CO2E involving ionic species (i.e., non-proton coupled electron transfer products).
An interesting point from analyzing Table 4 is that there is no net water consumption at the anode during CO2E for most products because the amount of water consumed is the same as the amount of water generated from the neutralization reaction between the OH−/CO32− and H+. The most notable exception is formate and acetate, due to their anionic nature. By using the parameters developed in Table 4, we can now analyze the total cathodic water consumption (Jw,CO(2)E) due to CO(2)E (i.e. including both reaction stoichiometry and the neutralization reaction between the OH− and CO2) as shown in eqn (15).
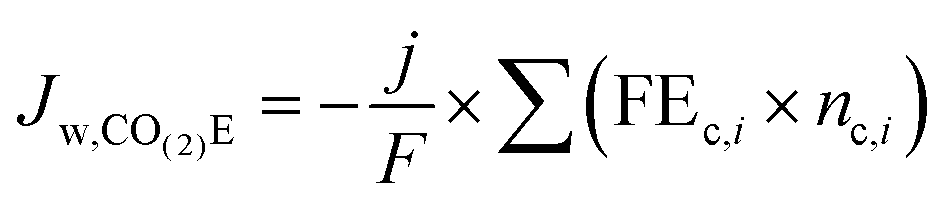 | (15) |
The question becomes whether this water consumed at the cathode can be supplied primarily by a humidified gas stream or whether a diffusion from the aqueous anode is necessary. A 100% humidified CO(2) stream at 60 °C has a water partial pressure of 0.20 bar, or in other words, the CO2:H2O ratio will be 4:1. However, the analysis of C2H4 (in Table 4) demonstrated the CO(2):H2O ratio for CO2E needs to be 1:1 or for COE 1:3 (note two CO's are consumed per C2H4 molecule). Thus at these temperatures (≥60 °C), the water provided from humidification is quite small, and with most experiments done at room temperature, this water provided is negligible. It should also be noted that if the amount of water carried in by the gaseous reactant stream is negligible then water transport away from the membrane via the gas phase should also be negligible.
However, the aforementioned analysis only holds for the case of 100% conversion or near 100% conversion, whereas at low conversions, water transport between the membrane and gas phase can be non-negligible. Recently, Wheeler et al.103 analyzed the changes in the source of the outlet cathodic water with humidification of inlet CO2. In the case of dry CO2 feed, water diffusing from the anode was obviously the sole source for humidifying the outlet CO2 and CO2E products at the cathode, however, even in the case of the wet feed (70% humidified at 25 °C), water diffusing from the anode remained the major source for humidifying the outlet cathodic gases. In both of these cases, the cathodic outlet gas actually gained water due to the fact that the chemical potential of water at the cathode/membrane interface is basically that of liquid water and even at 70% humidification, this still provides a small driving force for water penetration to further humidify the outlet stream. Thus, the Wheeler et al.103 work clearly demonstrated that a humidified cathode does not provide sufficient water for the stoichiometric reaction but simply helps to mitigate the cathode from drying out due to evaporated water leaving via the cathode outlet.
Quantitatively the variation in water content in the cathode inlet (Jw,inlet) and outlet flow rates (Jw,outlet) comes about from the diffusion of water from the cathode out to the cathode flow fields (Jw,c,ff) as follows:
| Jw,c,ff = Jw,outlet − Jw,inlet | (16) |
In the case of 100% relative humidity of incoming gas, this term will be zero, however at 0% relative humidity, Jw,c,ff will be a non-zero value that is a function of flow fields, gas diffusion layers, and any other aspect that will affect mass transport. In the set-up from the Wheeler et al.103 work, the Jw,c,ff was shown to be ca. 0.2 × 10−4 mol cm−2 s−1 at an incoming CO2 flowrate of 25 sccm cm−2. This value was taken at open-circuit conditions, because, as noted above, reaction stoichiometry substantially influences the water balance and would convolute this value.
Another way water transports through the AEM is by electro-osmotic drag (EOD), wherein anions migrating from the cathode to the anode carry water in their hydrated form. The total electroosmotic drag flux (JEOD) can be calculated from the following equation:
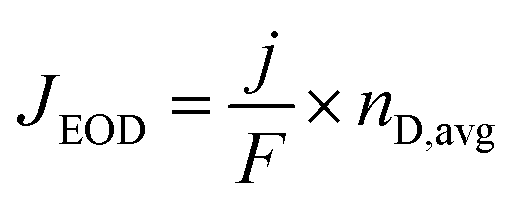 | (17) |
This topic has been lightly studied in the AEM fuel cell field with Jacobsen et al. showing that for a Tokuyama A201 AEM, each OH− carries ∼0.7 water molecules with them at 25 °C, but then this nD increases to approximately 1.3 at 50 °C.104 It should be noted that in this experiment, H2 was oxidized at the anode and H2O was reduced at the cathode. This prevented any potential gradient from influencing water transport but did provide a membrane that most probably had to vary λ across the membrane (see Fig. 10). Nevertheless, this should be quite relative to the conditions for CO2 electrolysis. With a set-up where both sides have vapor, Wang et al.105 showed an nD for OH− of 0.6 that was relatively independent of water content. More recently in a COE environment using a device with a catholyte (i.e. liquid on both sides of the AEM), we showed a water crossover value of 2.0 water per OH− using a Fumasep membrane (though this value does not deconvolute JEOD from JHP).106 It is interesting that in all these cases, the nD is actually less than the number of water molecules in the OH− hydration spheres, i.e., 4 in bulk water (Table 5).107
| Species | Hydration number | Hydrated radius [Å] | EOD coefficient (nD) in AEMa (@25 °C) | Ref. |
|---|---|---|---|---|
| a These values are for CO2 electrolysis for HCO3− and CO32− and for CO electrolysis for OH−. These values are also coupled with JHP but this term is expected to be minor. | ||||
| H2O | — | 1.46 | — | 108 |
| OH− | 4.0 ± 1.0 | 3.00 | 2.0 ± 0.1 | 105 and 109–112 |
| HCO3− | 5.3–6.9 | 3.64 | 6.7 ± 0.2 | 106, 113 and 114 |
| CO32− | 8.5–9.1 | 3.94 | 9.4 ± 0.7 | 106, 110 and 113–116 |
We also recently showed that operating under CO2 electrolysis conditions, with carbonate transfer leads to 9.4 water per carbonate (or 4.7 H2O/e−). While carbonates have a higher hydration shell than hydroxyl groups (see Table 5), this large water transfer is still relatively surprising. To demonstrate the importance of this, note that if for a 12 e− reduced molecule such as ethylene, ∼56 water molecules would transfer across the membrane per ethylene produced. Though experiments rarely operate with bicarbonate transfer through the AEM, bicarbonates transfer 6.7 water molecules per ion (or 6.7 H2O/e−) and would make this water crossover issue even more dramatic.
On the other hand, when looking at COE, a produced ethylene molecule should only result in ∼10 H2O per molecule. This substantial difference in EOD-based water transport during CO2E and COE significantly affect flooding in gas diffusion electrode-based devices and thus can affect performance significantly.
In addition to the water flux due to EOD, hydraulic pressure differences (also termed as back convection) affect the water flux at the cathode, and it can be expressed by the following equation:
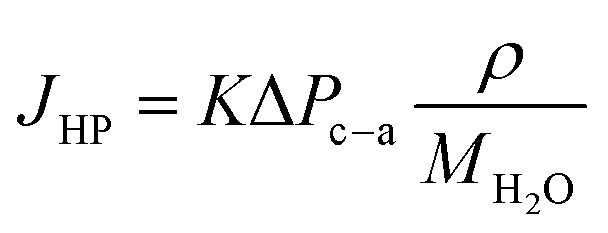 | (18) |
| ΔPc–a = Pc − Pa | (19) |
If we assume the membranes act as uniform cylindrical pores, we can also further describe K using the Hagen–Poiseuille equation as follows:
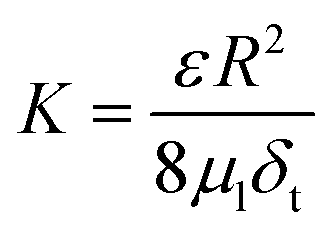 | (20) |
With the details of eqn (18) established, we can now analyze the pressure gradient across the membrane and what can influence that. Given CO(2) electrolyzers have a liquid on one side and a vapor on the other side, and the two sides are separated by a vertical separator, hydraulic pressure due to gravitation may provide a slight overpressure on the liquid side, most notably at the bottom of the cell. Since every 1 cm of water height corresponds to 0.98 mbar, this pressure may be notable for larger devices on the order of 100 cm high, however, for research-based test cells of <10 cm in height, it is much less of an issue. Pressure issues related to anolyte pumping may be a more prominent issue in these cases. In any case, a slight overpressure at the cathode allows for this to be balanced out. Another way to mitigate pressure-induced water penetration into the cathode is by using hydrophobic GDLs (which could be Teflon membranes) where the hydrophobicity of the GDL creates a capillary pressure (Pc) which can be determined using the following equation:
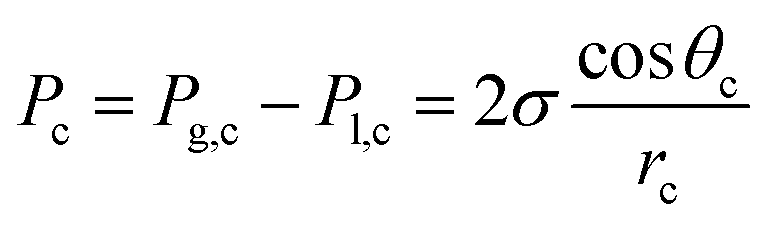 | (21) |
Since both Jdiff and JHP reduce with increasing the thickness of the membrane, for thicker membranes (such as Neosepta AMX ∼ 140 μm thickness),118 the water flux from EOD (JEOD) becomes a dominant force towards overall water flux. In this scenario, cathode dry-out could become a serious issue as the amount of water required for CO(2)E would not be sufficient even from a 100% humidified reactant stream. Therefore, thinner membranes are employed for CO(2)E, for example, Sustainion® AEM has a thickness of 50 μm.
Overall water balance in the electrolyzer
At this point, all the major terms influencing water have been denoted, allowing us to analyze the overall situation. The water buildup or drying out of the cathode, Jw@C, can be determined as:| Jw@C = JAEM,w − Jw,CO(2)E − Jw,c,ff | (22) |
Eqn (22) can be further analyzed by expanding the terms in Jw,c,ff and JAEM,w leading to the following:
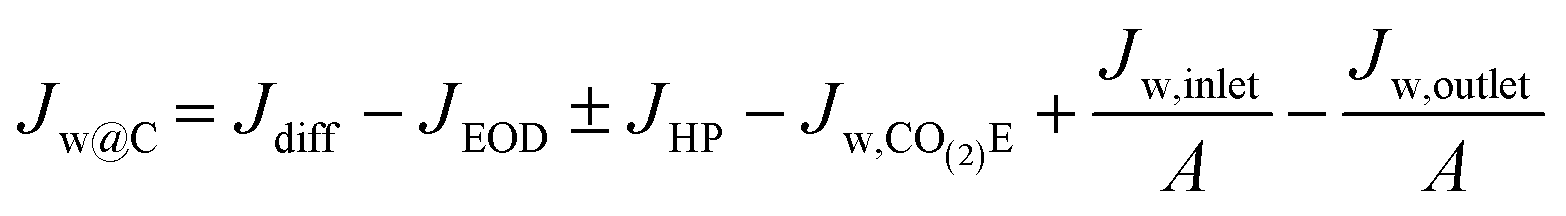 | (23) |
A further breakdown of eqn (23)viaeqn (7), (15), (17), (18) and rearrangement leads to:
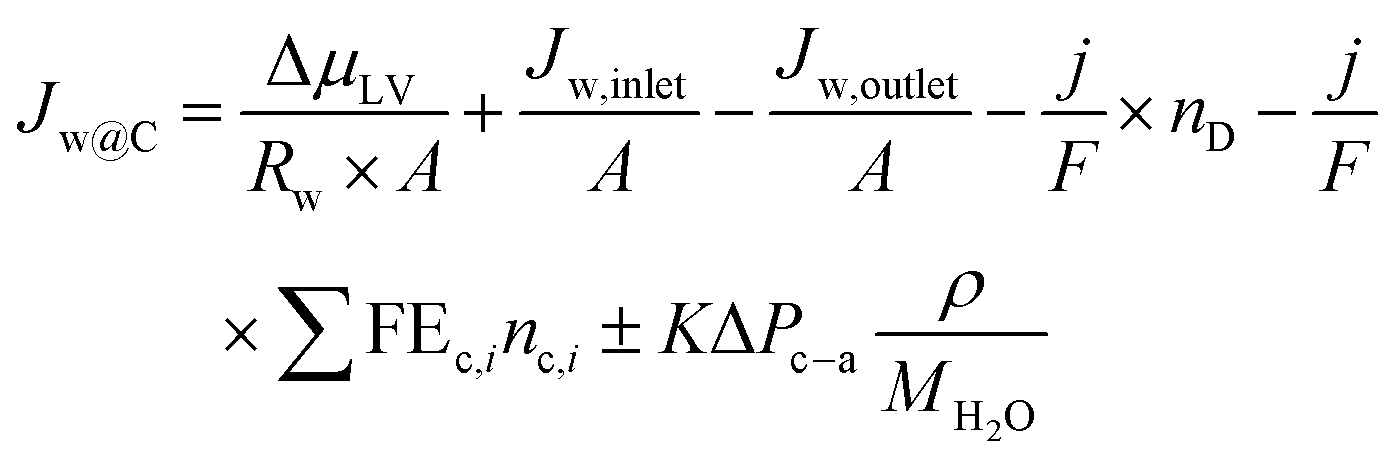 | (24) |
It should be noted in eqn (24), capitalized J relates to water fluxes whereas lowercase j is related to current density.
For eqn (24) a positive Jw@C entails the accumulation of water at the cathode, whereas a negative Jw@C entails drying out. As previously analyzed, Jw,inlet and Jw,outlet will only provide minimal control for the hydration content at the catalysts. However, the last term in eqn (24), which relates to hydraulic pressure, will play a more dominant role as this term will oppose the net value of all the other terms. As long as the hydraulic pressure is sufficiently large to oppose these other terms, this will allow Jw@C to be equal to zero. In other words, this hydraulic pressure flux term works to ensure that the catalyst is neither flooded nor dries out.
In light of eqn (23) and (24), we can examine how the correlation between different water fluxes affects the cathode flooding at different current densities. Even though the final term (i.e., the JHP term) is not a direct function of current density, it can still vary during electrolysis. This is because as the charge passed through the cathode increases, it starts to lose its hydrophobicity owing to electrowetting58 or loss of PTFE content in the MPL,119 which subsequently increases the hydraulic pressure gradient across the membrane (for more details on the role of electrode wettability in CO2E, see our previous review).31
By analyzing the steady-state situation in eqn (23) (i.e., Jw@C = 0) the net water flux across the membrane (JAEM,w) can be calculated as long as experimental data for Jw,c,ff (i.e., Jw,inlet & Jw,outlet) is available. Although we have not found reported data on Jw,c,ff for C2+ products in a CO(2) electrolyzer, Berlinguette and co-workers120 measured the Jw,c,ff values for CO formation in an MEA-based CO2 electrolyzer and showed that Jw,c,ff increases with the current density. From eqn (22) and (24), it is apparent that JEOD and Jw,CO(2)E increase with current density, which would cause Jw,c,ff to actually decrease. While JDiff could increase slightly to account for JEOD and Jw,CO(2)E providing a drier cathode, the net value of these three terms (JDiff, JEOD, and Jw,CO(2)R) would still be lead to a decrease in Jw,c,ff. Since Berlinguette and co-workers saw just the opposite, this would suggest JHP may be decreasing (from the loss of GDE hydrophobicity at higher currents), which would then lead to a higher Jw,c,ff. A decrease of JHP beyond a certain point would entail eqn (22) could not be kept at a steady state, which would lead to an accumulation at the cathode (i.e., Jw@C ≠ 0). While the authors observed a stable CO2E to CO formation until Jw,c,ff < 5 mg cm−2 h−1 (at 100 mA cm−2 using a high water uptake membrane, Fig. 11a), beyond this, it appears flooding took place as evidenced by decreased CO2E selectivity (Fig. 11b) and increased HER selectivity (Fig. 11c).
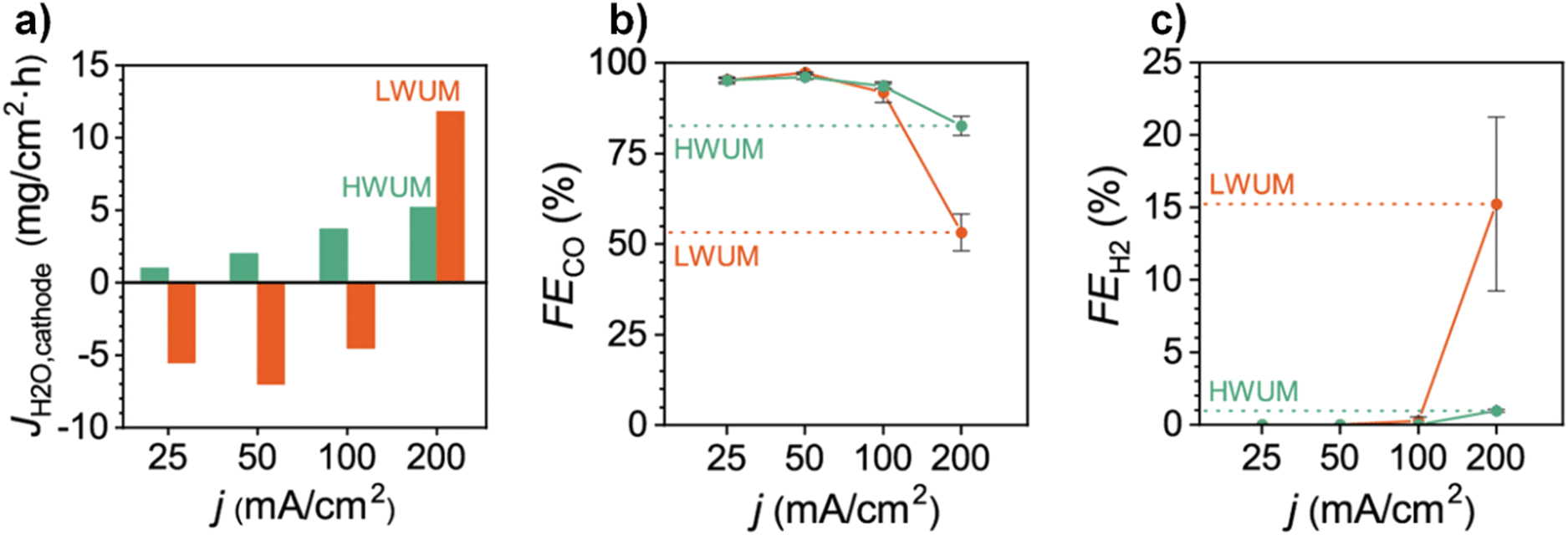 | ||
| Fig. 11 (a) Cathode water flux (Jw,c same as JH2O,cathode) at different current densities in an MEA-based CO2 electrolyzer having 55 μm thick high water uptake membrane (HWUM) and low water uptake membrane (LWUM). Differences in faradaic efficiencies of (b) CO and (c) H2 as a function of different current densities. Reproduced with permission.120 Copyright 2020, American Chemical Society. | ||
AEM's ionic conductivity in CO(2)E environments
While the dominant loss in CO(2)E devices relates to the cathodic and anodic overpotentials, Tafel kinetics entails these scale logarithmically with current, whereas membrane-based ion transfer losses scale linearly (Ohm's law). Thus, at commercially relevant current densities, membrane losses start to play an increasingly important role. This is further complicated by the fact that different ions may be transported through the AEM (OH−, CO32−, HCOO−etc.), each with their own ohmic resistance.When comparing anionic versus cationic species, the ionic mobility of OH− in dilute aqueous solutions is almost 43% lower than that of H+ (see Table 6).32 Furthermore, the dilute solution mobilities of HCO3−/CO32− are 65–77% lower (Table 6) than that of the OH− attributed to a combination of their larger size and inability to undergo structure diffusion (Grotthus).121 Similarly, the common anions' ionic conductivity (in aqueous solution) follows the same trend as their ionic mobilities (Table 6) albeit with less pronounced differences compared to the comparison of cations. Translating these results to ionic conductivity in AEMs, the Sustainion® membrane (X24) conduction at 60 °C is 64 mS cm−1 in OH− form, but only 24 mS cm−1 with HCO3−.122
| Ion | Ionic conductivity (Λ, 10−4, m2 S mol−1) | Diffusion coefficient (D, 10−5 cm2 s−1) | Mobility (μ, 10−8 m2 s−1 V−1) |
|---|---|---|---|
| H2O | — | 2.3 | — |
| H+ | 349.6 | 9.3 | 36.2 |
| Li+ | 38.7 | 1.0 | 4.0 |
| Na+ | 50.1 | 1.3 | 5.2 |
| K+ | 73.5 | 2.0 | 7.6 |
| Cs+ | 77.2 | 2.1 | 8.0 |
| OH− | 198.0 | 5.3 | 20.5 |
| HCO3− | 44.5 | 1.2 | 4.6 |
| CO32− | 69.3 | 0.9 | 7.2 |
| Cl− | 76.3 | 2.0 | 7.9 |
Given that AEMs and CEMs have vastly different functional groups and chemistries, AEM optimization does allow for AEMs to compete with CEMs in terms of conductivity. One optimization approach is to increase the charge density (or IEC) of the AEMs, which has allowed for similar levels of AEM conductivity (in OH− form) compared to CEMs.123 Recently, researchers have developed highly conductive AEMs even with low IEC by making changes in the polymeric backbone in a way that the densely and uniformly distributed cationic groups allow solvation spheres to overlap and thus provide enhanced ion transport.124–127 It should be noted that the AEM's conductivity is much more dependent on maintaining a high hydration level compared to CEM.
While increasing the IEC of an AEM is generally easy to achieve, introducing an excessive number of cationic functional groups into the polymer backbone leads to higher osmotic pressure,128 resulting in dramatically increased water uptakes, lower AEM conductivities and permselectivities, and reduced mechanical stability (without the employment of any mitigation measures).129 Consequently, many researchers have been successful in limiting water uptakes while increasing the charge density (IEC) by using several different approaches: (1) introducing crosslinks into the AEM;130 (2) attaching hydrophobic chains onto the cation functional groups (sometimes called extender chains);131 and (3) by employing a composite reinforcement component.71
The second area where some (particularly early generation) AEMs struggle in comparison to CEM is a lack of well-defined phase separation between the hydrophobic/hydrophilic microphases. This typically leads to tortuous ion pathways that reduce AEM conductivity.132 Subsequently, many strategies including block copolymerization,133,134 and introducing longer alkyl spacer/extender chains,135–138 have been used to develop AEMs with nanophase-separated morphologies in AEMs to achieve better OH− (or anion) transport efficiencies with less excessive levels of swelling.139–142 Additionally, substituting conventional benzyl-based side-chain quaternary ammonium cations with cyclic cationic functionality (e.g., piperidinium) in the polymer main-chain backbone has been shown to facilitate both anion conduction in AEMs and improved stability in concentrated KOH solutions.143–146 One such AEM, called PiperIon TP-85 (developed by W7 energy), demonstrated a higher OH−/HCO3−/CO32− conductivity and chemical stability over high alkaline solutions compared to the Sustainion membrane (Table 7). Recently, the Janáky group147 reported that PiperIon membranes (0.36 Ω cm2) showed almost half the membrane area specific resistance (ASR) compared to Sustainion (X37-50) membranes (0.85 Ω cm2) and were able to outperform the Sustainion membrane in their MEA-based CO2 electrolyzer by achieving over 57% higher current density on Ag cathodes at 3 V and 60 °C (0.1 M CsOH anolyte, with 12.5 mL min−1 cm−2 CO2 feed rate). Table 7 presents ionic conductivities of commonly used AEMs in different anion forms (but do note that not all OH− conductivities listed are exactly comparable or “true”80 due to the variety of the measurement methods used with varying levels of CO2 exclusion).
| Membrane | Conductivity (mS cm−1) | Ref. | ||
|---|---|---|---|---|
| OH− form | HCO3− form | CO32− form | ||
| HDPE stands for high-density polyethylene. | ||||
| FAA-3 | 30.8 | 1.5 | 2.8 | 90 |
| PAP-TP-85 | 58.2 | 5.0 | 6.3 | 90 |
| Tokuyama A201 | 32.8 | — | 9.0 | 156 |
| Quaternary AEMs with different IECs | 16.9, 16.0, 17.4, 24.6, 28.4 | — | 4.0, 2.5, 3.2, 4.3, 4.8 | 156 |
| PEEK-like quaternary ammonium AEM | 21.2 | — | 9.9 | 157 |
| PEEK-like AEMs | 27.8, 43.4, 62.0, 32.0 | — | 12.6, 20.5, 31.2, 15.2 | 158 |
| Sustainion® X24 | 64 (RT) and 102 (80 °C) | 24 (RT) and 66 (80 °C) | — | 122 |
| Sustainion® X37-50 | >130 (70 °C) | 7.9 (30 °C) | 7.8 (30 °C) | 147 and 159 |
| PiperIon TP-85 | >135 (70 °C, 50 μm) | 9.5 (30 °C, 32 μm) | 8.4 (30 °C, 32 μm) | 147 and 160 |
| Aemion | >80, no specified temperature | — | — | 161 |
| Quaternary ammonia poly(N-methyl.piperidine-co-p-terphenyl), QAPPT | 49 (30 °C) and 137 (80 °C) | — | — | 162 |
| Orion TM1 (m-TPN1) | 54 (30 °C) | — | — | 163 |
| HDPE-radiation-grafted benzyltrimethylamine (TMA) | 208 (80 °C and 80% RH) | 57 (80 °C and 80% RH) | — | 80 |
| HDPE-radiation-grafted benzyltriethylamine (TEA) | 54 (80 °C and 80% RH) | 14 (80 °C and 80% RH) | — | 80 |
| HDPE-radiation-grafted benzyl-N-methylpiperidine (MPIP) | 115 (80 °C and 80% RH) | 28 (80 °C and 80% RH) | — | 80 |
| HDPE-radiation-grafted benzyl-N-methylpyrrolidine (MPY) | 152 (80 °C and 80% RH) | 59 (80 °C and 80% RH) | — | 80 |
| HDPE-radiation-grafted benzyl-N,N-diethylmethylamine (DEMA) | 151 (80 °C and 80% RH) | 46 (80 °C and 80% RH) | — | 80 |
With CO2E often providing mixed ionic transport (OH−/HCO3−/CO32−), it is difficult to isolate effects due to ionic conductivity in an operational device, however, with COE only having OH− transporting through the membrane during steady-state operations, such analysis is simplified.
When comparing the state-of-the-art OH− conducting AEMs in Table 7versus an H+ conducting CEM such as Nafion (78 mS cm−1 @ room temp),148 there are competitive AEM conductivities. However, the use of HCO3−/CO32− AEM forms will entail a substantial increase in ohmic losses. For instance, Liu et al.149 showed that the conductivity of Sustainion (X37-50) AEM dropped from 80 mS cm−1 in 1 M KOH to almost 20 mS cm−1 in 1 M KHCO3 at 30 °C. Recently, Salvatore and Berlinguette150 demonstrated, using an AEM-based electrolyzer operating at 200 mA cm−2 (with 1 M KOH as anolyte and Sustainion X37-50 AEM), that membrane loss contributes significantly towards overall voltage consumption, accounting for ∼50% (i.e. 0.71 ± 0.10 V) of device overpotentials. This is more than expected given that Sustainion has an ex situ estimated ASR of 0.132 Ω cm2 in 1 M KOH at 40 °C (in situ ASR could be different due to differences in water content during operation and thus may affect conductivity).151 This ASR would be expected to lead to an ohmic loss of 0.026 V (a mere 3.7% of total membrane losses when performing CO2E in the MEA electrolyzer). However, the latter calculation considers pure OH− conductivity at 40 °C, while predominantly CO32− conduction through the AEM at 30 °C occurs in Salvatore and Berlinguette's experiments.150 The expected real ohmic losses through the Sustainion AEM would be higher (ca. 0.10 to 0.15 V) for CO32− conduction, but this leaves at least 0.56 V loss unaccounted for. This unaccounted voltage loss could be due to interfacial losses existing as an artifact from Schroeder's Paradox (as explained in Section 3.2). Although the interfacial resistance is minimal in the case where AEM is exposed to liquid on one side and saturated vapor on the other, the transport of CO32− will further reduce the water uptakes to OH− (see Fig. 8) at the cathode-membrane interface, leading to increased interfacial resistances.
An alternative to the AEM would be a bipolar membrane (BPM), however, with a Fumasep FBM, it was shown that at 200 mA cm−2 the overpotential due to the BPM was ∼2.6 V, owing to inefficient water dissociation reaction happening at the internal CEM/AEM interface of the BPM.152
The use of a catholyte in an AEM-based electrolyzer adds substantial additional resistance towards ion conduction attributed to the interface of the AEM/catholyte (the Donnan exclusion effect), and Burdyny and Smith40 modeled this loss for both 1 M KHCO3 and KOH (Fig. 12). Salvatore and Berlinguette150 showed that voltage at 200 mA cm−2 of a catholyte/AEM-based electrolyzer is much higher (4.61 ± 0.11 V) compared to a catholyte-free AEM analogue (2.73 ± 0.07 V). These additional voltage losses (1.88 V) using a catholyte-based configuration were attributed to the high interfacial losses (arising both from Schroeder's Paradox and Donnan exclusion)153–155 and ions conduction through the catholyte layer (0.69 ± 0.16 V). In contrast, Burdyny and Smith40 modeled an ohmic loss of around 0.27 V at 200 mA cm−2 in 1 M KOH (the same electrolyte used by Salvatore and Berlinguette). The differences between the ohmic losses in the two works could come from the fact that 1 M KOH catholyte in Salvatore's work will react with CO2 to form a mixture of KOH and KHCO3, and thus the observed ohmic losses in Salvatore's work are higher than 0.27 V (for 1 M KOH) and lower than 0.78 V (for 1 M KHCO3) (Fig. 12).
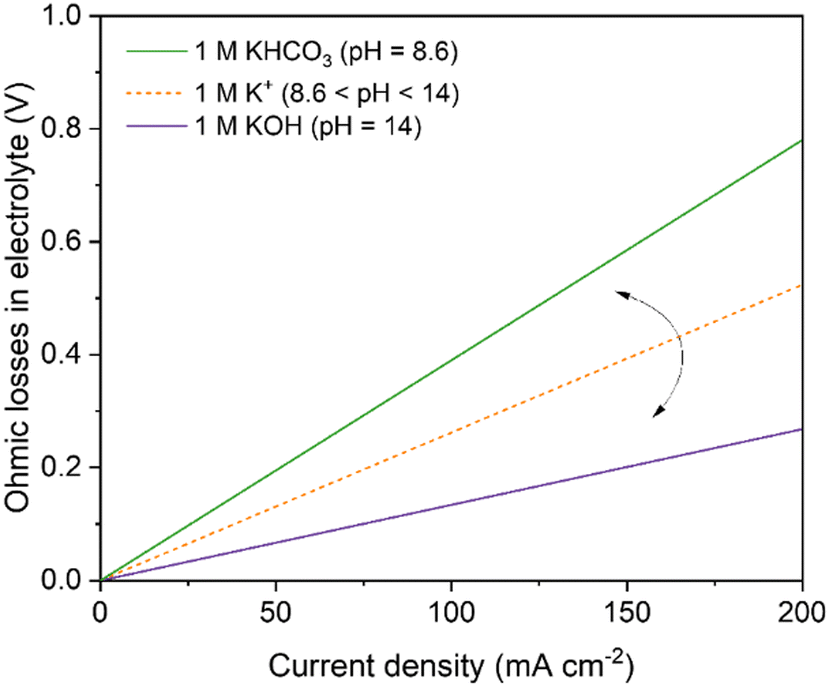 | ||
| Fig. 12 Projected ohmic losses in conventionally used electrolytes as a function of current density in flow-cell electrolyzer having a catholyte + anolyte layer thickness of 3 mm operating at 25 °C. The curved arrow in the figure shows that the ohmic losses for 1 M mixture of KOH + KHCO3 will vary between their respective ohmic losses at 1 M pure composition. | ||
Chemical stability of the AEMs
AEMs have traditionally exhibited poor chemical stabilities in highly alkaline conditions (especially when less than fully hydrated) compared to CEMs in highly acidic conditions, leading to CEMs having ca. one order of magnitude higher in-situ lifetimes (e.g., in fuel cells and electrolyzers).32,164–166 Since AEMs have been increasingly used in fuel cell and water electrolysis (green H2) fields, the mechanism behind the low chemical stability of AEMs in alkaline solutions is becoming more understood. The lifetimes of the AEMs are often limited by the innate instability of the membrane materials to the nucleophilic OH−,167 attacking both the anchored cation functional groups and the polymeric backbone of the AEM (the latter especially when containing heteroatom links), leading to multiple degradation pathways.32The OH− attack on the anchored cationic species in the AEM results in the removal of the positive charges and conductivity loss.32,168 The quaternary ammonium group, the most commonly used headgroups in the AEM literature, can be degraded by either nucleophilic substitution at the α-carbon (bonded to the functional group) or via Hoffmann elimination to the β-hydrogen (where there are hydrogens attached to the β-carbon) (Fig. 13). At lower membrane hydration levels, the energy barrier for Hoffmann elimination is significantly lower than the nucleophilic substitution.169 Studies have suggested that increasing the length of the alkyl functionalities can slow down the Hoffman elimination via steric hindrance.170,171 On the other hand, the OH− attack on imidazolium groups (normally present in Sustainion®) leads to the breakage of the imidazolium ring (Fig. 13).
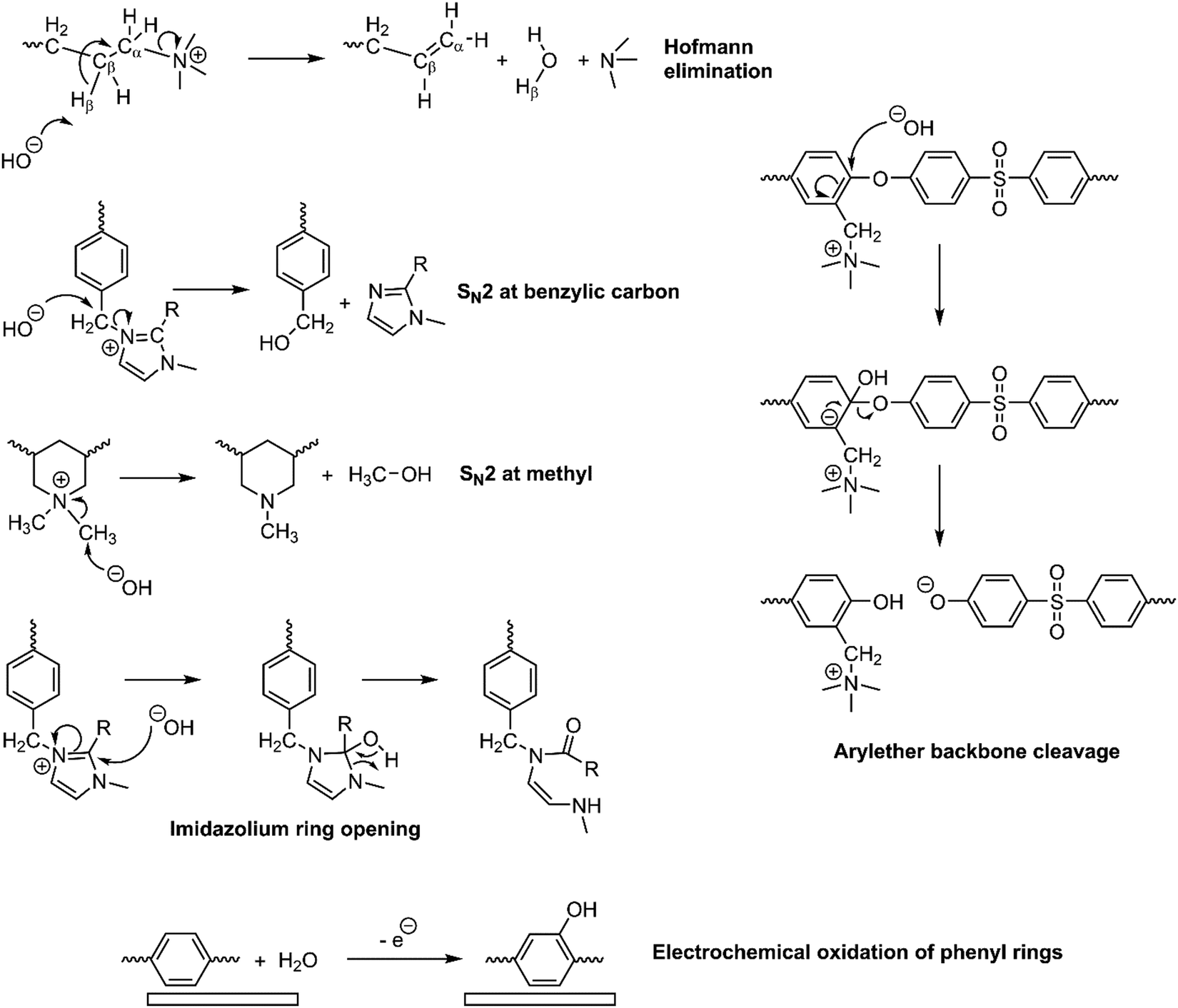 | ||
| Fig. 13 Potential chemical degradation pathways that can occur in widely used AEMs. Note: the degradation pathways shown are commonly discussed mechanisms (others can occur) and are not necessarily specific to the membrane type shown. | ||
Although most studies on AEM stability have focused on alkali-derived/chemical degradations (historically, because alkali degradations were so predominant and fast with older generations of AEMs), they are not the only degradation pathways. Now that later generations of “alkali-stable” AEMs are coming online,168 other radical and electrochemical oxidative degradation processes need to be considered, especially where polymer electrolytes are in contact with electrocatalysts at oxidative potentials. Non-precious-based OER/ORR catalysts (especially involving Fe) could degrade the AEM via radical oxidation reactions172 (also called Fenton oxidation), whereas platinum group metals (PGM) electrocatalysts can also cause oxidation of phenyl groups when in contact with polyaromatic polymer electrolytes, forming phenol groups (Fig. 13).173
The degradation of cationic groups in AEMs is not solely linked to the nature of the cation itself and can be affected by other factors.174 For example, weakly hydrated OH− (i.e., ultra high pH) are aggressively nucleophilic, resulting in faster degradation of AEMs when they are less than fully hydrated. The lower hydration of OH− can arise from low humidification of the AEM or when an AEM is in contact with a high concentration of aqueous OH−. Some of the initial breakthrough papers175–181 in the high current density CO2E field used a highly alkaline (up to 7 M KOH) catholyte to increase the C2+ production, meaning less hydration of OH− and reduced water uptake of the AEM due to lower activity of water in the concentrated electrolyte. While this approach in CO2E is commercially unsustainable because of the CO2 scavenging by the concentrated KOH, in the COE field (lacking the presence of carbonate species), highly alkaline electrolytes can be used. Having a high concentration of ions in the electrolyte allows for the formation of ion-aggregates through adsorption of co-ions and counter-ions (e.g., KOH), and thereby inhibits membrane swelling (i.e., low water content), which causes ionic channels to diminish for water to transport. Therefore, AEM degradation in flow cells having highly alkaline and concentrated electrolytes is anticipated to be higher than in MEA-based electrolyzers.
With most AEM in CO2E generally operates in carbonate form, degradation concerns in CO2E is less relevant due to lower concentration of highly reactive (aggressive) OH− nucleophile, while this could become a serious issue in COE. However, AEM ionomers (especially Aemion, Sustainion, and PiperIon) are less stable in the presence of HCO3−/CO32− as compared to OH−182 entailing that the use of AEM ionomers is less stable in CO2E than in COE.
Moreover, the production of liquid CO(2)E products, especially, ethanol could affect AEM stability. Owing to a lower dielectric constant of ethanol (ε = 24.30) compared to water (ε = 78.54), ethanol allows severe alkaline conditions.183,184 At a lower dielectric constant, the hydration of ions decreases, causing an enhancing reactivity between the ethoxide/OH− and the cationic group of the AEM, thus promoting the cation degradation rate in the AEM.
In terms of the polymer backbone, even a normally durable polymer can be compromised upon functionalization with quaternary ammonium groups. For example, the anchoring of electron-withdrawing effects of groups (such as trimethylammonium) to benzene rings in the polymer chains that are linked via heteroatoms (such as ether links – e.g., polysulfone) can lead to break down of the polymer backbone, even though such polyaromatic backbones are alkali-resistant when free of anchored quaternary ammonium groups.185 Discoloration along with embrittlement of an AEM after application in CO2/CO electrolysis could suggest degradation of the polymer backbone. Several studies in alkaline membrane fuel cells have demonstrated to reduce the effect of cation-induced destabilization of polymer backbone through removing heteroatom links, incorporating of aliphatic spacers,138,186,187 crosslinking,123,188 charge delocalization,189 and other means.190–193
Despite AEM demonstrating excellent CO(2)E efficiency and product selectivity,23 few studies focus on the chemical/alkaline stability of the AEMs from a CO(2)E perspective. At present, most of the CO(2)E studies are conducted for less than 200 h43,67,194–197 with only a few long-term studies available between 1000–4000 h.198,199 However, Yin et al.87 investigated this and found <5% degradation of cationic groups in their AEM (QAPPT) after a 100 h CO2E test on an Au/C-based MEA electrolyzer.
It should be appreciated that in situ degradation of an AEM in a device may be masked if it is in contact with aqueous alkaline solutions (at the anode and/or cathode). The AEM conductivity (ohmic resistance in the cell) or cell performance may degrade at a slower rate compared to chemical degradation as the degraded AEM may contain imbibed aqueous alkali and as such, still act as a separator that conducts anions. Long-term testing of durable electrolyzers (100–1000 h plus) should not rely purely on changes in device voltage and resistance (if testing AEM operando lifetimes but must include some form of post-mortem analysis of the components (e.g., spectroscopic analysis of the post-test AEM).
The current state-of-the-art of ion-selective membranes for CO(2)E
The following section presents an overview of the different ion-exchange membranes (CEM, BPM, and AEM) in the literature reported for CO(2)E at industrially relevant current densities. We show how AEM-based electrolyzers performed better in comparison to those using CEM and BPM for CO2E and COE in Fig. 14 and 15 respectively (detailed information on the source data is presented in the ESI†). The data plotted in Fig. 14 and 15 are based on studies performed under commercially relevant conditions (at or above 100 mA cm−2), with FE greater than 50% for C1 products, using predominantly flow cells or zero gaps. We excluded CO2E studies involving alkaline electrolytes (either catholyte or anolyte) such as KOH for CO2E since OH− forms (bi)carbonates with CO2, as mentioned previously. Without consistent KOH addition, this provides a non-steady state operation and additionally makes determining energy efficiency impossible due to non-steady state pH-induced biases. Similarly, we excluded COE studies involving neutral electrolytes (e.g., KHCO3), as the pH will gradually increase till it becomes stable at the steady state due to cathodically produced OH−s.200 For a list of CO2E data with different electrolytes (including hydroxide electrolytes), we refer the readers to Wakerley et al.25 review on CO2E in GDEs. When sufficient data were available, the energy efficiency was also analyzed for CO(2)E and presented in Fig. 14 and 15.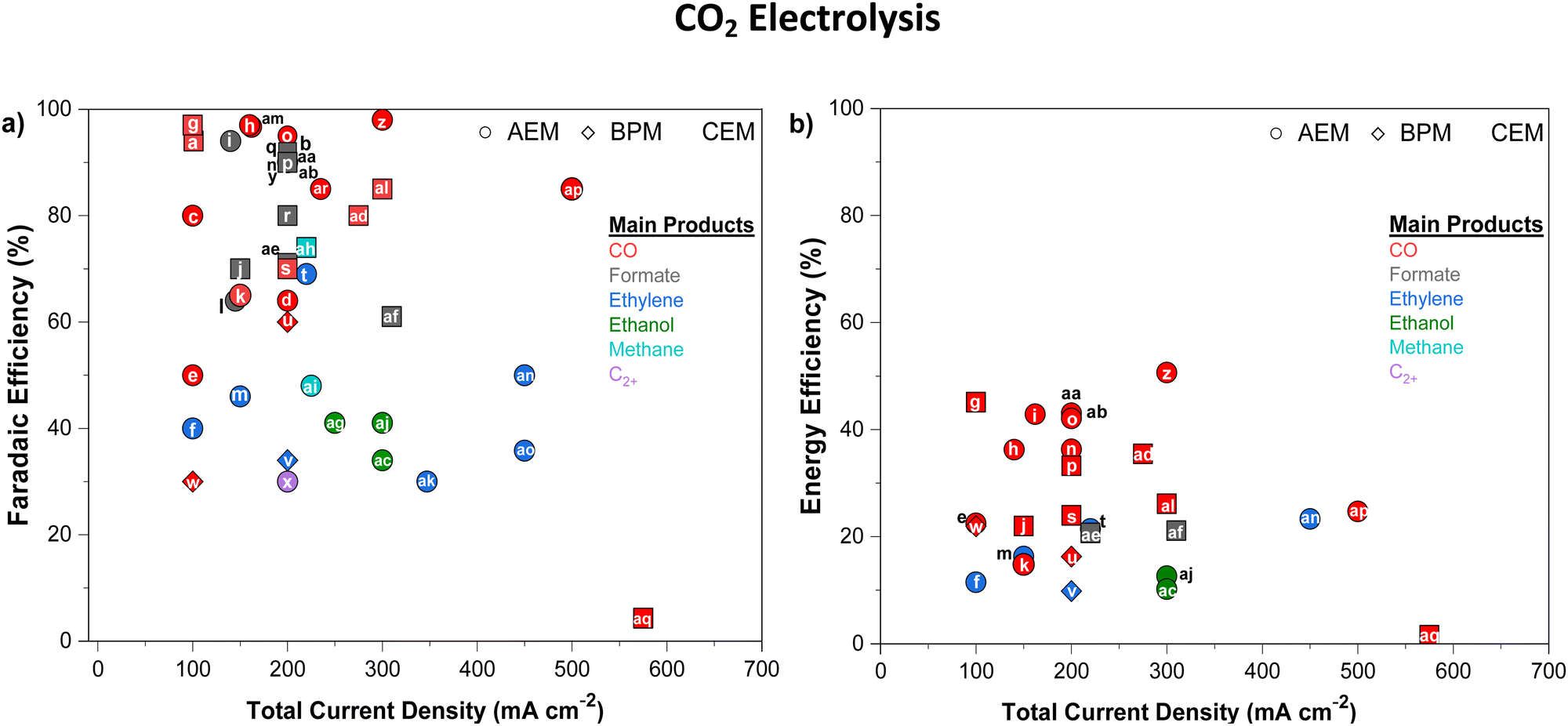 | ||
| Fig. 14 (a) Faradaic efficiency and (b) energy efficiency of different gaseous and liquid products for CO2E at different current densities. The symbols refer to the type of ion-selective membranes, while the different colors indicate the selectivity of the main product. The link of each data point (as shown in (a) and (b)) to the literature is located in ESI,† Table S1. | ||
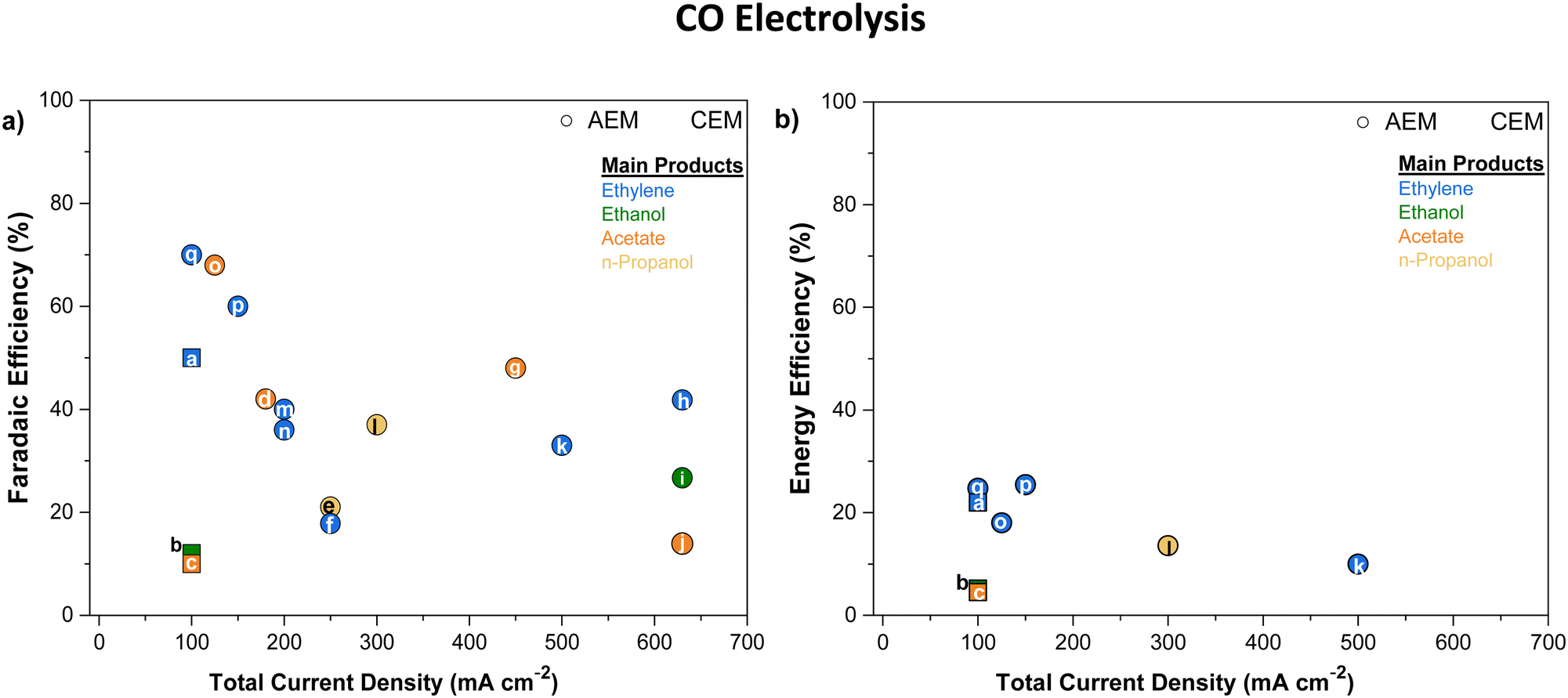 | ||
| Fig. 15 (a) Faradaic efficiency and (b) energy efficiency of different gaseous and liquid products for COE at different current densities. The symbols refer to the type of ion-selective membranes, while the different colors indicate the selectivity of the main product. The link of each data point (as shown in (a) and (b)) to the literature is located in ESI,† Table S2. | ||
CO2 electrolysis (CO2E)
| AEM commercial name | Company | Product | Counter ion (anion) | IEC (meq g−1) | Water uptake (wt%) | Tensile strength (MPa) | Thickness (μM) | Elongation at break (%) | ASR (Ω cm−2) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| N.A. stands for no data available and D.S. stands for data sheet (provided by the manufacturer). | ||||||||||
| Fumasep FAA3 | Fumatech | FAA-3-30 | Br− | 1.7–2.1 | <19 | 25–40 | 26–34 | 20–40 | 0.3–0.5 (Cl− form) | D.S. |
| FAA-3-50 | 1.85 | 10–25 | 25–40 | 45–55 | 15–60 | 0.6–1.5 (Cl− form) | ||||
| FAA-3-PK-75 | 1.39 | 10–20 | 30–60 | 75 | 10–30 | 1.2–2.0 (Cl− form) | ||||
| FAA-3-PK-130 | 1.1–1.4 (Cl−) | 10–25 | 40–80 | 110–130 | 15–40 | 1.8–4.0 (Cl− form) | ||||
| Selemion TMV | AGC Engineering Co. | AMVN | Cl− | 1.9 | 15 ± 2 | 0.3 | 100 | N.A. | 2.27 | D.S.,221 |
| ASVN | 2.1 | 15 ± 2 | 0.2 | 100 | N.A. | 4.76 | ||||
| DSV | 2.0 | 15 ± 2 | 0.15 | 95 | N.A. | 1.1 | ||||
| Sustainion | Dioxide Materials | X37-50 RT | Cl− | 2.52 | > 80 | N.A. | 50 | N.A. | 0.045 (1 M KOH) | 23, 48, 122 and 222 |
| X37-50 Grade 60 | 2.52 | 70–80 | N.A. | 50 | N.A. | N.A. | ||||
| PiperIon | W7-energy | PiperIon20 | HCO3− | 2.35 | 50 | >30 | 20 | >20 | N.A. | D.S. |
| AEMION | Ionomer | AF1-HNN8-25-X | I−/Cl− | 2.1–2.5 | 33–37 | 60 | 25 | 50–65 | 0.063 | D.S.223–225 |
| AF1-JMM5-25-X | 1.4–1.7 | 33–37 | 60 | 25 | 85 | 0.21–0.33 | ||||
| Orion TMI | Orion Polymer | m-TPN1 | Br− | 2.1 | 25 | 29 | 15–25 | 36 | 0.18 | D.S.,163,226 |
| A201 | Tokuyama | A201 | F− | 1.8 | 44 ± 5 | 96 (Cl− form) | 28 | 62 | N.A. | D.S.,227,228 |
Sustainion® AEMs have become a benchmark in this field and are utilized in many recent studies.216 For instance, Kutz et al.198 demonstrated a continuous stable operation of CO2E to CO (with FECO > 95%) featuring the zero-gap MEA for 3800 hours at 3 V. Similarly, Gabardo et al.211 performed a continuous 100 h CO2E operation while producing C2+ products at >100 mA cm−2, reporting the longest non-OH− mediated operation for C2H4 production with 40% selectivity. Targeting C2H5OH as the main product, Wang et al. used Sustainion-based AEMs with functionalized Cu cathode electrocatalysts in a zero-gap MEA-based CO2 electrolyzer, showing high selectivity up to 50% at 3.67 V (JC2H5OH ∼ 125 mA cm−2).217 Despite the extensive use of Sustainion AEMs in literature, these AEMs are unstable at a temperature above 60 °C over longer timeframes (particularly in less than fully hydrating conditions) and allow CO2/product crossover43 to the anode during CO2E.
The QAPPT-type AEMs have also shown promising results for CO(2)E, especially at higher temperatures (≥60 °C) regime due to their higher OH− conductivity and chemical stability.218 For example, Yin et al.87 used QAPPT-AEM operating at current densities of 500 mA cm−2 for CO2E to CO (at 60 °C), reaching a FECO > 90% at around 3 V. Recently, Endrődi et al.147 utilized a PiperIon-AEM (85% QAPPT-type and 15% CF3-based polymer backbone) in a zero-gap electrolyzer and achieved the highest ever reported current density to date (>1 A cm−2) in 0.1 M CsOH anolyte with a FECO of 90% and total cell voltage varying between 2.6–3.4 V. Even though these QAPPT-type AEMs showed better CO2E performance than Sustainion® AEM, the crossover of CO2 remains an issue.
In an effort to decrease CO2 crossover, McCallum et al.219 developed a multiphysics model for the MEA electrolyzer to investigate the effect of different parameters such as membrane thickness, CO2 partial pressure, and membrane charge on CO2 utilization at the cathode. Their model found that reducing the membrane thickness allowed greater back diffusion of H+ from the anode to the cathode. This helped shift the cathode side pH to less alkaline conditions and thus hamper the flux of HCO3−/CO32− to the anode. In contrast to McCallum's modeling work, Mardle et al.220 showed an increase in CO32− crossover rate (probably from increased CO32− diffusion through the AEM) when decreasing the membrane (AEMION) thickness from 50 to 25 μm, however, employing thinner AEM showed better CO2E stability.
From a product crossover perspective, McCallum et al.219 showed that elevating the electrolyzer temperature allows for increased evaporation and transportation of ethanol (or other volatile CO2E products) from the GDE into the outlet cathode stream. Earlier, Gabardo et al.211 experimentally showed ethanol cathode to anode ratio to increase by almost 4 times when increasing the temperature from 20 to 40 °C. On the other hand, reducing the CO2 partial pressure or CO2 feed flow rate only slightly decreases the ethanol crossover.219
In terms of CO2E using catholyte-based cells, the reported performances are similar to those obtained from the MEA-based cells using AEMs. For example, Kibria et al.209 reported a FEC2+ of 77% at 400 mA cm−2 and cathodic potential of −1.8 V versus RHE, using restructured Cu catalyst with 1 M KHCO3 as catholyte. Zhong et al.177 used Fumasep AEM and achieved an 80% selectivity for C2+ products over a modified de-alloyed Cu–Al (deposited on a PTFE substrate) at 600 mA cm−2. It is interesting to see that in most of the flow-cell studies, commercially developed AEMs (e.g., Fumatech) are implemented, presumably due to a better mechanical handling property compared to the newly developed Sustainion® and QAPPT-type AEMs.147
Cation exchange membranes (CEMs)
CEMs,102 such as the perfluorinated sulfonic acid (PFSA)-based Nafion have many benefits due to their high proton conductivities,229 impressive durability,230 and ability to prevent products and carbonates crossing over to the anode66,231 However, as noted previously, zero-gap CEM membranes tend to favor HER over CO(2)E due to either salting-out when operating in non-acidic anolytes or due to a lack of a sufficient homogenous electric field (caused by the lack of non-H+ cations) in acidic and pure water anolytes. However, incorporating a buffer layer between the CEM and the cathode assists in controlling the pH changes at the cathode interface and thereby decreasing the energy loss associated with the acid-base neutralization at the interface and tuning the product selectivity.232,233 For instance, Vennekoetter et al.233 compared different reactor designs for CO2E using CEM and reported a FECO of 56% over Ag GDEs after adding a buffer layer between the CEM and the Ag at 300 mA cm−2. Using the same approach, Delacourt et al.232 observed an increase in FECO from ∼ 0 to over 80% after inserting an aqueous KHCO3 buffering layer between a Nafion and Ag cathode operating at 20 mA cm−2. Similarly, by adding a 0.1 M KHCO3 buffer layer Kopljar et al.234 reported the highest formate selectivity (FEHCOO− = 90% over commercial Sn nanoparticles at 200 mA cm−2) reported in the literature, however, mass transfer issues decreased performance at higher current densities (<70% FE@250 mA cm−2).Schmid and co-workers199 resolved the cation crossover CEM issue by having a flowing catholyte and anolyte, which are mixed after the reactor and then recycled back into both the cathode and anode chambers. Provided there is sufficient flow and recycling in the reactor, this approach guarantees a constant pH and ion concentration on both sides. However, if liquid products are produced this electrolyte mixing may lead to product oxidation via the anode. With this approach, they demonstrated that using Ag GDEs and Nafion CEM at 300 mA cm−2 they were able to reach ∼60% FECO for 1200 hours of operation.199 As Ag only produces minimal amounts of formate, liquid product oxidation was not considered a serious issue in their work. Likewise, Jeanty et al.235 also used a similar approach with a stable 60% FECO for over 800 hours. Unfortunately, the post reactor mixing approach only works on flow-type cells, whereas for the zero-gap MEA cells, cation crossover will simply lead to salting-out as KHCO3 for CO2E or KOH for COE. For COE, increased KOH concentration may lead to corrosion issues before salting out of KOH would be reached.
The Sargent group took a different approach to suppress HER when using a CEM by adding a phosphate buffer layer (pH = 1–4) to the catholyte-based electrolyzer and reported high CO(2)E selectivity at current densities above 400 mA cm−2.63 While the buffer layer increased ohmic losses and still had 50% selectivity to H2, they provided an interesting proof of concept that CO2E and HER at high current densities can result in intense OH− production to neutralize the local environment of a bulk pH = 1 electrolyte. As expected, this approach only worked when there was a K+ cation in the catholyte to provide a sufficient electric field for CO2E to occur. It should be noted that though this work was done in a flow cell, the principle behind this approach should still hold in a zero-gap membrane. However, CEMs have an effective acidic concentration near pH = 0,61 which is an order of magnitude higher proton concentration compared to the Sargent work, and thus this would entail it would be significantly more challenging to neutralize the local environment based purely on hydroxyl formation from high current densities of CO2E/HER.
Effects of membrane parameters on the overall water balance
In general, it should be noted that AEM's water migration also depends on the water uptake (see Fig. 11a), membrane thickness, and operational temperature of the AEM. Primarily these membrane effects relate to varying resistance, Rw, in the JDiff term, and potentially JEOD since nD could be affected (see eqn (23) and (24)). By measuring the composite JAEM,w term, Reyes et al.120 demonstrated that AEMs having low water uptake (LWU) show lower net water flux through the AEM (from the anode to the cathode) in comparison to membranes having high water uptake (HWU), at current densities ≤ 100 mA cm−2. They attributed this change to HWU starting to swell more and thus allowing lower resistance for water to transport across the AEM. However, at 200 mA cm−2, JAEM,w became higher in the LWU than in HWU, causing the cathode to flood more with LWU and thus promoting the HER (see Fig. 11c). The increase in JAEM,w with LWU (at high current densities) might be due to a sharp increase in voltage at 200 mA cm−2 that could degrade GDE hydrophobicity,58,119 leading to a decreased JHP and thus an increased JAEM,w.In the same paper,120 the authors also suppressed HER at high current density CO2E operation in LWU AEMs by decreasing the AEM thickness. A thinner membrane should increase Jdiff by decreasing internal resistance, thus promoting flooding and an increase of HER. While it is unclear why the thinner membrane suppresses HER, we have seen a similar trend in our labs (unpublished) to validate this phenomenon.
Increasing temperature also modifies water transport. At higher temperatures, the polymeric backbone relaxes thus facilitating the increased formation of hydrated ionic channels, which causes the membrane to swell more. This effect allows hydrated species to carry water molecules more easily to the anode.102,236 In other words, this increases the nd value in the JEOD term. For example, Shafaque et al.237 showed a 38% increment in water uptake of CEM when increasing the temperature from 25 to 60 °C in an MEA-based CO2 electrolyzer. Moreover, water diffusivity in the AEM increases with the temperature, thus increasing JDiff. In addition, the temperature can affect the viscosity of water and the relative humidity (RH) of the feed CO2, which in turn can affect JEOD and JDiff,flow, respectively.103 Selectivity has also been shown to be a function of temperature,238 entailing Jw,CO(2)E is a function of temperature. Thus with all of the terms in eqn (23) as a function of temperature and minimal CO(2)E temperature experiments in literature, there is still a large number of unknowns with regards to how temperature affects water management with anion exchange membranes.
In summary, water transport in AEM involves multiple mechanisms via different driving forces in AEM channels during CO(2) electrolysis (see Fig. 16). At times, these mechanisms are frequently coupled, however, to the best of our knowledge, no studies have been conducted to deconvolute and understand the relative impact of each water transport mechanism. Given that there is a vast amount of literature available on water transport for fuel cells, there is already a path provided to develop and understand these underlying mechanisms from a CO(2)E perspective.
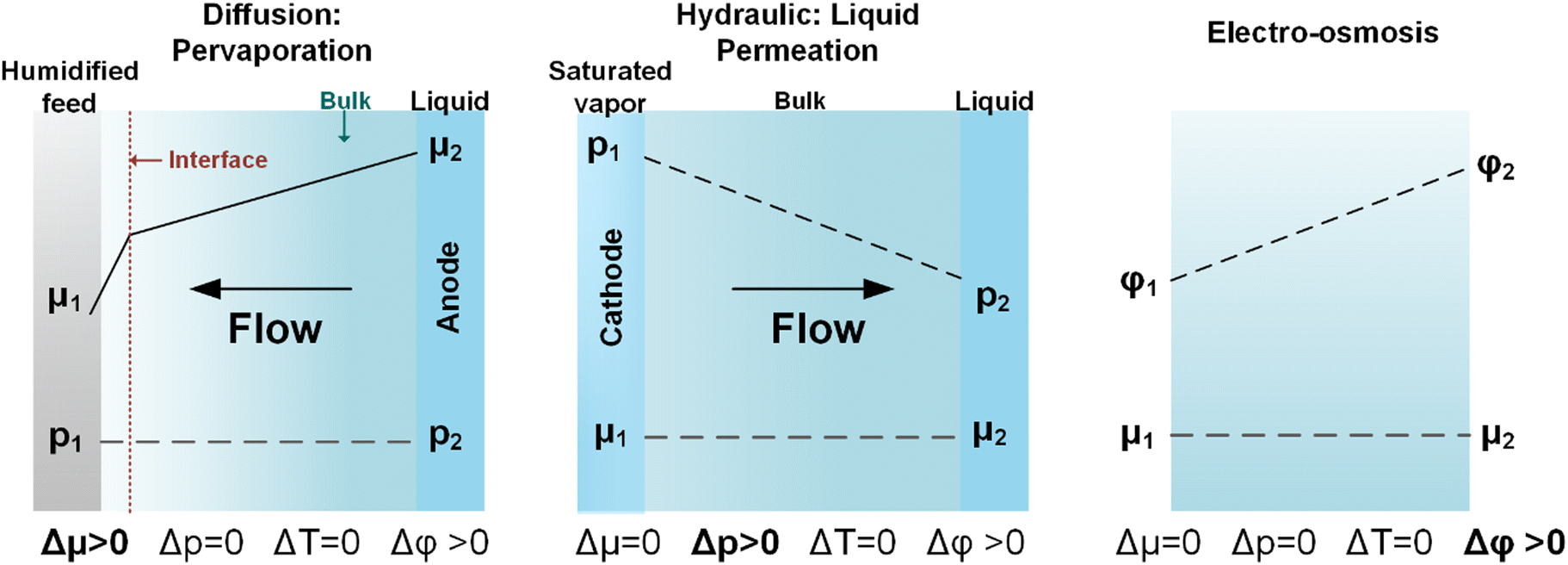 | ||
| Fig. 16 Schematic of the water transport mechanism in an AEM via different driving forces. | ||
Bipolar membranes (BPMs)
Bipolar membranes (BPM) can overcome some of the issues associated with AEMs and CEMs, albeit with an added potential increase due to the membrane's characteristics. Li et al.66 studied the use of the Fumasep BPMs (reverse bias) for syngas production, using an Ag-GDE or Bi/ionic-liquid cathode and an alkaline NiFeOx OER anode at various current densities. Although BPM-led electrolyzers showed lower pH-induced losses compared to the AEM and CEM counterparts, the BPMs allowed higher onset potentials and energy losses. These overpotentials are largely associated with the water dissociation reaction and further neutralization reactions between H+ and HCO3− at the interface (i.e., induced Donnan potential), affecting the overall electrolyzer performance as noted by the CO selectivity dropping after 1 h with apparent mass transport limitations above 200 mA cm−2. Salvatore et al.67 addressed the existing limitations of the BPM's cation exchange layer (CEL) part by incorporating a solid-supported liquid layer to regulate water management. This approach allowed for a stable FECO of 65% at 100 mA cm−2 for 24 h at 3.5–3.6 V.Recently, Blommaert et al.239 highlighted the importance of the BPM orientation in MEA-based CO2 electrolyzers, since the orientation determines the local reaction environment, the direction of the electric field at the interface, and the ionic transport mechanism. The BPM in forward-bias mode (where OH− and H+ meet at interface inside BPM and form water so no water splitting overpotential) over Ag-catalyst showed a lower FECO (decreased from 20% to 0% after 20 min of CO2E) due to higher water content and salt accumulation. The lack of water dissociation reaction in the membrane interface can decrease the cathode potential by reducing the chemical potential, but it facilitates the GDE flooding and the salt crossover through the AEL. In the case of the reverse bias, a stable FECO was reported (∼60%), despite the intrinsic limitations on this orientation associated with the acidic conditions at the cathode, significant CO2 crossover and higher cathodic potential (>2 times) were observed.
To reduce the ohmic losses from a thick catholyte layer and still maintain a suppressed HER, Yan et al.240 fabricated a BPM with a weak-acid CEL (<100 nm). This CEL was formed by alternatively introducing a strong CEL (of the BPM) to solutions of poly(acrylic acid) and poly(allylamine hydrochloride) via a layer-by-layer (LBL) assembly. This dilution of the CEL facilitated an improvement in the FECO as compared to a standard perfluorosulfonated-based BPM. However, the enhancement in FECO is lower than what was generally observed in alkaline conditions, and this was attributed to relatively low local pH at the LBL–BPM interface. In general, the design of BPM specifically for CO2/CO electrolyzers is currently limited due to fewer commercially available membranes with optimal control of swelling, high conductivity, and charge density for these applications, as current BPMs are mainly implemented in electrodialysis.241
A catholyte approach with an alkaline electrolyte helps reduce ohmic losses in the electrolyte and provides similar selectivities to a zero-gap approach. While the scavenging of CO2 by KOH entails this as an unsustainable process, in theory, this does allow a GDE to be more intimately analyzed. However, given that carbonates formed by CO2 equilibration with KOH reduce the pH, this non-steady-state pH makes the quantification of both product amounts and the thermodynamic driving forces quite complex.40 It should also be noted that operating in a buffered carbonate electrolyte can still lead to localized pH gradients. Burdyny and Smith40 showed in their model that the local pH can become ∼13 at both 0.1 and 1 M KHCO3 at current densities >100 mA cm−2. However, in contrast to alkaline catholytes, these reactors can reach an equilibrated state, allowing these local pH effects to be more easily and properly analyzed.200
Inspired by solid-state batteries, Xia et al.206 employed a solid-state electrolyte (SSE) in between the AEM (facing the cathode) and CEM (facing the anode) to suppress the liquid crossover and produced pure formic acid (0.1 M) stably for almost 100 h. Similarly, Miao et al.69 inserted a porous proton exchange layer (PPEL) in between the AEM and the CEM to increase proton conduction from the anode and reduce ethanol crossover from the incoming anode flow. Using PPEL, the authors restricted the ethanol crossover to the anode to less than 1% and further achieved a maximum ethanol concentration of 13.1 wt% (at the cathode) stably for over 20 h at 200 mA cm−2 by further reducing the PPEL layer from ∼2 to ∼0.75 mm.
To prevent CO2 crossover without significantly increasing in cell voltage, Xu et al.242 installed a very thin and highly porous micro-channel solid electrolyte layer between the AEM and the CEM. As a result, the authors successfully reduced the CO2 crossover and only lost between 3 to 4% CO2 to the anode while simultaneously regenerating CO2 internally in the porous solid electrolyte channel (CO2 recycled back to the cathode inlet stream). The authors also pursued an alkali-metal cation-free approach to prevent salt precipitation by employing fixed poly(aryl piperidinium) cations on the Cu catalyst layer and obtained a ∼43% C2H4 selectivity for over 200 h and with no signs of salt formation during and after CO2E.
CO electrolysis (COE)
As highlighted earlier, COE does not have the carbonate issues that CO2E has, meaning AEMs are optimally suited for this approach. However, AEMs are not as developed and durable as CEMs, so earlier works on COE used CEMs. For example, Schwartz et al.243 utilized a PFSA-based membrane and showed COE to C2+ products at 600 mA cm−2 and very high cathodic potential (−3.2 V vs. NHE) over Cu-based GDEs. Recently, researchers from Siemens244 used Nafion 117 and Cu NPs GDE to demonstrate an 89% C2+ selectivity at 300 mA cm−2, corresponding to an 18.6% single-pass CO conversion. Their 20 h long COE test at 200 mA cm−2 showed a 2% decrease in the selectivity of C2H4 (with initial FE ∼ 40%) and a ∼100 mV increase in cathodic potential. In this work, they used their previously mentioned approach of mixing catholyte and anolyte to maintain a stable pH environment.244 However, because of this, they could not quantify liquid products during their stability test.For COE with AEMs, Jiao and co-workers81 used a Sustainion® GDE electrolyzer in 2 M KOH capable of reaching 630 mA cm−2 with C2+ selectivity over 91% and a 26% CO single-pass conversion. Li et al.245 utilized a commercial Fumasep AEM for COE to alcohol production (ethanol and propanol combined FE ∼ 40%) at a partial current density of 277 mA cm−2 over a Pd-doped Cu catalyst. In another COE study by Kang and co-workers,246 30% acetate selectivity was achieved over atomically dispersed Cu catalysts at 144 mA cm−2. Ripatti et al.229 investigated COE using Cu-GDEs and interdigitated flow fields to carefully control gas and ion transport. By using a continuous-flow electrolyzer configuration, the study reported a stable electrolysis operation at current densities >100 mA cm−2, with a 68% single-pass CO conversion, FEC2+ of 75%, and cell potential of 2.5 V. While using a zero-gap approach, they demonstrated a direct production of 1.1 M potassium acetate at a cell potential of 2.4 V over 24 h. (Unfortunately, the KOH used to produce potassium acetate is typically of higher economic value.)
Given that this is such a new and rapidly developing field, there has been a lack of standardization as well as a focus on initial performance rather than durability. Because of this, data comparison between different studies is difficult and often not appropriate.247 These issues are exacerbated by the fact that this field is much more complex compared to the water electrolysis field from which it is often translating knowledge. Measuring selectivities of both gaseous and liquid products can be challenging, especially since slight variations can make a substantial difference in productivity. Furthermore, outlet volumetric flow measurements necessary to analyze gaseous products are complicated by the fact that varying product compositions make calibrating thermal conductivity or viscosity-derived flow meters extremely challenging, and thus favoring more non-traditional ways to measure outlet flow rates. High conversion rates and alkaline scavenging of CO2 to carbonate reactions make these outlet flow measurements essential).39 Additionally, product crossover to the anode, the humidity of CO(2) stream, cathodic working potential, and pH, bubble management, are all issues that have not only yet to be optimized or standardized, in many cases, they are not even analyzed. Thus, while the current literature relating to AEM, CEM, and BPM CO(2)E is informative, there is still a substantial amount of unknown trends and relationships that need to be discovered concomitantly to be able to fully isolate the influence of a given membrane on CO(2)E.
Future directions
In terms of membrane integration into CO2 and CO electrolysis, CEMs need a paradigm-shifting approach to resolving the poor CO(2)E selectivity versus H2 evolution and while BPMs can prevent CO2 crossover and maintain high CO(2)E selectivity, they can not do both simultaneously and have the additional issue of a substantially higher loss across the membrane compared to CEM or AEM. While both CEM and BPM can resolve their issues by adding a catholyte, this adds substantial ion transfer losses. Thus currently, there is not a clear future direction in terms of membrane integration as evidenced by the very diverse set of ideas that are currently being tested.The high selectivity and energy efficiency of AEM-based CO2 electrolysis have allowed this to be the front-runner technology, and we envision for this to continue to be the case. CO2 crossover is by far the largest bottleneck as this both loses CO2 and decreases membrane conductivity substantially. Designing AEM to selectivity conduct OH− over CO32− initially sounds appealing, but on a closer look will be highly challenging. This relates to the fact that once CO32− is formed anywhere there is a strong thermodynamic driving force for it to stay as a CO32−. With the pKa to CO2 (through a HCO3− intermediate) being at pH = 7.8, and pH within the membrane pH ≈ 14,248 this pH difference of 6 will create a Nernstian driving force of ∼ 360 mV (@298 K) favoring CO32− over OH−. If any membrane were to overcome this barrier to selectively conduct OH− over CO32− this 360 mV barrier would be incorporated into the ohmic resistance. Decreasing the local pH of the membrane could help lower this barrier, but this would entail reducing the AEM cationic functional groups, which would both hurt conductivity and increase product crossover. With pH a log scale in terms of OH−, and conductivity more linearly tied to OH− ions, a focus on OH− selective AEM, could easily do more harm than good.
Downstream CO2 separation may actually lead the future direction of the CO2E field for AEM-based approaches, or at least this issue needs to be more fully investigated to give a proper perspective into the issue of how troublesome CO2 crossover is. Note though that unlike direct air capture of CO2 that tries to up concentrate 420 ppm CO2 from an N2/O2 mixtures, traditional AEM-based CO2E produces a stream containing 50–75% CO2 in O2, thus allowing much easier separations. Analyzing and optimizing this approach falls traditionally into the domain of separation experts rather than electrochemists thus potential developments could take place in terms of separation techniques. Additionally, various anodic reactions beyond O2 evolution, such as Cl2 evolution or partial oxidation of biomass, could greatly simplify anodic CO2 separations while providing additional value to the electrolysis reactions.
In terms of pure AEM development, future AEMs for CO(2)E should have the following attributes:
• High OH− conductivity for COE and HCO3−/CO32− for CO2E
• Low area specific resistance
• High water uptake (between 50 to 80%) with low swelling (<10%)
• Stable in alkaline conditions (pH between 10–14)
• Stable at elevated temperatures (40–100 °C)
• Compatible with liquid CO(2)E products, for e.g., insoluble in 10 wt% alcohol
• Sufficient mechanical strength to allow high pressure and temperature
Thus new membrane chemistries are envisioned to find optimal chemistry that maximizes all these parameters. In addition to the aforementioned parameters, crosslinking and other techniques could allow narrow channels, thus preventing cation crossover to the cathode and CO(2)E crossover to the anode. From a simple ohm's law analysis, thinner membranes will reduce ohmic loss, but increase the diffusion gradient for undesired species crossing the membrane, thus optimizing this balance with every evolving membrane will be essential. However, from a deeper analysis, a substantial amount of ohmic loss relates to interfacial issues, thus mitigating these can also reduce losses.
Another under-investigated area is the pre-treatment/activation of AEMs. Excessive alkaline soaking may degrade the membrane, and any interactions with air will lead to some amounts of carbonate-based species within the membrane. Understanding these issues and developing protocols for consistent membrane testing is increasingly becoming important. Furthermore, as CO(2)E accelerates towards commercialization, even higher technology readiness level (TRL) issues need to be analyzed, such as cheap, scalable synthesis and processing techniques, long-term durability, and analysis of what will eventually degrade from the membrane and whether this could potentially contaminate the catalysts.
Conclusions and outlook
This work demonstrates that understanding and optimizing membranes for CO2 electrolysis is substantially more complex than that for water electrolysis, H2 fuel cells, or other commercialized electrochemical processes. The fact that CO2/CO catalysis demands an alkaline environment greatly restricts membrane flexibility. This has allowed for anion exchange membranes to dominate the field. Being much less developed than cation exchange membranes, this entails the literature and understanding of these membranes are much more restricted.With CO2 naturally forming negatively charged carbonates, this does create substantial issues in not only dealing with CO2 transferring from the cathode to the anode but also understanding the properties of a carbonate saturated membrane as opposed to the OH− form AEM used in H2 fuel cells and water electrolyzers. Furthermore, the need for cations to be at the catalyst interface for CO(2) catalysis also complicates and restricts our flexibility to design and operate membranes effectively. Building on these complications, water management at the cathode/membrane interface (at least in zero-gap cells) is an additional parameter to manage.
All of these parameters together demonstrate the complexity of the analysis that needs to be done on these AEM to fully characterize and optimize them for CO(2)E. Here, we tried to express these in a comprehensive framework that showed the core principles occurring as well as highlighting where no literature was available. While on the state-of-the-art CO(2)E section shows that these membranes can produce quite impressive results, the remainder of this work shows substantial understanding and optimizations can be made to push these devices to giving further improved performances.
Author contributions
Sahil Garg is the lead author who wrote the bulk of the manuscript, including the introduction, role of ion-exchange membrane types, ion and water transport in AEMs for CO(2)E, AEMs ionic conductivity in CO(2)E environment, and chemical stability of the AEMs. Carlos Giron Rodriguez wrote the part on the current state-of-the-art of ion-selective membrane for CO(2)E. Thomas Rufford gave input into editing various section involving AEMs and roles of AEMs in electrolysis. John Varcoe also provided input on the overall manuscript especially on AEMs in general. Brian Seger set the overall direction of the review and helped in editing the overall manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to this perspective/review has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 85144, (SELECT-CO2) as well as the Villum Center for the Science of Sustainable Fuels and Chemical grant 9455.References
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- D. Pletcher, Electrochem. Commun., 2015, 61, 97–101 CrossRef CAS.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- A. Demirbas and G. Arin, Energy Sources, 2002, 24, 471–482 CrossRef CAS.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- S. Verma, B. Kim, H.-R. M. Jhong, S. Ma and P. J. A. Kenis, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS.
- S. Shiva Kumar and V. Himabindu, Mater. Sci. Energy Technol., 2019, 2, 442–454 Search PubMed.
- I. Moussallem, J. Jörissen, U. Kunz, S. Pinnow and T. Turek, J. Appl. Electrochem., 2008, 38, 1177–1194 CrossRef CAS.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2019, 12, 1950–1968 RSC.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- M. Li, S. Garg, X. Chang, L. Ge, L. Li, M. Konarova, T. E. Rufford, V. Rudolph and G. Wang, Small Methods, 2020, 4, 2000033 CrossRef CAS.
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- F. Pan and Y. Yang, Energy Environ. Sci., 2020, 13, 2275–2309 RSC.
- X. Tan, C. Yu, Y. Ren, S. Cui, W. Li and J. Qiu, Energy Environ. Sci., 2021, 14, 765–780 RSC.
- Z.-Z. Wu, F.-Y. Gao and M.-R. Gao, Energy Environ. Sci., 2021, 14, 1121–1139 RSC.
- L. Sun, V. Reddu, A. C. Fisher and X. Wang, Energy Environ. Sci., 2020, 13, 374–403 RSC.
- M. Jouny, G. S. Hutchings and F. Jiao, Nat. Catal., 2019, 2, 1062–1070 CrossRef CAS.
- H. Zhang, J. Li, M.-J. Cheng and Q. Lu, ACS Catal., 2019, 9, 49–65 CrossRef CAS.
- D. A. Salvatore, C. M. Gabardo, A. Reyes, C. P. O’Brien, S. Holdcroft, P. Pintauro, B. Bahar, M. Hickner, C. Bae, D. Sinton, E. H. Sargent and C. P. Berlinguette, Nat. Energy, 2021, 6, 339–348 CrossRef CAS.
- R. A. Tufa, D. Chanda, M. Ma, D. Aili, T. B. Demissie, J. Vaes, Q. Li, S. Liu and D. Pant, Appl. Energy, 2020, 277, 115557 CrossRef CAS.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Nat. Rev. Mater., 2022, 7, 55–64 CrossRef CAS.
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2019, 4, 317–324 CrossRef CAS.
- M. König, J. Vaes, E. Klemm and D. Pant, iScience, 2019, 19, 135–160 CrossRef.
- M. Moura de Salles Pupo and R. Kortlever, Chem. Phys. Chem., 2019, 20, 2926–2935 CrossRef CAS.
- U. O. Nwabara, E. R. Cofell, S. Verma, E. Negro and P. J. A. Kenis, ChemSusChem, 2020, 13, 855–875 CrossRef CAS PubMed.
- M. Li, M. N. Idros, Y. Wu, T. Burdyny, S. Garg, X. S. Zhao, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2021, 9, 19369–19409 RSC.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- T. Zhou, R. Shao, S. Chen, X. He, J. Qiao and J. Zhang, J. Power Sources, 2015, 293, 946–975 CrossRef CAS.
- Q. Duan, S. Ge and C.-Y. Wang, J. Power Sources, 2013, 243, 773–778 CrossRef CAS.
- L. Liu, G. Huang and P. A. Kohl, J. Mater. Chem. A, 2018, 6, 9000–9008 RSC.
- Z. Lu, G. Polizos, D. D. Macdonald and E. Manias, J. Electrochem. Soc., 2008, 155, B163 CrossRef CAS.
- D. F. Alves-Lima, X. Li, B. Coulson, E. Nesling, G. A. H. Ludlam, R. Degl’Innocenti, R. Dawson, M. Peruffo and H. Lin, J. Membr. Sci., 2022, 647, 120329 CrossRef CAS.
- C. G. Arges, J. Parrondo, G. Johnson, A. Nadhan and V. Ramani, J. Mater. Chem., 2012, 22, 3733–3744 RSC.
- M. Ma, S. Kim, I. Chorkendorff and B. Seger, Chem. Sci., 2020, 11, 8854–8861 RSC.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J.-J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- K. G. Schulz, U. Riebesell, B. Rost, S. Thoms and R. E. Zeebe, Mar. Chem., 2006, 100, 53–65 CrossRef CAS.
- G. O. Larrazábal, P. Strøm-Hansen, J. P. Heli, K. Zeiter, K. T. Therkildsen, I. Chorkendorff and B. Seger, ACS Appl. Mater. Interfaces, 2019, 11, 41281–41288 CrossRef.
- Y. C. Li, Z. Yan, J. Hitt, R. Wycisk, P. N. Pintauro and T. E. Mallouk, Adv. Sustainable Syst., 2018, 2, 1700187 CrossRef.
- M. Lin, L. Han, M. R. Singh and C. Xiang, ACS Appl. Energy Mater., 2019, 2, 5843–5850 CrossRef CAS.
- J. Zhang, W. Luo and A. Züttel, J. Catal., 2020, 385, 140–145 CrossRef CAS.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377, 1–35 CrossRef CAS.
- H. Yang, J. J. Kaczur, S. D. Sajjad and R. I. Masel, J. CO2 Util., 2017, 20, 208–217 CrossRef CAS.
- Z. F. Pan, L. An, T. S. Zhao and Z. K. Tang, Prog. Energy Combust. Sci., 2018, 66, 141–175 CrossRef.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- H. Shin, K. U. Hansen and F. Jiao, Nat. Sustainable, 2021, 4, 911–919 CrossRef.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2020, 13, 977–985 RSC.
- Á. Vass, B. Endrődi, G. F. Samu, Á. Balog, A. Kormányos, S. Cherevko and C. Janáky, ACS Energy Lett., 2021, 3801–3808, DOI:10.1021/acsenergylett.1c01937.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2020, 13, 646–647 RSC.
- M. C. O. Monteiro, M. F. Philips, K. J. P. Schouten and M. T. M. Koper, Nat. Commun., 2021, 12, 4943 CrossRef CAS PubMed.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS.
- B. Endrődi, A. Samu, E. Kecsenovity, T. Halmágyi, D. Sebők and C. Janáky, Nat. Energy, 2021, 6, 439–448 CrossRef PubMed.
- M. E. Leonard, L. E. Clarke, A. Forner-Cuenca, S. M. Brown and F. R. Brushett, ChemSusChem, 2020, 13, 400–411 CrossRef CAS PubMed.
- B. Endrődi, E. Kecsenovity, A. Samu, F. Darvas, R. V. Jones, V. Török, A. Danyi and C. Janáky, ACS Energy Lett., 2019, 4, 1770–1777 CrossRef.
- B. Cermenek, J. Ranninger and V. Hacker, in Ethanol, ed. A. Basile, A. Iulianelli, F. Dalena and T. N. Veziroğlu, Elsevier, 2019, pp. 383–405 DOI:10.1016/B978-0-12-811458-2.00015-8.
- B. Seger, K. Vinodgopal and P. V. Kamat, Langmuir, 2007, 23, 5471–5476 CrossRef CAS PubMed.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC press, 2007 Search PubMed.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- D. A. Vermaas and W. A. Smith, ACS Energy Lett., 2016, 1, 1143–1148 CrossRef CAS.
- S. Z. Oener, M. J. Foster and S. W. Boettcher, Science, 2020, 369, 1099–1103 CrossRef CAS PubMed.
- Y. C. Li, D. Zhou, Z. Yan, R. H. Gonçalves, D. A. Salvatore, C. P. Berlinguette and T. E. Mallouk, ACS Energy Lett., 2016, 1, 1149–1153 CrossRef CAS.
- D. A. Salvatore, D. M. Weekes, J. He, K. E. Dettelbach, Y. C. Li, T. E. Mallouk and C. P. Berlinguette, ACS Energy Lett., 2018, 3, 149–154 CrossRef CAS.
- T. S. Zhao, Z. X. Liang and J. B. Xu, in Encyclopedia of Electrochemical Power Sources, ed. J. Garche, Elsevier, Amsterdam, 2009, pp. 362–369 DOI:10.1016/B978-044452745-5.00240-9.
- R. K. Miao, Y. Xu, A. Ozden, A. Robb, C. P. O’Brien, C. M. Gabardo, G. Lee, J. P. Edwards, J. E. Huang, M. Fan, X. Wang, S. Liu, Y. Yan, E. H. Sargent and D. Sinton, Joule, 2021, 5, 2742–2753 CrossRef CAS.
- M. Hren, M. Božič, D. Fakin, K. S. Kleinschek and S. Gorgieva, Sustainable Energy Fuels, 2021, 5, 604–637 RSC.
- S. Maurya, S.-H. Shin, Y. Kim and S.-H. Moon, RSC Adv., 2015, 5, 37206–37230 RSC.
- M. Mandal, G. Huang, N. U. Hassan, X. Peng, T. Gu, A. H. Brooks-Starks, B. Bahar, W. E. Mustain and P. A. Kohl, J. Electrochem. Soc., 2019, 167, 054501 CrossRef.
- K. Yassin, I. G. Rasin, S. Brandon and D. R. Dekel, J. Membr. Sci., 2020, 608, 118206 CrossRef CAS.
- V. Dubey, A. Maiti and S. Daschakraborty, Chem. Phys. Lett., 2020, 755, 137802 CrossRef CAS.
- T. Zelovich, L. Vogt-Maranto, M. A. Hickner, S. J. Paddison, C. Bae, D. R. Dekel and M. E. Tuckerman, Chem. Mater., 2019, 31, 5778–5787 CrossRef CAS.
- D. Dong, W. Zhang, A. C. T. van Duin and D. Bedrov, J. Phys. Chem. Lett., 2018, 9, 825–829 CrossRef CAS PubMed.
- D. Muñoz-Santiburcio and D. Marx, Nat. Commun., 2016, 7, 12625 CrossRef PubMed.
- T. J. Omasta and W. E. Mustain, in Anion Exchange Membrane Fuel Cells: Principles, Materials and Systems, ed. L. An and T. S. Zhao, Springer International Publishing, Cham, 2018, pp. 1–31 DOI:10.1007/978-3-319-71371-7_1.
- N. Ziv, W. E. Mustain and D. R. Dekel, ChemSusChem, 2018, 11, 1136–1150 CrossRef CAS PubMed.
- S. Haj-Bsoul, J. Varcoe and D. R. Dekel, J. Electroanal. Chem., 2022, 116112, DOI:10.1016/j.jelechem.2022.116112.
- M. Jouny, W. Luc and F. Jiao, Nat. Catal., 2018, 1, 748–755 CrossRef CAS.
- L. D. Chen, Nat. Catal., 2021, 4, 641–642 CrossRef CAS.
- K. Fukuta, H. Inoue, S. Watanabe and H. Yanagi, ECS Trans., 2009, 19, 23–27 CrossRef CAS.
- U. Krewer, C. Weinzierl, N. Ziv and D. R. Dekel, Electrochimica Acta, 2018, 263, 433–446 CrossRef CAS.
- J. Pan, L. Zhu, J. Han and M. A. Hickner, Chem. Mater., 2015, 27, 6689–6698 CrossRef CAS.
- P. Długołęcki, K. Nymeijer, S. Metz and M. Wessling, J. Membr. Sci., 2008, 319, 214–222 CrossRef.
- Z. Yin, H. Peng, X. Wei, H. Zhou, J. Gong, M. Huai, L. Xiao, G. Wang, J. Lu and L. Zhuang, Energy Environ. Sci., 2019, 12, 2455–2462 RSC.
- B. Eriksson, H. Grimler, A. Carlson, H. Ekström, R. Wreland Lindström, G. Lindbergh and C. Lagergren, Int. J. Hydrogen Energy, 2019, 44, 4930–4939 CrossRef CAS.
- P. v Schroeder, Z. Phys. Chem., 1903, 45U, 75–117 CrossRef.
- X. Luo, S. Rojas-Carbonell, Y. Yan and A. Kusoglu, J. Membr. Sci., 2020, 598, 117680 CrossRef CAS.
- K. M. Beers, S. Yakovlev, A. Jackson, X. Wang, A. Hexemer, K. H. Downing and N. P. Balsara, J. Phys. Chem. B, 2014, 118, 6785–6791 CrossRef CAS.
- A. Kusoglu, M. A. Modestino, A. Hexemer, R. A. Segalman and A. Z. Weber, ACS Macro Lett., 2012, 1, 33–36 CrossRef CAS PubMed.
- G. Alberti and R. Narducci, Fuel Cells, 2009, 9, 410–420 CrossRef CAS.
- A. Z. Weber and J. Newman, J. Electrochem. Soc., 2003, 150, A1008 CrossRef CAS.
- M. Bass and V. Freger, Desalination, 2006, 199, 277–279 CrossRef CAS.
- V. Freger, J. Phys. Chem. B, 2009, 113, 24–36 CrossRef CAS PubMed.
- M. Adachi, T. Navessin, Z. Xie, B. Frisken and S. Holdcroft, J. Electrochem. Soc., 2009, 156, B782 CrossRef CAS.
- T. D. Myles, A. M. Kiss, K. N. Grew, A. A. Peracchio, G. J. Nelson and W. K. S. Chiu, J. Electrochem. Soc., 2011, 158, B790 CrossRef CAS.
- M. G. Marino, J. P. Melchior, A. Wohlfarth and K. D. Kreuer, J. Membr. Sci., 2014, 464, 61–71 CrossRef CAS.
- X. Luo, A. Wright, T. Weissbach and S. Holdcroft, J. Power Sources, 2018, 375, 442–451 CrossRef CAS.
- M. Adachi, T. Navessin, Z. Xie, F. H. Li, S. Tanaka and S. Holdcroft, J. Membr. Sci., 2010, 364, 183–193 CrossRef CAS.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- D. G. Wheeler, B. A. W. Mowbray, A. Reyes, F. Habibzadeh, J. He and C. P. Berlinguette, Energy Environ. Sci., 2020, 13, 5126–5134 RSC.
- L. C. Jacobson, X. Ren and V. Molinero, J. Phys. Chem. C, 2014, 118, 2093–2103 CrossRef CAS.
- X. Wang, J. P. McClure and P. S. Fedkiw, Electrochim. Acta, 2012, 79, 126–132 CrossRef CAS.
- M. Ma, Z. Zheng, W. Yan, C. Hu and B. Seger, ACS Energy Lett., 2022, 7, 2595–2601 CrossRef CAS.
- W. E. Mustain, Curr. Opin. Electrochem., 2018, 12, 233–239 CrossRef CAS.
- J. M. Bockris and B. Conway, Modern Aspects of Electrochemistry: No. 12, Springer Science & Business Media, 2012 Search PubMed.
- A. L. Roy, J. Peng and T. A. Zawodzinski, 2018.
- E. R. Nightingale, J. Phys. Chem., 1959, 63, 1381–1387 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, Elsevier, Boston, 3rd edn, 2011, pp. 71–90 Search PubMed.
- J. J. Novoa, F. Mota, C. Perez del Valle and M. Planas, J. Phys. Chem. A, 1997, 101, 7842–7853 CrossRef CAS.
- K. Leung, I. M. B. Nielsen and I. Kurtz, J. Phys. Chem. B, 2007, 111, 4453–4459 CrossRef CAS PubMed.
- P. D. Dopieralski, A. Burakowski, Z. Latajka and I. Olovsson, Chem. Phys. Lett., 2011, 507, 89–95 CrossRef CAS.
- Y. Kameda, M. Sasaki, S. Hino, Y. Amo and T. Usuki, Physica B, 2006, 385-386, 279–281 CrossRef CAS.
- S. Yadav and A. Chandra, J. Phys. Chem. B, 2018, 122, 1495–1504 CrossRef CAS.
- A. L. Zydney, Membrane Science and Technology, ed. S. T. Oyama and S. M. Stagg-Williams, Elsevier, 2011, vol. 14, pp. 333–352 Search PubMed.
- D. García-Nieto and V. M. Barragán, Electrochim. Acta, 2015, 154, 166–176 CrossRef.
- S. Yu, X. Li, J. Li, S. Liu, W. Lu, Z. Shao and B. Yi, Energy Convers. Manage., 2013, 76, 301–306 CrossRef CAS.
- A. Reyes, R. P. Jansonius, B. A. W. Mowbray, Y. Cao, D. G. Wheeler, J. Chau, D. J. Dvorak and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 1612–1618 CrossRef CAS.
- M. E. Tuckerman, D. Marx and M. Parrinello, Nature, 2002, 417, 925–929 CrossRef CAS PubMed.
- Z. Liu, H. Yang, R. Kutz and R. I. Masel, J. Electrochem. Soc., 2018, 165, J3371–J3377 CrossRef CAS.
- A. Amel, S. B. Smedley, D. R. Dekel, M. A. Hickner and Y. Ein-Eli, J. Electrochem. Soc., 2015, 162, F1047–F1055 CrossRef CAS.
- Z. Yang, R. Guo, R. Malpass-Evans, M. Carta, N. B. McKeown, M. D. Guiver, L. Wu and T. Xu, Angew. Chem., Int. Ed., 2016, 55, 11499–11502 CrossRef CAS PubMed.
- X. Ge, Y. He, K. Zhang, X. Liang, C. Wei, M. A. Shehzad, W. Song, Z. Ge, G. Li, W. Yu, L. Wu and T. Xu, Research, 2021, 2021, 9762709 CrossRef CAS PubMed.
- H. M. Kim, C. Hu, H. H. Wang, J. H. Park, N. Chen and Y. M. Lee, J. Membr. Sci., 2022, 644, 120109 CrossRef CAS.
- W. Chen, X. Wang, T. Li, X. Yan, X. Wu, Y. Zhang, F. Zhang, S. Zhang and G. He, J. Membr. Sci., 2021, 640, 119815 CrossRef CAS.
- A. Jikihara, R. Ohashi, Y. Kakihana, M. Higa and K. Kobayashi, Membranes, 2013, 3, 1–15 CrossRef CAS PubMed.
- L. Zhu, J. Pan, Y. Wang, J. Han, L. Zhuang and M. A. Hickner, Macromolecules, 2016, 49, 815–824 CrossRef CAS.
- L. Zhu, T. J. Zimudzi, N. Li, J. Pan, B. Lin and M. A. Hickner, Polym. Chem., 2016, 7, 2464–2475 RSC.
- J. Han, S. Gong, Z. Peng, X. Cheng, Y. Li, H. Peng, Y. Zhu, Z. Ren, L. Xiao and L. Zhuang, J. Membr. Sci., 2021, 119096, DOI:10.1016/j.memsci.2021.119096.
- M. Zhang, C. Shan, L. Liu, J. Liao, Q. Chen, M. Zhu, Y. Wang, L. An and N. Li, ACS Appl. Mater. Interfaces, 2016, 8, 23321–23330 CrossRef CAS PubMed.
- F. Sepehr, H. Liu, X. Luo, C. Bae, M. E. Tuckerman, M. A. Hickner and S. J. Paddison, Macromolecules, 2017, 50, 4397–4405 CrossRef CAS.
- E. Kim, S. Lee, S. Woo, S.-H. Park, S.-D. Yim, D. Shin and B. Bae, J. Power Sources, 2017, 359, 568–576 CrossRef CAS.
- J. Pan, J. Han, L. Zhu and M. A. Hickner, Chem. Mater., 2017, 29, 5321–5330 CrossRef CAS.
- X. Gong, X. Yan, T. Li, X. Wu, W. Chen, S. Huang, Y. Wu, D. Zhen and G. He, J. Membr. Sci., 2017, 523, 216–224 CrossRef CAS.
- Z. Yang, J. Zhou, S. Wang, J. Hou, L. Wu and T. Xu, J. Mater. Chem. A, 2015, 3, 15015–15019 RSC.
- S. A. Nuñez, C. Capparelli and M. A. Hickner, Chem. Mater., 2016, 28, 2589–2598 CrossRef.
- X. Yan, B. Zhao, J. Liu, X. Zhang and G. He, J. Membr. Sci., 2018, 564, 436–443 CrossRef CAS.
- Y. He, J. Pan, L. Wu, Y. Zhu, X. Ge, J. Ran, Z. Yang and T. Xu, Sci. Rep., 2015, 5, 13417 CrossRef CAS.
- D. W. Shin, M. D. Guiver and Y. M. Lee, Chem. Rev., 2017, 117, 4759–4805 CrossRef CAS PubMed.
- C. H. Park, S. Y. Lee, D. S. Hwang, D. W. Shin, D. H. Cho, K. H. Lee, T.-W. Kim, T.-W. Kim, M. Lee, D.-S. Kim, C. M. Doherty, A. W. Thornton, A. J. Hill, M. D. Guiver and Y. M. Lee, Nature, 2016, 532, 480–483 CrossRef PubMed.
- D. R. Dekel, M. Amar, S. Willdorf, M. Kosa, S. Dhara and C. E. Diesendruck, Chem. Mater., 2017, 29, 4425–4431 CrossRef CAS.
- F. Gu, H. Dong, Y. Li, Z. Sun and F. Yan, Macromolecules, 2014, 47, 6740–6747 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, Macromolecules, 2017, 50, 2784–2793 CrossRef CAS.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Am. Chem. Soc., 2017, 139, 2888–2891 CrossRef CAS PubMed.
- B. Endrődi, E. Kecsenovity, A. Samu, T. Halmágyi, S. Rojas-Carbonell, L. Wang, Y. Yan and C. Janáky, Energy Environ. Sci., 2020, 13, 4098–4105 RSC.
- Y. Sone, P. Ekdunge and D. Simonsson, J. Electrochem. Soc., 1996, 143, 1254–1259 CrossRef CAS.
- Z. Liu, S. D. Sajjad, Y. Gao, H. Yang, J. J. Kaczur and R. I. Masel, Int. J. Hydrogen Energy, 2017, 42, 29661–29665 CrossRef CAS.
- D. Salvatore and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 215–220 CrossRef CAS.
- I. V. Pushkareva, A. S. Pushkarev, S. A. Grigoriev, P. Modisha and D. G. Bessarabov, Int. J. Hydrogen Energy, 2020, 45, 26070–26079 CrossRef CAS.
- M. B. McDonald, S. Ardo, N. S. Lewis and M. S. Freund, ChemSusChem, 2014, 7, 3021–3027 CrossRef CAS PubMed.
- G. M. Geise, L. P. Falcon, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2012, 423-424, 195–208 CrossRef CAS.
- A. R. Khare and N. A. Peppas, Biomaterials, 1995, 16, 559–567 CrossRef CAS PubMed.
- P. Długołęcki, P. Ogonowski, S. J. Metz, M. Saakes, K. Nijmeijer and M. Wessling, J. Membr. Sci., 2010, 349, 369–379 CrossRef.
- J. P. McClure, K. N. Grew and D. Chu, ECS Trans., 2015, 69, 35–44 CrossRef CAS.
- M. Unlu, J. Zhou and P. A. Kohl, Electrochem. Solid-State Lett., 2009, 12, B27 CrossRef CAS.
- N. Li, M. D. Guiver and W. H. Binder, ChemSusChem, 2013, 6, 1376–1383 CrossRef CAS PubMed.
- D. Materials, Sustainion anion exchange membranes, https://dioxidematerials.com/technology/sustainion-membranes/, (accessed 2022-01-07).
- J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. Ben Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. Xu and Y. Yan, Nat. Energy, 2019, 4, 392–398 CrossRef CAS.
- Ionomr, Ionomer Hydrogen Info Sheet, https://ionomr.com/wp-content/uploads/2018/12/Ionomr-Hydrogen-Info-Sheet.pdf, (accessed 2022-01-07).
- H. Peng, Q. Li, M. Hu, L. Xiao, J. Lu and L. Zhuang, J. Power Sources, 2018, 390, 165–167 CrossRef CAS.
- W.-H. Lee, E. J. Park, J. Han, D. W. Shin, Y. S. Kim and C. Bae, ACS Macro Lett., 2017, 6, 566–570 CrossRef CAS.
- N. Ul Hassan, M. Mandal, G. Huang, H. A. Firouzjaie, P. A. Kohl and W. E. Mustain, Adv. Energy Mater., 2020, 10, 2001986 CrossRef CAS.
- B. Motealleh, Z. Liu, R. I. Masel, J. P. Sculley, Z. Richard Ni and L. Meroueh, Int. J. Hydrogen Energy, 2021, 46, 3379–3386 CrossRef CAS.
- J. C. N. f. c. t. B.V.), Journal, 2014.
- T. H. Pham, A. Allushi, J. S. Olsson and P. Jannasch, Polym. Chem., 2020, 11, 6953–6963 RSC.
- C. G. Arges and L. Zhang, ACS Appl. Energy Mater., 2018, 1, 2991–3012 CrossRef CAS.
- H. Long, K. Kim and B. S. Pivovar, J. Phys. Chem. C, 2012, 116, 9419–9426 CrossRef CAS.
- H.-S. Dang and P. Jannasch, J. Mater. Chem. A, 2016, 4, 17138–17153 RSC.
- C. X. Lin, X. L. Huang, D. Guo, Q. G. Zhang, A. M. Zhu, M. L. Ye and Q. L. Liu, J. Mater. Chem. A, 2016, 4, 13938–13948 RSC.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef CAS.
- S. Maurya, A. S. Lee, D. Li, E. J. Park, D. P. Leonard, S. Noh, C. Bae and Y. S. Kim, J. Power Sources, 2019, 436, 226866 CrossRef CAS.
- J. Müller, A. Zhegur, U. Krewer, J. R. Varcoe and D. R. Dekel, ACS Mater. Lett., 2020, 2, 168–173 CrossRef PubMed.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. D. Arquer, A. Kiani, J. P. Edwards, P. D. Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- C. M. Gabardo, A. Seifitokaldani, J. P. Edwards, C.-T. Dinh, T. Burdyny, M. G. Kibria, C. P. O’Brien, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2018, 11, 2531–2539 RSC.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C.-S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S.-C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2016, 301, 219–228 CrossRef CAS.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. A. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS.
- T. T. H. Hoang, S. Ma, J. I. Gold, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2017, 7, 3313–3321 CrossRef CAS.
- R. A. Krivina, G. A. Lindquist, M. C. Yang, A. K. Cook, C. H. Hendon, A. R. Motz, C. Capuano, K. E. Ayers, J. E. Hutchison and S. W. Boettcher, ACS Appl. Mater. Interfaces, 2022, 14, 18261–18274 CrossRef CAS PubMed.
- Y. Yang, C. R. Peltier, R. Zeng, R. Schimmenti, Q. Li, X. Huang, Z. Yan, G. Potsi, R. Selhorst, X. Lu, W. Xu, M. Tader, A. V. Soudackov, H. Zhang, M. Krumov, E. Murray, P. Xu, J. Hitt, L. Xu, H.-Y. Ko, B. G. Ernst, C. Bundschu, A. Luo, D. Markovich, M. Hu, C. He, H. Wang, J. Fang, R. A. DiStasio, L. F. Kourkoutis, A. Singer, K. J. T. Noonan, L. Xiao, L. Zhuang, B. S. Pivovar, P. Zelenay, E. Herrero, J. M. Feliu, J. Suntivich, E. P. Giannelis, S. Hammes-Schiffer, T. Arias, M. Mavrikakis, T. E. Mallouk, J. D. Brock, D. A. Muller, F. J. DiSalvo, G. W. Coates and H. D. Abruña, Chem. Rev., 2022, 122, 6117–6321 CrossRef CAS PubMed.
- Z. Sun, J. Pan, J. Guo and F. Yan, Adv. Sci., 2018, 5, 1800065 CrossRef PubMed.
- C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2490–2495 CrossRef CAS.
- A. D. Mohanty and C. Bae, J. Mater. Chem. A, 2014, 2, 17314–17320 RSC.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS PubMed.
- N. J. Robertson, H. A. Kostalik, T. J. Clark, P. F. Mutolo, H. D. Abruña and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 3400–3404 CrossRef CAS PubMed.
- L.-C. Jheng, S. L.-C. Hsu, B.-Y. Lin and Y.-L. Hsu, J. Membr. Sci., 2014, 460, 160–170 CrossRef CAS.
- J. Pan, Y. Li, J. Han, G. Li, L. Tan, C. Chen, J. Lu and L. Zhuang, Energy Environ. Sci., 2013, 6, 2912–2915 RSC.
- J. Wang, S. Gu, R. Xiong, B. Zhang, B. Xu and Y. Yan, ChemSusChem, 2015, 8, 4229–4234 CrossRef CAS PubMed.
- W.-H. Lee, A. D. Mohanty and C. Bae, ACS Macro Lett., 2015, 4, 453–457 CrossRef CAS PubMed.
- G. S. Sailaja, S. Miyanishi and T. Yamaguchi, Polym. Chem., 2015, 6, 7964–7973 RSC.
- Z. Liu, R. I. Masel, Q. Chen, R. Kutz, H. Yang, K. Lewinski, M. Kaplun, S. Luopa and D. R. Lutz, J. CO2 Util., 2016, 15, 50–56 CrossRef CAS.
- C.-T. Dinh, F. P. García de Arquer, D. Sinton and E. H. Sargent, ACS Energy Lett., 2018, 3, 2835–2840 CrossRef CAS.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed.
- S. Garg, M. Li, T. Hussain, M. N. Idros, Y. Wu, X. S. Zhao, G. G. X. Wang and T. E. Rufford, ACS Appl. Mater. Interfaces, 2022, 14, 35504–35512 CrossRef CAS PubMed.
- R. B. Kutz, Q. Chen, H. Yang, S. D. Sajjad, Z. Liu and I. R. Masel, Energy Technol., 2017, 5, 929–936 CrossRef CAS.
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32–39 CrossRef CAS.
- M. Ma, W. Deng, A. Xu, D. Hochfilzer, Y. Qiao, K. Chan, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2022, 15, 2470–2478 RSC.
- J. Lee, J. Lim, C.-W. Roh, H. S. Whang and H. Lee, J. CO2 Util., 2019, 31, 244–250 CrossRef CAS.
- W. Zhu, S. Kattel, F. Jiao and J. G. Chen, Adv. Energy Mater., 2019, 9, 1802840 CrossRef.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef PubMed.
- T. Möller, W. Ju, A. Bagger, X. Wang, F. Luo, T. Ngo Thanh, A. S. Varela, J. Rossmeisl and P. Strasser, Energy Environ. Sci., 2019, 12, 640–647 RSC.
- S. R. Narayanan, B. Haines, J. Soler and T. I. Valdez, J. Electrochem. Soc., 2011, 158, A167 CrossRef CAS.
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. Liang, E. Stavitski, H. N. Alshareef and H. Wang, Nat. Energy, 2019, 4, 776–785 CrossRef CAS.
- C. Liang, B. Kim, S. Yang, L. Yang, C. Francisco Woellner, Z. Li, R. Vajtai, W. Yang, J. Wu, P. J. A. Kenis and P. M. Ajayan, J. Mater. Chem. A, 2018, 6, 10313–10319 RSC.
- W. Ma, J. Bu, Z. Liu, C. Yan, Y. Yao, N. Chang, H. Zhang, T. Wang and J. Zhang, Adv. Funct. Mater., 2021, 31, 2006704 CrossRef CAS.
- M. G. Kibria, C.-T. Dinh, A. Seifitokaldani, P. De Luna, T. Burdyny, R. Quintero-Bermudez, M. B. Ross, O. S. Bushuyev, F. P. García de Arquer, P. Yang, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, 1804867 CrossRef PubMed.
- F. P. G. De Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. P. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef.
- C. M. Gabardo, C. P. O’Brien, J. P. Edwards, C. McCallum, Y. Xu, C.-T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed.
- Y. C. Li, Z. Wang, T. Yuan, D.-H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. de Arquer, Y. Wang, C.-T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- D. Ren, J. Gao, L. Pan, Z. Wang, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Angew. Chem., Int. Ed., 2019, 58, 15036–15040 CrossRef CAS PubMed.
- J. J. Kaczur, H. Yang, Z. Liu, S. D. Sajjad and R. I. Masel, C, 2020, 6, 33 CAS.
- X. Wang, Z. Wang, F. P. García de Arquer, C.-T. Dinh, A. Ozden, Y. C. Li, D.-H. Nam, J. Li, Y.-S. Liu, J. Wicks, Z. Chen, M. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorović, F. Li, T.-T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S.-F. Hung, Y. Lum, M. Luo, Y. Min, A. Xu, C. P. O’Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS.
- E. J. Park and Y. S. Kim, J. Mater. Chem. A, 2018, 6, 15456–15477 RSC.
- C. McCallum, C. M. Gabardo, C. P. O’Brien, J. P. Edwards, J. Wicks, Y. Xu, E. H. Sargent and D. Sinton, Cell Rep. Phys. Sci., 2021, 2, 100522 CrossRef CAS.
- P. Mardle, S. Cassegrain, F. Habibzadeh, Z. Shi and S. Holdcroft, J. Phys. Chem. C, 2021, 125, 25446–25454 CrossRef CAS.
- S. M. Dischinger, S. Gupta, B. M. Carter and D. J. Miller, Ind. Eng. Chem. Res., 2020, 59, 5257–5266 CrossRef CAS.
- D. Henkensmeier, M. Najibah, C. Harms, J. Žitka, J. Hnát and K. Bouzek, J. Electrochem. Energy Convers. Storage, 2021, 18, 024001 CrossRef CAS.
- B. Mayerhöfer, D. McLaughlin, T. Böhm, M. Hegelheimer, D. Seeberger and S. Thiele, ACS Appl. Energy Mater., 2020, 3, 9635–9644 CrossRef.
- P. Fortin, T. Khoza, X. Cao, S. Y. Martinsen, A. Oyarce Barnett and S. Holdcroft, J. Power Sources, 2020, 451, 227814 CrossRef CAS.
- B. Shanahan, B. Britton, A. Belletti, S. Vierrath and M. Breitwieser, RSC Adv., 2021, 11, 13077–13084 RSC.
- S. Noh, J. Y. Jeon, S. Adhikari, Y. S. Kim and C. Bae, Acc. Chem. Res., 2019, 52, 2745–2755 CrossRef CAS PubMed.
- J. Peng, A. L. Roy, S. G. Greenbaum and T. A. Zawodzinski, J. Power Sources, 2018, 380, 64–75 CrossRef CAS.
- X. Ren, S. C. Price, A. C. Jackson, N. Pomerantz and F. L. Beyer, ACS Appl. Mater. Interfaces, 2014, 6, 13330–13333 CrossRef CAS PubMed.
- D. S. Ripatti, T. R. Veltman and M. W. Kanan, Joule, 2019, 3, 240–256 CrossRef CAS.
- G. Garg and S. Basu, Electrochim. Acta, 2015, 177, 359–365 CrossRef CAS.
- M. Ramdin, A. R. T. Morrison, M. de Groen, R. van Haperen, R. de Kler, L. J. P. van den Broeke, J. P. M. Trusler, W. de Jong and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2019, 58, 1834–1847 CrossRef CAS.
- C. Delacourt, P. L. Ridgway, J. B. Kerr and J. Newman, J. Electrochem. Soc., 2008, 155, B42 CrossRef CAS.
- J.-B. Vennekoetter, R. Sengpiel and M. Wessling, Chem. Eng. J., 2019, 364, 89–101 CrossRef CAS.
- D. Kopljar, A. Inan, P. Vindayer, N. Wagner and E. Klemm, J. Appl. Electrochem., 2014, 44, 1107–1116 CrossRef CAS.
- P. Jeanty, C. Scherer, E. Magori, K. Wiesner-Fleischer, O. Hinrichsen and M. Fleischer, J. CO2 Util., 2018, 24, 454–462 CrossRef CAS.
- T. A. Zawodzinski, C. Derouin, S. Radzinski, R. J. Sherman, V. T. Smith, T. E. Springer and S. Gottesfeld, J. Electrochem. Soc., 1993, 140, 1041–1047 CrossRef CAS.
- H. W. Shafaque, J. K. Lee, K. Krause, C. Lee, K. F. Fahy, P. Shrestha, M. Balakrishnan and A. Bazylak, Energy Convers. Manage., 2021, 243, 114302 CrossRef CAS.
- H. Yoshio, K. Katsuhei, M. Akira and S. Shin, Chem. Lett., 1986, 897–898 Search PubMed.
- M. A. Blommaert, R. Sharifian, N. U. Shah, N. T. Nesbitt, W. A. Smith and D. A. Vermaas, J. Mater. Chem. A, 2021, 9, 11179–11186 RSC.
- Z. Yan, J. L. Hitt, Z. Zeng, M. A. Hickner and T. E. Mallouk, Nat. Chem., 2021, 13, 33–40 CrossRef CAS.
- R. Pärnamäe, S. Mareev, V. Nikonenko, S. Melnikov, N. Sheldeshov, V. Zabolotskii, H. V. M. Hamelers and M. Tedesco, J. Membr. Sci., 2021, 617, 118538 CrossRef.
- Y. Xu, R. K. Miao, J. P. Edwards, S. Liu, C. P. O’Brien, C. M. Gabardo, M. Fan, J. E. Huang, A. Robb, E. H. Sargent and D. Sinton, Joule, 2022, 6, 1333–1343 CrossRef CAS.
- M. Schwartz, M. E. Vercauteren and A. F. Sammells, J. Electrochem. Soc., 1994, 141, 3119–3127 CrossRef CAS.
- N. S. Romero Cuellar, K. Wiesner-Fleischer, M. Fleischer, A. Rucki and O. Hinrichsen, Electrochim. Acta, 2019, 307, 164–175 CrossRef CAS.
- J. Li, A. Xu, F. Li, Z. Wang, C. Zou, C. M. Gabardo, Y. Wang, A. Ozden, Y. Xu, D.-H. Nam, Y. Lum, J. Wicks, B. Chen, Z. Wang, J. Chen, Y. Wen, T. Zhuang, M. Luo, X. Du, T.-K. Sham, B. Zhang, E. H. Sargent and D. Sinton, Nat. Commun., 2020, 11, 3685 CrossRef CAS PubMed.
- X. Fu, Y. Wang, H. Shen, Y. Yu, F. Xu, G. Zhou, W. Xie, R. Qin, C. Dun, C. W. Pao, J. L. Chen, Y. Liu, J. Guo, Q. Yue, J. J. Urban, C. Wang and Y. Kang, Mater. Today Phys., 2021, 19, 100418 CrossRef CAS.
- Z.-Z. Niu, L.-P. Chi, R. Liu, Z. Chen and M.-R. Gao, Energy Environ. Sci., 2021, 14, 4169–4176 RSC.
- Y. Zheng, T. J. Omasta, X. Peng, L. Wang, J. R. Varcoe, B. S. Pivovar and W. E. Mustain, Energy Environ. Sci., 2019, 12, 2806–2819 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee01818g |
| This journal is © The Royal Society of Chemistry 2022 |