
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-tortuous and dense single-particle-layer electrode for high-energy lithium-sulfur batteries†
Shuo
Feng
ab,
Rajesh Kumar
Singh
 a,
Yucheng
Fu
c,
Zhuo
Li
de,
Yulong
Wang
de,
Jie
Bao
a,
Zhijie
Xu
c,
Guosheng
Li
a,
Cassidy
Anderson
a,
Lili
Shi
a,
Yuehe
Lin
b,
Peter G.
Khalifah
de,
Wei
Wang
a,
Jun
Liu
a,
Jie
Xiao
a and
Dongping
Lu
*a
a,
Yucheng
Fu
c,
Zhuo
Li
de,
Yulong
Wang
de,
Jie
Bao
a,
Zhijie
Xu
c,
Guosheng
Li
a,
Cassidy
Anderson
a,
Lili
Shi
a,
Yuehe
Lin
b,
Peter G.
Khalifah
de,
Wei
Wang
a,
Jun
Liu
a,
Jie
Xiao
a and
Dongping
Lu
*a
aEnergy and Environmental Directorate, Pacific Northwest National Laboratory, Richland, WA, USA. E-mail: dongping.lu@pnnl.gov
bSchool of Mechanical and Materials Engineering, Washington State University, Pullman, WA, USA
cPhysical and Computational Science Directorate, Pacific Northwest National Laboratory, Richland, WA, USA
dDepartment of Chemistry, Stony Brook University, Stony Brook, NY, USA
eDepartment of Chemistry, Brookhaven National Laboratory, Upton, NY, USA
First published on 29th July 2022
Abstract
Reducing cathode porosity is essential to balancing the electrolyte distribution in lithium–sulfur (Li–S) cells, conserving more pore-filling electrolyte to extend cell cycle life. However, low-porosity electrodes built with nanosized sulfur/carbon (S/C) materials suffer from high tortuosity that significantly deteriorates electrode wetting and hence sulfur utilization. Enabling operation of high-loading sulfur electrodes under both low-porosity and lean-electrolyte conditions is still a challenge and is seldom discussed. In this study, we demonstrated a facile strategy for constructing low-tortuosity through-pores across both vertical and planar directions of electrodes by casting large particles into single-particle-layer electrodes. Through multi-scale characterizations and simulations, correlations between material/electrode structures, electrolyte permeability, polysulfide migration, and sulfur reactions were elucidated. The high-loading and dense sulfur cathode fabricated by this method delivers a high specific capacity (>1000 mA h g−1) at a very low electrolyte/sulfur (E/S) ratio of 4 μL mg−1. This study provides a practical approach to reducing the tortuosity of dense sulfur electrodes by manipulating the porosity distribution, which would be also applicable to improving the rate capability of other high-energy electrodes.
Broader contextThe lithium–sulfur (Li–S) battery holds great promise for vehicle electrification and grid energy storage due to its high theoretical energy density while maintaining a low cost. Existing barriers of the technology are the low cell-level volumetric energy density and limited cycle life; both are related to the use of highly porous sulfur electrodes (porosity ∼ 70%). However, for dissolution-reaction based Li–S batteries, reducing cathode porosity not only leads to a poor electrolyte wetting but a heterogeneous reaction throughout the entire cells, both of which will drastically affect batteries’ performance. Such consequences will be further exaggerated in practical conditions, with an electrolyte to sulfur ratio lower than 4 μL mg−1, for example. In this work, we determined that an optimized electrode architecture is extremely important to realize a high-energy Li–S battery. Combined with simulation and multiscale characterizations, a clear understanding of the effects that electrode structure has on electrolyte infiltration, sulfur reaction kinetics and failure mechanism is elucidated under realistic test conditions, bridging the material- and electrode-level discoveries for high-energy Li–S batteries. |
1. Introduction
The lithium–sulfur (Li–S) battery features a high theoretical energy density (∼2300 W h kg−1) and a very low cost, making it one of the most cost-effective ($ per kW h) battery technologies for vehicle electrification and grid energy storage.1–4 Development of a high-performance Li–S battery is plagued by the low electronic/ionic conductivities of S and Li2S, dissolution of lithium polysulfides (LiPSs), electrolyte consumption, and Li corrosion.5 To address these barriers, different strategies have been adopted to anchor soluble LiPSs generated during the electrochemical process and improve the associated cell cycling stability, including by using various carbon hosts,6–10 polymer backbones,11 or inorganic polar materials.10,12 However, promising performance is usually derived from cells with thin film electrodes and/or excessive amounts of electrolyte (electrolyte volume to sulfur weight ratio, E/S ratio >10 μL mg−1), which are not transferable to practical Li–S batteries.13–15 In recent years, efforts have increasingly been made to either improve sulfur loading in cathodes or reduce the electrolyte amount by developing polar materials or modifying material surfaces.16–19 Yet, sulfur cathode porosity, which is as high as 70%, is another important parameter that has been omitted from most of previous research.20 Compared with the low porosity of commercial Li nickel manganese cobalt oxide electrodes (NMCs) (20–30%), the sulfur cathode is occupied by a large fraction of voids or pores, which cause low cell-level energy density and a short cycle life.14,21 To realize a long cycle life of Li–S cells under lean-electrolyte conditions (E/S <4), it is essential to minimize the cathode porosity; otherwise most of the electrolyte will be trapped in the porous cathode, leaving only a small portion of electrolyte available to support cell cycling and compensate for the inevitable electrolyte consumption that occurs during lithium platting/stripping.22Nevertheless, reducing cathode porosity usually results in a highly tortuous electrode, in which electrolyte wetting becomes a significant challenge.23 For a Li–S cell based on sulfur dissolution–deposition reactions, lack of electrolyte wetting leads to both poor sulfur conversion kinetics and a low utilization rate.23–25 To build low tortuosity electrode architectures, many methods have been adopted1,26 including using magnetic templates,27 freeze drying,28,29 laser patterning30 or vertically-aligned-graphene based free-standing electrode.31 Open-throat pores can form a low-tortuosity structure and provide highways for electrolyte transport but these methods usually result in high porosity of electrode which lowers cell level energy density significantly. In addition, the complicated electrode processing or removal of the pore-forming templates decreases the feasibility of these approaches in practical applications. Moreover, for an Li–S battery which involves LiPS dissolution and diffusion, i.e., LiPS shuttling, an electrode structure with evenly distributed low tortuosity may accelerate the LiPS outflow and loss. So, the architecture of sulfur cathode needs to be optimized to balance the electrode wetting and LiPS shuttling. So far, practical sulfur cathodes that simultaneously fulfill high-sulfur-loading and low-porosity requirements have seldom been reported. Here, we report a facile preparation of low-tortuosity single-particle-layer electrode by aligning large-size secondary S/C particles and elucidate that (1) the low-tortuosity through pores of single-particle-layer electrode enhance electrolyte infiltration in dense electrode globally and (2) the high inside-tortuosity of large secondary S/C particles helps suppress LiPS shuttling locally. When porosity is reduced to as low as ∼45%, which is among the lowest porosities reported in recent literature,25,32 the cathode can still deliver a high discharge capacity of 4 mA h cm−2 (1001 mA h g−1), even at a very low E/S ratio of 4 μL mg−1, thereby providing a decent basis for the development of practical high-energy Li–S cells.
2. Results and discussion
2.1 Impacts of reducing sulfur cathode porosity
Electrode porosity describes the volume ratio between pores/voids and solid components in an electrode and is commonly controlled by electrode calendering. For the present sulfur electrode with a mass loading of ∼4 mg cm−2, its porosity can be reduced from ∼72% to ∼45% by decreasing its thickness from 120 to 60 μm (Table S1, ESI†). Sulfur cathodes used in previous studies usually had a very high porosity (>70%) due to the use of light and porous carbon hosts or additives.20,21 Given most of the pore volume in an Li–S cell is derived from the sulfur cathode when Li foil is used as anode, it is essential to decrease cathode porosity for improved cell-level volumetric energy density. As shown in Fig. S1a, (ESI†) with a porosity of 72%, the thick electrode has a volumetric capacity as low as 330 mA h cm−3 (corresponding to an electrode-level energy density of ∼693 W h L−1). If decreasing the porosity from 72% (120 μm) to 44.7% (60 μm), the corresponding volumetric capacity can be doubled to 661 mA h cm−3 (∼1388 W h L−1). In addition, reducing electrode porosity also benefits gravimetric energy density since a denser electrode requires less pore-filling electrolyte, which does not contribute to the capacity but decreases electrode's practical energy density (Fig. S1b, ESI†).In practice, enabling operation of a dense electrode under practical conditions (e.g., sulfur loading ≥ 4 mg cm−2, E/S ratio ≤ 4 μL mg−1) is quite challenging. First, decreasing the electrode porosity will result in more serious LiPS outflow due to the elevated LiPS concentration gradient of the dense electrode. For example, under flooded electrolyte conditions (E/S = 10 μL mgs−1), the high-porosity electrode (72%) can hold up to 21.8 v% of total electrolyte, which decreases remarkably to only 6.8 v% in a 44.7%-porosity electrode (Fig. S1c, (ESI†) the detailed calculation can be found in Table S2, ESI†). A consequence of such change is a higher LiPS concentration in the lower-porosity electrode. Driven by the elevated concentration gradient, more LiPS is prone to diffuse out of the electrode, exacerbating the shuttling effect. This phenomenon can be moderated by decreasing the electrolyte amount.33 For a 44.7%-porosity electrode, the electrolyte portion inside the electrode increases from 6.8 v% to 16.9 v% if the E/S ratio is decreased from 10 μL mgs−1 to 4 μL mgs−1 (Fig. S1d, ESI†). Second, for sulfur electrodes composed of nanocarbon materials, when reducing the electrode porosity, the loosely packed nanoparticles will intimately contact each other and form a high-tortuosity electrode with narrower or even disconnected channels.34 This will reduce the electrode's accessibility to electrolyte (especially under lean-electrolyte conditions)23,25,35 and lead to two more consequences: (1) only sulfur at the surface can quickly access electrolyte to participate in redox reactions, and (2) the generated Li2S and Li2S2 preferentially deposit on the electrode surface regions, blocking the electrode surface and accelerating irreversible capacity loss. Therefore, the fundamental challenges that need to be addressed in a low-porosity sulfur electrode are (1) the poor electrolyte accessibility caused by the highly tortuous electrode structure, (2) the exacerbated shuttling effect due to the increased LiPS concentration gradient, and (3) the early electrode surface passivation caused by Li2S deposition.
2.2 Design principles of low-porosity sulfur cathodes
Based on the preceding discussion, both material structure and electrode architecture should be rationally designed to simultaneously enhance electrolyte accessibility while relieving LiPS shuttling and electrode blocking. Accordingly, a clear understanding of the effects of S/C materials on electrolyte wetting and LiPS diffusion is required and these effects were first studied using computational fluid dynamics (CFD) simulations. Multiphase flow simulations using the volume of fluid (VOF) method were performed to investigate the electrolyte wetting process in porous electrodes at the same loading, thickness, and porosity but consisting of small (<20 μm) and large (>90 μm) particles. As shown in Fig. 1a and b, with the same electrode porosity and initial volume of electrolyte, the large-particle cathode (LPC) shows better wettability, and the electrolyte can reach the deeper part of the electrode. In contrast, electrolyte hardly penetrates the small-particle cathode (SPC) within the same time duration. Fig. S2a and b (ESI†) plot the detailed calculations of the degree of surface wetting and electrolyte absorption in the SPC and LPC during electrolyte infiltration. Under lean-electrolyte conditions, the wetting degree of SPC shows a plateau at the beginning, which means the electrolyte infiltration experiences significant repulsion due to the high capillary pressure developed in the small pores of the SPC, and only 20 v% of the electrode is wetted by electrolyte. While for LPC, both a quicker electrolyte infiltration and a higher wetting degree (31%) were observed. It is interesting to find that by adding more liquid load (i.e., creating a flooded electrolyte condition), the electrode wetting in both the LPC and SPC can be improved because the gravitational force overwhelms the pore-scale capillary pressure. This means a flooded electrolyte condition may obscure the electrode wetting issue observed under lean-electrolyte conditions. The multiphase flow simulation results suggest that an electrode structure composed of larger particles has quicker electrolyte infiltration and better wetting.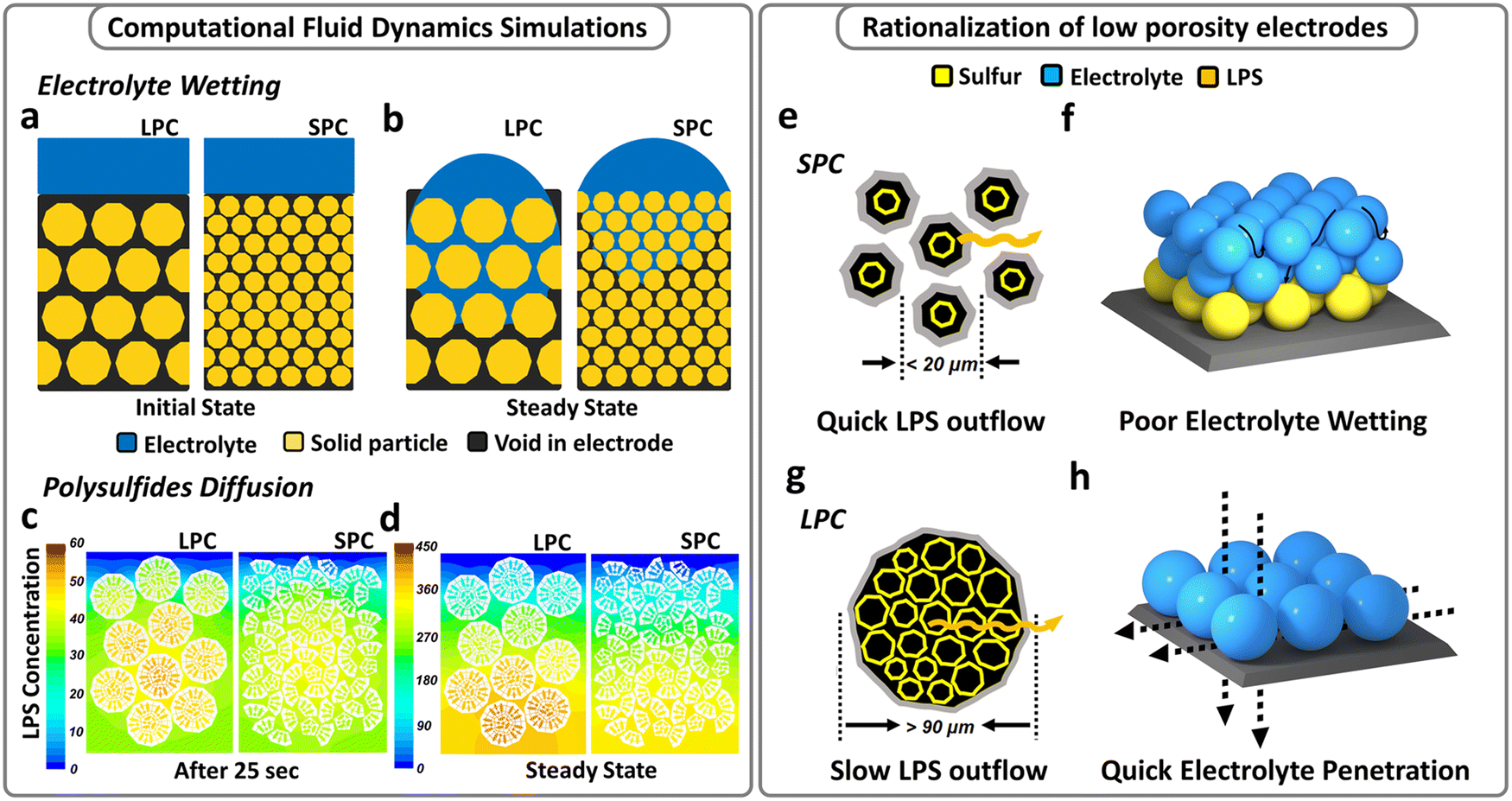 | ||
| Fig. 1 Simulations and design principles of different cathodes. (a) Initial state and (b) steady state of electrolyte infiltration in the LPC and SPC. (c) Intermediate state and (d) steady state of LiPS migration in the LPC and SPC. Schematics of (e) small S/C particles and (f) the resulting multi-particle-layer electrode structure. Schematics of (g) large secondary S/C particles and (h) the resulting single-particle-layer electrode structure. | ||
Further, a scalar transport simulation was performed to understand LiPS diffusion behaviors in LPC and SPC electrodes. It was revealed that the LPC does not necessarily lead to accelerated LiPS shuttling but better electrode wetting. As shown in Fig. 1c, the same basic structural units were integrated into secondary particles of different sizes, and the resulting LPC and SPC had the identical overall porosities. The initial states of the LPC and SPC are shown in Fig. S3 (ESI†) and both electrodes are fully wetted with electrolyte. Once the simulation started, the LiPS filled the pores inside the secondary particles. Driven by the concentration gradient, the LiPS will migrate outside the secondary particles in the electrode. Fig. 1c and d compare LiPS distribution in the LPC and SPC under intermediate (25 s) and the steady-state conditions, respectively. It is apparent that for large particles, a small proportion of LiPS diffuses out from large particles after 25 s, and a high polysulfide concentration still exists inside the particles. In contrast, within the same time period, small particles show an accelerated polysulfide outflow. Thus, after reaching steady state (the end of simulation, not the end of discharge), the polysulfide concentration in the LPC is higher than that in the SPC at each depth of electrodes (Fig. 1d), indicating suppressed polysulfide shuttling. The distinct polysulfide migration rate is due to the different effective diffusivities of LiPS in the large and small particles. Compared with larger particles, the effective diffusivity of LiPS in small particles is higher at each depth (Fig. S4a, ESI†), so that less time is needed for LiPS diffusion out of the electrode (Fig. S4b, ESI†), thereby resulting in a lower LiPS concentration inside (Fig. S4c, ESI†). In contrast, a larger secondary particle has a longer diffusion pathway from the center to the outer surface. Eventually, LiPS in the LPC has a higher chance to circulate inside the particles, suppressing LiPS shuttling and loss. As illustrated in Fig. 1g and h, the secondary particles form electrolyte diffusion channels, which determine the electrolyte infiltration rate; and the inner surfaces of secondary particles form internal LiPS diffusion pathways, which dictate the LiPS diffusion rate.
As suggested by the simulations, larger secondary particles are desired to enhance electrode wetting while reducing LiPS shuttling. Based on the preceding analysis, it is reasonable to fabricate a single-particle-layer electrode, a special case of LPC, if using the large particles whose sizes are comparable to or even larger than the target electrode thickness (Fig. 1h).
2.3 Materials for a low-porosity sulfur cathode
To validate the above mentioned design of single-particle-layer electrode, nanosized and porous Ketjen Black (KB) particles were used as basic structural units and integrated into secondary—integrated KB (IKB)—particles of different sizes by using a modified synthesis approach reported in our previous research.24 The morphologies of small (<20 μm) and large (>90 μm) secondary particles are exhibited in Fig. 2 and Fig. S5, (ESI†) and the measured Brunauer–Emmett–Teller (BET) surface areas are 1155 m2 g−1 and 1001 m2 g−1, respectively, before sulfur loading (Fig. S6a, ESI†). Although a lower surface area of larger particles, a similar pore volume is detected for large (3.86 cm3 g−1, Fig. S6b, ESI†) and small (3.88 cm3 g−1) particles. After sulfur loading, the surface areas of large and small integrated Ketjen Black/sulfur (IKB/S) particles are reduced to 15 and 24 m2 g−1, respectively. The large particles have a tap density (0.714 g cm−3) twofold larger than that of the smaller ones (0.41 g cm−3, Fig. S7, ESI†). | ||
| Fig. 2 Morphologies of S/C materials and corresponding electrode structures. Scanning electron microscopy (SEM) images of (a) an SPC surface and (b) smaller integrated KB/sulfur (IKB/S) particles. Micro-computed tomography (micro-CT) images of an SPC at low magnification (c and d) and high magnification (e–g). SEM images of (h) an LPC surface and (i) larger IKB/S particles. Micro-CT images of an LPC at low magnification (j and k) and high magnification (l–n). | ||
High-sulfur-loading electrodes (4 mg cm−2) can be easily tape-cast using both types of particles and calendered to 60 μm thick (Fig. S8, ESI†). The calculated electrode porosity is around 44.7%, which is among the lowest porosities reported in recent literatures.25,32 Scanning electron microscopy (SEM) characterization indicates that the SPC has a more compact and smoother surface (Fig. 2a); while in LPC, large pores are visible (Fig. 2h). Since shape of large particles is not normal sphere, after the electrode coating and calendaring, some of the large particles were deformed to fit the target electrode thickness of 60 μm. To visualize the electrode structures, X-ray micro-computed tomography (X-ray micro-CT) was used to scan the SPC and LPC. Three phases—S/C particle, binder/carbon additive, and voids—are separated based on the contrast and colored yellow, grey, and blue, respectively in Fig. 2(c–n). In the SPC, small particles tend to stack into a dense multi-particle-layer electrode during slurry coating and under calendering (Fig. 2c and e). In addition, with the spread of the particles along the plane direction under pressure, horizontally aligned pores are formed (Fig. 2d and f). As for the LPC, the cross-section micro-CT results indicate that it is composed of a single layer of particles (Fig. 2k and m) with through pores along both vertical and planar directions, which agrees with the design of single-particle-layer electrode.
To quantify electrode tortuosity in the LPC and SPC, the acquired CT images are reconstructed into 3D models (Fig. 2g and n) and the electrolyte flow patterns are analyzed using CFD simulations to identify the complete channels from electrode surface to current collector. By extracting the streamlines from simulated electrolyte flow paths, electrode tortuosity can be defined as follows: τ = averaged streamline length/electrode thickness. As shown in Fig. 2g, the SPC has an estimated tortuosity of 2.01 along the perpendicular direction. The high tortuosity suggests poor pore connectivity and a higher risk of electrolyte blockage in the SPC. In sharp contrast, the tortuosity in the LPC can be as low as 1.16, which is close to the lowest value (τ =1) in a porous medium. Hence, this proves that the large particles tend to form through-pores, which provides low-tortuosity channels for electrolyte infiltration starting from electrode surface to the bottom. In addition, from the reconstructed 3D models, more electrolyte flow-through channels are observed in the LPC than in the SPC. Besides, the single-particle-layer electrode composed of large particles also has better pore connectivity along the planar direction, thereby benefiting electrolyte transport along the plane direction (Fig. 2j and l).
2.4 Electrochemical performance of the low-porosity sulfur cathode
The effects of electrode architectures on sulfur reactions were first studied by testing the SPC and LPC at different porosities and electrolyte conditions. Fig. 3a–c compare the capacity retention of the SPC and LPC for 100 cycles at 0.1 C under flooded electrolyte conditions (E/S = 10 μL mg−1). At each level of porosity, the LPCs and SPCs have very similar capacities for the first discharge. This is consistent with the flow simulation results that flooded electrolyte conditions help relieve electrode wetting issues in dense electrodes. In subsequent cycles, the LPCs show much higher capacity and better capacity retention than the SPCs. The LPC electrode shows slightly improved cycling stability when porosity is reduced from 62% to 53% and maintains similar performance even at an extremely low porosity of 45%. After 30 cycles, the LPC electrodes with porosities of 62%, 53%, and 45% deliver reversible capacities of 932, 937, and 917 mA h g−1, corresponding to capacity retention of 87.9%, 88.1%, and 88.7%, respectively. It is important to note that for the LPC, most of the capacity loss occurs during the second discharge process, after which capacity stabilizes in the ensuing cycles (Fig. S9, ESI†). Apparently, at each porosity level, the 30th discharge curve is well overlapped with that of the 2nd cycle, showing a similar capacity and cell polarization. In contrast, performance degradation was observed in the SPCs as the porosity decreased. After 30 cycles, the reversible capacities/capacity retention of SPCs at 62%, 53%, and 45% porosity were 793 (76.1%), 742 (68.8%), and 728 mA h g−1 (71.1%), respectively. Another important observation is that particle size has a direct effect on the Coulombic efficiency under flooded electrolyte conditions. As shown in Fig. S9, (ESI†) at each porosity level, the LPC electrode always has higher Coulombic efficiency in both the first and subsequent cycles than the SPC electrodes. After 100 cycles, the LPC electrode at a low porosity of 45% delivers a capacity retention of ∼85% at an E/S = 10 μL mg−1 (Fig. 3c). This can be explained by the increased particle dimension and inner surface area of the large secondary particles, which reduce shuttling and hence irreversible loss of LiPS. To understand the effects of particle size on the reaction kinetics, we have tested both SPC and LPC at high C rate (1 C). As shown in Fig. S10, (ESI†) compared to SPC, the LPC shows a slightly increased polarization but a similar discharge capacity, which was observed only under flooded electrolyte conditions. If switched to lean electrolyte (E/S 4), poor electrolyte wetting of SPC starts to limit the reaction kinetics and lower the sulfur reactivity (Fig. S11, ESI†).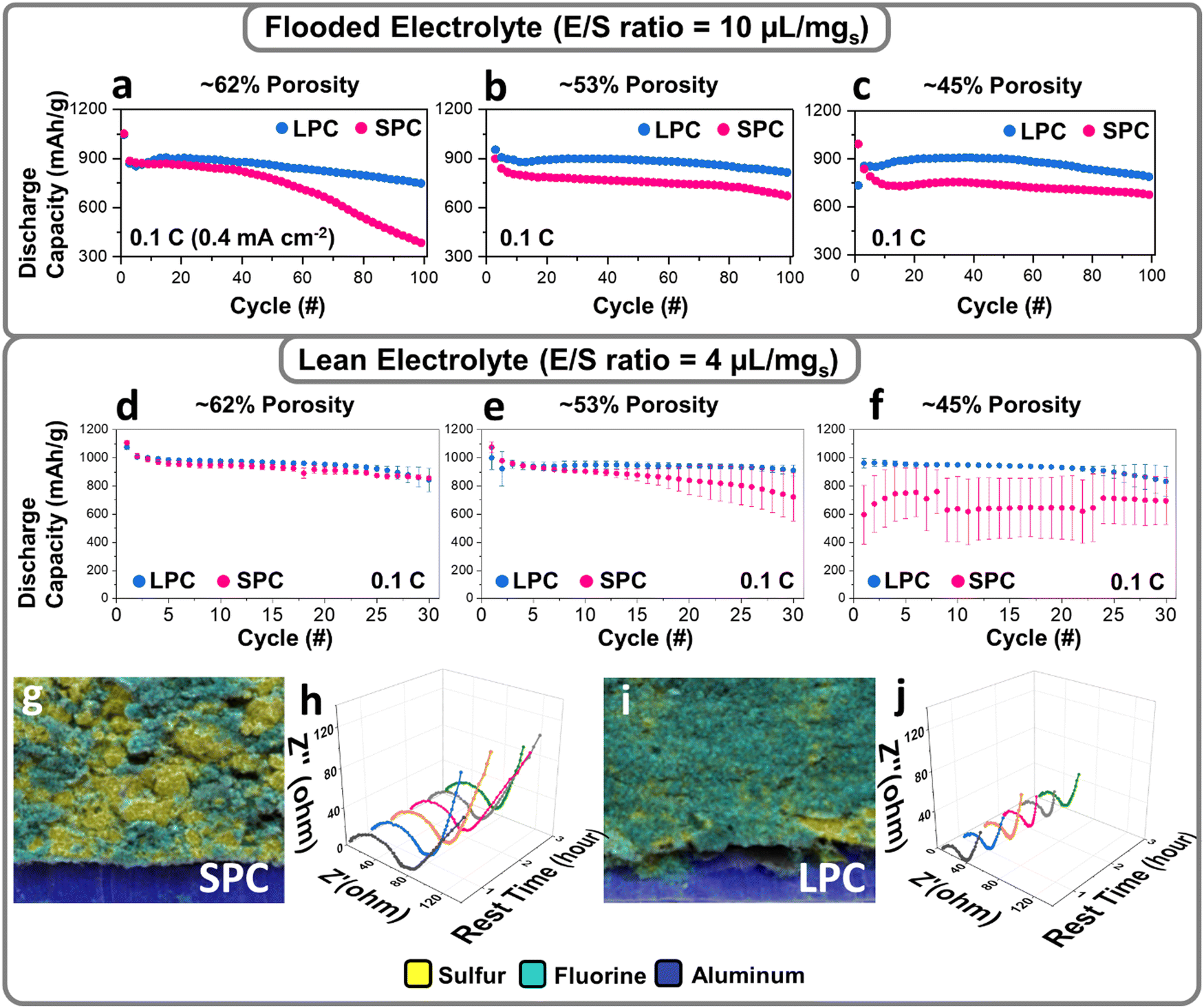 | ||
| Fig. 3 Electrochemical performances of the SPC and LPC under flooded (E/S ratio = 10 μL mgs−1) and lean (E/S ratio = 4 μL mgs−1) electrolyte conditions. Cycling performance of the SPC and LPC under flooded electrolyte condition (a–c) and lean electrolyte condition (d–f) at 0.1 C (1 C = 1000 mA g−1). Energy dispersive spectroscopy (EDS) images of SPC (g) and LPC (i) with 45% porosity after electrolyte infiltration. Electrochemical impedance spectroscopy (EIS) data of SPC (h and j) during cell rest, collected every 30 minutes. | ||
The LPC and SPC were further examined under lean-electrolyte conditions (E/S = 4 μL mg−1) where the electrolyte infiltration becomes more challenging, especially in a low-porosity cathode. At a relatively high porosity of 62%, the SPC electrodes have almost identical reversible capacities and capacity retention as those of the LPC electrodes (Fig. S11a, ESI†), although polarization is slightly higher in the SPC electrodes. With a decrease of electrode porosity, more deteriorated polarization and capacity decay were observed in the SPC when porosity was decreased to 53%. At an extremely low porosity of 45%, the first discharge capacity dropped to only 451 mA h g−1 (Fig. S11c, ESI†). In contrast, the LPC at 45% porosity still delivered a high specific capacity of 1001 mA h g−1. To avoid the bias among different cathodes, we included the data from six coin cells with the error bar provided (Fig. 3d–f), showing that the LPCs deliver a constant large discharge capacity with high reproducibility at each porosity level, while capacity fluctuation was usually observed in the SPCs, especially at 45% porosity. Such fluctuation indicates insufficient electrolyte wetting and varied wetting status in SPCs.
The electrolyte permeability in the SPC and LPC dense electrodes were studied by tracking the lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) distribution in energy dispersive spectroscopy (EDS) mapping, where fluorine from LiTFSI was used as the tracking reagent. As shown in Fig. 3g and i and Fig. S12, (ESI†) the LPC electrode exhibits a more uniform distribution of fluorine signal than the SPC electrode. Given the same material chemistry and electrolyte, the EIS data before cell cycling can also be an indicator of electrode wetting. Compared to the SPC (Fig. 3h), the LPC (Fig. 3j) shows much smaller resistances for both bulk and overall charge-transfer (Rct). This suggests a better wetting of the LPC electrode. The different electrolyte infiltration in SPC and LPC is not easy to demonstrate a significant impact on sulfur utilization at high porosity electrodes or flooded conditions but it will determine the electrochemical performance in low-porosity electrodes and lean-electrolyte conditions as approved in Fig. 3d–f. The benefits of using the LPC were demonstrated by comparing the energy density of the SPC and LPC under different electrolyte conditions (Fig. S13 and S14, ESI†). For SPC at flooded electrolyte conditions, the volumetric and gravimetric capacities follow the same increasing trend with decrease of porosity. This means if electrolyte wetting is not an issue at flooded electrolyte conditions, sulfur specific capacity increases with decreasing of porosity. However, at lean electrolyte conditions, the electrolyte wetting becomes worse with decrease of porosity, and sulfur specific capacity experiences a first increasing and then decreasing trend, especially at 45%. As a result, both volumetric and gravimetric capacity show a similar first increasing and then decreasing trend. For LPC, a similar increasing trend was observed for volumetric and gravimetric capacity at both flooded and lean electrolyte conditions. From above comparison, the LPC is superior versus SPC at practical lean electrolyte and low porosity conditions.
2.5 Sulfur reaction process in a low-porosity sulfur cathode
To understand the sulfur reactions in different dense SPCs and LPCs (porosity 45%) under lean-electrolyte conditions, a high-resolution synchrotron X-ray diffraction (XRD) study was performed. The cathodes were charged/discharged to the designed voltages and transmission ex situ XRD patterns were collected. Fig. 4a and c show the first discharge/charge profiles of the SPC and LPC electrodes under lean-electrolyte conditions (E/S = 4 μL mg−1), respectively. The dense LPC electrode delivers a high initial discharge capacity of ∼1000 mA h g−1 with two distinct discharge plateaus while the dense SPC electrode shows significantly suppressed discharge plateaus with an overall capacity of only 451 mA h g−1. The SPCs and LPCs were harvested and dried directly for XRD analysis without further washing. Due to the high brightness of hard X-ray, the difference in the phase evolutions of S, LiPS, and Li2S were clearly identified for the SPC and LPC (Fig. 4b and d). In this study, since LPC and SPC have the same electrode composition and are tested under the same conditions, any difference of the observed diffraction peaks would be ascribed to the different sulfur reaction extents and diffusion behaviors of the generated LiPS. For simplicity, the comparison is focused on the main phases of elemental S8, LiPS, and Li2S by tracking their respective characteristic diffraction peaks: 2.86°, 3.2° for S8; 1.2–2.6°, 2.4–2.6° and 2.95–3.15° for LiPS; and 3.38° and 3.89° for Li2S.36,37 Notably, the precise definition of the LiPS and the quantification of each phase during cell cycling require even higher-quality XRD patterns and will be reported in our follow-up publications.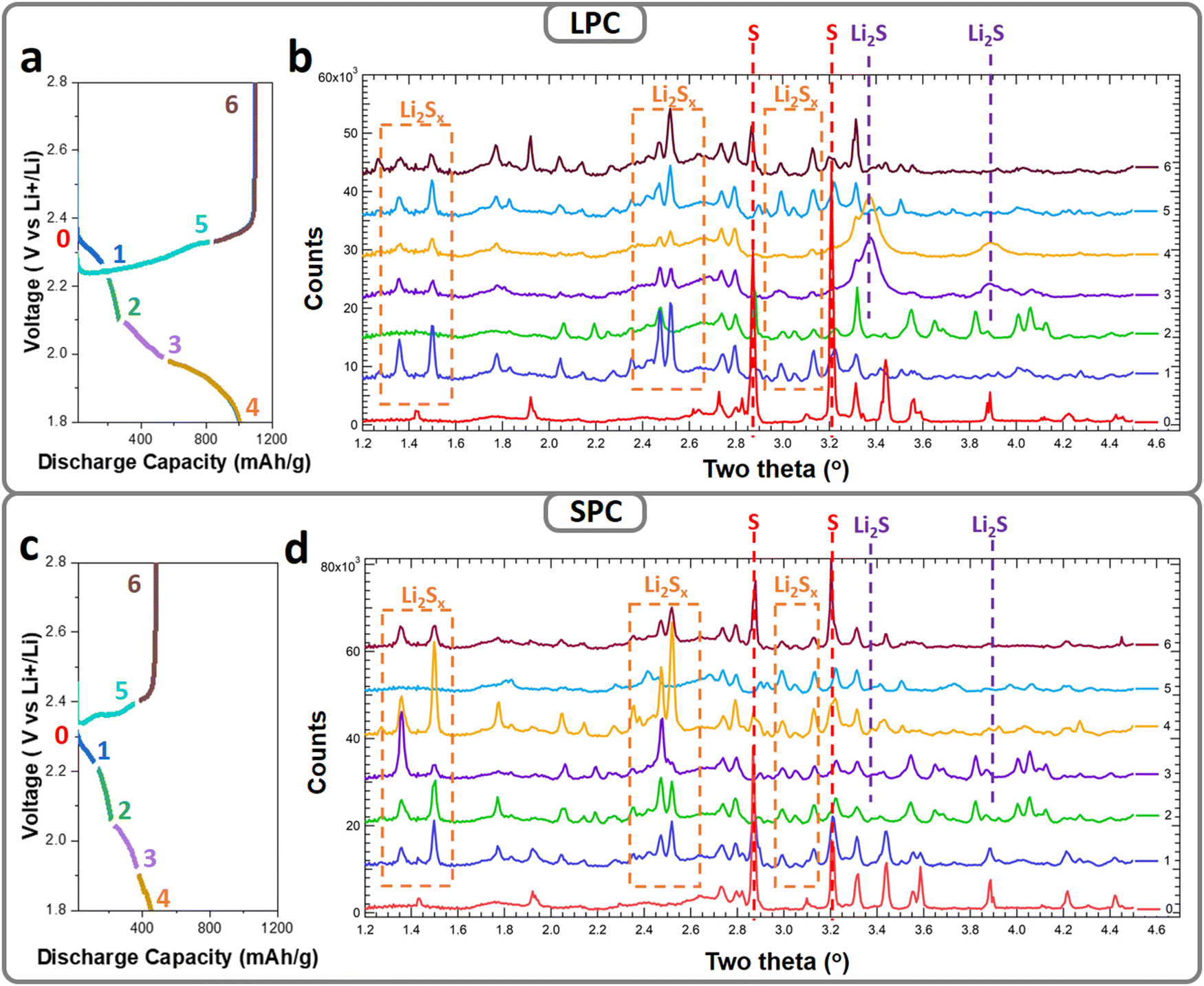 | ||
| Fig. 4 Ex situ synchrotron XRD characterization of the LPC and SPC at different depths of discharge. Typical first cycle discharge/charge curves and the corresponding XRD patterns (λ = 0.19316) for the LPC (a and b) and SPC (c and d). Successive diffraction patterns are vertically offset by 6000 relative to that of the pristine cathode (point 0, no offset). | ||
Both the pristine LPC and SPC have an α-S8 phase but in an amorphous or nanocrystalline state, as supported by the broad and low-intensity diffraction peaks at 2.86 and 3.2° (Fig. 4b-0 and d-0, respectively, labeled with red dashed lines). For the LPC electrode (Fig. 4b), once the discharge process starts (cutoff at 2.2 V), the α-S8 peaks become very weak, indicating fast reaction kinetics. Accompanying this, a new set of diffraction peaks was observed in the 2-theta ranges of 2.4–2.6° and 2.95–3.15° (labeled with orange dashed squares), suggesting conversions of S8 to LiPS. The densities of the new peaks decrease in subsequent discharging (cutoff: 2.1 V), indicating continuous reactions of LiPS. The XRD peaks observed in 1.2–1.6° were ascribed to the generated LiPS, which is a mixture of soluble Li2Sx with different chain lengths and relative ratio.38 Concentration of Li2Sx and ratio of each species are dependent on the depth of discharge and sulfur reactivity. With proceeding of cell reaction, total amount of Li2Sx, relative ratio, and their distribution inside the electrode evolve, resulting in the irregular changes of XRD peaks. In the voltage range of 2.1 to 1.9 V, with the decrease in LiPS diffraction intensity, a new set of peaks grow at 3.38° and 3.89° (labeled with violet dashed lines), corresponding to cubic-phase Li2S. The low and broad diffractions suggest the formed Li2S is amorphous or nanosized at the end of discharge. During the subsequent charging process, the Li2S peaks become weak and eventually disappear at 2.3 V, and the LiPS peaks reappear again, corresponding to the conversion of Li2S to LiPS. At end of charging (cutoff: 2.8 V), low-intensity diffraction peaks of S8 were observed, while in the SPC electrodes, distinct behaviors were identified for each voltage range. In contrast to the quick disappearance of S in the SPC electrode, the S8 phase still maintains at a high content after discharging to 2.2 V and coexists with the LiPS phase until 2.1 V. This suggests sluggish kinetics of the S-to-LiPS reaction. Different to the fast and complete phase transformation of LiPS to Li2S in the LPC electrode, the LiPS phase coexists during the whole discharging process (Fig. 4d), and only very weak diffractions of Li2S were found at the end of discharge (Fig. 4d). These observations suggest that transformation of LiPS to Li2S is suppressed in the SPC. One explanation for this would be that the large proportion of LiPS diffuses out of the secondary particles and even electrode after generation, which is consistent with the cell performance, i.e., lower capacity and a shorter second discharge plateau (Fig. 4c).
Electrochemical and ex situ XRD results indicate that distinct S reaction processes in the SPC and LPC originate from the second discharge plateau, i.e., LiPS-to-Li2S reactions. From the XRD results, for both cases, one can see that the soluble LiPS is generated during discharge but follows different pathways in the subsequent processes. Compared to the LPC electrode, the SPC electrode has much slower S-to-LiPS conversion kinetics, which may be caused by restricted electrode wetting. In addition, the LiPS diffuses out more quickly in the SPC and accumulates outside the electrode. During the next step, the LiPS-to-Li2S conversion, only part of the LiPS can re-access the active particle surface and form Li2S (or Li2S2) passivation layers, blocking the inflow of LiPS. A consequence of the blocked LiPS inflow would be the speed-up of sulfur irreversible loss (Fig. 4c), which would explain the very weak Li2S diffractions in the SPC at the end of discharge (Fig. 4d4). In the LPC electrode, longer diffusion time is needed for the LiPS to flow out of the LPC. This reduces the LiPS loss and improves the conversion rate to Li2S, as proved by XRD (Fig. 4b-3). Moreover, instead of forming a surface blocking layer, the LPC has larger and open pores to allow for the inflow of LiPS, which is also helpful for attaining high specific capacity.
The sulfur reactions in the dense SPC and LPC were further studied by in situ EIS and electrode morphology characterization. To decouple the interferences of Li metal while acquiring EIS spectra, a three-electrode cell configuration was used, where a tiny strip of LTO (Li4Ti5O12) reference electrode was wrapped with a polypropylene separator and placed between the S working electrode and Li metal counter electrode. Fig. 5a and c plot the first discharge curves of the LPC and SPC under lean-electrolyte conditions (E/S = 4 μL mg−1) during EIS analysis. EIS results (Fig. 5b and d) were acquired during cell discharging at intervals of 7000 seconds. Upon discharging, the overall Rct of the LPC electrode decreased slightly and remained stable from Phase I to Phase III. This is attributed to the combined contributions of polysulfide generation and enhanced wetting. An increase in Rct was observed during Phase IV and grew quickly at the end of Phase V due to the formation of solid Li2S/Li2S2. However, in the SPC electrode, the overall resistance increased very early starting from the end of Phase II and surged to a level as high as that of Phase V in the LPC electrode. These EIS results are consistent with the cell performance and ex situ XRD, confirming that the SPC electrode is blocked early and terminated. Accordingly, a mechanism illustration depicting the difference of reaction process between LPC and SPC is proposed in Fig. S16 (ESI†). This is supported by SEM/EDS characterization of the discharged cells. After the first discharge, compact and smooth coating layers composed of flower-like precipitations were observed on the SPC (Fig. 5g, h and Fig. S15, ESI†), while a cleaner surface and open pores were maintained on the LPC (Fig. 5e and f). It has been reported that when encapsulated in the carbon matrix, the LiPS will form amorphous or nanosized Li2S after discharge;39 otherwise, it tends to form flake-like Li2S particles. When examining the Li anode, the Li metal in an LPC cell maintained a relatively smooth surface after the first discharge (Fig. 5i and j), while more and larger particles of LiPS or Li2S were observed on the Li anode of the SPC cell (Fig. 5k and l), suggesting more serious LiPS outflow in the SPC.
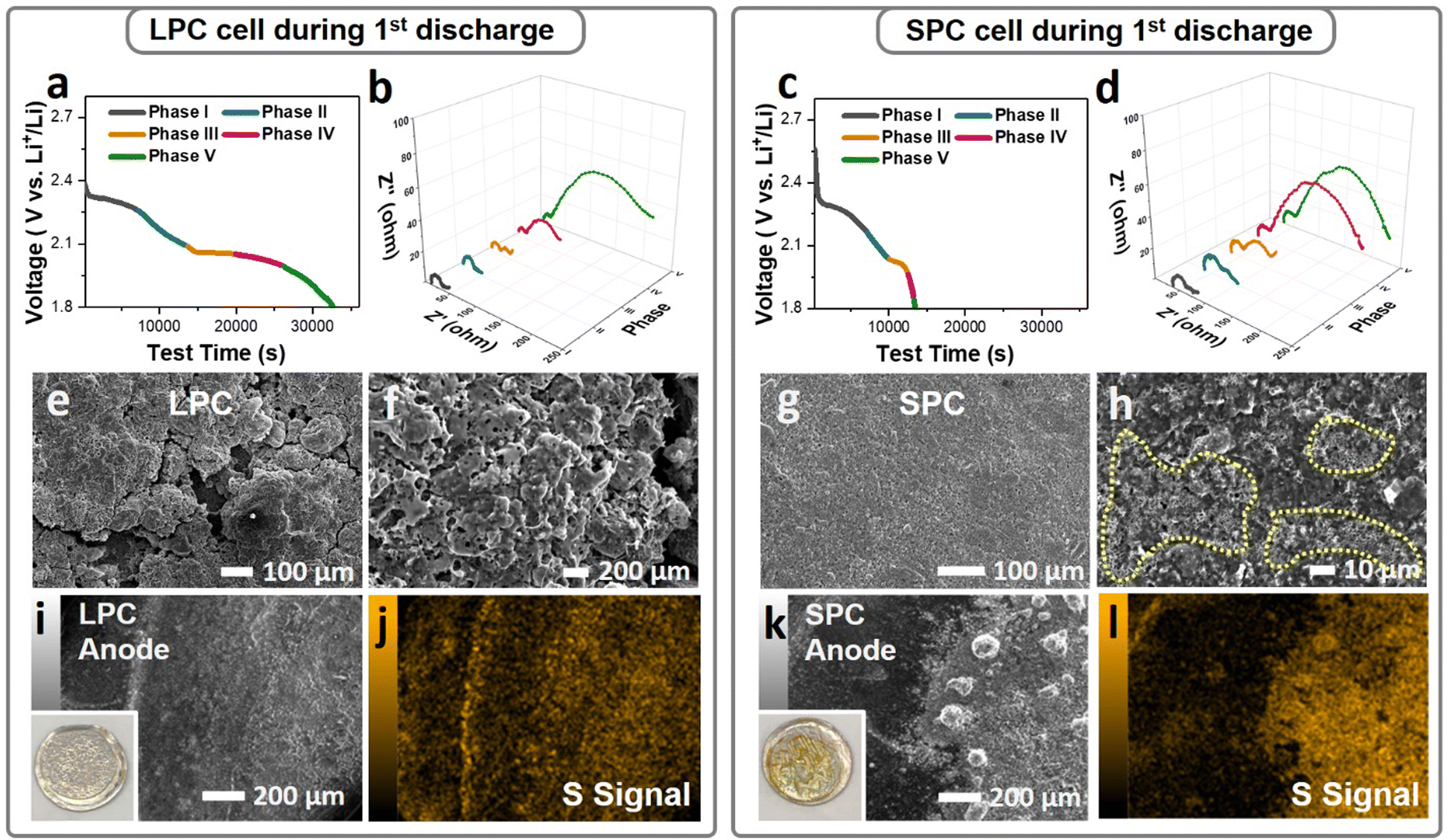 | ||
| Fig. 5 Characterizations of the LPC/SPC and the corresponding lithium anodes after first discharge under lean-electrolyte conditions. The first discharge curves and corresponding in situ EIS of the LPC (a and b) and SPC (c and d), respectively. SEM images of the LPC (e and f) and SPC (g and h). EDS analysis of the anodes in the LPC (i and j) and (k and l), the inset pictures are digital images of lithium chips after the first discharge. | ||
Conclusion
The design principle for the low-tortuous sulfur cathode was elucidated and validated by preparing a single-particle-layer electrode. The impacts of sulfur electrode architecture on cell performance were studied under practical conditions and were traced to material- and electrode-level understandings. The electrode porosity not only dominated the electrode-level volumetric and gravimetric capacity because of the large pore-volume and pore-filling electrolyte, but also altered the sulfur reaction and cell cycling by regulating the electrolyte infiltration. Reducing the electrode porosity without sacrificing the sulfur utilization rate is a key step toward development of realistic Li-S cells. By using a single-particle-layer cathode, we achieved a high sulfur-utilization rate (∼1001 mA h g−1) in high-loading (4 mg cm−2) and dense (∼45%) sulfur electrodes under very lean-electrolyte conditions (E/S = 4 μL mg−1). Using small (<20 μm) and large (>90 μm) IKB/S particles as example materials, we elucidated the critical impacts of secondary particle size on electrolyte permeability, LiPS shuttling, and sulfur reactions. For a given electrode porosity, the LPC comprising large particles with fewer outer surfaces but longer dimensions deliver superior performance over the SPC in terms of sulfur utilization, reaction kinetics, and capacity retention. The low-tortuosity through-pores of the LPC electrode are essential for quick electrode wetting and easy backflow of the LiPS during cycling. This study sheds new light on materials development and electrode design for practical high-energy Li-S batteries.Experimental section
Synthesis of IKB/S material
The integrated Ketjen Black/Sulfur (IKB/S) composite was prepared using a modified synthesis approach reported previously.24 Typically, KB powder (AkzoNobel) and poly(melamine-co-formaldehyde) methylated solution (Sigma-Aldrich) were thoroughly blended with a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and then the mixture was dried and carbonized at 900 °C under Ar atmosphere for 10 hours. The collected IKB was heat treated with sulfur (weight ratio IKB:S = 1:4) at 155 °C for 12 hours, resulting in IKB/S. The particles were then sieved and separated into different size ranges.
:1 and then the mixture was dried and carbonized at 900 °C under Ar atmosphere for 10 hours. The collected IKB was heat treated with sulfur (weight ratio IKB:S = 1:4) at 155 °C for 12 hours, resulting in IKB/S. The particles were then sieved and separated into different size ranges.
Electrode preparation
Carbon nanofiber (Sigma-Aldrich) was first dispersed in a polyacrylic acid (Sigma-Aldrich) dimethylformamide (DMF) solution (2 wt%) to form a uniform slurry. Then, the IKB/S particles were added and thoroughly mixed in a Thinky mixer for 15 minutes (ARE-310, Thinky). The weight ratio of IKB/S, carbon nanofiber, and polyacrylic acid was controlled at 8:1:1, and the solid content in the slurry was 20%. The obtained slurry was cast onto aluminum foil with a sulfur loading of 4 mg cm−2 and dried at a half vacuum and 60 °C for 12 hours. The porosities of the electrodes were further controlled by manipulating electrode thickness by calendering and were cut into disks with an area of 1.26 cm2. The details of electrode porosity calculation are listed in Tables S1 and S2 in ESI.† Generally, for a 4 mg cm−2 cathode with 64% sulfur content, the electrode porosity was estimated to be 72%, 63%, 52%, and 45% corresponding to an electrode thickness of 120, 90, 70, and 60 μm, respectively.
Characterization
The surface area of IKB was measured using nitrogen adsorption/desorption isotherms recorded by QUANTACHROME AUTOSORB 6-B gas sorption system. The calculation of surface area was based on the isotherms using the five points BET method. The morphologies of materials and electrode were observed using scanning electron microscopy (SEM, JEOL, JSM-IT800). For electrolyte distribution observation, the electrodes were assembled into coin cells with an E/S ratio of 4. After 3 hours at rest, the cells were disassembled, and the cathodes were taken out and dried under vacuum at 60 °C.For synchrotron XRD characterization, 14 cycled Li–S coin cells were disassembled inside an argon-filled glovebox. The cathode films were dried in glovebox overnight. In the glovebox, the 14 dried cathode discs were attached to the inner surface of a single large aluminum-lined pouch cell using a circle of Kapton tape slightly larger than the cathode diameter. The pouch was sealed under argon and was shipped to beamline 28-ID-2 of the National Synchrotron Light Source II (NSLS-II) at Brookhaven National Laboratory (BNL) for experiments. At the beamline, the pouch cell was attached to the window of a stiff aluminum frame (photograph and approximate dimensions provided in Fig. S17, ESI†). Calibration was carried out using a CeO2 powder (674b, NIST) sample in a Kapton capillary (1.1 mm in diameter) attached to the side of the frame. Diffraction data were collected using X-ray with a wavelength of 0.19316 Å on a PerkinElmer amorphous silicon-based area detector (2048 × 2048 pixels with 200 μm square pixel edges) at a distance of about 1.6 m using a 0.2 s subframe exposure time and a total acquisition time of 60 s per sample. Integration of the diffraction data was carried out over a 2θ range of 0.5–15° (d = 0.37–11.07 Å) with masks used to exclude the beam stop and the edges of the detector.
X-Ray micro computed tomography (X-ray micro-CT) method was employed to obtain the three-dimensional (3D) microstructure of sulfur electrodes. 3D-CT images of sulfur electrodes were reconstructed from a series of two-dimensional (2D) X-ray projection images obtained from a lab-based X-ray microscope (Zeiss, Versa 610). 2D images are measured at 20× (X-ray source energy and power: 65 kV, 6.5 W) and 40× (80 kV, 10 W) optical magnification in absorption-contrast mode. A total of 3202 2D projections were collected per 360° sample rotation with exposure times of 2 and 4 s for 20× and 40× optical magnifications, respectively.
Electrochemical characterization
2032-Type coin cells were assembled in an Ar-filled glovebox. The electrode area was 1.26 cm2. The thickness of the lithium anode was 250 μm. A Celgar-2400 separator with a 1.9 cm diameter was used in all coin cells. The electrolyte consists of 1 M lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, Gotion), 1,3-dioxolane (DOL, Gotion) and 1,2-dimethoxyethane (DME, Gotion) (1:1, v/v), 0.3 M LiNO3. The electrolyte to sulfur ratio was controlled as 10 μL mg−1 for the flooded electrolyte condition and 4 μL mg−1 for the lean-electrolyte condition. The coin cells were rested for 3 hours and then tested on a LANHE battery tester CT3001A at 0.1 C (1 C = 1000 mA g−1) at 30 °C in a voltage range of 1.8–2.8 V. The EIS study was performed on a Biologic SP-50 Potentiostat. The three-electrode-cell was assembled with the sulfur electrode and Li as working and counter electrodes, respectively, and a lithium titanate (Li4Ti5O12, LTO) coated Cu wire was used as reference electrode and placed between the working and counter electrodes.
Simulation method
Computational fluid dynamics (CFD) simulations were conducted to explain the species transport and wetting of the surface of micropores in the porous electrodes using a commercial CFD software STAR-CCM+. The simulations for the species transport were conducted to understand the transport of LiPS concentration in the electrode. Multiphase flow simulations using volume of fluid (VOF)40 explained the basic understanding of the electrolyte transport in the porous electrode comprising the smaller and larger pore particles. The wetting of the pore particles plays a critical role in the performance of an Li-S battery. Hence, an extensive simulation campaign comprising single phase flow and multiphase flow investigations was conducted in the porous media to understand the transport phenomenon inside the electrodes.Simulations for electrolyte wetting
The multiphase flow simulations using the VOF method were conducted first to understand the wetting of pore particles and electrolyte transport in the electrode. The governing equations were as follows:| ∇·u = 0, | (1) |
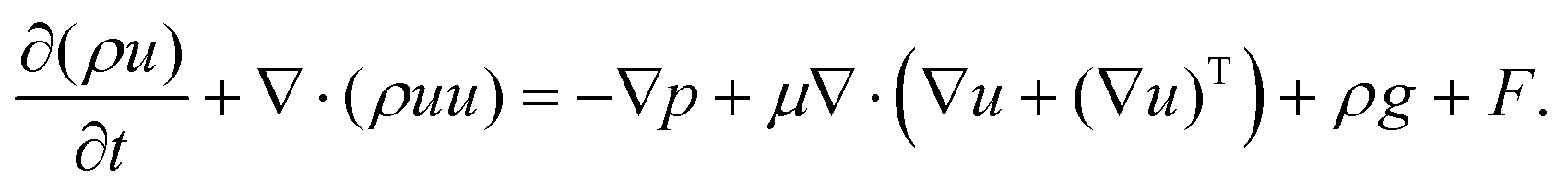 | (2) |
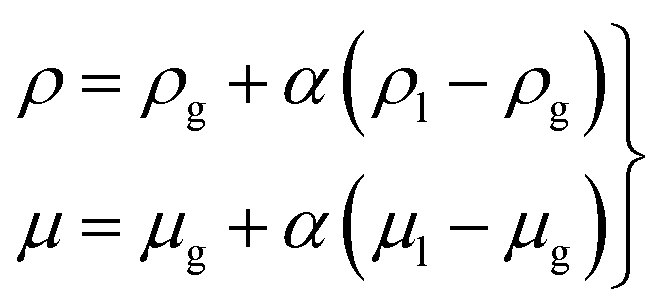 | (3) |
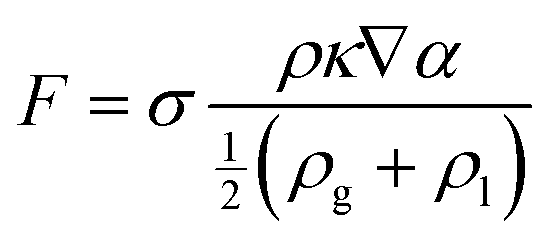 | (4) |
![[n with combining circumflex]](https://www.rsc.org/images/entities/i_char_006e_0302.gif) = ∇α/|∇α|):
= ∇α/|∇α|):| κ = ∇· | (5) |
According to the experimental observations, two sizes (72 and 32 μm) of particle were chosen in the flow simulations. The model of the computational flow domain was created by a random arrangement of larger and smaller particles (shown as the yellow regions in Fig. 2(a)). The electrolyte was poured into the flow domain through the top region above the electrode and flow was driven by the gravity. When adding electrolyte on the top of electrodes, gravity is the main driving force that initiates electrolyte infiltration. The flow is driven by gravity and the wetting is dictated by gravity, capillary force, and the surface characteristic of the solid substrate. In the pore-scale simulation, pressure drop is dictated by the pore-scale capillary pressure. In the simulation, the surface characteristics of the electrode were implemented by the contact angles at the surface of the particles, which is relative major of the adhesion and cohesion behavior. The wall of the particles was set as the no-slip wall with contact angle (γ). Both sides of the electrode were specified as the hydrophobic walls, whereas the wall of the pore particles was considered to be hydrophilic. The hydrophilic and hydrophobic conditions were defined by means of the value of the contact angle. The multiphase flow studies using the VOF method were conducted using air (ρg = 1.185 kg m−3 and μg = 0.0183 mPa s), and electrolyte was used in the Li–S battery. The electrolyte comprised a mixture of 1,3-dioxolane (DOL) and 1,2-dimethoxyethane (DME) with 1 M LiTFSI. The electrolyte had physical properties of (μl = 2.56 mPa s42 and ρl = 997 kg m−3), surface tension (σ = 34.4 mN m−1),41 and contact angle (γ = 20°).43,44 The implicit transient flow simulations were conducted with a very small timestep (Δt ∼ 10−7 s) for stability and convergence because the grid size was on the order of 0.1 μm.
Scalar transport simulations
In addition to conducting multiphase flow simulations for the wetting of pore particles, flow simulations were conducted to understand the transport phenomenon due to diffusion of LiPS in the electrode. The diffusion of LiPS in the electrode was studied by solving the passive scalar transport equation, which is based on the standard convection-diffusion equation: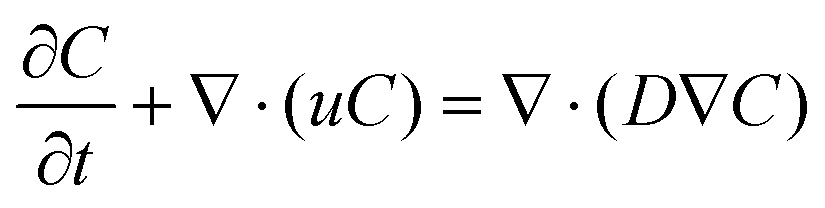 | (6) |
Here, C is the concentration, which is treated as a passive scalar, and D is its diffusion coefficient. As explained earlier, dissolution of the LiPS in the electrode occurs mainly due to diffusion, and internal and external LiPS concentration gradients exist. Note that the migration rate of the LiPS in the electrodes depends on the effective diffusion coefficient. In this regard, flow simulations for passive scalar transport were conducted to calculate the effective diffusivity in the electrodes. The passive scalar was representative of LiPS and the value of diffusion coefficient (D) of the scalar was kept constant for both electrodes. In contrast to solid pore particles used in two-phase flow studies, porous particles (see the porous zone inside the white region in Fig. S3(a)) were chosen in this case to understand the internal and external diffusion of LiPS in the electrode. The flow simulations were conducted by specifying the scalar flux rate at the internal pore surfaces of particles of the electrodes. The scalar flux specified at the internal pores deals the understanding the migration of LiPS in the electrodes. The top boundary of the flow domain was specified as a given value of the scalar concentration. Remaining boundaries of the flow domain were specified as zero flux. The unsteady flow simulations were performed and the scalar concentration at the internal pore surfaces was monitored. The simulations were continued until the scalar concentration at the internal pore wall achieved a steady-state value, and the results were further analyzed. Once the simulation achieved a steady state, the effective diffusivity (Deff) was computed as Deff = NΔy/S0ΔC, where N is flux at the internal wall of porous particles, Δy is distance from the electrode surface, ΔC is concentration difference, and S0 is the cross-sectional area.
Author contributions
Conceptualization: D. L. and S. F.; Data curation: S. F., R. S., Y. F., Z. L., Y. W., J. B., Z. X., G. L., C. A. and L. S.; Writing – original draft: S. F. and D. L.; Writing-review & editing: Y. L., P. K., W. W., J. L. and J. X. All authors contributed to the discussion and provide feedback on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Assistant Secretary for Energy Efficiency and Renewable Energy, Office of Vehicle Technologies of the U.S. Department of Energy through the Advanced Battery Materials Research Program (BMR and Battery500 Consortium under Contract No. DEAC02-05CH11231 and DEAC02-98CH10886). The modeling work was supported by the Energy Storage Materials Initiative (ESMI), under the Laboratory Directed Research and Development (LDRD) Program at Pacific Northwest National Laboratory (PNNL). PNNL is operated by Battelle under Contract No. DE-AC05-76RL01830 for the U.S. Department of Energy. Research was in part carried out at Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-SC0012704. Use of the National Synchrotron Light Source, Brookhaven National Laboratory II, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-SC0012704.References
- J. Xiao, Adv. Energy Mater., 2015, 5, 1501102 CrossRef.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed.
- S. S. Zhang, J. Power Sources, 2013, 231, 153–162 CrossRef CAS.
- K. Yuan, L. Yuan, J. Chen, J. Xiang, Y. Liao, Z. Li and Y. Huang, Small Struct., 2020, 2, 2000059 CrossRef.
- A. Manthiram, Y. Fu, S. H. Chung, C. Zu and Y. S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- L. Borchardt, M. Oschatz and S. Kaskel, Chem. – Eur. J., 2016, 22, 7324–7351 CrossRef CAS.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, Nat. Energy, 2016, 1, 16132 CrossRef CAS.
- H. J. Peng, J. Q. Huang, X. B. Cheng and Q. Zhang, Adv. Energy Mater., 2017, 7, 201700260 Search PubMed.
- J. Song, T. Xu, M. L. Gordin, P. Zhu, D. Lv, Y. B. Jiang, Y. Chen, Y. Duan and D. Wang, Adv. Funct. Mater., 2014, 24, 1243–1250 CrossRef CAS.
- C. Zhao, G. L. Xu, Z. Yu, L. Zhang, I. Hwang, Y. X. Mo, Y. Ren, L. Cheng, C. J. Sun, Y. Ren, X. Zuo, J. T. Li, S. G. Sun, K. Amine and T. Zhao, Nat. Nanotechnol., 2020, 16, 166 CrossRef PubMed.
- H. Yuan, J. Q. Huang, H. J. Peng, M. M. Titirici, R. Xiang, R. Chen, Q. Liu and Q. Zhang, Adv. Energy Mater., 2018, 8, 1802107 CrossRef.
- H. Wang, W. Zhang, J. Xu and Z. Guo, Adv. Funct. Mater., 2018, 28, 1707520 CrossRef.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X. Q. Yang and J. G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- A. Bhargav, J. He, A. Gupta and A. Manthiram, Joule, 2020, 4, 285–291 CrossRef.
- Y. Cao, M. Li, J. Lu, J. Liu and K. Amine, Nat. Nanotechnol., 2019, 14, 200–207 CrossRef CAS.
- P. Wang, B. Xi, M. Huang, W. Chen, J. Feng and S. Xiong, Adv. Energy Mater., 2021, 11, 2002893 CrossRef CAS.
- W. Deng, J. Phung, G. Li and X. Wang, Nano Energy, 2021, 82, 105761 CrossRef CAS.
- F. Qi, Z. Sun, X. Fan, Z. Wang, Y. Shi, G. Hu and F. Li, Adv. Energy Mater., 2021, 11, 2100387 CrossRef CAS.
- M. E. Zhong, J. Guan, Q. Feng, X. Wu, Z. Xiao, W. Zhang, S. Tong, N. Zhou and D. Gong, Carbon, 2018, 128, 86–96 CrossRef CAS.
- W. Xue, L. Miao, L. Qie, C. Wang, S. Li, J. Wang and J. Li, Curr. Opin. Electrochem., 2017, 6, 92–99 CrossRef CAS.
- S. Dörfler, H. Althues, P. Härtel, T. Abendroth, B. Schumm and S. Kaskel, Joule, 2020, 4, 539–554 CrossRef.
- G. Li, Z. Liu, Q. Huang, Y. Gao, M. Regula, D. Wang, L.-Q. Chen and D. Wang, Nat. Energy, 2018, 3, 1076–1083 CrossRef CAS.
- D. Lu, Q. Li, J. Liu, J. Zheng, Y. Wang, S. Ferrara, J. Xiao, J. G. Zhang and J. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 23094–23102 CrossRef CAS PubMed.
- D. Lv, J. Zheng, Q. Li, X. Xie, S. Ferrara, Z. Nie, L. B. Mehdi, N. D. Browning, J.-G. Zhang, G. L. Graff, J. Liu and J. Xiao, Adv. Energy Mater., 2015, 5, 1402290 CrossRef.
- N. Kang, Y. Lin, L. Yang, D. Lu, J. Xiao, Y. Qi and M. Cai, Nat. Commun., 2019, 10, 4597 CrossRef PubMed.
- J. U. Brackbill, D. B. Kothe and C. Zemach, J. Comput. Phys., 1992, 100, 335–354 CrossRef CAS.
- L. Li, R. M. Erb, J. Wang, J. Wang and Y. M. Chiang, Adv. Energy Mater., 2018, 9, 1802472 CrossRef.
- Y. Xie, G. Pan, Q. Jin, X. Qi, T. Wang, W. Li, H. Xu, Y. Zheng, S. Li, L. Qie, Y. Huang and J. Li, Adv. Sci., 2020, 7, 1903168 CrossRef CAS PubMed.
- Z. Han, S. Li, R. Xiong, Z. Jiang, M. Sun, W. Hu, L. Peng, R. He, H. Zhou, C. Yu, S. Cheng and J. Xie, Adv. Funct. Mater., 2021, 32, 2108669 CrossRef.
- K. H. Chen, M. J. Namkoong, V. Goel, C. Yang, S. Kazemiabnavi, S. M. Mortuza, E. Kazyak, J. Mazumder, K. Thornton, J. Sakamoto and N. P. Dasgupta, J. Power Sources, 2020, 471, 228475 CrossRef CAS.
- H. Chen, A. Pei, J. Wan, D. Lin, R. Vilá, H. Wang, D. Mackanic, H. G. Steinrück, W. Huang, Y. Li, A. Yang, J. Xie, Y. Wu, H. Wang and Y. Cui, Joule, 2020, 4, 938–952 CrossRef CAS.
- W. Xue, Z. Shi, L. Suo, C. Wang, Z. Wang, H. Wang, K. P. So, A. Maurano, D. Yu, Y. Chen, L. Qie, Z. Zhu, G. Xu, J. Kong and J. Li, Nat. Energy, 2019, 4, 374–382 CrossRef CAS.
- S. Zhang, Energies, 2012, 5, 5190–5197 CrossRef CAS.
- S. G. Lee, D. H. Jeon, B. M. Kim, J. H. Kang and C.-J. Kim, J. Electrochem. Soc., 2013, 160, H258–H265 CrossRef CAS.
- L. Shi, S.-M. Bak, Z. Shadike, C. Wang, C. Niu, P. Northrup, H. Lee, A. Y. Baranovskiy, C. S. Anderson, J. Qin, S. Feng, X. Ren, D. Liu, X. Q. Yang, F. Gao, D. Lu, J. Xiao and J. Liu, Energy Environ. Sci., 2020, 13, 3620–3632 RSC.
- J. Nelson, S. Misra, Y. Yang, A. Jackson, Y. Liu, H. Wang, H. Dai, J. C. Andrews, Y. Cui and M. F. Toney, J. Am. Chem. Soc., 2012, 134, 6337–6343 CrossRef CAS PubMed.
- N. A. Cañas, S. Wolf, N. Wagner and K. A. Friedrich, J. Power Sources, 2013, 226, 313–319 CrossRef.
- J. Conder, R. Bouchet, S. Trabesinger, C. Marino, L. Gubler and C. Villevieille, Nat. Energy, 2017, 2, 17069 CrossRef CAS.
- H. Pan, J. Chen, R. Cao, V. Murugesan, N. N. Rajput, K. S. Han, K. Persson, L. Estevez, M. H. Engelhard, J. G. Zhang, K. T. Mueller, Y. Cui, Y. Shao and J. Liu, Nat. Energy, 2017, 2, 813–820 CrossRef CAS.
- C. W. Hirt and B. D. Nichols, J. Comp. Phys., 1981, 39, 201–225 CrossRef.
- J. J. Hu, G. K. Long, S. Liu, G. R. Li and X. P. Gao, Chem. Commun., 2014, 50, 14647–14650 RSC.
- C. Sauter, R. Zahn and V. Wood, J. Electrochem. Soc., 2020, 167, 100546 CrossRef CAS.
- M. Rana, M. Li, Q. He, B. Luo, L. Wang, I. Gentle and R. Knibbe, J. Energy Chem., 2020, 44, 51–60 CrossRef.
- W. Shin, J. Lu and X. Ji, Carbon Energy, 2019, 1, 165–172 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee01442d |
| This journal is © The Royal Society of Chemistry 2022 |