
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA high-rate and high-efficiency molten-salt sodium–oxygen battery†
Yun Guang
Zhu
 ab,
Graham
Leverick
*ac,
Alessandra
Accogli
ad,
Kiarash
Gordiz
ac,
Yirui
Zhang
ac and
Yang
Shao-Horn
*abce
ab,
Graham
Leverick
*ac,
Alessandra
Accogli
ad,
Kiarash
Gordiz
ac,
Yirui
Zhang
ac and
Yang
Shao-Horn
*abce
aElectrochemical Energy Laboratory, Massachusetts Institute of Technology, Cambridge, MA, USA. E-mail: shaohorn@mit.edu; leverick@mit.edu
bResearch Laboratory of Electronics, Massachusetts Institute of Technology, Cambridge, MA, USA
cDepartment of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA
dDipartimento di Chimica, Materiali e Ingegneria Chimica Giulio Natta, Politecnico di Milano, 20131, Milano, Italy
eDepartment of Material Science, Massachusetts Institute of Technology, Cambridge, MA, USA
First published on 27th September 2022
Abstract
Alkali metal–oxygen batteries can provide greater specific energy than Li-ion batteries but often suffer from low power density, cycleability, and energy efficiency due to the sluggish kinetics of the oxygen electrode and parasitic reactions at both the oxygen and alkali metal electrodes. In this study, we demonstrated a molten-salt Na–O2 battery operating at 443 K with high areal energy (33 mW h cm−2geo) and power densities (19 mW cm−2geo), with high energy efficiency (∼90% at 5 mA cm−2geo), and stable cycling (400 cycles with no capacity loss). Raman, pressure tracking and titration measurements were used to show that the dominant discharge product is Na2O2. Moreover, the redox activity of nitrate anions in the molten salt was found to be critical to enable the formation of Na2O2. Through 18O-labeling experiments as well as discharging Na–Ar cells, we demonstrated that the discharge reaction occurs via the electrochemical reduction of NaNO3 to Na2O and NaNO2, where chemical reactions with O2 lead to the formation of Na2O2 from Na2O, and the regeneration of NaNO3 from NaNO2.
Broader contextAlkali metal–oxygen batteries have high theoretical energy densities and are promising electrochemical energy storage systems to enable the electrification of heavy-duty vehicles and aviation. Unfortunately, the practical realization of such metal–oxygen batteries is hindered by low power density, cycleability, and energy efficiency due to slow reacitons in the oxygen electrode and unwanted side reactions in both the oxygen and alkali metal electrodes. In this study, we introduce a novel molten-salt Na–O2 battery which operates at 170 °C featuring a liquid sodium metal negative electrode and Nickel-based oxygen electrode with a molten-salt electrolyte. By operating at increased temperature and using catalytically active nickel in the oxygen electrode, the rate capability of the battery is significantly enhanced. Moreover, the molten-salt electrolyte shows high stability; reducing parasitic reactions and enhancing cycle life. |
Introduction
Alkali metal–oxygen batteries have high theoretical energy densities and are promising electrochemical energy storage systems to enable the electrification of heavy-duty vehicles and aviation.1 For instance, Li–, Na–, and K–O2 batteries can deliver values of specific energy of 3458 W h kg−1 (Li, O2/active Li2O2), 1103 W h kg−1 (Na, O2/active NaO2), and 935 W h kg−1 (K, O2/active KO2) at room temperature, respectively (where calculation details can be found in the ESI†). However, the formation of dendrites2 and the high reactivity of alkali metals has hindered the development of metal-based batteries, including metal–oxygen batteries.3 Recently, research effort has focused on modifying the alkali metal negative electrode to increase rechargeability and stability, especially that of lithium metal.4,5 Moreover, the sluggish kinetics of the oxygen reduction and evolution reactions (ORR and OER)6 and slow diffusion of oxygen in the air electrode7 can increase the overpotentials at the positive electrode of metal–oxygen batteries, leading to poor energy efficiencies.8,9 To reduce the overpotentials of metal–oxygen batteries, solid electrochemical catalysts10 and redox mediators11,12 have been intensely investigated. Unfortunately, the (electro)chemical stability of the electrodes13 and electrolyte14–16 against reaction intermediates and products as well as the operating voltages remain a big challenge.Recently, a molten-salt Li–O2 battery has been reported by Giordani et al. in 2016, showing high electrolyte stability and high-energy efficiency (∼95%) at an operating temperature of 423 K.17 Unfortunately, the formation of Li2CO3 from the oxidation of carbon in the oxygen electrode led to poor cycling stability (<50 cycles, ∼2.6 mA h cm−2geo at ∼0.6 mA cm−2geo, normalized based on the geometric area of the positive electrode). Subsequently, Xia et al. have demonstrated a molten-salt Li–O2 battery with a four-electron conversion of O2 to Li2O using NiO in the oxygen electrode, showing stable cycling performance (150 cycles, 0.5 mA h cm−2geo at 0.2 mA cm−2geo) with a Coulombic efficiency of ∼100%.18 Koo and Kang have reported that iron(II, III) oxide can also serve as an effective catalyst to produce Li2O in nitrate molten-salt Li–O2 batteries.19 Most recently, we have shown that Li2O in nitrate molten-salt Li–O2 batteries is enabled by the redox activity of nitrate anions and by studying different 3d transition metal oxides have found that NiO has the lowest overpotential during discharge due to its optimal binding of nitrate and nitrite anions.20 Nitrate molten salts have also been used as the O2 reservoir for a closed Li–O2 battery system21 and molten-salt electrolytes have been used with other metal–oxygen batteries besides Li such as Fe,22,23 Zn24 and Mg,25 showing long cycling life and high rate capability. A schematic of the structure of a molten-salt Li–O2 battery is shown in Fig. 1a. Solid-state electrolytes are needed in molten-salt Li–O2 batteries to prevent crossover of soluble Li2O that can form due to the reaction between Li metal and the electrolyte.26 A molten-salt buffer layer was used to provide a highly Li+ conducting interface between the Li metal electrode and solid-state electrolyte in order to achieve high rate and energy efficiency in molten-salt Li–O2 batteries. Replacing Li by Na provides an opportunity to increase the power of the system due to low interfacial resistance between liquid Na and the solid-state electrolyte at an operating temperature of 443 K.
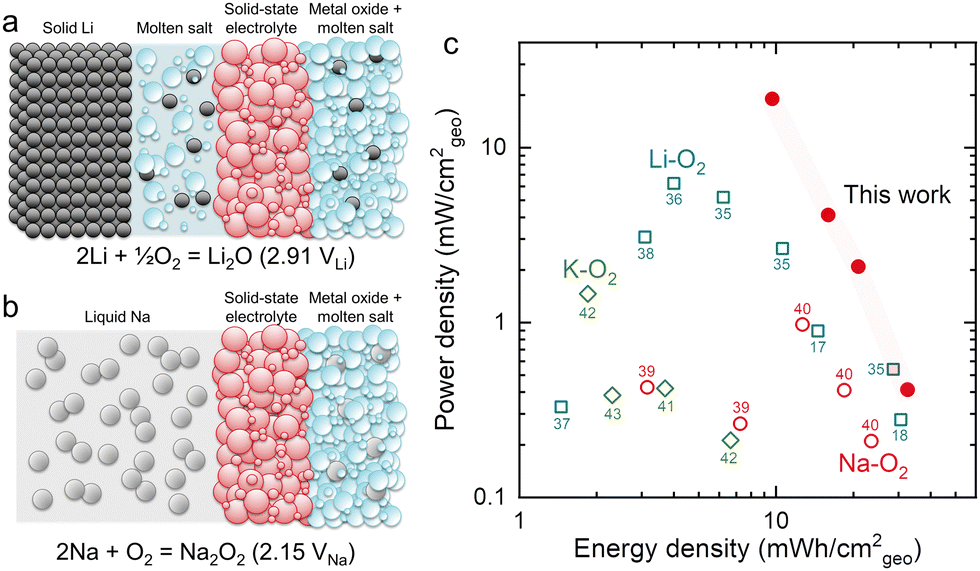 | ||
| Fig. 1 Schematic of a molten-salt Li–O2 battery18 (a) and the molten-salt Na–O2 battery demonstrated in this work (b). A conventional molten-salt Li–O2 battery contains Li metal, a buffer layer containing eutectic salts (for example, NaNO3 (33.3 w%)/KNO3 (66.7 w%)), a solid-state electrolyte, and a metal oxide-based oxygen positive electrode with eutectic salts. The molten-salt Na–O2 battery has a liquid Na metal negative electrode, solid-state electrolyte, and metal oxide-based oxygen positive electrode with eutectic salts (for example, NaNO3 (26.4 w%)/KNO3 (27.3 w%)/CsNO3 (46.3 w%) in Fig. S5, ESI†). (c) The comparison of energy and power densities based on the area of the positive electrode in a Ragone plot for alkali metal–oxygen batteries (Li–O2,17,18,35–38 Na–O2,39,40 K–O241–43 batteries). Detailed information on the reported performance is shown in Table S1 and Fig. S1 (ESI†). | ||
In this work, a molten-salt Na–O2 battery was developed with a liquid Na negative electrode and Ni oxygen electrode with a NaNO3/KNO3/CsNO3 eutectic salt electrolyte and a β-Al2O3 membrane, where the stable interface between liquid Na and β-Al2O3 can be advantageous relative to molten-salt Li–O2 batteries27 (Fig. 1b). Such molten-salt Na–O2 batteries showed high energy (33 mW h cm−2geo) and power densities (19 mW cm−2geo) as well as stable cycling (400 cycles, 0.5 mA h cm−2geo at 5 mA cm−2geo). Using Raman, pressure tracking and titration measurements, we show that the dominant discharge product is Na2O2. Moreover, the redox of nitrate anions is identified as being critical to enable the formation of Na2O2 upon discharge. Molten-salt Na–Ar cells show the electrochemical reduction of NaNO3 to Na2O and NaNO2. On the other hand, in an O2 environment the formed Na2O and NaNO2 can further react with O2 to yield Na2O2 and regenerate NaNO3, respectively. Finally, using 18O-labeling experiments, we demonstrate that the oxygen reduction reaction in molten-salt Na–O2 batteries occurs via a nitrate-mediated mechanism whereby NaNO3 facilitates an apparent 2e−/O2 overall reaction to form Na2O2. The presented nitrate-mediated molten-salt Na–O2 battery provides a novel approach to develop alkali metal–O2 batteries with high energy and power densities, with a cell architecture that can stabilize the alkali metal electrode.
Experimental
Materials
Nickel metal powder (325 mesh, 99.8%, Fisher Scientific Co. LLC.) was used for electrode preparation. NaNO3 (99.999%, Fisher Scientific Co. LLC.), KNO3 (99.99%, Fisher Scientific Co. LLC.), CsNO3 (99.99%, Fisher Scientific Co. LLC.), NaNO2 (>97%, Fisher Scientific), NaTFSI (sodium trifluoromethanesulfonimide, 97%, Sigma Aldrich), KTFSI (potassium trifluoromethanesulfonimide, 97%, Sigma Aldrich) were used to prepare the eutectic molten-salt electrolytes and electrodes. Na-β-Al2O3 discs (Ionotec Ltd.) were used as Na-ion conductors for molten-salt Na–O2 batteries. Na2O2 (97%, Sigma Aldrich), NaO2 (Thermo ScientificTM), Na2CO3 (99.5%, Sigma Aldrich) and K2CO3 (99.0%, Sigma Aldrich) were used as standard samples for Raman spectroscopy. Standardized titanium(IV) oxysulfate solution (Aldrich, ∼15 wt% in dilute sulfuric acid, 99.99% trace metals basis) was used to quantify Na2O2 in discharged electrodes.28 Hydrochloric acid (0.01 N, VWR) was used for acid–base titration experiments for Na2O and Na2O2 from discharged electrodes.29,30 A Griess reagent system (Promega) was used for nitrite titration for discharged electrodes.18Preparation of the Ni/NaNO3/KNO3/CsNO3 electrodes
2 g of Ni powder (325 mesh, 99.8%, Fisher Scientific Co. LLC.) was added into 5 mL of a NaNO3 (26.4 w%)/KNO3 (27.3 w%)/CsNO3 (46.3 w%) solution (0.25 g mL−1 in Deionized water, DIW). Then the above suspension was sonicated for 10 min and transferred into an oven for drying at 453 K for 2 hours. Next, the composite powder was ground for half an hour and pressed as an electrode (0.2 g and 12.7 mm of diameter) onto stainless steel mesh (120 mesh) with 2 tons of pressure for 1 min in an Ar-filled glove box. These electrodes were transferred into a vacuum Buchi glass oven at 473 K for two days and then stored in an argon-filled glove box. After preparation, there is a small amount of NO2− in the Ni/NaNO3/KNO3/CsNO3 electrodes, which can be attributed to at least one of the following reactions:31 Ni + NaNO3 → NiO + NaNO2 (ΔG0 = −128.7 kJ mol−1 from thermodynamic data in Table S3, ESI†), Ni + KNO3 → NiO + KNO2 (ΔG0 = −122.8 kJ mol−1 from thermodynamic data in Table S3, ESI†) or Ni + CsNO3 → NiO + CsNO2 (ΔG0 = −118.4kJ mol−1 from thermodynamic data in Table S3, ESI†).Preparation of Ni/NaTFSI/KTFSI electrodes
2 g of Ni powder (325 mesh, 99.8%, Fisher Scientific Co. LLC.) was added into 5 mL of a NaTFSI (31.9 w%)/KTFSI (68.1 w%) solution (0.25 g mL−1 in DIW). Then, the above suspension was sonicated for 10 min, transferred into an oven, and dried at 453 K for 2 hours. Next, the composite powder was ground for half an hour and compressed as a 12.7 mm electrode (0.2 g) on stainless steel mesh (120 mesh) with 2 tons of pressure for 1 min in an Ar-filled glove box. These electrodes were transferred into a vacuum Buchi glass oven at 473 K for two days and then stored in an Ar-filled glove box prior to use.Synthesis of Na3ONO2
Na2O (Fisher Scientific) and NaNO2 (>97%, Fisher Scientific) with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio was ground for 20 min, and then 50 mg of the mixture was pressed as a pellet. After that, the pellets were sealed in an air-tight stainless-steel (SS) reactor under an Ar environment. The reactor was put in the oven at 573 K for 20 h. The produced yellow pellet (Na3ONO2) was transferred into an Ar-filled glove box for characterization.
:1 molar ratio was ground for 20 min, and then 50 mg of the mixture was pressed as a pellet. After that, the pellets were sealed in an air-tight stainless-steel (SS) reactor under an Ar environment. The reactor was put in the oven at 573 K for 20 h. The produced yellow pellet (Na3ONO2) was transferred into an Ar-filled glove box for characterization.
Assembly of Na molten-salt cells
All parts of Na–O2 cells were dried in a vacuum oven at 353 K for 12 h before use. The liquid Na negative electrode and the Ni/salts positive electrode (12.7 mm in diameter) were separated by a piece of β-Al2O3 conductor. A schematic structure of the Na molten-salt cell is shown in the inset of Fig. 2a. After assembly, the cells were charged with O2 or Ar. The charged O2 or Ar pressure ranged from ∼70 to 280 kPa at room temperature. There was good wetting between liquid Na and β-Al2O3 membrane after resting for 2 hours in Na–O2 cells (Fig. S2, ESI†).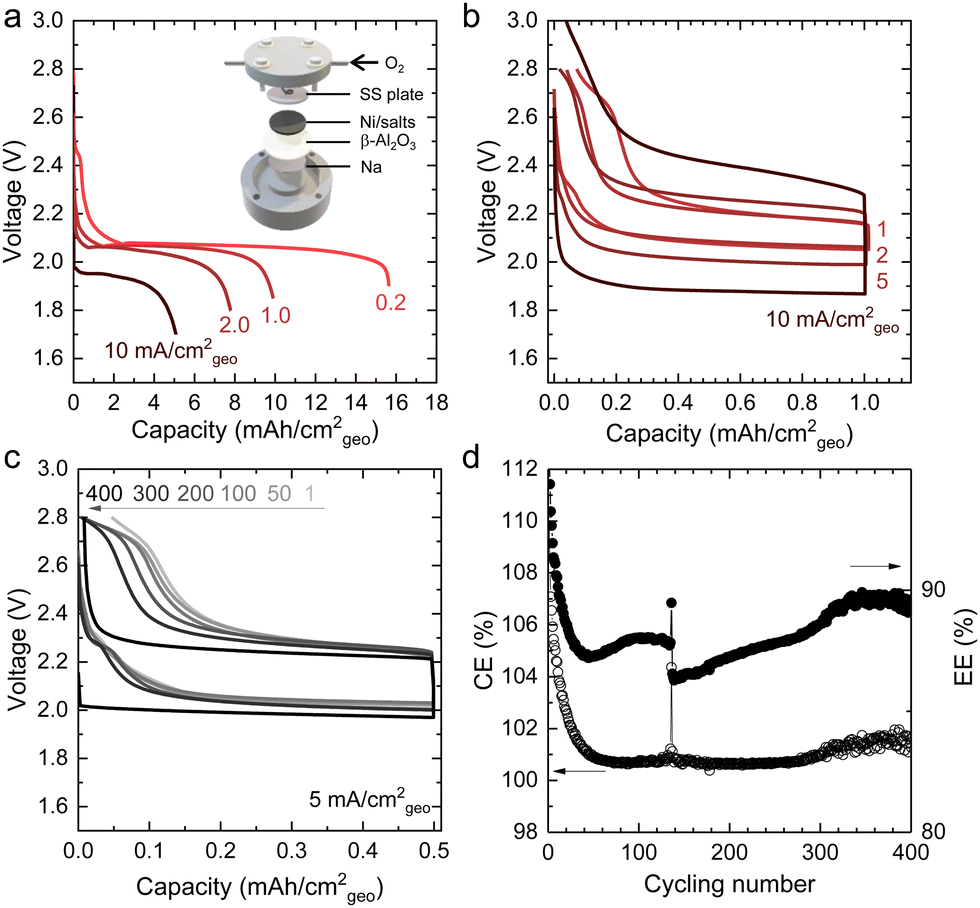 | ||
| Fig. 2 The electrochemical performance of molten-salt Na–O2 batteries. (a) Discharge profiles of molten-salt Na–O2 batteries at different current densities (0.2, 1.0, 2.0, 10.0 mA cm−2geo). The inset of (a) is the schematic structure of a molten-salt Na–O2 battery, including liquid Na negative electrode, β-Al2O3 membrane, Ni/NaNO3/KNO3/CsNO3/SS positive electrode, stainless steel (SS) mesh current collector, and O2. (b) Charge-discharge profiles of molten-salt Na–O2 batteries at different current densities (1, 2, 5, 10 mA cm−2geo) with a limited discharge capacity of 1.0 mA h cm−2geo (∼6% depth of discharge based on 16 mA h cm−2geo in (a)). (c) Charge–discharge profiles of a molten-salt Na–O2 battery at a current density of 5 mA cm−2geo (1st, 50th, 100th, 200th, 300th, and 400th cycles) with a limited discharge capacity of 0.5 mA h cm−2geo (∼3% depth of discharge based on 16 mA h cm−2geo in (a)). Cycling at deeper depths of discharge was limited by the β-Al2O3 membrane. (d) The Coulombic (CE) and energy efficiencies (EE) of a molten-salt Na–O2 battery at a current density of 5 mA cm−2geo for 400 cycles. All of the above cells were tested at 443 K on a hot plate with thermal insulation, where fluctuations during cycling are attributed to changes in the cell temperature. The molten salt is a mixture of NaNO3 (26.4 w%), KNO3 (27.3 w%), and CsNO3 (46.3 w%). | ||
Characterization of the reaction between Na2O and O2
10 mg of Na2O was added to 200 mg of Ni/salt powder (8/5 weight ratio) and then pressed as a pellet under a pressure of 2 tons. After that, the pellet was put into an air-tight cell with 275 kPa of O2 or Ar at 443 K for 48 h. After the reaction, the pellet was characterized in an air-tight cell using Raman spectroscopy.Quantification of the solubility of Na2O2 in molten salts
The solubility of Na2O2 in NaNO3 (26.4 w%)/KNO3 (27.3 w%)/CsNO3 (46.3 w%) eutectic molten salt was measured via an acid–base titration method. 2 w% of Na2O2 in the above molten salts was stirred at 443 K for two days, and then rest for three days at the same temperature. After that, the top, clear molten salt was collected for acid–base titration.Electrochemical measurements
Molten-salts Na–O2 cells were measured on a temperature-controlled hot plate. The temperature was set at 443 K. The operation voltage window was set between 1.8 to 2.8 V. The applied current density ranged from 0.1 to 10 mA cm−2geo. The battery tests were conducted using a Biologic VMP3 electrochemical workstation. The areal capacity, energy density and power density obtained from the electrochemical measurements were normalized by the area of the positive electrode.Differential electrochemical mass spectrometry (DEMS) measurements were conducted on a custom-made DEMS setup which has been detailed previously.3232O2 (16O16O), 34O2 (16O18O), and 36O2 (18O18O) were detected during charging with 10 min of accumulation time for each point. O2 pressure was measured during the discharge process to quantify the O2 consumption. Helium (Ultra High Purity 5.0 Grade, Airgas) was used as the carrier gas in DEMS measurements. The effective area of electrodes was 0.785 cm2 for DEMS measurement. The operation temperature was 443 K. The applied discharge and charge current densities ranged from 0.1 to 0.4 mA cm−2geo.
Quantification of discharge products
NO2− was quantified using the Griess method.18 NO2− titration was conducted on a UV-vis spectrophotometer (Genesys 180, Thermo Fisher Scientific). The NO2− calibration curve was generated using titration of the standard NO2− solutions (0, 0.01, 0.02, 0.05, 0.1, 0.5, 1 mM). 50 μL of standard NO2− solution and 50 μL of sulfanilic acid (10 mg mL−1 solution in 5% phosphoric acid, Promega) were added into a 1.2 mL plate deep well and then was kept in a dark environment for 3–5 min. Next, 50 μL of N-(1-naphthyl)ethylenediamine dihydrochloride (1 mg mL−1) solution (Promega) was added to the above solution and was kept in a dark environment for another 3–5 min. After that, 100 μL of the above solution was transferred into a quartz cuvette (10 mm path length, VWR) with 1.9 mL of deionized water (DIW). The solution in the cuvette was tested immediately using UV-vis with a scanning rate of 1 nm s−1 from 450 to 700 nm. The curve of absorbance vs. NO2− concentration was linearly fit, as shown in Fig. S3 (ESI†). For quantification of electrodes, the samples were dispersed in 50 mL of DIW, and then a clear solution was obtained by centrifugation. The clear solution was diluted ranging from 1/50 to 1/100. The diluted solution was then titrated using the above procedure.Na2O2 was quantified using titanium oxysulfate titration. Discharged electrodes were extracted from discharged molten-salt Na–O2 or Na–Ar cells in an Ar-filled glove box. Then, the electrode was removed from the glovebox and immediately dispersed in 50 mL of cooled DIW (stored in a refrigerator at 278 K) and stirred for 4 min. During this time, the following reaction occurred: Na2O2 + 2H2O → 2 NaOH + H2O2. There is a side reaction Na2O2 + H2O → 2NaOH + 0.5O2, which can be neglected based on literature findings.28 Next, 1 mL of the solution was filtered using a 0.2 μm filter and was added to 1 mL of cooled DIW and titrated with 0.5 mL standardized titanium(IV) oxysulfate solution (Aldrich, ∼15 wt% in dilute sulfuric acid, 99.99% trace metals basis). This step allows the fast reaction between H2O2 and Ti4+oxysulfate to form yellow pertitanic acid. The reaction is Ti4+ + H2O2 + 2H2O → H2TiO4 + 4H+. The concentration of the yellow pertitanic acid was determined using UV-vis spectroscopy (Genesys 180, Thermo Fisher Scientific) with a scanning rate of 1 nm s−1 from 350 to 650 nm. The UV-vis spectra of the titration of standardized H2O2 solutions (Certified ACS 31.7%, Fisher Chemical) at various concentrations (0.08, 0.2, 0.4, 0.8, 1.2, 1.6 mM) and the corresponding calibration curve are shown in Fig. S4 (ESI†).
Acid–based titrations for Na2O quantification were done using a pH meter (PH 700 meter, VWR) and 0.01 N of HCl standard solution (VWR). All discharged electrodes were first dispersed in 20 mL of DIW, and then clear solutions were collected via centrifugation and filtration using a 0.2 μm filter. The reaction of Na2O in the electrode and DIW is Na2O + H2O = 2NaOH.22 1 mL of the filtered solution was diluted to 10 ml for acid–base titration. The titration reaction is NaOH + HCl = NaCl + H2O.21 The end point of the titration was determined by the pH reaching ∼7 (6.5–7.5). In the event that Na2O2 was detected, the contribution from Na2O2 was deducted from the value determined from acid–base titration to determine the contribution from Na2O.
Characterization of electrodes
Ni electrodes were characterized through X-ray diffraction (XRD, Bruker D2), scanning electronic microscopy (SEM, Zeiss Merlin), and Raman spectroscopy (HORIBA Scientific LabRAM HR800). In the measurements of XRD and Raman spectra, the electrodes were sealed in air-tight cells. In XRD measurements, the applied voltage and current were 30 kV and 10 mA, respectively, using Cu-Kα radiation (λ = 1.54178Å). In Raman spectroscopy measurements, a red laser (λ = 632.8 nm) was used with 50-fold magnification. An exposure time of 15 s with a 600 grating was used, and each spectrum was accumulated five times.Results and discussion
Electrochemical performance of molten-salt Na–O2 cells
Molten-salt Na–O2 cells discharged at 443 K exhibited discharge voltages of 1.9–2.1 V at rates up to 10 mA cm−2geo, where the cells consisted of a liquid Na negative electrode, β-Al2O3 membrane and Ni/NaNO3/KNO3/CsNO3/stainless steel (SS) oxygen electrode (inset of Fig. 2a). A ternary NaNO3 (26.4 w%)/KNO3 (27.3 w%)/CsNO3 (46.3 w%) eutectic molten-salt electrolyte was selected due to its lower melting temperature (427 K in Fig. S5, ESI†) compared to NaNO3/KNO3 (494 K) and NaNO3/CsNO3 (464 K) binary eutectics (Table S2, ESI†). Critically, all three nitrate salts show thermal stability up to 600 K,33 which is significantly higher than the operating temperature of 443 K in this study. By investigating different weight ratios of Ni to NaNO3/KNO3/CsNO3 salts, an optimal ratio of 8/5 was identified, which yielded a discharge capacity of ∼16 mA h cm−2geo at 0.2 mA cm−2geo (Fig. S6, ESI†). Increasing current density from 0.2 to 10 mA cm−2geo was accompanied by an exponential decrease in areal capacity suggesting that the discharge process could be limited by O2 diffusion at high rates (Fig. 2a and Fig. S7, ESI†). On the other hand, the discharge voltages decreased linearly with increasing current densities in Fig. 2a and Fig. S7 (ESI†), which was consistent with the overpotential being governed by the cell's resistance of ∼10 Ω from the slope in Fig. S7 (ESI†). Further support came from electrochemical impedance spectroscopy (EIS) in Fig. S8 (ESI†) which revealed low Ohmic resistance (R1) of ∼7 Ω, charge transfer resistance between β-Al2O3 and the electrodes (R2) of ∼8 Ω, and low ionic resistances from β-Al2O3 (R3) of ∼2 Ω. These resistances add up to a total cell resistance of ∼17 Ω which is similar in magnitude to the slope of discharge voltage vs. current from Fig. S7 (ESI†) (∼10 Ω). Moreover, galvanostatic intermittent titration technique (GITT) measurements in Fig. S9a (ESI†), showed small overpotentials (<10 mV, the inset of Fig. S9a, ESI†) for the discharge plateau, with 3–4 mV of the overpotentials came from charge/discharge iR drop (Fig. S9, ESI†). Remarkably, these results suggest fast oxygen redox kinetics in molten-salt Na–O2 cells in contrast to reported room temperature Na–O2 batteries using organic aprotic solvents for the electrolytes and carbon nanotube electrodes34 which had a discharge overpotential of ∼1 V at a rate of 1 mA cm−2geo.Molten-salt Na–O2 cells also demonstrated low overpotentials during charging, as well as high cycling stability (400 cycles), and high Coulombic (∼100% at 5 mA cm−2geo) and energy (∼90% at 5 mA cm−2geo) efficiencies. Molten-salt Na–O2 cells were discharged and charged with capacities limited to 1 mA h cm−2geo at rates from 1 mA cm−2geo to 10 mA cm−2geo (Fig. 2b). Remarkably, even at high rates of 10 mA cm−2geo, the overpotential on charge remained small (<300 mV) for most of the charging process, only increasing sharply after 0.8 mA h cm−2geo. Moreover, molten-salt Na–O2 cells could be stably cycled at 5 mA cm−2geo to 0.5 mA h cm−2geo for 400 cycles (Fig. 2c) with negligible increase in overpotential, as well as stable Coulombic (CE) and energy efficiencies (EE) of ∼100% and ∼90%, respectively (Fig. 2d). Moreover, increasing the cut-off capacity during cycling to 1.0 mA h cm−2geo, the molten-salt Na–O2 cell still showed low overpotentials, long cycling life (100 cycles), and high CE (∼100%) and EE (∼90%) (Fig. S10, ESI†). Such molten-salt Na–O2 cells exhibited higher energy (33 mW h cm−2geo) and power densities (19 mW cm−2geo) as compared with even the highest performing nonaqueous Na–O2 cells with 24 mW h cm−2geo and 1.0 mW cm−2geo.40 Significantly, when compared to reported Li–O2 batteries,18,36 these molten-salt Na–O2 cells can also provide comparable areal energy density (33 mW h cm−2geovs. 30 mW h cm−2geo18) and higher areal power density (19 mW cm−2geovs. 6 mW cm−2geo36) than the highest performing cells reported to date. Please see Fig. 1c, Fig. S1, and Table S1 (ESI†) for a detailed comparison of the reported performance of alkali metal–oxygen batteries vs. the molten-salt Na–O2 cells reported in this work.
Discharge products of molten-salt Na–O2 cells
The discharge of molten-salt Na–O2 cells mainly produces Na2O2. Discharge of Ni/NaNO3/KNO3/CsNO3/SS electrodes was accompanied by the emergence of four Raman bands from 700 to 800 cm−1, which can be assigned to the O–O stretching vibration (Fig. 3a). For instance, the Raman bands at 737 and 792 cm−1 are consistent with those reported for the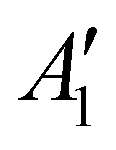 and
and 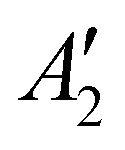 vibration modes of Na2O2,44 while those at 756 and 781 cm−1 can be attributed to O–O stretching of Cs2O2 (Ag mode)45 and K2O2 (Ag mode),44 respectively. The presence of K2O2 and Cs2O2 was unexpected because it is thermodynamically uphill to replace the Na+ ions in Na2O2 with K+ (Na2O2 + 2KNO3 → K2O2 + 2NaNO3, ΔG = 75.1 kJ mol−1) or Cs+ (Na2O2 + 2CsNO3 → Cs2O2 + 2NaNO3, ΔG = 199.7 kJ mol−1) from the molten nitrate electrolyte (please see ESI† for calculation details). We therefore hypothesize that the K2O2 and Cs2O2 observed in the discharged electrode came from soluble O22− in the molten-salt electrolyte that, upon cooling of the electrodes, became kinetically trapped out of equilibrium. This hypothesis is in agreement with the Raman spectra of an electrode that was cooled slowly (∼1 K min−1), which shows very weak Raman bands for K2O2 and Cs2O2 (Fig. S11, ESI†). Interestingly, Raman spectra of discharged electrodes that had not been washed in aprotic solvents between discharge and characterization (Fig. S12, ESI†) showed evidence of some Na2O formation, which will be discussed below. Further support that the dominant discharge product was Na2O2 came from XRD patterns of washed, discharged electrodes (Fig. 3b) that showed clear peaks from Na2O2 ((220), (112), and (300)), but none from K2O2 or Cs2O2 or Na2O, where small amounts of amorphous Cs2O2 and K2O2 formed during cooling of the electrode would not be detectable using XRD. SEM images (Fig. 3c and d) reveal Na2O2 large faceted crystals (5 to 30 μm) produced in the discharged electrode, which may be attributed to the high solubility of Na2O2 (65 mM, detailed method in ESI†) in NaNO3/KNO3/CsNO3 molten-salts.
vibration modes of Na2O2,44 while those at 756 and 781 cm−1 can be attributed to O–O stretching of Cs2O2 (Ag mode)45 and K2O2 (Ag mode),44 respectively. The presence of K2O2 and Cs2O2 was unexpected because it is thermodynamically uphill to replace the Na+ ions in Na2O2 with K+ (Na2O2 + 2KNO3 → K2O2 + 2NaNO3, ΔG = 75.1 kJ mol−1) or Cs+ (Na2O2 + 2CsNO3 → Cs2O2 + 2NaNO3, ΔG = 199.7 kJ mol−1) from the molten nitrate electrolyte (please see ESI† for calculation details). We therefore hypothesize that the K2O2 and Cs2O2 observed in the discharged electrode came from soluble O22− in the molten-salt electrolyte that, upon cooling of the electrodes, became kinetically trapped out of equilibrium. This hypothesis is in agreement with the Raman spectra of an electrode that was cooled slowly (∼1 K min−1), which shows very weak Raman bands for K2O2 and Cs2O2 (Fig. S11, ESI†). Interestingly, Raman spectra of discharged electrodes that had not been washed in aprotic solvents between discharge and characterization (Fig. S12, ESI†) showed evidence of some Na2O formation, which will be discussed below. Further support that the dominant discharge product was Na2O2 came from XRD patterns of washed, discharged electrodes (Fig. 3b) that showed clear peaks from Na2O2 ((220), (112), and (300)), but none from K2O2 or Cs2O2 or Na2O, where small amounts of amorphous Cs2O2 and K2O2 formed during cooling of the electrode would not be detectable using XRD. SEM images (Fig. 3c and d) reveal Na2O2 large faceted crystals (5 to 30 μm) produced in the discharged electrode, which may be attributed to the high solubility of Na2O2 (65 mM, detailed method in ESI†) in NaNO3/KNO3/CsNO3 molten-salts.
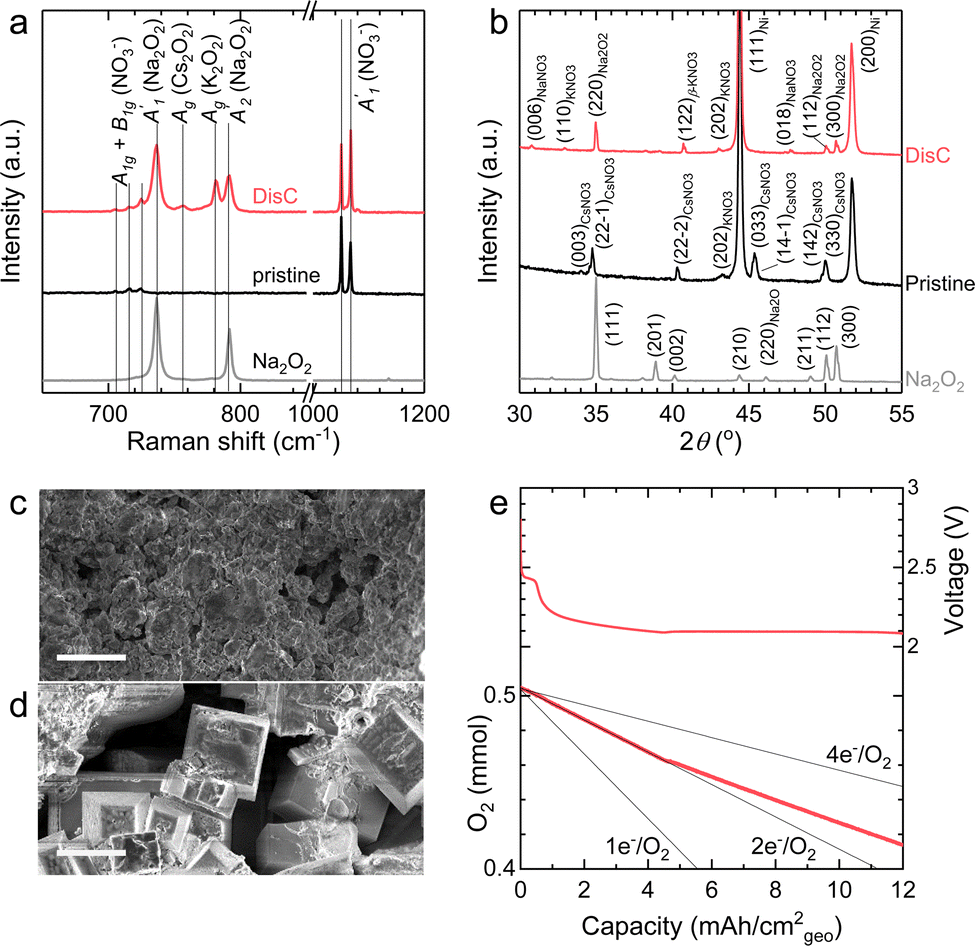 | ||
| Fig. 3 Characterization of Ni/NaNO3/KNO3/CsNO3/SS electrodes before and after discharge in molten-salt Na–O2 batteries. (a) The Raman spectra of Ni/NaNO3/KNO3/CsNO3/SS electrodes before and after discharge. Raman spectra of commercial Na2O2, NaNO3, KNO3, CsNO3 and prepared eutectic salts (Na/K/CsNO3) were used as the references (Fig. S14, ESI†). (b) The XRD patterns of Ni/NaNO3/KNO3/CsNO3/SS electrodes before and after discharge and that of commercial Na2O2 powder. Several standard XRD patterns of NaNO3 (#01-085-1466), KNO3 (#04-016-8139), CsNO3 (#04-012-0763), and Na2O2 (#01-074-0895) are references. β-KNO3 was formed in the quenching process46 and its XRD is from Materials Project.47 The discharged electrode was washed using dimethyl sulfoxide (DMSO) and then acetonitrile (AN) for the characterization of Raman spectra and XRD. (c and d) The morphology of pristine (c) and discharged (d) electrodes at current of 1 mA cm−2geo for ∼10 mA h cm−2geo (Fig. 2a). The scale bars in SEM images are 10 μm. (e) O2 pressure tracking during discharge of a molten-salt Na–O2 battery. The black lines represent the oxygen consumption with 1, 2, and 4 electrons per oxygen (e−/O2) processes. The effective geometric area of the Ni/NaNO3/KNO3/CsNO3/SS electrode in this measurement is 0.5 cm2. | ||
The formation of Na2O2, as well as some Na2O during discharge of molten-salt Na–O2 cells is further supported by the equilibrium potential from cyclic voltammogram (CV) measurements as well as pressure tracking and titration measurements. CVs collected using a molten-salt Na–O2 cell at 0.2 mV s−1 (Fig. S13, ESI†) showed a cathodic peak at 2.00 V, and anodic peak at 2.26 V. Significantly, the equilibrium potential estimated by E1/2 was 2.12 V, which is similar to that obtained from GITT measurements ∼2.09 V (Fig. S9a, ESI†), as well as the calculated thermodynamic potential of the 2Na+ + O2 + 2e− → Na2O2 at 443 K (2.15 VNa, see ESI† for details), but different from the formation NaO2 (2.07 VNa, see ESI† for details) and Na2O (1.86 VNa, see ESI† for details), in agreement with the overall reaction being the formation of Na2O2 from O2. Interestingly, pressure tracking measurements (Fig. 3e) of molten-salt Na–O2 cells during discharge showed two distinct regions corresponding to a 2.0 e−/O2 process at early discharge (0–4.5 mA h cm−2geo) and a 2.8 e−/O2 process later in discharge (4.5–12 mA h cm−2geo). The transition between the 2.0 e−/O2 and 2.8 e−/O2 processes at a discharge capacity of ∼4.5 mA h cm−2geo was accompanied by a transition from a sloped discharge voltage profile to a flat voltage profile once the discharge voltage reached ∼2.10 V. While the 2.0 e−/O2 in early discharge is consistent with the formation of Na2O2, the 2.8 e−/O2 process later in discharges suggests the formation of both Na2O2 (2 e−/O2) and Na2O (4 e−/O2). The origin of these two regions during cell discharge will be discussed later. Further evidence that the discharge product contained both Na2O2 and Na2O came from quantifications of a Ni/NaNO3/KNO3/CsNO3/SS electrode discharged to 6.0 mA h cm−2geovia the Griess method18 (Fig. S3, ESI†), Ti(IV) oxysulfate16 (Fig. S4, ESI†), and acid–based titrations29 (details in the Experimental section). As shown in Fig. 4a, the pristine Ni/NaNO3/KNO3/CsNO3/SS electrode showed only a small amount of NO2− (11.7 μmol), which may come from a chemical reaction between Ni and nitrate salts (please see details in the Experimental section), but no Na2O2 and Na2O. On the other hand, following discharge, there was no NO2−, but instead 22.2 μmol of Na2O and 99.9 μmol of Na2O2 were detected, which combined, accounts for 5.2 mA h cm−2geo comparable to the actual capacity of 6.0 mA h cm−2geo. The mechanism for the formation of both Na2O2 and Na2O will be discussed later in detail.
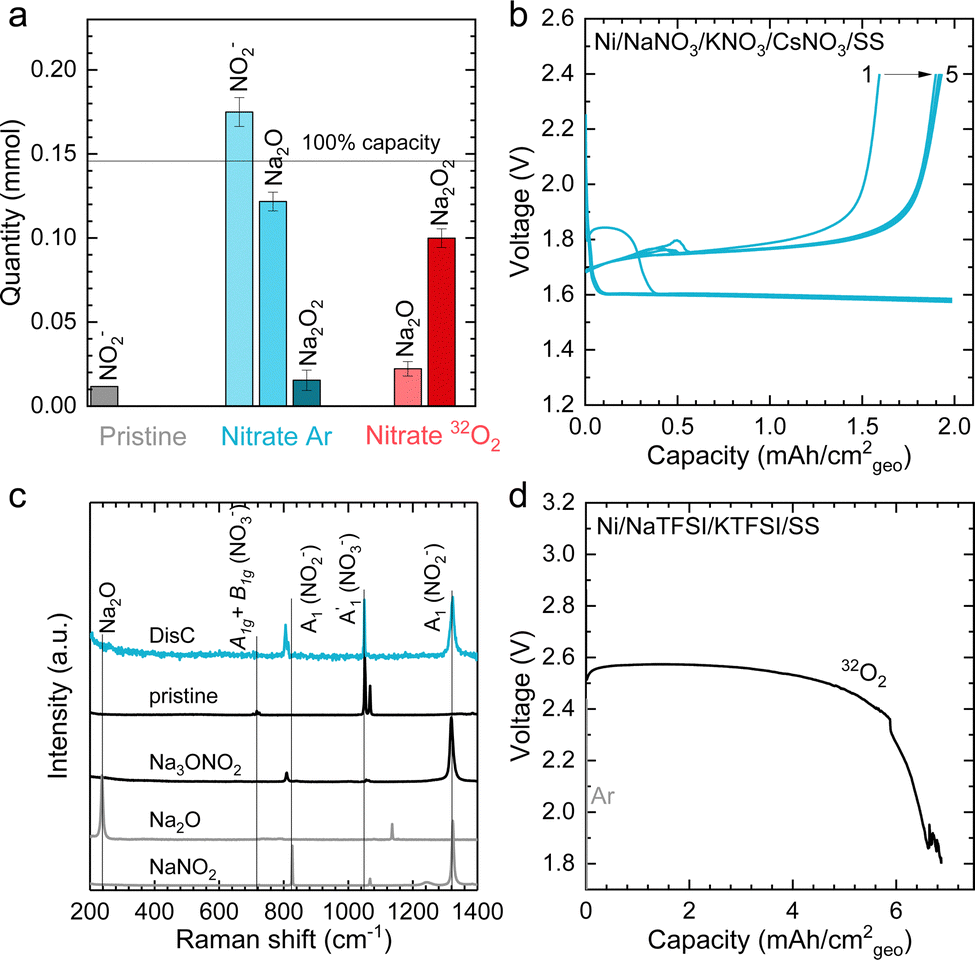 | ||
| Fig. 4 The electrochemical performance and characterization of molten-salt Na–Ar batteries. (a) Titration results of pristine and discharged Ni/NaNO3/KNO3/CsNO3/SS electrodes in O2 and Ar (see Fig. S15, ESI† for the discharge profiles of the electrode). The amounts of NO2− measured via the Griess method (Fig. S3, ESI†),18 Na2O via the acid–base method,29 and Na2O2via the titanium oxysulfate titration method (Fig. S4, ESI†).16 The pristine electrode showed a small amount of NO2−, which was attributed to the electrode preparation process (see Experimental section for details). (b) The charge–discharge profiles of a molten-salt Na–Ar cell at a current density of 1 mA cm−2geo (1st to 5th cycles) with a limited discharge capacity of 2.0 mA h cm−2geo at a temperature of 443 K. (c) The Raman spectra of positive electrodes before and after discharge from Na–Ar cells. Raman spectra of commercial Na2o and NaNO2, as well as synthetic Na3ONO2 as a reference (see Experimental section for preparation details). (d) The discharge profiles of molten-salt Na–O2 cells with eutectic salt of NaTFSI (31.9 w%)/KTFSI (68.1 w%) in 32O2 and Ar at current density of 0.2 mA cm−2geo at temperature of 483 K. | ||
Nitrate anions are redox active in molten-salt Na–O2 batteries
Discharging Ni/NaNO3/KNO3/CsNO3/SS electrodes in Na–Ar cells resulted in the formation of Na3ONO2 (from equimolar Na2O and NaNO2) with a discharge voltage of ∼1.6 V (Fig. S15 and S16, ESI†). Ni/NaNO3/KNO3/CsNO3/SS electrodes could be discharged to capacities of >12 mA h cm−2geo in Na–Ar cells (Fig. S17, ESI†). Na–Ar cells with Ni/NaNO3/KNO3/CsNO3/SS electrodes could be discharged stably at rates up to 5 mA cm−2geo (Fig. S17, ESI†) and after the first cycle, could be cycled with Coulombic efficiency (CE) of ∼104% for 5 cycles (Fig. 4b and Fig. S18, ESI†). Raman spectra of the discharged electrode (Fig. 4c) showed bands at 810, 1056, 1064, 1319 cm−1 which are consistent with those of Na3ONO2. Na3ONO2 is an adduct of equimolar Na2O and NaNO2, and can be readily formed from the reaction between Na2O and molten NaNO2 at elevated temperatures48 (details in the Experimental section). CV measurements of Na–Ar cells (Fig. S19, ESI†) showed a cathodic peak at 1.46 V and anodic peak at 1.89 V, corresponding to an equilibrium potential of ∼1.68 V, which is in good agreement with the thermodynamic potential of NaNO3 + 2Na+ +2e− → Na2O + NaNO2 at 443 K (E° = 1.49 V, see ESI† for details). Further support that the discharge of Ni/NaNO3/KNO3/CsNO3/SS electrodes in Na–Ar cells forms equimolar Na2O and NaNO2 came from titration measurements that showed 175.0 μmol of NaNO2, 121.7 μmol of Na2O and 15.4 μmol of Na2O2 (Fig. 4a), which combined, corresponds to a discharge capacity of 5.77 mA h cm−2geo and is close to the actual capacity of 6.0 mA h cm−2geo. The small amount of Na2O2 can be attributed to the direct formation of Na2O2 from NaNO3 given by NaNO3 + Na+ + e− → NaNO2 + 1/2Na2O2 with E° = 1.44 VNa at 443 K. The redox activity of nitrate anions in molten-salt Na–Ar cells is in agreement with recent work on molten-salt Li–Ar cells.49The presence of redox active nitrate anions in the electrolyte is essential to enable molten-salt Na–O2 cells that form Na2O2 during discharge. Given the observed redox activity of nitrate anions in molten-salt Na–Ar cells, molten-salt cells were constructed where nitrate anions were replaced with redox inactive bis(trifluoromethanesulfonyl)imide anions (TFSI−) by using NaTFSI/KTFSI (31.9/68.1 w%) eutectic salt50 with a melting temperature of 453 K as the electrolyte. The redox inactivity of TFSI− anions was confirmed by the negligible capacity of molten-salt Na–Ar cells with Ni/NaTFSI/KTFSI/SS electrodes (Fig. 4d). Molten-salt Na–O2 cells with Ni/NaTFSI/KTFSI/SS electrodes discharged at 483 K showed a much higher discharge voltage of ∼2.6 V (Fig. 4d) compared to Ni/NaNO3/KNO3/CsNO3/SS electrodes. Significantly, Raman spectra of the discharged Ni/NaTFSI/KTFSI/SS electrode (Fig. S20a, ESI†) indicated the formation of Na2CO3 (1080 cm−1, 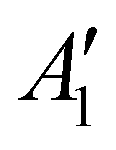 (CO32−)) and K2CO3 (1055 cm−1,
(CO32−)) and K2CO3 (1055 cm−1, 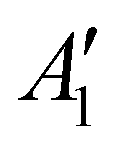 (CO32−)), but not Na2O2. The formation of Na2CO3 and K2CO3 could be attributed to the decomposition of the TFSI− anion, the only major source of carbon in the electrode. Acid–base titration measurements confirmed that negligible amounts of NaO2, Na2O2 or Na2O were present in discharged Ni/NaTFSI/KTFSI/SS electrodes. The formation of parasitic Na2CO3 and K2CO3 during the discharge of Ni/NaNO3/KNO3/CsNO3/SS electrodes is consistent with negligible capacity (<0.03 mA h cm−2geo) upon charging to 3 V (Fig. S20b, ESI†). Through comparison of NaNO3/KNO3/CsNO3 and NaTFSI/KTFSI electrolytes, it is clear that nitrate anions are critical for highly cyclable molten-salt Na–O2 batteries. We next examine the chemical reactions between O2 and the Na2O and NaNO2 that can form from the electrochemical reduction of NO3−.
(CO32−)), but not Na2O2. The formation of Na2CO3 and K2CO3 could be attributed to the decomposition of the TFSI− anion, the only major source of carbon in the electrode. Acid–base titration measurements confirmed that negligible amounts of NaO2, Na2O2 or Na2O were present in discharged Ni/NaTFSI/KTFSI/SS electrodes. The formation of parasitic Na2CO3 and K2CO3 during the discharge of Ni/NaNO3/KNO3/CsNO3/SS electrodes is consistent with negligible capacity (<0.03 mA h cm−2geo) upon charging to 3 V (Fig. S20b, ESI†). Through comparison of NaNO3/KNO3/CsNO3 and NaTFSI/KTFSI electrolytes, it is clear that nitrate anions are critical for highly cyclable molten-salt Na–O2 batteries. We next examine the chemical reactions between O2 and the Na2O and NaNO2 that can form from the electrochemical reduction of NO3−.
Chemical oxidation of Na2O and NaNO2 by O2
NaNO2 can be oxidized chemically by O2 to form NaNO3, while Na2O can react with O2 to form Na2O2 in Ni/NaNO3/KNO3/CsNO3/SS electrodes. In order to assess the reaction between Na2O and O2, Ni/NaNO3/KNO3/CsNO3/SS electrodes were prepared with added Na2O, and held at 443 K in both an O2 and Ar environment for 48 hours. Following 48 hours in an Ar environment, the Raman spectra of the Ni/NaNO3/KNO3/CsNO3/SS electrode with added Na2O was largely unchanged, retaining a strong signal at 237 cm−1 from Na2O (Fig. 5a). On the other hand, following the reaction in an O2 environment, the Raman peak for Na2O at 237 cm−1 disappeared and new Raman peaks at 733 (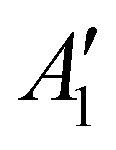 (Na2O2)), 754 (Ag (Cs2O2)), 773 (Ag (K2O2)), and 789 cm−1 (
(Na2O2)), 754 (Ag (Cs2O2)), 773 (Ag (K2O2)), and 789 cm−1 (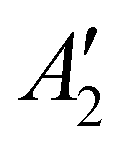 (Na2O2)) appeared.44,45 The disappearance of Na2O and appearance of Na2O2 can be attributed to the reaction given by Na2O + ½O2 → Na2O2, which is supported by the fact that Na2O2 is thermodynamically more stable than Na2O and NaO2 at 443 K, as shown in Fig. 5b. We also noticed that the two distinct Raman peaks at 1052 and 1068 cm−1 associated with the overlapping A1g modes of KNO351/CsNO352 and the A1g mode of NaNO3,53 respectively, in the pristine electrode became a single broad peak at 1054 cm−1 following the reactions in Ar and O2, where this change is attributed to the formation of a glassy NaNO3/KNO3/CsNO3 state54 following fast cooling of the cell. We further examined the chemicals reactions of Na2O and NaNO2 with O2 by exposing a Ni/NaNO3/KNO3/CsNO3/SS electrode discharged to 7.6 mA h cm−2geo in a He environment, to O2 at 443 K for 7 hours. Pressure tracking measurements upon the introduction of O2 showed an initial, very rapid decrease in pressure, following which the pressure slowly stabilized over ∼6 hours (Fig. S21, ESI†). By fitting the O2 consumption curve over the first 3 min we determined a rate of ∼0.18 mmol h−1, corresponding to a current density of 25 mA cm−2geo and indicating that the reactions of Na2O and/or NaNO2 with O2 can be very fast. This finding is in agreement with previous work55 that indicates that NO2− can be oxidized by O2 to form NO3− in bulk NaNO3/KNO3 molten salt at temperatures over 573 K.
(Na2O2)) appeared.44,45 The disappearance of Na2O and appearance of Na2O2 can be attributed to the reaction given by Na2O + ½O2 → Na2O2, which is supported by the fact that Na2O2 is thermodynamically more stable than Na2O and NaO2 at 443 K, as shown in Fig. 5b. We also noticed that the two distinct Raman peaks at 1052 and 1068 cm−1 associated with the overlapping A1g modes of KNO351/CsNO352 and the A1g mode of NaNO3,53 respectively, in the pristine electrode became a single broad peak at 1054 cm−1 following the reactions in Ar and O2, where this change is attributed to the formation of a glassy NaNO3/KNO3/CsNO3 state54 following fast cooling of the cell. We further examined the chemicals reactions of Na2O and NaNO2 with O2 by exposing a Ni/NaNO3/KNO3/CsNO3/SS electrode discharged to 7.6 mA h cm−2geo in a He environment, to O2 at 443 K for 7 hours. Pressure tracking measurements upon the introduction of O2 showed an initial, very rapid decrease in pressure, following which the pressure slowly stabilized over ∼6 hours (Fig. S21, ESI†). By fitting the O2 consumption curve over the first 3 min we determined a rate of ∼0.18 mmol h−1, corresponding to a current density of 25 mA cm−2geo and indicating that the reactions of Na2O and/or NaNO2 with O2 can be very fast. This finding is in agreement with previous work55 that indicates that NO2− can be oxidized by O2 to form NO3− in bulk NaNO3/KNO3 molten salt at temperatures over 573 K.
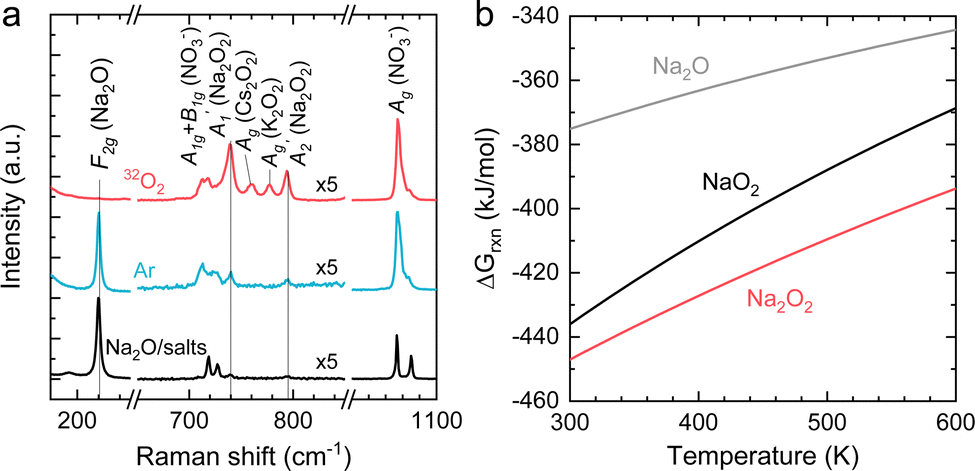 | ||
| Fig. 5 (a) The Raman spectra of Ni/NaNO3/KNO3/CsNO3/SS electrodes with the addition of Na2O before and after reaction in 412 kPa of Ar or 32O2 at 443 K. (b) The temperature dependence of the Gibbs free energies for the formation of NaO2, Na2O2, and Na2O. The Gibbs free energies are calculated based on the following reactions: 2Na + 2O2 → 2NaO2, 2Na + O2 → Na2O2 and 2Na + ½O2 → Na2O. Thermodynamic data used for the calculations are given in Table S3 (ESI†). | ||
Nitrate-mediated oxygen reduction reaction in Na–O2 batteries
The oxygen reduction reaction in molten-salt Na–O2 batteries occurs via a nitrate-mediated mechanism whereby NaNO3 is first reduced electrochemically to form Na2O and NaNO2 (reaction (1)), following which Na2O reacts with O2 to form Na2O2 (reaction (2)), and NaNO2 is oxidized by O2 to regenerate NaNO3 (reaction (3)), resulting in an apparent 2 e−/O2 overall reaction given by 2Na+ + O2 + 2e− → Na2O2 (reaction (4)). We note in the proposed reaction scheme that nitrate anions are not consumed during discharge such that the entire discharge capacity comes from the overall 2e−/O2 reduction of O2 to Na2O2. Each step of the nitrate-mediated 2 e−/O2 reaction has been demonstrated above, where the electrochemical reduction of NaNO3 to Na2O and NaNO2 was observed in Na–Ar cells (Fig. 4), and the oxidation of Na2O and NaNO2 by O2 was shown through chemical experiments (Fig. 5 and Fig. S21, ESI†). Further evidence that the formation of Na2O2 from O2 is facilitated by nitrate anions comes from 18O-isotopic labelling, where according to the purposed mechanism, discharge of Ni/NaN16O3/KN16O3/CsN16O3/SS electrodes in a 36O2 (18O18O) environment would proceed via:| 2Na+ + NaN16O3 +2e− → NaN16O2 + Na216O | (1) |
| Na216O + ½ 36O2 → Na216/18O2 | (2) |
| NaN16O2 + ½ 36O2 → NaN16/18O3 | (3) |
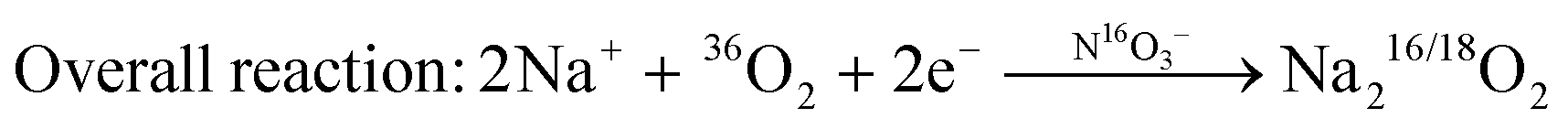 | (4) |
Raman spectroscopy of a Ni/NaN16O3/KN16O3/CsN16O3/SS electrode discharged in a 36O2 (18O18O) environment showed evidence of red-shifted Raman peaks associated with 18O-enrichment of both NaNO3 (reaction (2)) and Na2O2 (reaction (3)). As shown in Fig. 6a, 36O2 discharged electrodes showed several new red-shift peaks at 693, 713, and 768 cm−1 in the O–O stretching region relative to those of 16O-substituted peroxides at 733, 754, 773, and 789 cm−1, which could be attributed to 18O-substituted peroxides. Such red shifts of 18O-substituted Na2O2 are supported by the density functional theory (DFT) computed Raman spectra for Na218O2 and Na216O2, where the bands of Na218O2 are shifted by 46 cm−1 relative to Na216O2 (Fig. 6a). Of significance is to note that while red-shifted Raman bands appeared in the 36O2 discharged electrodes, the band at 789 cm−1 attributable to Na216O2 remained, suggesting the discharge product was a mixture of Na218O2 and Na216O2. In addition, in the NO3− symmetric stretching region, there are also red-shift peaks at 1010, 1013, 1018, 1029, 1033, 1037, 1045, and 1057 cm−1, consistent with the formation of 18O-substituted NaNO3, KNO3 and CsNO3 (Fig. 6b). Such red shifts in the NO3− symmetric stretching region due to 18O-enrichment is supported by simulated wavenumber of symmetric stretching as a function of 18O-substituted-N16O3− (Fig. 6b) obtained from DFT calculations of the isolated anion in vacuum at the B3LYP/6-31G(d,p) level, where greater red-shifts in the symmetric stretching were correlated with greater 18O substitution in N16O3−. The presence of 18O-substituted-N16O3− can be attributed to the oxidation of N16O2− by 36O2 given by reaction (3).
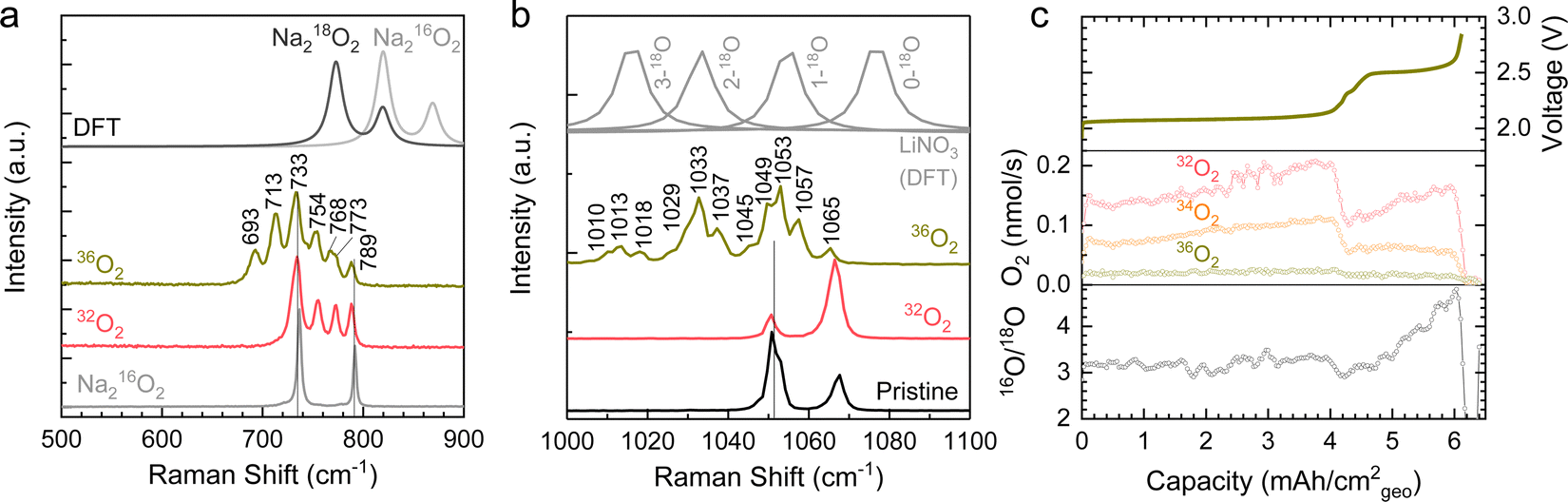 | ||
| Fig. 6 (a and b) The Raman spectra of discharged Ni/NaNO3/KNO3/CsNO3/SS electrodes of Na–O2 cells in 36O2 and 32O2 and DFT calculated Raman spectra. (c) The rate of oxygen evolution of 32O2, 34O2, and 36O2 as detected by DEMS measurements during charging a symmetric molten-salt Ni/NaNO3/KNO3/CsNO3/SS battery at 0.2 mA cm−2geo to 2.8 V at 443 K with He carrier gas. Prior to the DEMS measurements, a Na–O2 cell with Ni/NaNO3/KNO3/CsNO3/SS was discharged to 8 mA h cm−2geo at 0.2 mA cm−2geo and the positive electrode was collected to serve as the counter electrode for the DEMS measurement. In the DEMS experiments, the battery was kept at OCV for 5 hours at 443 K to ensure full melting of the mixture of NaNO3 (26 w%), KNO3 (27 w%), and CsNO3 (47 w%). The electrode geometric area was 0.785 cm2geo. The ratio of 16O/18O is ∼3 in the first charging plateau at ∼2.1 V and the ratio increased in the second charging plateau at 2.5 V. The redox potential of nitrate reduction in Ar was ∼1.65 V (vs. Na negative electrode) and was used to convert the measured cell potential to the voltage (vs. Na negative electrode) scale (Fig. S16, ESI†). | ||
Further evidence of a nitrate-mediated, apparent 2 e−/O2 oxygen reduction reaction came from differential electrochemical mass spectroscopy (DEMS) 18O-isotopic labelling experiments that showed that evolved oxygen came primarily from nitrate anions, as opposed to the O2 discharge environment. DEMS measurements were conducted using a symmetric molten-salt Na–O2 cell discharged in 36O2 (Fig. 6c). The negative electrode used in the DEMS cell was prepared in a Na–Ar cell with discharge capacity of 1.6 mA h cm−2geo to achieve a flat discharge plateau (Fig. S16, ESI†). Upon charging at 0.2 mA cm−2geo, three types of molecular oxygen (i.e., 32O2 (16O16O), 34O2 (16O18O), and 36O2 (18O18O)) were detected, consistent with the decomposition of Na216O16O, Na216O18O and Na218O18O, respectively. Remarkably, the amount of 36O2 evolved was very small (<2%), which indicates only a small amount of Na218O18O was formed during discharge in 36O2, consistent with a nitrate-mediated reaction, as opposed to the direct reduction of O2 to Na2O2. The high proportion (90%) of 32O2 (16O16O) detected during charge is in disagreement with reactions (1)–(4), which can be attributed to either (1) that the electrochemical reduction of NaNO3 to Na2O2 given by NaNO3 + Na+ + e− → NaNO2 + 1/2Na2O2 can contribute more significantly to the discharge process in Na–O2 cells as opposed to that observed in Na–Ar cells in Fig. 4a, (2) that the lower O2 pressure for 36O2 cells ∼100 kPa as opposed to 32O2 cells ∼410 kPa slowed reactions (2) and (3), resulting in a higher proportion of Na216O in the discharged electrode, which could evolve 32O2 (16O16O) or (3) that the nitrate-mediated reaction pathway is more complex than that given by reactions (1)–(4). Although this discrepancy motivates additional research into the detailed mechanism, we note that the high ratio of 16O/18O ∼3 in Fig. 6c strongly supports the hypothesis of a nitrate-mediated oxygen reduction reaction instead of direct O2 reduction in molten-salt Na–O2 cells.
In this work, we have reported a molten-salt Na–O2 battery where the formation of Na2O2 is mediated by the electrochemical activity of nitrate anions. Such findings are similar to our recent work on molten-salt Li–O2 batteries19 where the redox activity of nitrate anions can enable the formation of Li2O upon discharge at 423 K. However, in molten-salt Li–O2 cells, the Li2O formed from the reduction of NO3− to NO2− is thermodynamically preferred to Li2O2,18 whereas in Na–O2 cells, Na2O can further react with O2 to form Na2O2. The mediation of the oxygen reduction reaction by nitrate anions in molten-salt Li–O2 and Na–O2 cells has a number of interesting implications on the cell design and performance. First, the catalytic activity of the electrode surface towards nitrate redox is critical to enable high rates and low overpotentials. In molten-salt Li–O2 cells, we identified NiO as having high catalytic activity due to its optimum binding of NO3− and NO2−, where weaker binding catalysts like Cu2O were limited by NO3− adsorption whereas stronger binding catalysts like Mn3O4 were limited by the oxidation of NO2− by O2.19 The high performance of Ni-based catalysts for nitrate redox is in agreement with previous reports for Li–Ar cells.19,49 Second, the high weight of Ni-based electrodes limits the achievable specific energy of the positive electrode,18,19 necessitating the development of novel electrode materials with high catalytic activity for nitrate redox with lower weight, such as Ni-coated carbon.18 Third, while the electrolyte is not consumed in the overall reaction where nitrate anions can participate in multiple catalytic cycles over the span of a single discharge (reactions (1)–(3)), the role of nitrate's redox activity in the oxygen reduction reaction may introduce limitations in the lean-electrolyte regime needed to achieve high cell level specific energy, warranting additional research. Fourth, the blocking of O2 within the electrode due to accumulation of the discharge product may shift the discharge product towards the formation of Na2O at deeper discharges, which may be responsible for the presence of minor Na2O in addition to major Na2O2 in deep discharge observed in Fig. 3e. Finally, the temporary formation of NO2− in the electrolyte during discharge may locally alter the physicochemical properties of the molten-salt electrolyte, such as its melting point, viscosity or ionic conductivity. During charging, our recent work56 has shown that the 2 electron oxidation of bulk Na2O2 to O2 is limited by the last step (NaO2 → O2 + Na+ + e−), which is known to occur with small overpotentials in Na–O2 cells with aprotic solvents,39 rationalizing the small charging overpotentials observed in this work. However, further work is needed to confirm the reaction pathway upon charging in molten-salt Na–O2 batteries. While this work has shown that molten-salt Na–O2 batteries can achieve promising areal energy (33 mW h cm−2geo) and power densities (19 mW cm−2geo) as well as stable cycling (400 cycles) and high energy efficiencies ∼90%, further research efforts are needed to achieve commercially viable cell level performance.
Conclusions
In summary, we report high-performance molten-salt Na–O2 batteries with a simple structure, utilizing a nitrate-mediated reaction to achieve >2e−/O2. First, we investigated the electrochemical performance of these molten-salt Na–O2 batteries, showing high power density (19 mW cm−2geo) at 10 mA cm−2geo and high-energy efficiency (∼90%) at a high current density of 5 mA cm−2geo, with long cycle life (400 cycles). Next, using Raman, pressure tracking and titration measurements, we showed that the dominant discharge product was Na2O2. We further studied the redox activity of nitrate anions in Na–Ar cells, showing that NaNO3 could be electrochemically reduced to Na2O and NaNO2, where Na2O and NaNO2 could further react chemically with O2. Finally, using 18O-labeling experiments, we showed that the oxygen reduction reaction in molten-salt Na–O2 batteries occurred via a nitrate-mediated mechanism whereby NaNO3 was first electrochemically reduced to form Na2O and NaNO2, following which Na2O reacted with O2 to form Na2O2, while NaNO2 was oxidized by O2 to regenerate NaNO3, resulting in an apparent 2 e−/O2 overall reaction to form Na2O2. Such nitrate-mediated molten-salt Na–O2 batteries provide a novel approach to develop alkali metal–O2 batteries with high energy and power density.Author contributions
Y. G. Z., G. L. and Y. S. H. conceived and designed the study. Y. G. Z., G. L., A. A., K. G., Y. Z. and Y. S. H. conducted experiments, computation, and analysis. Y. G. Z., G. L. and Y. S. H. wrote the manuscript, and all authors edited the manuscript. There is no competing interest. All data and materials used in the work are available in the manuscript or supplementary materials.Conflicts of interest
Y. G. Z., G. L. and Y. S. H. have applied for a patent related to this work.Acknowledgements
This work is in part supported by Shell and Department of Navy award N00014-20-1-2221 issued by the Office of Naval Research. The United States Government has a royalty-free license throughout the world in all copyrightable material contained herein. This work made use of the MRSEC Shared Experimental Facilities at MIT, supported by the National Science Foundation under award number DMR-1419807. G. L. gratefully acknowledges partial support from a Natural Sciences and Engineering Research Council of Canada (NSERC) PGS-D and Siebel Scholarship (Class of 2020). We would like to thank Dr Shuting Feng for assistance with DFT calculations.Notes and references
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer and Y.-M. Chiang, Science, 2018, 360, eaas9793 CrossRef PubMed.
- H. Liu, X.-B. Cheng, Z. Jin, R. Zhang, G. Wang, L.-Q. Chen, Q.-B. Liu, J.-Q. Huang and Q. Zhang, EnergyChem, 2019, 1, 100003 CrossRef.
- H. Wang, D. Yu, C. Kuang, L. Cheng, W. Li, X. Feng, Z. Zhang, X. Zhang and Y. Zhang, Chemistry, 2019, 5, 313–338 CrossRef CAS.
- J. Xie, L. Liao, Y. Gong, Y. Li, F. Shi, A. Pei, J. Sun, R. Zhang, B. Kong and R. Subbaraman, Sci. Adv., 2017, 3, eaao3170 CrossRef.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194 CrossRef CAS PubMed.
- Y.-C. Lu, B. M. Gallant, D. G. Kwabi, J. R. Harding, R. R. Mitchell, M. S. Whittingham and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 750–768 RSC.
- J. Read, K. Mutolo, M. Ervin, W. Behl, J. Wolfenstine, A. Driedger and D. Foster, J. Electrochem. Soc., 2003, 150, A1351–A1356 CrossRef CAS.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS.
- Y.-C. Lu, H. A. Gasteiger and Y. Shao-Horn, J. Am. Chem. Soc., 2011, 133, 19048–19051 CrossRef CAS PubMed.
- G. V. Chase, S. Zecevic, T. W. Wesley, J. Uddin, K. A. Sasaki, V. P. Giordani, V. Bryantsev, M. Blanco and D. Addison, US Pat., 20120028137, 2012 Search PubMed.
- Y. Chen, S. A. Freunberger, Z. Peng, O. Fontaine and P. G. Bruce, Nat. Chem., 2013, 5, 489–494 CrossRef CAS PubMed.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng and P. G. Bruce, J. Am. Chem. Soc., 2012, 135, 494–500 CrossRef PubMed.
- Y. G. Zhu, F. T. Goh, R. Yan, S. Wu, S. Adams and Q. Wang, Phys. Chem. Chem. Phys., 2018, 20, 27930–27936 RSC.
- B. McCloskey, D. Bethune, R. Shelby, G. Girishkumar and A. Luntz, J. Phys. Chem. Lett., 2011, 2, 1161–1166 CrossRef CAS PubMed.
- S. Feng, M. Huang, J. R. Lamb, W. Zhang, R. Tatara, Y. Zhang, Y. G. Zhu, C. F. Perkinson, J. A. Johnson and Y. Shao-Horn, Chemistry, 2019, 5, 2630–2641 CrossRef CAS PubMed.
- V. Giordani, D. Tozier, H. Tan, C. M. Burke, B. M. Gallant, J. Uddin, J. R. Greer, B. D. McCloskey, G. V. Chase and D. Addison, J. Am. Chem. Soc., 2016, 138, 2656–2663 CrossRef CAS.
- C. Xia, C. Y. Kwok and L. F. Nazar, Science, 2018, 361, 777–781 CrossRef CAS PubMed.
- D. Koo and S. J. Kang, ACS Appl. Mater. Interfaces, 2021, 13, 47740–47748 CrossRef CAS PubMed.
- Y. G. Zhu, G. Leverick, L. Giordano, S. Feng, Y. Zhang, Y. Yu, R. Tatara, J. R. Lunger and Y. Shao-Horn, Joule, 2022, 6(8), 1887–1903 CrossRef CAS.
- K. Baek, J. G. Lee, A. Cha, J. Lee, K. An and S. J. Kang, J. Power Sources, 2018, 402, 68–74 CrossRef CAS.
- C. Peng, C. Guan, J. Lin, S. Zhang, H. Bao, Y. Wang, G. Xiao, G. Z. Chen and J. Q. Wang, ChemSusChem, 2018, 11, 1880–1886 CrossRef CAS PubMed.
- S. Zhang, Y. Yang, L. Cheng, J. Sun, X. Wang, P. Nan, C. Xie, H. Yu, Y. Xia and B. Ge, Energy Storage Mater., 2021, 35, 142–147 CrossRef.
- S. Liu, W. Han, B. Cui, X. Liu, F. Zhao, J. Stuart and S. Licht, J. Power Sources, 2017, 342, 435–441 CrossRef CAS.
- M. Shahabi, N. Masse, H. Sun, L. Wallace, A. Powell and Y. Zhong, in REWAS 2022: Energy Technologies and CO2 Management (Volume II), Springer; 2022, pp. 47–57 Search PubMed.
- S. Xia, X. Wu, Z. Zhang, Y. Cui and W. Liu, Chemistry, 2019, 5, 753–785 CrossRef CAS.
- X. Guo, L. Zhang, Y. Ding, J. B. Goodenough and G. Yu, Energy Environ. Sci., 2019, 12, 2605–2619 RSC.
- B. D. McCloskey, A. Valery, A. C. Luntz, S. R. Gowda, G. M. Wallraff, J. M. Garcia, T. Mori and L. E. Krupp, J. Phys. Chem. Lett., 2013, 4, 2989–2993 CrossRef CAS PubMed.
- L. S. Darken and H. F. Meier, J. Am. Chem. Soc., 1942, 64, 621–623 CrossRef CAS.
- J. Berkowitz, D. J. Meschi and W. A. Chupka, J. Chem. Phys., 1960, 33, 533–540 CrossRef CAS.
- P. D. Hooker and K. J. Klabunde, Chem. Mater., 1993, 5, 1089–1093 CrossRef CAS.
- J. R. Harding, PhD thesis, Massachusetts Institute of Technology, 2015.
- K. H. Stern, J. Phys. Chem. Ref. Data, 1972, 1, 747–772 CrossRef.
- N. Ortiz-Vitoriano, T. P. Batcho, D. G. Kwabi, B. Han, N. Pour, K. P. C. Yao, C. V. Thompson and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 2636–2643 CrossRef CAS PubMed.
- X. Gao, Y. Chen, L. Johnson and P. G. Bruce, Nat. Mater., 2016, 15, 882–888 CrossRef CAS PubMed.
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef CAS.
- Z. Zhu, A. Kushima, Z. Yin, L. Qi, K. Amine, J. Lu and J. Li, Nat. Energy, 2016, 1, 16111 CrossRef CAS.
- Y. Qiao, K. Jiang, H. Deng and H. Zhou, Nat. Catal., 2019, 2, 1035–1044 CrossRef CAS.
- P. Hartmann, C. L. Bender, M. Vračar, A. K. Dürr, A. Garsuch, J. Janek and P. Adelhelm, Nat. Mater., 2013, 12, 228–232 CrossRef CAS PubMed.
- H. Yadegari, M. N. Banis, B. Xiao, Q. Sun, X. Li, A. Lushington, B. Wang, R. Li, T.-K. Sham, X. Cui and X. Sun, Chem. Mater., 2015, 27, 3040–3047 CrossRef CAS.
- G. Cong, W. Wang, N.-C. Lai, Z. Liang and Y.-C. Lu, Nat. Mater., 2019, 18, 390–396 CrossRef CAS PubMed.
- W. Wang, N.-C. Lai, Z. Liang, Y. Wang and Y.-C. Lu, Angew. Chem., Int. Ed., 2018, 57, 5042–5046 CrossRef CAS PubMed.
- X. Ren and Y. Wu, J. Am. Chem. Soc., 2013, 135, 2923–2926 CrossRef CAS PubMed.
- H. Eysel and S. Thym, Z. Anorg. Allg. Chem., 1975, 411, 97–102 CrossRef CAS.
- T. Livneh, A. Band and R. Tenne, J. Raman Spectrosc., 2002, 33, 675–676 CrossRef CAS.
- R. Benages-Vilau, T. Calvet, M. A. Cuevas-Diarte and H. A. J. Oonk, Phase Transitions, 2016, 89, 1–20 CrossRef CAS.
- Persson, Data retrieved from the Materials Project for KNO3 (mp-5158) from database version v2021.11.10, Materials Project, 2022 DOI:10.17188/1263112.
- M. Jansen, Angew. Chem., Int. Ed. Engl., 1976, 15, 376–377 CrossRef.
- V. Giordani, D. Tozier, J. Uddin, H. Tan, B. M. Gallant, B. D. McCloskey, J. R. Greer, G. V. Chase and D. Addison, Nat. Chem., 2019, 11, 1133–1138 CrossRef CAS PubMed.
- R. Hagiwara, K. Matsumoto, K. Tamaki, T. Nohira and T. Goto, US Pat., 20090212743A1, 2009 Search PubMed.
- R. Carr, J. Vanos and B. H. Torrie, Chem. Phys. Lett., 1979, 65, 73–76 CrossRef CAS.
- M. Brooker, J. Chem. Phys., 1973, 59, 5828–5829 CrossRef CAS.
- D. Rousseau, R. Miller and G. Leroi, J. Chem. Phys., 1968, 48, 3409–3413 CrossRef CAS.
- M. K. Raju, Presented in part at the Proceedings of the Indian Academy of Sciences-Section A, 1945 Search PubMed.
- V. A. Sötz, A. Bonk, J. Forstner and T. Bauer, Thermochim. Acta, 2019, 678, 178301 CrossRef.
- J. R. Lunger, N. Lutz, J. Peng, M. Bajdich and Y. Shao-Horn, Chem. Mater., 2022, 34, 3872–3881 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee01774a |
| This journal is © The Royal Society of Chemistry 2022 |