
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances and perspectives in aqueous potassium-ion batteries
Xiao
Zhang†
ab,
Ting
Xiong†
b,
Bing
He
b,
Shihao
Feng
a,
Xuanpeng
Wang
 *c,
Lei
Wei
*b and
Liqiang
Mai
*a
*c,
Lei
Wei
*b and
Liqiang
Mai
*a
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, P. R. China. E-mail: mlq518@whut.edu.cn
bSchool of Electrical and Electronic Engineering, Nanyang Technological University, Singapore 639798, Singapore. E-mail: wei.lei@ntu.edu.sg
cDepartment of Physical Science & Technology, School of Science, Wuhan University of Technology, Wuhan 430070, P. R. China. E-mail: wxp122525691@whut.edu.cn
First published on 5th August 2022
Abstract
Aqueous potassium-ion batteries (AKIBs), utilizing fast diffusion kinetics of K+ and abundant electrode resources, are an emerging technology offering high power density and low cost. Many efforts have been made by far to enhance the electrochemical performances of AKIBs, and some encouraging milestones have been achieved. To provide a deep understanding of the progress, challenges, and opportunities of the emerging AKIBs, the recent advances in both cathode and anode materials, and electrolytes of the AKIB systems are comprehensively summarized and discussed. Additionally, the research efforts on the optimization of electrode material properties, the revealing of the reaction mechanism, the design of electrolytes, and the full cell fabrication for AKIBs are highlighted. Finally, insights into opportunities and future directions for achieving high-performance AKIBs and their applications are proposed.
 Xuanpeng Wang | Xuanpeng Wang is an Assistant Professor at the School of Science, Wuhan University of Technology (WUT). He received his PhD degree from WUT in 2019. His current research focuses on material synthesis, device design, in situ characterization of new-type potassium-ion batteries and sodium-ion batteries. |
 Lei Wei | Lei Wei is an Associate Professor at Nanyang Technological University in Singapore. He received the BE degree from the Wuhan University of Technology in 2005 and the PhD degree from the Technical University of Denmark in 2011. Then he joined the Massachusetts Institute of Technology as a postdoctoral associate. In 2014, he joined the Nanyang Technological University in Singapore as a Nanyang Assistant Professor. In 2019, he was promoted to Associate Professor with tenure. His main research interests are fiber-based devices, multi-functional fibers and fabrics, bio-fiber interfaces, and in-fiber energy generation and storage. He currently serves as the Director of Centre for Optical Fibre Technology (COFT) at Nanyang Technological University. He also serves as the chairman of the IEEE Photonics Society Singapore Chapter and the chair of Optica (formerly OSA) Singapore Section. |
 Liqiang Mai | Liqiang Mai is a Chair Professor of Materials Science and Engineering at the Wuhan University of Technology (WUT), Dean of the School of Materials Science and Engineering at WUT, and Fellow of the Royal Society of Chemistry. He received his PhD degree from WUT in 2004 and carried out his postdoctoral research at Georgia Institute of Technology in 2006–2007. He worked as an advanced research scholar at Harvard University in 2008–2011 and the University of California, Berkeley in 2017. His current research interests are focused on new nanomaterials for electrochemical energy storage and micro/nano energy devices. |
Broader contextCompared with traditional Li-ion batteries with toxic and flammable organic-based electrolytes, aqueous potassium-ion batteries (AKIBs) with mild electrolytes have shown great advantages in large-scale energy storage systems and wearable devices due to their good safety, low cost, and environmental friendliness. Despite recent breakthroughs in cathodes, anodes, and electrolytes, AKIBs still face significant challenges in energy density and cycle life, including narrow voltage windows, electrode dissolution, corrosion, and unexpected by-products. These fundamental problems may lead to irreversible capacity loss, poor cycling stability, and short circuit, severely limiting the efficient storage of K+ ions and the future applications of AKIBs. Here, a comprehensive and critical review on the recent advances on AKIBs is provided. The recent research efforts on the innovative electrode and electrolyte designs, the revealing of the reaction mechanism, and the full cell fabrication for AKIBs are highlighted. Based on the current developments, future research directions and perspectives toward high-performance AKIBs and their applications are proposed, guiding the development of this highly exciting field. |
1. Introduction
The depletion of fossil fuels, along with the severe environmental pollution, motivates us to explore renewable energy sources. While most renewable energy sources are intermittent, it is required to develop large-scale stationary energy storage systems (ESSs) to utilize them efficiently.1,2 Among various ESSs, the rechargeable battery technologies act as the primary energy storage solution due to their flexibility, low geographic requirements, and high energy conversion efficiency.3 Lithium-ion batteries (LIBs) have dominated the field for electric vehicles and portable electronic devices, while battery fire accidents occur frequently, mainly caused by the volatile and flammable organic electrolyte, which significantly increase the safety risks of LIBs.4–7Besides organic electrolytes, aqueous electrolytes could also be applied in batteries. Rechargeable LIBs based on aqueous electrolytes were firstly proposed in 1994 by Dahn et al.8 Since then, extensive research has been made toward developing various aqueous rechargeable metal-ion batteries, including non-Li+ (such as Na+, K+, Zn2+, Mg2+, Ca2+, Al3+, etc.) batteries.9–21 Aqueous batteries are particularly promising for solving current issues faced by the commercial LIBs, thanks to the following outstanding properties: (1) the safety of batteries is greatly improved by using aqueous electrolytes; (2) the cost of aqueous batteries is expected to be largely reduced due to the absence of strict assembly conditions and expensive organic solvents; and (3) the electrical conductivity of aqueous electrolytes is two orders of magnitude higher than that of organic electrolytes.
In this new family of aqueous rechargeable batteries, aqueous potassium-ion batteries (AKIBs) are garnering significant attention. AKIBs are rocking chair batteries similar to the traditional LIBs. The schematic configuration of a typical AKIB is shown in Fig. 1a, including a cathode (Prussian blue analog (PBA)), an anode (KTi2(PO4)3), and an aqueous electrolyte. During the charging process, K+ ions, as charge carriers, are released from the cathode material, then travel across the electrolyte, and are eventually inserted into the anode material, while electrons also transfer to the anode through the external circuit. While for the discharging process, the intercalated K+ ions are extracted from the anode and then re-intercalated into the cathode. At the same time, the electrons in the external circuit move from the anode to the cathode. It is well known that the AKIBs show the following advantages: (1) K shows a relatively low redox potential (−2.9 V vs. the standard hydrogen electrode (SHE)), leading to a high potential energy density; (2) K is 1000 times more abundant than Li in the Earth's crust, promising in lowing the battery cost; (3) low Lewis acidity of K+ results in the smallest hydrated ionic size (3.31 Å) in an aqueous solvent, which facilitates its fast diffusion (Fig. 1b and c).22 Therefore, AKIBs are considered one of the ideal electrochemical devices for large-scale ESSs. However, like many other aqueous batteries, AKIBs are hampered by two prevalent issues, namely limited energy density and suboptimal lifetime. Fundamentally, the inherent hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) of water enable AKIBs with a narrow electrochemical stable window (ESW) of 1.23 V, leading to insufficient energy density (Fig. 1d).23–26 In addition, side reactions occur between the electrode materials and H2O or precipitated O2 during the discharging/charging processes, which cause structural degeneration of the electrode material, thus affecting the cycle stability of the battery. Moreover, the dissolution of active electrode materials occurs in aqueous electrolytes due to the high dielectric property of water.
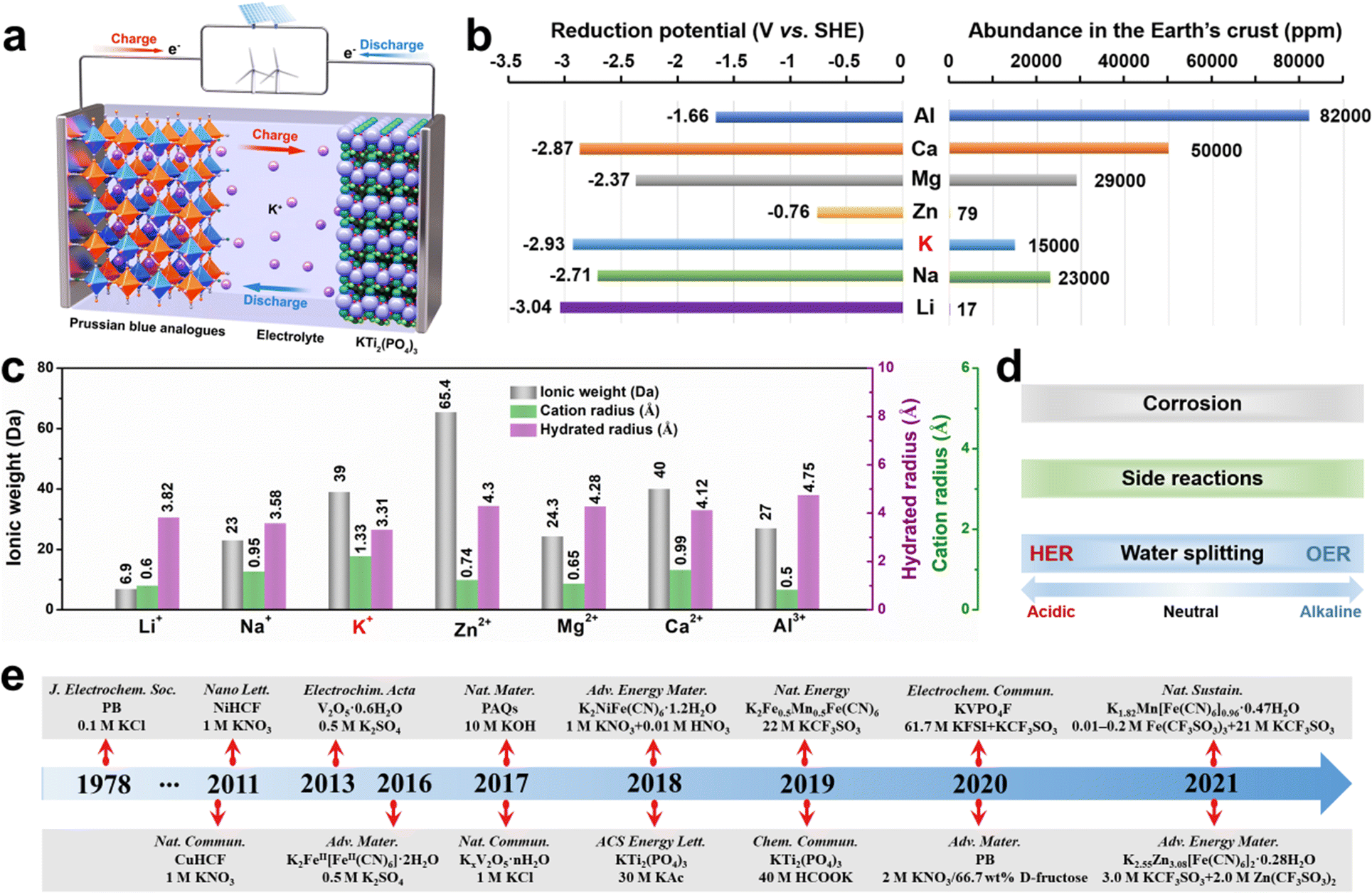 | ||
| Fig. 1 (a) Schematic illustration of AKIB. (b) Comparison of reduction potential and abundance in the earth of different metals. (c) Comparison of hydrated radius, cation radius, and ionic weight of metal-ion charge carriers. (d) The major challenges to be addressed in AKIBs. (e) Brief development history of the representative advancement in aqueous K+ storage. | ||
Pioneering work on the electrochemical behavior of Prussian blue (PB) for K+ storage was first performed by Neff et al. in 1978.10 Since then, researchers have made remarkable achievements in AKIBs via materials design and electrolyte optimization (see Fig. 1e). However, to date, a systematic summary on the latest development of AKIBs is still lacking. This review focuses on the latest advances and perspectives in electrode materials (cathodes and anodes) and electrolytes of AKIBs, then discusses current progress on the optimization of electrode material properties, the understanding of the reaction mechanism, the design of electrolytes, and the full cell fabrication. Finally, insights into the existing challenges and future development direction of high-performance AKIBs are summarized.
2. Electrodes for AKIBs
For the electrode materials for AKIBs, their redox potentials should be in or near the electrolysis potential range of water. The red dotted lines represent the electrolytic potentials of H2 and O2 generated under neutral pH conditions (Fig. 2). Electrode materials within this range can work normally, otherwise the electrode will continue to electrolyze water. A summary of the representative anode and cathode materials with redox potentials for AKIBs is given. As revealed, PB, PBAs, NASICON-type Na3V2(PO4)3, and MXenes exhibit desired potentials as cathodes for AKIBs. Some vanadium-based oxide materials offer desired potentials as both cathode and anode. KTi2(PO4)3 with low potential is the most widely-adopted anode for AKIBs. Organic materials and alloys can provide low redox potentials, while metal oxides and sulfides exhibit relatively high redox potentials. Therefore, in order to pursue state-of-the-art AKIBs, it is imperative to discover novel cathode materials with high redox potential and high-capacity anode materials, both of which could show stable electrochemical stability. | ||
| Fig. 2 Electrochemical stability windows of aqueous electrolytes and redox potentials of various electrode materials for AKIBs. | ||
2.1. Cathode materials
Similar to LIBs, the energy/power density and cost of AKIBs are dependent on the cathode material.27,28 Thus, developing and designing cathode materials with high discharge potential, high specific capacity, and robust crystal structure that allow intercalation and deintercalation of K+ is the major challenge for AKIBs. To date, materials that have been demonstrated as viable K+ host materials are limited, including PB, PBAs, vanadium-based materials, Na3V2(PO4)3, and MXenes. The cathode materials and corresponding electrochemical performances are summarized (Table 1). Furthermore, the K+ storage behavior, electrochemical performance, mechanisms, current issues, and optimization strategies of these cathode materials are discussed in this section.| Cathode materials | Electrolyte | Working potential range | Specific capacity/current | Capacity retention/cycles/current | Rate capability/current | Ref. |
|---|---|---|---|---|---|---|
| Fe4III[FeII(CN)6]3 3.4H2O | 3.75 M KNO3 | −0.25 to 0.85 V vs. Ag/AgCl | 84.7 mA h g−1/1C | 80.8%/1000/10C | 68.5 mA h g−1/6C | 29 |
| GR/PB | 0.1 M KCl | −0.2 to 1.0 V vs. Ag/AgCl | 141.2 mA h g−1/93 mA g−1 | — | ∼40 mA h g−1/2.3 A g−1 | 30 |
| PB-K//AC | 1 M KNO3 + 60.0 wt% maltose | 0–2 V vs. AC | 69.6 mA h g−1/0.5 A g−1 | 66.7%/2000/2 A g−1 | — | 31 |
| PB@PPy@Cloth | 1 M KCl | −0.2 to 1.1 V vs. SCE | 125 mA h g−1/100 mA g−1 | 89.5%/500/500 mA g−1 | 52 mA h g−1/2000 mA g−1 | 32 |
| PB | 0.1 M KCl | 0–0.6 V vs. SCE | — | — | — | 10 |
| PB-modified Pt electrode | 1 M KCl | −0.2 to 1.0 V vs. SCE | — | — | — | 33 |
| PB-Nf film | 1 M KCl | −0.3 to 1.2 V vs. SCE | — | — | — | 34 |
| CuHCF | 1 M KNO3 + 0.01 M HNO3 | 0.6–1.4 V vs. SHE | 59.1 mA h g−1/0.83C | 83%/40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000/17C 000/17C |
40.1 mA h g−1/83C | 35 |
| NiHCF | 1 M KNO3 + 0.01 M HNO3 | 0.4–1.0 V vs. SHE | 59 mA h g−1/10 mA g−1 | ∼93%/5000/498 mA g−1 | 38.9 mA h g−1/2502 mA g−1 | 36 |
| CuNiHCF | 1 M KNO3 | 0.4–1.3 V vs. SHE | 65 mA h g−1/50 mA g−1 | 91%/2000/500 mA g−1 | — | 37 |
| K2Co[Fe(CN)6] | 0.5 M K2SO4 | 0–1.2 V vs. Hg/HgO | 112 mA h g−1/400 mA g−1 | 95%/1000/4000 mA g−1 | 80 mA h g−1/4000 mA g−1 | 38 |
| K2FeII[FeII(CN)6]·2H2O | 0.5 M K2SO4 | 0–1.2 V vs. Ag/AgCl | 120 mA h g−1/200 mA g−1 | 96%/500/500 mA g−1 | 93 mA h g−1/3000 mA g−1 | 39 |
| ZnHCF | 0.5 M K2SO4 | −0.2 to 1.2 V vs. Ag/AgCl | 69.7 mA h g−1/300 mA g−1 | — | — | 40 |
| Ni2Zn1HCF | 0.6 M K2SO4 | 0–1 V vs. SCE | 66.0 mA h g−1/300 mA g−1 | 89.6%/30000/30 A g−1 |
43.8 mA h g−1/60 A g−1 | 41 |
| K1.93Fe[Fe(CN)6]0.97·1.82H2O | 1 M KNO3 | −0.2 to 1.0 V vs. SCE | 142 mA h g−1/75 mA g−1 | 88%/300/1500 mA g−1 | 40 mA h g−1/9000 mA g−1 | 42 |
| K2NiFe(CN)6·1.2H2O | 1 M KNO3 + 0.01 M HNO3 | 0–1 V vs. SCE | 77.4 mA h g−1/400 mA g−1 | 98.6%/5000/2400 mA g−1 | 42.1 mA h g−1/40 A g−1 | 43 |
| KFeMnHCF | 22M KCF3SO3 |
0–1.2V vs. Ag/AgCl |
135 mA h g−1/65 mA g−1 | 90%/10000/13 A g−1 |
85 mA h g−1/15.6 A g−1 | 44 |
| CoHCF | 22 M KCF3SO3 | 0–1.2 V vs. Ag/AgCl | 90.4 mA h g−1/20 mA g−1 | 70.0%/1000/600 mA g−1 | 65.6 mA h g−1/100 mA g−1 | 45 |
| K1.82Mn[Fe(CN)6]0.96·0.47H2O | 21 M KCF3SO3 + 0.01–0.2 M Fe(CF3SO3)3 | 0–1.25 V vs. Ag/AgCl | 160 mA h g−1/300 mA g−1 | ∼100%/130000/2500 mA g−1 |
95 mA h g−1/7000 mA g−1 | 46 |
| KMHCF | 20 M KAc gel | 0–1 V vs. Ag/AgCl | 57 mA h g−1/200 mA g−1 | ∼52.6%/400/200 mA g−1 | — | 47 |
| K2.55Zn3.08[Fe(CN)6]2·0.28H2O | 3.0 M KCF3SO3 + 2.0 M Zn(CF3SO3)2 | 0–1.2 V vs. SCE | 70.0 mA h g−1/1000 mA g−1 | 93.7%/10000/20 A g−1 |
26.5 mA h g−1/30 A g−1 | 48 |
| PG | 1 M KNO3 | 0–1 V vs. Ag/AgCl | 121.4 mA h g−1/111 mA g−1 | 44.7%/1100/500 mA g−1 | 83.7 mA h g−1/388 mA g−1 | 49 |
| PY/ITO | 0.1 M KCl | −0.32–1.2 V vs. Ag/AgCl | 142 mA h g−1/416 mA g−1 | 82%/500/416 mA g−1 | 57 mA h g−1/3333 mA g−1 | 50 |
| Na3V2(PO4)3 | 1 M K2SO4 | −0.2 to 0.9 V vs. SCE | — | — | — | 51 |
| K0.22V1.74O4.37·0.82 H2O | 1 M KCl | −0.1 to 0.9 V vs. Ag/AgCl | 183 mA h g−1/5 mV s−1 | ∼100%/5000/2 A g−1 (Full cell) | 93 mA h g−1/200 mV s−1 | 52 |
| V2O5·0.6H2O | 0.5 M K2SO4 | 0–1.0 V vs. SCE | 50 mA h g−1/100 mA g−1 | — | — | 53 |
| δ-K0.5V2O5 | 22 M KCF3SO3 | −0.4 to 0.8 V vs. SCE | 116 mA h g−1/100 mA g−1 | 88.2%/1000/100 mA g−1 | 65 mA h g−1/5 A g−1 | 54 |
| Nb2C | 1 M KCl | −0.2 to 0.8 V vs. SCE | 66.2 mA h g−1/0.1 A g−1 | 90.4%/20000/5 A g−1 |
32.7 mA h g−1/5 A g−1 | 55 |
| Ti2C | 1 M KCl | −0.2 to 0.8 V vs. SCE | 52.7 mA h g−1/0.1 A g−1 | 89.7%/20000/5 A g−1 |
26.4 mA h g−1/5 A g−1 | 55 |
| Ti3C2 | 1 M KCl | −0.2 to 0.8 V vs. SCE | 57.3 mA h g−1/0.1 A g−1 | 92.5%/20000/5 A g−1 |
21.4 mA h g−1/5 A g−1 | 55 |
The crystal structure of PB is a rigid cubic framework (a = b = c = 10.13 Å) with FeIII and FeII alternately occupying hexahedral vertex positions linked by C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N ligands, which can accommodate more K+ insertion/extraction (Fig. 3a). The electrochemical redox behavior of PB thin films in KCl-based electrolytes was investigated as early as 1978 by Neff et al.10 The electrode was bright blue when the scanning potential was 0.6 V vs. saturated calomel electrode (SCE), and then became colorless at 0.0 V vs. SCE. The color change of the electrode was caused by the reaction between PB and “Everitt's salt”. Subsequently, Itaya et al. discovered that the electrochemical behavior of PB-modified electrodes was related to the current density used to prepare PB films.33 The films prepared at higher current densities may contain incompatible Fe(CN)64−/Fe(CN)63− and Fe2+/Fe3+ ions, which showed sharp current peaks in the cyclic voltammetry (CV) curves. Higher charge loss indicated the existence of many non-coordinating ions in the films prepared at higher current densities. Honda et al. prepared a thin PB layer on Nafion (Nf) substrate by a surface complexation method.34 The composite electrode exhibited good electrochemical stability and a stable voltage of 0.68 V in the self-discharge test. They found that the battery discharge rate was significantly dependent on the amount of iron remaining in the Nf film. The iron ions in the matrix could act as electronic mediators between the PB electrodes and the surface of the matrix. Zarbin et al. developed a wet chemical method to prepare graphene (GR)/PB nanocomposites as cathode for AKIBs.30 Compared with pure PB film, the CV curves of GR/PB film exhibited sharper peaks, which benefited from the fast electron transfer provided by graphene in the film. Recently, commercially available PB as AKIB cathode was investigated in KNO3-based electrolyte by Shu's group.29 Based on the redox of Fe2+/Fe3+–N couple (0.2–0.4 V vs. Ag/AgCl), the PB cathode achieved a stable and high capacity of 84.7 mA h g−1 at 1C (Fig. 3b and c). Also, benefiting from the robust metal–organic framework, this cathode exhibited a stable cycle performance with 500 cycles for K+ storage (Fig. 3d). The slight shift (about 0.1°) of the ex situ XRD pattern indicated the subtle lattice shrinkage and expansion caused by the K+ intercalation and deintercalation of PB (Fig. 3e).
N ligands, which can accommodate more K+ insertion/extraction (Fig. 3a). The electrochemical redox behavior of PB thin films in KCl-based electrolytes was investigated as early as 1978 by Neff et al.10 The electrode was bright blue when the scanning potential was 0.6 V vs. saturated calomel electrode (SCE), and then became colorless at 0.0 V vs. SCE. The color change of the electrode was caused by the reaction between PB and “Everitt's salt”. Subsequently, Itaya et al. discovered that the electrochemical behavior of PB-modified electrodes was related to the current density used to prepare PB films.33 The films prepared at higher current densities may contain incompatible Fe(CN)64−/Fe(CN)63− and Fe2+/Fe3+ ions, which showed sharp current peaks in the cyclic voltammetry (CV) curves. Higher charge loss indicated the existence of many non-coordinating ions in the films prepared at higher current densities. Honda et al. prepared a thin PB layer on Nafion (Nf) substrate by a surface complexation method.34 The composite electrode exhibited good electrochemical stability and a stable voltage of 0.68 V in the self-discharge test. They found that the battery discharge rate was significantly dependent on the amount of iron remaining in the Nf film. The iron ions in the matrix could act as electronic mediators between the PB electrodes and the surface of the matrix. Zarbin et al. developed a wet chemical method to prepare graphene (GR)/PB nanocomposites as cathode for AKIBs.30 Compared with pure PB film, the CV curves of GR/PB film exhibited sharper peaks, which benefited from the fast electron transfer provided by graphene in the film. Recently, commercially available PB as AKIB cathode was investigated in KNO3-based electrolyte by Shu's group.29 Based on the redox of Fe2+/Fe3+–N couple (0.2–0.4 V vs. Ag/AgCl), the PB cathode achieved a stable and high capacity of 84.7 mA h g−1 at 1C (Fig. 3b and c). Also, benefiting from the robust metal–organic framework, this cathode exhibited a stable cycle performance with 500 cycles for K+ storage (Fig. 3d). The slight shift (about 0.1°) of the ex situ XRD pattern indicated the subtle lattice shrinkage and expansion caused by the K+ intercalation and deintercalation of PB (Fig. 3e).
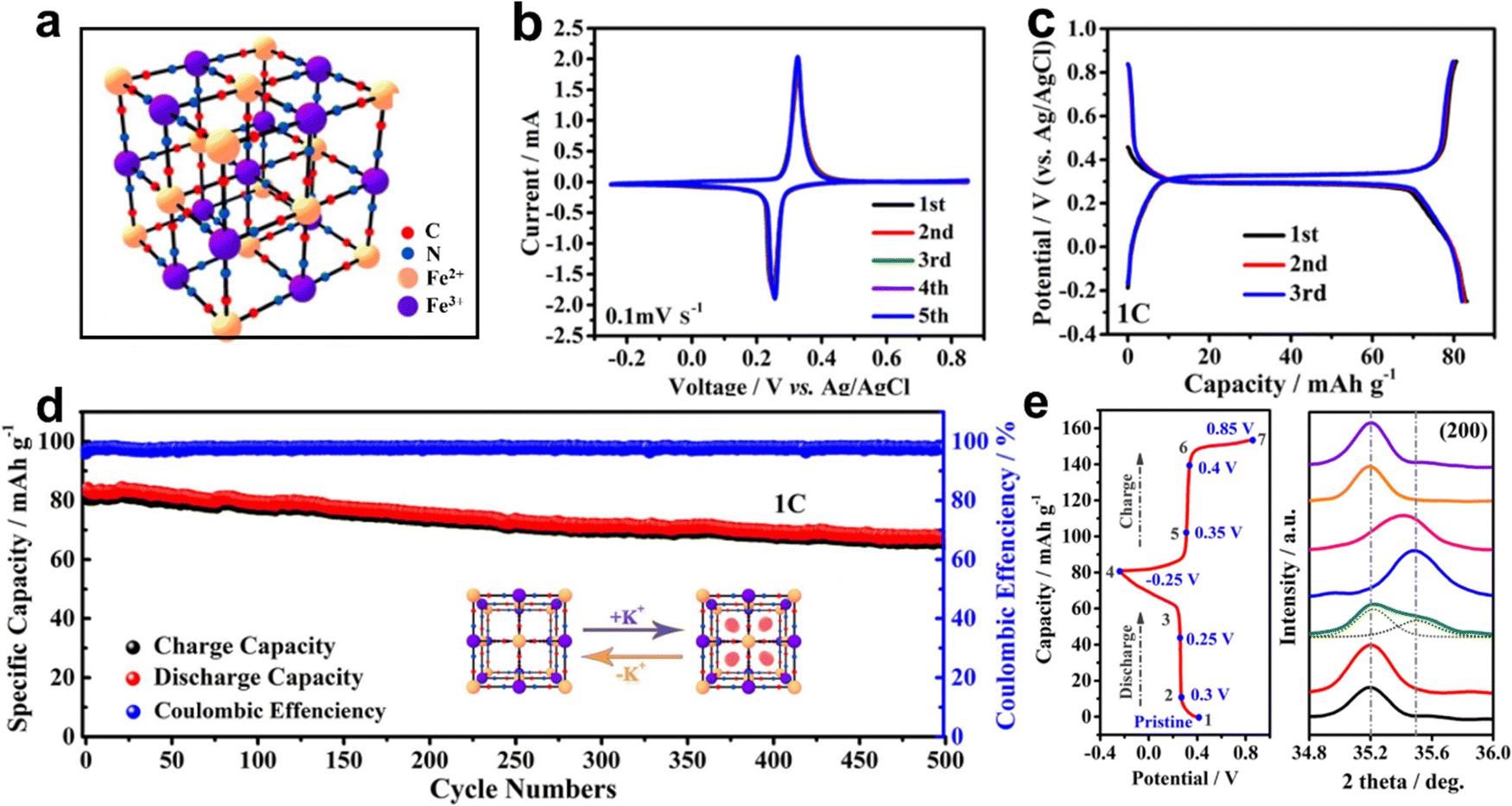 | ||
| Fig. 3 (a) Crystal structure of PB. (b) CV curves and (c) GCD profiles of PB. (d) Cyclability at 1C, and (e) ex situ XRD patterns of PB. Reproduced with permission from ref. 29. Copyright 2020, Elsevier Inc. | ||
Compared with PB, PBAs are more favored due to their diverse structures and excellent electrochemical performance. Recently, Cui et al. reported a series of PBAs, which showed excellent K+ storage behavior in AKIBs. Initially, nickel hexacyanoferrate (NiHCF) was synthesized using a precipitation method and then investigated for K+ storage.36,62 The structure of NiHCF is similar to that of PB, and Ni2+ occupies nitrogen coordination, replacing Fe3+ in PB (Fig. 4a). It was found that NiHCF reacted with K+ at 0.69 V vs. SHE in KNO3-based electrolyte (Fig. 4b). NiHCF possessed a discharge capacity of 59 mA h g−1 at C/6. Moreover, the excellent rate capability (with 66% capacity retention at 41.7C) and superior stability (93% capacity retention after 5000 cycles) were delivered. Subsequently, they synthesized copper hexacyanoferrate (CuHCF) using a similar method.35,62 The co-precipitation process of CuHCF was slower than that of PB. Due to the existence of excess Cu2+, highly crystalline and polydisperse nanoparticles could be grown in an orderly manner. The structure of CuHCF is also similar to that of PB, with Cu(II) occupying the nitrogen-coordinated P site and Fe(III) occupying the carbon-coordinated R site (Fig. 4c). CuHCF electrode realized a discharge potential of 0.946 V vs. SHE in KNO3-based electrolyte, which exceeded that of the NiHCF electrode (0.69 V) (Fig. 4d). A similar specific capacity of 59.14 mA h g−1 was recorded, indicating that high energy density could realize in the CuHCF electrode for AKIBs. In addition, the CuHCF electrode delivered higher rate capability (67% capacity retention of its initial capacity at 83C) and longer lifetime (17% capacity loss after 40000 cycles) compared to the NiHCF electrode. Additionally, hexacyanoferrate (ZnHCF) was utilized as cathode materials in Na2SO4, K2SO4, and ZnSO4 solutions.40 ZnHCF exhibits a rhombic structure, which is different from the cubic structure. FeC6 octahedra and ZnN4 tetrahedra are collected by CN ligands to generate a 3D open framework (Fig. 4e). In the K2SO4 electrolyte, a pair of redox peaks at 0.83/1.03 V vs. Ag/AgCl appeared higher than those of ZnSO4 and Na2SO4 electrolytes (Fig. 4f). The specific capacity of ZnHCF in K2SO4 electrolyte at 5C was 69.7 mA h g−1, which exceeded that of ZnSO4 electrolyte (64.4 mA h g−1), but lower than that of Na2SO4 electrolyte (71.4 mA h g−1).
 | ||
| Fig. 4 (a) Crystal structure and (b) typical discharge profiles of different rates of NiHCF. Reproduced with permission from ref. 36. Copyright 2011, American Chemical Society. (c) Crystal structure and (d) discharge profiles of different rates of CuHCF. Reproduced with permission from ref. 35. Copyright 2011, Springer Nature. (e) Crystal structure and (f) CV curves in K2SO4 (black), Na2SO4 (blue), and ZnSO4 (red) electrolytes of ZnHCF. Reproduced with permission from ref. 40. Copyright 2014, Wiley-VCH. (g) Crystal structure and (h) discharge profiles from 200 to 3000 mA g−1 of K2FeII[FeII(CN)6]·2H2O. Reproduced with permission from ref. 39. Copyright 2016, Wiley-VCH. (i) Crystal structure of K2Co[Fe(CN)6]. (j) GCD profiles of KNi[Co(CN)6] and K2Co[Fe(CN)6]. Reproduced with permission from ref. 38. Copyright 2021, The Royal Society of Chemistry. (k) Crystal structure and (l) rate performance from 5 to 500C of K2NiFe(CN)6·1.2H2O. Reproduced with permission from ref. 43. Copyright 2018, Wiley-VCH. | ||
Although the above-developed PBA-based AKIBs have shown satisfactory performance, their low K+ containing amount limits the reversible capacity. In this regard, Wang et al. reported K-rich iron hexacyanoferrate (K2FeIIFeII(CN)6·2H2O) nanocubes as cathode material for AKIBs.39 The structure of this material is orthogonally symmetric rather than the usual cubic structure (Fig. 4g). Two couples of charge/discharge plateaus (0.92/0.84 and 0.3/0.2 V) appeared in the galvanostatic charge/discharge (GCD) in 0.5 M K2SO4 electrolyte, which are related to the reversible K+ insertion/extraction. The electrode achieved a high capacity of 140 mA h g−1, manifesting the reaction of two-electron transfer. Discharge capacities of 111, 109, 104, 100, and 93 mA h g−1 were obtained at 0.2, 0.5, 1, 2, and 3 A g−1, respectively (Fig. 4h). It can be found that two-electron redox processes and high K content enabled high capacity of the electrode, and a robust open-framework structure facilitated fast kinetics of K+. Recently, K-poor KNi[Co(CN)6] and K-rich K2Co[Fe(CN)6] as AKIB cathodes were comparatively investigated by Chen et al.38 They simulated the atomic arrangement of K2Co[Fe(CN)6] with the square arrangement of atoms providing a large number of void sites to accommodate K+ (Fig. 4i). K2Co[Fe(CN)6] showed redox plateaus at 0.84/0.80 and 0.63/0.39 V, indicating two coordination sites for the intercalation/deintercalation of K+. While KNi[Co(CN)6] demonstrated a pair of discharge/charge plateaus at 0.94/0.54 V, suggesting one coordination site for K+ storage. Thus, K2Co[Fe(CN)6] delivered a discharge capacity of 112 mA h g−1 in K2SO4-based electrolytes, higher than that of KNi[Co(CN)6] (37 mA h g−1) (Fig. 4j). The higher capacity of K2Co[Fe(CN)6] can be ascribed to the abundant K element, abundant active sites, and two-electron redox processes. A mesoporous K-rich nickel ferrocyanide cathode (K2NiFe(CN)6·1.2H2O) for ultrafast AKIBs was developed by Zhao et al.43 Ni and Fe ions together with C-N bonds formed a double perovskite framework (Fig. 4k). At an ultrahigh current density of 500C, the electrode showed a high discharge capacity of 42.1 mA h g−1, equivalent to 54.4% at 5C. It is worth noting that it only took 4.1 s to complete one discharge/charge cycle at 500C, which was extremely high for the rate performance reported at that time (Fig. 4l). Based on the ex situ Raman spectroscopy characterization, the reversible transformation of FeIII and FeII during the redox process was demonstrated.
PBA compounds show controllable morphologies during preparation, and structural engineering plays an important role in promoting ion diffusion, inhibiting material dissolution, and improving electrochemical performance. Partial substitution of N-coordinated P ions with another metal ion is an efficient strategy to influence the properties of PBAs, which has been extensively studied in organic batteries.63,64 For example, the copper-nickel hexacyanoferrate (CuNiHCF) was synthesized by the ratio regulation of Cu to Ni in the material.37 Ni and Cu in the CuNiHCF crystal structure formed a fully miscible solution on the P site (Fig. 5a). The reaction potential of CuNiHCF with K+ was controllable from 0. 6 to 1.0 V by changing the content of Ni and Cu in the structure (Fig. 5b). NiHCF and CuHCF displayed 100% capacity retention after 2000 cycles, while Cu0.56Ni0.44HCF lost 9% of its initial capacity. The capacity loss of the Cu0.56Ni0.44HCF may be due to the dissolution of the electrode, and the relative concentrations of Ni and Cu also have an influence on the chemical stability of Cu0.56Ni0.44HCF. In recent work, a series of NixZnyHCF (x = 1, 1.5, or 2, y = 2, 1.5, or 1) was reported by Mai et al. as cathodes for AKIBs.41 NixZnyHCF was designed by the synergistic effect between high voltage Zn2+ and stable Ni2+. All three samples exhibited linear rigid coordination of Fe–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–M (M = Ni, Zn) mechanisms to form cubic frameworks. Among them, Ni2Zn1HCF showed the lowest polarization and the highest redox potential in the aqueous K2SO4 solution revealed by the CV curves (Fig. 5c). Ni2Zn1HCF exhibited the average equilibrium potentials of 0.6 V vs. SCE and the highest capacity retention of 85.5% at 166.7C (Fig. 5d). Impressively, Ni2Zn1HCF exhibited an ultrahigh rate capability at 1000C with 66% capacity retention and ultralong life span of 80000 cycles with 0.000385% capacity decay per discharge/charge cycle at 1000C. Such excellent performance was associated with the mechanism of near-pseudocapacitive intercalation, the large surface area, and the structural stability brought by the high Ni content. Hu et al. explored the electrochemical stability of K-rich K2MnFe(CN)6 affected by Fe substitution.44 They found that Mn ions replaced by the proper amount of Fe ions could effectively inhibit the dissolution of the material in the aqueous electrolyte during the discharge/charge process (Fig. 5e and f). Thus, the optimal K-rich sample, K2Fe0.35Mn0.65Fe(CN)6, achieved a high capacity of 135 mA h g−1 and high-rate performance with 5% capacity loss at 20C. In this regard, Fe substitution not only reduces the density of Mn3+ in the lattice, but also significantly changes the Mn2+/Mn3+–N redox reaction mechanism. Based on first principles calculations, K2Fe0.5Mn0.5Fe(CN)6 showed small band gap and low K+ ion diffusion energy when compared with that of K2MnFe(CN)6, suggesting the improved electronic conductivity and K+ diffusion dynamics (Fig. 5g and h).
N–M (M = Ni, Zn) mechanisms to form cubic frameworks. Among them, Ni2Zn1HCF showed the lowest polarization and the highest redox potential in the aqueous K2SO4 solution revealed by the CV curves (Fig. 5c). Ni2Zn1HCF exhibited the average equilibrium potentials of 0.6 V vs. SCE and the highest capacity retention of 85.5% at 166.7C (Fig. 5d). Impressively, Ni2Zn1HCF exhibited an ultrahigh rate capability at 1000C with 66% capacity retention and ultralong life span of 80000 cycles with 0.000385% capacity decay per discharge/charge cycle at 1000C. Such excellent performance was associated with the mechanism of near-pseudocapacitive intercalation, the large surface area, and the structural stability brought by the high Ni content. Hu et al. explored the electrochemical stability of K-rich K2MnFe(CN)6 affected by Fe substitution.44 They found that Mn ions replaced by the proper amount of Fe ions could effectively inhibit the dissolution of the material in the aqueous electrolyte during the discharge/charge process (Fig. 5e and f). Thus, the optimal K-rich sample, K2Fe0.35Mn0.65Fe(CN)6, achieved a high capacity of 135 mA h g−1 and high-rate performance with 5% capacity loss at 20C. In this regard, Fe substitution not only reduces the density of Mn3+ in the lattice, but also significantly changes the Mn2+/Mn3+–N redox reaction mechanism. Based on first principles calculations, K2Fe0.5Mn0.5Fe(CN)6 showed small band gap and low K+ ion diffusion energy when compared with that of K2MnFe(CN)6, suggesting the improved electronic conductivity and K+ diffusion dynamics (Fig. 5g and h).
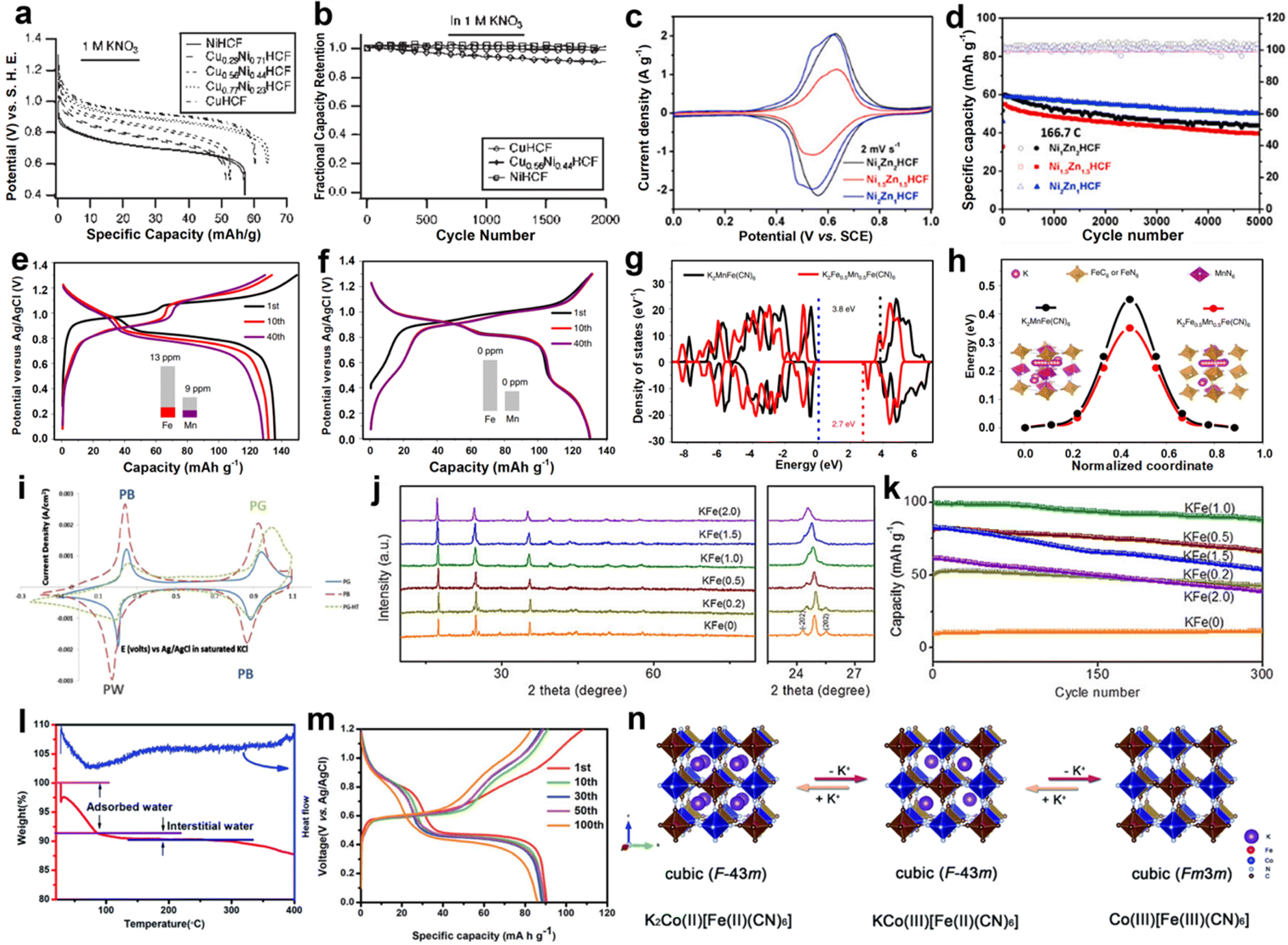 | ||
| Fig. 5 (a) GCD profiles of different rates and (b) cycling performance of CuNiHCF. Reproduced with permission from ref. 37. Copyright 2012, American Chemical Society. (c) CV curves and (d) cycling performance of NixZnyHCF. Reproduced with permission from ref. 41. Copyright 2020, Elsevier Inc. GCD profiles of (e) K2MnFe(CN)6 and (f) K2Fe0.35Mn0.65Fe(CN)6. (g) Density of states and (h) the K+ migration energy barriers in the K2Fe0.5Mn0.5Fe(CN)6 and K2MnFe(CN)6. Reproduced with permission from ref. 44. Copyright 2019, Springer Nature. (i) CV curves of PB, PG, and PG-HT electrodes in KNO3 electrolyte. Reproduced with permission from ref. 49. Copyright 2015, Elsevier Inc. (j) XRD patterns and (k) long-term cyclability at 1500 mA g−1 of K1.93Fe[Fe(CN)6]0.97·1.82H2O and other samples. Reproduced with permission from ref. 42. Copyright 2018, Wiley-VCH. (l) TG/DSC curves and (m) GCD profiles of K2CoFe(CN)6. (n) Schematic illustration of the structure and valence for K2CoFe(CN)6. Reproduced with permission from ref. 45. Copyright 2020, The Royal Society of Chemistry. | ||
In addition to the strategy of partial substitution, reducing grain size, increasing crystallinity, and reducing defects are also used to improve the electrochemical stability of PBAs. Very recently, Padigi and co-workers synthesized PB and Prussian green (PG) using different precursors (potassium ferricyanide and potassium ferrocyanide) as AKIB cathodes in KNO3-based electrolyte.49 Different precursors induced different reaction rates, resulting in much smaller PG particles (50–75 nm) than PB particles (2–10 μm). As revealed by the CV curves, the insertion and extraction of K+ were highly reversible for PG (Fig. 5i). K+ first intercalated at 0.89 V to form PB, and then K+ continued to intercalate at 0.2 V to generate Prussian white (PW). Compared to PG, PB exhibited similar K+ storage behavior. However, the specific capacity of PG exhibited a significant increase from 53 to 121 mA h g−1 compared with PB. This is due to the smaller particle size resulting in a shorter diffusion length for K+. Additionally, Li and co-workers synthesized a series of PW with different particle sizes and crystallinities by adjusting the acidity of hydrothermal conditions.42 As the increase of the concentration of hydrochloric acid, the structure of the material gradually transformed from the monoclinic phase of KFe (0) to the cubic phase of KFe (2.0), and the gradually broadened half-peak width indicated the decreased grain size of the material (Fig. 5j). When used as cathodes for AKIBs, KFe (1.0) electrode exhibited the highest capacity of 142 mA h g−1 at 75 mA g−1 and good stability with 88% capacity retention over 300 cycles at 1.5 A g−1 (Fig. 5k). The high crystallinity of the electrode alleviated its continuous dissolution in an aqueous solution, and the reduced grain size enhanced the ion diffusion kinetics, which synergistically improves the K+ storage stability. Moreover, Jiao et al. synthesized low-defect K2CoFe(CN)6 (CoHCF) using a coprecipitation method and investigated it as a cathode for AKIBs.45 CoHCF showed low water content of less than 10%, indicating low crystalline defects in the sample (Fig. 5l). The CV curves of the electrode demonstrated two pairs of redox peaks, suggesting the existence of two-electron transfer during the electrochemical process. It can deliver a high discharge capacity of 83.6 mA h g−1 at 20 mA g−1 in KCF3SO3-based electrolytes (Fig. 5m). Low defects could avoid the block of active sites, the inhibition of ion transport, and the reduction of electrochemical activity of P sites. During K+ intercalation/deintercalation, a solid solution reaction between K2Co(II)Fe(II) (CN)6 and KCo(III)Fe(II) (CN)6 in the low voltage range, and a phase transformation from F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m to Fm3m in the high voltage range were confirmed (Fig. 5n).
3m to Fm3m in the high voltage range were confirmed (Fig. 5n).
Apart from the above-mentioned improvement strategies, growing active materials on conductive substrates is also a useful strategy to enhance the electrochemical performance.65 For example, the Prussian yellow (PY) film on indium tin oxide (ITO) substrate was reported by Baioun and colleagues, and the obtained composite material exhibited superior electrochemical performances.50 A high capacity of 142 mA h g−1 and high capacity retention of 82% were demonstrated at 3C for 500 cycles, manifesting its good cycling stability. When this cathode material was assembled with the Zn anode to form a full cell, it achieved a high output potential of 1.9 V, indicating the promising application in AKIBs.
Recent research has demonstrated that PB and PBAs are promising K+ storage cathode materials due to their open framework, high voltage platform, simple preparation, and low cost. However, some challenges in practical applications still exist for PB and PBAs, which suffer from poor rate performance and fast capacity fading caused by low electronic conductivity, interstitial or coordinated water, excess lattice defects, and complex side reactions with electrolytes. Fortunately, a series of effective strategies have been adopted to modify the electrochemical property of these cathode compounds, including metal ion substitution or doping, particle size reduction, crystallinity enhancement, defect reduction, and conducting modification.66 Therefore, it is necessary to optimize the lattice structures through feasible structural design strategies, so that the electrode material can store K+ reversibly with high efficiency, and the cycle stability is not affected by unfavorable factors.
 | ||
| Fig. 6 (a) Crystal structure of Na3V2(PO4)3. (b) CV curves of Na3V2(PO4)3 at 5 mV s−1 in 1 M K2SO4, Na2SO4, and Li2SO4 electrolytes. Reproduced with permission from ref. 51. Copyright 2014, Wiley-VCH. | ||
In brief, polyanionic compounds are highly promising for high-energy AKIBs owing to their stable framework, high voltage, and high thermal stability. Compared with PB and PBAs, the relatively high density of polyanions makes them promising AKIB cathode materials. Although low electronic conductivity is a disadvantage that cannot be ignored, it can be well modified by nanocomposite design, carbon coating, the alkali ions partially substitution or other effective strategies. Therefore, the development of high-capacity polyanionic compounds is a promising research direction. Especially, through the investigation of new polyanion materials and structural engineering, the K+ storage capacity of such materials can be further improved.
Wu and co-workers investigated the electrochemical behavior of V2O5·0.6H2O in aqueous solutions containing three alkali metal sulfates (K2SO4, Na2SO4, and Li2SO4).53 The GCD profiles within 0–1 V (vs. SCE) exhibited a capacitor-type linear relationship (Fig. 7a). Interestingly, the specific capacity of V2O5·0.6H2O to storage K+ (50 mA h g−1) exceeded those of Li+ (37 mA h g−1) and Na+ (43 mA h g−1). Meanwhile, the charge-transfer resistance (Rct) value of V2O5·0.6H2O in K2SO4 electrolyte (2.4 Ω) was smaller than those in Na2SO4 (3.5 Ω) and Li2SO4 (6.1 Ω) solutions (Fig. 7b). The fast charge transfer dynamic in the K2SO4 solution validated the optimal electrochemical performance. In addition, the structural evolution of V2O5·0.6H2O during intercalation/deintercalation of Li+/Na+/K+ was presented (Fig. 7c). Compared with Li+ and Na+, the K+ insertion/extraction in the V2O5·0.6H2O interlayer was more feasible, which could be related to the moderate charge density of K+. However, V2O5·0.6H2O showed the rapid decay of capacity in the three electrolytes.
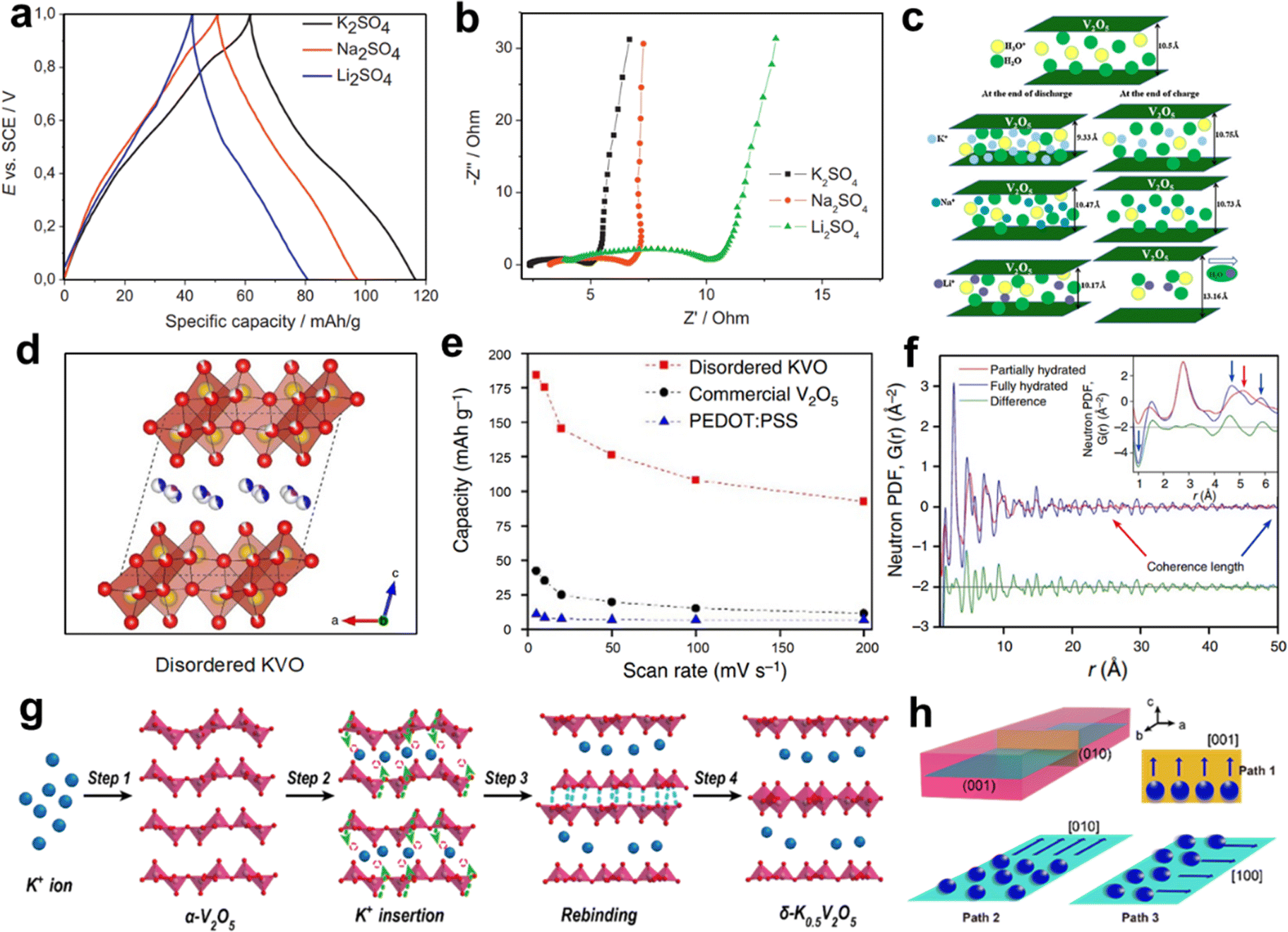 | ||
| Fig. 7 (a) GCD profiles at 100 mA g−1 and (b) Nyquist impedance plots of V2O5·0.6H2O in K2SO4, Na2SO4, and Li2SO4 electrolytes. (c) Schematic illustration of structural evolution of V2O5·0.6H2O in three electrolytes. Reproduced with permission from ref. 53. Copyright 2013, Elsevier Inc. (d) Crystal structure and (e) rate capability of disordered K0.22V1.74O4.37·0.82H2O. (f) Neutron PDFs of disordered K0.22V1.74O4.37·0.82H2O. Reproduced with permission from ref. 52. Copyright 2017, Springer Nature. Schematic illustration of (g) the reconstructing process from α-V2O5 to δ-K0.5V2O5 and (h) three possible pathways for K+ of δ-K0.5V2O5. Reproduced with permission from ref. 54. Copyright 2021, American Chemical Society. | ||
Pre-intercalation of alkali metal ions between the layers of V2O5 is a typical effective strategy to improve electrochemical performance. Teng et al. synthesized K-intercalated disordered vanadium oxide (K0.22V1.74O4.37·0.82H2O) as AKIB cathode.52 The electrode exhibited a bilayer structure that is composed of [VO6] octahedral units, and K+ and H2O existed between the V–O layers (Fig. 7d). When used as K+ storage cathode, K0.22V1.74O4.37·0.82H2O possessed a higher capacity of 183 mA h g−1 at 5 mV s −1 and higher rate performance than those of the commercial V2O5 material and PEDOT:PSS (Fig. 7e). Such good electrochemical stability was due to the strong coherence between the interlayer water and the V–O layer (Fig. 7f). In addition, the highly disordered nature of the material lowered the energy barrier for K+ transport. The synergistic effect endowed electrodes with high specific capacity and excellent rate performance. Similarly, Zhi et al. reconstructed α-V2O5 crystals into δ-K0.5V2O5 bronzes for K+ storage.54 The intercalated K+ was located between the V–O layers (Fig. 7g). The structural reconstructing strategy allowed the creation of more available K+ sites for δ-K0.5V2O5. At the same current density, the δ-K0.5V2O5 achieved a capacity of 116 mA h g−1, while α-V2O5 only showed a capacity of 17 mA h g−1. For δ-K0.5V2O5, three possible transport pathways for K+ in the (010) and (001) planes were suggested, and theoretical calculations demonstrated that the anisotropic storage mode of K+ in converges to the interlayer channel along the [100] direction was the dominant path for K+ (Fig. 7h).
To sum up, vanadium oxide and its derivatives are promising compounds in the field of AKIBs owing to multivalent and open frame structures. In general, vanadium-based materials exhibit both high specific capacity and good cycle stability. Their layered structure facilitates the reversible K+ intercalation/deintercalation. Nevertheless, the synthesis of materials requires strict hydrothermal conditions and high-temperature annealing, which hinders the commercialization to a certain extent. Furthermore, more research works should be paid on the regulation of interlayer spacing, the deployment of structural water, and defect engineering to improve the performance of such materials.
Recently, the exploration of MXenes (Nb2C, Ti2C, and Ti3C2) for aqueous K+ storage was reported by Zhi’s group.55 It is well known that MXenes are a huge family of which more than 30 have been synthesized so far.78 The crystal structure of MXene is a hexagonal close-packed structure, in which the C atoms are located in the octahedral gap. These three MXene species are typical layered structures with interlayer spacing of around 10 Å, which provides enough space for the transport of K+ ions (Fig. 8a–c). As AKIB cathodes, high discharge capacities of 66.2 mA h g−1 for Nb2C, 52.7 mA h g−1 for Ti2C, 57.3 mA h g−1 for Ti3C2 at 0.1 A g−1 and fast K+ transport kinetics at 5 A g−1 were displayed (Fig. 8d–f). No obvious plateaus in the GCD profiles are ascribed to the existence of pseudocapacitance. In addition, long-term cycling performance of the three cathodes with high capacity retentions was demonstrated. Based on ex situ XRD, Raman, and XPS characterizations, a highly reversible intercalation/deintercalation mechanism accompanied by only 1.8% variation of the Nb2C electrode was revealed (Fig. 8g–j).
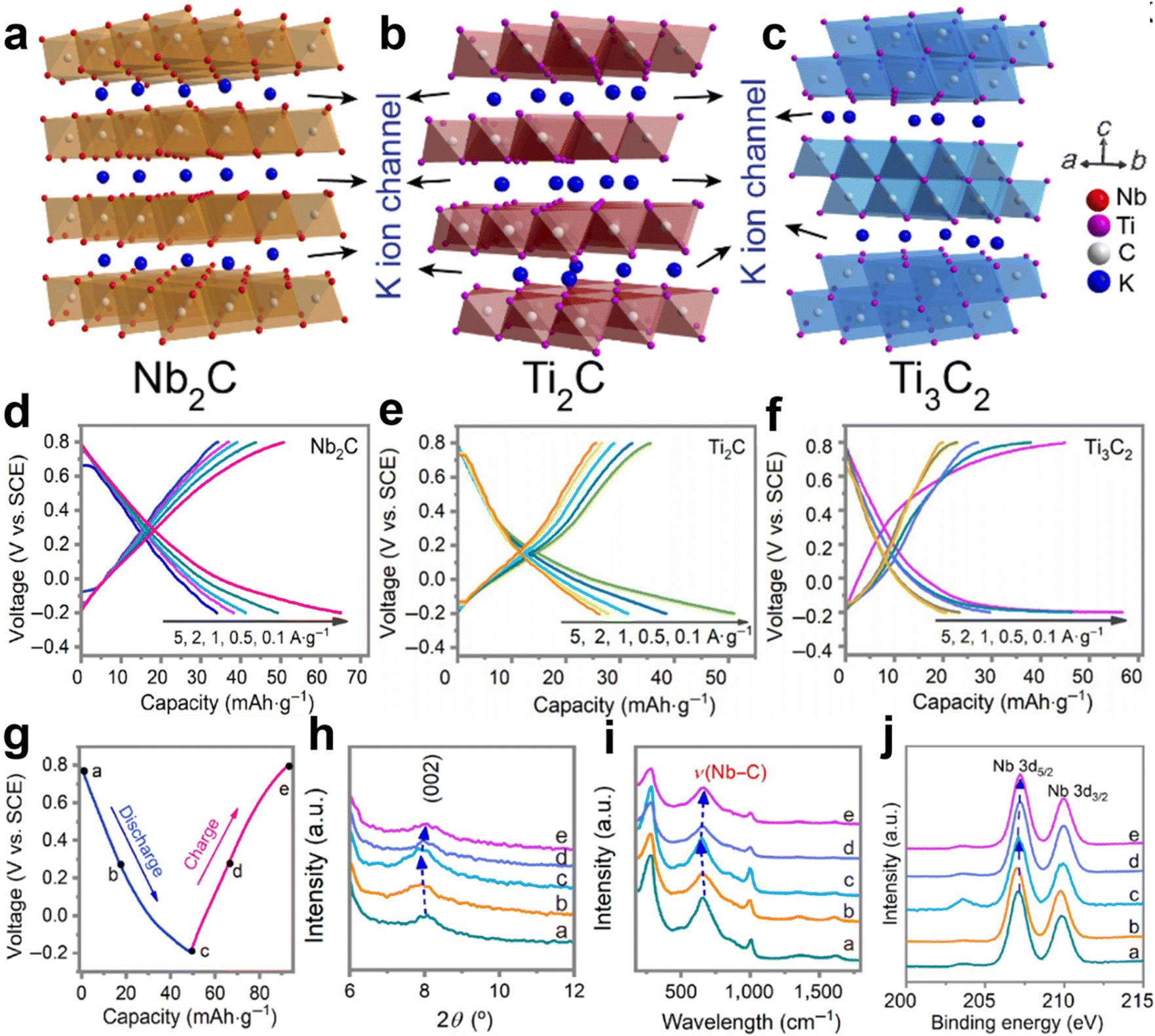 | ||
| Fig. 8 Crystal structures of (a) Nb2C, (b) Ti2C, and (c) Ti3C2. GCD profiles at different rates for (d) Nb2C, (e) Ti2C, and (f) Ti3C2. (g) GCD curves of Nb2C cathode at 0.5 A g−1. The corresponding (h) XRD, (i) Raman, and (j) XPS data during the cycling. Reproduced with permission from ref. 55. Copyright 2022, Tsinghua University Press. | ||
It should be pointed out that the research work on MXenes in aqueous K+ storage is still in its infancy, and the above literature shows that MXenes are a class of promising electrode materials for emerging AKIBs. Due to the large interlayer spacing and good electrical conductivity, MXene nanostructures deserve attention in future energy storage. More interesting works are expected to emerge as synthesis methods and improvement strategies continue to advance.
2.2. Anode materials
Although several typical cathode materials have demonstrated good K+ storage capacity so far, AKIB full cells are still infant, as their energy/power density and durability are also affected by anode materials. Since potassium metal cannot be directly used as the anode of AKIB, in order to meet the needs of rocking-chair batteries, the development of anode materials is equally important. This section will present the latest developments in AKIB anodes (Table 2). Examples of anode materials include NASICON-type materials, metal oxides/sulfides, alloy materials, and organic compounds.| Anode materials | Electrolyte | Working potential range | Specific capacity/current | Capacity retention/cycles/current | Rate capability/current | Ref. |
|---|---|---|---|---|---|---|
| KTi2(PO4)3/C | 21 M KCF3SO3 | −1.1 to 0 V vs. SCE | 78 mA h g−1/50 mA g−1 | ∼100%/40/0.05 A g−1 | 47.5 mA h g−1/2 A g−1 | 83 |
| KTi2(PO4)3 | 30 M KAc | −1.5 to −0.3 V vs. Ag/AgCl | ∼58 mA h g−1/0.1 A g−1 | 69%/11000/1 A g−1 |
∼12 mA h g−1/5 A g−1 | 84 |
| KTi2(PO4)3 | 40 M HCOOK | −1.5 to −0.3 V vs. Ag/AgCl | ∼21 mA h g−1/100 mA g−1 | — | — | 85 |
| KTi2(PO4)3@C | 2 M KFSI DMF/H2O | −1.5 to 0 V vs. SCE | 76 mA h g−1/1 A g−1 | ∼80%/10000/1 A g−1 (full cell) |
33 mA h g−1/20 A g−1 (Full cell) | 86 |
| K0.36(H2O)yWS2 | 0.5 M K2SO4 | −0.8 to 0.4 V vs. Ag/AgCl | 43.3 mA h g−1/0.2 A g−1 | 48.9%/250/0.5 A g−1 | 25.7 mA h g−1/5 A g−1 | 87 |
| K0.38(H2O)0.82MoS2 | 0.5 M K2SO4 | −0.8 to 0.4 V vs. Ag/AgCl | ∼57 mA h g−1/500 mA g−1 | 48.5%/500/5 A g−1 | ∼40 mA h g−1/8 A g−1 | 88 |
| α-MoO3@TiO2 | 0.5 M KPF6 | 0.05–1 V (Coin cell) | 41.27 mA h g−1/5C | 66.2%/20/5C | — | 89 |
| KxV2O5·nH2O | 0.5 M K2SO4 | −0.8 to 0 V vs. Ag/AgCl | 382 mA h g−1/5 mV s−1 | 80%/10000/500 mV s−1 (full cell) |
81 mA h g−1/1000 mV s−1 | 90 |
| Mo-based | 0.1 M KCl | −0.2 to 0.4 V vs. Ag/AgCl | 517 mA h g−1/7 A g−1 | — | ∼60 mA h g−1/112 A g−1 | 91 |
| Bi | 1 M KOH | −1.2 to 0 V vs. SCE | 254.3 mA h g−1/28.8 mA g−1 | 88.8%/1600/0.57 A g−1 | 147.3 mA h g−1/8.768 A g−1 | 92 |
| Bi | 1 M KOH | −1.2 to 0 V vs. SCE | ∼226 mA h g−1/0.2 A g−1 | 76.6%/2000/1 A g−1 | 220 mA h g−1/8 A g−1 | 93 |
| β-PTCDA | 30 M KFSI | −1.1 to 0.1 V vs. SCE | 145 mA h g−1/200 mA g−1 | 82%/500/2 A g−1 | 120.4 mA h g−1/8 A g−1 | 94 |
| PNTCDA | 3.75 M KNO3 | −1.0 to −0.2 V vs. SCE | 135 mA h g−1/360 mA g−1 | ∼79%/300/360 mA g−1 | 93.2 mA h g−1/5400 mA g−1 | 95 |
| PTCDI | 22M KCF3SO3 |
−1.3 to 0.3 V vs. Ag/AgCl | 125 mA h g−1/65 mA g−1 | 77%/1000/2.6 A g−1 | 110 mA h g−1/2.6 A g−1 | 44 |
| PAQS | 10 M KOH | −1.0 to 0.1 V vs. SHE | 200 mA h g−1/100 mA g−1 | 88%/1350/200 mA g−1 | 186 mA h g−1/2250 mA g−1 | 96 |
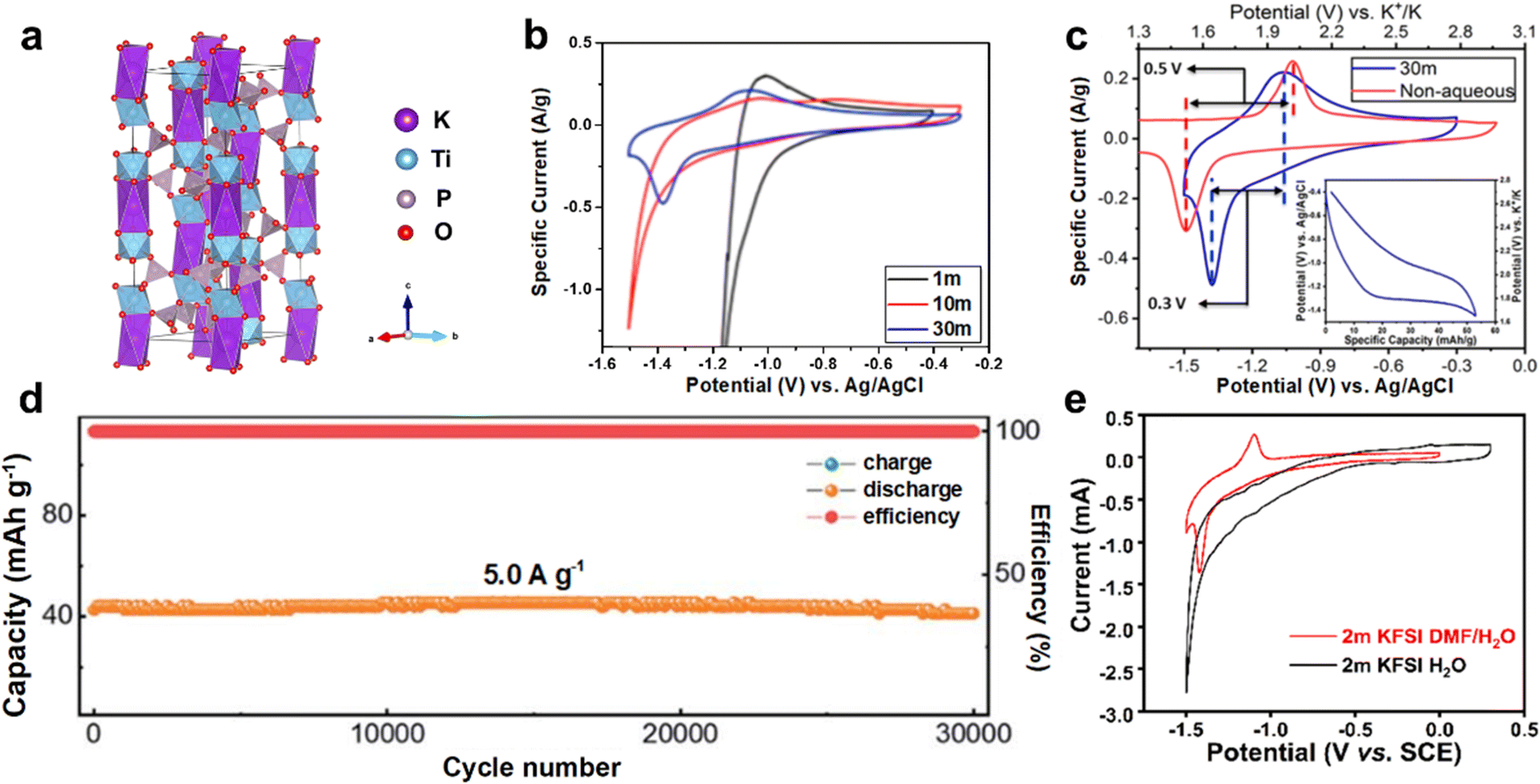 | ||
| Fig. 9 (a) Crystal structure of KTi2(PO4)3. (b) CV curves of KTi2(PO4)3 in different concentration KAc electrolytes at 0.5 mV s−1. (c) CV curves of KTi2(PO4)3 in aqueous and nonaqueous electrolytes. Reproduced with permission from ref. 84. Copyright 2018, American Chemical Society. (d) Long-term cycling performance of AKIB full cell consisted of K-FeHCF cathode and KTi2(PO4)3/C anode at 5.0 A g−1. Reproduced with permission from ref. 83. Copyright 2021, Royal Society of Chemistry. (e) CV curves of KTi2(PO4)3@C in 2 M KFSI electrolytes with and without DMF. Reproduced with permission from ref. 86. Copyright 2021, American Chemical Society. | ||
As an easy-to-handle technology, carbon coating or carbon composites shows great advantages in enhancing the electronic conductivity of anode materials. Recently, Li et al. synthesized KTi2(PO4)3/C nanoparticles that showed rapid delivery of K+.83 A full cell with KTi2(PO4)3/C anode, iron hexacyanoferrate (K2FeFe(CN)6) cathode, and 21 M KCF3SO3 WiSE electrolyte was constructed. The full cell achieved a discharging capacity of 73 mA h g−1 (based on KTi2(PO4)3/C anode) and an average discharge potential of 1.5 V. Even at 5 A g−1, the AKIB full cell exhibited little capacity loss of 3.3% after 30000 cycles (Fig. 9d). Besides, the KTi2(PO4)3/C showed fast reaction kinetics and maintained near-zero strain during K+ storage, thus achieving excellent performance.
To further enhance the electrochemical performance of KTi2(PO4)3@C, electrolyte additives are adopted. A hybrid electrolyte based on KFSI was designed by Li et al.86 DMF was introduced into the KFSI dilute electrolyte, which not only broadened the ESW of the electrolyte, but also modified the cycling stability of the electrode. Using 2 M KFSI DMF/H2O electrolyte, the KTi2(PO4)3@C anode exhibited distinct redox peaks in the CV curves, while almost no peaks were observed when using 2 M KFSI aqueous electrolyte (Fig. 9e). Therefore, the hybrid electrolyte stimulated the electrochemical activity of the KTi2(PO4)3@C due to the presence of DMF. Moreover, after 200 cycles, the KTi2(PO4)3@C anode showed a high capacity of 76 mA h g−1, indicating its good cycling stability.
In summary, titanium phosphate is still an ideal anode for AKIBs due to its small volume change during K+ storage, relatively good cycling stability, and nontoxicity. Note that the main drawbacks of titanium phosphate as anode for AKIBs are unsatisfactory discharge capacity and rate capability. Therefore, further improvements in capacity, cyclability, and rate performance are urgently needed. The optimization strategies, such as carbon coating, hybrid composites, and electrolyte engineering, are promising for the high performance of AKIBs.
Since Dahn et al. first used VO2 as an anode for aqueous LIBs in 1994, other vanadium-based electrode materials have been developed as anodes for AKIBs.8 Jiang et al. designed amorphous/crystalline K0.25V2O5·nH2O/o-V2O5 dual phases (ac-KxV2O5) anode for aqueous K+ micro-batteries (AKMBs).90 Amorphous K0.25V2O5·nH2O acted as a molecular pillar between layers, which not only enabled ac-KxV2O5 to possess new accommodation sites and ion diffusion paths, but also maintained the larger interfacial spacing of crystalline V2O5 bilayers (Fig. 10a). Such nanostructure was beneficial for the hydrated K+ intercalation/de-intercalation in ac-KxV2O5, whereas the K+-free ac-V2O5 was unfavorable for K+ storage, due to narrow interlayer spacing, sluggish diffusion kinetics, and drastic volume changes. Compared to c-KxV2O5 and a-KxV2O5, the capacity of ac-KxV2O5 was significantly improved due to its biphasic nanostructure, which provided more space for hydrated K+ and promoted K+ diffusion kinetics between layers (Fig. 10b). When coupled with the c-KxMnO2 cathode, the full microcells possessed a wide working voltage window of 1.6 V. Stable cyclability with ∼80% capacity retention over 10000 cycles was maintained at 500 mV s−1. Besides, the volumetric energy density (∼103 mW h cm−3) and power density (∼600 W cm−3) of AKMB far exceeded most reported aqueous systems at that time.
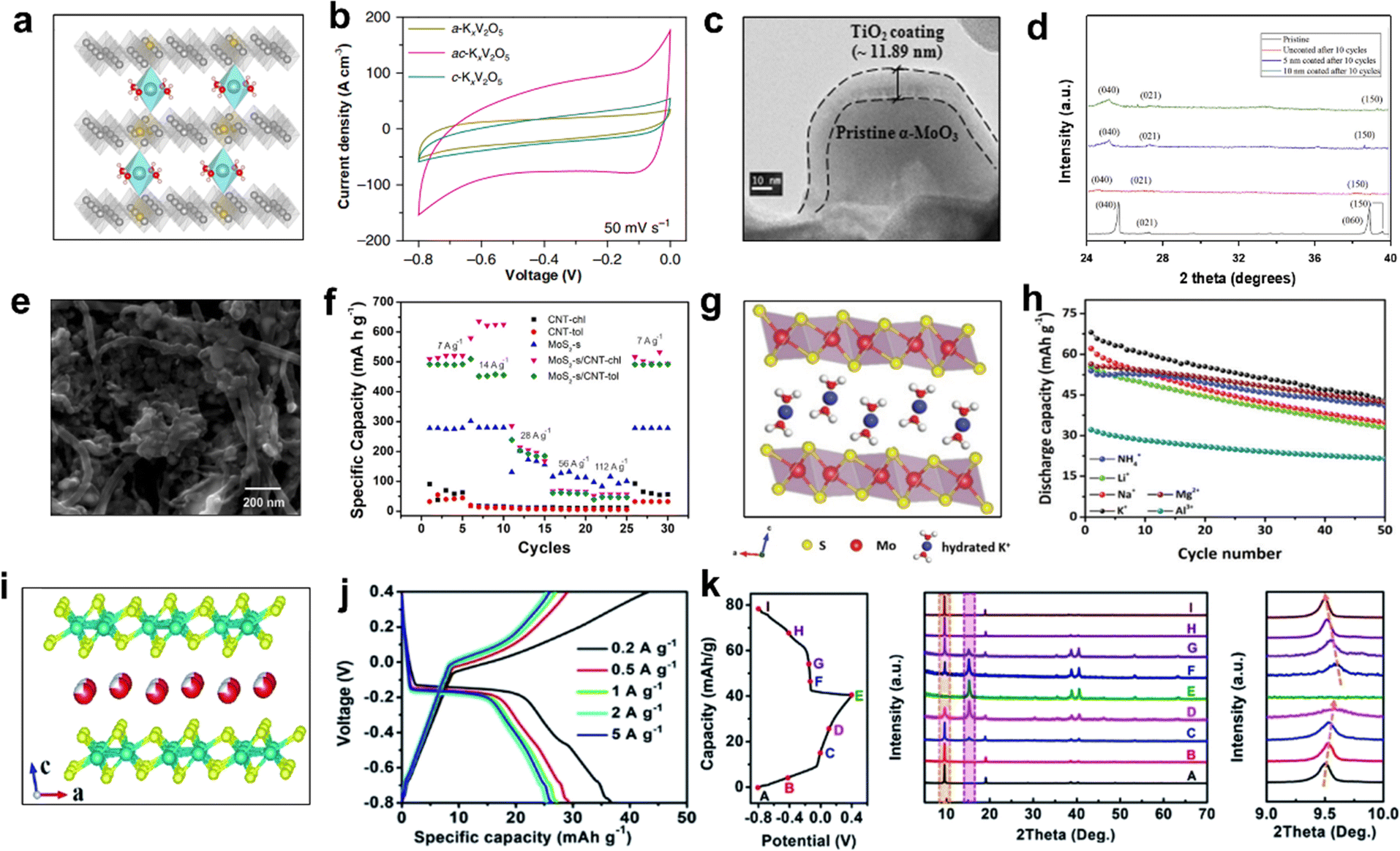 | ||
| Fig. 10 (a) Crystal structure of ac-KxV2O5. (b) CV curves of a-KxV2O5, ac-KxV2O5, and c-KxV2O5. Reproduced with permission from ref. 90. Copyright 2019, Springer Nature. (c) TEM image of α-MoO3 with TiO2 coating. (d) XRD patterns of α-MoO3 samples with and without coating after cycles. Reproduced with permission from ref. 89. Copyright 2016, Elsevier Inc. (e) SEM image of MoS2-s/CNT-chl. (f) Rate performance of CNT and Mo-based samples. Reproduced with permission from ref. 91. Copyright 2021, Elsevier Inc. (g) Crystal structure and (h) cycling performance with different cations at 0.5 A g−1 of K0.38(H2O)0.82MoS2. Reproduced with permission from ref. 88. Copyright 2020, The Royal Society of Chemistry. (i) Crystal structure, (j) GCD profiles at different current densities, and (k) ex situ XRD patterns of K0.36(H2O)yWS2. Reproduced with permission from ref. 87. Copyright 2019, The Royal Society of Chemistry. | ||
Molybdenum (Mo)-based materials (such as MoO3 and MoS2) have been studied extensively as the intercalation anodes for LIBs and SIBs.103,104 Park et al. demonstrated the effect of material strain on α-MoO3 anode for AKIBs with TiO2 film deposited on the surface using the ex situ XRD method.89 The TiO2 layer was 11 nm thick after 10 nm ALD deposition, confirming the approximate coating thickness applied (Fig. 10c). The XRD patterns of α-MoO3 and α-MoO3 coated with 5 nm and 10 nm TiO2 after 10 discharge/charge cycles revealed the improved structure stability with the increase of TiO2 coating thickness (Fig. 10d). As a result, the 10 nm-coated sample exhibited the highest capacity retention, attributed to the low strain at the interface of amorphous TiO2/crystalline α-MoO3. Moreover, Zarbin et al. prepared several Mo-based/carbon nanotube (CNT) nanocomposites by a simple liquid/liquid interfacial route (LLIR)-based method.91 It can be noticed that MoS2 and CNT-chl mixed homogeneously (Fig. 10e). Most of the GCD profiles showed plateaus at 0.1 V, indicating that K+ was intercalated in MoS2 bulk. The ultrahigh specific capacity of 517 mA h g−1 made it promising for AKIBs (Fig. 10f). In addition, Huang et al. discovered that the hydrated Mo-based ternary sulfide K0.38(H2O)0.82MoS2 reversibly stored a variety of hydrated cations (K+, Li+, Na+, Mg2+, Al3+, NH4+).88 Similar to 2H MoS2, K0.38(H2O)0.82MoS2 exhibits a typical layered structure, but the intercalation of hydrated K+ increases the interlayer spacing from 6.2 Å to 9.3 Å (Fig. 10g). The higher electrical conductivity of K0.38(H2O)0.82MoS2 is due to the presence of twisted [MoS6] octahedra in the [Mo–S] layer. The redox peaks of CV curves were mainly located between −0.4 and 0.4 V. Interestingly, K0.38(H2O)0.82MoS2 possessed a discharge capacity of ∼68 mA h g−1 for K+ storage, which exceeded other guest ions (Fig. 10h). Based on the results of ex situ XRD and XPS characterizations, a typical two-phase reaction mechanism was revealed, with the first phase transition between K0.38(H2O)0.82MoS2 and 1T′ MoS2, followed by the reversible intercalation and deintercalation between 1T′ MoS2 and Ax(H2O)yMoS2.
Tungsten disulfide (WS2) has also been regarded as a promising electrode material due to the large lattice spacing of 6.2 Å and the weak van der Waals interactions.105 Recently, Huang et al. synthesized K0.36(H2O)yWS2 layered compound as a novel anode material for AKIBs.87 The structure of K0.36(H2O)yWS2 consisted of WS2 layers and interlayer hydrated K+, where the WS2 layers were connected by shared twisted [WS6] octahedra (Fig. 10i). This electrode exhibited large interlayer spacing of 9.258 Å and high conductivity of 35.8 S m−1. When used as anode for AKIBs, two obvious plateaus at ∼−0.15 V in the discharge profile and ∼0.05 V in the charge profile were observed, corresponding to the K+ intercalation/deintercalation process (Fig. 10j). The K0.36(H2O)yWS2 obtained a high capacity of 43.3 mA h g−1 at 0.2 A g−1 within −0.8 to 0.4 V vs. Ag/AgCl. The good rate performance of K0.36(H2O)yWS2 was ascribed to the high ionic conductivity and large interlayer spacing. Based on ex situ XRD results, the highly reversible storage mechanism of hydrated K+ in K0.36(H2O)yWS2 electrode was revealed (Fig. 10k).
Note that metal oxides/sulfides are mainly divided into two categories: intercalation mechanism and conversion mechanism. Most of the intercalated anodes exhibit stable electrochemical performance. However, these materials show low specific capacities and high operating voltages, resulting in low energy densities. For conversion materials, large volume changes caused by conversion reaction will lead to pulverization of the active material, resulting in capacity decay. In addition, large voltage hysteresis and poor first Coulombic efficiency (CE) lead to capacity loss. Therefore, despite the promise of high capacity for this class of materials, these major issues cannot be ignored. Metal oxides/sulfides are still in the primary stages of development as AKIB anode materials, so advanced structural design (nanoengineering or coating) and tuning of electrode/electrolyte interfaces are efficient strategies to achieve highly stable oxides/sulfides anodes.
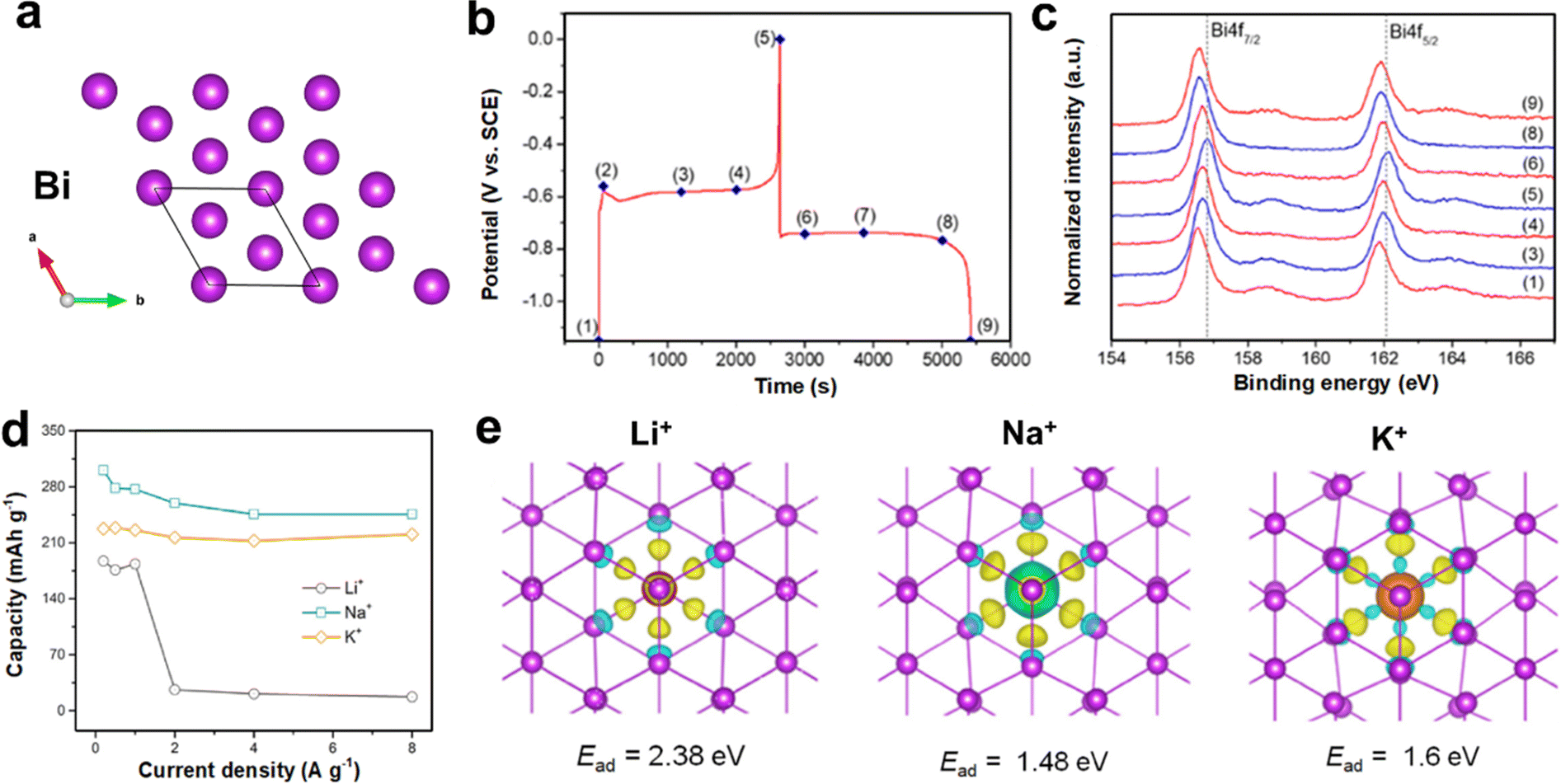 | ||
| Fig. 11 (a) Crystal structure of Bi. (b) The second GCD profiles of Bi anode at 1 mA cm−2. (c) Corresponding ex situ XPS spectrum. Reproduced with permission from ref. 92. Copyright 2020, Elsevier Inc. (d) Rate performance of Bi anode in K+/Na+/Li+ electrolytes. (e) The charge density difference induced by three ions absorption, respectively. Reproduced with permission from ref. 93. Copyright 2022, Elsevier Inc. | ||
In order to deepen the understanding of the energy storage mechanism of Bi anode, Zheng et al. carried out a systematic work about Bi storage of different alkali metal ions (Li+, Na+, and K+).93 The overpotential and voltage polarization in Li+ electrolyte were the largest, followed by K+ electrolyte, and the smallest in Na+ electrolyte. This means that the dealloying reaction of Bi with Li+ was the most difficult during the entire discharge process, while Na+ was the easiest. As a result, the Bi anode exhibited the highest capacity and the best rate performance for Na+ storage, followed by K+ storage, while it exhibited the lowest capacity and the fastest decay in terms of Li+ storage (Fig. 11d). Based on the charge density difference, the adsorption energy of Li+ in Bi layers was highest, manifesting the strongest interaction between Li+ and Bi (Fig. 11e). In addition, 8 Li and 8 Na atoms transferred charges to Bi, which were two atoms more than that of K, probably due to the large radius of K+. Larger volume expansion would cause the capacity of K+ storage to decline more seriously than that of Na+ storage.
In summary, alloy material exhibits good electrochemical performance in AKIBs. However, the research on the emerging alloy anode materials for AKIBs is still in its infancy. The main disadvantages of these materials are large volume expansion, various side reactions, and high voltage hysteresis, resulting in poor cycling performance and CEs. With the further development of electrode structure design, as well as in-depth insights into the K+ storage mechanism and advanced electrolytes, alloy anodes are promising to become one of the stable and high-capacity anodes for AKIBs.
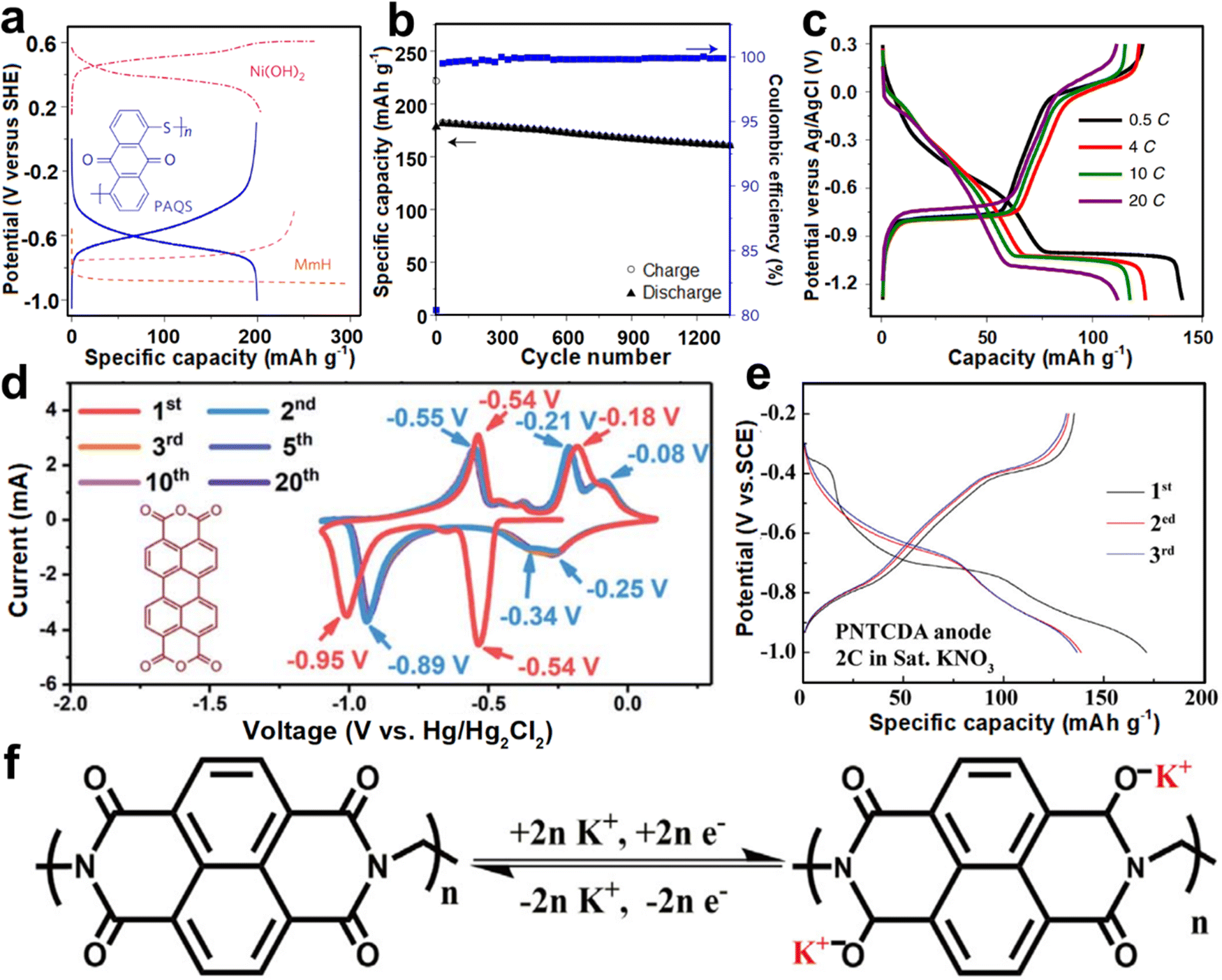 | ||
| Fig. 12 (a) GCD profiles of PAQS, MmH, and Ni(OH)2. (b) Cycling performance of a PAQS–Ni(OH)2 cell at 1C. Reproduced with permission from ref. 96. Copyright 2017, Springer Nature. (c) GCD profiles of PTCDI at different rates. Reproduced with permission from ref. 44. Copyright 2019, Springer Nature. (d) CV curves of β-PTCDA at 10 mV s−1. Reproduced with permission from ref. 94. Copyright 2020, The Royal Society of Chemistry. (e) GCD profiles of PNTCDA. (f) Schematic illustration of the electrochemical mechanism of PNTCDA. Reproduced with permission from ref. 95. Copyright 2020, Elsevier Inc. | ||
Hu et al. proposed 3,4,9,10-perylenetetracarboxylic diimide (PTCDI) as an anode for aqueous K+ storage.44 PTCDI anode showed two couples of redox voltage plateaus at −0.45/−0.77 V and −1.05/0.13 V (vs. Ag/AgCl) and achieved a discharging capacity of 125 mA h g−1 at 0.5C (Fig. 12c). Superior rate performance (110 mA h g−1 at 20C) and good cyclability (77% capacity retention over 1000 cycles) were also obtained. The common dissolution of PTCDI anode during the discharge/charge cycle was suppressed when using the concentrated electrolyte. When assembled with the KFeMnHCF cathode, the AKIB full cell possessed a discharge voltage of 1.27 V and remarkable cycling stability. Soon after, Wang et al. utilized concentrated electrolyte to suppress the HER of β-perylene-3,4,9,10-tetracarboxylic dianhydride (β-PTCDA) during the discharge/charge process.94 The redox peaks associated with reversible potassiation/de-potassiation with the carbonyl groups occurred within −1.1 to 0.1 V (vs. Hg/Hg2Cl2) (Fig. 12d), which may be ascribed to the reversible transition reaction between the C–O–K and CO bonds. β-PTCDA possessed an initial capacity of 145 mA h g−1 at 0.2 A g−1 and subsequently a stable capacity of 130 mA h g−1, which were induced by the two-electron reaction from β-PTCDA to K2PTCDA. Besides, Li and co-workers reported another redox-active polyimide, namely, 1,4,5,8-naphthalenetetracarboxylic dianhydride-derived polyimide (PNTCDA).95 PNTCDA anode exhibited high reversibility with a discharge capacity of 135 mA h g−1 at 2C within −1 to −0.2 V (vs. SCE) in KNO3 electrolyte (Fig. 12e). Compared with the K2SO4 electrolyte, PNTCDA showed higher rate performance in the KNO3 electrolyte. Besides, an enolization process mainly occurred when combined with K+ insertion/extraction, where charge redistribution in the conjugated structure promoted the reaction (Fig. 12f).
In a word, the main challenge of organic electrode materials is the dissolution in electrolytes. This problem could be greatly reduced with some engineering techniques (high-concentration electrolytes and additives). However, further in-depth research is needed to improve the electronic conductivity and energy density in order to meet the needs of practical applications. One of the common strategies to improve electrical conductivity is to incorporate conductive carbon materials when preparing electrodes, but this method will reduce the tap density of electrodes to a certain extent.
3. Electrolytes for AKIBs
Electrolytes, as ion transport media between cathode and anode, are especially critical in battery systems.117–123 The liquid electrolyte of AKIBs is to dissolve K salt in water in a certain proportion. The most representative K salts for AKIBs are listed in Table 3 and their physical and chemical properties are summarized. Compared to organic solvents, water is an attractive alternative owing to its fundamental safety, higher ionic conductivity, and low cost.124 The electrolytes used in AKIBs can be classified into two catalogs, including conventional liquids and hydrogels.| K Salts | Solubility (g per 100 g H2O) | Molality (mol kg−1) | Ionic Conductivity (maximum value, mS cm−1) | Cost ($ per kg) | Toxicity | Ref. |
|---|---|---|---|---|---|---|
| K2SO4 | 12.0 | 0.7 | 78.01 (0.48 M) | 95.5 | Low | 125 |
| KCl | 35.5 | 4.8 | 108.6 (1 M) | 132 | Nontoxic | 125 |
| KNO3 | 38.3 | 3.8 | 74.28 (0.79 M) | 122 | Low | 125 |
| KOH | 121 | 21.6 | 626.6 (6 M) | 62.1 | Moderate | 126 |
| KPF6 | 9.3 | 0.5 | — | 363 | Low | 89 |
| KFSI | 657.7 | 30 | ∼120 (∼5 M) | 323 (100 g) | Nontoxic | 94 |
| KCF3SO3 | 414.0 | 22 | ∼140 (∼6 M) | 104 (25 g) | Nontoxic | 44 |
| HCOOK | 331 (18 °C) | 39.4 | 130 (10 M) | 104 | Nontoxic | 85 |
| KAc | 269 | 27.4 | ∼112 (∼5 M) | 330 (2.5 kg) | Low | 84 |
3.1. Aqueous liquid electrolytes
WiSE electrolytes have been successively used in AKIBs, including 22 M KCF3SO3, 27.8 M K(PTFSI)0.12(TFSI)0.08(CF3SO3)0.8·2H2O, 30 M KAc, 30 M KFSI, 40 M HOOCK, and 61.7 M KFSI + KCF3SO3.44,84,85,94,131,132 High concentration electrolytes show several advantages as follows: (1) concentrated electrolytes can widen the ESW. Ji et al. first investigated KAc-based WiSE electrolytes in AKIBs, and found that the ESW could be widened to 3.2 V (Fig. 13a).84 Hu et al. reported an AKIB full cell with KCF3SO3-based WiSE electrolytes.44 The 22 M KCF3SO3 electrolyte exhibited a wider ESW (3 V, from −1.3 to 1.7 V vs. Ag/AgCl), and its redox potential is lower than that in 1 M KCF3SO3 electrolyte. Cheng et al. proposed 40 M HCOOK-based WiSE electrolytes, showing a wide ESW up to 4 V.85 Wang et al. explored 30 M KFSI-based WiSE electrolyte with the ESW of 3.97 V.94 (2) Concentrated electrolytes can improve the electronic conductivity. At the same concentration, the conductivity of KAc-based electrolytes is higher than that of other reported fluorinated imide salts such as LiTFSI.84 The higher conductivity may be ascribed to the dominance of acetate salt or the Lewis weak acidity of K+. Compared with LiCF3SO3 and NaCF3SO3, the high concentration of KCF3SO3 exhibited a series of excellent properties, high ionic conductivity (76 mS cm−1 at room temperature), low viscosity, and wide voltage window (Fig. 13b).44 The conductivity of HOOCK-based electrolyte reached about 130 mS cm−1 at 10 M and still maintained 46 mS cm−1 even at 40 M, which exceeded other fluorinated imine-based WiSE electrolytes and previously reported KAc.85 The electrical conductivity of KFSI-based electrolytes also surpasses those of LiTFSI-based electrolytes and KAc-based electrolytes at the same concentrations.94 (3) Concentrated electrolytes can effectively prohibit the dissolution of electrode. UV-vis spectra showed the existence of strong adsorption of V3+ ions in the cycled 1 M KCF3SO3-based electrolyte, while no adsorption occurred in the 22 M KCF3SO3-based electrolyte, indicating that the high concentration electrolyte inhibited the dissolution of the δ-K0.5V2O5 (Fig. 13c).54 Similarly, the electrolyte turned pink after 5 cycles for PTCDI anode in 1 M KCF3SO3 electrolyte, while it was still transparent in 22 M KCF3SO3 electrolyte, indicating that the dissolution in the concentrated electrolyte was effectively suppressed.44 A similar phenomenon was also observed in 30 M KFSI-based electrolyte, which ensured the integrity of β-PTCDA and achieved long-term cycling performance.94
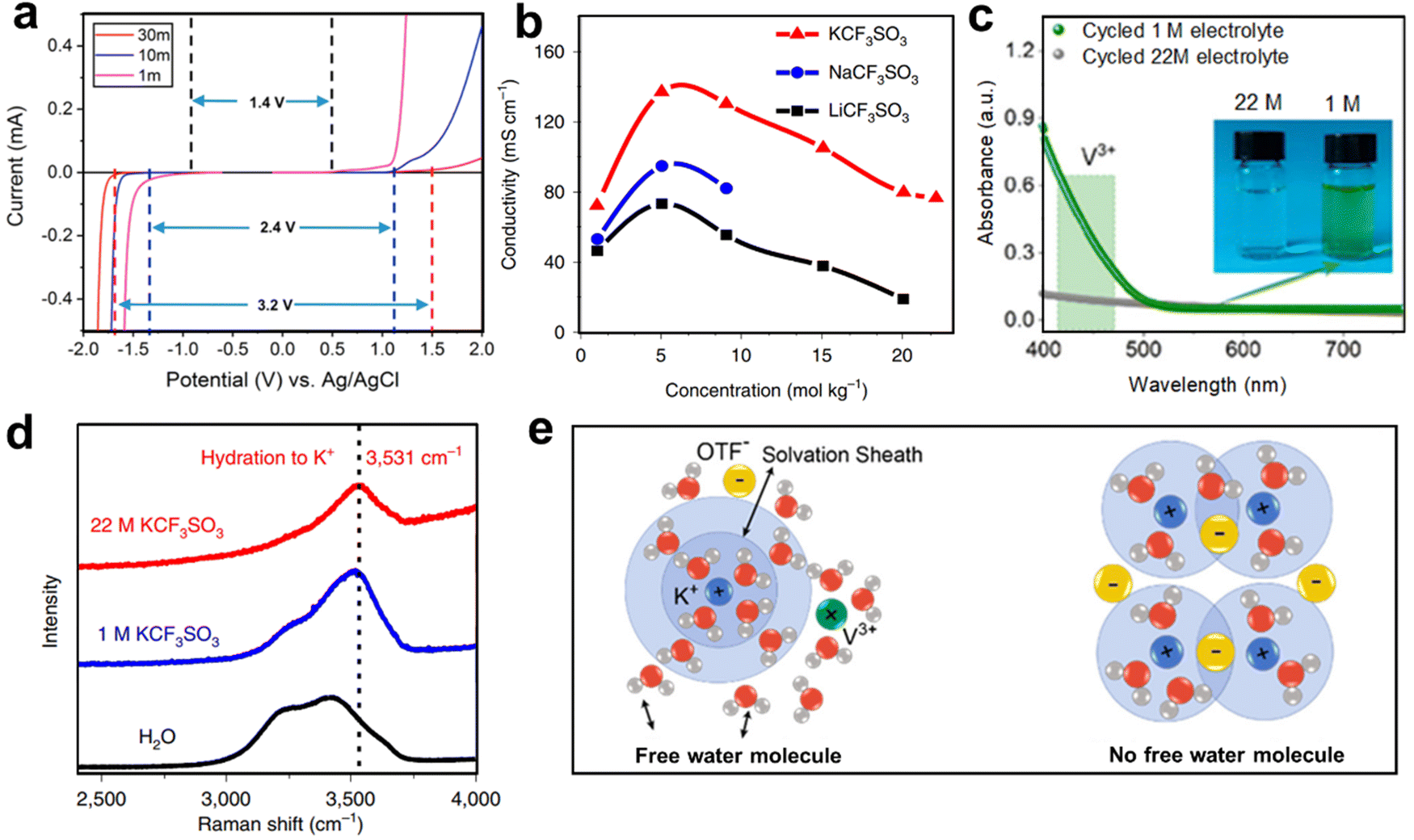 | ||
| Fig. 13 (a) Linear sweep voltammetry of KAc-based electrolytes. Reproduced with permission from ref. 84. Copyright 2018, American Chemical Society. (b) Ionic conductivity of different concentration LiCF3SO3-, NaCF3SO3- and KCF3SO3-based electrolytes. Reproduced with permission from ref. 44. Copyright 2019, Springer Nature. (c) UV-vis spectroscopy of the cycled 1 and 22 M KCF3SO3-based electrolytes. Reproduced with permission from ref. 54. Copyright 2021, American Chemical Society. (d) The Raman spectrum of two electrolytes and H2O. Reproduced with permission from ref. 44. Copyright 2019, Springer Nature. (e) Schematic illustration of the solvation structure in two electrolytes. Reproduced with permission from ref. 54. Copyright 2021, American Chemical Society. | ||
It is worth mentioning that the widened voltage window and suppressed dissolution properties are associated with the reduction of free water in concentrated electrolytes. As shown in Fig. 13d, the water state in 1 M electrolyte was similar to that of pure water, indicating that a large amount of free molecular water existed in the electrolyte with low concentration, so that the V3+ species could be dissolved in the electrolyte.44 In the 22 M electrolyte, weak hydrogen bonds remained and shifted to the right, while strong hydrogen bonds gradually disappeared, indicating that the strong K+ solvation effect confines water molecules. The confined free water in WiSE only dissolved the limited V3+, which hindered the dissolution of the active electrode (Fig. 13e).54
Sugar, as an organic additive, can decrease the content of free water and change the tetrahedral structure of water. Xue et al. explored inexpensive, readily available sugar-concentrated aqueous electrolytes for AKIBs.31 The amount of free water molecules decreased with the increase of sugar concentration. The tetrahedral structure of water was destroyed by the super-concentrated sugar, which broke the hydrogen bonds to reduce the binding degree of water molecules. These sugar-based electrolytes exhibited a wide ESW of ∼2.812 V, wide operating temperature range, and good ionic conductivity. Ultra-concentrated maltose significantly improved the first discharge capacity, which was increased from 29.2 mA h−1 to 69.6 mA h−1 after adding super-concentrated maltose (Fig. 14a and b). Besides, the capacity retention over 2000 cycles in 1 M KNO3/60.0 wt% maltose electrolyte was higher than that of in 1 M KNO3 electrolyte.
 | ||
| Fig. 14 GCD profiles of PB-K//AC battery in (a) KNO3 electrolyte and (b) KNO3/60.0 wt% maltose electrolyte. Reproduced with permission from ref. 31. Copyright 2020, Wiley-VCH. (c) ESW of KFSI in aqueous electrolytes with and without DMF. (d) Ex situ XPS spectrum of F1s in 2 M KFSI DMF/H2O electrolyte. Reproduced with permission from ref. 86. Copyright 2021, American Chemical Society. (e) In situ XRD patterns and the corresponding GCD profiles in 1.0 M KCF3SO3 and 3.0 M KCF3SO3 + 2.0 M Zn(CF3SO3)2 electrolytes. Reproduced with permission from ref. 48. Copyright 2021, Wiley-VCH. (f) Schematic illustration of the electrochemical behavior of KMnF electrode in 21 M KCF3SO3 aqueous electrolyte with and without Fe(CF3SO3)3. Reproduced with permission from ref. 46. Copyright 2021, Springer Nature. | ||
DMF, as another organic additive, can promote the anions decomposition and promote the formation of robust solid electrolyte interfaces (SEI). A 2 M KFSI diluted electrolyte using a mixed solvent of N,N-dimethylformamide/water (DMF/H2O) was developed by Li et al.86 The addition of DMF enabled the electrolyte with a window of 2.89 V, which is wider than the bare water electrolyte (2.66 V), along with low viscosity, high conductivity, and high safety (Fig. 14c). Also, DMF solvent showed a lower LUMO energy than that of H2O solvent, which could facilitate the decomposition of FSI− to form –CFx and KF, generating a robust solid electrolyte interphase (Fig. 14d).
Zn(CF3SO3)2, as an ionic additive, can change the K+ storage mechanism of electrode materials through the liquid phase. Mai et al. found different electrochemical energy storage mechanisms of K2.55Zn3.08[Fe(CN)6]2·0.28H2O (KZnHCF) electrode material in different electrolytes.48 Both solid solution and two-phase mechanisms were found in 0.5 M K2SO4 and 1.0 M KCF3SO3 electrolytes, while only the solid solution mechanism was found in 5.0 M KCF3SO3 and 3.0 M KCF3SO3 + 2.0 M Zn(CF3SO3)2 electrolytes (Fig. 14e). Zn2+ acted as a replacement for K+ in the electrolyte, resulting in the significantly enhanced structural stability of KZnHCF during K+ insertion/extraction. This liquid-phase-induced solid-solution-phase ion storage mechanism enabled stable cycling of the KZnHCF electrode material for 10000 cycles.
Fe(CF3SO3)2, as another ionic additive, can inhibit the dissolution of the material by surface modification. Lu et al. developed an in situ electrochemical cation substitution method by adding a certain amount of iron ions (Fe3+) into the electrolyte.46 The surface of K1.82Mn[Fe(CN)6]0.96·0.47H2O (KMnF) was modified and converted into a KFexMn1−xF electrode by the electrolyte (Fig. 14f). Although the initial dissolution of manganese was not suppressed, the introduced Fe3+ filled the vacancies generated by the manganese dissolution during the discharge process, so that the entire framework of the PB material was stabilized. The substitution reaction occurred on the electrode surface, so that a film was formed, which could inhibit the further dissolution of Mn and absorption of Fe.
3.2. Hydrogel electrolytes
Liquid electrolytes are common in aqueous energy storage devices due to their high ionic conductivity. However, liquid electrolyte devices may suffer from electrolyte leakage and unsatisfactory dislocations when stressed. To avoid these issues, hydrogel electrolytes are introduced, i.e., some hydrophilic polymers can be introduced into the K salt aqueous solution. Hydrogel electrolytes exhibit high physical flexibility and good electrochemical performance.148–152Polyvinyl alcohol (PVA) and carboxymethyl cellulose sodium (CMC) based hydrogel has been used in AKIBs. Ma et al. assembled a flexible solid-state symmetric AKIB with PB electrode and KCl-PVA hydrogel electrolyte (Fig. 15a).32 It is worth noting that an ESW of ∼1.3 V and an ionic conductivity of 3.7 × 10−2 S cm−1 could be achieved using PVA-based hydrogel electrolyte, which provided satisfactory feasibility to fabricate flexible batteries. The flexible full cell exhibited an average potential of ∼0.6 V with a high capacity of 58 mA h g−1 at 100 mA g−1 (Fig. 15b). An outstanding capacity retention of 98.5% at the bending angle of 60° was delivered. However, the following capacity decayed during the cycle test may be caused by the evaporation of water that reduces the ionic conductivity and increases the interfacial overpotential. Soon after, Jiang et al. assembled an all-solid-state flexible AKMB on a poly(methyl methacrylate) (PMMA) substrate using KCl-PVA hydrogel electrolyte to meet the specific micropower requirements of wearable electronic devices (Fig. 15c).90 The all-solid-state AKMB exhibited almost no capacity fading when bent to 90° due to its outstanding mechanical flexibility (Fig. 15d). In addition, AKIB was assembled with thin-film solar cells into a self-powered system, powering the LED lights when sunlight was available, showing potential applications in electronic microdevices (Fig. 15e).
 | ||
| Fig. 15 (a) Schematic illustration of symmetric AKIB with KPB bipolar electrodes and PVA hydrogel electrolyte. (b) GCD profiles of a full cell at 100 mA g−1. Reproduced with permission from ref. 32. Copyright 2018, The Royal Society of Chemistry. (c) Photograph of flexible AKMBs with PVA hydrogel electrolyte. (d) Capacity retention of flexible AKMBs under different bending angles. (e) Schematic illustration of a self-powered system assembled with flexible AKMBs and thin-film solar cells. Reproduced with permission from ref. 90. Copyright 2019, Springer Nature. (f) Diffusion coefficient of electrolyte component with and without CMC. (g) Cycling performance of KMHCF with the liquid and gel electrolytes. Reproduced with permission from ref. 47. Copyright 2020, Elsevier Inc. | ||
Recently, Varzi et al. developed another hydrogel electrolyte by adding CMC to a highly concentrated aqueous electrolyte.47 The instability issue of KMHCF in the alkaline environment caused by the acetate-based electrolyte was solved. Molecular dynamics simulation calculations showed that the presence of CMC lowered the movement of water molecules and acetate ions by ∼2 times, while the movement of K+ was 2.2 times faster when the electrolyte viscosity increased (Fig. 15f). The hydrogel electrolyte possessed a high ionic conductivity of 22.3 mS cm−1 and a wide ESW. As a result, KMHCF with gel electrolyte showed better cyclability and higher CEs than liquid electrolyte due to the reduced Fe and Mn dissolution (Fig. 15g).
In conclusion, the electrochemical performance of a battery is closely associated with electrolytes. In different electrolyte systems, electrode materials exhibit completely different electrochemical behaviors. Despite these advances, hydrogel electrolytes still lack some properties suitable for commercial applications, such as lower ionic conductivity, poor electrode/electrolyte interface, and higher overpotentials compared with liquid electrolytes. Therefore, further research is needed in the field of electrolytes to match the cathode and anode to make this potential system useful for practical applications.
4. Full cells for AKIBs
The significant challenges that AKIB still faces are not limited to improving the operating voltage and capacity of half-cells. More importantly, the cathode and anode materials can be combined to work together to ensure that the full cells meet the energy density requirements. Ensuring the economic sustainability of AKIB full cells has significant implications for large-scale energy storage needs. AKIB full cells consist of cathodes, anodes, separators, and electrolytes, and these components can be used to design full-cells in different configurations as needed. In the laboratory, components for full-cell designs are typically assembled in coin cell-type devices (CR2016, CR2025, or CR2032). Currently commonly used cathode materials for AKIB full cells include PBA, layered vanadium oxide, and hydroxide. The anode materials matched with them include organics, KTi2(PO4)3, alloy materials, and Zn flakes. It is worth noting most of the cathodes and anodes of full cells usually require an activation process, i.e., pre-potassiation, in the half-cell to eliminate the detrimental effects during initial cycling. In addition, before full-cell assembly, it is often necessary to control the mass ratio of anode and cathode to optimize their charge balance.Fig. 16 shows the electrochemical performance of representative AKIB full cells. PBAs and organic materials were the most frequently used cathode and anode materials, respectively. KZnHCF//Zn and KMnF//PTCDI exhibited the highest voltage output (∼1.87 V) and the highest specific capacity (75 mA h g−1), respectively, however, these two satisfactory combinations still need further improvement (Fig. 16a).46,48 As shown in Fig. 16b, some systems, such as K-FeHCF//KTi2(PO4)3/C and δ-K0.25V2O5//PTCDI, still displayed considerable capacity retention even after tens of thousands of cycles.54,83 The energy densities of most systems were less than 100 W h kg−1, indicating that the cyclability and power output may be sufficient, while the energy densities were still lower than aqueous batteries using other metal carriers (Fig. 16c). It is noteworthy that most AKIB full-cell systems (K-FeHCF//KTi2(PO4)3/C, KFHCF//β-PTCDA, δ-K0.5V2O5//PTCDI, and KFeMnHCF//PTCDI) were assembled based on high-concentration electrolytes, which would increase the cost and reduce the energy density of the battery.44,54,83,94 In addition, pouch cells (KFeMnHCF//PTCDI, KMnF//PTCDI, and δ-K0.25V2O5//PTCDI) exhibited excellent wide temperature performance, showing application potential under extreme conditions.44,46,54
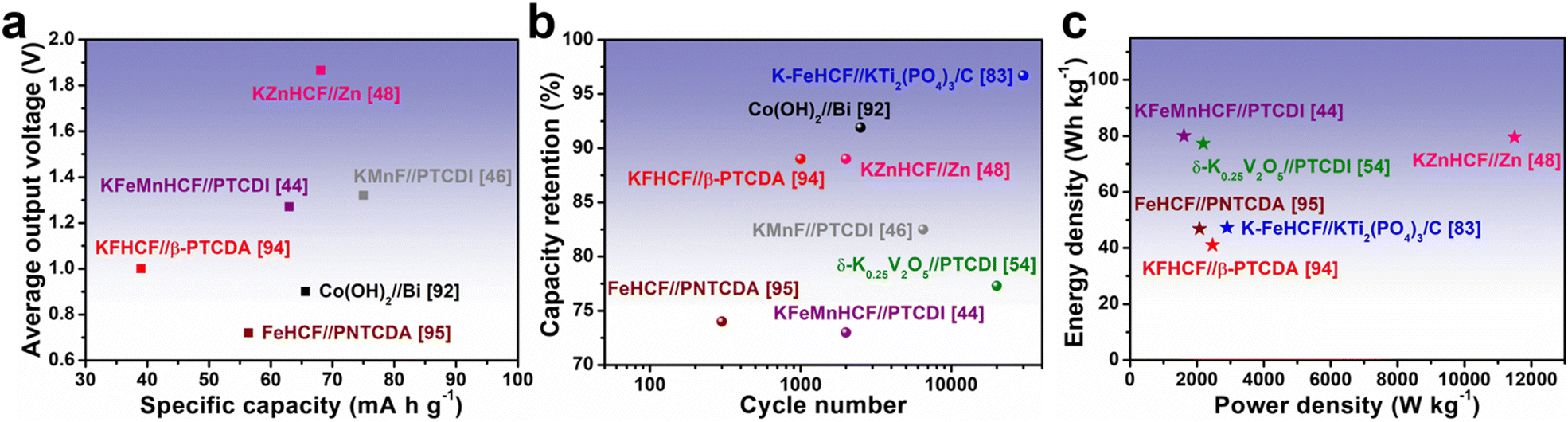 | ||
| Fig. 16 (a) Average output voltage versus specific capacity, (b) capacity retention versus cycle number, and (c) energy density versus power density of selected AKIB full cells reported in the literature. | ||
To sum up, the operating voltage, cycle performance, and cost of the cathode are the bottlenecks restricting its further practical application. The electrochemical behavior of cathodes and anodes of AKIBs, such as capacity utilization, balance, and electrolyte compatibility, requires in-depth study. In particular, electrolyte compatibility between electrode and electrolyte or between cathode and anode electrodes is an important factor.
5. Summary and perspectives
AKIBs have received increasing attention in the field of large-scale ESSs thanks to their low price, high power density, environmental friendliness, and high safety. This review systematically overviewed the recent advances in AKIB research and discussed strategies for improving electrochemical performance, including cathodes, electrolytes, and anodes. Although some remarkable advances have been achieved in AKIBs, further research is needed to fulfill the requirements of practical applications of high-performance AKIBs. This section will evaluate the current key electrode materials, characterization techniques, theoretical calculations, and applications (Fig. 17). | ||
| Fig. 17 The opportunities and future directions for achieving high-performance AKIBs. | ||
5.1 Key materials
Designing high-performance electrode materials for AKIBs is more challenging than ALIBs and ASIBs, because K+ is much larger than Na+ and Li+. Current cathode materials for AKIBs still exhibit limited weight/volume energy capacity. PB and PBAs show superior cycling stability and high voltage platform, which are common cathode materials for AKIBs. However, the capacities of PB and PBAs need to be further boosted before successful commercialization. Vanadium-based oxides are important electrodes due to their reversibility, but the output voltage is slightly inferior to PB. Polyanionic compounds and MXenes as K+ storage cathode materials exhibit high working voltage, but low capacity.For the anode materials for AKIBs, the current research is still mainly focused on organic materials and polyanionic compounds, and attempts to use metal compounds/sulfides and alloy-based materials, etc. Although the developed organic electrode materials exhibit excellent K+ storage ability, most of them benefit from the concentrated electrolytes, and the issue of easy dissolution requires extra strategies to improve. NASICON-type KTi2(PO4)3 shows relatively low potential but good cycling stability, and their low capacity limits their practical applications. Despite the high capacity and energy density of metal oxide/sulfide and alloyed anodes, the active anode particles experience large volume changes, resulting in poor cycling stability with further pulverization and aggregation. Thus, to facilitate the commercial viability of AKIBs by matching with cathode materials, fundamental breakthroughs of anode materials with high performance must be achieved.
The current situation is that the choice of cathode and anode materials is limited. Strategies adopted to modify the electrochemical properties of cathode and anode materials for AKIBs are far from adequate compared to the organic systems. Generally speaking, the ideal electrode material needs to meet the following requirements: (1) suitable redox potential (high redox potential for the cathode, low redox potential for the anode), (2) high specific K+ storage capacity, (3) good compatibility with electrolyte, (4) high electronic and ionic conductivity, (5) excellent structural stability, (6), high thermal and chemical stability, (7) environmentally friendly, and (8) low cost. To realize high-performance AKIBs, it is urgently needed to continuously discover and develop novel cathode and anode materials with the above properties. More strategies to improve the electrochemical performance of AKIB electrode materials are needed, including composites with highly conductive materials, morphology design, surface modification, element doping, and electrolyte optimization.
AKIB electrolytes, especially the traditional liquid electrolytes, have taken a leap forward in recent years. One type of aqueous electrolytes is the diluted electrolyte, which can be easily prepared by using K2SO4, KCl, KNO3, and KOH salts. In recent years, other concentrated aqueous electrolytes based on KAc, KCF3SO3, HOOCK, and KFSI have been developed, and the voltage range could be greatly extended to 3–4 V, which is a significant research direction for AKIBs. In addition, a certain amount of additives in the electrolyte could stabilize the electrode material by utilizing the common ion effect. Hydrogel electrolytes are obtained by adding polymers such as PVA and CMC to the conventional aqueous electrolytes, which may advance the application of flexible K+ storage devices. In general, an ideal electrolyte should demonstrate the following major properties: (1) high ionic conductivity for fast K+ transport, (2) stable and wide electrochemical window with no parasitic side reactions (HER, OER, or electrode dissolution, etc.), (3) good wettability, (4) excellent wide temperature application capability, (5) environmental friendliness, and (6) low cost. In order to achieve the above goals of high-performance electrolytes, strategies such as adding additives, adjusting concentrations, and using gel electrolytes may be expected to be realized. In addition, the investigation on AKIB separators is lacking.
5.2 Advanced characterization techniques
During the electrochemical process of an alkali metal ion battery, all components (cathode, anode, and electrolyte) are relatively dynamic. The electrochemical stability of the battery is closely associated with these changes in the internal structure or composition. Therefore, there is an increasing need to apply in situ characterization techniques to collect electrochemical information in real time, especially for those transient processes that generate unstable phases. In situ characterization has been widely used in LIB research, but its application in AKIB research is still lacking. Advanced in situ characterization techniques (such as XRD, XPS, cryo-electron microscopy, TEM, STEM, Raman spectroscopy, and Fourier transform infrared microscopy) can help analyze the K+ insertion/extraction process, interfacial reaction, the transport of K+ ions, and gain more details about the side reactions. In addition, the combined technology of in situ characterization system is the trend of future development, such as spectroscopy-electrical in situ characterization system.5.3 Theoretical calculations
Combined with advanced characterization techniques, theoretical calculations, and machine learning can be used as auxiliary tools to deepen the basic understanding of the AKIB mechanism. For example, molecular dynamic simulations and first-principles calculations can provide detailed information on the behavior of redox reactions at the molecular and atomic levels, respectively. Moreover, according to DFT calculations, the adsorption energies of intermediates can be calculated and analyzed to reveal the preferred reaction paths of electrodes in specific electrolytes. In addition, artificial intelligence and machine learning will play an equally important role in predicting and optimizing the most reasonable material combination and battery design.5.4 Applications
AKIBs are expected to be applied to large-scale ESSs in the future, including tiny electronic devices, sensor devices, and flexible wearable electronic devices. The industrialization of AKIBs will comprehensively consider these key components, including cathode, anode, electrolyte, separator, current collector, battery packaging and manufacturing, cost, and performance. Furthermore, when a relatively expensive and high-concentration electrolyte is applied to AKIBs, its cost should be evaluated. Overall, a timely assessment of the existing problems and solutions in AKIBs will facilitate the translation of laboratory-based research battery designs into the industry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51832004, 21905218), the Key Research and Development Program of Hubei Province (2021BAA070), the Natural Science Foundation of Hubei Province (2019CFA001, 2020CFB519), the Sanya Science and Education Innovation Park of Wuhan University of Technology (2021KF0019, 2020KF0019), and the Fundamental Research Funds for the Central Universities (WUT: 2020IVB034, 2020IVA036). This work was supported by the Singapore Ministry of Education Academic Research Fund Tier 2 (MOE2019-T2-2-127 and MOE-T2EP50120-0002), A*STAR under AME IRG (A2083c0062), and the Singapore National Research Foundation Competitive Research Program (NRF-CRP18-2017-02). This work was supported by A*STAR under its IAF-ICP Programme I2001E0067 and the Schaeffler Hub for Advanced Research at NTU. This work was also supported by NTU-PSL Joint Lab collaboration. X. Z. gratefully acknowledges financial support from the Chinese Scholarship Council.References
- D. Larcher and J.-M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed
.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed
- O. Schmidt, A. Hawkes, A. Gambhir and I. Staffell, Nat. Energy, 2017, 2, 1–8 Search PubMed
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S.-Z. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS PubMed
-
J. M. Tarascon and M. Armand, Materials for sustainable energy: a collection of peer-reviewed research and review articles from Nature Publishing Group, World Scientific, 2011, pp. 171–179 Search PubMed
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Sci. Adv., 2018, 4, eaas9820 CrossRef PubMed
- W. Li, J. R. Dahn and D. S. Wainwright, Science, 1994, 264, 1115–1118 CrossRef CAS PubMed
- Z. Li, D. Young, K. Xiang, W. C. Carter and Y. M. Chiang, Adv. Energy Mater., 2013, 3, 290–294 CrossRef CAS
- V. D. Neff, J. Electrochem. Soc., 1978, 125, 886 CrossRef CAS
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., Int. Ed., 2012, 124, 957–959 CrossRef
- R. Y. Wang, C. D. Wessells, R. A. Huggins and Y. Cui, Nano Lett., 2013, 13, 5748–5752 CrossRef CAS PubMed
- S. Liu, J. J. Hu, N. F. Yan, G. L. Pan, G. R. Li and X. P. Gao, Energy Environ. Sci., 2012, 5, 9743–9746 RSC
- H. Zhang, X. Tan, H. Li, S. Passerini and W. Huang, Energy Environ. Sci., 2021, 14, 5788–5800 RSC
- M. Li, Z. Li, X. Wang, J. Meng, X. Liu, B. Wu, C. Han and L. Mai, Energy Environ. Sci., 2021, 14, 3796–3839 RSC
- H. Kim, J. Hong, K.-Y. Park, H. Kim, S.-W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed
- S. Gheytani, Y. Liang, F. Wu, Y. Jing, H. Dong, K. K. Rao, X. Chi, F. Fang and Y. Yao, Adv. Sci., 2017, 4, 1700465 CrossRef PubMed
- X. Tang, D. Zhou, B. Zhang, S. Wang, P. Li, H. Liu, X. Guo, P. Jaumaux, X. Gao, Y. Fu, C. Wang, C. Wang and G. Wang, Nat. Commun., 2021, 12, 2857 CrossRef CAS PubMed
- F. Wang, X. Fan, T. Gao, W. Sun, Z. Ma, C. Yang, F. Han, K. Xu and C. Wang, ACS Cent. Sci., 2017, 3, 1121–1128 CrossRef CAS PubMed
- X. Sun, V. Duffort, B. L. Mehdi, N. D. Browning and L. F. Nazar, Chem. Mater., 2016, 28, 534–542 CrossRef CAS
- X. Wu, Y. Qi, J. J. Hong, Z. Li, A. S. Hernandez and X. Ji, Angew. Chem., Int. Ed., 2017, 56, 13026–13030 CrossRef CAS PubMed
- W. Zhang, Y. Liu and Z. Guo, Sci. Adv., 2019, 5, eaav7412 CrossRef CAS PubMed
- G. Liang, F. Mo, X. Ji and C. Zhi, Nat. Rev. Mater., 2021, 6, 109–123 CrossRef CAS
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC
- L. Xia, L. Yu, D. Hu and G. Z. Chen, Mater. Chem. Front., 2017, 1, 584–618 RSC
- M. R. Lukatskaya, B. Dunn and Y. Gogotsi, Nat. Commun., 2016, 7, 1–13 CrossRef PubMed
- M. Huang, M. Li, C. Niu, Q. Li and L. Mai, Adv. Funct. Mater., 2019, 29, 1807847 CrossRef
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef
- M. Xia, X. Zhang, T. Liu, H. Yu, S. Chen, N. Peng, R. Zheng, J. Zhang and J. Shu, Chem. Eng. J., 2020, 394, 124923 CrossRef CAS
- L. C. Lopes, S. Husmann and A. J. G. Zarbin, Electrochim. Acta, 2020, 345, 136199 CrossRef CAS
- H. Bi, X. Wang, H. Liu, Y. He, W. Wang, W. Deng, X. Ma, Y. Wang, W. Rao and Y. Chai, Adv. Mater., 2020, 32, 2000074 CrossRef CAS PubMed
- K. Lu, H. Zhang, S. Gao, Y. Cheng and H. Ma, Nanoscale, 2018, 10, 20754–20760 RSC
- K. Itaya, T. Ataka and S. Toshima, J. Am. Chem. Soc., 1982, 104, 4767–4772 CrossRef CAS
- K. Honda and H. Hayashi, J. Electrochem. Soc., 1987, 134, 1330 CrossRef CAS
- C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2011, 2, 1–5 Search PubMed
- C. D. Wessells, S. V. Peddada, R. A. Huggins and Y. Cui, Nano Lett., 2011, 11, 5421–5425 CrossRef CAS PubMed
- C. D. Wessells, M. T. McDowell, S. V. Peddada, M. Pasta, R. A. Huggins and Y. Cui, ACS Nano, 2012, 6, 1688–1694 CrossRef CAS PubMed
- X. Wei, J. Wei, Y. Song, D. Wu, X. D. Liu, H. Chen, P. Xiao and Y. Zhang, Chem. Commun., 2021, 57, 7019–7022 RSC
- D. Su, A. McDonagh, S. Z. Qiao and G. Wang, Adv. Mater., 2017, 29, 1604007 CrossRef PubMed
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Adv. Energy Mater., 2015, 5, 1400930 CrossRef
- M. Huang, X. Wang, J. Meng, X. Liu, X. Yao, Z. Liu and L. Mai, Nano Energy, 2020, 77, 105069 CrossRef CAS
- C. Li, X. Wang, W. Deng, C. Liu, J. Chen, R. Li and M. Xue, ChemElectroChem, 2018, 5, 3887–3892 CrossRef CAS
- W. Ren, X. Chen and C. Zhao, Adv. Energy Mater., 2018, 8, 1801413 CrossRef
- L. Jiang, Y. Lu, C. Zhao, L. Liu, J. Zhang, Q. Zhang, X. Shen, J. Zhao, X. Yu and H. Li, Nat. Energy, 2019, 4, 495–503 CrossRef CAS
- K. Zhu, Z. Li, T. Jin and L. Jiao, J. Mater. Chem. A, 2020, 8, 21103–21109 RSC
- J. Ge, L. Fan, A. M. Rao, J. Zhou and B. Lu, Nat. Sustain., 2021, 1–10 Search PubMed
- J. Han, A. Mariani, H. Zhang, M. Zarrabeitia, X. Gao, D. V. Carvalho, A. Varzi and S. Passerini, Energy Storage Mater., 2020, 30, 196–205 CrossRef
- M. Huang, J. Zhu, R. Yu, Y. Liu, X. Liu, J. Wu, Q. An and L. Mai, Adv. Energy Mater., 2021, 11, 2102342 CrossRef CAS
- P. Padigi, J. Thiebes, M. Swan, G. Goncher, D. Evans and R. Solanki, Electrochim. Acta, 2015, 166, 32–39 CrossRef CAS
- A. Baioun, H. Kellawi and A. Falah, Curr. Nanosci., 2018, 14, 227–233 CrossRef CAS
- W. Song, X. Ji, Y. Zhu, H. Zhu, F. Li, J. Chen, F. Lu, Y. Yao and C. E. Banks, ChemElectroChem, 2014, 1, 871–876 CrossRef CAS
- D. S. Charles, M. Feygenson, K. Page, J. Neuefeind, W. Xu and X. Teng, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed
- Q. T. Qu, L. L. Liu, Y. P. Wu and R. Holze, Electrochim. Acta, 2013, 96, 8–12 CrossRef CAS
- G. Liang, Z. Gan, X. Wang, X. Jin, B. Xiong, X. Zhang, S. Chen, Y. Wang, H. He and C. Zhi, ACS Nano, 2021, 15, 17717–17728 CrossRef CAS PubMed
- G. Liang, X. Li, Y. Wang, S. Yang, Z. Huang, Q. Yang, D. Wang, B. Dong, M. Zhu and C. Zhi, Nano Res. Energy, 2022, 1, e9120002 CrossRef
- A. Zhou, W. Cheng, W. Wang, Q. Zhao, J. Xie, W. Zhang, H. Gao, L. Xue and J. Li, Adv. Energy Mater., 2021, 11, 2000943 CrossRef CAS
- R. Y. Wang, B. Shyam, K. H. Stone, J. N. Weker, M. Pasta, H. W. Lee, M. F. Toney and Y. Cui, Adv. Energy Mater., 2015, 5, 1401869 CrossRef
- H. J. Buser, D. Schwarzenbach, W. Petter and A. Ludi, Inorg. Chem., 1977, 16, 2704–2710 CrossRef CAS
- F. Herren, P. Fischer, A. Ludi and W. Haelg, Inorg. Chem., 1980, 19, 956–959 CrossRef CAS
- K. Itaya, I. Uchida and V. D. Neff, Acc. Chem. Res., 1986, 19, 162–168 CrossRef CAS
- K. Hurlbutt, S. Wheeler, I. Capone and M. Pasta, Joule, 2018, 2, 1950–1960 CrossRef CAS
- C. D. Wessells, S. V. Peddada, M. T. McDowell, R. A. Huggins and Y. Cui, J. Electrochem. Soc., 2011, 159, A98 CrossRef
- D. Yang, J. Xu, X.-Z. Liao, Y.-S. He, H. Liu and Z.-F. Ma, Chem. Commun., 2014, 50, 13377–13380 RSC
- Y. Moritomo, S. Urase and T. Shibata, Electrochim. Acta, 2016, 210, 963–969 CrossRef CAS
- G. Li, B. Huang, Z. Pan, X. Su, Z. Shao and L. An, Energy Environ. Sci., 2019, 12, 2030–2053 RSC
- Y. Zhong, X. Xu, J.-P. Veder and Z. Shao, iScience, 2020, 23, 100943 CrossRef CAS PubMed
- C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113, 6552–6591 CrossRef CAS PubMed
- X. Zhang, X. Rui, D. Chen, H. Tan, D. Yang, S. Huang and Y. Yu, Nanoscale, 2019, 11, 2556–2576 RSC
- Q. Wang, J. Xu, W. Zhang, M. Mao, Z. Wei, L. Wang, C. Cui, Y. Zhu and J. Ma, J. Mater. Chem. A, 2018, 6, 8815–8838 RSC
- G. Chen, Q. Huang, T. Wu and L. Lu, Adv. Funct. Mater., 2020, 30, 2001289 CrossRef CAS
- X. Xu, F. Xiong, J. Meng, X. Wang, C. Niu, Q. An and L. Mai, Adv. Funct. Mater., 2020, 30, 1904398 CrossRef CAS
- Y.-L. Ding, Y. Wen, C. Wu, P. A. van Aken, J. Maier and Y. Yu, Nano Lett., 2015, 15, 1388–1394 CrossRef CAS PubMed
- X. Xu, F. Xiong, J. Meng, Q. An and L. Mai, Mater. Today Nano, 2020, 10, 100073 CrossRef
- H. Tang, Z. Peng, L. Wu, F. Xiong, C. Pei, Q. An and L. Mai, Electrochem. Energy Rev., 2018, 1, 169–199 CrossRef CAS
- S. Zhang, H. Tan, X. Rui and Y. Yu, Acc. Chem. Res., 2020, 53, 1660–1671 CrossRef CAS PubMed
- Y. Zhang, E. H. Ang, K. N. Dinh, K. Rui, H. Lin, J. Zhu and Q. Yan, Mater. Chem. Front., 2021, 5, 744–762 RSC
- X. Li, Z. Huang, C. E. Shuck, G. Liang, Y. Gogotsi and C. Zhi, Nat. Rev. Chem., 2022, 6, 389–404 CrossRef
- F. Ming, H. Liang, G. Huang, Z. Bayhan and H. N. Alshareef, Adv. Mater., 2021, 33, 2004039 CrossRef CAS PubMed
- X. Li, X. Ma, Y. Hou, Z. Zhang, Y. Lu, Z. Huang, G. Liang, M. Li, Q. Yang, J. Ma, N. Li, B. Dong, Q. Huang, F. Chen, J. Fan and C. Zhi, Joule, 2021, 5, 2993–3005 CrossRef CAS
- Y. Tian, Y. An, J. Feng and Y. Qian, Mater. Today, 2022, 52, 225–249 CrossRef CAS
- R. Zhao, A. Elzatahry, D. Chao and D. Zhao, Matter, 2022, 5, 8–10 CrossRef CAS
- X. Li, Q. Li, Y. Hou, Q. Yang, Z. Chen, Z. Huang, G. Liang, Y. Zhao, L. Ma, M. Li, Q. Huang and C. Zhi, ACS Nano, 2021, 15, 14631–14642 CrossRef CAS PubMed
- Y. Li, W. Deng, Z. Zhou, C. Li, M. Zhang, X. Yuan, J. Hu, H. Chen and R. Li, J. Mater. Chem. A, 2021, 9, 2822–2829 RSC
- D. P. Leonard, Z. Wei, G. Chen, F. Du and X. Ji, ACS Energy Lett., 2018, 3, 373–374 CrossRef CAS
- T. Liu, L. Tang, H. Luo, S. Cheng and M. Liu, Chem. Commun., 2019, 55, 12817–12820 RSC
- X. Yuan, Y. Li, Y. Zhu, W. Deng, C. Li, Z. Zhou, J. Hu, M. Zhang, H. Chen and R. Li, ACS Appl. Mater. Interfaces, 2021, 13, 38248–38255 CrossRef CAS PubMed
- Y. Mao, M. Xie, W. Zhao, K. Yuan, Y. Fang and F. Huang, RSC Adv., 2019, 9, 32323–32327 RSC
- M. Xie, W. Zhao, Y. Mao and F. Huang, Dalton Trans., 2020, 49, 3488–3494 RSC
- N. D. Schuppert, S. Mukherjee, A. M. Bates, E.-J. Son, M. J. Choi and S. Park, J. Power Sources, 2016, 316, 160–169 CrossRef CAS
- Y.-Q. Li, H. Shi, S.-B. Wang, Y.-T. Zhou, Z. Wen, X.-Y. Lang and Q. Jiang, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed
- A. Schmidt, M. K. Ramos, C. M. Ferreira, B. A. Braz and A. J. G. Zarbin, Electrochim. Acta, 2021, 387, 138500 CrossRef CAS
- T. Qin, X. Chu, T. Deng, B. Wang, X. Zhang, T. Dong, Z. Li, X. Fan, X. Ge and Z. Wang, J. Energy Chem., 2020, 48, 21–28 CrossRef
- T. Qin, W. Zhang, Y. Ma, W. Zhang, T. Dong, X. Chu, T. Li, Z. Wang, N. Yue and H. Liu, Energy Storage Mater., 2022, 45, 33–39 CrossRef
- H. Chen, Z. Zhang, Z. Wei, G. Chen, X. Yang, C. Wang and F. Du, Sustainable Energy Fuels, 2020, 4, 128–131 RSC
- M. Wang, H. Wang, H. Zhang and X. Li, J. Energy Chem., 2020, 48, 14–20 CrossRef
- Y. Liang, Y. Jing, S. Gheytani, K.-Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841–848 CrossRef CAS PubMed
- J. Han, Y. Niu, S.-J. Bao, Y.-N. Yu, S.-Y. Lu and M. Xu, Chem. Commun., 2016, 52, 11661–11664 RSC
- Z. Wei, D. Wang, M. Li, Y. Gao, C. Wang, G. Chen and F. Du, Adv. Energy Mater., 2018, 8, 1801102 CrossRef
- J. Mei, T. Liao, L. Kou and Z. Sun, Adv. Mater., 2017, 29, 1700176 CrossRef PubMed
- H. Tan, Y. Feng, X. Rui, Y. Yu and S. Huang, Small Methods, 2020, 4, 1900563 CrossRef CAS
- X. Liu, J.-Q. Huang, Q. Zhang and L. Mai, Adv. Mater., 2017, 29, 1601759 CrossRef PubMed
- Q. Pan, Z. Tong, Y. Su, S. Qin and Y. Tang, Adv. Funct. Mater., 2021, 31, 2103912 CrossRef CAS
- Y. Jiang, Y. Wang, J. Ni and L. Li, InfoMat, 2021, 3, 339–352 CrossRef CAS
- S. H. Lee, Y. H. Kim, R. Deshpande, P. A. Parilla, E. Whitney, D. T. Gillaspie, K. M. Jones, A. H. Mahan, S. Zhang and A. C. Dillon, Adv. Mater., 2008, 20, 3627–3632 CrossRef CAS
- M. Zheng, H. Tang, Q. Hu, S. Zheng, L. Li, J. Xu and H. Pang, Adv. Funct. Mater., 2018, 28, 1707500 CrossRef
- K. Song, C. Liu, L. Mi, S. Chou, W. Chen and C. Shen, Small, 2021, 17, 1903194 CrossRef CAS PubMed
- X. Wang, S. Tang, W. Guo, Y. Fu and A. Manthiram, Mater. Today, 2021, 50, 259–275 CrossRef CAS
- J. Wang, Z. Liu, J. Zhou, K. Han and B. Lu, ACS Mater. Lett., 2021, 3, 1572–1598 CrossRef CAS
- H. Wang, D. Yu, X. Wang, Z. Niu, M. Chen, L. Cheng, W. Zhou and L. Guo, Angew. Chem., Int. Ed., 2019, 58, 16451–16455 CrossRef CAS PubMed
- S. Ji, C. Song, J. Li, K. S. Hui, W. Deng, S. Wang, H. Li, D. A. Dinh, X. Fan, S. Wu, J. Zhang, F. Chen, Z. Shao and K. N. Hui, Adv. Energy Mater., 2021, 11, 2101413 CrossRef CAS
- Y. Chen, W. Luo, M. Carter, L. Zhou, J. Dai, K. Fu, S. Lacey, T. Li, J. Wan, X. Han, Y. Bao and L. Hu, Nano Energy, 2015, 18, 205–211 CrossRef CAS
- S. Xu, Y. Chen and C. Wang, J. Mater. Chem. A, 2020, 8, 15547–15574 RSC
- C. Han, J. Zhu, C. Zhi and H. Li, J. Mater. Chem. A, 2020, 8, 15479–15512 RSC
- Y. Liang, C. Luo, F. Wang, S. Hou, S.-C. Liou, T. Qing, Q. Li, J. Zheng, C. Cui and C. Wang, Adv. Energy Mater., 2019, 9, 1802986 CrossRef
- A. V. Desai, R. E. Morris and A. R. Armstrong, ChemSusChem, 2020, 13, 4866–4884 CrossRef CAS PubMed
- H. Cui, L. Ma, Z. Huang, Z. Chen and C. Zhi, SmartMat, 2022 DOI:10.1002/smm2.1110
- X. Zhang, J. Meng, X. Wang, Z. Xiao, P. Wu and L. Mai, Energy Storage Mater., 2021, 38, 30–49 CrossRef
- S. Chen, M. Zhang, P. Zou, B. Sun and S. Tao, Energy Environ. Sci., 2022, 15, 1805–1839 RSC
- J. Huang, Z. Guo, Y. Ma, D. Bin, Y. Wang and Y. Xia, Small Methods, 2019, 3, 1800272 CrossRef
- H. Ao, Y. Zhao, J. Zhou, W. Cai, X. Zhang, Y. Zhu and Y. Qian, J. Mater. Chem. A, 2019, 7, 18708–18734 RSC
- J. Yan, J. Wang, H. Liu, Z. Bakenov, D. Gosselink and P. Chen, J. Power Sources, 2012, 216, 222–226 CrossRef CAS
- J. Zhang, Z. Cao, L. Zhou, G. Liu, G.-T. Park, L. Cavallo, L. Wang, H. N. Alshareef, Y.-K. Sun and J. Ming, ACS Energy Lett., 2020, 5, 2651–2661 CrossRef CAS
- Y. Lv, Y. Xiao, L. Ma, C. Zhi and S. Chen, Adv. Mater., 2022, 34, 2106409 CrossRef CAS PubMed
- Y. Wang, J. Yi and Y. Xia, Adv. Energy Mater., 2012, 2, 830–840 CrossRef CAS
- R. B. McCleskey, J. Chem. Eng. Data, 2011, 56, 317–327 CrossRef CAS
- R. J. Gilliam, J. W. Graydon, D. W. Kirk and S. J. Thorpe, Int. J. Hydrogen Energy, 2007, 32, 359–364 CrossRef CAS
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed
- M. R. Lukatskaya, J. I. Feldblyum, D. G. Mackanic, F. Lissel, D. L. Michels, Y. Cui and Z. Bao, Energy Environ. Sci., 2018, 11, 2876–2883 RSC
- Y. Zhu, J. Yin, X. Zheng, A.-H. Emwas, Y. Lei, O. F. Mohammed, Y. Cui and H. N. Alshareef, Energy Environ. Sci., 2021, 14, 4463–4473 RSC
- H. Gao, K. Tang, J. Xiao, X. Guo, W. Chen, H. Liu and G. Wang, J. Energy Chem., 2022, 69, 84–99 CrossRef
- S. Ko, Y. Yamada and A. Yamada, Electrochem. Commun., 2020, 116, 106764 CrossRef CAS
- Q. Zheng, S. Miura, K. Miyazaki, S. Ko, E. Watanabe, M. Okoshi, C. P. Chou, Y. Nishimura, H. Nakai and T. Kamiya, Angew. Chem., Int. Ed., 2019, 131, 14340–14345 CrossRef
- S. S. Zhang, J. Power Sources, 2006, 162, 1379–1394 CrossRef CAS
- S. Guo, L. Qin, T. Zhang, M. Zhou, J. Zhou, G. Fang and S. Liang, Energy Storage Mater., 2021, 34, 545–562 CrossRef
- G. G. Eshetu, M. Martinez-Ibañez, E. Sánchez-Diez, I. Gracia, C. Li, L. M. Rodriguez-Martinez, T. Rojo, H. Zhang and M. Armand, Chem. – Asian J., 2018, 13, 2770–2780 CrossRef CAS PubMed
- F. Wang, Y. Lin, L. Suo, X. Fan, T. Gao, C. Yang, F. Han, Y. Qi, K. Xu and C. Wang, Energy Environ. Sci., 2016, 9, 3666–3673 RSC
- Y. Du, Y. Li, B. B. Xu, T. X. Liu, X. Liu, F. Ma, X. Gu and C. Lai, Small, 2021, 2104640 CrossRef PubMed
- A. M. Haregewoin, A. S. Wotango and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 1955–1988 RSC
- H. Zhang, G. G. Eshetu, X. Judez, C. Li, L. M. Rodriguez-Martínez and M. Armand, Angew. Chem., Int. Ed., 2018, 57, 15002–15027 CrossRef CAS PubMed
- L. Li, S. Zhao, Z. Hu, S.-L. Chou and J. Chen, Chem. Sci., 2021, 12, 2345–2356 RSC
- J. Cao, D. Zhang, X. Zhang, Z. Zeng, J. Qin and Y. Huang, Energy Environ. Sci., 2022, 15, 499–528 RSC
- F. Wan, L. Zhang, X. Dai, X. Wang, Z. Niu and J. Chen, Nat. Commun., 2018, 9, 1656 CrossRef PubMed
- N. Li, G. Li, C. Li, H. Yang, G. Qin, X. Sun, F. Li and H.-M. Cheng, ACS Appl. Mater. Interfaces, 2020, 12, 13790–13796 CrossRef CAS PubMed
- W. Xu, K. Zhao, W. Huo, Y. Wang, G. Yao, X. Gu, H. Cheng, L. Mai, C. Hu and X. Wang, Nano Energy, 2019, 62, 275–281 CrossRef CAS
- S. J. Banik and R. Akolkar, Electrochim. Acta, 2015, 179, 475–481 CrossRef CAS
- K. E. K. Sun, T. K. A. Hoang, T. N. L. Doan, Y. Yu and P. Chen, Chem. – Eur. J., 2018, 24, 1667–1673 CrossRef CAS PubMed
- T. Otani, Y. Fukunaka and T. Homma, Electrochim. Acta, 2017, 242, 364–372 CrossRef CAS
- Z. Wang, H. Li, Z. Tang, Z. Liu, Z. Ruan, L. Ma, Q. Yang, D. Wang and C. Zhi, Adv. Funct. Mater., 2018, 28, 1804560 CrossRef
- S. Huang, F. Wan, S. Bi, J. Zhu, Z. Niu and J. Chen, Angew. Chem., Int. Ed., 2019, 131, 4357–4361 CrossRef
- Y. Xia, N. Xu, L. Du, Y. Cheng, S. Lei, S. Li, X. Liao, W. Shi, L. Xu and L. Mai, ACS Appl. Mater. Interfaces, 2020, 12, 22930–22938 CrossRef CAS PubMed
- Y. Cheng, J. Shu, L. Xu, Y. Xia, L. Du, G. Zhang and L. Mai, Adv. Energy Mater., 2021, 11, 2100026 CrossRef CAS
- H. Yin, C. Han, Q. Liu, F. Wu, F. Zhang and Y. Tang, Small, 2021, 17, 2006627 CrossRef CAS PubMed
Footnote |
| † X. Z. and T. X. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |