
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeological storage of hydrogen in deep aquifers – an experimental multidisciplinary study
P. G.
Haddad
a,
M.
Ranchou-Peyruse
 abc,
M.
Guignard
c,
J.
Mura
a,
F.
Casteran
ab,
L.
Ronjon-Magand
c,
P.
Senechal
d,
M.-P.
Isaure
c,
P.
Moonen
de,
G.
Hoareau
e,
D.
Dequidt
f,
P.
Chiquet
bg,
G.
Caumette
bh,
P.
Cezac
ab and
A.
Ranchou-Peyruse
*bc
abc,
M.
Guignard
c,
J.
Mura
a,
F.
Casteran
ab,
L.
Ronjon-Magand
c,
P.
Senechal
d,
M.-P.
Isaure
c,
P.
Moonen
de,
G.
Hoareau
e,
D.
Dequidt
f,
P.
Chiquet
bg,
G.
Caumette
bh,
P.
Cezac
ab and
A.
Ranchou-Peyruse
*bc
aUniversité de Pau et Pays de l’Adour, E2S UPPA, LaTEP, Pau, France
bJoint Laboratory SEnGA, UPPA-E2S-Teréga, 64000, Pau, France
cUniversité de Pau et Pays de l’Adour, E2S UPPA, CNRS, IPREM, Pau, France. E-mail: anthony.ranchou-peyruse@univ-pau.fr; Tel: +33 540 145 164
dUniversité de Pau et Pays de l’Adour, E2S UPPA, CNRS, DMEX, Pau, France
eUniversité de Pau et Pays de l’Adour, E2S UPPA, CNRS, TOTAL, LFCR, Pau, France
fSTORENGY – Geosciences Department, Bois-Colombes, France
gTeréga – Geosciences Department, Pau, France
hTeréga – Environment Department, Pau, France
First published on 12th July 2022
Abstract
Dihydrogen (H2) is a promising source of energy in the field of energy transition. Similarly to natural gas, identifying storage solutions for large volumes of H2 is essential. Geological storage of H2 and methane mixtures in underground gas storage (UGS) such as deep aquifers is universally promising in particular in our current energy avid world. That said, interactions between water formation, reservoir rock, gas mixture and the microbial ecosystems remain poorly defined and further clarifications on this issue remain fundamental. Our study aims at identifying the effect of H2 injection on the aforementioned milieu and at clarifying those interactions in UGS. The aquifer was reproduced experimentally in a reactor: water and rock phases sampled from the actual aquifer and a synthetic gas phase representing the gaseous mix to store. Since the beginning of the experiment (at a pressure of 85 bar methane – 1% carbon dioxide, 47 °C), sulfate was consumed continuously until its depletion from the liquid phase. As soon as H2 was injected (10% H2 at 95 bar), formate was produced in the aqueous phase and CO2 was consumed from the gas phase. Once sulfate was depleted, the microbial activities were based on the consumption of H2 and CO2, indicating a switch in the microbial ecosystems towards Subsurface Lithoautotrophic Microbial Ecosystems (SLiMEs). Transcriptomic diversity analyses subsequently confirmed the increased activity of methanogens after H2 injection. Moreover, methanogenic archaea became the majority in the ecosystem. Once the CO2 was depleted in the gas phase, H2 consumption and formate production instantly stopped. In less than 90 days, nearly 40% of injected H2 transformed either into H2S, formate and methane. This suggests that microbial life harbored in a deep aquifer has a major impact on the evolution of H2 storage especially on sulfate, CO2, calcite and H2 concentrations in the system.
1. Introduction
Dihydrogen (H2) is receiving interest for its use as a green energy vector in the context of energy transition. Nowadays, H2 is mainly produced artificially (96%) from fossil fuels, in particular steam reforming of natural gas.1 The growing interest toward using H2 as an alternative for fossil fuels involves the increase in its carbon-free production. Green H2 can be produced by electrolysis of water from surplus renewable energy (solar, wind) and thus compensate for their intermittent production.2,3 This H2 can then be stored locally or injected into the natural gas grid (green Power-to-Gas). Natural gas networks are complex, often interconnected between several countries and require geological storage (saline cavities, deep aquifers and depleted hydrocarbon reservoirs) to compensate for periods of peak consumption.Used for their large capacities and capability to store natural gas, deep aquifers represent attractive underground storage systems.4 However, H2 properties must be taken into consideration since H2 is different from the usual stored gases such as methane, air and carbon dioxide.5,6 H2 injection with natural gas in the underground storage sites may promote reactions between the subsurface minerals and fluids, thus altering the storage properties.2,6,7 Many studies are conducted to identify the geochemical changes that may occur after H2 injection in the underground. For instance, the comparison of mudstones from different storage formations in Germany in the context of the H2STORE project showed that clay-rich reservoir rocks may be more influenced since these minerals may strongly increase the pore-exposed reaction surface.8 Moreover, another H2STORE study showed that pore-filling anhydrite and carbonate cements tend to dissolve when widely present in the system under high pressure and high temperature H2 storage.9 Thus, mineralogical reactions and dissolution may alter the formation's porosity and permeability, risking the underground storage.2,8–11 In the case of anhydrite and carbonate cement dissolution, changes will be observed in the composition of the formation water and promote salt precipitation. In addition to mineral dissolution or precipitation, changes in the thermodynamic equilibrium conditions induced by the injection of H2 can significantly modify the chemical reactivity of the gas-rock-brine system. The latter is also greatly affected by microbial activity.12
In detail, deep aquifers are well known to harbor deep underground ecosystems with much diversified microbial metabolisms.13 H2 can be found in the deep underground from many inorganic reactions such as the graphitization of methane into graphite, alteration of iron-rich minerals, water radiolysis or even magmatic degassing.14–17 H2 is an energy source for many microorganisms hosted in such deep ecosystems as underground gas storage (UGS) aquifers.6 Ecosystems defined by Stevens and McKinley as SLiMEs (Subsurface Lithoautotrophic Microbial Ecosystems) are based on H2 and carbon dioxide as energy and carbon sources, respectively.18 Subsequently, and although the existence of SLiMEs was questioned by Anderson et al.,19 many examples of SLiMEs have been described for different geological environments.20–27 In deep aquifers used for geological storage, H2 is supposed to be mainly produced from the fermentation of organic molecules and consumed by some sulfate-reducers, homoacetogens and methanogens.4,28–30 It has long been known that these three functional groups may compete for this resource,25,31–33 along with other compounds found in underground systems. But the consequences for the storage can be significant: consumption of H2 (and CO2), production of methane and/or sulfide with modification of the gas quality, pyrite precipitates, toxicity, and even tubing corrosion.12 For example, during town gas storage in Lobodice (Czechoslovakia), 54% vol. of H2 was injected into an underground reservoir at 40 bar, 25–45 °C. After 7 months, the gas mixture contained 37% vol. of H2 while the fraction of methane increased from 21.9 to 40% vol.34 Due to in situ chemical reactions and microbial activities, fluctuations in the storage gas composition can occur.6,10,35,36
In order to understand the effect of H2 injection on such diversified ecosystems, a multidisciplinary study was conducted. The impact of H2 on an operated natural gas storage site was explored experimentally under in situ conditions. Multi-phase aquifers were reproduced in a reactor. Water formation and the autochthonous microbial community were sampled from the specified aquifer with a downhole sampler and used as the aqueous phase. A sample of reservoir rock represented the solid phase. At the beginning of the experiment, the gas phase was composed of methane and 1% of carbon dioxide representing the current stored natural gas in the aquifer. The pressure and the temperature were 85 bar and 47 °C, corresponding to the temperature and the pressure of the UGS. H2 was injected after 21 days with a proportion corresponding to 10% of the gas mixture, increasing the total pressure to 95 bar, at the same temperature of 47 °C. Aqueous and gas phases were sampled throughout the experiment for chemical analyses and microbial studies. The solid phase was analyzed before and after the experiment to evaluate the evolution of the mineralogical phases. Then, results were correlated to identify interactions between liquid, solid, gas phases and the microbial ecosystems.
2. Materials and methods
2.1 Site description and water formation characteristics
The studied reservoir is a Parisian aquifer (989 m depth) with a geological formation belonging to the Late Triassic sedimentary Layer (Rhaetian-Norian). The formation water was sampled from a monitoring well of an operated underground natural gas storage (Fig. 1). The well is close to the water–gas interface. Therefore, the sampled liquid has the highest exposure to gas interactions in the underground. The selected well was identified in a recent study and was assigned the code name Pb_T_1.37 The sampling was accomplished from the well under pressure with a downhole sampler (1.088 L), equipped with a PDS Sampler (Leutert Bottom Hole Positive Displacement Sampler) and from the wellhead.38 Before taking the samples, the well was flushed with formation water (production of ten times the well's volume). Water sampling was also carried out from the wellhead. All formation water samples were stored in sealed bottles at 30 °C until use. The formation water's composition and its physicochemical characteristics (CARSO Laboratory, Lyon, France) are presented in Table 1. Under anoxic conditions, 0.612 L of wellhead sampled water was filter-sterilized at 0.1 μm (PES 47 mm membranes, 0.1 μm, Sartorius) and added to underground sampled formation water. Before injection into the reactor, a volume of 0.1 L was taken from the final mixture for microbial diversity analyses. The remaining 1.46 L from the rest of the final mixture was injected into the sterilized reactor.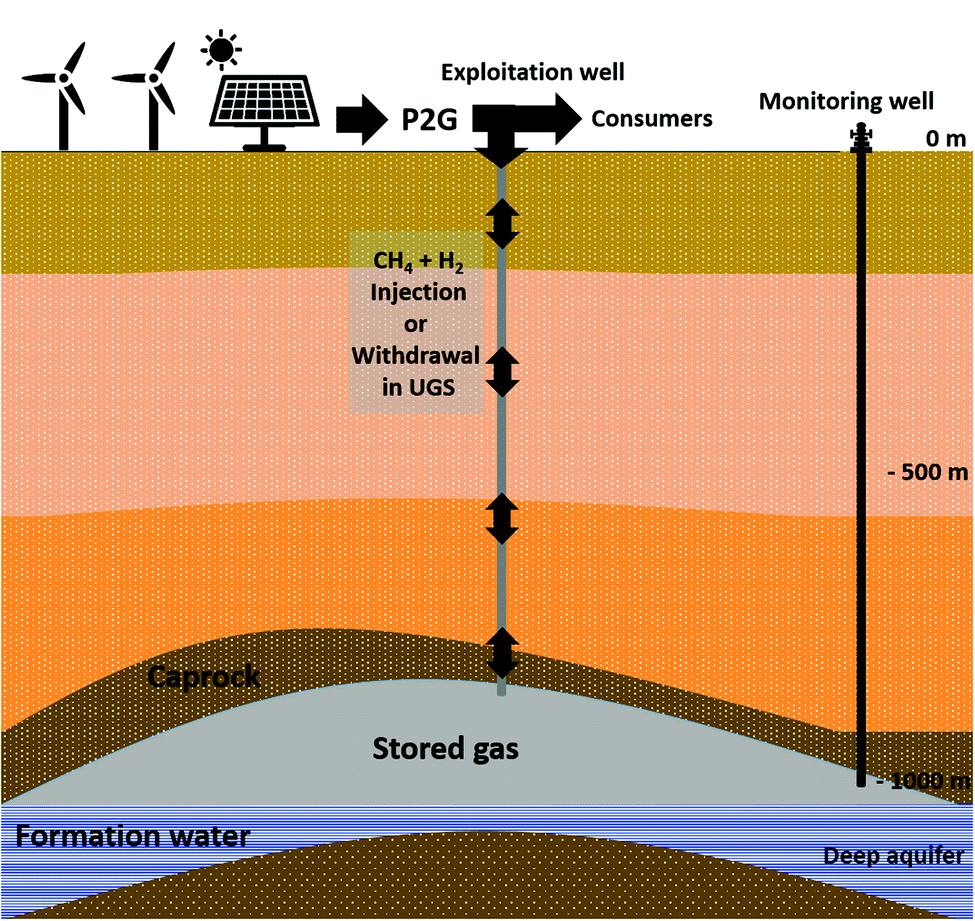 | ||
| Fig. 1 Scheme of an aquifer used for gas storage. | ||
| Physico-chemical parameters | Value | Unit |
|---|---|---|
| Pressure | 95 | Bar |
| Temperature | 47 | °C |
| pH | 7.9 | |
| Redox potential | −365.6 | mV |
| Conductivity at 25 °C | 1256 | μS cm |
| Organic Carbon | <1 | mg L−1 |
| Compounds | ||
| Chloride | 7.10 | mM |
| Nitrate | <0.0016 | mM |
| Nitrite | <0.0004 | mM |
| Sulfate | 0.15 | mM |
| Carbonate | 0 | mM |
| Bicarbonate | 8.51 | mM |
| Calcium | 0.26 | mM |
| Ferrous iron | <0.89 | μM |
| Total Iron | 6.88 | μM |
| Magnesium | 0.14 | mM |
| Potassium | 0.27 | mM |
| Sodium | 14.89 | mM |
The solid phase was a mixture of rock cuttings and core samples recovered from two drillings carried out in the reservoir. The reservoir rock is known to contain sandstone with clays and calcite.37
2.2 Treatment of the solid phase
Before use, the reservoir rock was respectively rinsed with isopropanol and sterilized demineralized water in order to eliminate potential hydrocarbons and drilling brine salts. Afterwards, it was dried out overnight in an oven at 90 °C. For X-ray tomography, capillaries filled with the reservoir rock samples were used.2.3 Abiotic experiment
This experiment helped to identify if any chemical compound would be released from the solid sample into the aqueous phase in a simplified protocol. Thereby, variations of the aqueous phase composition observed before and after its injection in the HP-reactor were correlated to dissolved compounds from the solid phase. This preliminary experiment consisted of mixing 50 g of the treated solid phase with 500 g of distilled water in a sealed bottle at ambient pressure and temperature. The bottle was flushed with N2 gas to make an inert atmosphere. After 15 days, the aqueous phase composition was determined.2.4 Biotic experiment
 | ||
| Fig. 2 Scheme and photography of the reactor used during the experiment. | ||
Inside the reactor, a basket made of Teflon was used to maintain the solid phase. The bottom of the basket was a Hastelloy C-276 metallic disc with 10 μm pores in order to prevent particles from falling into the bottom of the reactor. A mobile piston operated by an external pump allowed pressure adjustment after liquid phase sampling. When the piston was at its lowest position, the available volume inside the cell was 4.2 L. The aqueous and gas phases were mixed using a Rushton turbine and a double disc stirrer with four vertical blades, respectively.
At the end of the experiment, the remaining aqueous phase was recovered under anoxic and sterilized conditions and stored at 4 °C until use. The gas phase was evacuated and the reactor was flushed with filtered N2 gas (Classic Filters, Rochester England, 0.3 μm filter). Afterwards, the basket containing the solid phase and the capillaries was quickly and carefully removed from the reactor and then placed in a sealed anaerobic jar that contained an anaerobiosis generator and indicator pockets (Dutscher Ref 0260001). Afterwards the jar was moved into an anaerobic glove box where the solid phase was sampled for analyses.
In order to assess the variations in the gas phase, an in line gas chromatography with a micro-thermal conductivity detector (GC-μTCD; Micro GC Fusion; Chemlys; France) was used in agreement with the specifications detailed previously.4 The measurements were performed in triplicate and the measurement uncertainty was 5%.
The primers used were 515F and 928R (400 nM of each;40), DSR 2060F and DSR 4R (300 nM of each;42,43), mlas and mcrA-rev (400 nM of each;44,45) for the ARNr16S, dsrB and mcrA genes, respectively. All reactions were in 20 μL final reaction volume in accordance with the supplier's instructions. The number of copies was calculated by using a standard with serially 10-fold diluted pCRTM 2.1-TOPO plasmid (TOPO TA cloning kit, Invitrogen).
In order to determine the number of moles in the gas phase, the ideal gas law adjusted to the compressibility factor was used. The total pressure was measured by the pressure sensor that was installed in the reactor and the gas temperature by the thermocouple respectively. The volume of the gas phase was calculated by subtracting the volumes of the piston, solid basket and remaining aqueous phase in the reactor from the total volume of the reactor. The gas phase composition was given by μGC analysis (2.4.c-4.c). The PhreeqC model was used to calculate the compressibility factor of the gas mixture based on Peng–Robinson's equation. The compressibility was therefore considered to be 0.88 for the mixture CH4–CO2 and 0.9 for the gas mixture CH4–CO2–H2.
3. Results
3.1 Monitoring of the liquid phase composition during the abiotic experiment
Even after the rinsing process, chemical compounds can still be released from the solid phase into formation water. Moreover, the reservoir rock can still contain chemical compounds coming from the drilling brines. The abiotic experiment was conducted in order to identify and to quantify those products. After 15 days of mixture with the solid sample, the water was analyzed by ionic chromatography. Acetate (0.24 ± 0.012 mM), chloride (0.15 ± 0.008 mM), sulfate (0.5 ± 0.025 mM), sodium (0.99 ± 0.049 mM), calcium (0.12 ± 0.006 mM), magnesium (0.25 ± 0.013 mM) and potassium (0.71 ± 0.036 mM) were measured, indicating the potential dissolution of these ions from the solid phase and/or ions coming from drilling brines.3.2 Monitoring of the physico-chemical evolution in the liquid phase during biotic experiment
The formation water sampled from the Parisian aquifer (989 m depth) contained the autochthonous microorganisms harbored in the storage reservoir. Physico-chemical parameters (Table 1) showed that the aqueous phase was highly reduced (Eh around −365 mV), which is expected for a deep aquifer formation water. The concentration of dissolved elements was not very high (31.3 mM or 1152.6 mg L−1) and the salinity was estimated at 3% of seawater salinity.51 Chloride (7.10 mM) and bicarbonate (8.51 mM) had the highest concentrations among anions whereas sodium (1.89 mM), potassium (0.27 mM) and calcium (0.26 mM) had the highest concentrations among cations. The nature of the reservoir rock and the concentrations of dissolved calcium and bicarbonate indicated that those two ions resulted from an interaction between formation water and minerals. Compounds like barite which is present in the solid phase can potentially be the source of sulfate (0.15 mM) which was analyzed in the liquid phase.Three days before the experiment, the water mixture prepared for the biotic experiment contained initially 6.81 × 10−2 ± 3.4 × 10−3 mmol of sulfate. During this time, the microbial community has been revitalized. After liquid injection into the reactor, some solid phase compounds might get dissolved due to interactions between the aqueous phase and the reservoir rock. Thereby and as observed during the abiotic experiment, the sulfate quantity increased in the aqueous phase to attain 2.13 ± 0.11 mmol (Fig. 3(a)). On day 15, sulfate started to remarkably decrease indicating that the sulfate-reduction metabolism was active. Sulfate consumption continued even after H2 injection on day 21, and the sulfate quantity bottomed at 3.51 × 10−2 ± 1.7 × 10−3 mmol. In order to maintain the experiment, the aqueous phase was refilled on day 52. The added 0.854 L volume was composed of formation water sterilized-filtered with a 0.1 μm filter to avoid any microbial interference. The water injection induced a 3.15 × 10−2 ± 1.6 × 10−3 mmol increase of sulfate. On day 64, sulfate was completely depleted from the aqueous phase. The total sulfate quantity consumed during the experiment was around 2.51 ± 1.2 × 10−1 mmol.
 | ||
| Fig. 3 Variations in the aqueous phase composition during the biotic high-pressure experiment. The vertical line on day 10 represents the CH4 + CO2 reinjection. The vertical dotted lines on day 21 represent hydrogen injection. The vertical line on day 52 represents the water formation injection. (a) Sulfate variation in the liquid phase; (b) acetate variation in the liquid phase; (c) formate variation in the liquid phase. Stage 1: with sulfate and CO2, stage 2: with sulfate, CO2 and H2, stage 3: with CO2, H2 without sulfate, stage 4: with H2, without sulfate and CO2. | ||
The quantity of acetate increased in the reactor at the beginning of the experiment (day 1) (Fig. 3(b)). On day 21 and after H2 injection, acetate reached a plateau that persisted until the injection of formation water. The injected water induced a small increase of 2.80 × 10−1 ± 1.4 × 10−2 mmol in acetate quantity. Afterwards, no remarkable variation in acetate was noted until the end of the experiment.
After H2 injection in the gas phase, a remarkable detection of formate was noted in the aqueous phase (Fig. 3(c), stage 2). Formate was not detected in the formation water used in the experiment (Table 1). The increase in formate persisted until reaching a peak of 12.13 ± 6.1 × 10−1 mmol on day 71. From day 78 and until the end of the experiment, the formate quantity remained stable at around 10.58 ± 0.5 × 10−1 mmol.
As observed during the abiotic experiment, calcium increased in the aqueous phase due to solid–liquid interactions. Calcium quantity attained 3.15 ± 1.6 × 10−1 mmol on day 21. After the water injection on day 52, calcium increased to around 4.55 ± 2.3 × 10−1 mmol and remained constant afterwards.
3.3 Monitoring of the physico-chemical evolution in the gas phase during a biotic experiment
The gas phase was mainly composed of methane. Initially, 85.8 bar ± 0.8 bar of CH4 + 1% CO2 were injected into the reactor at a temperature of 47 °C. Thereby, 6.28 ± 2.51 × 10−1 moles of methane were injected at the beginning of the experiment. On day 10, the quantity of 1.57 × 10−1 ± 7.8 × 10−3 moles of CH4 were lost from the gas phase (Fig. 4(a)). The calculated solubility via PhreeqC under the experiment's conditions was 7.28 × 10−2 mol Kgw−1 confirming that the quantity of methane that was lost is attributed to the gas–liquid thermodynamic equilibrium. On day 10, the piston level was lowered so that 1 cm of the solid phase was immersed in the aqueous phase. After stabilizing the piston's position, a gas mixture was added in order to reestablish the desired experimental conditions of pressure. Consequently the injected gas mixture induced a 4.08 × 10−1 ± 2.0 × 10−2 mole increase in CH4 quantity. No variations in CH4 were later observed until the injection of filtered formation water (day 52) when 4.92 × 10−2 ± 2.5 × 10−3 moles were lost from the gas phase due to solubility. Afterwards no remarkable changes in CH4 quantity were noted. Theoretical thermodynamic CH4 evolution calculated with PhreeqC is presented in the curve Fig. 4(a).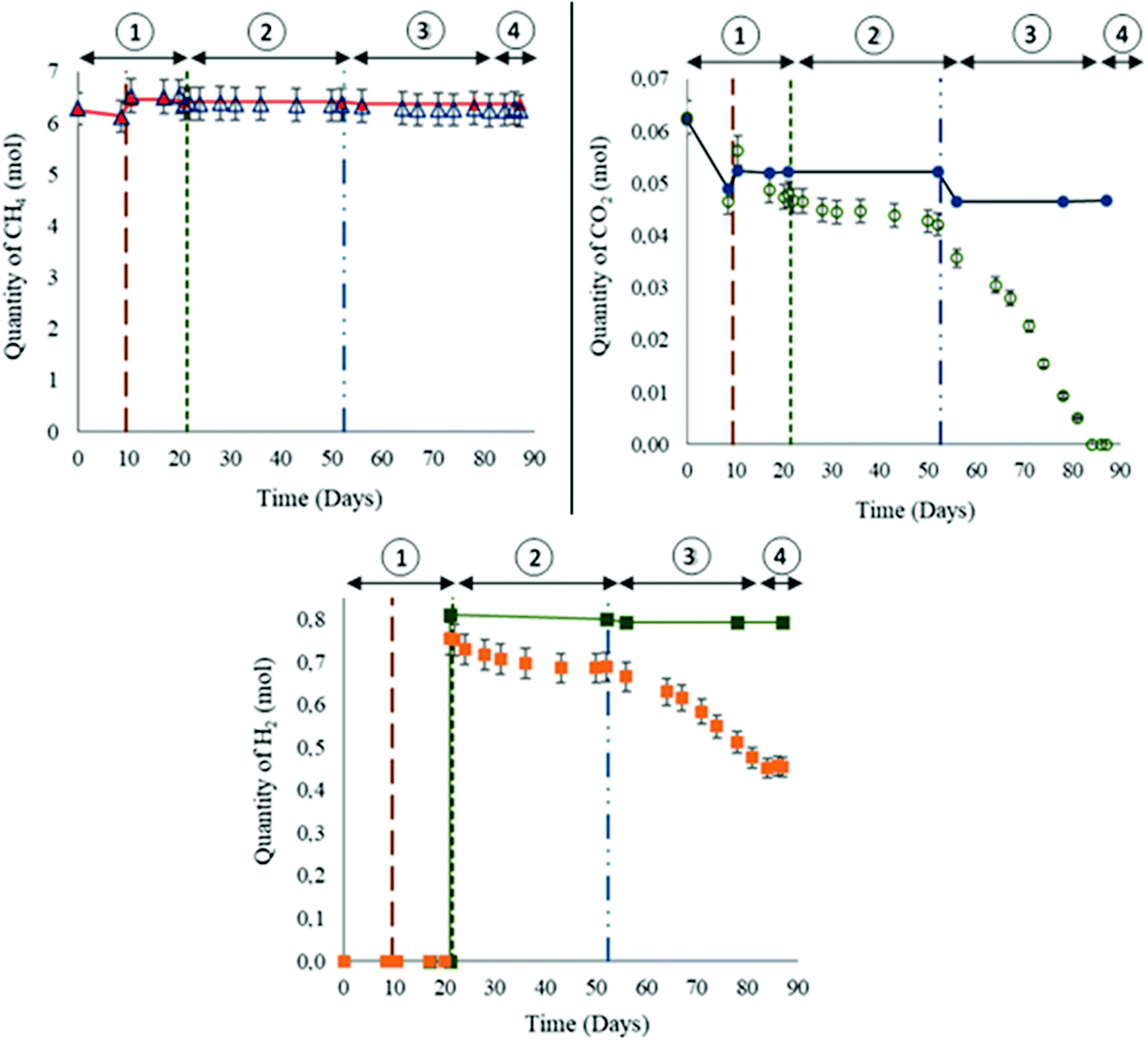 | ||
| Fig. 4 Variations in the gas composition during the biotic high-pressure experiment. The vertical line on day 10 represents the CH4 + CO2 reinjection. The vertical dotted lines on day 21 represent hydrogen injection. The vertical line on day 52 represents the water formation injection. (a) Methane variation in the gas phase. The red triangles and line represent the calculated values. (b) Carbon dioxide variation in the gas phase. The blue dots and line represent the calculated values. (c) Hydrogen variation in the gas phase. The green squares and line represent the calculated values. Stage 1: with sulfate and CO2, stage 2: with sulfate, CO2 and H2, stage 3: with CO2, H2 without sulfate, stage 4: with H2, without sulfate and CO2. | ||
Initially, CO2 constituted 1% of the gas mixture. At the beginning of the experiment, 6.26 × 10−2 ± 3.1 × 10−3 moles were injected (Fig. 4(b)). A decrease of 1.61 × 10−2 ± 8.1 × 10−4 moles was observed on day 10 due to a solubility similar to the observations noted with CH4. Following the reinjection of the gas mixture CH4–CO2, 9.81 × 10−3 ± 4.9 × 10−4 moles of CO2 were added. The variations calculated with PhreeqC showed that the liquid–gas thermodynamic equilibrium was reached and established before day 17. Note that the calculated values do not correspond to modelling of the reactor, but to a simulation of a simplified system in order to estimate the gas–liquid thermodynamic equilibrium. Before H2 injection, the remaining CO2 quantity was less than expected. Even after H2 injection, the quantity of CO2 continued to decrease. However as of day 53 and until CO2 depletion from the gas phase, the rate of CO2 consumption was higher than the one noted between days 10 to 52. On day 84, the CO2 was not detected anymore from the gas phase.
H2 injection was performed on day 21. A quantity of 7.52 × 10−1 ± 3.8 × 10−2 moles was injected with a resulting total system pressure of 95.2 ± 9.5 × 10−1 bar (Fig. 4(c)). H2 is poorly soluble (7.63 × 10−3 mol kg w−1via PhreeqC). On day 52, 6.89 × 10−1 ± 3.4 × 10−2 moles of H2 remained in the gas phase. The variations between the calculated and the measured values suggested possible consumption. The decline in H2 quantity persisted after water formation injection. However, similar to the above mentioned observation of heightened CO2 consumption, the rate of H2 decline between days 53 and 84 was remarkably greater than before day 52. From day 84 and until the end of the experiment, H2 consumption completely stopped and the quantity remained stable at 4.54 × 10−1 ± 2.3 × 10−2 moles.
3.4 Evolution of the microbial community during the experiment
One of the main challenges related to H2 storage in deep aquifers is the remaining uncertainty of its potential effects on microbial life. The formation water containing the autochthonous microorganisms was sampled using two down hole samplers in order to avoid the non-contamination of the microbial flora by biofilms located in the well and at the well head. The microorganisms were under anoxic conditions at 30 °C, a lower temperature than on site in order to halt microbial activities, until use in the reactor.Throughout the experiment, extractions of nucleic acids from liquid samples taken from the reactor were performed. The dsrB gene encodes for the disulfite reductase whereas the mcrA gene encodes for the methyl coenzyme M reductase; enzymes that are characteristics of sulfate-reducers and methanogens, respectively. The obtained partial 16S rRNA, dsrB and mcrA genes and transcripts were sequenced using MiSeq and quantified using qPCR and qRT-PCR in order to follow the evolution and expression of the overall microorganisms, sulfate-reducers and methanogens (percentage efficiency mean of 96% ± 8%); (Fig. 5 and 6). The concentration of the total microorganisms in the formation water injected into the reactor was 3.5 × 105 ± 2.8 × 104 copies of 16S rRNA genes per mL. Following the injection and the rise in pressure of the reactor, the microbial concentration was 3.71 × 105 ± 4.26 × 104 copies of 16S rRNA genes per mL, suggesting a successful transfer. This concentration varied slightly during the 87 days of the experiment with values ranging between 6.5 × 104 ± 6.1 × 103 and 1.7 × 106 ± 6.1 × 103 copies of 16S rRNA genes per mL. Their overall activity as measured by the quantification of 16S rRNA transcripts (cDNA) was the highest during the first 56 days of incubation. Sulfate-reducers concentration peaked at 3.1 × 106 ± 4.4 × 105 copies of the dsrB gene per mL on day 22, the day following H2 injection. Their concentration steadily decreased afterwards reaching the value of 7.9 × 103 ± 9.0 × 102 copies of the dsrB gene per mL at the end of incubation. As expected, the dsrB transcripts decreased in parallel to the decrease of the sulfate, with noted concentrations ranging between 5.2 × 101 ± 1.1 × 101 and 2.8 × 101 ± 4.1 × 100 copies dsrB transcripts per mL. However as of day 51 when the sulfate quantity was less than 0.08 mmol, the methanogenic activity increased reaching a peak of 1.8 × 104 ± 2.8 × 102 copies of mcrA gene per mL transcripts. After complete CO2 depletion from the system, the methanogenic activity declined. If sulfate-reducing was a key metabolism of the microbial community at the start of the experiment, the decrease in sulfate in the presence of H2 and CO2 allowed an increase in methanogenesis with a peak of mcrA transcripts of 1.8 × 104 ± 2.8 × 102 copies per mL at day 78.
 | ||
| Fig. 5 Evolution of the microbial community during the incubation at 47 °C, at 85 bar (CH4, 1% CO2) before and after hydrogen injection (95 bar with 10% H2). The taxonomic diversity is monitored by 16S rRNA gene sequencing performed in an aqueous phase sampled from the reactor throughout the experiment. | ||
Several microbial families were identified in the reactor after incubation under the system's temperature of 47 °C including Thermodesulfovibrionaceae, Thermicanaceae, Methanothermobacteriaceae, Desulfotomaculaceae and Thermacetogeniaceae (Fig. 5). These families are well known in the literature to include bacterial strains with thermophilic characteristics. During the first few days of incubation under simulated storage conditions with a gas phase composed of CH4 + 1% CO2 at 85 bar, the microbial community was largely dominated by the Spirochaetaceae family. Under similar conditions, the Spirochaetaceae family most certainly functioned via the fermentation of organic molecules trapped in the solid phase or fermentation of necromass. Sulfate-reducers, dominated by the Desulfurisporaceae and Thermodesulfovibrionaceae families, were less numerous at the beginning of the incubation. Nevertheless, monitoring of 16S rRNA and dsrB transcript concentrations demonstrated an increased activity of sulfate-reducers. This enhanced activity explained the increase in sulfate-reducers growth and their enlarging proportion in the microbial community (Fig. 5 and 6). After injection of 10% of H2 into the gas phase under a total pressure of 95 bar and with a sulfate quantity between 1.33 and 0.08 mmol, the Thermodesulfovibrionaceae family continued to proliferate forming up to 55% of the relative diversity and 83% of the relative active diversity on day 51. The phylum Acidobacteria was maintained until the 78th day of incubation. An increase in its transcripts started after H2 injection and coincided with the onset of formate production. A water sample taken from the reactor on day 56 was used to culture a simplified community dominated by the Acidobacteriota phylum (data not shown). In the presence of H2 and CO2, the bacterial strain produced formate. When CO2 started to decrease in the reactor, formate production slowed down, eventually stopping when CO2 was completely depleted whereas Methanothermobacteriaceae continued to be active. When sulfate became less than 0.08 mmol (less than 0.08 mmol L−1), Desulfotomaculaceae proportion went from 2% to 20% of the total microbial community and that of Desulfitibacteraceae from 2.5% to 19%. At the same time, the Methanothermobacteriaceae became more abundant constituting up to half of the microbial community between days 56 and 78 of incubation. With the disappearance of sulfate on day 65, the activity of Thermodesulfovibrionaceae, Desulfotomaculaceae, and Desulfitibacteraceae decreased in favor of proliferation of families known as chemo-organotrophic such as Caldicoprobacteraceae, Lentimicrobiaceae, Eubacteriaceae or Hungateiclostridiaceae. At the end of the experiment (days 84 and 87), the microbial diversity was stabilized from the point of view of the microorganisms present (Fig. 5(A)) but not the microbial activity with in particular an increase of the 16S rRNA transcripts of the Methanothermobacteriaceae (Fig. 5(B)).
 | ||
| Fig. 6 Quantitative evolution of the microbial community during the incubation at 47 °C, at 85 bar (CH4, 1% CO2) before and after hydrogen injection (95 bar with 10% H2). The quantification is estimated by qPCR of 16S rDNA gene copies for each mL of sampled water through the experiment. | ||
Analysis of the diversity of the microbial community was carried out on the solid phase as well. Three samples of reservoir rock were taken from the basket at the end of the experiment and involved a sample from the bottom of the basket which was always immersed in the liquid phase, another sample from the middle at the gas–liquid interface and a last sample from the top of the basket where the solid was in contact with the gas phase (Fig. 7). Results showed a net dominance of Methanothermobacteriaceae at all three depths but more notably at the top and middle of the solid phase. The major families in the liquid phase at the end of the incubation (Fig. 5) were similar to the solid phase and comprised Methanothermobacteriaceae, Thermodesulfovibrionaceae, Spirochaetaceae, Hungateiclostridiaceae, Desulfotomaculaceae and Caldicoprobacteraceae.
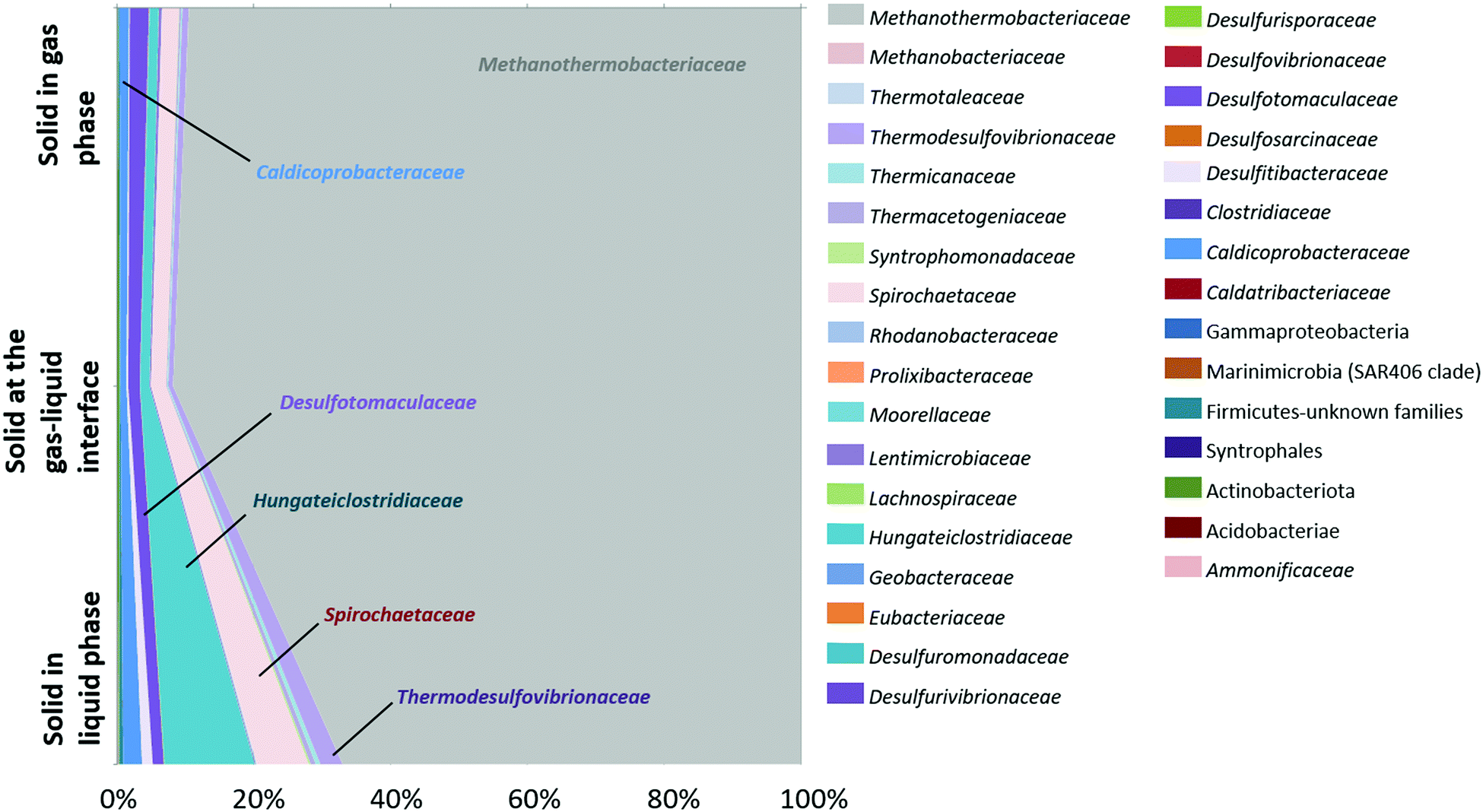 | ||
| Fig. 7 Microbial community after the incubation at 47 °C, at 85 bar (CH4, 1% CO2) followed by hydrogen injection (95 bar with 10% H2). The taxonomic diversity was monitored by 16S rRNA gene sequencing performed on different heights of the solid phase at the end of the experiment. | ||
3.5 Evolution of the solid phase during the experiment
Once the experiment came to its end and the solid basket was removed from the reactor, the solid phase was analyzed through X-ray tomography and XRD.XRD performed on the reservoir rock sample prior to the experiment (t0) showed that it was mainly composed of quartz with barite and calcite (Fig. 8(A)) along with minor amounts of clay. At the end of the experiment (tf), barite and calcite were not detected anymore which indicates their dissolution. Some residual barite and calcite were observed by SEM-EDX (Fig. 8(B) and (C)) but at significantly lower levels than prior to the experiment. In particular, the residual barite grains show morphologies that clearly indicate significant dissolution (Fig. 8(C)). On the contrary, clay minerals slightly increased compared to the initial solid phase, and newly formed vermicular kaolinite was observed by SEM-EDX (Fig. 8(C)). Pyrite was also identified as a trace mineral and its presence as framboidal pyrite was attested by SEM-EDX, suggesting that some pyrite was formed during the experiment.
 | ||
| Fig. 8 Powder XRD patterns for the solid phase (A) before incubation (t0) and after 87 days of incubation in the reactor (tf). SEM images of the solid phase at tf (B) and (C). XRD patterns at tf were collected at the surface of the reactor (tf-surface), at mid-height (tf-middle) and at the bottom (tf-bottom). K: kaolinite, Q: quartz, C: calcite, Py: pyrite. The peak labelled ‘clays’ originated from (020) and (110) of clay minerals. | ||
Two capillaries (CAP1 and CAP2) filled with reservoir rock and positioned in the solid basket were scanned by X-ray tomography before and after the experiment (Fig. 9(A1)). At each capillary, two zones were targeted: the middle zone (position 1 or “P1”, situated at 1.5 cm from the top of the 3 cm capillary) and the bottom zone (P2, 2.38 cm from the top) as shown on Fig. 9(A2) for CAP1. P1 was situated at the interface between the unsaturated and the saturated regions in the sample whereas P2 was submerged throughout the entire experiment. As the results were similar for both capillaries, we only presented the results obtained for CAP1. The grains (in yellow color) constituted 63–65% ± 2% of the sample volume and this ratio remained globally stable throughout the experiment (compare Fig. 9(B2) and (C2)). The porosity (in blue color) at the end of the experiment was 9% ± 2% lower than prior and was accompanied by the appearance of a deposit (in green color) estimated to be 8% ± 3% of the total volume in the P2 zone (not shown). The visualized volume at P1 showed 10% ± 3% of deposits with a notable heterogeneous distribution as the deposit proportion decreased from 12% ± 2% in the saturated zone to 7% ± 2% in the unsaturated zone. During the experiment and after every liquid sampling, the piston was elevated to immerse 1 cm of the capillary. The liquid movement might have affected the formation of deposits and mobilized them in the lower half of the capillaries. Additionally, biomineralization processes might have also contributed to the observed reduction in porosity via production of iron sulfide or clays among other mechanisms. Analysis of the water phase showed a decrease of grey image level from the saturated zone to the unsaturated zone (Fig. 9(C4) and (C5)). The grey image level was linked to the attenuation coefficient and reflected variations in water composition. Variations in chemical composition (higher water mineralization in a saturated zone) or of microorganism concentrations (more significant in an unsaturated zone) could explain these observations.
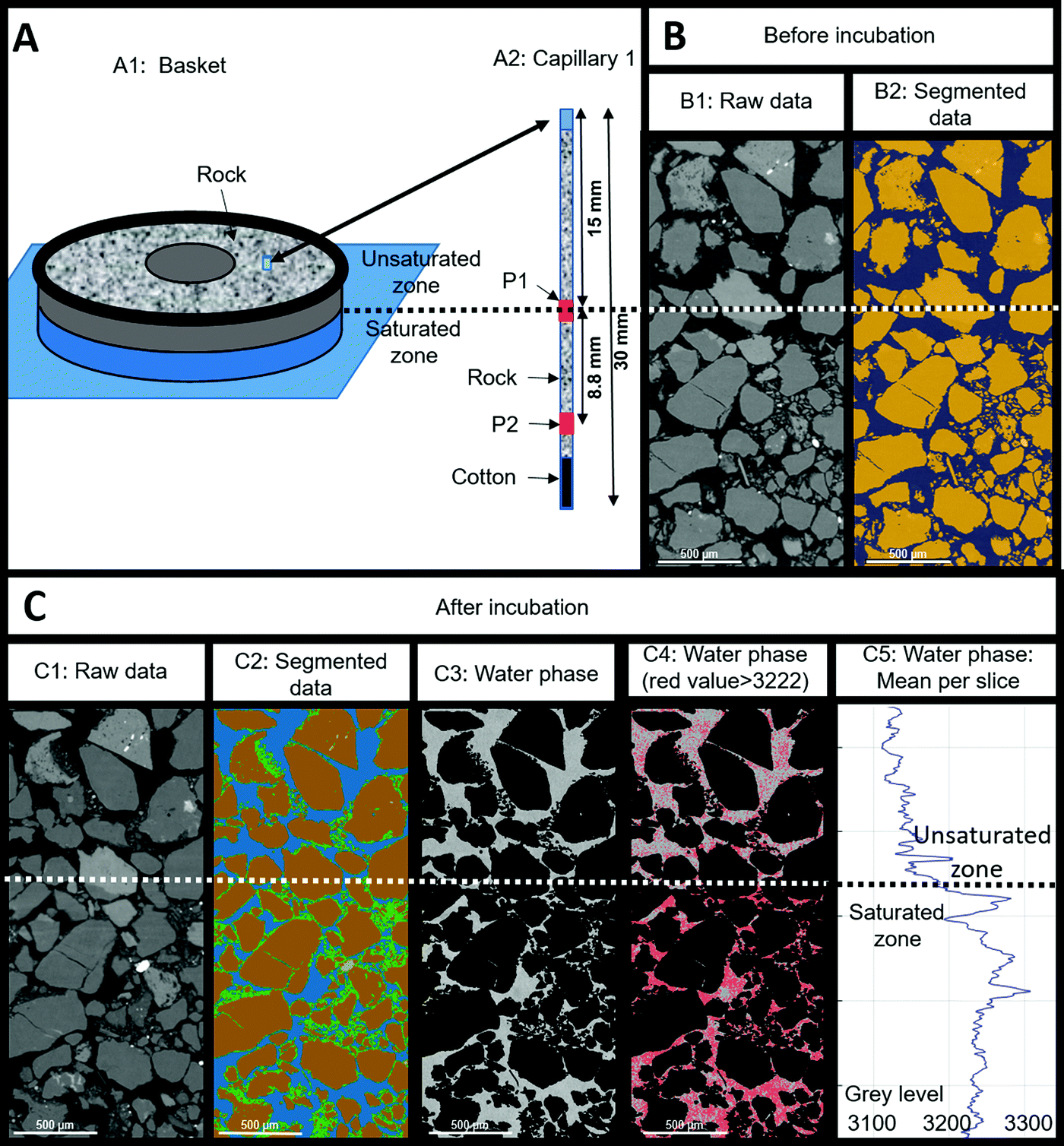 | ||
| Fig. 9 X-Ray tomography results obtained for capillary 1 before and after incubation. (A1): basket containing the reservoir rock and capillaries, the lower part (in blue) was water saturated while the upper part (in grey) was unsaturated. Capillaries were positioned vertically in the basket and were filled with reservoir rock. The blue cylinder in the basket shows the position of the capillary 1. (A2): capillary 1 with the P1 and P2 zones scanned by X-ray tomography before and after incubation (in red). The scan before incubation was performed on the capillary filled with dry reservoir rock. (B1): longitudinal image obtained by X-ray tomography (raw data) at P1 position before incubation and (B2) the associated segmentation of the air phase (in grey) and the grain phase (in orange). (C1): longitudinal image obtained by X-ray tomography (raw data) at P1 position after incubation and (C2) the associated segmentation of the water phase in grey level from the raw data. (C3): proportion of deposit per slice. (C4): extraction of the water phase in grey level from the raw data. Water phase in grey levels and pixels (in red) with a grey level greater than 3222 (mean value obtained for the water phase on the studied volume). (C5): mean value obtained per slice along the vertical axes. The horizontal dotted line corresponds to the limit between the saturated and unsaturated zones. | ||
4. Discussion
In order to decrease green gas emissions, dihydrogen (H2) emerged as a credible alternative for fossil fuels. In fact, H2 is a well-known green multi-use energy vector that can make a difference in the energy transition scenario.52,53 However, in order for H2 to play this key role in the future energy mix, large-scale storage of this gas in diverse geological reservoirs such as salt caves, depleted hydrocarbon reservoirs and deep aquifers is indispensable.54,55 In order to decrease the use of fossil fuels, the European objective targets to reach 10% of H2 in the gas mixture injected into the natural gas grid.56 In this context, H2 mixed with natural gas, biomethane or methane produced via (green-)power-to-gas makes H2 transport and storage more cost-effective and environmentally friendly.3 Nevertheless, the interconnection of surface gas networks with geological storage implicates that a massive influx of H2 is to be expected in the deep aquifers which are home to an indigenous microbial community capable of using this molecule as an energy source. The elucidation of the characteristics and results of H2-microbial interactions is crucial to avoid unexpected and harmful environmental outcomes.6,57,58 H2 is a well described energy source and electron donor for many of those prokaryotic microorganisms.5,35,59–61 Moreover, many studies highlighted the existence of microbial communities that thrive on the consumption of H2 and CO2 in such deep environments. Those communities are known by the term SLiMEs (Subsurface Lithoautotrophic Microbial Ecosystems) created by Stevens and McKinley (1995). Understanding the effect of H2 injection in these ecosystems is a fundamental step towards establishing an effective and comprehensive energetic transition.In this study, the injection of 10% of H2 with methane – 1% carbon dioxide gas phase in a Parisian basin aquifer was experimentally simulated. The analyses of the reservoir rock showed a disappearance of calcite and barite at the end of the incubation. The well Pb_T_1 that was selected for sampling was close to the water–gas interface,37 which implicates that interactions between water formation and stored gas were the highest at this location. The aqueous phase had a low salinity estimated at around 3% of seawater salinity. The rock sample obtained from the studied site by drilling operations was defined as rock cutting. It was also rich in barite (BaSO4) and calcite (CaCO3). Results from the abiotic experiment showed the release of several elements from the solid phase. In fact, high concentrations of calcium and sodium found in Pb_T_1 could be related to solid–liquid interactions in the deep aquifer, such as calcite dissolution. Calcium and chloride could be attributed to brines used during drilling operations. Sulfate ions detected during the abiotic experiment could originate from the dissolution of barite detected in the rock cuttings (Storengy, industrial operator, personal communication) and as observed by XRD (Fig. 8). Interestingly in anoxic and saline conditions the anaerobic halophilic bacteria increased barite dissolution compared to the abiotic process.62 Consequently the developing microbial activity in the reactor is likely to be involved in this process.
4.1 Physicochemical and bacterial evolution before H2 injection (Step 1)
The first phase of the experiment was to reproduce the aquifer's conditions before H2 injection. Once the solid and aqueous phases were placed in the reactor, the gas phase was injected. The latter was composed of CH4 with 1% CO2 in order to simulate the natural gas storage. During the first 10 days, the solid basket was completely immersed in the aqueous phase in order to ensure its colonization by microorganisms which replicates the ecosystem of the deep aquifer. Before injection into the reactor, the formation water containing the autochthonous microorganisms was stored in an anoxic flask with a N2 atmosphere and limited in terminal electron acceptors (sulfate, CO2) explaining the dominance of fermenters at the start of the experiment. The simple organic acids, CO2 and H2 known to be produced by the fermenters then allowed the other members of the community to be maintained, in particular sulfate-reducers and methanogens. On day 10, the water phase level was lowered using the piston in order to get only 1 cm of the solid immersed. In order to readjust the pressure, one last reinjection of CH4–CO2 was performed. Modeling via PhreeqC showed that after the reinjection of CH4–CO2, the thermodynamic equilibrium was established on day 17 (Fig. 4(a)–(c)). On day 21 and before H2 injection (Fig. 4(b)), CO2 decreased significantly in the gas. The calculated values correspond to an estimation of the gas–liquid thermodynamic equilibrium of the abiotic system. Thus, these results suggested that the decrease of CO2 was not solely attributed to the gas solubility in the aqueous phase but to an additional CO2 consumption that could possibly be related to microbial activities. Under a pressure of 85 bar and a temperature of 47 °C, microorganisms were able to develop and be active. The studied activity of bacteria belonging to the Spirochaetaceae and Anaerolinaceae families (Fig. 5) suggests a fermentation activity likely generating limited quantities of H24,63 which can be instantaneously consumed through litho-autotrophy by other microorganisms such as those belonging to the Thermodesulfovibrionaceae family, for example.64 Sulfate reduction manifested through the decrease in sulfate in the aqueous phase and the presence of active sulfate-reducers identified as the members of Thermodesulfovibrionaceae and Desulfurisporaceae families. Acetate increased up to a level of 1.30 mmol L−1 during the experiment. This presence of acetate could be explained by the microbial fermentative and sulfate-reducing activities with regard to incomplete oxidizers.65 Interestingly, this acetate accumulation suggests the absence of acetoclastic microorganisms in the community.4.2 Physicochemical and microbial evolution after H2 injection before sulfate depletion (Step 2)
H2 was injected in the gas phase on day 21. Sulfate consumption continued even after H2 injection until a complete sulfate depletion on day 60. Meanwhile in the gas phase, CO2 and H2 sustained a minor reduction in their quantity.66 We expected an increase in sulfate reduction66 which did not occur on the view of the sulfate decrease curve. This suggests that the sulfate reducers had mainly a heterotrophic metabolism, perhaps on the end products of fermentation and the fermenters necromass. Thermodynamic simulations of gas–liquid equilibrium showed that H2 reduction was not secondary to thermodynamic equilibrium as the latter was established 3 days after the injection (Fig. 4(c)). Surprisingly, formate quantities appeared in the liquid phase immediately after H2 injection (Fig. 3(c)). Formate production persisted between day 21 and day 60. The cultivation of a simplified bacterial community able to produce formate allowed us to attribute formate production in the reactor to members of the Acidobacteria phylum. A 16S rDNA gene sequence was closely related to an environmental sequence retrieved in subsurface lithoautotrophic ecosystems (KM410334;67). The Acidobacteria phylum is abundant and ubiquitous and its members were particularly found in aquifers.68 Despite their wide distribution, the members of this phylum are technically difficult to grow and culture on conventional media making their identification challenging.69 Therefore only 56 cultivable species had been described among 6500 identified taxa. This was in part due to their oligotrophic nature which served them up when rich growing media favored other microorganisms. Acidobacteria gathered together (homo)-acetogens70,71 were described as versatile heterotrophs.68 Although energetically disadvantaged facing hydrogenotrophic methanogens, especially at low H2 concentrations, (homo)-acetogens have a kinetic advantage under certain physicochemical conditions.72 In the presence of H2 and CO2, the production of formate is usually an intermediate step in the energy metabolism; however it can be a terminal result via certain microorganisms belonging to the chemolithotrophic homoacetogenic bacteria.73 Although this part of the work was never published, formate production was also highlighted during the simulation of subsurface H2 storage in Márton Berta's PhD Thesis.74 Although there is no competition between sulfate reducers, formate producers and hydrogenotrophic methanogens for H2 and CO2, in excess, methanogens appeared to be inhibited by the sulfate reducing activity, probably through sulfide emission. When sulfate was depleted from the system (Step 3), the Methanothermobacteriaceae proportion increased in the community. The conditions under these circumstances were more proliferation namely H2 and CO2. We hypothesize, although aquifers are not acidic, the formation of biofilms could allow local acidification,75 which favors the development of Acidobacteriota which are a potential source of formate.4.3 Physicochemical and microbial evolution after H2 injection and sulfate depletion (Step 3)
On day 60, sulfate was completely consumed by sulfate-reducing bacteria. Hydrogen sulfide (H2S) was not detected in the gas phase. This finding is likely related to the minimal quantity of produced sulfide compared to the total number of moles in the gas phase and to the interaction of sulfide with the solid phase. In fact, the total quantity of sulfate consumed was 2.51 ± 1.25 × 10−1 mmol. If all the sulfate was transformed into hydrogen sulfide, a portion of H2S would be estimated to be 0.04% of the total gas mixture at the end of the experiment. The observed reduction in porosity, the formation of deposits observed through X-ray tomography (Fig. 9) and framboidal pyrite detected by XRD (Fig. 8) strongly suggested that sulfide might have reacted with iron that was present in the system resulting in pyrite production in the solid phase.Even after sulfate depletion from the aqueous phase, formate production persisted. In the gas phase, a decrease in CO2 and H2 quantities was noted. However, rates of consumption of these two gases were higher than those in the previous periods. H2 and CO2's fast consumptions suggested a lithoautotrophic growth. Studies suggested that such metabolisms were developed in order to maintain the sustainability of deep underground ecosystems such as SLiMEs. Hydrogenotrophic methanogens were detected in the microbial diversity studies (Fig. 5 and 6). Despite that methanogenic activity significantly increased after reduction of sulfate quantities to less than 0.08 mmol, no methane production was identified in the gas phase. Of course, this observation does not imply that methane was not produced but is rather explained by the fact that the initial high concentration of methane in the gas phase (90% methane, 95 bar) made it impossible to detect subsequent small methane production. In fact, if the lost quantities of CO2 from day 56 till total depletion were transformed into methane, the estimated produced quantity of CH4 would be equal to 3.57 × 10−2 ± 1.78 × 10−3 moles. Considering the quantity of CH4 at that time in the gas phase (6.33 ± 3.17 × 10−1 moles), produced CH4 due to methanogenic archaea was too minimal to be detected. Nevertheless, the activity of methanogens was clearly demonstrated via the quantification of mcrA transcripts by RTqPCR, and the sequencing of cDNAs from 16S rRNA transcripts (Fig. 5 and 6). Methanogens metabolism continued until the complete depletion of CO2 from the gas phase on day 84.
4.4 Physicochemical and microbial evolution after H2 injection, sulfate and carbon dioxide depletion (Step 4)
During the last few days of the experiment, the gas phase was depleted in CO2. H2 consumption and formate production, which was observed after the H2 injection until day 84, stopped simultaneously with the depletion of CO2 from the gas phase. This observation confirmed the relationship between H2, formate and CO2. Analysis of the composition of the mineral phase by XRD at the end of incubation showed the absence of calcite (CaCO3) which was initially detected prior to the experiment (Fig. 8). The increase in calcium observed in the aqueous phase could be attributed to these mineralogical results. Actually, it is obvious that the CO2, carbonates/bicarbonates and calcite were in equilibrium in the reactor and that the disappearance of CO2via the various autotrophic metabolisms (sulfate-reduction, (homo)-acetogenesis and methanogenesis) coupled with hydrogenotrophy contributed to the depletion of these minerals. At the very end of the incubation, the Methanothermobacteriaceae still maintained metabolic activity (Fig. 5 and 6) while the formate producers seemed to have stopped producing formate (Fig. 3). These results suggested a better affinity of these methanogens for CO2 compared to formate producers.Some microorganisms have the potential to survive in the rock bathed in the gas phase, taking into account the humidity of the latter and the residual formation water trapped in part of the porosity of the rock as in the actual conditions of the studied aquifer. Therefore at the end of the experiment, a microbial community dominated by Methanothermobacteriaceae was detected (Fig. 7). These hydrogenotrophic methanogenic archaea present in the residual water were found in close topographical proximity to CO2 and H2, the former serving as a carbon and electron acceptor source and the latter as an energy and electron donor source. Based on this observation, we hypothesize that the remaining volume of water supposedly reduced is subjected to be quickly depleted in sulfate and other molecules which are substantial for the survival of other functional groups such as sulfate-reducing microorganisms or fermenters.
5. Conclusion
In this study, the UGS aquifer Pb_T_1 situated in the Parisian basin region was simulated in a high pressure reactor. Effects of 10% H2 stored with methane were studied through the identification of interactions between reservoir rock, formation water, autochthonous microbial life and the new gas mixture. In the beginning, the actual storage conditions were reproduced with CH4 and CO2 forming the gas mixture. The fraction of H2 was injected on day 21 after the development of the system. Although these XRD analyses showed a depletion of calcite and barite in the solid phase indicating mineral dissolution, the solid matrix showed a decrease in its porosity likely originating from clay deposits and iron sulfide precipitation. At a constant pH solution, H2 injection probably caused those mineral alterations as well as the activity of the microbial ecosystem. In less than 90 days after simulation of an aquifer storage of natural gas, nearly 40% of injected H2 transformed either into H2S, formate and methane due to microbial activities and dissolved into the aqueous phase. This decrease in H2 significantly decelerated with the disappearance of the original 1% of CO2 from the gas phase and a large part of the calcite present in the solid phase. It is likely that on a smaller scale, H2 continued to be consumed by hydrogenotrophic microorganisms associated with consumption of CO2 produced by degradation of organic matter by heterotrophic microorganisms.In order to ensure the safety and efficacy of H2 injection in the underground, many parameters are to be taken into consideration. This study's results showed that H2 storage might have a considerable impact on the reservoir rock particularly if it is rich in calcite, since calcite can dissolve secondary to carbon dioxide for bioconsumption. Mineral dissolution can cause variations in reservoir petrophysical characteristics such as porosity and permeability. Moreover, H2 which is known to be a crucial energy source for microbial life, is consumed. These phenomena impact the composition of the stored gas and contribute to the loss of H2. Moreover, in sulfate rich aquifers, hydrogen sulfide produced by sulfate-reducing microorganisms can sour the stored gas unless enough quantities of dissolved iron are present to mineralize sulfides.
In 1995, Stevens and McKinley theorized the concept of ecosystems that are able to evolve independently from organic matter and energy produced via photosynthesis. These H2-based deep environmental ecosystems had been named SLiMEs for Subsurface Lithoautotrophic Microbial Ecosystems. Subsequently, dozens of studies have attempted to invalidate, confirm or refine this concept, sometimes with a redefinition of terminologies such as True-SliMEs and HyperSLiME.76,77 The massive influx of H2 of anthropogenic origin in deep aquifers can potentially set the basis for the advent of Artificial-SLiMEs and therefore the creation of new microbial ecosystems.
Studies of different aquifers under various conditions of pressure, temperature, gas and water compositions can help optimize the practice of H2 storage in the underground. Moreover, in order to model the evolution of future storage in deep aquifers that are housing life, and perhaps in salt caverns with potential microorganisms in the residual brines, this study showed that it is fundamental to take into consideration sulfate concentrations, the CO2 and microbial activities in order to avoid unfavorable outcomes.
Author contributions
FC, MRP, PCe and ARP co-conceived the study. PH, JM, FC, and PCe carried out the high pressure and simulation experiments. MG, MRP and ARP applied the microbiological approaches. PS, M-PI, PM and GH used the imaging and mineralogical characterization. All authors contributed in the interpretation of the results and paper writing.Declaration of interests
DD, GC and PCh are employed by two French companies specialized in geological natural gas storage which are Storengy and Teréga respectively.Conflicts of interest
No conflict of interest is declared in relation to the work described.Acknowledgements
Enagas, SNAM, Storengy and Teréga are acknowledged for funding this research project. We are grateful to the Genotoul platform (genotoul.fr) for the sequencing analyses. MRP salary was supported by E2S-UPPA. We would also like to thank TotalEnergies for providing UMS 3360 DMEX with the Zeiss Xradia Versa 510 which carried out the tomographic acquisitions reported in this article.References
- World Energy Council, Innovation Insights Brief – New Hydrogen Economy – Hype or Hope?, https://www.worldenergy.org/publications/entry/innovation-insights-brief-new-hydrogen-economy-hype-or-hope, (accessed February 9, 2022).
- N. Heinemann, J. Alcalde, J. M. Miocic, S. J. T. Hangx, J. Kallmeyer, C. Ostertag-Henning, A. Hassanpouryouzband, E. M. Thaysen, G. J. Strobel, C. Schmidt-Hattenberger, K. Edlmann, M. Wilkinson, M. Bentham, R. Stuart Haszeldine, R. Carbonell and A. Rudloff, Energy Environ. Sci., 2021, 14, 853–864 RSC
.
- A. Kovač, M. Paranos and D. Marciuš, Int. J. Hydrogen Energy, 2021, 46, 10016–10035 CrossRef
- P. G. Haddad, J. Mura, F. Castéran, M. Guignard, M. Ranchou-Peyruse, P. Sénéchal, M. Larregieu, M.-P. Isaure, I. Svahn, P. Moonen, I. Le Hécho, G. Hoareau, P. Chiquet, G. Caumette, D. Dequidt, P. Cézac and A. Ranchou-Peyruse, Sci. Total Environ, 2022, 806, 150690 CrossRef CAS PubMed
- R. Tarkowski, Renewable Sustainable Energy Rev., 2019, 105, 86–94 CrossRef CAS
- D. Zivar, S. Kumar and J. Foroozesh, Int. J. Hydrogen Energy, 2021, 46, 23436–23462 CrossRef CAS
- S. Bauer, C. Beyer, F. Dethlefsen, P. Dietrich, R. Duttmann, M. Ebert, V. Feeser, U. Görke, R. Köber, O. Kolditz, W. Rabbel, T. Schanz, D. Schäfer, H. Würdemann and A. Dahmke, Environ. Earth Sci., 2013, 70, 3935–3943 CrossRef
- S. Henkel, D. Pudlo and R. Gaupp, Energy Procedia, 2013, 40, 25–33 CrossRef CAS
- S. Flesch, D. Pudlo, D. Albrecht, A. Jacob and F. Enzmann, Int. J. Hydrogen Energy, 2018, 43, 20822–20835 CrossRef CAS
-
L. Ganzer, V. Reitenbach, D. Pudlo, M. Panfilov, D. Albrecht and R. Gaupp, All Days, SPE, London, UK, 2013 Search PubMed
- L. Hashemi, M. Blunt and H. Hajibeygi, Sci. Rep., 2021, 11, 8348 CrossRef CAS PubMed
- C. Hemme and W. van Berk, Appl. Sci., 2018, 8, 2282 CrossRef CAS
- C. Magnabosco, L.-H. Lin, H. Dong, M. Bomberg, W. Ghiorse, H. Stan-Lotter, K. Pedersen, T. L. Kieft, E. van Heerden and T. C. Onstott, Nat. Geosci., 2018, 11, 707–717 CrossRef CAS
-
M. Cannat, F. Fontaine and J. Escartín, in Geophysical Monograph Series, ed. P. A. Rona, C. W. Devey, J. Dyment and B. J. Murton, American Geophysical Union, Washington, D. C., 2010, vol. 188, pp. 241–264 Search PubMed
- I. G. Draganić, Radiat. Phys. Chem., 2005, 72, 181–186 CrossRef
- F. Klein, J. D. Tarnas and W. Bach, Elements, 2020, 16, 19–24 CrossRef CAS
- S. L. Worman, L. F. Pratson, J. A. Karson and E. M. Klein, Geophys. Res. Lett., 2016, 43, 6435–6443 CrossRef CAS
- T. O. Stevens and J. P. McKinley, Science, 1995, 270, 450–455 CrossRef CAS
- R. T. Anderson, F. H. Chapelle and D. R. Lovley, Science, 1998, 281, 976–977 CrossRef CAS PubMed
- O. Basso, J.-F. Lascourreges, F. Le Borgne, C. Le Goff and M. Magot, Res. Microbiol., 2009, 160, 107–116 CrossRef CAS PubMed
- F. H. Chapelle, K. O’Neill, P. M. Bradley, B. A. Methé, S. A. Ciufo, L. L. Knobel and D. R. Lovley, Nature, 2002, 415, 312–315 CrossRef PubMed
- M. Crespo-Medina, K. I. Twing, M. D. Y. Kubo, T. M. Hoehler, D. Cardace, T. McCollom and M. O. Schrenk, Front. Microbiol., 2014, 5, 604 Search PubMed
- N. K. Fry, J. K. Fredrickson, S. Fishbain, M. Wagner and D. A. Stahl, Appl. Environ. Microbiol., 1997, 63, 1498–1504 CrossRef CAS PubMed
- S. A. Haveman and K. Pedersen, FEMS Microbiol. Ecol., 2002, 39, 129–137 CrossRef CAS PubMed
- S. Kotelnikova and K. Pedersen, FEMS Microbiol. Rev., 1997, 20, 339–349 CrossRef CAS
- L.-H. Lin, J. Hall, J. Lippmann-Pipke, J. A. Ward, B. Sherwood Lollar, M. DeFlaun, R. Rothmel, D. Moser, T. M. Gihring, B. Mislowack and T. C. Onstott, Geochem., Geophys., Geosyst., 2005, 6, 7 CrossRef
- K. Takai, M. R. Mormile, J. P. McKinley, F. J. Brockman, W. E. Holben, W. P. Kovacik Jr and J. K. Fredrickson, Environ. Microbiol., 2003, 5, 309–320 CrossRef CAS PubMed
- T. Aüllo, S. Berlendis, J.-F. Lascourrèges, D. Dessort, D. Duclerc, S. Saint-Laurent, B. Schraauwers, J. Mas, D. Patriarche, C. Boesinger, M. Magot and A. Ranchou-Peyruse, Front. Microbiol., 2016, 7, 122 Search PubMed
- P. Bombach, A. Chatzinotas, T. R. Neu, M. Kästner, T. Lueders and C. Vogt, FEMS Microbiol. Ecol., 2010, 71, 237–246 CrossRef CAS PubMed
- M. Ranchou-Peyruse, C. Gasc, M. Guignard, T. Aüllo, D. Dequidt, P. Peyret and A. Ranchou-Peyruse, Microb. Biotechnol., 2017, 10, 469–479 CrossRef CAS PubMed
- S. Shimizu, M. Akiyama, T. Naganuma, M. Fujioka, M. Nako and Y. Ishijima, Geobiology, 2007, 5, 423–433 CrossRef CAS
- J. Weijma, F. Gubbels, L. W. Hulshoff Pol, A. J. M. Stams, P. Lens and G. Lettinga, Water Sci. Technol., 2002, 45, 75–80 CrossRef CAS PubMed
- G. Muyzer and A. Stams, Nat. Rev. Microbiol., 2008, 6, 441–454 CrossRef CAS PubMed
- P. Smigan, M. Greksäk, J. Kozänkovä, F. Buzek, V. Onderka and I. Wolf, FEMS Microbiol. Lett., 1990, 73, 221–224 CrossRef CAS
- M. Panfilov, Transp. Porous Media, 2010, 85, 841–865 CrossRef CAS
- M. Panfilov, G. Gravier and S. Fillacier, in ECMOR X - 10th European Conference on the Mathematics of Oil Recovery, European Association of Geoscientists & Engineers, Amsterdam, Netherlands, 2006.
- M. Ranchou-Peyruse, J.-C. Auguet, C. Mazière, C. X. Restrepo-Ortiz, M. Guignard, D. Dequidt, P. Chiquet, P. Cezac and A. Ranchou-Peyruse, Environ.
Microbiol., 2019, 21, 3953–3964 CrossRef CAS PubMed
- N. Kampman, M. J. Bickle, A. Maskell, H. J. Chapman, J. P. Evans, G. Purser, Z. Zhou, M. F. Schaller, J. C. Gattacceca, P. Bertier, F. Chen, A. V. Turchyn, N. Assayag, C. Rochelle, C. J. Ballentine and A. Busch, Chem. Geol., 2014, 369, 51–82 CrossRef CAS
- K. Fukuzawa, Y. Saitoh, K. Akai, K. Kogure, S. Ueno, A. Tokumura, M. Otagiri and A. Shibata, Biochim. Biophys. Acta, Biomembr., 2005, 1668, 145–155 CrossRef CAS PubMed
- Y. Wang and P.-Y. Qian, PLoS One, 2009, 4, e7401 CrossRef PubMed
- F. Escudié, L. Auer, M. Bernard, M. Mariadassou, L. Cauquil, K. Vidal, S. Maman, G. Hernandez-Raquet, S. Combes and G. Pascal, Bioinformatics, 2018, 34, 1287–1294 CrossRef PubMed
- J. Geets, B. Borremans, L. Diels, D. Springael, J. Vangronsveld, D. van der Lelie and K. Vanbroekhoven, J. Microbiol. Methods, 2006, 66, 194–205 CrossRef CAS PubMed
- M. Wagner, A. J. Roger, G. Brusseau and D. A. Stahl, J. Bacteriol., 1998, 180, 2975–2982 CrossRef CAS PubMed
- L. M. Steinberg and J. M. Regan, Appl. Environ. Microbiol., 2009, 75, 4435–4442 CrossRef CAS PubMed
- L. M. Steinberg and J. M. Regan, Appl. Environ. Microbiol., 2008, 74, 6663–6671 CrossRef CAS PubMed
- D. L. Parkhurst and C. A. J. Appelo, Description of input and examples for PHREEQC version 3-A computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations, U.S. Geological Survey Water-Resources Investigations, 2013.
- C. A. J. Appelo, Appl. Geochem., 2015, 55, 62–71 CrossRef CAS
- R. D. Lindberg and D. D. Runnells, Science, 1984, 225, 925–927 CrossRef CAS PubMed
- D. C. Thorstenson, Geochim. Cosmochim. Acta, 1970, 34, 745–770 CrossRef CAS
- A. Stefánsson, S. Arnórsson and Á. E. Sveinbjörnsdóttir, Chem. Geol., 2005, 221, 289–311 CrossRef
- D. Banks, V. V. Kadnikov and Franck, Hydrochemical data report from sampling of two deep abandoned hydrocarbon exploration wells: Byelii Yar and Parabel’, Tomsk oblast’, western Siberia, Russian Federation., Geological Survey of Norway, 2014.
- L. Barreto, A. Makihira and K. Riahi, Int. J. Hydrogen Energy, 2003, 18 Search PubMed
- B. Johnston, M. C. Mayo and A. Khare, Technovation, 2005, 25, 569–585 CrossRef
- A. M. Elberry, J. Thakur, A. Santasalo-Aarnio and M. Larmi, Int. J. Hydrogen Energy, 2021, 46, 15671–15690 CrossRef CAS
- RAG, Underground SUN Storage - chemical storage of renewable energy in porous subsurface reservoirs with exemplary testbed, https://www.underground-sun-storage.at/, (accessed July 12, 2021).
- T. Trouvé, E. Sauvage, D. Mockly, P. Chambon, J. M. Noé, S. Roche Vu Quang, M. Mack, F. Ferré and B. Schnell, Conditions techniques et économiques d’injection d’hydrogène dans les réseaux de gaz naturel, GRTgaz - GRDF - Téréga - Storengy - Géométhane - Elengy - GDS - Régaz-Bordeaux - SPEGNN, 2019.
- N. Eddaoui, M. Panfilov, L. Ganzer and B. Hagemann, Transp. Porous Media, 2021, 139, 89–108 CrossRef CAS
- R. Tarkowski, Int. J. Hydrogen Energy, 2017, 42, 347–355 CrossRef CAS
- B. Hagemann, M. Rasoulzadeh, M. Panfilov, L. Ganzer and V. Reitenbach, Comput. Geosci., 2016, 20, 595–606 CrossRef
- V. Reitenbach, L. Ganzer, D. Albrecht and B. Hagemann, Environ. Earth Sci., 2015, 73, 6927–6937 CrossRef CAS
- G. Strobel, B. Hagemann, T. M. Huppertz and L. Ganzer, Renewable Sustainable Energy Rev., 2020, 123, 109747 CrossRef CAS
- B. Ouyang, D. M. Akob, D. Dunlap and D. Renock, Appl. Geochem., 2017, 76, 51–59 CrossRef CAS
- X. Dong, C. Greening, T. Brüls, R. Conrad, K. Guo, S. Blaskowski, F. Kaschani, M. Kaiser, N. A. Laban and R. U. Meckenstock, ISME J., 2018, 12, 2039–2050 CrossRef CAS PubMed
- O. Haouari, M.-L. Fardeau, J.-L. Cayol, G. Fauque, C. Casiot, F. Elbaz-Poulichet, M. Hamdi and B. Ollivier, Syst. Appl. Microbiol., 2008, 31, 38–42 CrossRef CAS PubMed
- Y. A. Frank, V. V. Kadnikov, A. P. Lukina, D. Banks, A. V. Beletsky, A. V. Mardanov, E. I. Sen’kina, M. R. Avakyan, O. V. Karnachuk and N. V. Ravin, Front. Microbiol., 2016, 7, 2000 Search PubMed
- M. Berta, F. Dethlefsen, M. Ebert, D. Schäfer and A. Dahmke, Environ. Sci. Technol., 2018, 52, 4937–4949 CrossRef CAS PubMed
- T. L. Hamilton, D. S. Jones, I. Schaperdoth and J. L. Macalady, Front. Microbiol., 2015, 5, 756 Search PubMed
- S. A. Eichorst, D. Trojan, S. Roux, C. Herbold, T. Rattei and D. Woebken, Environ. Microbiol., 2018, 20, 1041–1063 CrossRef CAS PubMed
- S. Kalam, A. Basu, I. Ahmad, R. Z. Sayyed, H. A. El-Enshasy, D. J. Dailin and N. L. Suriani, Front. Microbiol., 2020, 11, 580024 CrossRef PubMed
-
H. L. Drake, K. Küsel and C. Matthies, Acetogenic Prokaryotes, in The Prokaryotes, ed. M. Dworkin, S. Falkow, E. Rosenberg, K. H. Schleifer and E. Stackebrandt, Springer, New York, NY, 2006, pp. 354–420 Search PubMed
- W. Liesack, F. Bak, J.-U. Kreft and E. Stackebrandt, Arch. Microbiol., 1994, 162, 85–90 CAS
- R. Conrad, F. Bak, H. J. Seitz, B. Thebrath, H. P. Mayer and H. Schütz, FEMS Microbiol. Ecol., 1989, 5, 285–293 CrossRef
- V. Peters, P. H. Janssen and R. Conrad, Curr. Microbiol., 1999, 38, 285–289 CrossRef CAS
- M. Berta, 2017.
- R. Jia, T. Unsal, D. Xu, Y. Lekbach and T. Gu, Int. Biodeterior. Biodegrad., 2019, 137, 42–58 CrossRef CAS
- K. H. Nealson, F. Inagaki and K. Takai, Trends Microbiol., 2005, 13, 405–410 CrossRef CAS PubMed
- K. Takai, K. Nakamura, K. Suzuki, F. Inagaki, K. H. Nealson and H. Kumagai, Paleontol. Res., 2006, 10, 269–282 CrossRef
| This journal is © The Royal Society of Chemistry 2022 |