
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in triplet–triplet annihilation upconversion and singlet fission, towards solar energy applications
Andrew J.
Carrod
 a,
Victor
Gray
*b and
Karl
Börjesson
*a
a,
Victor
Gray
*b and
Karl
Börjesson
*a
aUniversity of Gothenburg, Department of Chemistry and Molecular Biology, Kemivägen 10, 41296, Gothenburg, Sweden. E-mail: karl.borjesson@gu.se
bDepartment of Chemistry - Ångström Laboratory, Uppsala University, Box 523, 751 20, Uppsala, Sweden. E-mail: victor.gray@kemi.uu.se
First published on 2nd November 2022
Abstract
Solar energy is an ample renewable energy resource, with photovoltaic (PV) technology enabling a direct route from light to electricity. Currently, PVs are limited in photon conversion efficiency, due in major part to spectral losses. Mitigation of these losses is therefore important, economically and environmentally. Two processes that aim to increase solar light utilisation are described herein. The first is triplet–triplet annihilation upconversion (TTA-UC), through which two incoherent photons of low energy can produce one of higher energy, reducing below bandgap losses. Secondly, singlet fission (SF), through which two triplet states may be obtained from one initial singlet excited state, in theory allowing two electrons per photon in a PV, reducing thermalisation losses. These fields are often covered seperately, despite being the reverse processes of one another. This work aims to consolidate research in the two fields and highlight their similarities and common challenges, specifically those relevant to PV applications. Herein, we cover systems primarily based on organic small molecules (anthracene, rubrene, tetracene, pentacene), and detail the fabrication of functional materials containing them (MOFs, gels, SAMs on TiO2, thin evaporated and solution cast films, and cavities). We further offer our recommendations for the focus of future work in both the TTA and SF fields, and discuss the need to address current limitations such as poor triplet diffusion, limited charge injection to PVs, and material stability. Specifically, one could do this by cherry picking ideas from other research fields, for example photosensitisers for photodynamic therapy could be used as TTA sensitisers, and molecules having a considerable excited state aromaticity could be considered as SF materials. We hope this review may aid development towards the end goal of an efficient PV, incorporating either, or both, SF and TTA-UC materials.
 Andrew J. Carrod | Andrew J. Carrod graduated from the University of Hull, earning a degree in chemistry with honors in 2016. He later earned a PhD degree in chemistry from the University of Birmingham in 2021, with a thesis on the subject of photoactive iridium(III) complexes, under the supervision of Professor Zoe Pikramenou. After working as a postdoctoral researcher for Professor Karl Börjesson at the University of Gothenburg, he has been awarded a MSCA postdoctoral fellowship to remain at the same institution. His current research interests focus on chemical synthesis of photoactive molecules, and further studying their utility in triplet–triplet annihilation upconversion systems. |
 Victor Gray | Victor Gray is a researcher at Uppsala University, Department of Chemistry – Ångström in Sweden. He obtained his PhD from Chalmers University of Technology in Sweden with Kasper Moth Poulsen (2017). He spent 3 years at the Cavendish Laboratory, University of Cambridge (UK), as a postdoctoral researcher with Akshay Rao working on organic–inorganic hybrid systems for photon conversion. In 2022 he joined Uppsala University as an independent researcher studying photophysical processes in solar energy harvesting materials. |
 Karl Börjesson | Karl Börjesson is Professor in physical chemistry at the University of Gothenburg. He obtained his PhD from Chalmers University of Technology in Sweden under the supervision of Bo Albinsson in 2011, and where after that a postdoctoral fellow at the University of Strasbourg with Paolo Samori and at Chalmers University of Technology with Kasper Moth-Poulsen. He started his independent career in 2015 at the University of Gothenburg and has been there since. His research is focused on photochemistry and photophysics, strong light-matter interactions, and materials chemistry. His prime methodologies include optical spectroscopy and physical organic chemistry. |
Broader contextRenewable energy is positioned to take centre-stage in future decarbonisation efforts, such as those agreed at the Paris and COP26 summits. One renewable energy source, solar light, holds the potential to mitigate the growing energy and environmental crisis. Researchers have continuously tried to increase the efficiency of solar harvesting devices, yet limitations have been unavoidable since some of the energy from the solar spectrum is inaccessible. If solar cells were more efficient, the cost per unit of electricity produced in PVs would drastically decrease, leading to an increased uptake of this technology. Subsequently, a reduction in fossil fuel reliance will be observed. To reduce spectral losses, and therefore increase efficiencies in PVs, one could imagine the monochromation of solar light. Two of the most relevant candidates for this are triplet–triplet annihilation upconversion (TTA-UC) and singlet fission (SF) materials. We here provide a detailed overview of both techniques, critically assessing progress and state of the art in the development of TTA-UC and SF materials. This will guide the future development of new TTA-UC and SF materials that may be incorporated into PVs, and is further of interest to other optoelectronic devices such as OLEDs. |
Introduction
Sunlight is a near infinite source of energy, and assuming ideal conditions, sunlight reaches the Earth's surface with an irradiance of 1000 W m−2.1 To put the number in perspective, if all sunlight could be turned into electricity with no losses, an area of around 7 m2 would provide the same energy per hour as combusting 1 kg of coal.2 With the global call for decarbonisation,3 and renewed commitment from the world's leading energy consumers to an increased use of renewable energy,4–6 it is important to create solar energy harvesting technologies with the highest possible efficiency.Energy production through photovoltaic (PV) devices has grown nearly exponentially in the previous 10 years,7 as a result of a radical decrease in the levelised cost of electricity (LCOE) from solar cells. One large factor in this decrease is the decline in production costs with increasing manufacturing scale, to a point at which production cost is only a small factor in PV price.8 Further increases to the efficiency of these devices, which are currently limited in maximum theoretical efficiency, can drive the LCOE even lower and would be of great scientific interest, with large economic and environmental impact.9 Furthermore, an increase in PV module efficiency leads not only to cheaper prices for energy consumers but also to a decrease in the space needed to provide a similar energy output. In turn efficiency increases will allow the integration of PV devices into increasingly innovative locations, as in urban design with building integrated photovoltaics, reducing the need for land space sacrifice in energy production from PV's.10
In this review we describe the existing limitations of single junction photovoltaics and describe the loss mechanisms inherent to these PV devices. We further detail two distinct yet complimentary photophysical processes; triplet–triplet annihilation upconversion (TTA-UC) and singlet fission (SF), as well as assessing their practical use as strategies for the enhancement of solar cell efficiency. Currently TTA-UC and singlet fission are seen as viable chemical approaches to PV efficiency enhancement. They achieve this through monochromation of the solar spectrum. It is therefore prudent to consolidate this research, detailing the recent progress in the field and its future perspectives.
Limitations in current photovoltaic devices
The limit of theoretical efficiency for single junction non-concentrated solar cells has been determined as 33.16% reached at a bandgap of 1.34 eV. Known as the Shockley–Queisser (SQ) limit, this value was calculated using some fundamental assumptions and approximations.11,12 Assumptions made were that all photons with energies above the bandgap (Eg) create free electrons and holes, which will yield one electron per photon. Furthermore, that re-emission of light would occur upon each hole–electron recombination. As it is a maximum theoretical limit, the SQ value was also calculated based on perfect materials containing no defects, and the end of the absorption window is estimated as a step function. These assumptions do not hold true in real-world conditions; however it is possible to assess real-world cells under true laboratory conditions and contrast them to the SQ value, assessing their energy loss channels so as to elucidate methods to improve their efficiency.A detailed look at energy loss in solar cells
In this section we will aim to detail the major energy loss channels in PV cells. Energy loss processes in solar cells are broadly classified into two terms, intrinsic13 and extrinsic losses.14 These umbrella terms are used to denote processes which can be eliminated (extrinsic) or that are fundamental limitations (intrinsic). As we will later consider energy enhancement assuming perfect cells, we will not describe in detail extrinsic losses, as these can in theory be eliminated. We will consider each of the intrinsic factors in turn then, and briefly describe their physical mechanism and magnitude.Firstly, some energy loss in any solar cell can be attributed to the deliberate tuning of the solar cell Eg to a sizeable value. The Eg value must be large enough to produce a reasonable voltage, however this does not allow for the capture of the whole range of the solar spectrum. A sizeable proportion of the solar irradiation at ground level appears in the near infrared region (Fig. 1a).1 Many of the solar cell materials have Eg larger than 1.1 eV (1130 nm), in which case the loss of energy through an inability to absorb infrared light is significant.15 Thermalisation is the second major loss of energy; due to the absorption of photons with energy larger than Eg. Relaxation to the bandgap energy is realised through a release of heat.16 Other less substantial but still measurable energy loss pathways are emission losses, Carnot losses and Boltzmann losses. Emission losses are due to the radiative electron–hole recombination events occurring within the single bandgap PV's.17 Some energy loss is inherent upon the transfer of heat energy to electrical work, the voltage drop due to this exchange in photovoltaic device is known as the Carnot factor.18 Entropy is increased due to an angular discrepancy between absorption and emission. This in turn leads to a voltage decrease known as the Boltzmann loss.15 This review focuses on strategies to mitigate the first two loss processes; reducing sub-bandgap losses and thermalisation, through utilisation of known photophysical processes. The next section will discuss these methods, and how they operate to enable the improvement of the efficiency of known solar cells.
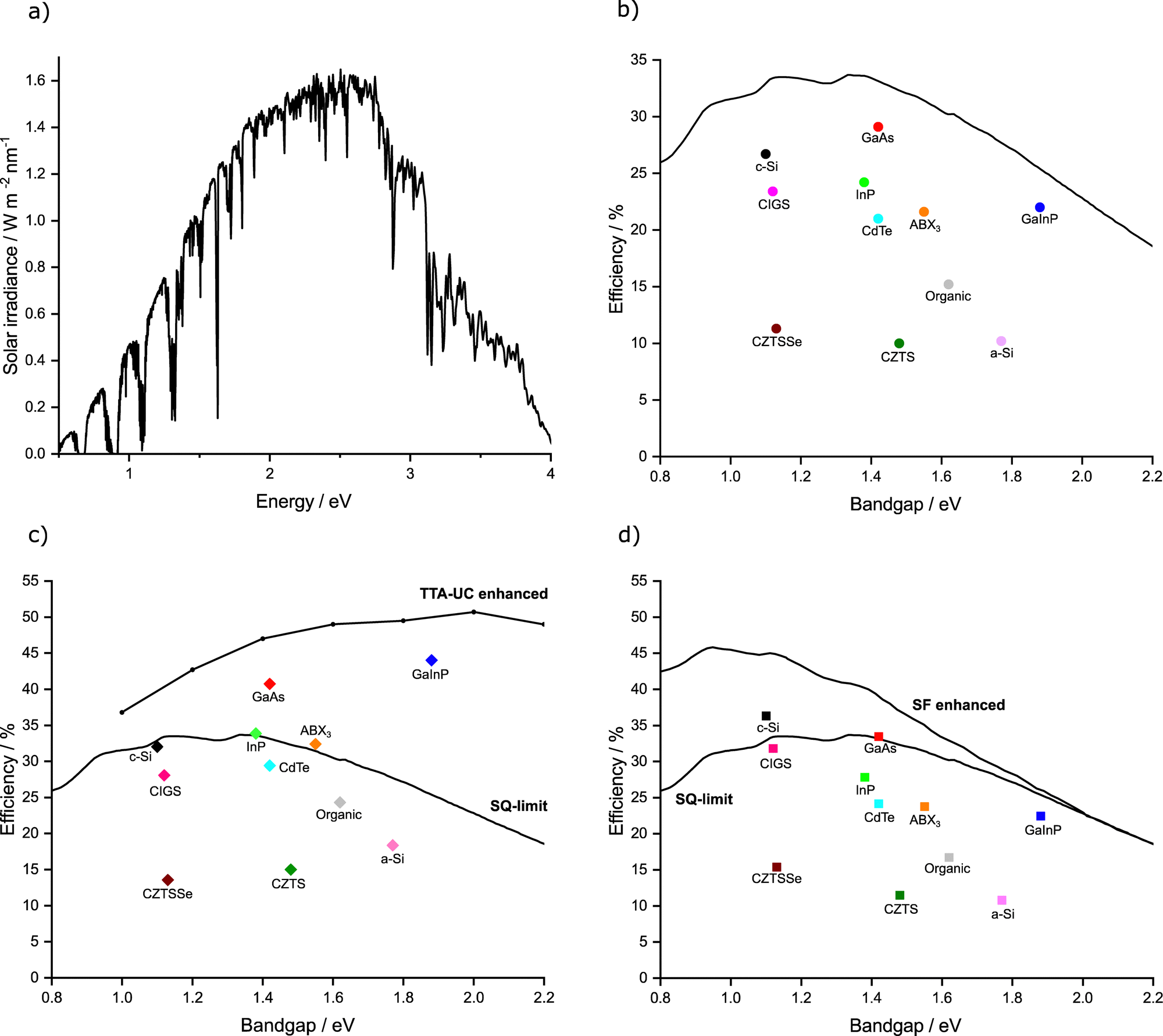 | ||
| Fig. 1 Graphical representation of (a) the AM 1.5 solar spectrum. (b) Shockley–Queisser limit of efficiency (black line) plotted alongside the cells of highest confirmed efficiency in each major class of solar cell. Where c-Si = crystalline silicon, GaAs = gallium arsenide, and GaInP = gallium indium phosphide, InP = indium phosphide, CIGS = copper indium gallium selenide, CdTe = cadmium telluride, ABX3 = perovskite, CZTS = Copper zinc tin sulphide, CZTSSe = Copper zinc tin sulphide selenide, a-Si = amorphous silicon. (c and d) SQ and TTA-UC/SF enhanced efficiency limits (black lines) plotted alongside current highest maximum efficiency possible in each major class of solar cell assuming ideal TTA-UC/SF enhancement. We use the data from Fig. 1b and increases are calculated as per ref. 46 and 52, for TTA-UC and SF respectively. | ||
Overcoming the SQ limit: mitigating losses through photon conversion
Overcoming the SQ limit for solar cells can be achieved in a seemingly facile manner, by eliminating some or all of the losses described above. One way to do this would be to extend the optical window in which a PV device may operate without compromise on the bandgap of the material. This is possible in multi-junction photovoltaics where current efficiencies have been experimentally measured at 47% in six junction concentrator systems.19 These are theorised to give a possible maximum efficiency of around 87% at an infinite number of junctions.20 Multi-junction solar cells are however not widely used in terrestrial energy production, due to the complexity of fabrication and scarcity of raw materials required in multi junction PVs.21,22To improve single junction photovoltaics, one could consider monochromation of all or part of the solar spectrum. Two methods for doing so are upconversion and singlet fission (SF). Sub-bandgap photons may be converted to a photon with energy above that of Eg in a process known as upconversion. This process has been observed in a variety of molecules e.g. those containing lanthanide ions,23,24 and organic chromophores,25 albeit through differing mechanisms. Lanthanide based upconversion has been covered in depth by other reviews,26–29 and readers are referred to this literature for a full overview of the mechanism and properties. In terms of use in solar harvesting the main challenge for lanthanide-based UC is the requirement of high intensity excitation light, due to narrow and weak absorption bands. Strategies are being developed to counter these drawbacks, mainly focussed on imaging applications.30 In this review we will focus on systems using small organic molecules as annihilators, in which the most described upconversion process relevant to solar cells is triplet–triplet annihilation photon upconversion (TTA-UC). The benefit of the TTA-UC process is that high conversion efficiencies can be reached using a low-intensity and non-coherent excitation light source.
To mitigate the loss of energy through thermalisation, multiexciton generation (MEG) has been proposed. Through this method, two excitons are generated from one excitation event. It has been observed in a variety of materials, such as semiconducting quantum dots,31,32 lanthanide ions33 and organic molecules.34 For inorganic materials (QDs and lanthanide nanoparticles) the topic has been extensively covered by others.32,35–37 We will not discuss these materials in depth, but recognise that the challenges in QD based MEG are related to modest yield, as well as how to efficiently extract the multiple excited states due to their often ultrashort lifetimes.38–40 Instead, we will focus on singlet fission (SF), a multiexciton generation process in organic molecules. SF is a spin-allowed process, in which the production of two triplet excited states occurs from one initial excited singlet state. It thus offers the possibility of creating two electron–hole pairs from one high-energy photon when in conjunction to a solar cell. This process is widely observed in polycyclic aromatic hydrocarbons, either on the same chromophore (homofission) or on different chromophores (heterofission).34 SF has been observed with efficiencies of unity,41 and has seen implementation in PV devices to raise their external quantum efficiency.42,43 To summarise, by including SF and/or TTA-UC capabilities into PV devices, thermalisation and below bandgap energy losses can be mitigated.
Prospect of photon conversion in different classes of PV devices
The maximum measured efficiency as of this review using each kind of PV material (in single junction, non-concentrated cells) is given in Fig. 1b.44 It can be observed that many cells are nearly reaching the theoretical limit, with crystalline silicon (c-Si), gallium arsenide (GaAs), and gallium indium phosphide (GaInP) all reaching above 80% of their theoretical maximum value. The champion cell remains GaAs,44,45 but concerns have been raised about the toxicity of the material.The relative benefits of TTA-UC to solar cell technology depends on the Eg of the solar cell in question. Trupke et al. have calculated a solar cell efficiency limit based on the theoretical increases provided by an upconversion process.46Fig. 1c displays this TTA-UC enhanced limit alongside the traditional SQ limit, both under AM 1.5 irradiation. Assumptions were made by the original authors, such as a near constant refractive index over the range of bandgaps, which was used to be representative of several types of PVs (Si and GaAs).47 The values were based on ideal upconversion systems for each Eg. Nonetheless, it gives an estimation of maximum efficiency when applying upconversion to PVs. We have also predicted and plotted in this figure the maximum efficiency value that would be possible for the record device in each class of PV if an ideal TTA-UC system were applied. We do this by taking the ratio between the traditional SQ limit and the TTA enhanced limit, then multiplying by the current record percentage efficiency.
The map of solar cell potential is redrawn by using TTA-UC. Now, the potential of large Eg systems is considerably increased, while small Eg systems are only marginally increased. By measure of efficiency increase, GaAs, InP or GaInP, and ABX3 show high performance potential when utilising TTA-UC. GaAs, InP and GaInP cells are all expensive technologies that see little use in commercial PV's today.48 Perovskite solar cells have seen a remarkable amount of progress in recent years and whilst stability issues remain, the rapid progress is encouraging.49 The high Eg of perovskite materials make this a noteworthy material to investigate for TTA-UC enhancement. Whilst slightly lower in potential efficiency than the aforementioned perovskites, the next best predicted material would be CdTe. This PV class already possesses a sizeable market share, with fast and cheap manufacturing processes. Moreover, studies have concluded that this system has negligible environmental toxicity as the technology is resistant to normal environmental conditions.50
Similar to the case of TTA-UC, the benefit of SF applied to a solar cell depends on the Eg. The revised solar cell efficiency limit of a SF solar cell was calculated by Lee et al. and Tayebjee et al.51,52 As can be seen in Fig. 1d, the benefit of SF contrasts that of TTA-UC with the largest benefit for low Eg solar cells, such as c-Si and CIGS solar cells, approaching theoretical efficiencies of roughly 45%. Furthermore, a combined TTA-UC and SF solar cell would have the potential to reach high power conversion efficiencies across the entire range of Eg relevant to known photovoltaic materials.53
The triplet pair, the commonality of triplet–triplet annihilation and singlet fission
In essence, TTA and SF are reverse equilibrium processes, where the common denominator is the so-called triplet pair. It is therefore constructive to discuss the triplet pair from both a TTA and SF point of view. A simplistic way of viewing these processes is through the Johnson–Merrifield model (eqn (1)) that describe TTA and SF as a two-step process,54 with an intermediate correlated triplet pair state (1(TT)):| S1S0 ⇄ 1(TT) ⇄ T1 + T2 | (1) |
| S1S0 ⇄ 1(TT) ⇄ [T1⋯T1] ⇄ T1+ T1 | (2) |
The nature of triplet pair states from a SF point of view has been extensively discussed in recent reviews.59,61–63 Although the triplet pair states in eqn (2) are considered intermediates, it should be noted that in the literature it is not always clear which of the steps are referred to as SF. I.e. is rapid and efficient formation of 1(TT) enough to label a system efficient for SF, even if no further separation of 1(TT) occurs? Zhu and co-workers argue that the better and most relevant definition of SF in terms of yield and rates should be the formation of [T1⋯T1].61 However, it is often difficult to experimentally distinguish between each of the 1(TT), [T1⋯T1] and T1 + T1 states. For TTA the definition is more clear-cut, as measurable emission is what is typically used to observe TTA. On the other hand, the intermediate states are more challenging to study, due to their fleeting lifetime compared to their often diffusion limited formation time.
In the TTA literature, the widely accepted mechanism is based on a coupling between two triplet states with strong exchange coupling (a parameter describing the strength of the electronic and magnetic interactions between triplets).64,65 If we consider the two initial triplets in TTA (T1 + T1), and not taking the weakly bound triplet pair state into account, triplet pair formation with nine possible outcomes is possible (Fig. 2).66–69 Where one is singlet 1(TT), three are triplets 3(TT) and five are quintets 5(TT). The lack of possibility for quintet formation on a single chromophore (for energetic reasons) would mean that 5(TT) dissociation is the only fate possible and therefore four recombination outcomes are possible and not nine. Moreover, TTA to a higher excited triplet state is plausible, with the consequence of the formation of one ground state and an excited state that will relax and reform T1, following Kashas rule.66 Only, TTA of 1(TT) leads to S1 formation and the spin statistical limit for the process is therefore 40%.
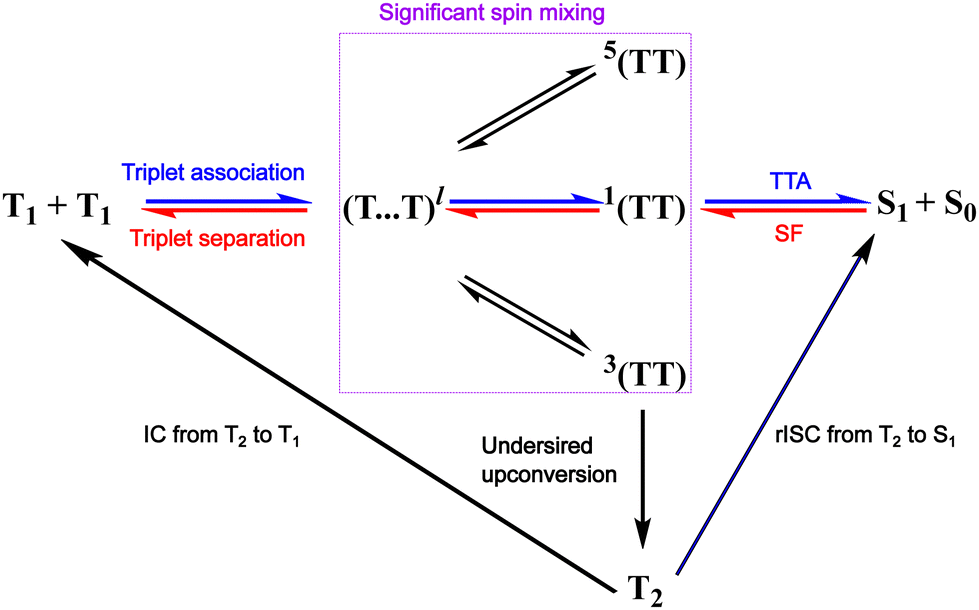 | ||
| Fig. 2 Model of the TTA and SF processes in accordance with eqn (2), we consider triplet pairs of pure spin states as coupled to one another through the intermediate (T⋯T)l. If the molecules forming the triplet pair are only weakly exchange coupled, then are triplet states no longer considered as eigenstates and significant spin mixing occurs. Processes directly leading to TTA-UC and SF are marked with blue and red colour, respectively. | ||
However in recent literature it was proposed that it is more appropriate in some cases to consider the triplet pair as weakly exchange coupled.69 For such cases, spin is not a good quantum number and states having contributions of different spins form. Now, the quintet states from the strong exchange coupled model gets a fraction of singlet spin state character, enabling TTA to some degree. This changes the theoretical spin statistical limit to 67%.69 It should be noted that these investigations were done for rubrene in the solid-state, and to date it is unclear if these results are translatable to other materials and conditions. One particular example, annihilation of perylene tolerates bulky substituents, indicating that annihilation does not require a very close contact pair.70,71
Theoretical calculations have also played an important role to deepen our understanding of the triplet pair state and their formation, in particular in terms of SF. For theoretical calculations it is most common to consider the coupling between the initial S1S0 state and the 1(TT) state when estimating the SF rate.72 However, the coupling mechanism between chromophores involved in SF has been a topic of recent debate.73–84 The different coupling mechanisms are presented in Fig. 3. Using first order perturbation theory, coupling occurs directly between S1S0 and 1(TT), analogously to internal conversion (direct mechanism, Fig. 3).72,74 This coupling is, however, often quite small. Using higher order perturbation theory, the coupling of S1S0 and 1(TT) to charge transfer (CT) states can be considered, often resulting in a larger coupling.72,74,77,82,85–87 If the CT states are higher in energy than S1S0, they only participate ‘virtually’ in a super exchange mechanism, and are never populated (mediated mechanism, Fig. 3).72,77,79,82,83,85,86,88–96 However, in some materials the CT state can be energetically accessible from S1S0 and might then be populated as an intermediate in the transition to 1(TT) (two step mechanism, Fig. 3).97–99 The coupling, and hence the rate of 1(TT) formation, in SF materials varies greatly depending on for example the molecular structure,81 molecular packing in the solid-state,75,81,100–106 and solvent for solution phase chromophores.83,93,94,96,98,99,107–113 If the electronic coupling is weak, SF is often described within a non-adiabatic picture (Fig. 4a), treating the localised region where the diabatic potential curves cross as non-adiabatic. Here the rate increases with increasing coupling as described by conventional Marcus theory for electron transfer.81,114 With sufficiently large coupling, the non-adiabatic picture is no longer appropriate, instead an adiabatic picture where S1S0 continuously evolves to 1(TT) is more suitable (Fig. 4b). It has been shown that in the adiabatic regime the rate of singlet fission becomes independent of electronic coupling.81,87,92 However, debate is still ongoing as to the exact nature of the coupling responsible for ultrafast SF. For example, in contrast to super-exchange mediated coupling, vibronic coherence between S1 and 1(TT), along with entropic effects have recently been suggested to drive 1(TT) formation.99,115–121 The required coupling between chromophores in terms of TTA is a much less explored aspect that likely will become more important as more solid-state TTA systems are developed.
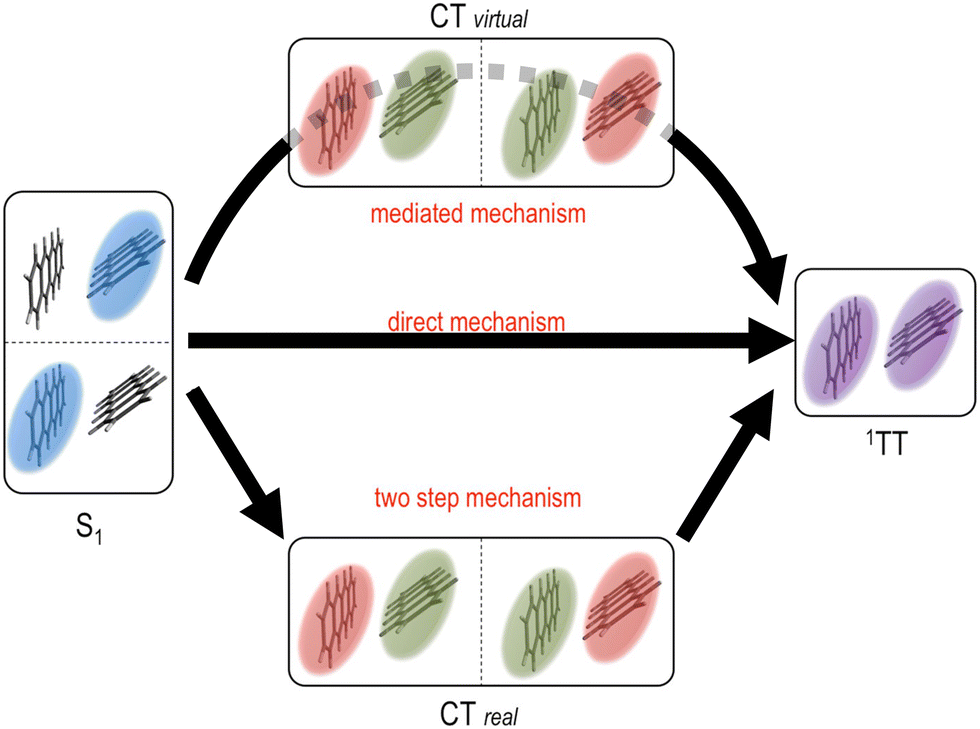 | ||
| Fig. 3 Possible mechanisms for the S1S0 to 1(TT) transition in singlet fission. Top path: The mediated super exchange mechanism where S1S0 and/or 1(TT) couple via virtual CT states. Middle path: The direct mechanism where S1S0 couples directly to 1(TT). Bottom path: The two-step mechanism where an intermediate CT state is formed prior to 1(TT) formation. Figure adapted from ref. 72, with permission from the American Chemical Society. Copyright 2018. | ||
 | ||
| Fig. 4 (a) Non-adiabatic picture of SF for weakly coupled SF chromophores. (b) Adiabatic picture of SF for strongly coupled chromophores. (c) The rate of SF as a function of electronic coupling. Figures reprinted from ref. 81 with permission. Copyright 2014 Nature Springer. | ||
Energetic and entropic requirements
In TTA, the energy of 1(TT) (or by first approximation twice T1) must be larger or equal to that of S1 to allow TTA to occur efficiently (2 × ET1 ≥ ES1). The opposite is true for SF, where the initially formed singlet excited state must be higher or equal in energy to the energy of the two formed triplets (ES1 ≥ 2 × ET1). It is noteworthy that some materials can show both SF and TTA, as the energetic barriers are overcome by kBT.For SF, this requirement can be further relaxed in some systems. For example, thermally activated triplet dissociation has been observed in systems having a rapid and exothermic relaxation from the S1 state to 1(TT).121–124 Another important consideration that relaxes the energetic requirement is the role of entropy in SF. As has been discussed extensively recently, entropy can play a determining role in the efficiency of endothermic SF.61,73,125,126 Both the step from S1 to the triplet pair state 1(TT), as well as the step from the triplet pair state to the weakly bound pair state [T1⋯T1] are associated with a gain in entropy as described by the equations below.
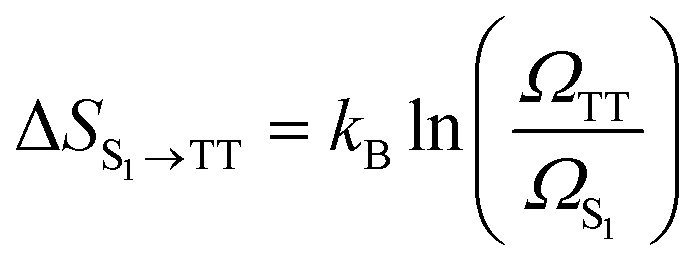 | (3) |
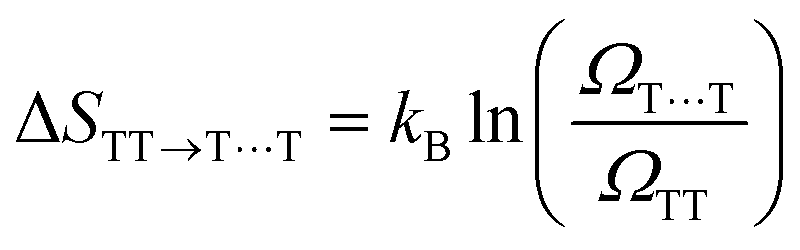 | (4) |
It is important to also consider the energy of the second triplet (T2) state when studying new SF or TTA chromophores.76,131,132 Ideally T2 lies higher in energy than the combined energy of the two T1 states (ET2 > 2 × ET1), to minimise undesired triplet–triplet annihilation to T2 (Fig. 2).34 It is known that reverse intersystem crossing from T2 to S1 is possible, which in theory allows for zero wastage of photons if rapid.133 However, relying on reverse intersystem crossing to kinetically outcompete T2 to T1 internal conversion is not a general advisable strategy when designing new materials. Whether considering a model based on a weakly or strongly exchange coupled triplet pair, the energetics of annihilator molecules play a crucial role in limiting TTA yields. Thus, energetics can turn off loss mechanisms from the 3(TT) state, allowing in principle for unity efficiency.134,135
Key existing challenges and limitations in TTA-UC and SF
We have discussed fundamental mechanisms and limitations in both the TTA and SF regimes including spin state-coupling dynamics. We will briefly discuss here some of the current material challenges in maximising both yield and/or functionality of TTA or SF in PV applications. We will later reference these challenges in the relevant sections, and how current progress has helped or failed to address them.Energetic matching to PVs
When designing TTA and SF systems, particular attention must be paid to the absorption energy vs. PV absorption edges. We refer to Fig. 1, which highlights the issue. To achieve meaningful solar cell efficiency improvements, it is important that the photon converting system operates far enough below or above the absorption edge. For example, A TTA-UC system that operates with sensitiser absorption at 1.12 eV could improve ABX3 but not c-Si solar cells due to energetic matching. We will therefore discuss some strategies that are being developed to address energetic requirements and their advantages and drawbacks.Energy and charge injection
To use the created excited states from TTA or SF, PVs need to extract them in one way or another. It is therefore important to consider the organic–inorganic interface between the photon converting material and PV material. Charge injection kinetics have been studied for both SF and TTA materials on metal oxide surfaces with various degrees of success. Excitonic energy transfer across the organic–inorganic interface is another aspect that either enables the use of nanocrystals as spin-mixers in TTA and SF systems or allows for direct use of the excitons in a PV. This review will cover strategies that are being used to overcome hurdles unique to the organic–inorganic interfaces in TTA and SF.Material stability
Relevant to both SF and TTA, is the photostability of acenes, which are commonly used in both processes.136,137 These compounds degrade under photoirradiation, most commonly by reacting with singlet oxygen.138,139 Oftentimes, acenes are used in SF due to their favourable energetics and planar stacking, making the fabrication of crystalline solids facile. Whilst some groups have modified the acene core to make more photostable derivatives, it is observed that the morphology changes influence the SF efficiency to a significant extent.140,141 Whilst some strategies such as polymer encapsulation and oxygen scavenging have been used with varied levels of success in TTA-UC,142–145 these are not always relevant to the solid-state. We will therefore discuss some of the methods being used to negate oxygen quenching in solid-state SF and TTA-UC. We also consider the viability of these strategies in light of morphological requirements.Triplet diffusion
One of the largest difficulties in solid-state photon conversion is the transport of triplets: either to an interface for charge/energy injection in the case of SF; or for TTA, from two sensitisers to the point of annihilation. Molecular diffusion drives the latter process in solution, however in the solid-state this must be achieved by exciton diffusion. It is well known that the orientation, crystallinity and packing of molecules play a large role in the triplet exciton diffusivity of solid materials for SF and OLEDs.76,146–150 This means that there are two interlinked hurdles common to TTA and SF related to triplet diffusion. One is the reversibility of both processes leading to inherent losses if the energies of excited states are not properly tuned to favour one direction.132 The other is that molecular structure is highly important to the stacking and packing morphology.149–151 We will highlight in this review strategies that are being used to overcome morphological hurdles, and equally those that are used to aid exciton diffusion.Triplet–triplet annihilation based photon upconversion (TTA-UC)
The TTA-UC field started fairly modestly with an initial description of the process given by Parker and Hatchard in the early 1960s.152 The field faced a renewal of interest in the 2000s,46,153–155 porphyrin and coordination complexes extend the observed anti-Stokes shift and have led to huge TTA-UC efficiency enhancement over early examples. The field has seemingly progressed to a point that the focus is no longer on understanding of the process, but on the efficiency optimisation and application of TTA-UC systems to functional materials. It can be argued that it has been a pre-mature turn of focus, as there are still many fundamental aspects of TTA that are not well understood (e.g. spin selection). In the following sections we will first describe the component photophysical processes governing TTA and the efficiency thereof. We will further detail the figures of merit generally applied in the TTA field, and discuss these in the context of PVs and their efficiency.Forming annihilator triplets in TTA-UC
We have so far considered the nature of the TTA event, focussing on the interaction of the triplet states. Now we will describe the typical processes used to generate these triplet states. In sensitised TTA, the type focussed on in this work, the overall event is a bimolecular process requiring a sensitiser and annihilator to function. The presence of a sensitiser with a high intersystem crossing yield is necessary, due to the extremely low oscillator strength of direct singlet to triplet transitions for annihilators.156 The overall photophysical process is described schematically in Fig. 5 together with example spectra and a typical visual observation.134 The idealised TTA-UC process forms one high energy photon from two low energy photons, and the process can be broken down into stages based on independent processes having their own requirements.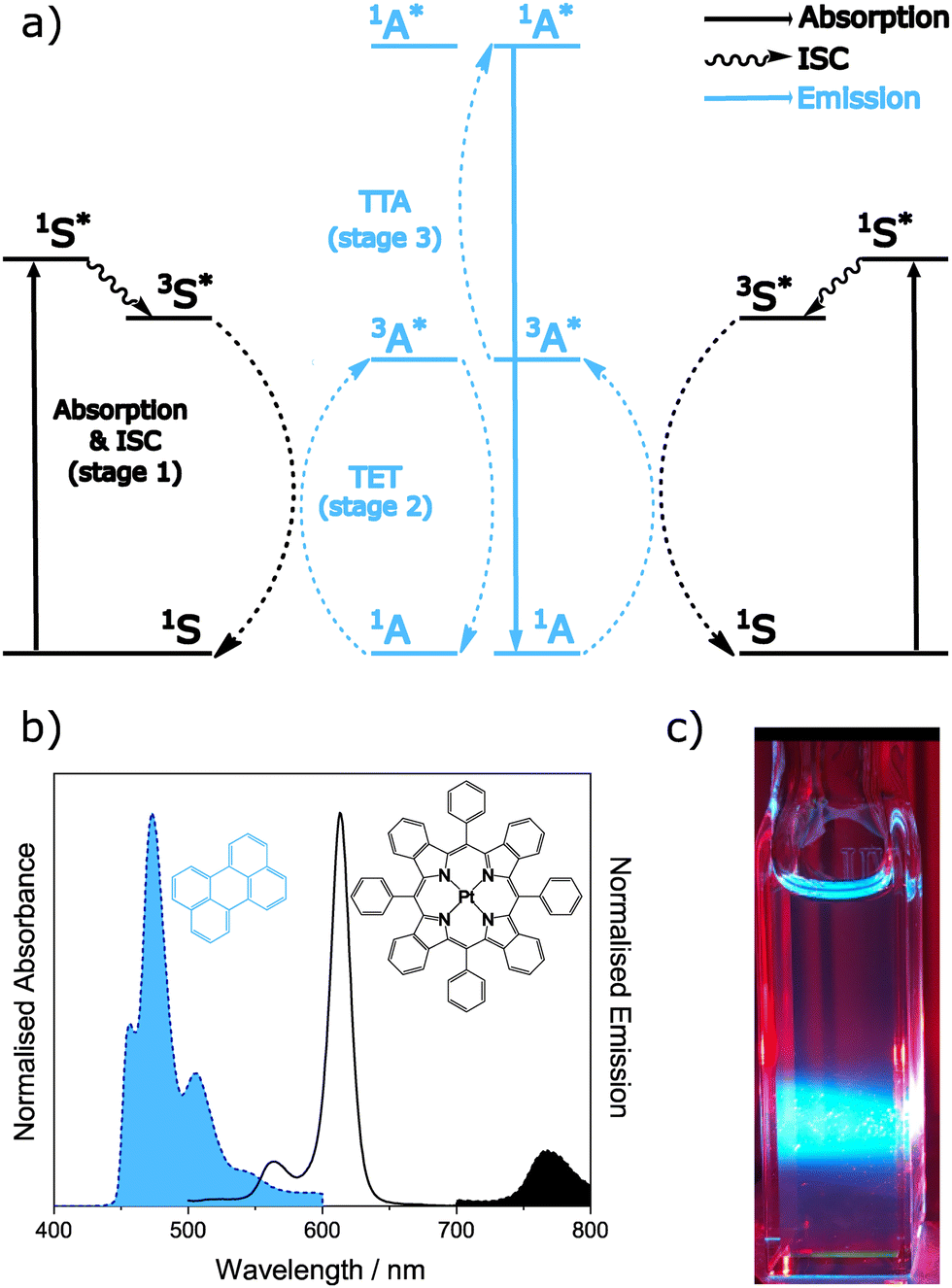 | ||
| Fig. 5 (a) Energy level diagram for the TTA-UC process. The triplet energy transfer (TET) occurs from the triplet excited state of the sensitiser to the triplet excited state of the annihilator. Two annihilators in their triplet excited states then meet, allowing triplet–triplet annihilation (TTA) to occur, and the subsequent emission from the singlet excited state to be seen. Colours are used to denote the species involved in each process. (b) Representative absorption (black unfilled) and emission (filled blue for annihilator component and filled black for sensitiser component) spectra collected from a solution containing 3 μM of sensitiser (black) and 1 mM of annihilator (blue) excited at 627 nm. (c) Optical camera image taken of the aforementioned solution under excitation from a 617 nm LED source. Data is adapted from ref. 134. | ||
The TTA-UC quantum yield
The upconversion efficiency (Φuc) in TTA-UC systems is based on the IUPAC definition of the quantum yield (Φ; eqn (5)).168 In the case where emission is desired, each count of an emitted photon is used to define an event. The maximal Φuc is therefore 50%, because two absorbed photons generate one emitted photon.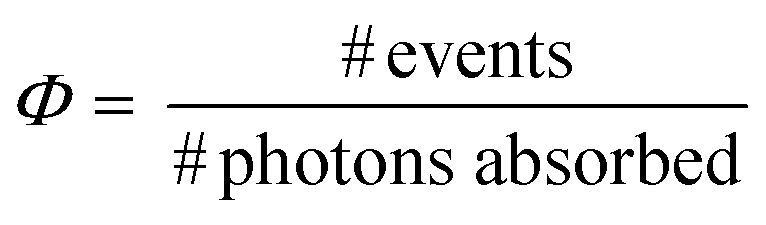 | (5) |
The upconversion efficiency is a function of the individual efficiencies of each step in the process. Eqn (6) defines the yield of the annihilator singlet states theoretically produced (Φmax). This relies on the efficiency of intersystem crossing (ΦISC), the sensitiser to annihilator triplet energy transfer efficiency (ΦTET), and efficiency of the triplet–triplet annihilation process (ΦTTA). In eqn (6) the value of ΦTTA incorporates the spin statistical factor, and by definition then has a maximum quantum efficiency of 50%.
| Φmax = ΦISCΦTETΦTTA | (6) |
| Φsing = ΦsmaxΦf(1 − ΦBET) | (7) |
| ΦUC = ΦsingΦexp | (8) |
The threshold intensity
In the context of PV devices, upconversion is driven by sunlight. It is therefore important that the maximum quantum yield of the upconversion process is reached at equal to or sub-solar photon flux (Fig. 1). The excitation density at which the resultant upconversion intensity is described by equal contribution of first and second order processes is called the threshold intensity (Ith).To mathematically define this term, we separate first order and a second order processes. The first-order decay is a combination of phosphorescence, non-radiative decay and quasi first-order quenching processes related to oxygen (this effect should be eliminated in ideal systems). The second-order decay solely originates from triplet–triplet annihilation. We can express the time-based decay of annihilator triplet concentration, [3A*], as a function of the rates of first (kT) and second order (kTTA) decay. Eqn (9) details the overall equation.71,172
 | (9) |
 | (10) |
 | ||
| Fig. 6 (left) Typical plot of Φuc, as a function of excitation intensity. (right) log–log plot of UC emission intensity against excitation intensity. Areas where the weak and strong annihilation limits are valid are shaded in red and blue respectively. The threshold intensity is identified, Ith, where the lines with slope 1 (blue) and slope 2 (red) cross. Reproduced with permission from ref. 68. Copyright 2018 Elsevier B.V. | ||
Red/near infrared to visible TTA-UC systems
It is the energy of the sensitiser that governs which type of PV device that the UC system can be linked to. In general, TTA-UC systems have suffered from a poor match between the sensitiser absorption and the PV Eg. We will, therefore, here discuss TTA-UC, with a focus on the sensitiser species. The absorption wavelengths are considered in the context of differing PV materials and a summary of all the molecular sensitisers covered in this section is given in Fig. 7, in which we detail their structure and properties.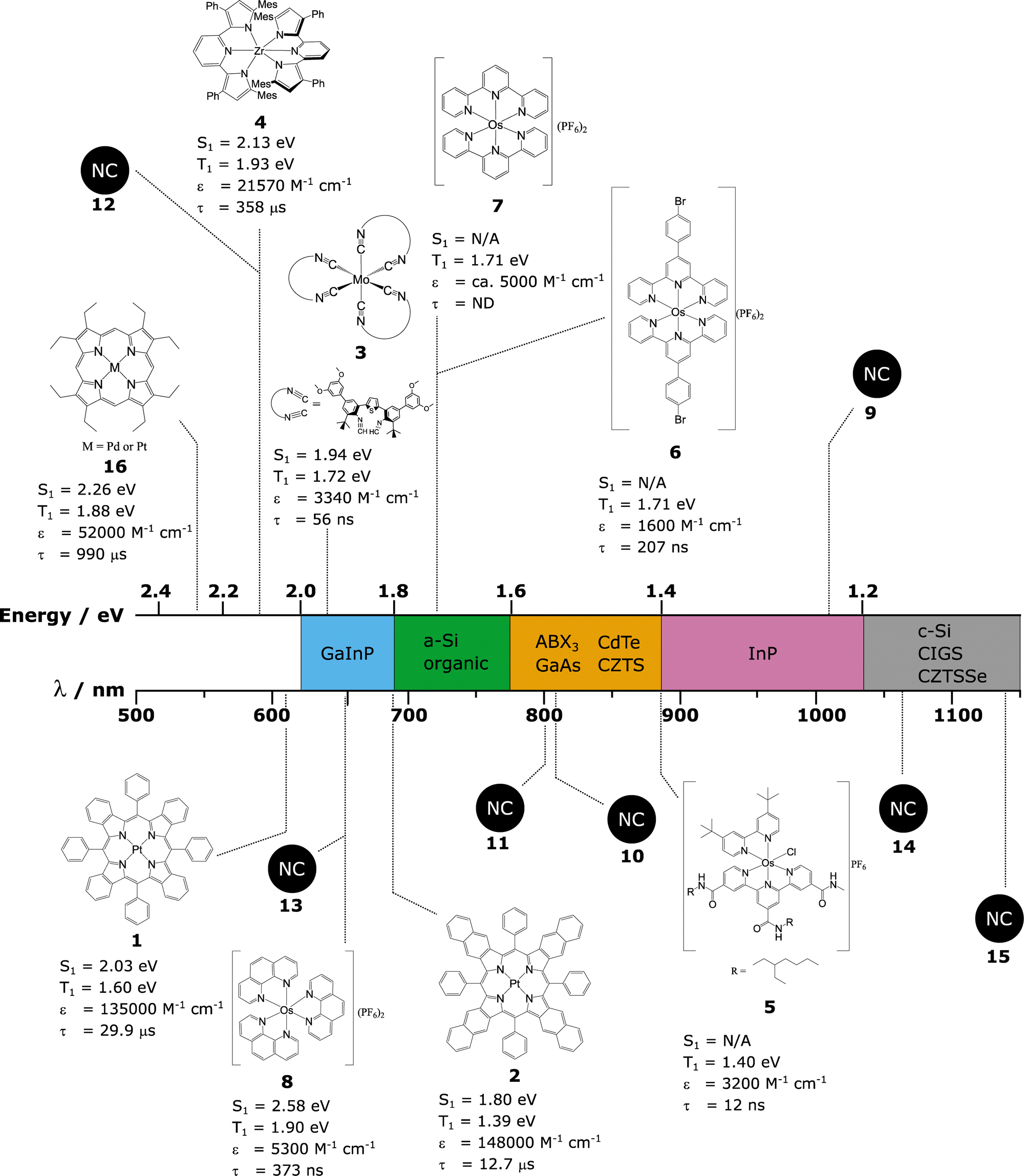 | ||
| Fig. 7 Structures of the molecular sensitisers discussed in this work, also shown are the excitonic peaks of nanocrystal sensitisers used in TTA-UC discussed in this work. The types of PV material to which the sensitisers could be applied to in order to monochromise sufficient amounts of the solar spectrum to increase device efficiency are roughly split up into coloured areas, white areas inside the central bar indicates energies that are not relevant to any type of current PV material. The S1 and T1 energies given are estimates from absorption and emission spectral maxima where no other data is presented in the original works. Lifetimes are the phosphorescence lifetimes of the molecules shown. | ||
Coordination complexes in NIR to visible TTA-UC
Coordination complexes are a useful class of chemical compounds, allowing low energy transitions due to metal to ligand (or ligand to metal) transitions.175,176 Even if the metals are not directly involved in the transition, a more efficient ISC can still be observed.177 The earliest examples of near infrared to visible TTA-UC utilise metalloporphyrin complexes,155 having a high triplet yield.178–181 Indeed many groups continue using porphyrin complexes as sensitisers (1, 2),182–186 although due to the breadth of literature concerning porphyrins we will not specifically cover them in this section. Instead, we will address here recent works related to other classes of organometallic and coordination complexes.Platinum-group elements has been seen ubiquitously in TTA-UC. However, the scarcity of such metals prompted investigation into alternative photosensitisers based on earth abundant metal centres. Wenger et al. presented an example of a Mo(0) complex that shows potential as a sensitiser for upconversion with excitation in the visible red region (3).187 The synthesised Mo(0) complex offers an earth abundant core that possesses high photostability under 500 mW irradiation at 532 nm. Red to blue upconversion using the Mo(0) complex in tandem with 9,10-diphenylanthracene (DPA), was achieved using 635 nm (1.95 eV) excitation light. Castellano et al. later used a similar strategy to avoid precious metals, using the ligand-to-metal charge transfer of a Zr(IV) complex (4) to sensitise DPA.188 Importantly, the Ith value (0.115 mW cm−2) lies below that of the integrated solar flux through the Zr(IV) photosensitiser's absorption band (26.7 mW cm−2). Both complexes unfortunately suffer from high singlet energies (1.94 eV and 2.13 eV respectively) rendering these complexes less than ideal for PV applications. Referring to Fig. 1C, to be a relevant sensitiser for upconversion assisted energy conversion in the majority of PV cell types, the singlet excited state energy would ideally be somewhere around 1.3–1.7 eV or below. Significant synthetic modifications of their ligands would be necessary to tune the energy levels of these complexes to be favourable in PV applications, however this might be achieved in a similar manner to other coordination complexes.189,190
To circumvent the energy loss when sensitisers relax from the initially excited singlet state to the triplet state, direct singlet to triplet excitation can be performed. Such transitions facilitate a gain in the observed anti-Stokes shift of the TTA-UC system. The first example of this phenomena used in TTA-UC was demonstrated by Kimizuka et al. wherein an Os(II) complex (5) was used as a triplet sensitiser for rubrene.191 NIR to visible upconversion was observed with an excitation at 938 nm and upconverted emission around 570 nm. Complex 5 has a suitable triplet excitation energy to capture solar light at lower energies than the majority of PV types (Fig. 7). However, the system shows an extremely poor ΦUC of 0.0007% in deaerated dichloromethane. Many further derivatisations have been carried out in order to improve efficiency, or increase the anti-Stokes shift (6, 7, 8).192–194 The leading Os(II) complex (7) was reported together with anthracene based annihilators, with ΦUC of 5.5% and an anti-Stokes shift of 1.28 eV. One caveat of direct S0 to T1 transitions are their small ε values and fast relaxation. It therefore seems unlikely that this type of material will achieve high enough molar absorptivities to get an Ith in the solar spectral range. Also, since the absorptivity is fundamentally linked to the rate of emission, mitigation strategies would need to be developed.
Nanocrystal and nanoparticle assisted TTA-UC
Addressing both energetic matching with PV bandgaps and triplet exciton diffusion, lead and cadmium-based nanocrystals (NCs) have been shown to be effective in sensitised upconversion from low intensity and low energy light. Baldo et al. reported NIR to visible upconversion sensitised by colloidal NC's with excitation wavelengths above 1 μm (9), sparking an interest in NC sensitised TTA-UC.195 The Ith values for this system were higher than the solar photon flux however, due to inefficient TET. To address this issue the group of Tang et al. introduced a mediator ligand.196–198 The role of the mediator was to facilitate TET. This was achieved by covalently attaching a molecule having the appropriate triplet energy, forming a link between the PbS-Cd NC (10) and the rubrene annihilator (Fig. 8). This system allowed for the first example of a TTA-UC system based on nanocrystals to operate below the solar photon flux.196–198 | ||
| Fig. 8 Working principle of the energy transfer process in acene capped nanocrystals. Here, an excitonic state of a PbS–CdS nanocrystal is excited with long wavelength light (black solid arrow) transfers energy to the triplet state T1t of a bound tetracene-5-carboxylic acid (TET1, black dashed arrow), which in turn will transfer energy to the triplet state T1r of rubrene (TET2, pink dashed arrow). Finally, the triplet states of two diffusing rubrene molecules will recombine (TTA-UC, blue dashed arrows) to produce an upconverted photon (blue solid line). | ||
Lian et al. later set out to study the factors governing the overall ΦUC in tetracene mediated UC using NC's.199 They used a PbS NC (11) which was coated with one of three mediators, having different lengths of linker unit. The results showed that the effect of the overall upconversion quantum yield can be broken down into its reliance on two processes, the first and second TET (NC to mediator as TET1, then mediator to annihilator as TET2) such that ΦUC ∝ ΦTET1ΦTET2. There were two key findings in this work. Firstly, the energy transfer rate from NC to tetracene decreases rapidly with increasing spacer length. Secondly, the energy transfer rate from tetracene to annihilator increases with increasing spacer length. Taken as an overall effect, the longer spacer lengths were found to have a detrimental impact on the ΦUC due to the hugely reduced ΦTET1.199 These results demonstrate the importance of rational mediator design to drive the desired energy transfer process.
The toxicity of nanocrystals fabricated from Cd and Pb has been highlighted as a potential risk. Just as for PV materials containing these elements, there is a drive to demonstrate NC sensitised upconversion with more benign elements.162,200–202 Wu et al. recently proposed InP-based nanocrystals as sensitisers for solution based TTA-UC using anthracene as a mediator and DPA as an annihilator.203 InP NCs are well known to possess trap states,204 limiting their use as sensitisers. However, in this work creating a 0.6 nm shell of ZnSe/ZnS (12) was sufficient to eliminate the trap states.203 The highly emissive zinc coated NCs were subsequently coated with 9-anthracene carboxylic acid, and displayed an anti-Stokes shift of 0.98 eV, and a ΦUC of 5%. The non-toxic NCs in this work provide a route for elimination of toxicity in TTA-UC.203
Whilst all NC examples discussed above are zero dimensional structures, Nienhaus et al. have presented the first example of a 1-dimensional nanorod sensitiser for TTA-UC, i.e. where quantum-confinement is present in two dimensions.163 The study aimed to increase not only the breadth of quantum-confined materials used in TTA-UC but also improve the anti-Stokes shifts shown by nanocrystals. The work used the mediator/annihilator couple of 9-anthracene carboxylic acid (ACA)/DPA sensitised by cadmium telluride nanorods (13). The authors demonstrate TTA-UC using a material with well-defined facets and unique anisotropic shape that allows self-assembly into macrostructures. As discussed above, control over macroscopic structure is a key determinant in triplet energy transfer, crucial in TTA-UC and SF.
To be relevant to c-Si solar cells the absorption energy of the sensitiser must lie below 1.12 eV (Fig. 1). In 2020 a PbS nanocrystal (15) based TTA-UC system that operates below the bandgap of c-Si was proposed by Schmidt et al.,205 PbS NC's were used as sensitisers, violanthrone derivative (V79) as annihilator and singlet oxygen as mediator. Oxygen's use as a triplet mediator for V79 was previously documented,206 and relied on the triplet state of the annihilator lying lower in energy than the singlet state of oxygen (0.98 eV). This is the case for V79 where the triplet level was estimated around 0.94 eV. PbS NC's were excited at 1140 nm, and upconversion was observed. Although this occurs at low efficiency due to inefficient TET to oxygen.206 Rao et al. also presented an example of an upconversion system that could operate below the 1.12 eV bandgap of crystalline silicon.207 The system combined the mediator and annihilator functions into one molecule, 5,11-bis(triethylsilylethynyl)anthradithiophene (TES-ADT). This was achieved by forming a reversible bond to the NP on the timescale of the triplet excited state.207,208 The material has a triplet level of 1.08 eV and when coupled to a nanocrystal (14), displayed upconverted emission after excited at 1125 nm. Whilst the ΦUC is small (∼0.047%), the work avoided the triplet mediator energy limitation of tetracene and allowed an anti-Stokes shift of 0.86 eV.207 The same TES-ADT system was recently used in a binary solid-state system, using higher energy PbS NCs.209 The ΦUC determined to 0.2% with 975 nm excitation. One limiting factor is likely the propensity of TES-ADT chromophores to undergo SF in the solid-state.124,210,211 Both Rao and Schmidt argued that these systems are a step towards relevance of TTA-UC in c-Si solar cells. While this is true, we refer to Fig. 1 to critically assess the importance of upconversion on c-Si solar cells. In c-Si the potential efficiency gain from upconversion is around 5%. This may therefore not be an ideal pairing of UC system to a PV type, we ask would it not be better to use the UC system with a GaAs solar cell for instance? We therefore regard these works as a significant intellectual achievement, stretching the sensitiser absorption edge as well as the annihilator triplet energy towards lower energies. In principle achieving quite optimal annihilator triplet energies for pairing with a high bandgap solar cell.
This section has broadly discussed sensitisers for TTA-UC, which remains an active field of research. We have described above the strategies being developed to address key loss factors. Some of these include singlet–triplet direct energy transfer, and mediated energy transfer from metal nanostructures to TTA annihilators. Both of which simultaneously address low energy absorption and can show efficient TET, and today the structure to function relationship is understood.199,212 Furthermore, works have addressed wide ranging limitations, from toxicity to earth abundance of the metal centre. For upconversion to be relevant to a majority of PVs, the annihilator emission should be at around 1.7–2.0 eV (the annihilator emission should be slightly lower than the PV bandgap). This translates to a lowest possible annihilator triplet energy of 0.85–1 eV (for exothermic TTA). It is an interesting observation, that when being around these triplet energies, singlet oxygen becomes irrelevant as a quencher. Thus, removing the need of encapsulating upconversion systems within PV applications.
Solid-state TTA-UC
Incompatibilities between safety or stability when utilising solution TTA-UC can be difficult to overcome. The solvents used are often flammable, toxic, or volatile. Solid-state TTA-UC offers an alternative that is easier to apply to rigid photovoltaics, and indeed any application where a solid phase is desirable, such as heterogenous catalysis.213 Triplet exciton diffusion has fundamentally limited development of high efficiency solid-state TTA-UC. In principle, molecular diffusion must be replaced with exciton diffusion when going from solution to solid-state.214,215 Further major loss factors include back energy transfer and aggregation of sensitisers and/or annihilators, especially tied to the stacking and packing morphology.68,216 In this section we categorise efforts to reduce losses by material type and detail their outlook in photovoltaics.Soft-matrix based solid-state TTA-UC
In soft systems, such as gels, the inside of micelles, and in polymers below their glass transition temperature, molecular diffusion is allowed in an otherwise apparent solid-state system.154,217 The quasi-solid nature of these materials is an intermediate between solution based and fully rigid systems, and therefore they have upconversion efficiencies approaching those in solution.218Gels are liquid in solid emulsions. These materials have been used as a matrix for upconversion systems,219–221 and temperature responsive systems have further been developed.222–224 To investigate the existence of a structure-upconversion relationship in gel systems, a low molecular weight gelator was covalently attached to the annihilator DPA with different attachment positions.225 They were combined with sensitiser 16 (Fig. 7). The Ith ≈ 400–1000 mW cm−2 and ΦUC were found to be attachment position dependent. Morphological effects on upconversion efficiency are however currently unclear in gel systems.
TTA-UC system's working in liquid conditions can be compartmentalised inside micelles. The micelle provides molecular diffusion, and oxygen exclusion. They can further be incorporated into a solid polymer host for a variety of practical applications.226 A general structure of a solid polymer host incorporating upconversion micelles is given in Fig. 9a. Protein matrices encapsulating micelles formed from SDS were synthesised by Kumar et al.227 Particularly efficient TTA-UC with a ΦUC of 10% was observed from 16 and DPA. However, Ith values were in the range of 400–700 mW cm−2. TTA-UC in biopolymeric materials has been further demonstrated by Kimizuka et al., using Triton X-100 micelles containing the 16-DPA sensitiser–annihilator couple.228 By changing to a biopolymer, the Ith value was lowered to 14 mW cm−2. Furthermore, samples showed stability over 2 years of storage in ambient conditions. Another recent example,229 uses crosslinked hydrophilic polymers to encapsulate organic hydrophobic solvent containing sensitiser 16 and DPA. Upconversion efficiency (ΦUC = 23%) and stability of the system is high. Whilst the ΦUC is high, so is the Ith value (500 mW cm−2). All systems described above have used 16 as a sensitiser and from a UC in PV perspective this is problematic. This is because sensitiser 16 has a singlet energy higher than the absorption edge of all PVs (Fig. 1). We appreciate however that high stabilities were observed, that upconversion efficiencies close to that of dilute solutions were observed and that the concept could be transferred to more PV relevant materials.
 | ||
| Fig. 9 Graphical representation of the working principle of (a) micelle or pore encapsulated solid gel for TTA-UC, and (b) rubbery polymer matrices with Tg below room temperature. | ||
Soft polymers with glass transition temperatures below room temperature have also been used as a matrix for TTA-UC. The working principle is graphically demonstrated in Fig. 9b and is based on a relatively high diffusion of dopants in a polymer above Tg. Initial investigations by Castellano et al.,230 sparked many future studies.231–234 In a more recent example, PVA was used as an oxygen blocking barrier for a thin layer of a soft polymer containing 1 and perylene. This dual polymeric structure was then backed with glass to provide rigidity and an Ith of 70 mW cm−2 and a ΦUC of 7% was recorded in oxygenated conditions.235 Following early work with WO3 photocatalysts,236,237 a soft polymer matrix containing 1 and perylene was applied to the photocatalyst BiVO4. This aided in the capture of photons below the bandgap of the catalyst, increasing the H2 and O2 evolution by around 17% against bare BiVO4.238 Non-heavy metal based upconversion has also been demonstrated in soft polymers, with thiosquaraine and rubrene doped into a polymer matrix. The formed film was excited at 685 nm under aerated conditions, and upconverted emission was observed. The authors however did not report a ΦUC.239 Soft polymer matrices can be regarded as a transition from the solution-based gels and micelles to a truly solid-state system. Efficient systems exist, but questions could be raised as to the appropriateness of incorporating rubbery materials within PV technology.
MOF based TTA-UC
Metal–organic-frameworks (MOFs) are porous rigid structures, which are organised based on coordination bonding of the constituent molecules (Fig. 10). This gives them a much higher order than gel based systems, and can be considered entirely solid-state as they do not require any solvent to be present in the pores.240 MOFs avoid aggregation of the molecular components, and allow for some level of oxygen exclusion. MOFs have been demonstrated to function as upconverters when dispersed in solution.241–245 Although they have also been investigated in entirely solid-state systems. A highly crystalline zirconium MOF was synthesised containing linkers of both sensitiser and annihilator in a controlled ratio.246 The osmium sensitiser is a carboxylic acid derivative of 7 and the annihilator is based on anthracene. MOFs synthesised with a sensitiser![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :annihilator ratio of 1:94 were encapsulated inside a polymer blend and Ith and ΦUC values were measured at ∼10 W cm−2 and 0.006% respectively. The low upconversion quantum yield was reasoned to be due to trap sites from locally disordered structures within the framework. We would argue that MOFs for upconversion applications may benefit when made using a templated surface reactions in a continuous flow approach. This method produces frameworks with lower numbers of defects and a high surface smoothness, which should benefit optical investigations.247,248
:annihilator ratio of 1:94 were encapsulated inside a polymer blend and Ith and ΦUC values were measured at ∼10 W cm−2 and 0.006% respectively. The low upconversion quantum yield was reasoned to be due to trap sites from locally disordered structures within the framework. We would argue that MOFs for upconversion applications may benefit when made using a templated surface reactions in a continuous flow approach. This method produces frameworks with lower numbers of defects and a high surface smoothness, which should benefit optical investigations.247,248
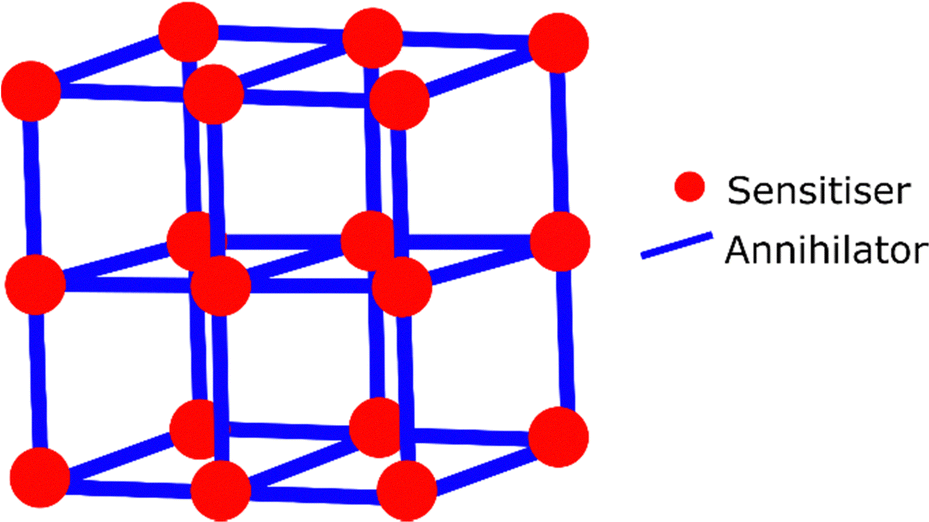 | ||
| Fig. 10 General structure of a MOF with coordination bonded annihilators and sensitisers in an ordered network. | ||
It was recently shown that by tuning the geometry between chromophores in MOFs the triplet exchange coupling could be controlled to direct the spin-mixing of singlet and quintet triplet pair states.249 If controlled correctly this could result in improved upconversion yields compared to random oriented materials.
Matrix free solid-state upconversion
We define matrix free TTA-UC as sensitiser and annihilator materials being in their solid-state and no molecular diffusion being present. This may be seen as the most facile processing method for solid films, and easiest to apply to PVs. However, neat conditions can give a multitude of problems such as aggregation-induced energy loss, FRET back to the sensitiser, non-radiative triplet decay, and annihilator quenching such as excimer formation.71,250–252 In this section we describe recent literature that attempts to understand and find fabrication methods to avoid these losses.Within bulk solid-state upconversion materials, Kazlauskas et al. investigated a method to reduce detrimental aggregation of the annihilator.253 Two analogues, rubrene and t-butyl-substituted rubrene, were doped into polystyrene (Fig. 11a). It was shown that t-butyl-substitution greatly increased Φf, as the substituted rubrene exhibited reduced SF. To raise upconversion yields, tetraphenyldibenzoperiflanthene (DBP) was used at 0.5% weight in the polystyrene film. A study by Nienhaus et al. further highlighted the importance of annihilator aggregation in rubrene containing systems.254 In spin coated disordered films, SF is negligible and DBP doping at any concentration did not lead to increased upconversion yields (Fig. 11b). Later work by Clark et al. suggested that the initial singlet fission event from S1 to 1(TT) actually happens with equal efficiency regardless of the presence of DBP. However, the DBP inhibits further triplet dissociation and leads to the higher yields.255 The described studies indicate the importance of material design in matrix free systems, with annihilator aggregation and structural order observed to be highly influential on solid-state upconversion efficiency.
 | ||
| Fig. 11 (a) Structures of the rubrene and t-butyl substituted analogue, along with a cartoon depiction of the mechanism behind SF inhibition. (b) Structures of the rubrene annihilator and DBP dopant used to investigate the effect of dopant in crystalline and amorphous films. (c) Sensitiser annihilator bilayer structure for increasing photocurrent in TiO2. (d) Unmodified bilayer and back energy transfer (BET) preventing DMPPP doped bilayer structures. | ||
Grozema et al. described a trilayer TTA-UC system for increasing photocurrent generation. The layers consist of a crystalline zinc phthalocyanine sensitiser layer, and a crystalline perylenetetracarboxylic acid diimide annihilator layer. The layers were made on a polycrystalline TiO2 support (Fig. 11c).256 Excitation of the sensitiser at 700 nm resulted in a photoconductance signal. The maximum photon to electron injection quantum yield was 0.92%. Baldo et al. later investigated whether sensitiser–annihilator bilayer or blended structures are the most efficient.257 Several materials based upon 16 and an anthracene based annihilator were made. Spatial separation of sensitiser and annihilator was tested, to observe the effects of reducing back energy transfer (Fig. 11d). The ΦUC for the blocker-doped device (2.5%) was around an order of magnitude higher than for the worst performing bilayer device. Similarly positive results were observed in the value of Ith, where the spatially separated blocker-doped, and bilayer devices display values of 238 and 1404 mW cm−2, respectively. This study shows that overcoming back energy transfer to the sensitiser and aggregation, by means of selective doping, provide a pathway to achieving higher performance solid-state TTA upconversion devices. More recently the group of Hiramoto et al. have used the concept of charge recombination at a semiconductor interface to facilitate TTA-UC.258,259 The group used non-fullerene acceptors as bilayers with rubrene, which was used as the annihilator. This energy transfer mechanism does not rely on ISC and therefore differs significantly from the traditional mechanism in Fig. 5. Fig. 12 graphically displays triplet formation at a semiconductor interface in the context of TTA-UC. The first step relies on diffusion of singlet excitons to the sensitiser/annihilator interface, where the energy is relaxed to charge transfer states of singlet character. Triplet annihilators are then created through dynamics between free charges and charge transfer states. Finally, two annihilator triplets form a singlet through TTA, leading to UC emission from the emitter layer.
 | ||
| Fig. 12 Energy diagram for the mechanisms of TTA-UC at an organic semiconductor interface, green represents processes happening in the sensitiser layer, black represents processes happening at the interface and blue represents processes happening in the annihilator layer. | ||
The work of Hiramoto et al. was the first to utilise singlet diffusion followed by charge recombination at an interface to produce efficient TTA-UC. As it was a novel route to realise TTA in the solid-state it is worth significant discussion. Firstly, the influence of the sensitiser ISC on the UC yield is eliminated as CT states are efficiently formed at the interface. Secondly, diffusion in the sensitiser layer is realised by singlet excitons, with triplets formed only near the interface. This prevents non-radiative triplet recombination, which is a major loss process in conventional sensitiser-annihilator bilayer upconverters. Another advantage of this system is that it can be modified to work under both electrical and light stimulus. We would argue that research in this direction should take inspiration from lessons drawn in the field of organic photovoltaics.260,261 Specifically, it is difficult to make a planar layer absorb all incoming light and at the same time have an efficient exciton diffusion to the interface. Therefore, the use of these materials in bulk heterojunctions should be considered.
Nanophotonic resonators are a lesser explored approach to increase ΦUC and reduce Ith in solid-state TTA-UC. In an attempt to reduce Ith for NIR to visible upconversion, Baldo et al. constructed a Fabry–Pérot microcavity (Fig. 13a).262 PbS nanocrystals with an excitonic peak tuned to 980 nm were used as the sensitiser and rubrene was used as the annihilator doped with DBP at 1%. For previous solid-state systems using PbS nanocrystals, ΦUC was unmeasurable due to the low absorbance of PbS monolayers.195,263 Using a Fabry–Pérot cavity, a 74 fold increase of excitons in the PbS monolayer was achieved. The emission intensity of the microcavity is increased by a factor of 227, with a ΦUC of 0.06%.262 This small value of ΦUC can be ascribed to inefficient TET in the solid-state, high back energy transfer from rubrene to the NCs, and singlet and triplet trap states in the material. The Ith value for the microcavity is 13 mW cm−2. The low Ith and far increased ΦUC clearly demonstrate the advantages of applying Fabry–Pérot cavities to solid-state TTA-UC. When the coupling between the electromagnetic field in the Fabry–Pérot cavity and the molecular transition dipole moment gets sufficiently large, the system enters the strong coupling regime.264 Börjesson et al. used Fabry–Pérot cavities strongly coupled to DPP(PhCl)2 to “turn-on” TTA-UC (Fig. 13b).265 Whilst DPP(PhCl)2 shows poor TTA-UC in solution, owing to ES1 − 2ET1 > 0, the triplet pair from two triplet excitons could directly convert into exciton–polaritons by TTA inside the microcavity. This is due to a lowered polariton energy compared to the ES1 facilitating the rate of TTA. The most efficient cavities show a small anti-Stokes shift of <0.1 eV (613 nm to 590 nm), therefore this system is currently lacking applicability to PVs. The work however demonstrates the ability to utilise optical cavities rather than chemical modifications to influence TTA-UC parameters. One particular challenge this may address could be the combination of UC and strong-coupling in solar NIR driven catalysis.266–268
 | ||
| Fig. 13 (a) Microcavity fabricated by Baldo et al. to vastly increase absorption through Bragg diffraction, (b) strong coupling microcavity fabricated by Börjesson et al. to enable turn-on TTA-UC. Images are adapted from (a) ref. 262, and (b) ref. 265, with permission from the American Chemical Society. | ||
This section has covered solid-state systems, from encapsulation of liquids to neat solid films. Encapsulated systems provide high upconversion efficiencies and have already found applications.226 However, they contain solvent, limiting applicability in PV technology. Neat solid-state film upconversion has higher applicability in PV technology, but has so far not reached high enough upconversion efficiencies. Particularly, limiting factors in solid-state upconversion include aggregation induced processes and low triplet diffusion. We encourage continued structure optimisation of annihilator molecules with regards to packing and energy transfer. This has been somewhat less explored than sensitiser modification. Equally, new methods of exciton diffusion must be sought, and we particularly envisage interest in the semiconductor type setup introduced by Hiramoto et al. Further to this, new classes of functional materials are being explored, such as MOFs and strong-coupling materials.
Singlet fission based energy conversion
In 1965 Singh et al. reported SF for the first time when studying the laser excitation of anthracene crystals.269 A few years later Swenberg and Stacy explained the temperature dependent fluorescence quenching of tetracene by invoking this newly rationalised photophysical process.270 About a decade later, in 1979, Dexter proposed that SF in tetracene could be used to improve the efficiency of solar cells.271 However, it wasn’t until the early 2000's that interest in SF for solar energy harvesting really started to grow.34,74 Since then, the focus has been on understanding the SF process in detail, often through the help of ultrafast optical spectroscopy and magnetic field dependent measurements.59,61–63,76,80,272–274 Another related research focus has been the design of new SF chromophores.34,41,74,275 Above we introduced the details of the SF process and its relation to TTA. Here we will start by discussing the efficiency of the SF process followed by a brief overview of SF active materials and their relevance for PV applications. The following section will then summarise and discuss the many examples and approaches towards incorporating photon conversion with PVs.The singlet fission quantum yield
As with TTA-UC, the reporting of quantum yields is not always consistent across the literature, with the discrepancy arising from how the authors choose to define SF.61 As briefly mentioned above, it is often unclear if SF refers to the overall process of forming two free triplets from the initial singlet state, or if SF is complete when one of the intermediate triplet pair states is formed (1(TT) or [T1⋯T1]). The quantum yield of free triplet formation can reach a maximum of 200% using the IUPAC definition of quantum yields (eqn (1)), as two triplets can be formed for every absorbed photon. However, if the strongly bound correlated triplet pair state 1(TT) is considered as one state (e.g. similar the optically dark Ag state in polyenes),62 then the maximum quantum yield of 1(TT) formation should be reported with a maximum of 100%, which is not always the case. We therefore agree with the discussion of Zhu and co-workers that the most relevant definition of SF is the formation of [T1⋯T1].61Measuring the quantum yield of SF can be difficult, first one has to decide how to define SF, and secondly, one has to be able to distinguish between the triplet pair states and free triplet states, as well as measure these states quantitatively. As triplet states are non-emissive, one typically has to rely on transient absorption to follow the SF dynamics. The SF quantum yield can be estimated in a number of ways. It can be determined from the rate of singlet quenching and triplet formation.276 Singlet quenching can be measured using time-resolved fluorescence measurements. However, this method determines the yield of the state of which S1 decays into, i.e., 1(TT) and not necessarily free triplet yields. Further, depending on how the singlet quenching is determined this method might overlook other deactivation pathways and overestimate the yield. Hence one should ensure that the singlet quenching rate matches the triplet rise time.
Alternatively, one could use transient absorption spectroscopy. If the ground state bleach is not significantly overlapped the quantum yield can be obtained from the ratio of the ground state bleach at initial excitation to those at longer delay times. The ground state bleach at these times correspond to S1 and S1 + T1 populations, respectively.107,277 Unfortunately, many molecules have singlet and/or triplet spectra that significantly overlap the ground state bleach, complicating the analysis. Another popular approach requiring transient absorption spectroscopy, is to quantify the triplets generated using the triplet state molar absorptivity.127,277–281
In solution, a lower limit of the SF triplet yield can be determined from singlet oxygen emission.282 The SF yield can also be inferred by measuring the external or internal quantum efficiency of optoelectronic devices such as photovoltaics or photodetectors incorporating SF active materials.42,81,283,284 Another more recent method is to measure the photoluminescence enhancement of a NC-SF mixture.285–289 By measuring the triplet transfer efficiency to the NC, the SF yield can be determined. This method has been used in both solution and solid-state, and the yields correspond well with those determined by other means.285–289
Which method is better can depend on the actual system and rate of SF. Ultrafast SF might be difficult to resolve accurately using transient absorption spectroscopy setups with a large pulse width. Furthermore, often free triplet states are only fully resolvable on the ns–ms time scale, whereas the initial S1 population must be determined after initial excitation on the fs timescale. Hence separate setups might be required, complicating the comparison of the population of these states. It is therefore better to estimate the QY in multiple ways, e.g. from transient absorption measurements, and if applicable a device/photon enhancement to ensure these values align reasonably.
Singlet fission materials
To fulfil the energetic requirements discussed above, a SF material should have a large S1–T1 splitting. There are different ways to guarantee a large singlet–triplet splitting in molecular materials and it has been an ongoing debate on how to best design SF chromophores to fulfil this requirement.34,74,290,291 Below we cover some of the most common materials that have been studied for SF.Initially, the only types of chromophores that were studied for SF were even-carbon alternant hydrocarbons, such as anthracene, tetracene, pentacene, and polyenes etc. (Fig. 14).34 In these types of structures the HOMO and LUMO overlap significantly, resulting in a large exchange integral Kexchange and hence a large S1–T1 splitting (Δ(S1–T1) ≈ 2Kexchange).34,74 Pentacene and tetracene, and their derivatives, are probably the most studied SF materials to date.34,41,74,274,275
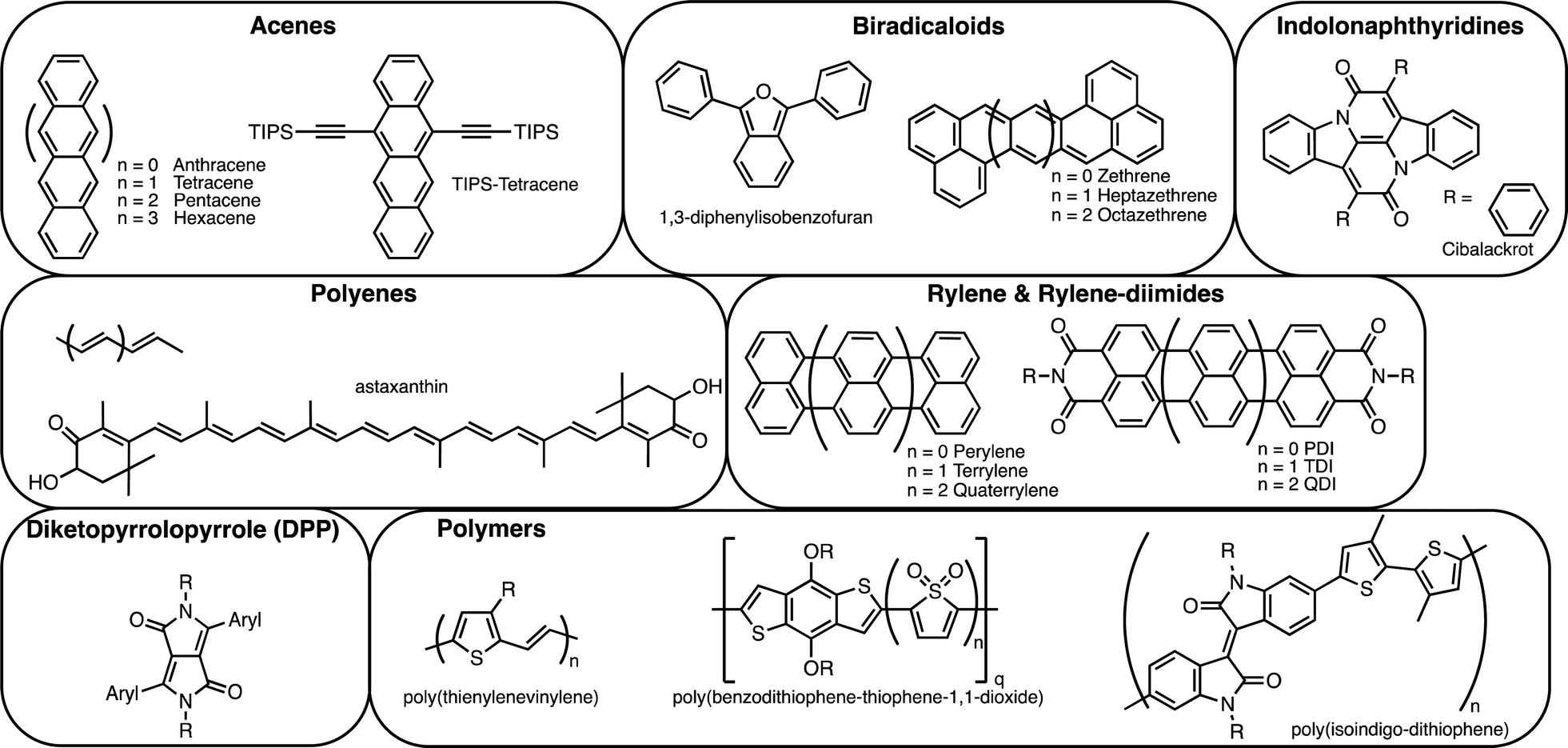 | ||
| Fig. 14 Molecular structures of chromophore families shown to undergo SF. | ||
In 2006 Michl and co-workers introduced another class of molecules that tend to have large S1–T1 separations, biradicals and biradicaloids.292 With two singly occupied degenerate orbitals, biradicals can have triplet ground states, and by tuning the biradical character of the molecule, the singlet–triplet gap can be tuned (Fig. 15). Michl and co-workers later also demonstrated efficient SF in the biradicaloid 1,3-diphenylisobenzofuran with 200 ± 30% triplet yield (Fig. 14).277 Nakano and co-workers later refined the biradical approach, by correlating the multiple biradical characters y0 and y1 with the energetic requirements for SF (where y0 and y1 corresponds to the occupation number of the lowest unoccupied natural orbital LUNO and LUNO + 1, respectively).293–296 They concluded that materials with a small y1 and a non-zero y0 can all fulfil the energy matching criteria for SF, and that molecules with a y1/y0 < 0.2, are a good starting point for new SF capable materials.296 Smith and Michl's reviews on SF offers an extensive summary of the even-carbon alternant hydrocarbon and biradicaloid materials that had been investigated up until their publication in 2010 and 2013.34,74 Since, the quest for more SF materials has continued. Michl and co-workers investigated a series of small molecule biradicaloids, using quantum chemical calculations.297,298 In 2017, Lukman et al. reported SF in the biradicaloid family of zethrene compounds.123
 | ||
| Fig. 15 Energy levels of closed shell (left) and open shell (right). By tuning the biradical character the singlet and triplet gap can be tuned (center). Reprinted from ref. 292 with permission from American Chemical Society, copyright 2006. | ||
Another family of materials similar to the acenes that has large S1–T1 separation is rylenes and their diimide derivatives (Fig. 14). Rylene-diimides have been widely studied for optoelectronic applications thanks to their highly versatile and tunable properties.299–303 SF has also been observed in rylene-diimide crystalline films,97,101,140,278,304–310 self-assembled nanocrystals and aggregates,311,312 as well as in some dimer structures.83,96,127,313,314 Triplet yields have been observed to approach 200% in solid films.
Diketopyrrolopyrroles (DPP, Fig. 14) are another class of molecules that have been investigated extensively for optoelectronic applications, including SF. Since the S1 energy of DPPs can be tuned without significantly altering the T1 energy, the energetics can be tuned to fulfil the requirements for SF and/or TTA.315 SF has been observed in some DPP materials including films, dimers and nanoparticles, with triplet yields exceeding 150%.104,109,316–320 Guldi and co-workers summarised the many works on SF chromophores in two recent reviews.41,275
Besides intermolecular SF, as observed in the materials discussed above, intramolecular SF has been observed in dimer versions of the same materials.76,83,94–96,98,99,105,109–111,132,274,276,279,313,314,321–331 Dimers are an interesting class of SF materials as the coupling between the monomers can be systematically engineered. Hence, dimers are useful when probing and addressing fundamental questions of SF. Furthermore, dimers can possibly have an advantage also in devices, where an optimized chromophore coupling could be designed into the structure, removing morphology and packing dependent issues. Although, in many dimers the question remains if complete SF is actually observed as the 1(TT) state does not separate further, contrasting the definition of SF in eqn (2).63,276 As the triplet excited states are forced to maintain close proximity in the dimers, it is not surprising that free triplets are seldom observed. To overcome this issue, Campos and co-workers developed structures where triplet separation was energetically downhill.56 Another approach is to extend the dimers to oligomers or polymers to allow the individual triplets to physically separate.127,331,332 However, it is not always enough to extend the dimers to oligomers to obtain free triplets, for example Sanders et al. reported that in polypentacene the triplet pair never separated into free triplets.326 Other polymers that have been reported to undergo SF are poly(thienylenevinylene),333 benzodithiophene-thiophene-1,1-dioxide donor–acceptor polymers,108,334 and isoindigo based donor–acceptor polymers.335 Understandably, most work on SF dimers has been performed in solution, as it allows for a cleaner system to study and address specific questions related to e.g. intermolecular coupling. However, it is highly relevant from a device perspective to also study dimers in the solid-state. For example, it is unclear if some of the limits observed in dimer solutions (i.e. poor dissociation of 1(TT)) can be overcome in solid-state.
Recently new design strategies to develop new SF materials have emerged. Tuning of the S1 and T1 energies in materials can be achieved by altering the degree of aromatic/antiaromatic character in the ground and excited states, as explored by Fallon et al. and El Bakouri et al.290,291,336 Fallon et al. and Zeng et al. showed that Cibalackrot and other indolonaphthyridines can be suitable SF chromophores (Fig. 14).290,291 Wang et al. also used the tuning of aromaticity and developed a fused dipyrrolonaphthyridinedione chromophore with a triplet energy around 1.2 eV and triplet yield of 173%.337 Another approach to tuning the singlet–triplet gap is introducing strain to the molecular structure.305 Furthermore, Padula et al. searched through the Cambridge Structural Database and evaluated over 40000 structures to find around 200 possible SF chromophores, many not previously considered.338 The development of SF materials is in its infancy, many more will undoubtedly be experimentally demonstrated in the future. As will be evident from the next section, the development of new SF chromophores has a huge potential impact on photovoltaic applications, as today the proof-of-principle devices have all used the same three types of molecules (or derivatives thereof): pentacene, tetracene and 1,3-diphenylisobenzofuran.
Incorporation of TTA and SF with photovoltaics
In recent years the progress in overcoming the fundamental limitations of TTA and SF has further allowed progress in applying the systems to photovoltaics and other devices. In this section we will discuss the incorporation of photon conversion materials to functional devices. We will first detail the work carried out and then finish with a critical overview of continuing challenges and limitations to commercial uptake.TTA-UC in solar cells
TTA-UC has cemented its place in many research fields related to photochemistry.216,339–347 Although, none is as prevalent as photovoltaics.67,348–356 In the following section, we will discuss the incorporation of TTA-UC into photovoltaics. Even though initially most examples of TTA-UC enhanced devices relied on solution phase systems,340,350,351,357–362 we will limit our discussion to the more recent and practically relevant solid-state systems.We have discussed above (see discussion around Fig. 1) the theoretical improvements that can be made to the efficiencies of PVs when utilising TTA-UC. In practicality these improvements are harder to realise. Device architecture, dye sensitisation and the upconversion process all play a role in the improvements to short circuit current densities (JSC) seen.363 An earlier perspective by Schmidt et al. remarked that to be relevant to PV use, a significant increase in short circuit current density when using upconversion must be observed over a suitable reference material. I.e. (ΔJuc > 0.1 mA cm−2 under one sun excitation density).364 In this section we will highlight recent examples of solar cells assisted by upconversion, and where possible assess them against the ΔJuc figure of merit.
Much effort has been made in applying upconverting layers to TiO2 dye sensitised solar cells. In 2018 Nagata et al. immobilised both a platinum sensitiser complex (PtTPO), and a DPA carboxylic acid derivative onto TiO2 nanoparticles at an FTO electrode (Fig. 16a).158 Immobilisation of the DPA or platinum complex alone led to no significant current density increases. On immobilisation of the two dyes concurrently (10:1, annihilator:sensitiser), a small ΔJuc of 3.6 × 10−2 mA cm−2 was observed under 1 sun irradiation. This led to a 0.72% power conversion efficiency for the upconverting device compared to 0.60% with annihilator immobilised alone. Immobilised molecular layers were again used by Hanson et al. in 2019.365 Supramolecular assemblies were constructed, consisting of a singlet sensitiser, a triplet sensitiser and an annihilator (Fig. 16b). The addition of the singlet sensitiser broadened the absorption envelope of the upconversion layer and allowed the filling of a so-called transparency window in the triplet sensitiser–annihilator bilayer. A record JSC for a TTA-UC solar cell was reported (0.315 mA cm−2). According to the definition above however, the ΔJuc should be the increase offered only by upconversion and not by direct dye sensitisation of the solar cell. This value was not directly reported. CdSe NCs were later used by the same group in the fabrication of TiO2 solar cells with upconversion assisted efficiencies.366 CdSe NCs were coated with a DPA derivative before being implanted between the TiO2 and Pt electrodes together with an electron mediator (Fig. 16c). The cells showed, however, lower Jsc values (29 μA cm−2) than their previously reported systems. Unfortunately, there are several drawbacks regarding the use of CdSe NCs in solar cells. Below unity energy transfer yields from the NC to the DPA were observed, along with slow electron regeneration, and competitive excited state quenching by the redox mediator. These are all challenges that need addressing.
 | ||
| Fig. 16 (a)–(d) Structures of the PV devices discussed in this review, all devices except for (c) were tested under 1 sun conditions. This figure has been adapted with permission from references (a) ref. 158, copyright 2019 American Chemical Society; (b) ref. 365, copyright 2019 American Chemical Society; (c) ref. 366, copyright 2020 American Chemical Society; (d) ref. 370, copyright 2020 Wiley-VCH GmbH; and (e) ref. 371, copyright 2021 Royal Society of Chemistry. | ||
We will now turn our attention to some recent examples of perovskite based PVs, which have been explored in upconversion applications.367–369 Kimizuka et al. applied a known singlet to triplet sensitiser (5) as an add-on technology to increase the number of photons absorbed (Fig. 16d).370 The sensitiser was immobilised in a PVA matrix, along with rubrene as an annihilator, and DPB as a singlet sink. The PVA film was applied below the solar cell material and a 938 nm laser was used as an excitation source. Measuring the current density with and without the PVA layer achieved a ΔJuc ∼ 0.1 mA cm−2, however, this value was only achieved at an excitation density of 4 W cm−2, around 40 times higher than the entire energy of the solar spectrum. Chen et al. used perovskite and rubrene:DPB mixed layers, which were spin coated into the device creating a charge transfer complex through coulombic interactions.371 The perovskite was described as the sensitiser, the rubrene as the annihilator and the DPB as the emitter (Fig. 16e). Increased power conversion efficiency and a high ΔJsc were measured. The conclusions of this work have, however, been subject to some criticism from others within the field.372 Earlier work demonstrated the complex nature, and non-optimised procedure of using perovskite as a sensitiser, and rubrene as an annihilator in the TTA-UC process.172,367–369 The efficiency gains are likely to be valid, but due to improvements made to the continuity and crystallinity of the perovskite surface. It is unlikely that in the material proposed by Chen et al., TTA-UC is the main mechanism of efficiency improvement, although we note the authors explanation that it is possible for these mechanisms to operate synergistically.373
Singlet fission in solar cells
There are numerous ways of incorporating SF materials with solar cells. A common approach is to incorporate the SF materials as part of the active material in the solar cell, for example as one of the dyes in a dye sensitised solar cell (DSSC),34,292 or organic photovoltaic (OPV).51 Perhaps the most intuitive way to incorporate SF with solar cells is to directly couple the SF material on top of a solar cell material. Here the triplet states could either transfer to the SC through triplet energy transfer or dissociate into free charges at the SC/SF interface. This approach was first proposed by Dexter over 40 years ago,271 but it has turned out to be extremely challenging to achieve efficient sensitisation of the SC in this way.43,271,374 A more recent approach that has been proposed, but not yet implemented in photovoltaics, is to convert the SF process into a photon multiplication process. Photon multiplication requires the dark triplet states generated by SF to be converted into emissive states, so that two low energy photons can be emitted for every high energy photon absorbed.72,273 In the next few paragraphs we will discuss the progress in applying SF to photovoltaic devices and the future prospect of the various approaches. We will start with devices where SF materials are part of the active material (DSSC, Organic PV, Inorganic PVs) and continue with devices where the SF material instead is coupled to the solar cell.DSSC
As the prospect of using SF in solar cells re-emerged as a possible and practical way to improve the efficiency of solar cells in the early 2000's, Michl and co-workers pursued the idea of applying SF to DSSCs. To effectively achieve the theoretical efficiency gain of around 45%, a SF dye with ET1 of 0.9–1.1 eV and ES1 of 1.8–2.2 eV would have to be combined with a conventional dye with ES1 of 0.9–1.1 eV.292 Michl, Johnson and co-workers initially used a 1,3-diphenylisobenzofuran derivative that could anchor to the TiO2 electrode of an DSSC.375 The main challenge with this approach is that electron injection from the singlet state out-competes SF in these surface bound chromophores.112,375–377 To overcome this issue, a surface passivation strategy has been pursued, where a thin insulating layer of ZrO2 or AlO2 is deposited to slow down electron injection allowing SF to occur (Fig. 17).112,375–377 Kunzmann et al. used a pentacene dimer where SF occurred intra-molecularly, and electron injection up to 130% was reported.378 By using a dimer structure the chromophore coupling can be directly designed compared to surface bound monomer structures where the non-ideal coupling compared to solid-state crystals has led to a slowing of the SF process.377 Another advantage with a dimer approach is that SF would not be directly influenced by adding in a conventional low bandgap dye. Banerjee et al. recently demonstrated an alternative approach to adding a low bandgap dye to a surface anchored SF dye by metal–ion assisted self-assembly.376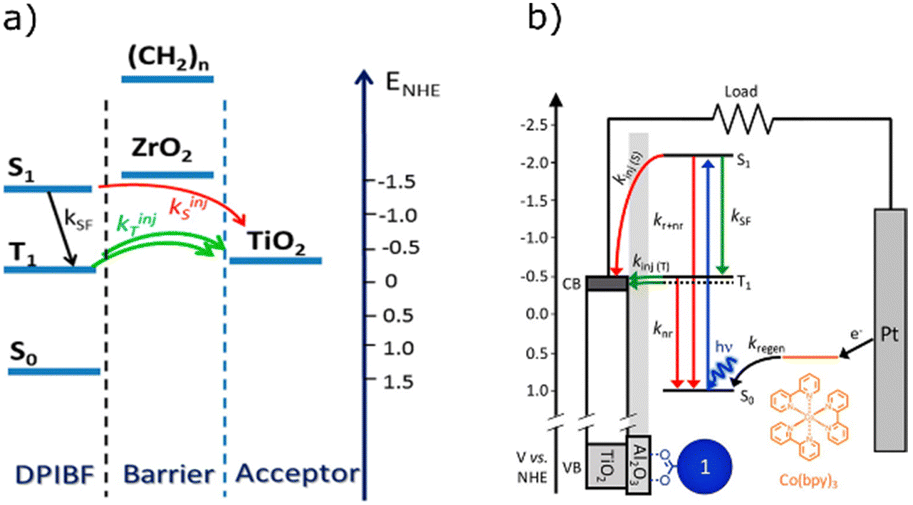 | ||
| Fig. 17 Energy level alignment and device configuration of the two 1,3-diphenylisobenzofuran (DPIBF, 1) DSSCs studied by (a) Schrauben et al. and (b) Banerjee et al. 1,3-diphenylisobenzofuran functions as the SF sensitizer and an insulating layer of ZrO2 or Al2O3 is required to minimize direct electron transfer from S1 to the TiO2. (a) Reprinted from ref. 375, with permission from the American Chemical Society. Copyright 2015. (b) Reprinted from ref. 376 with permission from the American Chemical Society. Copyright 2018. | ||
To achieve highly efficient SF sensitised DSSC the focus should lie in finding a suitable SF dye, one that is efficient in dimer form, has a suitable triplet energy relative to the conventional TiO2 electrodes and is stable enough for long-term illumination. As new SF materials are developed, these conditions can hopefully be met, as the DSSC structure, already relying on dye incorporation, is particularly suitable for SF sensitisation.
Organic PVs
Another PV type that already uses molecular dyes is organic photovoltaics (OPVs). It is perhaps not surprising that the first examples of solar cells with EQEs and IQEs exceeding 100% were reported in OPVs containing SF dyes.283,379–381 Careful molecular engineering is required, to obtain the correct morphology and minimise charge recombination and triplet-charge annihilation, whilst maintaining an optimal interface and energetics for electron transfer.382–389 In all cases to date, even though SF is operational, the benefit in these OPV is minimal on overall performance compared to an optimised OPV without SF.390,391 One reason for this poor enhancement is related to the morphology of the device. For efficient SF to occur, bilayer OPVs must be used,42,283,379,381,386,388,389,392 which sets strict limits to device thickness. This is due to the short triplet diffusion lengths,393 and hence total photon absorption.380 In traditional OPVs, bulk heterojunctions are more common, but these structures have proved less efficient for SF materials.386,388 However, in some instances SF can be compatible in mixed blends if the SF chromophores aggregate appropriately, as observed by Jadhav et al. for tetracene in a C60 copper-phthalocyanine (CuPc) blend.394Many of the above-mentioned proof-of-principle SF-OPVs also have another significant short coming, to obtain any gain from SF a low energy absorber is also needed in the device. This has been addressed by Jadhav et al.394 and Tritsch et al.389 where CuPc has been incorporated as a low bandgap chromophore (Fig. 18).
 | ||
| Fig. 18 (a) Schematic illustration of a tetracene/CuPc/C60 OPV where the CuPc low bandgap donor and C60 acceptor constitute the traditional OPV and tetracene functions as an additional singlet fission sensitizer for high energy photons. (b) energy alignment of the materials in the device in (a). Reprinted from ref. 394, with permission from American Chemical Society. Copyright 2011. | ||
An alternative to adding in a low bandgap chromophore to the OPV device is to use the SF OPV as a top cell in a voltage matched parallel connected tandem cell, with a low bandgap bottom cell (Fig. 19).395,396 This approach has been successfully used to demonstrate an overall EQE exceeding 100%. An additional benefit of a voltage matched, rather than current matched series connected tandem cell is their better spectral stability, and smaller sensitivity to diffuse rather than direct lighting (Fig. 19).395,396
 | ||
| Fig. 19 (a) Conventional series connected tandem which requires current matching to operate efficiently. (b) A singlet fission-based voltage matched tandem connected in parallel. (c) theoretical maximum efficiency of a Si single junction device, a parallel and series connected tandem as a function of the ratio between diffuse and direct lighting. Reprinted from ref. 396, with permission from American Chemical Society, Copyright 2020. | ||
Challenges remain in bringing SF OPVs to real life practical PV applications. One must obtain a suitable morphology for SF, charge separation and electron/hole migration in one device. Furthermore, the device must constitute a SF molecule, a low bandgap chromophore and an acceptor molecule. Collating everything into a single device is one of the major hurdles in this field. Conventional OPVs struggle with morphology optimisation as is and blending in another dye that also has stringent requirements on morphology results in a difficult puzzle to solve. Moreover, moving away from acene based SF chromophores would be required for photostable SF chromophores that can be used in devices. However, parallel connected voltage matched tandem cells by the use of SF top cells is an interesting and highly promising implementation of SF warranting further efforts to achieving efficient SF sensitised OPVs.
Inorganic PVs
SF chromophores have also been combined with inorganic PV materials. For example, Ehrler et al. combined pentacene with PbS NCs and PbSe NC/amorphous silicon in functioning devices.397,398 Even though the IQE did not exceed 100%, they concluded that triplet excitons could be dissociated into free charges at the organic/inorganic interface.397,398 They later improved the device performance by using a solution processable pentacene derivative (TIPS-Pc), together with PbS NCs. This system was able to achieve a 170% IQE and 4.8% overall device efficiency.399The potential for SF to sensitise perovskite solar cells has been investigated recently. Even though no devices were prepared, the demonstration of ultrafast charge injection from the triplet pair state or SF generated triplet states to perovskite films is a promising first step.400,401 In these two studies TIPS-Pc was used as the SF material, and a MAPbI3 perovskite film was used as the device relevant electron acceptor.400,401 These studies call for evaluating SF systems in full devices. However, as discussed above (Fig. 1), the most SF relevant devices are those with low bandgap (<1.4 eV) material. Hence the future direction of SF sensitised inorganic PVs should build on this advantage compared to organic PVs, and exploit the many low bandgap materials available, such as PbS/PbSe NCs and NIR perovskite materials.
Directly coupled singlet fission solar cell devices
A drawback to the SF active material PVs described above, is that a completely new device has to be designed, optimised and fabricated. By adding a SF material to an already optimised and efficient solar cell, part of the wheel does not have to be re-invented. Ideally, SF could be combined with a c-Si cell, as c-Si is the dominating PV technology today and has a suitable bandgap for SF enhancement (Fig. 1d). However, adding a layer of SF material to a c-Si cell, as proposed by Dexter almost half a century ago, has not resulted in any significant gains.43,374 In 2018 MacQueen et al. evaporated 100 nm tetracene on top of a hydrogen passivated n-doped c-Si bottom cell. PEDOT:PSS was used as a hole transporting layer on top of the tetracene. This design was chosen to achieve triplet harvesting from tetracene, either through exciton dissociation or triplet energy transfer at the tetracene-silicon interface (Fig. 20). The Si substrate remained well passivated, but the tetracene acted to shadow the device and slightly lower the overall current.43 They concluded that the SF material did not improve the device performance, likely due to a potential mismatch between the tetracene triplet ionisation energy and the Si conduction band edge (Fig. 20a). The triplet yield was not measured directly, but modelling suggested about 8% exciton to photocurrent generation from tetracene.43 It has been reported previously that Si can have a quenching effect of tetracene singlet emission with negligible triplet injection, and that an insulating interlayer can alleviate the quenching.402,403 Importantly, an insulating layer on the c-Si surface is often employed in solar cells already to passivate the Si surface. Moreover, one might envision that a properly designed interlayer might even participate and facilitate energy transfer from a SF material. Einzinger et al. used an 8 Å layer of hafnium oxynitride on Si to enable efficient triplet injection into the semiconductor.374 The insulating layer allowed for an overall 133% exciton transfer from tetracene to Si, with 56% being singlet transfer and 76% triplet transfer. However, the overall power conversion efficiency was recorded as 5.1%, significantly lower than conventional c-Si PVs. The poor efficiency was limited by carrier collection from the front surface of the cell, as the device structure employed was a interdigitated back contacted cell, (Fig. 20c).374 Future directions will require understanding and optimisation of the surface passivation required for efficient solar cell performance, while maintaining efficient triplet and/or charge injection from the SF material. Daiber et al. recently introduced an optical screening method to test triplet quenching at the organic–silicon interface, to quickly evaluate the effect of different surface treatments.404 Another significant hurdle for these types of SF devices is the small absorption afforded by the 30–100 nm thin SF layer deposited on the solar cell surface.43,374 This is limited by the triplet diffusion length of the SF material to ensure efficient triplet migration to the interface.405–407 | ||
| Fig. 20 (a) Band alignment of tetracene in a c-Si PV device. (b) Schematic illustration of the device and the processes SF, triplet energy transfer and charge transfer in the device studied by MacQueen et al. (a) and (b) Reprinted from ref. 36 with permission from Royal Society of Chemistry. (c) Device structure of the tetracene sensitized c-Si interdigitated back contacted solar cell used by Einzinger et al. Reprinted with permission from ref. 374. Copyright Nature Springer 2019. | ||
Any developed SF material must be photostable over years, while also meeting the energetic requirements for SF and charge/triplet injection to the solar cell. If these challenges can be overcome there are a significant gains to expect for silicon PVs.404 Depending on if the triplet transfer occurs through charge transfer or Dexter transfer, Daiber et al. estimated that the efficiency limit of a SF-silicon solar cell could reach 34.6% and 32.9%, respectively.404 An additional advantage of using SF, or any other type of multi exciton generation process, with c-Si PVs is that the operating temperature of the device is decreased as thermalisation is minimised.408 The decreased operating temperature in turn leads to a longer device lifetime.408
Singlet fission based photon multiplication
Ideally, a SF material could be added to an optimised solar cell as is, without the need to modify the underlying device. However, this ambition has turned out to be challenging, as discussed above. By converting the SF process into a photon multiplication process (PM), i.e. a high energy photon is split into two low energy photons, a solar cell could be optimised for what it does best, absorbing photons to generate current, and the SF material could be optically coupled to the device (Fig. 21a). This approach would overcome the requirements of direct and close contact between the solar cell and SF material, as charge or electron transfer would be exchanged for photon emission and re-absorption.273,409 A photon multiplier could enhance c-Si cells above 30% efficiency under real-world conditions (Fig. 21d–f). | ||
| Fig. 21 (a) Schematic of a photon multiplier material coupled to a silicon PV. The SF material first absorbs a high energy photon and undergoes SF to generate two triplet states. These non-emissive triplet states are transferred via triplet energy transfer (TET) to a NIR emitter, for example a semiconducting NC (QD). The NIR emitters emits two low energy photons for every high energy photon absorbed. The low energy photons are subsequently absorbed by the PV device. Adapted from ref. 133 with permission from AIP publishing. (b) and (c) Detailed illustration of the photon multiplier process with a PbS NC NIR emitter and TIPS-tetracene carboxylic acid mediator ligands. A high energy photon is absorbed by the SF material TIPS-tetracene. After triplet generation from SF the two triplets migrate and transfer to NCs via a mediator ligand. The NCs then relaxes radiatively emitting NIR photons. Reprinted from ref. 288, with permission from American Chemical Society. Copyright 2020. (d–f) Estimated PV efficiencies using a photon multiplier approach or tandem device applied to silicon cells of different base efficiencies for standard test conditions (d), sun light and temperature conditions in Netherlands (e) and Colorado (f). Reprinted from ref. 409, with permission from the American Chemical Society, Copyright 2018. | ||
The question is then, how to convert the SF process into a photon multiplication process? There are in principle two strategies: the SF process could be engineered to generate two singlet excited states instead of triplet states.410,411 Alternatively, the two generated triplets could be transferred to an emissive material not sensitive to the spin of the exciton, such as semiconducting nanocrystals (NCs).289,412 The former approach has not gained much interest and there remains many uncertainties about how viable this approach is.410 The latter approach has recently been demonstrated practically in solution,286 and in films.285 The overall process (Fig. 21b and c) occurs as follows: in a solution or film with high density of SF materials, a high energy photon is first absorbed by the SF material to initiate SF. The two formed triplet states are eventually transferred to a NC of appropriate energy, via a surface anchored triplet mediator ligand. The NC then relaxes radiatively emitting a low energy photon. With this approach Allardice et al. demonstrated how the emission of PbS NCs could be enhanced 1.25 times when exciting the SF material in solution.286 With close to quantitative triplet transfer to the NC the emission enhancement could also be used to calculate the actual free triplet yield in the SF material according to eqn (11).286,287,289
 | (11) |
In thin films, as the SF yield is usually higher than in solution, Allardice et al. later showed a 190% exciton multiplication factor using TIPS-tetracene films with PbS NCs emitters and TIPS-tetracene carboxylic acid mediator ligands. The overall photon multiplication was limited by the low emission quantum yield of the PbS NCs.285 However, one key demonstration was that the photon multiplier approach was compatible with thick films of the SF material TIPS-tetracene. Thick films allowed >95% of the incoming high energy light to be absorbed by the SF material while minimal NC absorption across the spectra was maintained.285
Gray et al. worked out the energetic requirement for efficient triplet transfer to ligated NCs. They concluded that SF materials with triplet energies around 1.4 eV would be required for NCs with emission relevant for c-Si.287 This triplet energy is slightly higher than most common SF chromophores, hence warranting further research to develop new SF materials.
In 2018 Nagata et al. used a SF-PM approach in OLEDs to improve the light emission efficiency.413 Instead of NCs as the low energy emitter they used an erbium(III) complex to harvest the triplets generated in rubrene, resulting in a 100.8% exciton generation efficiency.413 The Er emission at 1530 nm is too low in energy to be combined with c-Si, but other lanthanide ions could be considered with more suitable emission wavelengths.414
The photon multiplication approach is relatively new and has not been applied to PVs yet. Two key challenges remain: firstly, finding a combination of materials (SF chromophores and NCs) that can operate at energies relevant for c-Si. Secondly, developing NCs (or other emitters) with emission quantum yields exceeding 80–90% that can still efficiently accept triplets. Ultimately, a combination of materials that can be mixed and processed into a suitable film morphology that allows high yields of triplet generation, triplet transfer and NC emission will be required.
Challenges & opportunities in TTA-UC and SF application to PVs
Radiative transfer of photons to PVs
In most TTA-UC systems, and in SF-PM, the end product is photons. In many aspects this is a unique advantage of TTA-UC and SF-PM compared to other approaches to improve solar cells such as multi junction cells, which are hampered by the need to current match the individual PV materials. It reduces the complexity and, at least theoretically, decouples the device and photon conversion material, allowing for the separate optimisation of each part. It also might allow for incorporating these materials with already existing devices in the future, greatly enhancing the potential impact. However, for the generated photons to benefit a solar cell they have to be transferred to the device. There are numerous ways one can imagine this to be done with varying degrees of complexity and associated cost.First, the simplest and probably the most desirable approach is to add the TTA-UC or SF-PM layer underneath or on top of the PV device, respectively. With a realistic refractive index of the photon conversion material of 1.7 theoretical modelling suggest that only 10% of the emitted light in a SF-PM is lost due to emission leaving the layer away from the solar cell.409 Most of the light is in fact directed to the solar cell directly or through total internal reflection.409 Since SF and TTA materials are very similar it is reasonable to believe that similar high coupling efficiencies can be reached for a TTA-UC layer.
Most TTA-UC PV devices in literature have used the simple back layer approach,370,415 even when employing liquid TTA-UC systems.350,351,357,359,360,416 For TTA-UC this requires the PV to have transparent back electrodes (for the relevant low energy photons). This might require a change in device structure for some types of PVs, accompanied with a decreased efficiency.339 Hence, any gain from TTA-UC must outperform any losses due to change in device structure. Furthermore, to maximise the photon flux into the TTA-UC material, the layer is often capped by a reflecting back surface.350,351,359,370,416 A TTA-UC back layer might compete with the implementation of bifacial PV modules.339 It has been argued that these bifacial modules can conservatively estimate a 10% efficiency gain, compared to a monofacial module, any TTA-UC layer must be able to compete with this gain to be implemented. However, bifacial modules is a technique mostly relevant for high efficiency Si devices used in solar farms. As discussed in Fig. 1, Si devices have less to gain from TTA-UC and hence a competitive comparison between TTA-UC and bifacial modules is not the most relevant. Instead, considering that bifacial modules are not possible for building mounted or building integrated PVs, and that these types of PVs are proposed to benefit from next generation PV materials such as the higher bandgap perovskites, OPVs and DSSC, we believe that TTA-UC can still play an important role in improving these systems.
Another possibility is to develop luminescent solar concentrators (LSC) that incorporate TTA-UC and/or SF-PM and use these with relevant PV devices. A LSC is a device that absorbs sunlight in luminescent chromophores dispersed in a polymer or glass layer.417 Both direct and diffuse sunlight is concentrated at the edges by re-emission from the chromophores followed by total internal reflection, effectively waveguiding the light to the edges.417 Hence the PV device is attached to the edges of the LSC. In LSCs, light is lost by emission that escapes the emissive layer. Typically ∼25% of the absorbed photons are lost in this way.417 Furthermore, for LSCs to effectively concentrate light, there has to be a free energy loss to compensate for the loss in entropy associated with the light concentration.418 This free energy loss is usually achieved by a Stokes shift between the absorbed and re-emitted photons. Hence, some energy will be lost as heat with the LSC approach. However, it was recently shown that for SF-PM LSCs this loss requirement is relaxed, and at low intensities a SF-PM based LSC might outperform standard LSCs.418 How the fundamental limits of a LSC change for a TTA-UC based LSC has not yet been discussed, but would be a useful way to evaluate the potential of this approach. Ha et al. experimentally demonstrated a TTA-UC LSC coupled to a DSSC.419 Although, the reported efficiency has been questioned.420 Still, much is left to explore for the LSC approach. For example, in some LSC designs, the light concentration could also be used to ensure that the TTA-UC system operates above its Ith.339
Regardless of the approach to couple TTA-UC and SF-PM to a device, the emissive material would require a high QY to minimise losses. This is particularly important as in a thick film or LSC, substantial amounts of reabsorption can occur. The simplicity of the layer approach makes it particularly attractive for incorporating photon conversion materials with devices. Proof-of-principle devices for TTA-UC and theoretical modelling of SF-PM materials indicate that optical coupling these materials to PVs is feasible, and the bottle neck is now associated with inefficient systems or materials limitations.
Stability and cost considerations
Both TTA-UC and SF materials research has been heavily reliant on acene materials. Efforts have been made to develop more stable SF materials. However, the photo instability of TTA-UC and SF materials is a common challenge that remains.Considering that commercial solar cell modules have degradation rates of approximately 0.5% per year,421 and lifetimes exceeding 25 years, it would be interesting to consider the maximum degradation rate of the photon conversion material that still allows for economic viability. For example, it was recently estimated for perovskite-silicon tandem solar cell with an PCE of 28% would still be economically viable if the perovskite material degraded at a rate of 2% per year.422 A possible advantage with some of the organic photon conversion materials is that the degradation product is mostly transparent to visible light, hence not affecting the intrinsic PV device performance. This might mean that a higher degree of degradation is acceptable.408
Besides degradation, materials toxicity and scarcity should be considered. Recent work by Wickerts et al. highlighted that photon upconversion materials using NC sensitisers need to achieve a 0.05–2 percentage points increase of module efficiency per applied gram of NC for it to be preferable over installing more PVs, from a climate perspective.
Finally, for TTA-UC and SF to become practically relevant they must provide efficiency gains that overcome any additional cost of adding the photon conversion material to the PV module. Richards et al. concluded that it was unlikely that an NIR upconversion material (TTA-UC or rare earth based) would be economically viable to improve the commercially dominant c-Si PV modules.339 Even with the most optimistic estimates, like sensitiser absorption in the 1400–1650 nm region and 50% UC yield, the low cost and high efficiency of current c-Si modules makes it unfeasible.339 However, as shown in Fig. 1, c-Si is not the most optimal PV material to benefit from photon upconversion. Efforts should instead focus on complementing next generation solar cells with higher band gaps, like OPVs and perovskite devices. Moreover, other applications, such as solar driven photocatalysis, could be more economically relevant.339
A cost and life cycle analysis of SF coupled PVs would be highly relevant. Especially as several approaches towards incorporating SF materials with PVs are approaching practical feasibility. A number of theoretical papers indicate that SF enhanced solar cells have many properties that can compete with silicon based tandem devices.396,408,409 Considering that tandem devices are on the verge of commercialisation there is much hope that SF will also be feasible in the near future.
Concluding remarks and future requirements in solving fundamental TTA and SF limitations
TTA-UC and SF have been growing in prevalence within the scientific literature, but have remained largely separated and discussed without a clear connection. This is unfortunate owing to the intertwined nature of the photophysical processes in both techniques. We have discussed herein how SF and TTA-UC systems have been thoroughly investigated, understood, and some level of optimisation has occurred. However, we also note there is a long away to go before TTA-UC and SF can be practically used in devices. We provide an analysis of the future challenges and prospects with regards to utilisation of TTA-UC and SF in photovoltaic technology.Specifically, we started this review with an introduction to the major existing challenges in SF and TTA. It was clear that challenges common to both techniques include, but are not limited to poor triplet diffusion in solid state, reversibility of TTA and SF, and poor matching of the energy levels to PV materials. It should be noted that both the TTA and SF processes are very efficient under optimised conditions, such as in solution. The challenge then is to retain these high efficiencies when applying them to photovoltaic relevant conditions. In tandem we must improve the transfer of energy to the PV through improved energy/electron/photon transfer pathways. No challenge within photon conversion for PV applications is completely solved. The most direct path forward might be to look over the shoulder of other research areas.
Increasing relevance of solar energy capture
Solar capture with focus towards defined PV bandgaps (Fig. 1) must remain a vital research area.67,184 Here, the selection of PV must be carefully considered. For TTA-UC, the PV materials with large bandgaps would benefit most. These include GaAs, InP, GaInP, CdTe, Organic and ABX3. Many of these are expensive technologies with little commercial viability, although promising research continues into perovskite, organic and CdTe PVs and efforts of combining TTA-UC with these devices would therefore be most relevant. The rapidly moving research field of thermally activated delayed fluorescence could serve as a source of inspiration when designing new low-loss sensitisers. QD sensitizers is another promising direction in this regard, where there still remain fundamental questions to address in terms of mediating efficient triplet transfer. Conversely, SF may be applied to low bandgap PV materials, such as c-Si and CIGS, for maximal benefit. Given the huge number of commercial c-Si systems in place around the world, it would seem as though these are the natural target. From a material perspective, the increased understanding of excited state aromaticity could provide a pathway towards new molecular motifs for SF.336,423Improving triplet exciton diffusion
Especially in the solid-state this is crucial. The number of studies targeting triplet diffusion could increase in the field of TTA-UC. Here, the field of SF is considerably ahead, and many lessons has been learned that could be implemented in TTA-UC systems. An alternative approach to self-assembled systems are framework materials such as MOFs, where there is a scope of engineering long distance conjugation and thus diffusion.249Material stability and abundancy
Both are important factors for commercial implementation. The material stability in photon conversion remains underreported in the literature, and it would seem beneficial to see a rise in reporting of this data. Furthermore, scarce transition metals must be replaced in triplet sensitisers. Here, CT mediated ISC can play a role, and inspiration can further be taken from the research of photodynamic therapy, which have similar requirements on sensitisers.Ultimately, both SF and TTA-UC have potential to become practical technologies for PV applications. While the efficiency enhancements observed in TTA-UC and SF PV devices currently remain below commercially relevant values, a pathway exists to increase efficiency gains and improve cost viability. Currently, there is a lack of efficient and stable materials and solid-state morphology tuning is complicated. However, the mechanisms are understood much better today, and therefore the future is bright for photon conversion.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
KB gratefully acknowledge financial support from the Knut and Alice Wallenberg Foundation (KAW 2017.0192). VG acknowledges funding from the Swedish research council, Vetenskapsrådet 2018-00238 and 2021-03744.Notes and references
- A. International, ASTM G173 - 03(2020), (accessed 27 May 2021).
- EIA, What is the heat content of U.S. coal? https://www.eia.gov/tools/faqs/faq.php?id=72&t=2, (accessed 04 November 2021).
- S. R. Kurtz, A. M. Leilaeioun, R. R. King, I. M. Peters, M. J. Heben, W. K. Metzger and N. M. Haegel, MRS Bull., 2020, 45, 159–164 CrossRef.
- S. Department, U.S.-China Joint Statement Addressing the Climate Crisis, https://www.state.gov/u-s-china-joint-statement-addressing-the-climate-crisis/, (accessed 20 May 2021, 2021).
- Lancet, The Lancet, 2019, 393, 1911 CrossRef.
- A. Jäger-Waldau, I. Kougias, N. Taylor and C. Thiel, Renewable Sustainable Energy Rev., 2020, 126, 109836 CrossRef.
- I. E. Agency, Snapshot 2021, 2021 Search PubMed.
- IRENA, Renewable Power Generation Costs in 2019, IRENA, 2020 Search PubMed.
- A. Jäger-Waldau, PV Status Report 2019, Publications Office of the European Union, 2020 Search PubMed.
- H. Gholami and H. Nils Røstvik, Energies, 2021, 14, 2531 CrossRef.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- S. Rühle, Solar Energy, 2016, 130, 139–147 CrossRef.
- L. Hirst and N. Ekins-Daukes, Quantifying intrinsic loss mechanisms in solar cells: Why is power efficiency fundamentally limited?SPIE, 2010 Search PubMed.
- C. H. Henry, J. Appl. Phys., 1980, 51, 4494–4500 CrossRef CAS.
- L. C. Hirst and N. J. Ekins-Daukes, Prog. Photovolt. Res. Appl., 2011, 19, 286–293 CrossRef.
- O. Dupré, R. Vaillon and M. A. Green, Solar Energy, 2016, 140, 73–82 CrossRef.
- A. Wang and Y. Xuan, Energy, 2018, 144, 490–500 CrossRef.
- P. Baruch and J. E. Parrott, J. Phys. D: Appl. Phys., 1990, 23, 739–743 CrossRef CAS.
- J. F. Geisz, M. A. Steiner, N. Jain, K. L. Schulte, R. M. France, W. E. McMahon, E. E. Perl and D. J. Friedman, IEEE J. Photovoltaics, 2018, 8, 626–632 Search PubMed.
- A. D. Vos, J. Phys. D: Appl. Phys., 1980, 13, 839–846 CrossRef.
- S. Sundaram, D. Benson and T. K. Mallick, in Solar Photovoltaic Technology Production, ed. S. Sundaram, D. Benson and T. K. Mallick, Academic Press, 2016, pp. 7–22 DOI:10.1016/B978-0-12-802953-4.00002-0.
- X. Li, Q. Xu, L. Yan, C. Ren, B. Shi, P. Wang, S. Mazumdar, G. Hou, Y. Zhao and X. Zhang, Nanophotonics, 2021, 10, 2001–2022 CrossRef CAS.
- M. Safdar, A. Ghazy, M. Lastusaari and M. Karppinen, J. Mater. Chem. C, 2020, 8, 6946–6965 RSC.
- B. Zhou, L. Yan, J. Huang, X. Liu, L. Tao and Q. Zhang, Nat. Photonics, 2020, 14, 760–766 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- B. Mehrdel, A. Nikbakht, A. A. Aziz, M. S. Jameel, M. A. Dheyab and P. M. Khaniabadi, Nanotechnology, 2021, 33, 082001 CrossRef PubMed.
- A. Nadort, J. Zhao and E. M. Goldys, Nanoscale, 2016, 8, 13099–13130 RSC.
- D. Kang, E. Jeon, S. Kim and J. Lee, BioChip J., 2020, 14, 124–135 CrossRef CAS.
- S. Wen, J. Zhou, K. Zheng, A. Bednarkiewicz, X. Liu and D. Jin, Nat. Commun., 2018, 9, 2415 CrossRef PubMed.
- M. V. DaCosta, S. Doughan, Y. Han and U. J. Krull, Anal. Chim. Acta, 2014, 832, 1–33 CrossRef CAS.
- M. Li, R. Begum, J. Fu, Q. Xu, T. M. Koh, S. A. Veldhuis, M. Grätzel, N. Mathews, S. Mhaisalkar and T. C. Sum, Nat. Commun., 2018, 9, 4197 CrossRef.
- H. Goodwin, T. C. Jellicoe, N. J. L. K. Davis and M. L. Böhm, Nanophotonics, 2018, 7, 111–126 Search PubMed.
- D. Zhou, D. Liu, G. Pan, X. Chen, D. Li, W. Xu, X. Bai and H. Song, Adv. Mater., 2017, 29, 1704149 CrossRef.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- R. Datt, S. Bishnoi, D. Hughes, P. Mahajan, A. Singh, R. Gupta, S. Arya, V. Gupta and W. C. Tsoi, Solar RRL, 2022, 6, 2200266 CrossRef CAS.
- R. T. Wegh, H. Donker, K. D. Oskam and A. Meijerink, Science, 1999, 283, 663–666 CrossRef CAS.
- W. G. J. H. M. V. Sark, A. Meijerink and R. E. I. Schropp, in Third Generation Photovoltaics, ed. F. Vasilis, IntechOpen, Rijeka, 2012, Ch. 1 DOI:10.5772/39213.
- K. J. Karki, F. Ma, K. Zheng, K. Zidek, A. Mousa, M. A. Abdellah, M. E. Messing, L. R. Wallenberg, A. Yartsev and T. Pullerits, Sci. Rep., 2013, 3, 2287 CrossRef.
- M. C. Beard, J. M. Luther, O. E. Semonin and A. J. Nozik, Acc. Chem. Res., 2013, 46, 1252–1260 CrossRef CAS PubMed.
- H. Goodwin, T. C. Jellicoe, N. J. L. K. Davis and M. L. Böhm, Nanophotonics, 2018, 7, 111–126 Search PubMed.
- R. Casillas, I. Papadopoulos, T. Ullrich, D. Thiel, A. Kunzmann and D. M. Guldi, Energy Environ. Sci., 2020, 13, 2741–2804 RSC.
- P. D. Reusswig, D. N. Congreve, N. J. Thompson and M. A. Baldo, Appl. Phys. Lett., 2012, 101, 113304 CrossRef.
- R. W. MacQueen, M. Liebhaber, J. Niederhausen, M. Mews, C. Gersmann, S. Jäckle, K. Jäger, M. J. Y. Tayebjee, T. W. Schmidt, B. Rech and K. Lips, Mater. Horiz., 2018, 5, 1065–1075 RSC.
- M. Green, E. Dunlop, J. Hohl-Ebinger, M. Yoshita, N. Kopidakis and X. Hao, Prog. Photovolt. Res. Appl., 2021, 29, 3–15 CrossRef.
- B. M. Kayes, H. Nie, R. Twist, S. G. Spruytte, F. Reinhardt, I. C. Kizilyalli and G. S. Higashi, 2011 37th IEEE Photovoltaic Specialists Conference, 2011, 000004–000008 Search PubMed.
- T. Trupke, A. Shalav, B. S. Richards, P. Würfel and M. A. Green, Sol. Energy Mater. Sol. Cells, 2006, 90, 3327–3338 CrossRef CAS.
- T. Trupke, M. A. Green and P. Würfel, J. Appl. Phys., 2002, 92, 4117–4122 CrossRef CAS.
- J. Ramanujam, A. Verma, B. González-Díaz, R. Guerrero-Lemus, C. del Cañizo, E. García-Tabarés, I. Rey-Stolle, F. Granek, L. Korte, M. Tucci, J. Rath, U. P. Singh, T. Todorov, O. Gunawan, S. Rubio, J. L. Plaza, E. Diéguez, B. Hoffmann, S. Christiansen and G. E. Cirlin, Prog. Mater. Sci., 2016, 82, 294–404 CrossRef CAS.
- H. J. Snaith, Nat. Mater., 2018, 17, 372–376 CrossRef CAS PubMed.
- V. M. Fthenakis, Renewable Sustainable Energy Rev., 2004, 8, 303–334 CrossRef CAS.
- J. Lee, P. Jadhav, P. D. Reusswig, S. R. Yost, N. J. Thompson, D. N. Congreve, E. Hontz, T. Van Voorhis and M. A. Baldo, Acc. Chem. Res., 2013, 46, 1300–1311 CrossRef CAS PubMed.
- M. J. Y. Tayebjee, D. R. McCamey and T. W. Schmidt, J. Phys. Chem. Lett., 2015, 6, 2367–2378 CrossRef CAS PubMed.
- J. Y. T. Murad, R. Akshay and W. S. Timothy, J. Photonics Energy, 2018, 8, 1–9 Search PubMed.
- R. C. Johnson and R. E. Merrifield, Phys. Rev. B: Condens. Matter Mater. Phys., 1970, 1, 896–902 CrossRef.
- M. W. B. Wilson, A. Rao, J. Clark, R. S. S. Kumar, D. Brida, G. Cerullo and R. H. Friend, J. Am. Chem. Soc., 2011, 133, 11830–11833 CrossRef CAS PubMed.
- A. B. Pun, A. Asadpoordarvish, E. Kumarasamy, M. J. Y. Tayebjee, D. Niesner, D. R. McCamey, S. N. Sanders, L. M. Campos and M. Y. Sfeir, Nat. Chem., 2019, 11, 821–828 CrossRef CAS PubMed.
- G. D. Scholes, J. Phys. Chem. A, 2015, 119, 12699–12705 CrossRef CAS PubMed.
- P. M. Zimmerman, Z. Zhang and C. B. Musgrave, Nat. Chem., 2010, 2, 648–652 CrossRef CAS PubMed.
- A. J. Musser and J. Clark, Annu. Rev. Phys. Chem., 2019, 70, 323–351 CrossRef CAS PubMed.
- R. D. Pensack, E. E. Ostroumov, A. J. Tilley, S. Mazza, C. Grieco, K. J. Thorley, J. B. Asbury, D. S. Seferos, J. E. Anthony and G. D. Scholes, J. Phys. Chem. Lett., 2016, 7, 2370–2375 CrossRef CAS PubMed.
- K. Miyata, F. S. Conrad-Burton, F. L. Geyer and X. Y. Zhu, Chem. Rev., 2019, 119, 4261–4292 CrossRef CAS PubMed.
- H. Kim and P. M. Zimmerman, Phys. Chem. Chem. Phys., 2018, 20, 30083–30094 RSC.
- S. N. Sanders, A. B. Pun, K. R. Parenti, E. Kumarasamy, L. M. Yablon, M. Y. Sfeir and L. M. Campos, Chem, 2019, 5, 1988–2005 CAS.
- W. Heisenberg, Zeitschrift für Physik, 1926, 38, 411–426 CrossRef.
- S. L. Bayliss, L. R. Weiss, A. Rao, R. H. Friend, A. D. Chepelianskii and N. C. Greenham, Phys. Rev. B, 2016, 94, 045204 CrossRef.
- T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103–125 RSC.
- V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345–10352 RSC.
- V. Gray, K. Moth-Poulsen, B. Albinsson and M. Abrahamsson, Coord. Chem. Rev., 2018, 362, 54–71 CrossRef CAS.
- D. G. Bossanyi, Y. Sasaki, S. Wang, D. Chekulaev, N. Kimizuka, N. Yanai and J. Clark, JACS Au, 2021, 1, 2188–2201 CrossRef CAS PubMed.
- C. Ye, V. Gray, K. Kushwaha, S. Kumar Singh, P. Erhart and K. Börjesson, Phys. Chem. Chem. Phys., 2020, 22, 1715–1720 RSC.
- C. Ye, V. Gray, J. Mårtensson and K. Börjesson, J. Am. Chem. Soc., 2019, 141, 9578–9584 CrossRef CAS PubMed.
- D. Casanova, Chem. Rev., 2018, 118, 7164–7207 CrossRef CAS PubMed.
- W.-L. Chan, M. Ligges and X. Y. Zhu, Nat. Chem., 2012, 4, 840–845 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS.
- J. C. Johnson, A. J. Nozik and J. Michl, Acc. Chem. Res., 2013, 46, 1290–1299 CrossRef CAS PubMed.
- J. J. Burdett and C. J. Bardeen, Acc. Chem. Res., 2013, 46, 1312–1320 CrossRef CAS PubMed.
- W.-L. Chan, T. C. Berkelbach, M. R. Provorse, N. R. Monahan, J. R. Tritsch, M. S. Hybertsen, D. R. Reichman, J. Gao and X. Y. Zhu, Acc. Chem. Res., 2013, 46, 1321–1329 CrossRef CAS PubMed.
- P. M. Zimmerman, C. B. Musgrave and M. Head-Gordon, Acc. Chem. Res., 2013, 46, 1339–1347 CrossRef CAS PubMed.
- D. Beljonne, H. Yamagata, J. L. Brédas, F. C. Spano and Y. Olivier, Phys. Rev. Lett., 2013, 110, 226402 CrossRef CAS PubMed.
- G. B. Piland, J. J. Burdett, R. J. Dillon and C. J. Bardeen, J. Phys. Chem. Lett., 2014, 5, 2312–2319 CrossRef CAS PubMed.
- S. R. Yost, J. Lee, M. W. B. Wilson, T. Wu, D. P. McMahon, R. R. Parkhurst, N. J. Thompson, D. N. Congreve, A. Rao, K. Johnson, M. Y. Sfeir, M. G. Bawendi, T. M. Swager, R. H. Friend, M. A. Baldo and T. Van Voorhis, Nat. Chem., 2014, 6, 492–497 CrossRef CAS PubMed.
- N. Monahan and X. Y. Zhu, Annu. Rev. Phys. Chem., 2015, 66, 601–618 CrossRef CAS PubMed.
- E. A. Margulies, C. E. Miller, Y. Wu, L. Ma, G. C. Schatz, R. M. Young and M. R. Wasielewski, Nat. Chem., 2016, 8, 1120–1125 CrossRef CAS PubMed.
- N. R. Monahan, D. Sun, H. Tamura, K. W. Williams, B. Xu, Y. Zhong, B. Kumar, C. Nuckolls, A. R. Harutyunyan, G. Chen, H.-L. Dai, D. Beljonne, Y. Rao and X. Y. Zhu, Nat. Chem., 2017, 9, 341–346 CrossRef CAS PubMed.
- R. W. A. Havenith, H. D. de Gier and R. Broer, Mol. Phys., 2012, 110, 2445–2454 CrossRef CAS.
- K. Aryanpour, A. Shukla and S. Mazumdar, J. Phys. Chem. C, 2015, 119, 6966–6979 CrossRef.
- T. C. Berkelbach, M. S. Hybertsen and D. R. Reichman, J. Chem. Phys., 2014, 141, 074705 CrossRef PubMed.
- E. C. Greyson, J. Vura-Weis, J. Michl and M. A. Ratner, J. Phys. Chem. B, 2010, 114, 14168–14177 CrossRef CAS PubMed.
- P. M. Zimmerman, F. Bell, D. Casanova and M. Head-Gordon, J. Am. Chem. Soc., 2011, 133, 19944–19952 CrossRef CAS PubMed.
- T. Zeng, R. Hoffmann and N. Ananth, J. Am. Chem. Soc., 2014, 136, 5755–5764 CrossRef CAS PubMed.
- D. Casanova, J. Chem. Theory Comput., 2014, 10, 324–334 CrossRef CAS PubMed.
- T. C. Berkelbach, M. S. Hybertsen and D. R. Reichman, J. Chem. Phys., 2013, 138, 114103 CrossRef PubMed.
- M. Chen, J. Y. Shin, R. M. Young and M. R. Wasielewski, J. Chem. Phys., 2020, 153, 094302 CrossRef CAS PubMed.
- B. S. Basel, J. Zirzlmeier, C. Hetzer, S. R. Reddy, B. T. Phelan, M. D. Krzyaniak, M. K. Volland, P. B. Coto, R. M. Young, T. Clark, M. Thoss, R. R. Tykwinski, M. R. Wasielewski and D. M. Guldi, Chem, 2018, 4, 1092–1111 CAS.
- R. M. Young and M. R. Wasielewski, Acc. Chem. Res., 2020, 53, 1957–1968 CrossRef CAS PubMed.
- Y. Hong, J. Kim, W. Kim, C. Kaufmann, H. Kim, F. Würthner and D. Kim, J. Am. Chem. Soc., 2020, 142, 7845–7857 CrossRef CAS PubMed.
- E. A. Margulies, J. L. Logsdon, C. E. Miller, L. Ma, E. Simonoff, R. M. Young, G. C. Schatz and M. R. Wasielewski, J. Am. Chem. Soc., 2017, 139, 663–671 CrossRef CAS PubMed.
- S. Lukman, K. Chen, J. M. Hodgkiss, D. H. P. Turban, N. D. M. Hine, S. Dong, J. Wu, N. C. Greenham and A. J. Musser, Nat. Commun., 2016, 7, 13622 CrossRef CAS PubMed.
- A. M. Alvertis, S. Lukman, T. J. H. Hele, E. G. Fuemmeler, J. Feng, J. Wu, N. C. Greenham, A. W. Chin and A. J. Musser, J. Am. Chem. Soc., 2019, 141, 17558–17570 CrossRef CAS PubMed.
- J. N. Schrauben, J. L. Ryerson, J. Michl and J. C. Johnson, J. Am. Chem. Soc., 2014, 136, 7363–7373 CrossRef CAS PubMed.
- S. W. Eaton, L. E. Shoer, S. D. Karlen, S. M. Dyar, E. A. Margulies, B. S. Veldkamp, C. Ramanan, D. A. Hartzler, S. Savikhin, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2013, 135, 14701–14712 CrossRef CAS PubMed.
- Y. J. Bae, G. Kang, C. D. Malliakas, J. N. Nelson, J. Zhou, R. M. Young, Y.-L. Wu, R. P. Van Duyne, G. C. Schatz and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 15140–15144 CrossRef CAS PubMed.
- E. A. Buchanan and J. Michl, J. Am. Chem. Soc., 2017, 139, 15572–15575 CrossRef CAS PubMed.
- P. E. Hartnett, E. A. Margulies, C. M. Mauck, S. A. Miller, Y. Wu, Y.-L. Wu, T. J. Marks and M. R. Wasielewski, J. Phys. Chem. B, 2016, 120, 1357–1366 CrossRef CAS PubMed.
- T. Nagami, S. Ito, T. Kubo and M. Nakano, ACS Omega, 2017, 2, 5095–5103 CrossRef CAS PubMed.
- A. Zaykov, P. Felkel, E. A. Buchanan, M. Jovanovic, R. W. A. Havenith, R. K. Kathir, R. Broer, Z. Havlas and J. Michl, J. Am. Chem. Soc., 2019, 141, 17729–17743 CrossRef CAS PubMed.
- D. Guzmán, I. Papadopoulos, G. Lavarda, P. R. Rami, R. R. Tykwinski, M. S. Rodríguez-Morgade, D. M. Guldi and T. Torres, Angew. Chem., Int. Ed., 2021, 60, 1474–1481 CrossRef PubMed.
- G. He, E. Busby, K. Appavoo, Q. Wu, J. Xia, L. M. Campos and M. Y. Sfeir, J. Chem. Phys., 2020, 153, 244902 CrossRef CAS PubMed.
- C. M. Mauck, Y. J. Bae, M. Chen, N. Powers-Riggs, Y.-L. Wu and M. R. Wasielewski, ChemPhotoChem, 2018, 2, 223–233 CrossRef CAS.
- R. Montero, V. Martínez-Martínez, A. Longarte, N. Epelde-Elezcano, E. Palao, I. Lamas, H. Manzano, A. R. Agarrabeitia, I. López Arbeloa, M. J. Ortiz and I. Garcia-Moreno, J. Phys. Chem. Lett., 2018, 9, 641–646 CrossRef CAS PubMed.
- I. Papadopoulos, M. J. Álvaro-Martins, D. Molina, P. M. McCosker, P. A. Keller, T. Clark, Á. Sastre-Santos and D. M. Guldi, Adv. Energy Mater., 2020, 10, 2001496 CrossRef CAS.
- E. Sundin, R. Ringström, F. Johansson, B. Küçüköz, A. Ekebergh, V. Gray, B. Albinsson, J. Mårtensson and M. Abrahamsson, J. Phys. Chem. C, 2020, 124, 20794–20805 CrossRef CAS.
- L. Xue, X. Song, Y. Feng, S. Cheng, G. Lu and Y. Bu, J. Am. Chem. Soc., 2020, 142, 17469–17479 CrossRef CAS PubMed.
- T. W. Schmidt, J. Chem. Phys., 2019, 151, 054305 CrossRef.
- A. A. Bakulin, S. E. Morgan, T. B. Kehoe, M. W. B. Wilson, A. W. Chin, D. Zigmantas, D. Egorova and A. Rao, Nat. Chem., 2016, 8, 16–23 CrossRef CAS PubMed.
- R. Tempelaar and D. R. Reichman, J. Chem. Phys., 2017, 146, 174703 CrossRef PubMed.
- R. Tempelaar and D. R. Reichman, J. Chem. Phys., 2017, 146, 174704 CrossRef PubMed.
- Y. Fujihashi, L. Chen, A. Ishizaki, J. Wang and Y. Zhao, J. Chem. Phys., 2017, 146, 044101 CrossRef PubMed.
- H.-G. Duan, A. Jha, X. Li, V. Tiwari, H. Ye, K. Nayak Pabitra, X.-L. Zhu, Z. Li, J. Martinez Todd, M. Thorwart and R. J. D. Miller, Sci. Adv., 2020, 6, eabb0052 CrossRef CAS PubMed.
- A. J. Musser, M. Liebel, C. Schnedermann, T. Wende, T. B. Kehoe, A. Rao and P. Kukura, Nat. Phys., 2015, 11, 352–357 Search PubMed.
- H. L. Stern, A. Cheminal, S. R. Yost, K. Broch, S. L. Bayliss, K. Chen, M. Tabachnyk, K. Thorley, N. Greenham, J. M. Hodgkiss, J. Anthony, M. Head-Gordon, A. J. Musser, A. Rao and R. H. Friend, Nat. Chem., 2017, 9, 1205–1212 CrossRef CAS PubMed.
- T. S. Lee, Y. L. Lin, H. Kim, R. D. Pensack, B. P. Rand and G. D. Scholes, J. Phys. Chem. Lett., 2018, 9, 4087–4095 CrossRef CAS PubMed.
- S. Lukman, J. M. Richter, L. Yang, P. Hu, J. Wu, N. C. Greenham and A. J. Musser, J. Am. Chem. Soc., 2017, 139, 18376–18385 CrossRef CAS PubMed.
- C. K. Yong, A. J. Musser, S. L. Bayliss, S. Lukman, H. Tamura, O. Bubnova, R. K. Hallani, A. Meneau, R. Resel, M. Maruyama, S. Hotta, L. M. Herz, D. Beljonne, J. E. Anthony, J. Clark and H. Sirringhaus, Nat. Commun., 2017, 8, 15953 CrossRef CAS PubMed.
- A. B. Kolomeisky, X. Feng and A. I. Krylov, J. Phys. Chem. C, 2014, 118, 5188–5195 CrossRef CAS.
- H. Nagashima, S. Kawaoka, S. Akimoto, T. Tachikawa, Y. Matsui, H. Ikeda and Y. Kobori, J. Phys. Chem. Lett., 2018, 9, 5855–5861 CrossRef CAS PubMed.
- N. V. Korovina, C. H. Chang and J. C. Johnson, Nat. Chem., 2020, 12, 391–398 CrossRef CAS PubMed.
- P. Irkhin and I. Biaggio, Phys. Rev. Lett., 2011, 107, 017402 CrossRef PubMed.
- G. M. Akselrod, P. B. Deotare, N. J. Thompson, J. Lee, W. A. Tisdale, M. A. Baldo, V. M. Menon and V. Bulović, Nat. Commun., 2014, 5, 3646 CrossRef CAS PubMed.
- G. He, L. M. Yablon, K. R. Parenti, K. J. Fallon, L. M. Campos and M. Y. Sfeir, J. Am. Chem. Soc., 2022, 144, 3269–3278 CrossRef CAS PubMed.
- R. P. Groff, P. Avakian and R. E. Merrifield, Phys. Rev. B: Condens. Matter Mater. Phys., 1970, 1, 815–817 CrossRef.
- S. Lukman, A. J. Musser, K. Chen, S. Athanasopoulos, C. K. Yong, Z. Zeng, Q. Ye, C. Chi, J. M. Hodgkiss, J. Wu, R. H. Friend and N. C. Greenham, Adv. Funct. Mater., 2015, 25, 5452–5461 CrossRef CAS.
- X. Tang, R. Pan, X. Zhao, H. Zhu and Z. Xiong, J. Phys. Chem. Lett., 2020, 11, 2804–2811 CrossRef CAS PubMed.
- A. J. Carrod, A. Cravcenco, C. Ye and K. Börjesson, J. Mater. Chem. C, 2022, 10, 4923–4928 RSC.
- S. Hoseinkhani, R. Tubino, F. Meinardi and A. Monguzzi, Phys. Chem. Chem. Phys., 2015, 17, 4020–4024 RSC.
- V. Gray, J. R. Allardice, Z. Zhang and A. Rao, Chem. Phys. Rev., 2021, 2, 031305 CrossRef.
- A. J. Baldacchino, M. I. Collins, M. P. Nielsen, T. W. Schmidt, D. R. McCamey and M. J. Tayebjee, arXiv, 2022, preprint, arXiv:2202.01326.
- O. Cvrčková and M. Ciganek, Polycyclic Aromat. Compd., 2005, 25, 141–156 CrossRef.
- S. Dong, A. Ong and C. Chi, J. Photochem. Photobiol., C, 2019, 38, 27–46 CrossRef CAS.
- A. K. Le, J. A. Bender, D. H. Arias, D. E. Cotton, J. C. Johnson and S. T. Roberts, J. Am. Chem. Soc., 2018, 140, 814–826 CrossRef CAS PubMed.
- P. J. Budden, L. R. Weiss, M. Müller, N. A. Panjwani, S. Dowland, J. R. Allardice, M. Ganschow, J. Freudenberg, J. Behrends, U. H. F. Bunz and R. H. Friend, Nat. Commun., 2021, 12, 1527 CrossRef CAS PubMed.
- P. Bharmoria, N. Yanai and N. Kimizuka, Gels, 2019, 5, 18 CrossRef CAS PubMed.
- P. Bharmoria, S. Hisamitsu, H. Nagatomi, T. Ogawa, M.-A. Morikawa, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2018, 140, 10848–10855 CrossRef CAS PubMed.
- D. Dzebo, K. Moth-Poulsen and B. Albinsson, Photochem. Photobiol. Sci., 2017, 16, 1327–1334 CrossRef CAS PubMed.
- Q. Liu, M. Xu, T. Yang, B. Tian, X. Zhang and F. Li, ACS Appl. Mater. Interfaces, 2018, 10, 9883–9888 CrossRef CAS PubMed.
- A. D. Poletayev, J. Clark, M. W. B. Wilson, A. Rao, Y. Makino, S. Hotta and R. H. Friend, Adv. Mater., 2014, 26, 919–924 CrossRef CAS PubMed.
- D. Rai and R. J. Holmes, J. Mater. Chem. C, 2019, 7, 5695–5701 RSC.
- M. A. Baldo, C. Adachi and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 10967–10977 CrossRef CAS.
- M. Wakasa, T. Yago, Y. Sonoda and R. Katoh, Commun. Chem., 2018, 1, 9 CrossRef.
- R. J. Hudson, D. M. Huang and T. W. Kee, J. Phys. Chem. C, 2020, 124, 23541–23550 CrossRef CAS.
- V. K. Thorsmølle, R. D. Averitt, J. Demsar, D. L. Smith, S. Tretiak, R. L. Martin, X. Chi, B. K. Crone, A. P. Ramirez and A. J. Taylor, Phys. Rev. Lett., 2009, 102, 017401 CrossRef.
- C. A. Parker and C. G. Hatchard, Proc. Chem. Soc., 1962, 386–387, 10.1039/PS9620000373.
- A. Monguzzi, J. Mezyk, F. Scotognella, R. Tubino and F. Meinardi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 195112 CrossRef.
- R. R. Islangulov, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2007, 129, 12652–12653 CrossRef CAS PubMed.
- S. Baluschev, V. Yakutkin, T. Miteva, Y. Avlasevich, S. Chernov, S. Aleshchenkov, G. Nelles, A. Cheprakov, A. Yasuda, K. Müllen and G. Wegner, Angew. Chem., Int. Ed., 2007, 46, 7693–7696 CrossRef CAS PubMed.
- R. H. Clarke and R. M. Hochstrasser, J. Mol. Spectrosc., 1969, 32, 309–319 CrossRef CAS.
- S. Chen, F. Chen, P. Han, C. Ye, S. Huang, L. Xu, X. Wang and Y. Song, RSC Adv., 2019, 9, 36410–36415 RSC.
- T. Morifuji, Y. Takekuma and M. Nagata, ACS Omega, 2019, 4, 11271–11275 CrossRef CAS PubMed.
- Y. Kageshima, S. Tateyama, F. Kishimoto, K. Teshima, K. Domen and H. Nishikiori, Phys. Chem. Chem. Phys., 2021, 23, 5673–5679 RSC.
- K. Chen, M. Hussain, S. S. Razi, Y. Hou, E. A. Yildiz, J. Zhao, H. G. Yaglioglu and M. D. Donato, Inorg. Chem., 2020, 59, 14731–14745 CrossRef CAS PubMed.
- C. E. Elgar, H. Y. Otaif, X. Zhang, J. Zhao, P. N. Horton, S. J. Coles, J. M. Beames and S. J. A. Pope, Chem. – Eur. J., 2021, 27, 3427–3439 CrossRef CAS PubMed.
- P. Xia, E. K. Raulerson, D. Coleman, C. S. Gerke, L. Mangolini, M. L. Tang and S. T. Roberts, Nat. Chem., 2020, 12, 137–144 CrossRef CAS PubMed.
- Z. A. VanOrman, C. R. Conti, G. F. Strouse and L. Nienhaus, Chem. Mater., 2021, 33, 452–458 CrossRef CAS.
- R. P. Groff, R. E. Merrifield, A. Suna and P. Avakian, Phys. Rev. Lett., 1972, 29, 429–431 CrossRef CAS.
- K. Chen, Y. Dong, X. Zhao, M. Imran, G. Tang, J. Zhao and Q. Liu, Front. Chem., 2019, 7, 821 CrossRef CAS PubMed.
- Z. Wang and J. Zhao, Org. Lett., 2017, 19, 4492–4495 CrossRef CAS PubMed.
- N. Kiseleva, M. A. Filatov, J. C. Fischer, M. Kaiser, M. Jakoby, D. Busko, I. A. Howard, B. S. Richards and A. Turshatov, Phys. Chem. Chem. Phys., 2022, 24, 3568–3578 RSC.
- S. E. Braslavsky, Pure Appl. Chem., 2007, 79, 293–465 CrossRef CAS.
- Y. Zhou, F. N. Castellano, T. W. Schmidt and K. Hanson, ACS Energy Lett., 2020, 5, 2322–2326 CrossRef CAS.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CrossRef CAS.
- U. Resch-Genger and K. Rurack, Pure Appl. Chem., 2013, 85, 2005–2013 CrossRef CAS.
- Y. Y. Cheng, B. Fückel, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, J. Phys. Chem. Lett., 2010, 1, 1795–1799 CrossRef CAS.
- A. Haefele, J. Blumhoff, R. S. Khnayzer and F. N. Castellano, J. Phys. Chem. Lett., 2012, 3, 299–303 CrossRef CAS.
- F. Edhborg, A. Olesund and B. Albinsson, Photochem. Photobiol. Sci., 2022, 21, 1143–1158 CrossRef CAS PubMed.
- K. K.-W. Lo, in Photophysics of Organometallics, ed. A. J. Lees, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, pp. 73–114 DOI:10.1007/3418_2009_3.
- A. J. Carrod, F. Graglia, L. Male, C. Le Duff, P. Simpson, M. Elsherif, Z. Ahmed, H. Butt, G.-X. Xu, K. Kam-Wing Lo, P. Bertoncello and Z. Pikramenou, Chem. – Eur. J., 2022, 28, e202103541 CrossRef CAS PubMed.
- A. F. Rausch, H. H. H. Homeier and H. Yersin, in Photophysics of Organometallics, ed. A. J. Lees, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, pp. 193–235 DOI:10.1007/3418_2009_6.
- A. Harriman, J. Chem. Soc., Faraday Trans. 2, 1981, 77, 1281–1291 RSC.
- E. l G. Azenha, A. C. Serra, M. Pineiro, M. M. Pereira, J. Seixas de Melo, L. G. Arnaut, S. J. Formosinho and A. M. d A. Rocha Gonsalves, Chem. Phys., 2002, 280, 177–190 CrossRef CAS.
- R. C. Evans and P. Douglas, ACS Appl. Mater. Interfaces, 2009, 1, 1023–1030 CrossRef CAS PubMed.
- A. Gorski, M. Kijak, E. Zenkevich, V. Knyukshto, A. Starukhin, A. Semeikin, T. Lyubimova, T. Roliński and J. Waluk, J. Phys. Chem. A, 2020, 124, 8144–8158 CrossRef CAS PubMed.
- T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. Lett., 2010, 1, 195–200 CrossRef CAS.
- T. N. Singh-Rachford, A. Nayak, M. L. Muro-Small, S. Goeb, M. J. Therien and F. N. Castellano, J. Am. Chem. Soc., 2010, 132, 14203–14211 CrossRef CAS PubMed.
- F. Deng, J. R. Sommer, M. Myahkostupov, K. S. Schanze and F. N. Castellano, Chem. Commun., 2013, 49, 7406–7408 RSC.
- J. R. Sommer, A. H. Shelton, A. Parthasarathy, I. Ghiviriga, J. R. Reynolds and K. S. Schanze, Chem. Mater., 2011, 23, 5296–5304 CrossRef CAS.
- J. K. Gallaher, K. M. Wright, L. Frazer, R. W. MacQueen, M. J. Crossley, F. N. Castellano and T. W. Schmidt, Energy Environ. Sci., 2021, 14, 5541–5551 RSC.
- J. B. Bilger, C. Kerzig, C. B. Larsen and O. S. Wenger, J. Am. Chem. Soc., 2021, 143, 1651–1663 CrossRef CAS PubMed.
- M. Yang, S. Sheykhi, Y. Zhang, C. Milsmann and F. N. Castellano, Chem. Sci., 2021, 12, 9069–9077 RSC.
- K. Hasan, A. K. Bansal, I. D. W. Samuel, C. Roldán-Carmona, H. J. Bolink and E. Zysman-Colman, Sci. Rep., 2015, 5, 12325 CrossRef CAS PubMed.
- A. F. Henwood and E. Zysman-Colman, Chem. Commun., 2017, 53, 807–826 RSC.
- S. Amemori, Y. Sasaki, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2016, 138, 8702–8705 CrossRef CAS PubMed.
- Y. Sasaki, S. Amemori, H. Kouno, N. Yanai and N. Kimizuka, J. Mater. Chem. C, 2017, 5, 5063–5067 RSC.
- R. Haruki, Y. Sasaki, K. Masutani, N. Yanai and N. Kimizuka, Chem. Commun., 2020, 56, 7017–7020 RSC.
- Y. Wei, M. Zheng, L. Chen, X. Zhou and S. Liu, Dalton Trans., 2019, 48, 11763–11771 RSC.
- M. Wu, D. N. Congreve, M. W. B. Wilson, J. Jean, N. Geva, M. Welborn, T. Van Voorhis, V. Bulović, M. G. Bawendi and M. A. Baldo, Nat. Photonics, 2016, 10, 31–34 CrossRef CAS.
- Z. Huang, D. E. Simpson, M. Mahboub, X. Li and M. L. Tang, Chem. Sci., 2016, 7, 4101–4104 RSC.
- M. Mahboub, Z. Huang and M. L. Tang, Nano Lett., 2016, 16, 7169–7175 CrossRef CAS PubMed.
- Z. Huang, Z. Xu, M. Mahboub, X. Li, J. W. Taylor, W. H. Harman, T. Lian and M. L. Tang, Angew. Chem., Int. Ed., 2017, 56, 16583–16587 CrossRef CAS PubMed.
- Z. Xu, Z. Huang, C. Li, T. Huang, F. A. Evangelista, M. L. Tang and T. Lian, ACS Appl. Mater. Interfaces, 2020, 12, 36558–36567 CrossRef CAS PubMed.
- S. T. Hossain and S. K. Mukherjee, J. Hazard. Mater., 2013, 260, 1073–1082 CrossRef CAS PubMed.
- M. Ayoubi, P. Naserzadeh, M. T. Hashemi, M. Reza Rostami, E. Tamjid, M. M. Tavakoli and A. Simchi, Sci. Rep., 2017, 7, 12896 CrossRef PubMed.
- L. Huang, E. Kakadiaris, T. Vaneckova, K. Huang, M. Vaculovicova and G. Han, Biomaterials, 2019, 201, 77–86 CrossRef CAS PubMed.
- R. Lai, Y. Sang, Y. Zhao and K. Wu, J. Am. Chem. Soc., 2020, 142, 19825–19829 CrossRef CAS PubMed.
- R. J. Ellingson, J. L. Blackburn, P. Yu, G. Rumbles, O. I. Mićić and A. J. Nozik, J. Phys. Chem. B, 2002, 106, 7758–7765 CrossRef CAS.
- E. M. Gholizadeh, S. K. K. Prasad, Z. L. Teh, T. Ishwara, S. Norman, A. J. Petty, J. H. Cole, S. Cheong, R. D. Tilley, J. E. Anthony, S. Huang and T. W. Schmidt, Nat. Photonics, 2020, 14, 585–590 CrossRef CAS.
- B. Fückel, D. A. Roberts, Y. Y. Cheng, R. G. C. R. Clady, R. B. Piper, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, J. Phys. Chem. Lett., 2011, 2, 966–971 CrossRef.
- N. Nishimura, J. R. Allardice, J. Xiao, Q. Gu, V. Gray and A. Rao, Chem. Sci., 2019, 10, 4750–4760 RSC.
- N. Tripathi, M. Ando, T. Akai and K. Kamada, ACS Appl. Nano Mater., 2021, 4, 9680–9688 CrossRef CAS.
- N. Tripathi, M. Ando, T. Akai and K. Kamada, J. Mater. Chem. C, 2022, 10, 4563–4567 RSC.
- D. G. Bossanyi, M. Matthiesen, S. Wang, J. A. Smith, R. C. Kilbride, J. D. Shipp, D. Chekulaev, E. Holland, J. E. Anthony, J. Zaumseil, A. J. Musser and J. Clark, Nat. Chem., 2021, 13, 163–171 CrossRef CAS PubMed.
- G. Mayonado, K. T. Vogt, J. D. B. Van Schenck, L. Zhu, G. Fregoso, J. Anthony, O. Ostroverkhova and M. W. Graham, J. Phys. Chem. C, 2022, 126, 4433–4445 CrossRef CAS.
- Z. Xu, Z. Huang, T. Jin, T. Lian and M. L. Tang, Acc. Chem. Res., 2021, 54, 70–80 CrossRef CAS PubMed.
- B. Pfund, D. M. Steffen, M. R. Schreier, M.-S. Bertrams, C. Ye, K. Börjesson, O. S. Wenger and C. Kerzig, J. Am. Chem. Soc., 2020, 142, 10468–10476 CrossRef CAS PubMed.
- A. Ronchi and A. Monguzzi, J. Appl. Phys., 2021, 129, 050901 CrossRef CAS.
- D. Dzebo, K. Börjesson, V. Gray, K. Moth-Poulsen and B. Albinsson, J. Phys. Chem. C, 2016, 120, 23397–23406 CrossRef CAS.
- C. E. McCusker and F. N. Castellano, Top. Curr. Chem., 2016, 374, 19 CrossRef PubMed.
- S. Raišys, S. Juršėnas, Y. C. Simon, C. Weder and K. Kazlauskas, Chem. Sci., 2018, 9, 6796–6802 RSC.
- C. Healy, L. Hermanspahn and P. E. Kruger, Coord. Chem. Rev., 2021, 432, 213756 CrossRef CAS.
- X. Liu, J. Fei, P. Zhu and J. Li, Chem. – Asian J., 2016, 11, 2700–2704 CrossRef CAS PubMed.
- K. Sripathy, R. W. MacQueen, J. R. Peterson, Y. Y. Cheng, M. Dvořák, D. R. McCamey, N. D. Treat, N. Stingelin and T. W. Schmidt, J. Mater. Chem. C, 2015, 3, 616–622 RSC.
- R. Vadrucci, C. Weder and Y. C. Simon, Mater. Horiz., 2015, 2, 120–124 RSC.
- C. Ye, J. Ma, S. Chen, J. Ge, W. Yang, Q. Zheng, X. Wang, Z. Liang and Y. Zhou, J. Phys. Chem. C, 2017, 121, 20158–20164 CrossRef CAS.
- P. Duan, D. Asthana, T. Nakashima, T. Kawai, N. Yanai and N. Kimizuka, Faraday Discuss., 2017, 196, 305–316 RSC.
- P. Duan, N. Yanai, H. Nagatomi and N. Kimizuka, J. Am. Chem. Soc., 2015, 137, 1887–1894 CrossRef CAS PubMed.
- D. F. Barbosa de Mattos, A. Dreos, M. D. Johnstone, A. Runemark, C. Sauvée, V. Gray, K. Moth-Poulsen, H. Sundén and M. Abrahamsson, J. Chem. Phys., 2020, 153, 214705 CrossRef CAS PubMed.
- S. N. Sanders, T. H. Schloemer, M. K. Gangishetty, D. Anderson, M. Seitz, A. O. Gallegos, R. C. Stokes and D. N. Congreve, Nature, 2022, 604, 474–478 CrossRef CAS PubMed.
- A. M. Oddo, T. Mani and C. V. Kumar, ACS Appl. Mater. Interfaces, 2020, 12, 39293–39303 CrossRef CAS PubMed.
- P. Bharmoria, S. Hisamitsu, Y. Sasaki, T. S. Kang, M.-A. Morikawa, B. Joarder, K. Moth-Poulsen, H. Bildirir, A. Mårtensson, N. Yanai and N. Kimizuka, J. Mater. Chem. C, 2021, 9, 11655–11661 RSC.
- F. Saenz, A. Ronchi, M. Mauri, R. Vadrucci, F. Meinardi, A. Monguzzi and C. Weder, Adv. Funct. Mater., 2021, 31, 2004495 CrossRef CAS.
- T. N. Singh-Rachford, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2009, 131, 12007–12014 CrossRef CAS PubMed.
- A. Turshatov, D. Busko, N. Kiseleva, S. L. Grage, I. A. Howard and B. S. Richards, ACS Appl. Mater. Interfaces, 2017, 9, 8280–8286 CrossRef CAS PubMed.
- A. Monguzzi, M. Mauri, A. Bianchi, M. K. Dibbanti, R. Simonutti and F. Meinardi, J. Phys. Chem. C, 2016, 120, 2609–2614 CrossRef CAS.
- M. J. Bennison, A. R. Collins, B. Zhang and R. C. Evans, Macromolecules, 2021, 54, 5287–5303 CrossRef CAS PubMed.
- R. Vadrucci, A. Monguzzi, F. Saenz, B. D. Wilts, Y. C. Simon and C. Weder, Adv. Mater., 2017, 29, 1702992 CrossRef PubMed.
- A. L. Hagstrom, H.-L. Lee, M.-S. Lee, H.-S. Choe, J. Jung, B.-G. Park, W.-S. Han, J.-S. Ko, J.-H. Kim and J.-H. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 8985–8992 CrossRef CAS PubMed.
- A. Monguzzi, A. Oertel, D. Braga, A. Riedinger, D. K. Kim, P. N. Knüsel, A. Bianchi, M. Mauri, R. Simonutti, D. J. Norris and F. Meinardi, ACS Appl. Mater. Interfaces, 2017, 9, 40180–40186 CrossRef CAS PubMed.
- A. Monguzzi, F. Bianchi, A. Bianchi, M. Mauri, R. Simonutti, R. Ruffo, R. Tubino and F. Meinardi, Adv. Energy Mater., 2013, 3, 680–686 CrossRef CAS.
- D. Choi, S. K. Nam, K. Kim and J. H. Moon, Angew. Chem., Int. Ed., 2019, 58, 6891–6895 CrossRef CAS PubMed.
- S. R. Pristash, K. L. Corp, E. J. Rabe and C. W. Schlenker, ACS Appl. Energy Mater., 2020, 3, 19–28 CrossRef CAS.
- Q. Wang and D. Astruc, Chem. Rev., 2020, 120, 1438–1511 CrossRef CAS PubMed.
- C. Ye, S. Chen, J. Liao, Y. S. Zhang, X. Wang and Y. Song, J. Phys. Chem. C, 2020, 124, 18482–18489 CrossRef CAS.
- I. Roy, S. Goswami, R. M. Young, I. Schlesinger, M. R. Mian, A. E. Enciso, X. Zhang, J. E. Hornick, O. K. Farha, M. R. Wasielewski, J. T. Hupp and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 5053–5059 CrossRef CAS PubMed.
- F. Meinardi, M. Ballabio, N. Yanai, N. Kimizuka, A. Bianchi, M. Mauri, R. Simonutti, A. Ronchi, M. Campione and A. Monguzzi, Nano Lett., 2019, 19, 2169–2177 CrossRef CAS PubMed.
- S. Gharaati, C. Wang, C. Förster, F. Weigert, U. Resch-Genger and K. Heinze, Chem. – Eur. J., 2020, 26, 1003–1007 CrossRef CAS PubMed.
- P. Mahato, A. Monguzzi, N. Yanai, T. Yamada and N. Kimizuka, Nat. Mater., 2015, 14, 924–930 CrossRef CAS PubMed.
- B. Joarder, A. Mallick, Y. Sasaki, M. Kinoshita, R. Haruki, Y. Kawashima, N. Yanai and N. Kimizuka, ChemNanoMat, 2020, 6, 916–919 CrossRef CAS.
- Y. Yang, S. Mallick, F. Izquierdo-Ruiz, C. Schäfer, X. Xing, M. Rahm and K. Börjesson, Small, 2021, 17, 2103152 CrossRef CAS PubMed.
- M. Ratsch, C. Ye, Y. Yang, A. Zhang, A. M. Evans and K. Börjesson, J. Am. Chem. Soc., 2020, 142, 6548–6553 CrossRef CAS PubMed.
- D.-G. Ha, R. Wan, C. A. Kim, T.-A. Lin, L. Yang, T. Van Voorhis, M. A. Baldo and M. Dincă, Nat. Mater., 2022, 21, 1275–1281 CrossRef CAS PubMed.
- M. Hosoyamada, N. Yanai, T. Ogawa and N. Kimizuka, Chem. – Eur. J., 2016, 22, 2060–2067 CrossRef CAS PubMed.
- V. Gray, K. Börjesson, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, J. Phys. Chem. C, 2016, 120, 19018–19026 CrossRef CAS.
- S. Raišys, K. Kazlauskas, S. Juršėnas and Y. C. Simon, ACS Appl. Mater. Interfaces, 2016, 8, 15732–15740 CrossRef PubMed.
- E. Radiunas, M. Dapkevičius, S. Raišys, S. Juršėnas, A. Jozeliūnaitė, T. Javorskis, U. Šinkevičiūtė, E. Orentas and K. Kazlauskas, Phys. Chem. Chem. Phys., 2020, 22, 7392–7403 RSC.
- S. Wieghold, A. S. Bieber, Z. A. VanOrman, A. Rodriguez and L. Nienhaus, J. Phys. Chem. C, 2020, 124, 18132–18140 CrossRef CAS.
- D. G. Bossanyi, Y. Sasaki, S. Wang, D. Chekulaev, N. Kimizuka, N. Yanai and J. Clark, J. Mater. Chem. C, 2022, 10, 4684–4696 RSC.
- K. M. Felter, M. C. Fravventura, E. Koster, R. D. Abellon, T. J. Savenije and F. C. Grozema, ACS Energy Lett., 2020, 5, 124–129 CrossRef CAS PubMed.
- T.-A. Lin, C. F. Perkinson and M. A. Baldo, Adv. Mater., 2020, 32, 1908175 CrossRef CAS PubMed.
- S. Izawa and M. Hiramoto, Nat. Photonics, 2021, 15, 895–900 CrossRef CAS.
- S. Izawa, M. Morimoto, S. Naka and M. Hiramoto, Adv. Opt. Mater., 2022, 10, 2101710 CrossRef CAS.
- S. Izawa, N. Shintaku, M. Kikuchi and M. Hiramoto, Appl. Phys. Lett., 2019, 115, 153301 CrossRef.
- Z. Zhang, J. Yuan, Q. Wei and Y. Zou, Front. Chem., 2018, 6, 414 CrossRef PubMed.
- M. Wu, T.-A. Lin, J. O. Tiepelt, V. Bulović and M. A. Baldo, Nano Lett., 2021, 21, 1011–1016 CrossRef CAS PubMed.
- M. Wu, J. Jean, V. Bulović and M. A. Baldo, Appl. Phys. Lett., 2017, 110, 211101 CrossRef.
- M. Hertzog, M. Wang, J. Mony and K. Börjesson, Chem. Soc. Rev., 2019, 48, 937–961 RSC.
- C. Ye, S. Mallick, M. Hertzog, M. Kowalewski and K. Börjesson, J. Am. Chem. Soc., 2021, 143, 7501–7508 CrossRef CAS PubMed.
- M. Hertzog, B. Munkhbat, D. Baranov, T. Shegai and K. Börjesson, Nano Lett., 2021, 21, 1320–1326 CrossRef CAS PubMed.
- J. Lather, P. Bhatt, A. Thomas, T. W. Ebbesen and J. George, Angew. Chem., Int. Ed., 2019, 58, 10635–10638 CrossRef CAS PubMed.
- K. Nagarajan, A. Thomas and T. W. Ebbesen, J. Am. Chem. Soc., 2021, 143, 16877–16889 CrossRef CAS PubMed.
- S. Singh, W. J. Jones, W. Siebrand, B. P. Stoicheff and W. G. Schneider, J. Chem. Phys., 1965, 42, 330–342 CrossRef CAS.
- C. E. Swenberg and W. T. Stacy, Chem. Phys. Lett., 1968, 2, 327–328 CrossRef CAS.
- D. L. Dexter, J. Lumin., 1979, 18–19, 779–784 CrossRef CAS.
- J. J. Burdett, G. B. Piland and C. J. Bardeen, Chem. Phys. Lett., 2013, 585, 1–10 CrossRef CAS.
- A. Rao and R. H. Friend, Nat. Rev. Mater., 2017, 2, 17063 CrossRef CAS.
- C. Hetzer, D. M. Guldi and R. R. Tykwinski, Chem. – Eur. J., 2018, 24, 8245–8257 CrossRef CAS PubMed.
- T. Ullrich, D. Munz and D. M. Guldi, Chem. Soc. Rev., 2021, 50, 3485–3518 RSC.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. T. Trinh, B. Choi, J. Xia, E. J. Taffet, J. Z. Low, J. R. Miller, X. Roy, X. Y. Zhu, M. L. Steigerwald, M. Y. Sfeir and L. M. Campos, J. Am. Chem. Soc., 2015, 137, 8965–8972 CrossRef CAS PubMed.
- J. C. Johnson, A. J. Nozik and J. Michl, J. Am. Chem. Soc., 2010, 132, 16302–16303 CrossRef CAS PubMed.
- S. Masoomi-Godarzi, C. R. Hall, B. Zhang, M. A. Gregory, J. M. White, W. W. H. Wong, K. P. Ghiggino, T. A. Smith and D. J. Jones, J. Phys. Chem. C, 2020, 124, 11574–11585 CrossRef CAS.
- T. Sakuma, H. Sakai, Y. Araki, T. Mori, T. Wada, N. V. Tkachenko and T. Hasobe, J. Phys. Chem. A, 2016, 120, 1867–1875 CrossRef CAS PubMed.
- H. L. Stern, A. J. Musser, S. Gelinas, P. Parkinson, L. M. Herz, M. J. Bruzek, J. Anthony, R. H. Friend and B. J. Walker, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7656 CrossRef CAS PubMed.
- B. J. Walker, A. J. Musser, D. Beljonne and R. H. Friend, Nat. Chem., 2013, 5, 1019–1024 CrossRef CAS PubMed.
- Y. Jue Bae, M. D. Krzyaniak, M. B. Majewski, M. Desroches, J.-F. Morin, Y.-L. Wu and M. R. Wasielewski, ChemPlusChem, 2019, 84, 1432–1438 CrossRef PubMed.
- N. Congreve Daniel, J. Lee, J. Thompson Nicholas, E. Hontz, R. Yost Shane, D. Reusswig Philip, E. Bahlke Matthias, S. Reineke, T. Van Voorhis and A. Baldo Marc, Science, 2013, 340, 334–337 CrossRef CAS PubMed.
- J. Lee, P. Jadhav and M. A. Baldo, Appl. Phys. Lett., 2009, 95, 033301 CrossRef.
- J. Allardice, V. Gray, S. Dowland, D. T. Toolan, M. P. Weir, J. Xiao, Z. Zhang, J. F. Winkel, A. J. Petty II and J. Anthony, arXiv, 2020, preprint, arXiv:2009.05764.
- J. R. Allardice, A. Thampi, S. Dowland, J. Xiao, V. Gray, Z. Zhang, P. Budden, A. J. Petty, N. J. L. K. Davis, N. C. Greenham, J. E. Anthony and A. Rao, J. Am. Chem. Soc., 2019, 141, 12907–12915 CrossRef CAS PubMed.
- V. Gray, J. R. Allardice, Z. Zhang, S. Dowland, J. Xiao, A. J. Petty, J. E. Anthony, N. C. Greenham and A. Rao, ACS Nano, 2020, 14, 4224–4234 CrossRef CAS PubMed.
- V. Gray, Z. Zhang, S. Dowland, J. R. Allardice, A. M. Alvertis, J. Xiao, N. C. Greenham, J. E. Anthony and A. Rao, J. Phys. Chem. Lett., 2020, 11, 7239–7244 CrossRef CAS PubMed.
- N. J. Thompson, M. W. B. Wilson, D. N. Congreve, P. R. Brown, J. M. Scherer, T. S. Bischof, M. Wu, N. Geva, M. Welborn, T. V. Voorhis, V. Bulović, M. G. Bawendi and M. A. Baldo, Nat. Mater., 2014, 13, 1039–1043 CrossRef CAS PubMed.
- K. J. Fallon, P. Budden, E. Salvadori, A. M. Ganose, C. N. Savory, L. Eyre, S. Dowland, Q. Ai, S. Goodlett, C. Risko, D. O. Scanlon, C. W. M. Kay, A. Rao, R. H. Friend, A. J. Musser and H. Bronstein, J. Am. Chem. Soc., 2019, 141, 13867–13876 CrossRef CAS PubMed.
- W. Zeng, O. El Bakouri, D. W. Szczepanik, H. Bronstein and H. Ottosson, Chem. Sci., 2021, 12, 6159–6171 RSC.
- I. Paci, J. C. Johnson, X. Chen, G. Rana, D. Popović, D. E. David, A. J. Nozik, M. A. Ratner and J. Michl, J. Am. Chem. Soc., 2006, 128, 16546–16553 CrossRef CAS PubMed.
- S. Ito, T. Minami and M. Nakano, J. Phys. Chem. C, 2012, 116, 19729–19736 CrossRef CAS.
- T. Minami and M. Nakano, J. Phys. Chem. Lett., 2012, 3, 145–150 CrossRef CAS.
- T. Nagami, K. Okada, H. Miyamoto, W. Yoshida, T. Tonami and M. Nakano, J. Phys. Chem. C, 2020, 124, 11800–11809 CrossRef CAS.
- M. Nakano, The Chemical Record, 2017, 17, 27–62 CrossRef CAS PubMed.
- A. Akdag, Z. Havlas and J. Michl, J. Am. Chem. Soc., 2012, 134, 14624–14631 CrossRef CAS PubMed.
- J. Wen, Z. Havlas and J. Michl, J. Am. Chem. Soc., 2015, 137, 165–172 CrossRef CAS PubMed.
- S. V. Bhosale, M. Al Kobaisi, R. W. Jadhav, P. P. Morajkar, L. A. Jones and S. George, Chem. Soc. Rev., 2021, 50, 9845–9998 RSC.
- A. Nowak-Król, K. Shoyama, M. Stolte and F. Würthner, Chem. Commun., 2018, 54, 13763–13772 RSC.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed.
- G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118, 3447–3507 CrossRef CAS PubMed.
- Y. V. Aulin, K. M. Felter, D. D. Günbas, R. K. Dubey, W. F. Jager and F. C. Grozema, ChemPlusChem, 2018, 83, 230–238 CrossRef CAS PubMed.
- F. S. Conrad-Burton, T. Liu, F. Geyer, R. Costantini, A. P. Schlaus, M. S. Spencer, J. Wang, R. H. Sánchez, B. Zhang, Q. Xu, M. L. Steigerwald, S. Xiao, H. Li, C. P. Nuckolls and X. Zhu, J. Am. Chem. Soc., 2019, 141, 13143–13147 CrossRef CAS PubMed.
- S. W. Eaton, S. A. Miller, E. A. Margulies, L. E. Shoer, R. D. Schaller and M. R. Wasielewski, J. Phys. Chem. A, 2015, 119, 4151–4161 CrossRef CAS PubMed.
- M. H. Farag and A. I. Krylov, J. Phys. Chem. C, 2018, 122, 25753–25763 CrossRef CAS.
- A. K. Le, J. A. Bender and S. T. Roberts, J. Phys. Chem. Lett., 2016, 7, 4922–4928 CrossRef CAS PubMed.
- L. Ma, K. J. Tan, H. Jiang, C. Kloc, M.-E. Michel-Beyerle and G. G. Gurzadyan, J. Phys. Chem. A, 2014, 118, 838–843 CrossRef CAS PubMed.
- K. Nagarajan, A. R. Mallia, V. S. Reddy and M. Hariharan, J. Phys. Chem. C, 2016, 120, 8443–8450 CrossRef CAS.
- C. M. Mauck, K. E. Brown, N. E. Horwitz and M. R. Wasielewski, J. Phys. Chem. A, 2015, 119, 5587–5596 CrossRef CAS PubMed.
- C. Schierl, A. Niazov-Elkan, L. J. W. Shimon, Y. Feldman, B. Rybtchinski and D. M. Guldi, Nanoscale, 2018, 10, 20147–20154 RSC.
- M. Chen, Y. J. Bae, C. M. Mauck, A. Mandal, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 9184–9192 CrossRef CAS PubMed.
- W. Ni, G. G. Gurzadyan, J. Zhao, Y. Che, X. Li and L. Sun, J. Phys. Chem. Lett., 2019, 10, 2428–2433 CrossRef CAS PubMed.
- A. B. Pun, L. M. Campos and D. N. Congreve, J. Am. Chem. Soc., 2019, 141, 3777–3781 CrossRef CAS PubMed.
- M. Chakali, H. Mandal, M. Venkatesan, B. Dyaga, V. J. Rao and P. R. Bangal, J. Photochem. Photobiol., A, 2021, 406, 113017 CrossRef CAS.
- A. M. Levine, C. Schierl, B. S. Basel, M. Ahmed, B. A. Camargo, D. M. Guldi and A. B. Braunschweig, J. Phys. Chem. C, 2019, 123, 1587–1595 CrossRef CAS.
- S. Masoomi-Godarzi, M. Liu, Y. Tachibana, V. D. Mitchell, L. Goerigk, K. P. Ghiggino, T. A. Smith and D. J. Jones, Adv. Energy Mater., 2019, 9, 1901069 CrossRef.
- C. M. Mauck, P. E. Hartnett, E. A. Margulies, L. Ma, C. E. Miller, G. C. Schatz, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2016, 138, 11749–11761 CrossRef CAS PubMed.
- C. M. Mauck, P. E. Hartnett, Y.-L. Wu, C. E. Miller, T. J. Marks and M. R. Wasielewski, Chem. Mater., 2017, 29, 6810–6817 CrossRef CAS.
- E. G. Fuemmeler, S. N. Sanders, A. B. Pun, E. Kumarasamy, T. Zeng, K. Miyata, M. L. Steigerwald, X. Y. Zhu, M. Y. Sfeir, L. M. Campos and N. Ananth, ACS Cent. Sci., 2016, 2, 316–324 CrossRef CAS PubMed.
- E. Kumarasamy, S. N. Sanders, M. J. Y. Tayebjee, A. Asadpoordarvish, T. J. H. Hele, E. G. Fuemmeler, A. B. Pun, L. M. Yablon, J. Z. Low, D. W. Paley, J. C. Dean, B. Choi, G. D. Scholes, M. L. Steigerwald, N. Ananth, D. R. McCamey, M. Y. Sfeir and L. M. Campos, J. Am. Chem. Soc., 2017, 139, 12488–12494 CrossRef CAS PubMed.
- A. M. Müller, Y. S. Avlasevich, W. W. Schoeller, K. Müllen and C. J. Bardeen, J. Am. Chem. Soc., 2007, 129, 14240–14250 CrossRef PubMed.
- K. R. Parenti, G. He, S. N. Sanders, A. B. Pun, E. Kumarasamy, M. Y. Sfeir and L. M. Campos, J. Phys. Chem. A, 2020, 124, 9392–9399 CrossRef CAS PubMed.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, K. Appavoo, M. L. Steigerwald, L. M. Campos and M. Y. Sfeir, J. Am. Chem. Soc., 2016, 138, 7289–7297 CrossRef CAS PubMed.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. L. Steigerwald, M. Y. Sfeir and L. M. Campos, Angew. Chem., Int. Ed., 2016, 55, 3373–3377 CrossRef CAS PubMed.
- M. J. Y. Tayebjee, S. N. Sanders, E. Kumarasamy, L. M. Campos, M. Y. Sfeir and D. R. McCamey, Nat. Phys., 2017, 13, 182–188 Search PubMed.
- M. T. Trinh, A. Pinkard, B. Pun Andrew, N. Sanders Samuel, E. Kumarasamy, Y. Sfeir Matthew, M. Campos Luis, X. Roy and X. Y. Zhu, Sci. Adv., 2017, 3, e1700241 Search PubMed.
- J. D. Cook, T. J. Carey, D. H. Arias, J. C. Johnson and N. H. Damrauer, J. Phys. Chem. A, 2017, 121, 9229–9242 CrossRef CAS PubMed.
- S. Nakamura, H. Sakai, H. Nagashima, Y. Kobori, N. V. Tkachenko and T. Hasobe, ACS Energy Lett., 2019, 4, 26–31 CrossRef CAS.
- Z. Wang, H. Liu, X. Xie, C. Zhang, R. Wang, L. Chen, Y. Xu, H. Ma, W. Fang, Y. Yao, H. Sang, X. Wang, X. Li and M. Xiao, Nat. Chem., 2021, 13, 559–567 CrossRef CAS PubMed.
- L. M. Yablon, S. N. Sanders, H. Li, K. R. Parenti, E. Kumarasamy, K. J. Fallon, M. J. A. Hore, A. Cacciuto, M. Y. Sfeir and L. M. Campos, J. Am. Chem. Soc., 2019, 141, 9564–9569 CrossRef CAS PubMed.
- A. J. Musser, M. Al-Hashimi, M. Maiuri, D. Brida, M. Heeney, G. Cerullo, R. H. Friend and J. Clark, J. Am. Chem. Soc., 2013, 135, 12747–12754 CrossRef CAS PubMed.
- E. Busby, J. Xia, Q. Wu, J. Z. Low, R. Song, J. R. Miller, X. Y. Zhu, L. M. Campos and M. Y. Sfeir, Nat. Mater., 2015, 14, 426–433 CrossRef CAS PubMed.
- J. Hu, K. Xu, L. Shen, Q. Wu, G. He, J.-Y. Wang, J. Pei, J. Xia and M. Y. Sfeir, Nat. Commun., 2018, 9, 2999 CrossRef PubMed.
- O. El Bakouri, J. R. Smith and H. Ottosson, J. Am. Chem. Soc., 2020, 142, 5602–5617 CrossRef CAS PubMed.
- L. Wang, L. Lin, J. Yang, Y. Wu, H. Wang, J. Zhu, J. Yao and H. Fu, J. Am. Chem. Soc., 2020, 142, 10235–10239 CrossRef CAS PubMed.
- D. Padula, Ö. H. Omar, T. Nematiaram and A. Troisi, Energy Environ. Sci., 2019, 12, 2412–2416 RSC.
- B. S. Richards, D. Hudry, D. Busko, A. Turshatov and I. A. Howard, Chem. Rev., 2021, 121, 9165–9195 CrossRef CAS PubMed.
- M. Barawi, F. Fresno, R. Pérez-Ruiz and V. A. de la Peña O’Shea, ACS Appl. Energy Mater., 2019, 2, 207–211 CrossRef CAS.
- H.-i Kim, S. Weon, H. Kang, A. L. Hagstrom, O. S. Kwon, Y.-S. Lee, W. Choi and J.-H. Kim, Environ. Sci. Technol., 2016, 50, 11184–11192 CrossRef CAS PubMed.
- T. Yu, Y. Liu, Y. Zeng, J. Chen, G. Yang and Y. Li, Chem. – Eur. J., 2019, 25, 16270–16276 CrossRef CAS PubMed.
- S. M. Borisov, C. Larndorfer and I. Klimant, Adv. Funct. Mater., 2012, 22, 4360–4368 CrossRef CAS.
- N. A. Kukhta, T. Matulaitis, D. Volyniuk, K. Ivaniuk, P. Turyk, P. Stakhira, J. V. Grazulevicius and A. P. Monkman, J. Phys. Chem. Lett., 2017, 8, 6199–6205 CrossRef CAS PubMed.
- D. Y. Kondakov, Philos. Trans. R. Soc., A, 2015, 373, 20140321 CrossRef PubMed.
- J. Yao, S. Xiao, S. Zhang, Q. Sun, Y. Dai, X. Qiao, D. Yang, J. Chen and D. Ma, J. Mater. Chem. C, 2020, 8, 8077–8084 RSC.
- L. Xing, Z.-L. Zhu, J. He, Z. Qiu, Z. Yang, D. Lin, W.-C. Chen, Q. Yang, S. Ji, Y. Huo and C.-S. Lee, Chem. Eng. J., 2020, 421, 127748 CrossRef.
- T. Dilbeck, S. P. Hill and K. Hanson, J. Mater. Chem. A, 2017, 5, 11652–11660 RSC.
- C. Li, C. Koenigsmann, F. Deng, A. Hagstrom, C. A. Schmuttenmaer and J.-H. Kim, ACS Photonics, 2016, 3, 784–790 CrossRef CAS.
- Y. Y. Cheng, A. Nattestad, T. F. Schulze, R. W. MacQueen, B. Fückel, K. Lips, G. G. Wallace, T. Khoury, M. J. Crossley and T. W. Schmidt, Chem. Sci., 2016, 7, 559–568 RSC.
- A. Nattestad, Y. Y. Cheng, R. W. MacQueen, T. F. Schulze, F. W. Thompson, A. J. Mozer, B. Fückel, T. Khoury, M. J. Crossley, K. Lips, G. G. Wallace and T. W. Schmidt, J. Phys. Chem. Lett., 2013, 4, 2073–2078 CrossRef CAS PubMed.
- J. de Wild, A. Meijerink, J. K. Rath, W. G. J. H. M. van Sark and R. E. I. Schropp, Energy Environ. Sci., 2011, 4, 4835–4848 RSC.
- A. Monguzzi, D. Braga, M. Gandini, V. C. Holmberg, D. K. Kim, A. Sahu, D. J. Norris and F. Meinardi, Nano Lett., 2014, 14, 6644–6650 CrossRef CAS PubMed.
- T. Dilbeck and K. Hanson, J. Phys. Chem. Lett., 2018, 9, 5810–5821 CrossRef CAS PubMed.
- A. J. Robb, E. S. Knorr, N. Watson and K. Hanson, J. Photochem. Photobiol., A, 2020, 390, 112291 CrossRef CAS.
- Y. L. Lin, M. Koch, A. N. Brigeman, D. M. E. Freeman, L. Zhao, H. Bronstein, N. C. Giebink, G. D. Scholes and B. P. Rand, Energy Environ. Sci., 2017, 10, 1465–1475 RSC.
- Y. Y. Cheng, B. Fückel, R. W. MacQueen, T. Khoury, R. G. C. R. Clady, T. F. Schulze, N. J. Ekins-Daukes, M. J. Crossley, B. Stannowski, K. Lips and T. W. Schmidt, Energy Environ. Sci., 2012, 5, 6953–6959 RSC.
- W. M. Rowan, F. S. Tim, K. Tony, C. Yuen Yap, S. Bernd, L. Klaus, J. C. Maxwel and S. Timothy, 2013.
- T. F. Schulze, Y. Y. Cheng, B. Fückel, R. W. MacQueen, A. Danos, N. J. L. K. Davis, M. J. Y. Tayebjee, T. Khoury, R. G. C. R. Clady, N. J. Ekins-Daukes, M. J. Crossley, B. Stannowski, K. Lips and T. W. Schmidt, Aust. J. Chem., 2012, 65, 480–485 CrossRef CAS.
- T. F. Schulze, J. Czolk, Y.-Y. Cheng, B. Fückel, R. W. MacQueen, T. Khoury, M. J. Crossley, B. Stannowski, K. Lips, U. Lemmer, A. Colsmann and T. W. Schmidt, J. Phys. Chem. C, 2012, 116, 22794–22801 CrossRef CAS.
- C. Simpson, T. M. Clarke, R. W. MacQueen, Y. Y. Cheng, A. J. Trevitt, A. J. Mozer, P. Wagner, T. W. Schmidt and A. Nattestad, Phys. Chem. Chem. Phys., 2015, 17, 24826–24830 RSC.
- F. S. Tim, C. Yuen Yap, H. K. Tony, C. Maxwell, S. Bernd, L. Klaus and W. S. Timothy, J. Photonics Energy, 2013, 3, 1–14 Search PubMed.
- J. Alves, J. Feng, L. Nienhaus and T. W. Schmidt, J. Mater. Chem. C, 2022, 10, 7783–7798 RSC.
- L. Frazer, J. K. Gallaher and T. W. Schmidt, ACS Energy Lett., 2017, 2, 1346–1354 CrossRef CAS.
- Y. Zhou, C. Ruchlin, A. J. Robb and K. Hanson, ACS Energy Lett., 2019, 4, 1458–1463 CrossRef CAS.
- D. Beery, J. P. Wheeler, A. Arcidiacono and K. Hanson, ACS Appl. Energy Mater., 2020, 3, 29–37 CrossRef CAS.
- S. Wieghold, A. S. Bieber, J. Lackner, K. Nienhaus, G. U. Nienhaus and L. Nienhaus, ChemPhotoChem, 2020, 4, 704–712 CAS.
- S. Wieghold, A. S. Bieber, Z. A. VanOrman, L. Daley, M. Leger, J.-P. Correa-Baena and L. Nienhaus, Matter, 2019, 1, 705–719 CrossRef CAS.
- L. Nienhaus, J.-P. Correa-Baena, S. Wieghold, M. Einzinger, T.-A. Lin, K. E. Shulenberger, N. D. Klein, M. Wu, V. Bulović, T. Buonassisi, M. A. Baldo and M. G. Bawendi, ACS Energy Lett., 2019, 4, 888–895 CrossRef CAS.
- M. Kinoshita, Y. Sasaki, S. Amemori, N. Harada, Z. Hu, Z. Liu, L. K. Ono, Y. Qi, N. Yanai and N. Kimizuka, ChemPhotoChem, 2020, 4, 5271–5278 CrossRef CAS.
- W. Sheng, J. Yang, X. Li, G. Liu, Z. Lin, J. Long, S. Xiao, L. Tan and Y. Chen, Energy Environ. Sci., 2021, 14, 3532–3541 RSC.
- L. Nienhaus and T. W. Schmidt, Energy Environ. Sci., 2021, 14, 6050–6052 RSC.
- W. Sheng, J. Yang, X. Li, S. Xiao, L. Tan and Y. Chen, Energy Environ. Sci., 2021, 14, 6053–6054 RSC.
- M. Einzinger, T. Wu, J. F. Kompalla, H. L. Smith, C. F. Perkinson, L. Nienhaus, S. Wieghold, D. N. Congreve, A. Kahn, M. G. Bawendi and M. A. Baldo, Nature, 2019, 571, 90–94 CrossRef CAS PubMed.
- J. N. Schrauben, Y. Zhao, C. Mercado, P. I. Dron, J. L. Ryerson, J. Michl, K. Zhu and J. C. Johnson, ACS Appl. Mater. Interfaces, 2015, 7, 2286–2293 CrossRef CAS PubMed.
- T. Banerjee, S. P. Hill, M. A. Hermosilla-Palacios, B. D. Piercy, J. Haney, B. Casale, A. E. DePrince, M. D. Losego, V. D. Kleiman and K. Hanson, J. Phys. Chem. C, 2018, 122, 28478–28490 CrossRef CAS.
- N. A. Pace, D. H. Arias, D. B. Granger, S. Christensen, J. E. Anthony and J. C. Johnson, Chem. Sci., 2018, 9, 3004–3013 RSC.
- A. Kunzmann, M. Gruber, R. Casillas, J. Zirzlmeier, M. Stanzel, W. Peukert, R. R. Tykwinski and D. M. Guldi, Angew. Chem., Int. Ed., 2018, 57, 10742–10747 CrossRef CAS PubMed.
- M. Tabachnyk, B. Ehrler, S. Bayliss, R. H. Friend and N. C. Greenham, Appl. Phys. Lett., 2013, 103, 153302 CrossRef.
- N. J. Thompson, D. N. Congreve, D. Goldberg, V. M. Menon and M. A. Baldo, Appl. Phys. Lett., 2013, 103, 263302 CrossRef.
- T. C. Wu, N. J. Thompson, D. N. Congreve, E. Hontz, S. R. Yost, T. Van Voorhis and M. A. Baldo, Appl. Phys. Lett., 2014, 104, 193901 CrossRef.
- A. V. Akimov and O. V. Prezhdo, J. Am. Chem. Soc., 2014, 136, 1599–1608 CrossRef CAS PubMed.
- W.-L. Chan, J. R. Tritsch and X. Y. Zhu, J. Am. Chem. Soc., 2012, 134, 18295–18302 CrossRef CAS PubMed.
- P. J. Jadhav, P. R. Brown, N. Thompson, B. Wunsch, A. Mohanty, S. R. Yost, E. Hontz, T. Van Voorhis, M. G. Bawendi, V. Bulović and M. A. Baldo, Adv. Mater., 2012, 24, 6169–6174 CrossRef CAS PubMed.
- S. Kawata, Y.-J. Pu, A. Saito, Y. Kurashige, T. Beppu, H. Katagiri, M. Hada and J. Kido, Adv. Mater., 2016, 28, 1585–1590 CrossRef CAS PubMed.
- Y. L. Lin, M. A. Fusella, O. V. Kozlov, X. Lin, A. Kahn, M. S. Pshenichnikov and B. P. Rand, Adv. Funct. Mater., 2016, 26, 6489–6494 CrossRef CAS.
- A. N. Stuart, P. C. Tapping, E. Schrefl, D. M. Huang and T. W. Kee, J. Phys. Chem. C, 2019, 123, 5813–5825 CrossRef CAS.
- N. J. Thompson, E. Hontz, D. N. Congreve, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Adv. Mater., 2014, 26, 1366–1371 CrossRef CAS PubMed.
- J. R. Tritsch, W.-L. Chan, X. Wu, N. R. Monahan and X. Y. Zhu, Nat. Commun., 2013, 4, 2679 CrossRef PubMed.
- G. Bernardo, T. Lopes, D. G. Lidzey and A. Mendes, Adv. Energy Mater., 2021, 11, 2100342 CrossRef CAS.
- A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Chem. Soc. Rev., 2019, 48, 1596–1625 RSC.
- W.-L. Chan, M. Ligges, A. Jailaubekov, L. Kaake, L. Miaja-Avila and X. Y. Zhu, Science, 2011, 334, 1541–1545 CrossRef CAS PubMed.
- T. Zhang, D. Rai and R. J. Holmes, J. Phys. Chem. Lett., 2021, 12, 966–972 CrossRef CAS PubMed.
- P. J. Jadhav, A. Mohanty, J. Sussman, J. Lee and M. A. Baldo, Nano Lett., 2011, 11, 1495–1498 CrossRef CAS PubMed.
- J. M. Lee, M. H. Futscher, L. M. Pazos-Outón and B. Ehrler, Prog. Photovolt. Res. Appl., 2017, 25, 936–941 CrossRef.
- L. M. Pazos-Outón, J. M. Lee, M. H. Futscher, A. Kirch, M. Tabachnyk, R. H. Friend and B. Ehrler, ACS Energy Lett., 2017, 2, 476–480 CrossRef PubMed.
- B. Ehrler, K. P. Musselman, M. L. Böhm, R. H. Friend and N. C. Greenham, Appl. Phys. Lett., 2012, 101, 153507 CrossRef.
- B. Ehrler, M. W. B. Wilson, A. Rao, R. H. Friend and N. C. Greenham, Nano Lett., 2012, 12, 1053–1057 CrossRef CAS PubMed.
- L. Yang, M. Tabachnyk, S. L. Bayliss, M. L. Böhm, K. Broch, N. C. Greenham, R. H. Friend and B. Ehrler, Nano Lett., 2015, 15, 354–358 CrossRef CAS PubMed.
- D. Guo, L. Ma, Z. Zhou, D. Lin, C. Wang, X. Zhao, F. Zhang, J. Zhang and Z. Nie, J. Mater. Chem. A, 2020, 8, 5572–5579 RSC.
- S. Lee, D. Hwang, S. I. Jung and D. Kim, J. Phys. Chem. Lett., 2017, 8, 884–888 CrossRef CAS PubMed.
- T. Hayashi, T. G. Castner and R. W. Boyd, Chem. Phys. Lett., 1983, 94, 461–466 CrossRef CAS.
- G. B. Piland, J. J. Burdett, T.-Y. Hung, P.-H. Chen, C.-F. Lin, T.-L. Chiu, J.-H. Lee and C. J. Bardeen, Chem. Phys. Lett., 2014, 601, 33–38 CrossRef CAS.
- B. Daiber, S. P. Pujari, S. Verboom, S. L. Luxembourg, S. W. Tabernig, M. H. Futscher, J. Lee, H. Zuilhof and B. Ehrler, J. Chem. Phys., 2020, 152, 114201 CrossRef CAS PubMed.
- Y. Wan, Z. Guo, T. Zhu, S. Yan, J. Johnson and L. Huang, Nat. Chem., 2015, 7, 785–792 CrossRef CAS PubMed.
- Y. Wan, G. P. Wiederrecht, R. D. Schaller, J. C. Johnson and L. Huang, J. Phys. Chem. Lett., 2018, 9, 6731–6738 CrossRef CAS PubMed.
- T. Zhu and L. Huang, J. Phys. Chem. Lett., 2018, 9, 6502–6510 CrossRef CAS PubMed.
- Y. Jiang, M. P. Nielsen, A. J. Baldacchino, M. A. Green, D. R. McCamey, M. J. Y. Tayebjee, T. W. Schmidt and N. J. Ekins-Daukes, Prog. Photovolt. Res. Appl., 2021, 29, 899–906 CrossRef CAS.
- M. H. Futscher, A. Rao and B. Ehrler, ACS Energy Lett., 2018, 3, 2587–2592 CrossRef CAS PubMed.
- D. Casanova, J. Chem. Theory Comput., 2015, 11, 2642–2650 CrossRef CAS PubMed.
- G. Klein and R. Voltz, Int. J. Radiat. Phys. Chem., 1975, 7, 155–174 CrossRef CAS.
- M. Tabachnyk, B. Ehrler, S. Gélinas, M. L. Böhm, B. J. Walker, K. P. Musselman, N. C. Greenham, R. H. Friend and A. Rao, Nat. Mater., 2014, 13, 1033–1038 CrossRef CAS PubMed.
- R. Nagata, H. Nakanotani, W. J. Potscavage Jr and C. Adachi, Adv. Mater., 2018, 30, 1801484 CrossRef PubMed.
- T. J. Milstein, D. M. Kroupa and D. R. Gamelin, Nano Lett., 2018, 18, 3792–3799 CrossRef CAS PubMed.
- K. Kim, S. K. Nam and J. H. Moon, ACS Appl. Energy Mater., 2020, 3, 5277–5284 CrossRef CAS.
- J.-E. Bernice Mae Yu, W. Haibin, E. E. Achilles, K. Takaya, S. Hiroshi, A. Nazmul and O. Yoshitaka, J. Photonics Energy, 2020, 10, 1–20 Search PubMed.
- J. Roncali, Adv. Energy Mater., 2020, 10, 2001907 CrossRef CAS.
- T. K. Baikie, A. Ashoka, A. Rao and N. C. Greenham, arXiv, 2022, preprint, arXiv:2203.06736.
- S.-J. Ha, J.-H. Kang, D. H. Choi, S. K. Nam, E. Reichmanis and J. H. Moon, ACS Photonics, 2018, 5, 3621–3627 CrossRef CAS.
- C. Yang and R. R. Lunt, ACS Photonics, 2021, 8, 678–681 CrossRef CAS.
- D. C. Jordan and S. R. Kurtz, Prog. Photovolt. Res. Appl., 2013, 21, 12–29 CrossRef.
- J. Qian, M. Ernst, N. Wu and A. Blakers, Sustainable Energy Fuels, 2019, 3, 1439–1447 RSC.
- W. Zeng, D. W. Szczepanik and H. Bronstein, J. Phys. Org. Chem., 2022, e4441 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |