
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA carbonization/interfacial assembly-driven electroplating approach for water-splitting textile electrodes with remarkably low overpotentials and high operational stability†
Jeongmin
Mo‡
 a,
Younji
Ko‡
a,
Young Soo
Yun
b,
June
Huh
a and
Jinhan
Cho
*ab
a,
Younji
Ko‡
a,
Young Soo
Yun
b,
June
Huh
a and
Jinhan
Cho
*ab
aDepartment of Chemical and Biological Engineering, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea. E-mail: jinhan71@korea.ac.kr
bKU-KIST Graduate School of Converging Science and Technology, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
First published on 15th July 2022
Abstract
A key requirement for realizing highly efficient commercial water-splitting devices is to develop non-noble metal-based electrodes that can generate a large amount of hydrogen fuels with low overpotentials and high operational stability. Herein, we introduce high-performance water-splitting electrodes (WSEs) with extremely low overpotentials and unprecedently high operation stability via a carbonization/interfacial assembly-induced electroplating approach. To this end, silk textiles were first converted to carboxylic acid-functionalized conductive textiles using carbonization and subsequent acid treatment. Then, amine linkers were assembled onto the conductive textiles to achieve favorable interfacial interactions with electrocatalysts. For a hydrogen evolution reaction (HER) electrode, Ni was electroplated onto the interface-modified textile, while to prepare an oxygen evolution reaction (OER) electrode, NiFeCo was additionally electroplated onto the Ni-electroplated textile. These HER and OER electrodes exhibited extremely low overpotentials in alkaline media (12 mV at 10 mA cm−2 for the HER and 186 mV at 50 mA cm−2 for the OER), outperforming the conventional non-noble metal-based electrodes. Additionally, the overall-water-splitting reaction of full-cell electrodes was stably maintained at a remarkably high current density of 2000 mA cm−2 and a low cell voltage of 1.70 V. We believe that our approach can provide a basis for developing commercially available high-performance WSEs.
Broader contextAn electrochemical water-splitting reaction offers an effective pathway to generate hydrogen fuels and store electricity from various intermittent but renewable energy sources. Recently, substantial efforts have been devoted to fabricating high-performance and low-cost water-splitting electrodes that can generate a large amount of hydrogen fuels per unit area with low overpotentials and long-term stability under alkaline conditions. To achieve this goal, non-noble metal-based catalysts have been introduced onto porous substrates with large surface area using solution processes. However, non-uniform coating of electrocatalysts onto porous substrates, unfavorable interfacial interactions between electrocatalysts and substrates, and/or relatively low electrical conductivity of electrocatalysts notably increased the overpotentials of electrodes, and simultaneously induced unstable operation at high current density. To address these problems, a carbonization/interfacial assembly-driven electroplating approach was applied to highly porous silk textiles. Benefiting from the fine control of the electrocatalytic deposition on the carbonized silk fibrils, insulating silks were almost perfectly converted to high-performance water-splitting electrodes with bulk metal-like conductivity, large electrocatalytic area, extremely low overpotentials, and unprecedently high operation stability. Our approach can provide a promising tool for developing high-performance electrodes for water electrolyzers and other electrochemical energy devices. |
1. Introduction
Exploiting and securing sustainable/renewable fuel sources for clean energy are the first prerequisite to meet the energy needs of the upcoming societies aiming at carbon neutrality (i.e., net-zero carbon dioxide (CO2) emissions). Among various fuels, hydrogen (H2) is one of the most representative clean energy sources, which is also characterized by a high energy density and convenient energy carrier for energy storage.1–6 However, in spite of such notable advantages, most of the hydrogen gas produced at present is generated from fossil fuel-based chemical processes such as the steam-reforming process and coal gasification, incurring large amounts of CO2 as a by-product.5–7Therefore, many research efforts have been devoted to the development of an efficient water-splitting process that can produce hydrogen gas without any by-product, most of which are concentrated on the development of high-performance water-splitting electrodes (WSEs). In particular, it is desirable that these electrodes should be composed of non-precious metal/metal oxide-based electrocatalysts instead of expensive and rare noble metals such as Pt, Ir, and Ru.8–13 Additionally, the formed WSEs should have low overpotentials for the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER).14–18 However, regrettably, low activity of conventional non-noble metal electrocatalysts has imposed a limit on lowering the overpotentials of HER and OER electrodes. For example, the overpotentials of most Ni-based composite electrodes prepared using chemical reduction and/or hydrothermal methods generally exceed 40 mV at 10 mA cm−2 for the HER and 200 mV at 10 mA cm−2 for the OER in alkaline media, which are much higher than those of noble metal-based HER and OER electrodes.19–23 In addition, the cell voltage of overall WSEs based on non-noble metal catalysts (i.e., full-cell electrodes composed of HER and OER electrodes) is above approximately 1.5–1.6 V even at a relatively low current density of 10 mA cm−2,24 and these WSEs have had much difficulty in stably maintaining the water-splitting reaction with the above-mentioned cell voltage at higher current density.
Considering that the amount of the generated hydrogen gas and the operation stability of electrodes are also related to the magnitude of the externally applied current density, the limited performance of conventional WSEs, which can mainly be attributed to undesirable structure properties and the intrinsic properties of the electrodes (i.e., low active surface area, electrical conductivity, and electrocatalytic activity), is a very large obstacle in realizing the mass production of hydrogen gas. Furthermore, the formation of poor interfacial interactions between substrates and electrocatalysts accelerates the detachment of electrocatalysts during the water-splitting reaction, which result in a poor overall reaction and operation stability. In particular, when the overall WSEs are operated at a high current level for producing a large amount of hydrogen gas, the pressure of the generated gas bubbles increases significantly, which has a fatal effect on the weakly adsorbed electrocatalysts on the host electrodes.25–27 Given that commercial WSEs in alkaline media should require stable operation at a current density of 200–400 mA cm−2 in the cell voltage range of approximately 1.8–2.4 V,14,28 to achieve a lower cell voltage at a higher current density for long operation time remains as a tremendous challenge.
To resolve the abovementioned issues and develop more efficient WSEs, a variety of binary or ternary metal composites for electrocatalysts have been deposited onto three dimensional (3D) porous conductive supports (metal foams, carbon clothes, or carbon papers) through chemical reduction,29,30 chemical reduction-induced electroplating,31 or hydrothermal synthesis of electrocatalytic metal precursor ions.30,32,33 However, most approaches (i.e., painting and electroless deposition) reported to date face challenges in controlling the interfacial interactions between substrates and active materials as well as lowering the internal resistance of electrocatalysts, thus resulting in unsatisfactory electrical conductivity, a non-uniform coating, and unfavorable interfacial adhesion of active materials throughout the 3D porous substrate.34,35 Furthermore, there have not been deep considerations about the interfacial interaction between the 3D porous support and electrocatalytic layer. As mentioned earlier, although unfavorable interfacial interaction leads to unstable operation (by adhesion failure) at high current density as well as the agglomeration and/or non-uniform coating (i.e., a decrease of accessible electrocatalytic area), little experimental consensus has existed on the design of interfaces that can enhance the electrocatalytic performance of WSEs. Therefore, the development of a unique approach for efficiently resolving such critical issues has been strongly demanded for commercially available high-performance WSEs.
Herein, we introduce carbonization/interfacial assembly-driven WSEs with remarkably low overpotentials, low cell voltage, and high operation stability. We also highlight that the favorable interaction formed at the interface between numerous fibrils within carbonized silk textile (CST) and the electrocatalytic layer can have significant influence on the electrical conductivity, active surface area, electrocatalytic structure, and operation stability of water-splitting electrodes. In this study, silk textiles were first carbonized for the preparation of a conductive support. Notably, these carbonized protein-based silk textiles exhibited higher electrical conductivity and more stable mechanical properties than other carbonized cellulose-based textiles including cotton because the β-sheet structure aligned along the axis of silk fibers could easily induce the formation of well-aligned polyaromatic molecules at relatively low pyrolysis temperature (<1000 °C). The formed CST was further modified with amine (NH2) groups that can have a high affinity for the surfaces of various metals or transition metal oxides via coordination bonding. Thus, when an electroplating process for the formation of an electrocatalytic metal and/or metal oxide layer was performed onto the conductive NH2-CST support, highly uniform and robust electrocatalytic layers could be formed onto the NH2-CST without metal aggregation. Additionally, an electroplated Ni layer onto the NH2-CST could generate thermodynamically stable crystalline β-Ni(OH)2 with higher HER performance than α-Ni(OH)2 with a metastable structure and relatively low HER performance. A subsequent electroplating of NiFeCo onto the Ni-electroplated CST induced the formation of NiFeCo (oxy)hydroxide with a wrinkled amorphous structure, enabling more efficient OER performance than the crystalline structure.36–38
The formed electrocatalytic textile electrodes (HER and OER electrodes) exhibited extremely low overpotentials (∼12 mV at 10 mA cm−2 for the HER and 186 mV at 50 mA cm−2 for the OER), showing a bulk metal-like sheet resistance value of approximately 0.05 Ω sq−1 and a highly porous structure. Furthermore, the overall WSEs maintained a low cell voltage of approximately 1.70 V even at an unprecedently high current density of 2000 mA cm−2 for long operation time (>1640 h), outperforming the non-noble metal-based WSEs reported to date. We believe that our approach can provide a basis for developing and designing high-performance textile electrodes with high electrical conductivity, large surface area, and high activity.
2. Results and discussion
2.1. Preparation of amine-functionalized carbonized silk textiles
To prepare a NH2-functionalized conductive support for textile-based WSEs (Scheme 1), natural textiles composed of silk protein fibers were first carbonized at high temperature in an inert atmosphere. The temperature-dependent microstructural characteristics of the CSTs were investigated by Raman spectroscopy. As shown in Fig. 1(a), the Raman spectra of the CSTs measured in the given temperature range exhibited two distinct peaks centered at ∼1338 and 1587 cm−1, corresponding to the D and G bands, respectively. The D band originates from the disordered A1g vibration mode of hexagonal carbon structures, and the G band arises from the E2g vibration mode of six-membered aromatic rings.39 Therefore, with increasing carbonization temperature, the notable growth in the G and D bands indicated an increase in the formation rate of hexagonal ring structures having sp2-hybridized carbon atoms. The presence of these structures suggested that the electrical conductivity gradually increased with the carbonization temperature.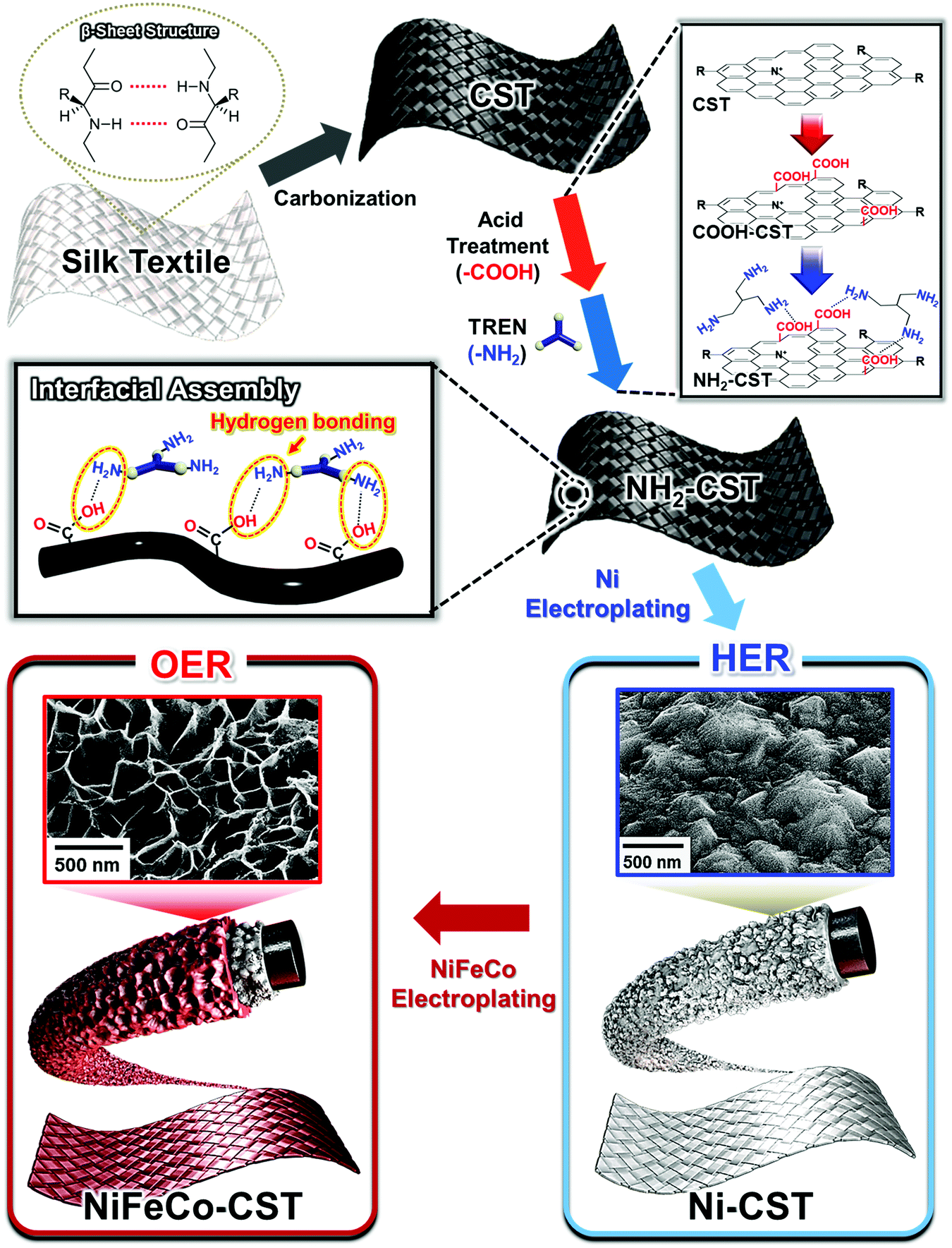 | ||
| Scheme 1 Schematic illustration of the carbonization/interfacial assembly-driven electroplating approach to fabricate water-splitting electrodes. | ||
 | ||
| Fig. 1 Preparation of the amine (NH2)-functionalized carbonized silk textile. (a) Raman spectra of the carbonized silk textile (CST) measured in the 350–950 °C temperature range. (b) Electrical properties of CST as a function of the carbonization temperature. (c) Relative electrical conductivity (σ/σ0) of CST and a carbonized cotton textile as a function of the bending cycling number (bending radius of ∼0.6 cm). (d) Planar field-emission scanning electron microscopy (FE-SEM) images of NH2-CST (the inset shows the thickness of the CST fiber). (e) Fourier transform infrared (FTIR) spectra and schematic illustration of CST, COOH-CST, and NH2-CST. (f) N 1s XPS spectra of NH2-CST. (g) Water contact angle of NH2-CST. | ||
Additionally, the X-ray diffraction (XRD) patterns of the CSTs exhibited a graphitic carbon (002) peak at approximately 22°, indicating partial graphitic ordering (Fig. S1a, ESI†).40 Moreover, X-ray photoelectron spectroscopy (XPS) measurement was performed to further confirm the chemical state of CST. The N 1s spectra of the CST can be deconvoluted by four different peaks (by pyridinic-N-oxide N, graphitic N, pyrrolic N, and pyridinic N), indicating the transformation from N elements within pristine silk to various N substituents in graphene and/or graphite (Fig. S1b, ESI†).41
It has been reported that the structural and chemical configurations of silk proteins dramatically change at temperatures above the onset of thermal degradation (T > 250 °C).42 Specifically, the β-sheet structure of the CST formed at 300 °C was transformed into a sp2-hybridized carbon hexagonal structure (T ≈ 350–1000 °C) and then a highly ordered graphitic structure (T > 2000 °C). This unique pyrolysis behavior of the silk textile as a β-sheet-rich protein resulted in a higher electrical conductivity and lower sheet resistance than those of natural cotton, which is composed of cellulose fibrils, at the same carbonization temperature (Fig. 1(b) and Fig. S2, ESI†). As a result, when the carbonization temperature was increased from 550 to 950 °C, the electrical conductivity of the CSTs increased from 4.2 × 10−6 to 4.3 S cm−1, and its sheet resistance notably decreased from 6.8 × 106 to 6.6 Ω sq−1. Although a higher carbonization temperature could induce the formation of a more conductive CST, we chose a carbonization temperature of 950 °C, which could grant sufficient electrical conductivity for electroplating an electrocatalytic layer onto the CST.
Next, we confirmed that the CST could maintain high electrical conductivity under external mechanical stress such as bending (Fig. 1(c) and Fig. S3, ESI†). Specifically, as the radius of curvature decreased from 1.5 to 0.2 cm, the electrical conductivity of the CST remained constant, which was in stark contrast to that of the carbonized cotton textile, which retained only 23.5% of its initial conductivity at a radius of curvature of approximately 0.6 cm. In addition, bending cycling tests were performed on the CST and carbonized cotton textile with a radius of curvature of 0.6 cm. Even after 5000 cycles, the electrical conductivity of the CST retained 98% of its initial conductivity. However, the carbonized cotton textile maintained only 9.5% of its initial conductivity. These results evidently show that the CST possessed high mechanical and electrical stability.
After the carbonization process, an additional acid treatment and an amine group attachment process using tris(2-aminoethyl)amine (TREN, Mw ∼ 146 g mol−1) molecules converted the CST to the NH2-functionalized CST (NH2-CST) to achieve a high affinity for electrocatalytic components. We confirmed that the NH2-CST maintained a highly porous fibril structure without any morphological deformation after the surface treatments (Fig. 1(d)). In this case, the diameters of the NH2-CST fibrils were estimated to be approximately 7 μm. Fig. 1(e) shows the Fourier transform infrared (FTIR) spectra of the bare CST, acid-treated (COOH-CST), and NH2-CST. The spectra of these samples all exhibited C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching peaks originating from aromatic rings at 1560 cm−1. In particular, the spectrum of the COOH-CST sample showed a characteristic stretching peak of CO on an aromatic ring at 1700 cm−1. After the subsequent amine attachment process, the FTIR spectrum of the resultant NH2-CST displayed broad N–H stretching (3300–3500 cm−1) and bending absorption peaks (1700 cm−1) arising from the TREN molecules.43 Although the N–H bending peak and CO stretching peak overlapped exactly, the observed increase in the peak intensity at 1700 cm−1 was attributed to the attachment of the TREN molecules. The chemical compositions (i.e., CO peak and –NH/–NH2 peaks) of COOH-CST and NH2-CST were further confirmed using X-ray photoelectron spectroscopy (XPS) (Fig. 1(f), Fig. S4 and Table S1, ESI†). Additionally, the hydrophilicity of the NH2-CST was confirmed by the complete wetting of water droplets (pH 5.8) onto the NH2-CST (Fig. 1(g)). These results implied that the outermost surface of the CST was converted to the hydrophilic NH2-functionalized CST due to the deposition of TREN molecules that can have hydrogen-bonding interaction with the COOH-CST.
C stretching peaks originating from aromatic rings at 1560 cm−1. In particular, the spectrum of the COOH-CST sample showed a characteristic stretching peak of CO on an aromatic ring at 1700 cm−1. After the subsequent amine attachment process, the FTIR spectrum of the resultant NH2-CST displayed broad N–H stretching (3300–3500 cm−1) and bending absorption peaks (1700 cm−1) arising from the TREN molecules.43 Although the N–H bending peak and CO stretching peak overlapped exactly, the observed increase in the peak intensity at 1700 cm−1 was attributed to the attachment of the TREN molecules. The chemical compositions (i.e., CO peak and –NH/–NH2 peaks) of COOH-CST and NH2-CST were further confirmed using X-ray photoelectron spectroscopy (XPS) (Fig. 1(f), Fig. S4 and Table S1, ESI†). Additionally, the hydrophilicity of the NH2-CST was confirmed by the complete wetting of water droplets (pH 5.8) onto the NH2-CST (Fig. 1(g)). These results implied that the outermost surface of the CST was converted to the hydrophilic NH2-functionalized CST due to the deposition of TREN molecules that can have hydrogen-bonding interaction with the COOH-CST.
The favorable interfacial interaction between conductive textile and electrocatalytic components (including the electrocatalytic metal and/or metal oxide components) and the high electrical conductivity of the textile play a pivotal role in realizing the conformal deposition of active components onto the numerous fibrils within the textile. Taking these factors into consideration, the surface modification of electrically conductive CST using TREN molecules was expected to provide the possibility that an electrocatalytic metal (and/or metal oxide) layer with a high affinity (i.e., covalent-bonding) for the NH2 groups can be uniformly deposited onto the entire regions, ranging from the interior to the exterior of the carbonized textile. Furthermore, when electrocatalytic components such as Ni or NiFeCo were electroplated onto the NH2-CST, the outermost NH2 groups could accelerate the reduction of the metal ions in the electrocatalytic components.
2.2. Preparation of a HER electrode using a Ni electroplated-CST
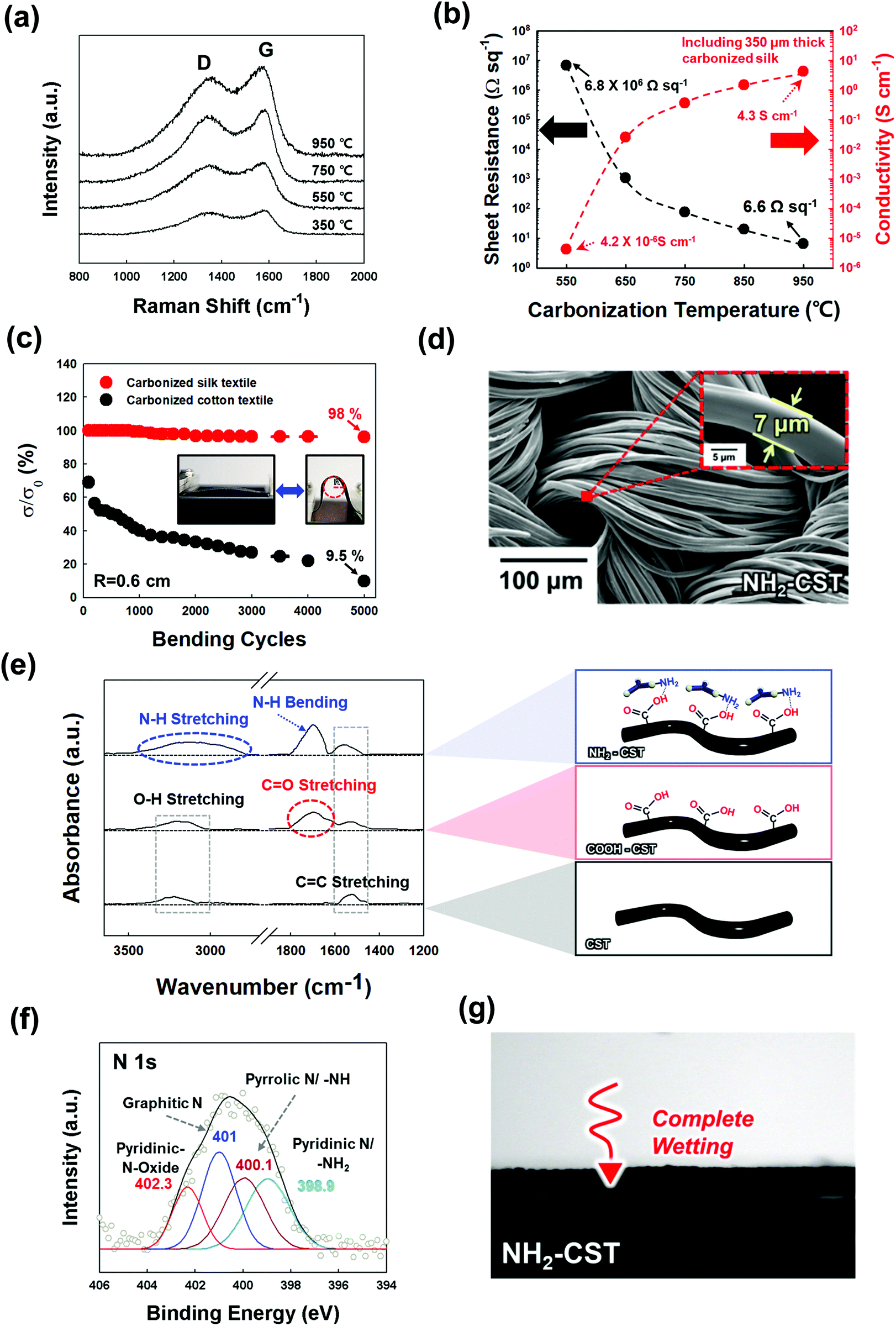
To prepare a water-splitting electrode, an electrocatalytic Ni layer was first electroplated onto the NH2-CST with a sheet resistance of ∼6.6 Ω sq−1 (by pyrolysis at 950 °C) (Fig. 2(a)). During electroplating at a current density of 360 mA cm−2, the size of the electroplated Ni (EP Ni) seeds rapidly increased during the initial 5 min, and continuing the electroplating for 20 min induced the formation of a protuberant Ni layer through fusion of the Ni seeds. In addition to these structural transformations, the electrical conductivity of the EP Ni-CST increased from 13.68 (electroplated for 1 min) to 1566.4 S cm−1 (electroplated for 20 min), and the sheet resistance decreased from 1.74 (electroplated for 1 min) to 0.015 Ω sq−1 (electroplated for 20 min) (Fig. 2(b)). Although the electrical properties of EP Ni-CST could be continuously improved by increasing the electroplating time, they nearly plateaued (0.05 Ω sq−1) after 5 min. Therefore, for process efficiency, the electroplating time for the preparation of the HER electrode was fixed at 5 min. Furthermore, we also confirmed that EP Ni-CST could retain 99.5% of the initial electrical conductivity after 5000 bending cycles at a radius of curvature of approximately 0.6 cm (Fig. S5, ESI†). These results implied that CST was completely converted to electrocatalytic textile with extremely low sheet resistance and good mechanical flexibility.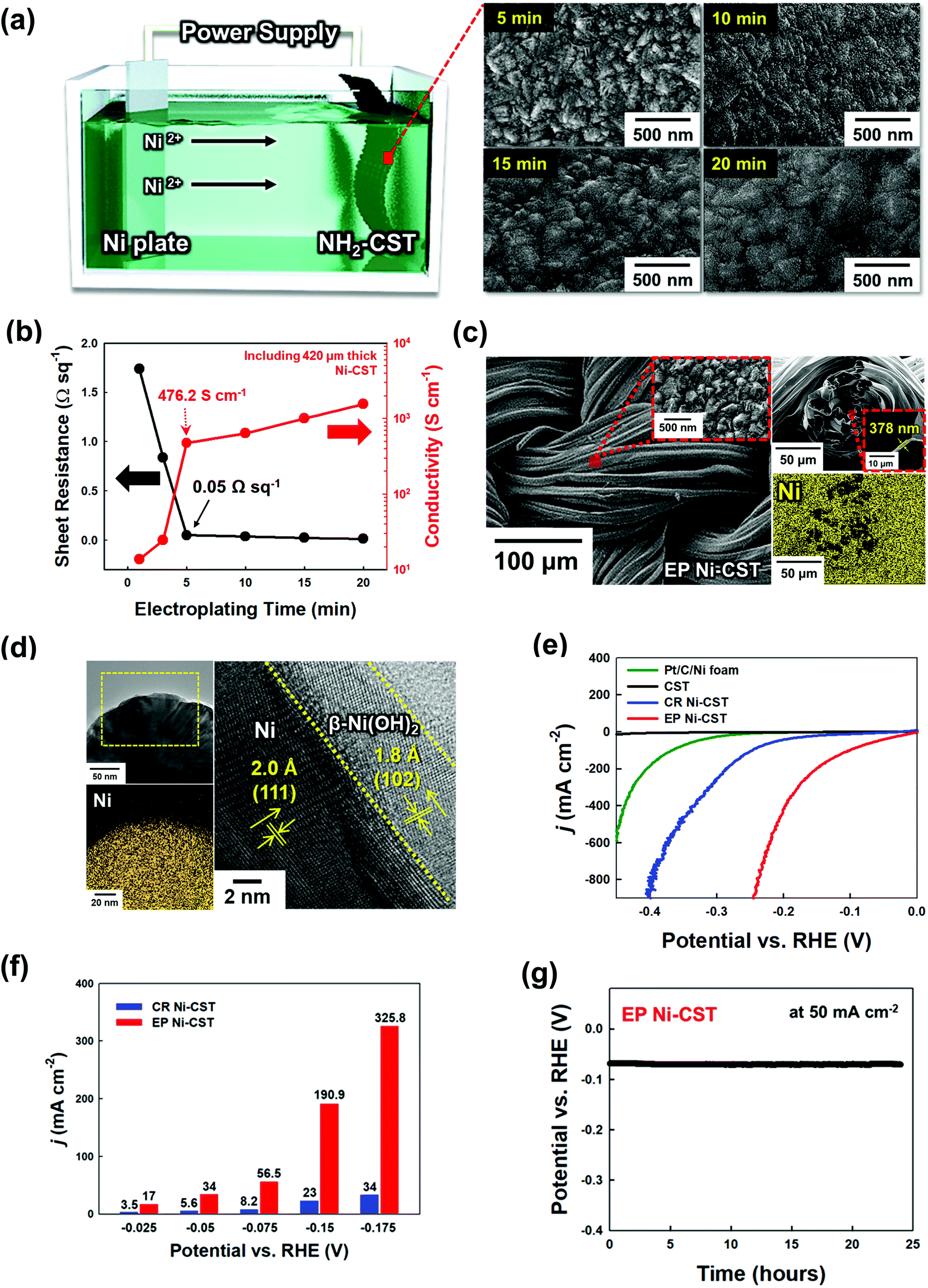 | ||
| Fig. 2 Structural analysis and performance of the EP Ni-CST electrode for the hydrogen evolution reaction (HER). (a) Schematic illustration of the Ni electroplating process (left) and FE-SEM images (right) of the EP Ni layer as a function of electroplating time. (b) Sheet resistance and electrical conductivity of EP Ni-CST as a function of electroplating time at a current density of 360 mA cm−2. (c) FE-SEM images and energy-dispersive X-ray spectroscopy (EDX) mapping image of EP Ni-CST (the inset shows the surface morphology (left) and thickness of the EP Ni layer (right)). (d) HR-TEM image (left top and right panels) and its corresponding EDX mapping image (left bottom panel) of EP Ni-CST. (e) HER polarization curves recorded with Pt/C/Ni foam, CST, CR Ni-CST, and EP Ni-CST electrodes in a 1 M KOH electrolyte at a scan rate of 2 mV s−1. Herein, j is defined as the geometric areal current density. (f) Change in the current densities |j| of CR Ni-CST and EP Ni-CST with increasing potential. (g) Chronopotentiometry curves of EP Ni-CST at 50 mA cm−2 for 24 h. | ||
More importantly, the EP Ni components were homogeneously and uniformly deposited onto all the NH2-CST fibrils, ranging from the outer surface to the central region of NH2-CST without any metal agglomeration, and the samples exhibited a highly protuberant surface morphology with a size of a few hundred nanometers and a layer thickness of approximately 378 nm, as confirmed by field-emission scanning electron microscopy (FE-SEM) images and energy-dispersive X-ray spectroscopy (EDX) mappings (Fig. 2(c)). Furthermore, we highlight that our assembly approach using NH2-functionalized molecules could also be applied to commercial hydrophobic carbon cloth as well as to CST, as mentioned earlier (Fig. S6, ESI†). That is, in the case of electroplating Ni onto a hydrophobic carbon cloth, the formed EP Ni layer was concentrated on the outermost surface of the carbon cloth. However, our approach of using an amine molecule-based interfacial assembly after acid treatment induced the formation of a highly uniform EP Ni layer over the entire NH2-functionalized carbon cloth.
As another approach for the preparation of Ni-based textiles, a chemical reduction (CR) process has been widely used.29,30,32,33 However, it should be noted here that the perfect conversion from only conductive CST or NH2-CST to Ni-CST was not easily accomplished by a CR process. The deposition of CR Ni onto the NH2-CST generated nonuniformly coated regions (without Ni components) and partially aggregated Ni components (Fig. S7, ESI†). Specifically, in the case of CR Ni deposition, the Ni precursor ions were preferentially deposited onto the NH2-CST and then were chemically reduced by a reducing agent. However, the Ni ions were not densely adsorbed onto the NH2-CST during the first deposition stage because of the electrostatic repulsion between the same charged Ni ions without a strong electric driving force. Additionally, the Ni seeds concentrically grew onto the selected area through the nucleation growth mechanism, which resulted in non-uniform Ni deposition onto NH2-CST. The sheet resistance (0.27 Ω sq−1) of CR Ni-CST was approximately 5 times higher than that of EP Ni-CST because of the abovementioned non-uniform Ni coating and the relatively large number of impurities (i.e., nonmetallic components) stemming from CR deposition (Fig. S8, ESI†). Furthermore, the active surface area of EP Ni-CST (surface area ∼4.45 m2 g−1) was much larger than those of CR Ni-CST (0.8 m2 g−1) and commercial Ni foam (0.53 m2 g−1), as confirmed by Brunauer–Emmett–Teller (BET) analysis (Fig. S9, ESI†). All of these observations clearly indicated that the EP Ni layer on the NH2-CST completely governs the electrical conductivity, and furthermore contributes to the increase of active surface area. Therefore, it is reasonable to expect that EP Ni-CST (specifically EP Ni on NH2-CST) will exhibit higher performance than those prepared using the different approaches and/or textiles mentioned above.
The detailed nanostructure of the EP Ni-CST could be further confirmed by high-resolution transmission electron microscopy (HR-TEM). The HR-TEM and energy dispersive X-ray (EDX) mapping images of EP Ni-CST evidently showed that a 3.5–5.4 nm-thick β-Ni(OH)2 layer with the (102) plane and a d-spacing of 1.8 Å was formed onto the EP Ni-CST (Fig. 2(d)).44,45 Considering that thermodynamically stable crystalline β-Ni(OH)2 exhibits high HER activity, these results show that the overall HER performance of EP Ni-CST strongly depends on the outermost β-Ni(OH)2 layer-coated Ni fibrils with ultrathin thickness and large surface area.
In addition, the crystalline structures of the EP Ni-CST and CR Ni-CST were investigated using XRD analysis. The XRD pattern of the EP Ni layer exhibited an evident (102) reflection peak of β-Ni(OH)2, as well as the (111), (200), and (220) peaks indicative of face-centered cubic (fcc) Ni (Fig. S10, ESI†). These crystal structures of the EP Ni layer were nearly identical to those of the bulk Ni foam (Fig. S11, ESI†). In contrast, the XRD pattern of the CR Ni layer exhibited the characteristic peaks (i.e., (006) and (018) peaks) of metastable α-Ni(OH)2 (JCPDS 38-0715) and the (111) peak of fcc Ni without any β-Ni(OH)2 (Fig. S12, ESI†).46 It has been reported that the Ni deposition using CR or the Ni electroplating at a low current density (∼a few mA cm−2) can increase the amount of α-Ni(OH)2 with a metastable structure, and on the other hand thermodynamically stable β-Ni(OH)2 (or mixed α/β-phase materials) can be preferentially formed by electroplating at a high current.47 Therefore, considering that our approach employed a relatively high current density of 360 mA cm−2, it was quite reasonable to conclude that the EP Ni layer on the NH2-CST could produce a larger amount of thermodynamically stable β-Ni(OH)2 than the CR Ni layer.
We further investigated the chemical structures of EP Ni- and CR Ni-CST using XPS (Fig. S13, ESI†). The Ni 2p spectra taken from both EP Ni-CST and CR Ni-CST exhibited a strong peak intensity originating from Ni(OH)2. However, it should be noted here that the binding energy (∼855.3 eV) of Ni(OH)2 is slightly higher than that (∼853.7 eV) of NiO.48,49 That is, the binding energies of EP Ni-CST were negatively shifted by 0.1–0.3 eV relative to those of CR Ni-CST. These phenomena indicated that the EP Ni-CST contains weaker Ni–OH bonds within β-Ni(OH)2 than the CR Ni-CST. Particularly, given that weaker Ni–OH bonds have stronger hydrogen bonding interactions with water molecules in alkaline media, the EP Ni-CST with relatively weak Ni–OH bonds could exhibit a lower overpotential for the HER compared to the CR Ni-CST with strong Ni–OH bonds.50 Yu et al. reported that on the basis of the high catalytic activity obtained from density functional theory (DFT) calculations,51 β-Ni(OH)2 was more active than α-Ni(OH)2. They also reported that the adsorption of water molecules onto β-Ni(OH)2 was energetically more favorable than that onto α-Ni(OH)2, with a more negative adsorption energy. As a result, considering that the EP Ni-CST can predominantly generate the β-Ni(OH)2 structure with high HER activity without the formation of a less active α-Ni(OH)2 structure, our approach is very effective in preparing high-performance HER electrodes.
To test the applicability of our newly developed electrodes, we examined the HER performance of EP Ni-CST (Ni electroplating time of ∼5 min), CR Ni-CST, and Pt/C/Ni foam electrodes in a three-electrode electrochemical cell using 1 M KOH electrolyte (Fig. 2(e) and Fig. S14, ESI†). First, the EP Ni-CST electrode exhibited an overpotential of 12 mV at a current density of 10 mA cm−2 (68 mV at 50 mA cm−2), which was considerably lower than those of the CR Ni-CST electrode (overpotentials of 90 and 199 mV at 10 and 50 mA cm−2, respectively), and Pt/C/Ni foam (253 and 331 mV at 10 and 50 mA cm−2, respectively) electrodes. It should also be noted that the overpotentials of EP Ni-CST were remarkably lower than those of previously reported non-noble metal-based HER electrodes (Table S2, ESI†). Moreover, the EP Ni-CST electrode exhibited high current densities of 190.9 mA cm−2 and 325.8 mA cm−2 at potentials of −0.150 V and −0.175 V, respectively, which were approximately 8.3 and 9.6 times higher than those of the CR Ni-CST electrode at the same potentials (Fig. 2(f)). In addition to this performance, the Tafel slope in the low overpotential range (Fig. S15 and S16, ESI†) and charge transfer resistance (Rct) of EP Ni-CST measured by electrochemical impedance spectroscopy (EIS) were approximately 31.8 mV dec−1 and 9.5 Ω cm−2, respectively, which were much lower than those (Tafel slope of ∼135.8 mV dec−1 and Rct of ∼29.1 Ω cm−2) of CR Ni-CST. These results demonstrate that EP Ni-CST exhibited faster HER kinetics and more efficient charge transfer between the electrode surface and electrolyte than CR Ni-CST.
Additionally, the series resistance (Rs) value is determined by the sum of the resistance generated from cell components (i.e., electrolytes, active materials, and current collectors) and their interfaces (active materials/current collectors).52,53 In this case, the EP Ni-CST exhibited smaller Rs (∼3.9 Ω cm−2 at 100 kHz) than the CR Ni-CST (Rs ∼5.7 Ω cm−2 at 100 kHz), which evidently implied better electron/ion transfer of EP Ni-CST based on an extremely large surface area and unique nanostructures (Fig. S17, ESI†). The correlation among electrical conductivity, charge transfer, and HER performance could be further confirmed by the electroplating time-controlled EP Ni-CST. That is, upon increasing the electroplating time of Ni onto the CST from 1 to 5 min, the sheet resistance and Rct of the formed EP Ni-CST were notably decreased, which could resultantly enhance the HER performance (Fig. S18, ESI†).
We also examined the electric double-layer capacitance of EP Ni-CST and CR Ni-CST using scan rate-dependent cyclic voltammetry (CV) (Fig. S19, ESI†) because the catalytic activity of electrocatalysts is significantly influenced by the electrochemically active surface area (ECSA).54 Because Ni has a specific capacitance of 0.040 mF cm−2, the ECSA of EP Ni-CST was estimated to be approximately 2418 cm2, which was 6.5 times higher than that of CR Ni-CST (373 cm2). To investigate the intrinsic activity of the Ni catalysts, the current densities were further normalized with respect to the ECSA (Fig. S20, ESI†). In this case, the EP Ni-CST showed an approximately 1.4-fold enhancement in its intrinsic activity relative to the CR Ni-CST at −0.15 V vs. a reversible hydrogen electrode (RHE). These results implied that EP Ni-CST had a higher water dissociation capability, greater charge transfer efficiency, and larger ECSA than CR Ni-CST.
Furthermore, we investigated the electrochemical catalytic stability of EP Ni-CST and CR Ni-CST through chronopotentiometry tests (at 50 mA cm−2 for 24 h). During the test, the EP Ni-CST stably maintained its initial potential without any notable decay for 24 h. Additionally, Ni ions were not detected after the HER test, as confirmed by inductively coupled plasma-mass spectrometry (ICP-MS) (Fig. 2(g) and Fig. S21, ESI†). However, in the case of CR Ni-CST, the potential gradually increased upon increasing the operation time. Furthermore, after the HER test, large numbers of Ni ions (approximately 17.7 μg L−1) were detected by ICP-MS. These results evidently implied that the electrocatalyst on CR Ni-CST could easily detach from the electrode and/or dissolved in the electrolyte during the HER. Particularly, when a high current level was applied to the CR Ni-CST, the high pressure of the generated gas bubbles strongly induced the detachment of the electrocatalytic aggregates on CR Ni-CST, which seriously degraded the performance of the WSEs. These results suggest that our approach can produce a highly electrocatalytically active and stable HER electrode.
2.3. Preparation of an OER electrode using a NiFeCo electroplated-CST
Generally, the overall kinetics of a water-splitting device composed of HER and OER electrodes is strictly limited by the sluggish OER rather than the HER.6,9,15 Thus, it is critical to prepare OER electrodes with a low overpotential and long-term stability in addition to preparing high-performance HER electrodes. It is well known that the trimetal-based catalyst NiFeCo, among various non-noble metal-based OER catalysts, possesses high OER activity.55 Although there has been controversy regarding the true active sites on NiFeCo-based electrocatalysts for the OER,56 it is evident that the OER performance of NiFe-based hydroxides can be considerably enhanced by the introduction of Co ions into the NiFe system.57 Recently, Zhao et al. reported that Fe doping could pull partial electrons from the Ni/Co active sites in NiFeCo mixed hydroxides, which increased the electron affinity of the Ni and Co sites to the adsorption of OH−, thus improving the charge transfer from the adsorbed OH− to the Ni and Co sites.58 In addition to these previous results, the high OER activity of NiFeCo could be further predicted using density functional theory (DFT) calculations (Fig. S22–S24, ESI†). In this case, NiFeCo had a lower Gibbs free energy of the rate determining step (RDS) and a narrower bandgap (between the valence and conduction bands) than Ni and NiFe, which almost coincided with the DFT results reported by other research groups.57,58 These results also support the fact that the introduction of Fe and Co ions into the Ni-based system can alter the electronic structure, which can resultantly enhance the catalytic activity and electrical conductivity of active materials (more details are provided in Fig. S22–S24, ESI†).Based on these results, a NiFeCo layer was subsequently electroplated onto the EP Ni-CST. We first investigated the surface morphology of the electroplated NiFeCo layer on the EP Ni-CST by FE-SEM measurements (Fig. S25, ESI†). In this case, the optimal current density and the electroplating time were determined to be 30 mA cm−2 and 10 min, respectively. As shown in Fig. 3(a) and Fig. S26, ESI,† the electroplated NiFeCo layer had a highly uniform and wrinkled nanostructure with a thickness of approximately 142 nm (i.e., the total EP layer thickness of EP NiFeCo/Ni was ∼520 nm) and a loading mass of 2 mg cm−2. Despite this unique structure, the formed OER electrode (i.e., EP NiFeCo-CST) retained the overall porous network of pristine CST and EP Ni-CST. That is, NiFeCo did not block the pores (Fig. S27, ESI†). We also confirmed that EP NiFeCo-CST could preserve 96% of its initial electrical conductivity after a 5000 bending cycles test at a radius of curvature of approximately 0.6 cm (Fig. S28, ESI†). These phenomena clearly implied that EP NiFeCo-CST possessed high mechanical and electrical stability as a result of the formation of favorable interfaces between EP Ni and EP NiFeCo layers as well as between NH2-CST and EP Ni layer.
 | ||
| Fig. 3 Structural analysis and performance of the EP NiFeCo-CST electrode for the oxygen evolution reaction (OER). (a) Planar FE-SEM images of EP NiFeCo-CST (the inset shows the surface morphology of the EP NiFeCo layer). (b) HR-TEM (top) and EDX mapping images (bottom) of EP NiFeCo-CST. (c) XRD patterns of EP NiFeCo-CST and EP Ni-CST. (d) Raman spectra of EP NiFeCo-CST before and after the OER test. (e) OER polarization curves recorded with IrO2/Ni foam, EP Ni-CST, hydrothermal NiFeCo-CST, and EP NiFeCo-CST electrodes in a 1 M KOH electrolyte at a scan rate of 2 mV s−1. (f) Tafel plots of EP Ni-CST, hydrothermal NiFeCo-CST, and EP NiFeCo-CST. (g) Electrochemical impedance spectroscopy (EIS) plots of the hydrothermal NiFeCo-CST and EP NiFeCo-CST electrodes recorded at an applied potential of 1.5 V vs. RHE. (h) Chronopotentiometry curve of EP NiFeCo-CST at 1000 mA cm−2 for 100 h. | ||
We also investigated the chemical composition and structure of the NiFeCo layer using XPS, HR-TEM, XRD, and Raman spectroscopy. First, as observed from XPS measurements, the Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe:Co atomic ratio was estimated to be approximately 1:0.98:0.97. The Ni, Fe, and Co in the NiFeCo layer mainly existed in the form of Ni2+/Ni3+, Fe2+/Fe3+, and Co2+ (with some Co3+), respectively, implying that the EP NiFeCo-CST was composed of a mixture of the Ni, Fe, and Co oxides/hydroxides (Fig. S29, ESI†).55 We further investigated the change of the electronic configuration after the subsequent electroplating of NiFeCo. Interestingly, the Ni 2p spectrum peaks of EP NiFeCo-CST were shifted to lower binding energies (∼0.4 eV) compared to those of EP Ni-, and EP NiFe-CST. On the other hand, in the case of the Fe 2p spectrum, the peaks of EP NiFeCo-CST were shifted to higher binding energies (∼0.35 eV) compared to those of EP NiFe-CST. These peak shifts were mainly due to the charge redistribution at the Ni and Fe atomic sites, implying that the strong electronic interactions among Ni, Fe, and Co could lead to the optimal adsorption energy of reaction intermediates for improving catalyst performance (more details are provided in Fig. S30, ESI†).17,59
:Fe:Co atomic ratio was estimated to be approximately 1:0.98:0.97. The Ni, Fe, and Co in the NiFeCo layer mainly existed in the form of Ni2+/Ni3+, Fe2+/Fe3+, and Co2+ (with some Co3+), respectively, implying that the EP NiFeCo-CST was composed of a mixture of the Ni, Fe, and Co oxides/hydroxides (Fig. S29, ESI†).55 We further investigated the change of the electronic configuration after the subsequent electroplating of NiFeCo. Interestingly, the Ni 2p spectrum peaks of EP NiFeCo-CST were shifted to lower binding energies (∼0.4 eV) compared to those of EP Ni-, and EP NiFe-CST. On the other hand, in the case of the Fe 2p spectrum, the peaks of EP NiFeCo-CST were shifted to higher binding energies (∼0.35 eV) compared to those of EP NiFe-CST. These peak shifts were mainly due to the charge redistribution at the Ni and Fe atomic sites, implying that the strong electronic interactions among Ni, Fe, and Co could lead to the optimal adsorption energy of reaction intermediates for improving catalyst performance (more details are provided in Fig. S30, ESI†).17,59
Additionally, the HR-TEM and its EDX mapping images indicated that the NiFeCo layer possessed a homogeneously mixed structure (Fig. 3(b)), which also coincided with the EDX mapping images measured using FE-SEM (see Fig. S26, ESI†). Moreover, the XRD pattern of NiFeCo displayed no additional peaks except the (111), (200), and (220) (for fcc-Ni), and (102) (for β-Ni(OH)2) peaks originating from the buried EP Ni layer (Fig. 3(c)). These results suggest that the NiFeCo layer has an amorphous structure enabling high electrocatalytic activity.
The Raman spectrum displayed two peaks at 475 and 530 cm−1, which were assigned to γ-NiOOH and α-FeOOH, respectively (Fig. 3(d)).60–63 In this case, Ni and Fe could interact with each other, which could slightly shift their Raman spectrum in the NiOOH–FeOOH system.60 Additionally, it is well known that γ-NiOOH exhibits the highest electrocatalytic ability among all Ni-based electrocatalysts,63 and on the other hand α-FeOOH has the highest thermodynamic stability among all FeOOH electrocatalysts.64
Previous studies have mainly used the hydrothermal process as a method of preparing OER electrodes.30,32,33 However, it should be noted that using this process it is much difficult to uniformly deposit the NiFeCo layer on EP Ni-CST due to the aggregation in micro-/nanoscale regions, resulting in much smaller surface areas than electroplating-induced NiFeCo layers (Fig. S31, ESI†). In particular, the hydrothermal process performed at high temperature and pressure increases the crystallinity within Ni-mixed oxides to some degree,65 which induces a relatively low OER performance compared to that of an amorphous structure (Fig. S32, ESI†). These phenomena imply that the EP NiFeCo layer formed onto EP Ni-CST can exhibit higher OER performance than hydrothermal NiFeCo-CST.
On the basis of these results, the OER performance of EP NiFeCo-CST (specifically EP NiFeCo/EP Ni-CST) was evaluated in 1 M KOH using a three-electrode cell, and the results were compared with those of EP Ni-CST, hydrothermal NiFeCo-CST (i.e., hydrothermal NiFeCo/EP Ni-CST), and IrO2/Ni foam electrodes. To minimize the redox reaction effect, we investigated the overpotentials of all OER electrodes to be 50 and 100 mA cm−2 instead of conventional 10 mA cm−2 (Fig. S33, ESI†). In this case, the overpotentials of EP NiFeCo-CST were estimated to be approximately 186 and 196 mV at 50 and 100 mA cm−2, respectively (Fig. 3(e)). These extremely low overpotentials outperformed those of hydrothermal NiFeCo-CST (279.5 mV at 50 mA cm−2 and 297 mV at 100 mA cm−2), and IrO2/Ni foam (326.3 mV at 50 mA cm−2 and 342.1 mV at 100 mA cm−2) obtained at the same current densities. Additionally, when the EP Ni-CST and CR Ni-CST were used as OER electrodes instead of the HER electrodes, their overpotentials were approximately 353 and 403 mV at 50 mA cm−2, respectively, which clearly indicated that the NiFeCo layer had a decisive effect on the OER performance (Fig. S34, ESI†). It should also be noted that the overpotentials of EP NiFeCo-CST were remarkably reduced compared to those of previously reported non-noble metal-based OER electrodes, including NiFeCo electrodes (Table S3, ESI†). Furthermore, the EP NiFeCo-CST electrode also exhibited faster OER kinetics, showing a considerably smaller Tafel slope (30.9 mV dec−1) than other electrodes (Fig. 3(f)).
In addition to the overpotentials, the detachment of electrocatalysts from electrodes during the water-splitting reaction strongly affects the performance (particularly the operational stability) of overall WSEs and OER electrodes. Therefore, the formation of a robust NiFeCo layer and a favorable interface between the NiFeCo layer and the EP Ni-CST is critical for achieving WSEs with long-term stability. Therefore, we investigated the chemical structures of the NiFeCo layer before and after the OER test by using Raman spectroscopy and ICP-MS (Fig. 3(d) and Fig. S35, ESI†).
First, the Raman spectra of EP NiFeCo-CST showed that there were no meaningful changes in the peaks originating from γ-NiOOH and α-FeOOH before and after the OER test. As mentioned earlier, among various Ni-based electrocatalysts, γ-NiOOH exhibits considerably high electrocatalytic ability, and α-FeOOH possesses better stability than γ-FeOOH. In this case, we did not observe any leakage of Ni, Fe, and Co ions during the OER, as confirmed by ICP-MS, demonstrating the robustness of the formed EP NiFeCo layer. On the other hand, the Raman spectrum of hydrothermal NiFeCo-CST before the OER had several peaks attributed to γ-NiOOH, α-FeOOH, and γ-FeOOH. Particularly, the γ-FeOOH of hydrothermal NiFeCo-CST completely disappeared after the OER test, and large amounts of Fe ions (approximately 158 μg L−1) were detected by ICP-MS, which implied that γ-FeOOH electrocatalysts detached from the OER electrode and/or dissolved in the electrolyte. More importantly, Ni (approximately 2.88 μg L−1) and Co (approximately 14.5 μg L−1) ions were also detected after the OER test of hydrothermal NiFeCo-CST, which was in stark contrast with the results obtained after the OER test of EP NiFeCo-CST. Thus, it was reasonable to conclude that our EP approach could produce highly active γ-NiOOH and α-FeOOH with a robust structure, resulting in an electrode with high electrocatalytic activity and excellent operational stability.
Furthermore, the EIS results showed that EP NiFeCo-CST exhibited fast OER kinetics (Fig. 3(g)). Specifically, the Rs and Rct of EP NiFeCo-CST were estimated to be approximately 4.8 Ω cm−2 (at 100 kHz) and 3.8 Ω cm−2, respectively. This is consistent with previous reports that when Fe and Co ions are incorporated into a Ni hydroxide lattice, they can operate as n-type dopants that enhance the electrical conductivity of Ni hydroxides by forming a unique nanosheet structure (Fig. S24, ESI†).66,67 In addition, the extremely large surface area (the uniform and wrinkled nanostructures) of the EP NiFeCo-CST electrode without organic impurities could significantly decrease the charge transfer resistance. However, the Rs and Rct of the hydrothermal NiFeCo-CST electrode were higher (Rs ∼7.3 and Rct ∼14.2 Ω cm−2) than those of the EP NiFeCo-CST electrode due to the abovementioned properties of the hydrothermal NiFeCo layer. That is, the reduced resistance of EP NiFeCo-CST was closely related to the improvement of charge transfer efficiency between the OER electrode and electrolyte as a result of enhanced electrical conductivity and increased surface area. To further confirm these phenomena, we measured the ECSAs of the OER electrodes (Fig. S36, ESI†). In this case, the Cdl values of the EP NiFeCo-CST and hydrothermal NiFeCo-CST electrodes were calculated to be approximately 58.6 mF and 1.3 mF, respectively. Thus, the ECSA of EP NiFeCo-CST was estimated to be approximately 1465 cm2, which was approximately 45 times higher than that of hydrothermal NiFeCo-CST (ECSA of 32.5 cm2). These results indicate that EP NiFeCo-CST has more uniform and larger surface area with more exposed active sites than hydrothermal NiFeCo-CST.
We also investigated the catalytic durability of EP NiFeCo-CST through chronopotentiometry tests (at 1000 mA cm−2 for 100 h) and observed excellent durability (Fig. 3(h)). Our electrodes could stably maintain high electrocatalytic activity without notable decay. However, in the case of hydrothermal NiFeCo-CST, its overpotential gradually increased upon increasing the operation time at the same current density, indicating that the electrocatalyst of hydrothermal NiFeCo-CST detached from the electrode and/or dissolved in the electrolyte during the reaction (Fig. S37, ESI†). These results suggest that our EP electrodes can simultaneously exhibit considerably high OER performance and excellent operational stability.
2.4. Water electrolyzer using EP Ni-CST and EP NiFeCo-CST electrodes
A full-cell device composed of EP Ni-CST (for the HER cathode) and EP NiFeCo-CST (for the OER anode) was prepared to investigate its overall water-splitting performance (Fig. 4(a)). The cell voltages of the EP Ni-CST‖EP NiFeCo-CST device were measured to be approximately 1.37, 1.48, 1.53, 1.68, and 1.7 V at 10, 50, 100, 1000, and 2000 mA cm−2, respectively, which were much lower than those of the CR Ni‖hydrothermal NiFeCo-based device (i.e., CR Ni-CST‖hydrothermal NiFeCo-CST, 1.45 V at 10 mA cm−2 and 1.95 V at 1000 mA cm−2), and the Pt‖IrO2-based device (i.e., Pt/C/Ni foam‖IrO2/Ni foam, 1.49 V at 10 mA cm−2 and 1.92 V at 1000 mA cm−2) (Fig. S38, ESI†). Moreover, when the amounts of experimentally generated hydrogen and oxygen gases were compared with the theoretically calculated amounts of these gas products, the faradaic efficiency of the EP Ni-CST‖EP NiFeCo-CST device was estimated to be approximately 99.8% at 10 mA cm−2 (Fig. 4(b) and Fig. S39, ESI†). In particular, the cell voltages of our full-cell device at various current densities were much lower than those of previously reported non-noble metal-based full-cell devices (Fig. 4(c) and Table S4, ESI†).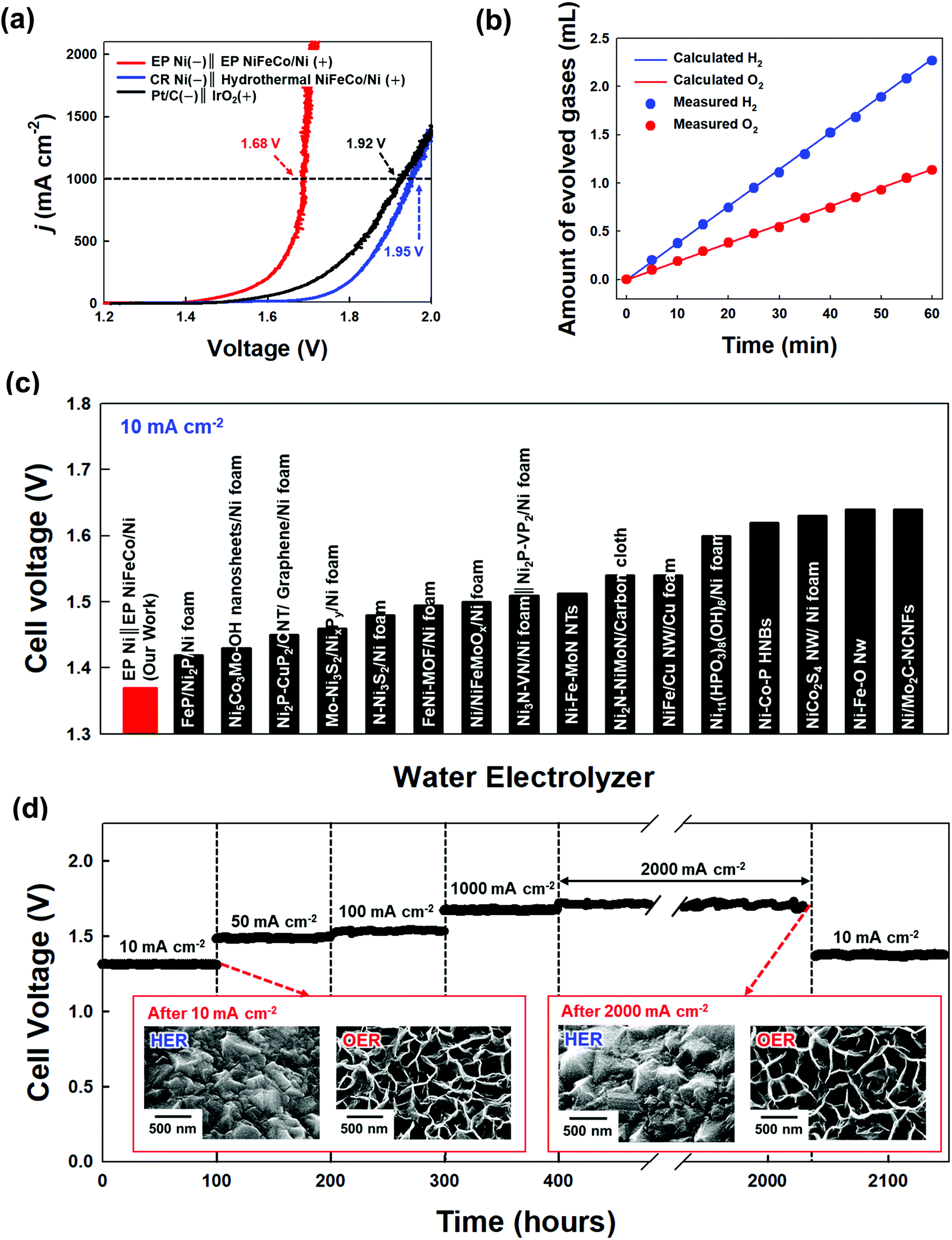 | ||
| Fig. 4 Overall performance of the water-splitting electrodes. (a) Polarization curves of the EP Ni-CST‖EP NiFeCo-CST electrodes, CR Ni-CST‖hydrothermal NiFeCo-CST electrodes, and Pt/C/Ni foam‖IrO2/Ni foam electrodes. (b) Theoretically calculated and experimentally measured amounts of H2 and O2 gases from EP Ni-CST‖EP NiFeCo-CST as a function of the interval time at a constant current density of 10 mA cm−2. (c) Comparison of cell voltages (at 10 mA cm−2) between the EP Ni-CST‖EP NiFeCo-CST electrodes (in our work) and other non-noble metal-based water-splitting electrodes (reported by other research groups). (d) Chronopotentiometry curves of the EP Ni-CST‖EP NiFeCo-CST electrodes recorded at 10 mA cm−2, 50 mA cm−2, 100 mA cm−2, 1000 mA cm−2, and 2000 mA cm−2 (the inset shows the planar FE-SEM images of the EP Ni-CST‖EP NiFeCo-CST electrodes after the stability test). | ||
We further measured the long-term operational stability of the EP Ni-CST‖EP NiFeCo-CST cell during the bulk electrolysis of water in 1 M KOH by chronopotentiometry at current densities of 10, 50, 100, 1000, and 2000 mA cm−2 for a long operation time (total 2140 h) (Fig. 4(d) and Fig. S40, ESI†). In this case, we observed that the corresponding cell voltages were maintained nearly constant, and furthermore the full-cell operation could be maintained for long operation time (at least >1640 h) even at an extremely high current density of 2000 mA cm−2. In particular, the cell voltage at 10 mA cm−2 after chronopotentiometry for 1640 h at 2000 mA cm−2 was nearly identical to the initial cell voltage (at 10 mA cm−2). Moreover, after the harsh accelerated durability tests (particularly the test at 2000 mA cm−2), it should be noted that the EP Ni-CST and EP NiFeCo-CST electrodes did not have serious performance damage, implying the excellent operational stability of the EP Ni-CST‖EP NiFeCo-CST. On the other hand, the electrocatalytic activities of the CR Ni-CST‖hydrothermal NiFeCo-CST cell and Pt/C/Ni foam‖IrO2/Ni foam cell gradually decayed even at a relatively low current density (Fig. S41, ESI†).
To further confirm the high electrocatalytic activity and operational stability of the EP Ni-CST‖EP NiFeCo-CST device, we investigated the surface morphologies and chemical structures after long-term stability tests by using SEM and the XPS analysis. After the stability tests, the EP Ni-CST‖EP NiFeCo-CST did not exhibit the morphological changes such as the delamination and/or crack of the electroplated electrocatalytic layer (the inset of Fig. 4(d)). Additionally, in the case of the HER electrode (EP Ni-CST), there was no notable electronic structure change in the Ni 2p XPS spectrum before and after stability tests (Fig. S42, ESI†). These results evidently imply that the electronic structure of Ni(OH)2, which act as active sites for the HER electrode, is maintained well without any meaningful change after the stability test. On the other hand, in the case of the OER electrode, it was observed that the relative intensity for the M3+ (M = Ni, Fe, Co) peak (particularly the 2p3/2 peaks) was slightly increased, and additionally the entire peaks were slightly shifted to the higher binding energy after the stability test (Fig. S43, ESI†). That is, the M2+ were partially converted to M3+, which implied the formation of highly active M-OOH phase during the OER process.68,69 However, it should be noted that such a slight change observed in EP NiFeCo can increase the OER activity. As a result, these phenomena demonstrate that the EP Ni-CST‖EP NiFeCo-CST can preserve the optimal electronic structures for both the HER and OER without any significant deterioration of the electrocatalytic performance even under extremely harsh water-splitting conditions. Furthermore, these results suggest that the ability to control the interfacial interaction between substrates and catalyst layers can further improve the electrocatalytic activity and operational stability of electrodes. More importantly, it should be noted that the operational stability test performed at an extremely high current density of 2000 mA cm−2 has never been reported by other research groups (Table S5, ESI†).
To ensure the performance reliability of electrodes, more electrode samples (5 samples) were additionally examined. In this case, their activities were almost identically measured within a negligible error range (Fig. S44, ESI†), implying that the electrodes prepared from our approach were highly reproducible. We also highlight that our approach is effective in preparing the large-scale HER and OER electrodes in that the carbonization/interfacial assembly and electroplating process can be easily carried out irrespective of the substrate size and shape (Fig. S45, Table S6, and Video, ESI†). Furthermore, it was demonstrated that the EP Ni-CST‖EP NiFeCo-CST electrodes could have high catalytic stability as well as excellent electrocatalytic activities (cell voltage: 1.45 V at 50 mA cm−2) even in 30 wt% KOH solution at 60 °C (i.e., widely-adopted electrolyte used in a commercial alkaline electrolyzer) (Fig. S46, ESI†).
These excellent performances (specifically, the low cell voltages, high operational stability, and high faradaic efficiency) were mainly attributed to the following three main reasons: (i) hierarchical micro-/nanostructures with interconnected channels that can efficiently promote charge, ion, and gas transport; (ii) formation of robust interfacial structures among all electrode components; and (iii) highly porous CST-based electrodes with bulk metal-like electrical conductivity and high electrocatalytic activity. Thus, it was concluded that the performance exhibited by our devices was superior to those of conventional non-noble metal-based full-cell devices under the same or similar experimental conditions.
3. Conclusions
We demonstrated that carbonization/interfacial assembly could effectively convert insulating silk textiles into porous conductive textiles and enable favorable interfacial interactions; additionally, subsequent Ni and NiFeCo electroplating onto these conductive textiles generated high-performance HER and OER electrodes with extremely low overpotentials, fast reaction kinetics, and unprecedentedly high operational stability. In particular, the protein-induced CSTs exhibited higher electrical conductivity and mechanical properties than cellulose-based textiles at relatively low carbonization temperatures due to the β-sheet structure aligned along the axis of the silk fiber; the abovementioned properties were highly advantageous for preparing water-splitting electrodes. The formed EP Ni-CST (for the HER) and EP NiFeCo-CST (for the OER) electrodes exhibited overpotentials of 12 mV at 10 mA cm−2 for the HER and 186 mV at 50 mA cm−2 for the OER in alkaline media, respectively. In a full-cell device composed of these electrodes, the cell voltages were measured to be approximately 1.37 and 1.7 V at 10 and 2000 mA cm−2, respectively. In particular, our full-cell device maintained stable operation at an unprecedentedly high current density of 2000 mA cm−2 for at least 1640 h in 1 M KOH. Given that our approach can also be easily applied to various textile substrates, retaining the highly porous structure of the textile while avoiding pore blockage, we believe that our strategy can provide a basis for designing a variety of other electrochemical electrodes along with water-splitting electrodes requiring a large active surface area and facile charge transport.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (NRF-2021R1A2C3004151 and NRF-2021M3H4A3A01062964). This work was supported by the KU-KIST School Program and a Korea University Grant.References
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, P. Paulikas Arvydas, V. Stamenkovic and M. Markovic Nenad, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- J. A. Turner, Science, 1999, 285, 687–689 CrossRef CAS PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- C.-H. Chen, D. Wu, Z. Li, R. Zhang, C.-G. Kuai, X.-R. Zhao, C.-K. Dong, S.-Z. Qiao, H. Liu and X.-W. Du, Adv. Energy Mater., 2019, 9, 1803913 CrossRef.
- S. Y. Tee, K. Y. Win, W. S. Teo, L.-D. Koh, S. Liu, C. P. Teng and M.-Y. Han, Adv. Sci., 2017, 4, 1600337 CrossRef PubMed.
- Q. Xu, J. Zhang, H. Zhang, L. Zhang, L. Chen, Y. Hu, H. Jiang and C. Li, Energy Environ. Sci., 2021, 14, 5228–5259 RSC.
- S. Chen, C. Pei and J. Gong, Energy Environ. Sci., 2019, 12, 3473–3495 RSC.
- D. Y. Chung, P. P. Lopes, P. Farinazzo Bergamo Dias Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik, Y. Zhu, S. Seifert, S. Lee, V. R. Stamenkovic and N. M. Markovic, Nat. Energy, 2020, 5, 222–230 CrossRef.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- G. Zhang, G. Wang, Y. Liu, H. Liu, J. Qu and J. Li, J. Am. Chem. Soc., 2016, 138, 14686–14693 CrossRef CAS PubMed.
- Y. Wu, F. Li, W. Chen, Q. Xiang, Y. Ma, H. Zhu, P. Tao, C. Song, W. Shang, T. Deng and J. Wu, Adv. Mater., 2018, 30, 1803151 CrossRef PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef.
- F. Yu, H. Zhou, Y. Huang, J. Sun, F. Qin, J. Bao, W. A. Goddard, S. Chen and Z. Ren, Nat. Commun., 2018, 9, 2551 CrossRef PubMed.
- K. Xu, H. Ding, H. Lv, S. Tao, P. Chen, X. Wu, W. Chu, C. Wu and Y. Xie, ACS Catal., 2017, 7, 310–315 CrossRef CAS.
- L. Yu, B. Y. Xia, X. Wang and X. W. Lou, Adv. Mater., 2016, 28, 92–97 CrossRef CAS PubMed.
- Y. Sun, K. Xu, Z. Wei, H. Li, T. Zhang, X. Li, W. Cai, J. Ma, H. J. Fan and Y. Li, Adv. Mater., 2018, 30, 1802121 CrossRef PubMed.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS PubMed.
- B. Fei, Z. Chen, J. Liu, H. Xu, X. Yan, H. Qing, M. Chen and R. Wu, Adv. Energy Mater., 2020, 10, 2001963 CrossRef CAS.
- S. Li, Y. Wang, S. Peng, L. Zhang, A. M. Al-Enizi, H. Zhang, X. Sun and G. Zheng, Adv. Energy Mater., 2016, 6, 1501661 CrossRef.
- Y. Jia, L. Zhang, G. Gao, H. Chen, B. Wang, J. Zhou, M. T. Soo, M. Hong, X. Yan, G. Qian, J. Zou, A. Du and X. Yao, Adv. Mater., 2017, 29, 1700017 CrossRef PubMed.
- H. Yan, Y. Xie, A. Wu, Z. Cai, L. Wang, C. Tian, X. Zhang and H. Fu, Adv. Mater., 2019, 31, 1901174 CrossRef PubMed.
- A. Wu, Y. Xie, H. Ma, C. Tian, Y. Gu, H. Yan, X. Zhang, G. Yang and H. Fu, Nano Energy, 2018, 44, 353–363 CrossRef CAS.
- C. Hu, L. Zhang and J. Gong, Energy Environ. Sci., 2019, 12, 2620–2645 RSC.
- R. Iwata, L. Zhang, K. L. Wilke, S. Gong, M. He, B. M. Gallant and E. N. Wang, Joule, 2021, 5, 887–900 CrossRef CAS.
- Z. Lu, W. Zhu, X. Yu, H. Zhang, Y. Li, X. Sun, X. Wang, H. Wang, J. Wang, J. Luo, X. Lei and L. Jiang, Adv. Mater., 2014, 26, 2683–2687 CrossRef CAS PubMed.
- N. Han, K. R. Yang, Z. Lu, Y. Li, W. Xu, T. Gao, Z. Cai, Y. Zhang, V. S. Batista, W. Liu and X. Sun, Nat. Commun., 2018, 9, 924 CrossRef PubMed.
- K. Zeng and D. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS.
- Y. Jiang and Y. Lu, Nanoscale, 2020, 12, 9327–9351 RSC.
- H. Yang, M. Driess and P. W. Menezes, Adv. Energy Mater., 2021, 11, 2102074 CrossRef CAS.
- A. Sahasrabudhe, H. Dixit, R. Majee and S. Bhattacharyya, Nat. Commun., 2018, 9, 2014 CrossRef PubMed.
- H. Sun, Z. Yan, F. Liu, W. Xu, F. Cheng and J. Chen, Adv. Mater., 2020, 32, 1806326 CrossRef CAS PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- C. Huang, L. Kang, J. Zhang, J. Li, S. Wan, N. Zhang, H. Xu, C. Wang, Y. Yu, C. Luo and X. Wu, ACS Appl. Energy Mater., 2018, 1, 7182–7190 CrossRef CAS.
- G. Zhou, W. He, S. Wang, C.-y Chen and C. P. Wong, Mater. Lett., 2013, 108, 75–78 CrossRef CAS.
- A. Indra, P. W. Menezes, N. R. Sahraie, A. Bergmann, C. Das, M. Tallarida, D. Schmeißer, P. Strasser and M. Driess, J. Am. Chem. Soc., 2014, 136, 17530–17536 CrossRef CAS PubMed.
- D. L. Smith Rodney, S. Prévot Mathieu, D. Fagan Randal, Z. Zhang, A. Sedach Pavel, J. Siu Man Kit, S. Trudel and P. Berlinguette Curtis, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- W. Kanan Matthew and G. Nocera Daniel, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- Y. Wang, D. C. Alsmeyer and R. L. McCreery, Chem. Mater., 1990, 2, 557–563 CrossRef CAS.
- Z. Q. Li, C. J. Lu, Z. P. Xia, Y. Zhou and Z. Luo, Carbon, 2007, 45, 1686–1695 CrossRef CAS.
- C. Wang, X. Li, E. Gao, M. Jian, K. Xia, Q. Wang, Z. Xu, T. Ren and Y. Zhang, Adv. Mater., 2016, 28, 6639 CrossRef CAS PubMed.
- S. Y. Cho, Y. S. Yun, S. Lee, D. Jang, K.-Y. Park, J. K. Kim, B. H. Kim, K. Kang, D. L. Kaplan and H.-J. Jin, Nat. Commun., 2015, 6, 7145 CrossRef PubMed.
- W. Chidawanyika and T. Nyokong, Carbon, 2010, 48, 2831–2838 CrossRef CAS.
- Z. Zhu, H. Yin, C.-T. He, M. Al-Mamun, P. Liu, L. Jiang, Y. Zhao, Y. Wang, H.-G. Yang, Z. Tang, D. Wang, X.-M. Chen and H. Zhao, Adv. Mater., 2018, 30, 1801171 CrossRef PubMed.
- R. Majee, A. Kumar, T. Das, S. Chakraborty and S. Bhattacharyya, Angew. Chem., Int. Ed., 2020, 59, 2881–2889 CrossRef CAS PubMed.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Proc. R. Soc. A, 2015, 471, 20140792 CrossRef PubMed.
- A. Dutta and J. Datta, J. Mater. Chem. A, 2014, 2, 3237–3250 RSC.
- S. Tao, Q. Wen, W. Jaegermann and B. Kaiser, ACS Catal., 2022, 12, 1508–1519 CrossRef CAS.
- Z. Zeng, K.-C. Chang, J. Kubal, N. M. Markovic and J. Greeley, Nat. Energy, 2017, 2, 17070 CrossRef CAS.
- X. Yu, J. Zhao, L.-R. Zheng, Y. Tong, M. Zhang, G. Xu, C. Li, J. Ma and G. Shi, ACS Energy Lett., 2018, 3, 237–244 CrossRef CAS.
- Z. He and F. Mansfeld, Energy Environ. Sci., 2009, 2, 215–219 RSC.
- L. Yu, H. Zhou, J. Sun, F. Qin, F. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2017, 10, 1820–1827 RSC.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- L. Qian, Z. Lu, T. Xu, X. Wu, Y. Tian, Y. Li, Z. Huo, X. Sun and X. Duan, Adv. Energy Mater., 2015, 5, 1500245 CrossRef.
- X. Bo, R. K. Hocking, S. Zhou, Y. Li, X. Chen, J. Zhuang, Y. Du and C. Zhao, Energy Environ. Sci., 2020, 13, 4225–4237 RSC.
- M. K. Bates, Q. Jia, H. Doan, W. Liang and S. Mukerjee, ACS Catal., 2016, 6, 155–161 CrossRef CAS.
- Q. Zhao, J. Yang, M. Liu, R. Wang, G. Zhang, H. Wang, H. Tang, C. Liu, Z. Mei, H. Chen and F. Pan, ACS Catal., 2018, 8, 5621–5629 CrossRef CAS.
- H. Liang, A. N. Gandi, C. Xia, M. N. Hedhili, D. H. Anjum, U. Schwingenschlögl and H. N. Alshareef, ACS Energy Lett., 2017, 2, 1035–1042 CrossRef CAS.
- A. P. Lee, J. Webb, D. J. Macey, W. van Bronswijk, A. R. Savarese and G. C. de Witt, J. Biol. Inorg. Chem., 1998, 3, 614–619 CrossRef CAS.
- S. Niu, Y. Sun, G. Sun, D. Rakov, Y. Li, Y. Ma, J. Chu and P. Xu, ACS Appl. Energy Mater., 2019, 2, 3927–3935 CrossRef CAS.
- L. Bai, S. Lee and X. Hu, Angew. Chem., Int. Ed., 2021, 60, 3095–3103 CrossRef CAS PubMed.
- Z. Qiu, C.-W. Tai, G. A. Niklasson and T. Edvinsson, Energy Environ. Sci., 2019, 12, 572–581 RSC.
- W. Luo, C. Jiang, Y. Li, S. A. Shevlin, X. Han, K. Qiu, Y. Cheng, Z. Guo, W. Huang and J. Tang, J. Mater. Chem. A, 2017, 5, 2021–2028 RSC.
- A. Das and A. K. Ganguli, RSC Adv., 2018, 8, 25065–25078 RSC.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- H. Cheng, Y.-Z. Su, P.-Y. Kuang, G.-F. Chen and Z.-Q. Liu, J. Mater. Chem. A, 2015, 3, 19314–19321 RSC.
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Nat. Commun., 2019, 10, 5106 CrossRef PubMed.
- Q. He, H. Xie, Z. u Rehman, C. Wang, P. Wan, H. Jiang, W. Chu and L. Song, ACS Energy Lett., 2018, 3, 861–868 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee01510b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |