
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh performance Li-, Na-, and K-ion storage in electrically conducting coordination polymers†
Jiande
Wang
a,
Xiaolong
Guo
a,
Petru
Apostol
a,
Xuelian
Liu
a,
Koen
Robeyns
 a,
Loïk
Gence
b,
Cristian
Morari
c,
Jean-François
Gohy
a and
Alexandru
Vlad
*a
a,
Loïk
Gence
b,
Cristian
Morari
c,
Jean-François
Gohy
a and
Alexandru
Vlad
*a
aInstitute of Condensed Matter and Nanosciences, Université catholique de Louvain, Louvain-la-Neuve, Belgium. E-mail: alexandru.vlad@uclouvain.be
bInstituto de Física, Pontificia Universidad Católica de Chile, Santiago, Chile
cInstitutul National de Cercetare-Dezvoltare pentru Tehnologii Izotopice şi Moleculare Cluj-Napoca, Cluj-Napoca, Romania
First published on 8th August 2022
Abstract
Coordination polymers (CPs) made of redox-active organic moieties and metal ions emerge as an important class of electroactive materials for battery applications. However, the design and synthesis of high voltage alkali-cation reservoir anionic CPs remains challenging, hindering their practical applications. Herein, we report a family of electrically conducting alkali-cation reservoir CPs with the general formula of A2-TM-PTtSA (wherein A = Li+, Na+, or K+; TM = Fe2+, Co2+, or Mn2+; and PTtSA = benzene-1,2,4,5-tetra-methylsulfonamide). The incorporation of transition metal centers not only enables intrinsic high electrical conductivity, but also shows an impressive redox potential increase of as high as 1 V as compared to A4-PTtSA analogues, resulting in a class of organometallic cathode materials with a high average redox potential of 2.95–3.25 V for Li-, Na- and K-ion batteries. A detailed structure – composition – physicochemical properties – performance correlation study is provided relying on experimental and computational analysis. The best performing candidate shows excellent rate capability (86% of the nominal capacity retained at 10C rate), remarkable cycling stability (96.5% after 1000 cycles), outstanding tolerance to low carbon content (5 wt%), high mass loading (50 mg cm−2), and extreme utilisation conditions of low earth orbit space environment tests. The significance of the disclosed alkali-ion reservoir cathodes is further emphasized by utilizing conventional Li-host graphite anode for full cell assembly, attaining a record voltage of 3 V in an organic cathode Li-ion proof-of-concept cell.
Broader contextOrganic battery materials rise as potential alternatives for post-Li batteries. Storage of sodium, potassium, as well as divalent and even trivalent cations has been shown to proceed more efficiently within organic battery materials given the higher robustness and resilience of molecular crystals phase changes. However, chemistries with alkali-ion reservoir and high voltage operation remains a challenge in the field, with few developments only recently proposed. Herein, we make use of coordination chemistry to design and synthesize a new class of electrically conducting anionic coordination polymers for all practically relevant alkali-cation storage. The working voltage of these chemistries is nearly by 1 V higher than of state of art organic or coordination polymer chemistries, reaching values of 2.9–3.5 V in Li-, Na-, or K- based cells. The reported phases are insoluble in battery electrolytes, have high gravimetric density, and can be cycled with low carbon content and high mass loading. These results provide guidelines for higher capacity materials by designing systems with also reversible redox of transition metals, while also provide opportunities for exploring divalent or trivalent cations in this class of conducting coordination polymers. |
Introduction
Coordination polymers (CPs, also including the sub-class of MOFs) constructed from redox-active organic linkers and transition-metal centers have flourished in recent years as potential electrode materials for the next-generation electrochemical energy storage.1–5 These materials are generally insoluble in non-aqueous electrolytes given the rigid and extended coordination structure, unlike the organometallic complexes6 and small organic compounds,7–9 thus guaranteeing high cycling stability in an electrochemical cell. Compared to their constituent organic building blocks, CPs can be also designed to offer enhanced electronic conductivity benefiting from the extended conjugation system,10 an essential property for energy storage application.3,11 In addition, redox potential of the redox unit can also be tuned by transition metal cations through inductive polarizing effect.12 All these advantages stimulate further quest for higher performance CPs especially as cathode materials for alkali cation electrochemical storage.Pioneering studies of CPs as cathode materials were reported by Tarascon et al. on Fe-MIL-53 electrodes.13,14 The charge storage was enabled by the redox activity of only Fe(III) centers, with the terephthalate units not participating given their redox activity at lower than applied redox potentials.15 Subsequently, a variety of metal–organic coordination chemistries, including MOFs, have been designed by introducing redox-active functional groups on the organic ligands, either of n-type storage mechanism (cations are required for charge compensation),16–20 or p-type (anions are required for charge compensation).21,22 Whereas p-type chemistries are suitable for dual-ion batteries or anion storage batteries, the n-type cation storing redox materials are the mainstream of current research and commercial battery application.23
Despite the extensive developments of n-type organic chemistries, most CP cathode materials studied so far are prepared and tested in the oxidized form, requiring an initial battery discharge protocol (unlike the conventional Li-ion cathodes that work on charge first).4,5,12,24,25 The consequences are generally the low voltage attained, and the limited compatibility with conventional Li-ion cell constructs that require the cathode material to be a Li-reservoir (i.e., in the reduced Li-containing state). Alkali and alkali-earth cation containing MOFs with ionic conduction properties have been developed recently26–29 with however no reversible redox, and electronic insulator properties, thus not ideal as electrode material for batteries. Further to the rareness of Li-ion CPs as battery electrode materials,12 no redox active Na-ion and K-ion CPs have been synthesized and tested so far (Table S1, ESI†). In addition to being cation-reservoir cathode materials suitable to couple with traditional anode materials for metal–ion cells, another intrinsic advantage of cation-reservoir cathode chemistries is their feasibility for anode-free cells,30 implying thus increased practicality, safety and energy density.
Depending on metal – linker coordination modes, CPs can result in 1D (one dimensional, generally non-porous), 2D or 3D structural topologies (i.e., MOFs or porous CPs). MOFs have high porosity and large surface area, a double-faced property. On one side, high porosity endows MOFs with remarkable properties for various applications, such as gas separation or storage, catalysis,31 as well as high energy density supercapacitive storage.3,32 On the opposite side, this results in low volumetric energy densities, a major constrains for many portable and mobile electric applications. Non-porous CPs, on the other hand, have a better application prospect as battery electrode materials than MOFs, given their higher material density, and thus higher energy stored per volume unit.
Few developments on non-porous, electrically conducting CPs have been initially reported by Wang group, including nickel 1,2,4,5-benzenetetraamine33 and nickel tetrathiooxalate.34 As electrode materials for sodium ion storage, these displayed relatively low average redox potentials of 1.2 V and 2.0 V vs. Na+/Na, respectively. An aluminum-coordinated poly(tetra-hydroxybenzoquinone) material was also proposed as a potential candidate for Na-ion storage, with an average discharge potential of 2 V vs. Na+/Na.35 However, large redox potential window required for full material utilization makes these chemistries more suitable as anode rather than cathode electrode materials. Making CPs with higher redox potential, ideally >3 V (vs. Li+/Li, for example), has thus become a well-acknowledged urgent necessity in the field. Other than enabling high voltage cells, this endows redox stability towards molecular oxygen and such phases could be prepared and handled in ambient atmosphere conditions and at low costs. The alkali cation-reservoir form, alike the commercial inorganic cathode materials (e.g. LiFePO4), could be thus used for direct full cell assembly and upon future development, suitable for organic cathode anode free design.36
In this work, we present a new family of alkali-ion contained Coordination Polymers (CPs) that are directly synthesized and handled in their reduced alkali-ion reservoir form (Scheme 1 and Table S1, ESI†). The interest in designing the CPs with the composition of A2-TM-PTtSA (A = Li+, Na+, K+; TM = Fe2+, Co2+, Mn2+; PTtSA4− = benzene-1,2,4,5-tetra-methylsulfonamide) is manyfold. First, the TM center considerably increases the redox potential of the PTtSA linker, rationalized through the high polarizing power of the spectator TM cation. The immediate effect of this is the increase of redox potential by as much as 1 V when compared to A4-PTtSA parent alkali cation-based chemistries. The redox potential values attained with these chemistries is also among the highest attained compared to previously reported organic cathode materials (Table S1, ESI†). Additionally, two orders of magnitude gain in electronic conductivity are being observed upon inclusion of TM centers, reaching values in the range of 10−6–10−7 S cm−1 at room temperature, metrics comparable to those of inorganic battery cathode materials.37–42 The studied A2-TM-PTtSA CPs display a sequential two-electron organic linker centered redox, with a reversible capacity of ∼100 mA h g−1. Notably, the Li2-TM-PTtSA electrodes show excellent rate capability and cycling stability, with also remarkable electrode performance even at low carbon content (5 wt%) and high mass loading (50 mg cm−2), assigned to the high electrical conductivity and material density. The Na- and K-ion analogues show similar electrochemical properties, consolidating the universal feasibility of the reported chemistries for any relevant alkali-cation battery chemistry. Overall, these results not only extend the landscape of CP cathode materials based on sulfonamide linkers, but also provide valuable guidelines of finely tuning the electroactive properties via the coordination chemistry of redox active organic materials.
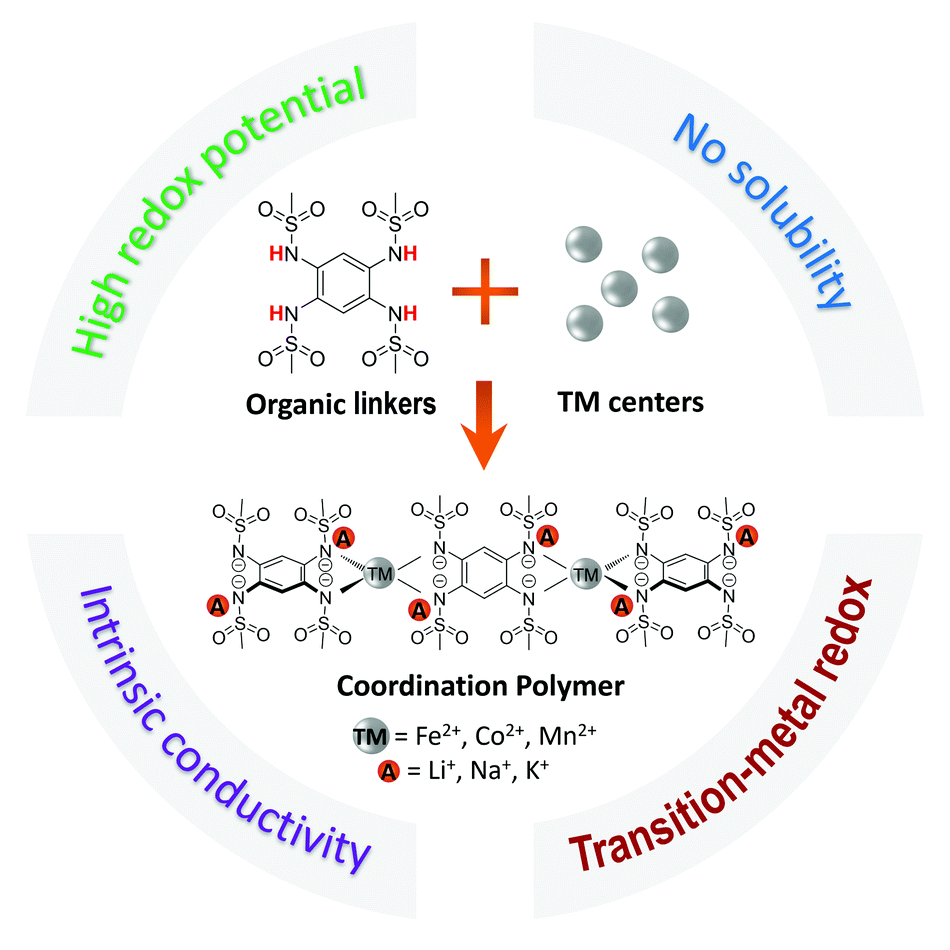 | ||
| Scheme 1 Design principle and synthesis route of the alkali-ion contained coordination polymers with the general formula of A2-TM-PTtSA, wherein: A = Li+, Na+, K+; TM = Fe2+, Co2+, Mn2+, and PTtSA = benzene-1,2,4,5-tetra-methylsulfonamide. | ||
Result and discussion
The synthesis of A2-TM-PTtSA CPs follows a one-step approach consisting in one-pot room temperature reaction of equimolar amounts of deprotonated ligand A4-PTtSA (A = Li+, Na+, K+) with the corresponding divalent transition-metal salt (refer to SI for detailed synthesis information). Various combinations of A+ and TM2+ have been screened and according to the best performances attained so far, five main compositions are selected and discussed in this work (Scheme 1 and Fig. 1A): Li2-TM-PTtSA (TM2+ = Fe2+, Co2+, Mn2+) along with A2-Co-PTtSA (A+ =Na+, K+). The chemical and physicochemical properties were analyzed by various techniques (Fourier-Transform Infrared Spectroscopy, Powder X-ray Diffraction, Elemental Analysis, ICP-OES and ion-chromatography) all confirming the formation of the desired materials (Fig. 1B and Fig. S1–S5, ESI†).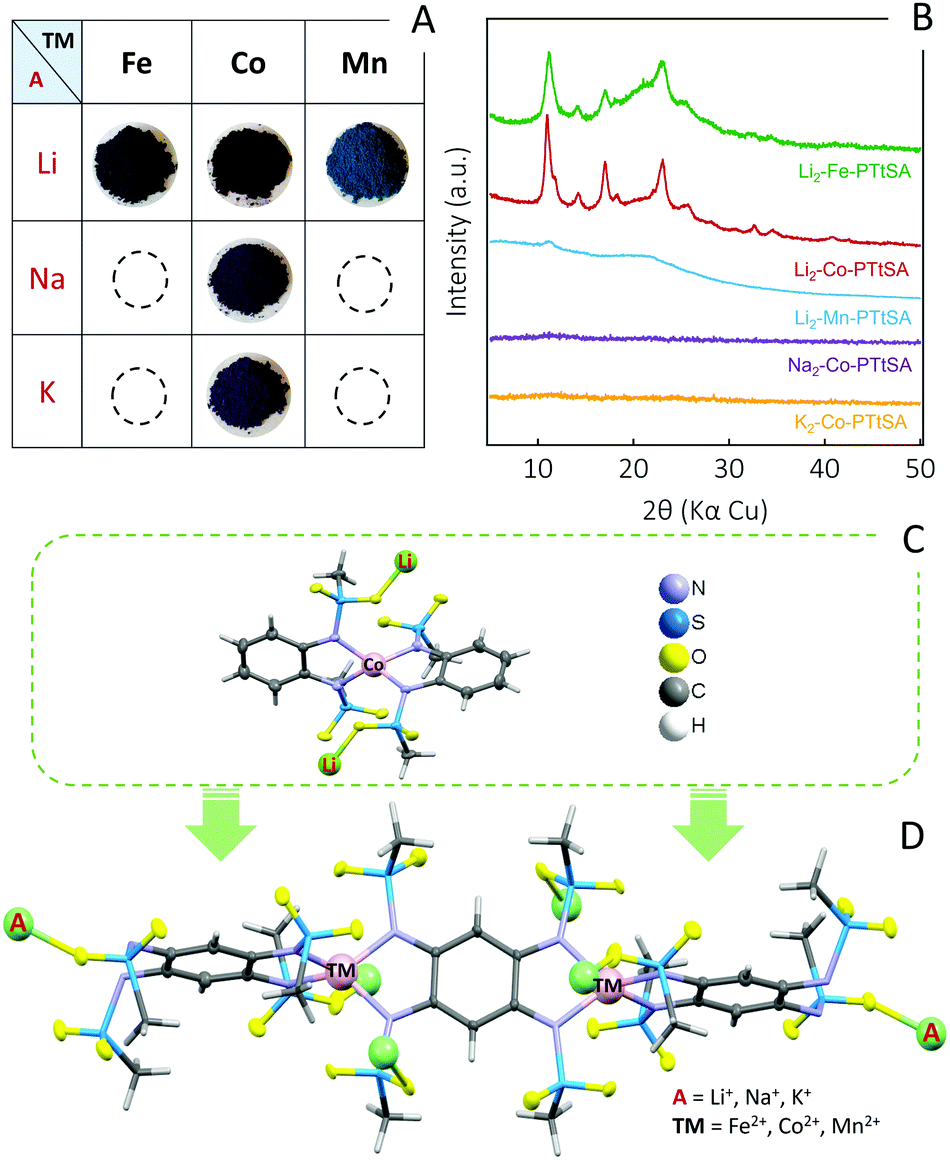 | ||
| Fig. 1 Overview of the A2-TM-PTtSA CPs studied in this work. (A) Photographs of prepared A2-TM-PTtSA powders with different transition-metal nodes and alkali-cations; (B) PXRD data highlighting the impact of transition-metal and alkali-cation nature on the crystallinity of the materials; (C) Crystal structure of Li2-Co(II)-[o-PDSA]2 complex. (D) The proposed schematic structure for the A2-TM-PTtSA CPs. | ||
All the synthesized materials have been found prone to produce amorphous (Na2/K2-Co-PTtSA) or poorly crystalline (Li2-Fe/Co/Mn-PTtSA) phases, largely accounting on coordination framework distortion due to the dislocation of alkali cations12 (Fig. 1B). The quality of the PXRD data was not enough to resolve the crystal structure for the Li2-Fe-PTtSA and Li2-Co-PTtSA phases, and any attempt in improving the crystallinity thus far failed. It is worth however being noted that the Li2-Fe/Co-PTtSA phases (and to a lesser extent Li2-Mn-PTtSA) are iso-structural, with a series of peaks located at similar diffraction angles (i.e., at 11.0°, 14.2°, 17.0° and 23.1°).
To determine the coordination environment of TM, as well as the location of alkaline metal ions, a model quasi-isostructural complex was prepared and analyzed. The selected ligand, N,N′-(1,2-phenylene)dimethanesulfonamide (H2-o-PDSA) emulates half of the PTtSA reactive sites and it is reasonable to assume similar binding modes of the o-PDSA and PTtSA ligands with TM(II) and A(I).43 A single crystal of a model Li2-Co(II)-[o-PDSA]2 complex was prepared, and structure was determined from single-crystal X-ray data (Fig. 1C, refer to ESI† for synthesis and analysis details). According to the obtained structure, Co(II) is tetrahedrally coordinated to the four nitrogen atoms of two o-PDSA molecules (Fig. S6, ESI†). Each of the two Li-cations required for charge compensation, bind to the two oxygen atoms of different mesyl groups, and to two MeOH molecules retained in the crystal. Accordingly, the structure of A2-TM-PTtSA CPs can be regarded as a 1D extension of the Li2-Co(II)-[o-PDSA]2 model complex (Fig. 1D), although disorder or formation of branching coordination modes cannot be excluded, explaining also the poor crystallinity of many studied phases. The formation of a compact material is also confirmed by the measured apparent density of the A2-TM-PTtSA CP powders, all in the range of 1.6–2.0 g cm−3.
Next, we evaluated the electrical conductivity of the prepared A2-TM-PTtSA CPs given that the molecular design fulfills the requirements for intrinsic charge conduction properties. Variable-temperature conductivity measurements for all studied A2-TM-PTtSA were conducted on pressed-pellets using the two-probe d.c. current–voltage technique (for experimental details refer to ESI†). The temperature dependence of the conductivity for all phases follows the linear relationship of the logarithm conductivity with respect to the reciprocal of the temperature in the Arrhenius plot (Fig. 2A). It is found that Li2-Fe-PTtSA and Li2-Co-PTtSA show the highest conductivity, whereas Na2-Co-PTtSA and K2-Co-PTtSA display nearly one magnitude lower conductivity values. Higher crystallinity and long-range order, as evidenced by PXRD data (Fig. 1B), corroborate the observed electronic conductivity trend. A prerequisite for electrical conductivity in coordination polymers is delocalization of charge, either via “through-space” π–π stacking interactions between organic moieties or through the formation of large delocalized systems between the metal d-orbital and the π level of the bridging ligand.10 Since all A2-TM-PTtSA indicate weak π–π stacking interactions (absence of the characteristic diffraction peak around 25.8°),44 the charge conduction can be presumed to proceed preferentially via a through-bond type mechanism, with electron self-exchange between the redox active TM centers and organic linkers. Electron hopping mechanism also possibly exists in the coordination polymers. This is further supported by the significative loss in conductivity (of more than two orders of magnitude) for redox innocent metal derivatives (e.g. for the A4-PTtSA wherein A = Li+, Na+, or K+, Fig. S7, ESI†).45 A disordered (disrupted) CP chain structure remains thus a plausible explanation for lower conductivity values found for Li2-Mn, as well as Na2-Co and K2-Co versions.
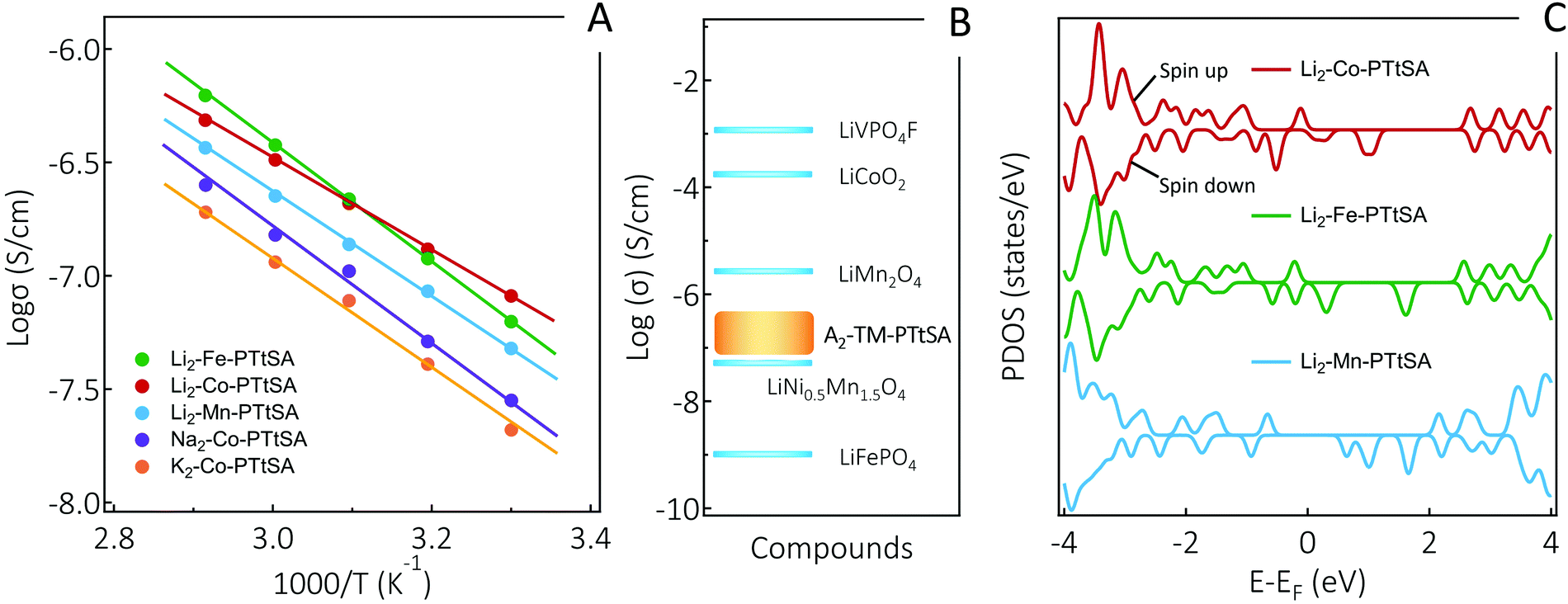 | ||
| Fig. 2 Electrical conductivity analysis of the studied A2-TM-PTtSA CPs. (A) Arrhenius plot of electrical conductivity for the prepared materials; (B) electrical conductivity comparison of selected commercial inorganic Li-ion electrodes and the studied CPs; (C) total density of states (DFT calculation) for Li2-TM-PTtSA coordination polymers (positive/negative values for spin up/down); at room temperatures the states around Fermi level (about ±0.1 eV) will participate to electronic transport. | ||
Stronger ligand–metal interaction and electron self-exchange (π–d orbitals overlap) are additional aspects that corroborate higher conductivities measured for Co and Fe derivatives. DFT calculation shows that the length of the periodic CPs along the chain as well as the metal–nitrogen distances are on inverse proportion to atomic numbers (for details, refer to DFT section: Fig. S16 and Table S1, ESI†), indicating lower values for the Co and higher for the Mn CPs. Given the fact that longer bond is an indication for weaker interaction, the ligand–metal interaction as well as π–d orbital overlap between periodic units follows a trend of Co > Fe > Mn, which correlates with the conductivity trend found for the three Li2-TM-PTtSA (TM = Co, Fe, Mn) CPs (Fig. 2A). This trend can also be explained by band gap differences between these CPs (Fig. 2C), where the DOS of 3d orbitals of transition metals follows a relatively homogeneous trend, with a gradually widened gap form Co to Fe and Mn CPs. These trends are also reflected in the electrochemical data analysis, which will be discussed in the subsequent.
From practical perspective, the room temperature electrical conductivity of A2-TM-PTtSA materials all display high values in the range of 10−7 to 10−6 S cm−1 (Fig. 2A). To put this in perspective, when compared to the inorganic Li-ion cathodes (Fig. 2B), these values are certainly lower than of LiVPO4F,37 LiCoO240 and LiMn2O4,42 however surpassing those of LiNi0.5Mn1.5O441 and LiFePO4.39 These are important considerations as it enable electrode formulations with low amount of conducting carbon additives. The combined effect of high electrical conductivity, material density (1.6–2.0 g cm−3), redox potential, and the versatility for various cations storage, place the A2-TM-PTtSA family of cathodes materials on top of reported organic battery materials, with key parameters for practical batteries.46
Electron delocalization theory explains the structure, stability, and reactivity in many classes of compounds including organo–metallic complexes.6 Intermolecular delocalization can additionally result in intrinsic electron conducting properties, as discussed earlier. Redox potential change through secondary coordination interactions is another effect of metal–ligand interaction. Many studies so far have been focused on redox changes of the metal centers, owing to the fact that most commonly studied ligands do not have a reversible redox.4,5 However, the opposite, or the combined effect of both metal and ligand redox are also known, with some exemplary developments in the field of energy storage achieved recently. Namely, ligand–metal center redox potential changes have been studied in distinct coordination complexes6, and in MOFs,12,47 with notable significative redox potential raise of organic linkers explained by the inductive effect of the coordinated cations. This was found to induce significant electron density variation on the ligands, increasing their redox potentials.
According to these rationales, we also monitored the redox behavior and potential change of the A2-TM-PTtSA CP series (Fig. 3), with the parent organic anionic unit (Li4-PTtSA) as reference. First noticeable change is that, compared to Li4-PTtSA with only one simultaneous two-electron process (i.e., one redox plateau, or one redox peak pair in the corresponding dQ/dV plot), the CPs show two sequential one-electron redox plateaus (Fig. S8, ESI†), with additional features observed in the dQ/dV plots (Fig. 3A). The second interesting feature is the significant potential increase, with Li2-Co-PTtSA displaying the highest values, with two redox plateaus averaging at 2.85 and 3.40 V vs. Li+/Li. This is 700 mV higher than in parent Li4-PTtSA, (Fig. 3A and D). The high redox voltage of the A2-TM-PTtSA materials (in average >3 V vs. Li+/Li) additionally implies air-stability of these material, an important asset for handling and processing of the electrodes in air. Chemical and electrochemical analyses of the air-exposed powders show minimal oxidation, with the battery galvanostatic charge–discharge showing highly reversible redox in the subsequent cycles (Fig. S9, ESI†).
 | ||
| Fig. 3 The impact of the spectator cation on the electrochemistry of A2-TM-PTtSA CP family. (A) Differential capacity plots for Li2-Co-PTtSA, Li2-Fe-PTtSA, and Li2-Mn-PTtSA as compared to parent Li4-PTtSA. (B) Na2-Co-PTtSA as compared to parent Na4-PTtSA and (C) K2-Co-PTtSA as compared to parent K4-PTtSA. (D) Quantitative analysis of the redox potential increase in A2-Co-PTtSA as compared to A4-PTtSA. | ||
The used divalent spectator cations (Fe2+, Mn2+ and Co2+) have a higher polarizing power (Z/r2) than of the alkali cations48 (Fig. S10, ESI†). This translates into higher charge delocalization and the decrease of the electron density on the organic redox center, resulting in higher redox potential of the PTtSA4− unit. The polarizing power of the transition metal cations follows the trend of Co2+ (3.86) > Fe2+ (3.37) > Mn2+ (3.12), explaining the higher average redox potential for Li2-Co-PTtSA (3.13 V vs. Li+/Li) than Li2-Fe-PTtSA (3.07 V) and Li2-Mn-PTtSA (3.05 V). The redox potential is also affected by the metal–ligand interaction, with the strongest interaction found in Li2-Co-PTtSA and weakest in Li2-Mn-PTtSA (refer to DFT section in ESI:† Table S1). A significant redox potential increase is also observed for Na- and K-ion derivatives (Fig. 3B and C), with an average of 450 mV and 540 mV higher as compared to parent Na4-PTtSA and K4-PTtSA derivatives, respectively. The redox potential increase for the second plateau is as high as 730 mV and 1 V for Na4-PTtSA and K4-PTtSA, respectively (Fig. 3D). This results in high working redox potentials averaging at 2.93 V vs. Na+/Na and 3.25 V vs. K+/K, for the Na2-Co-PTtSA and K2-Co-PTtSA electrodes, which are the highest (to the best of our knowledge) values reported so far in organic or coordination complex redox materials for Na- and K-ion batteries.
Amongst the analyzed series of CPs, the Co-based CPs provide the best performances so far – i.e., highest voltage and electronic conductivity (Fig. 2A, 3A and Fig. S11, ESI†). The Li2-Co-PTtSA is also the one showing the most distinct separation between the two redox plateaus, which makes this phase also suitable to investigate the one-electron oxidized intermediate state (Fig. S11, ESI†). From a sustainability point of view, it is certainly by far not the optimal composition as Co-based battery materials are progressively being banned from the battery technology. The Fe and Mn(II) versions do fully comply with the sustainability developments, and these have been found as good as the Co(II) versions in terms of cycling stability and power performances, albeit the slightly lower energy density metrics inherent of lower redox potential. Given the large library of chemistries and properties disclosed, yet driven by the working voltage attained so far, for the sake of consistency we opted to focus on the Co version for our lab-scale fundamental analyses and testing. The Fe(II) and Mn(II) derivatives certainly worth further exploration from fundamental but also practical point of view, with clearly routes open also for other divalent cation explorations like Ni(II), Zn(II), or alkali-earth metals.
The charge storage performances of the A2-Co-PTtSA CP family as cathode materials are summarized in Fig. 4. The galvanostatic charge/discharge curves of the Li2-Co-PTtSA display two pairs of sloping plateaus in the ranges of 2.7–3.0 V and 3.3–3.6 V vs. Li+/Li, which point to multiple redox reactions during the charge/discharge process, and the highly reversible nature of charge storage (Fig. 4A). The initial discharge capacity of ∼101 mA h g−1 (Fig. S12, ESI†), is close to theoretical 2-electron value of 103 mA h g−1 under a cycling rate of C/5 (corresponding to 1 Li+ exchange in 2.5 h). The electrode retains 97 mA h g−1 of capacity after 100 cycles with over 96% capacity retention (Fig. 4A). The Li2-Co-PTtSA electrode also shows excellent rate capability, and even at a high rate of 10C, the electrode can deliver a capacity of 83.5 mA h g−1 (86% of the initial capacity, Fig. 4A inset). The excellent rate performance can be attributed to the relatively higher material density and intrinsic electronic conductivity. Long-term cycling at 5C shows excellent capacity retention of 96.5% over 1000 cycles, corresponding to 0.0035% capacity decay per cycle (Fig. 4B). The stable cycling stability and no voltage slippage corroborate the highly stable and robust of the CPs for Li storage.
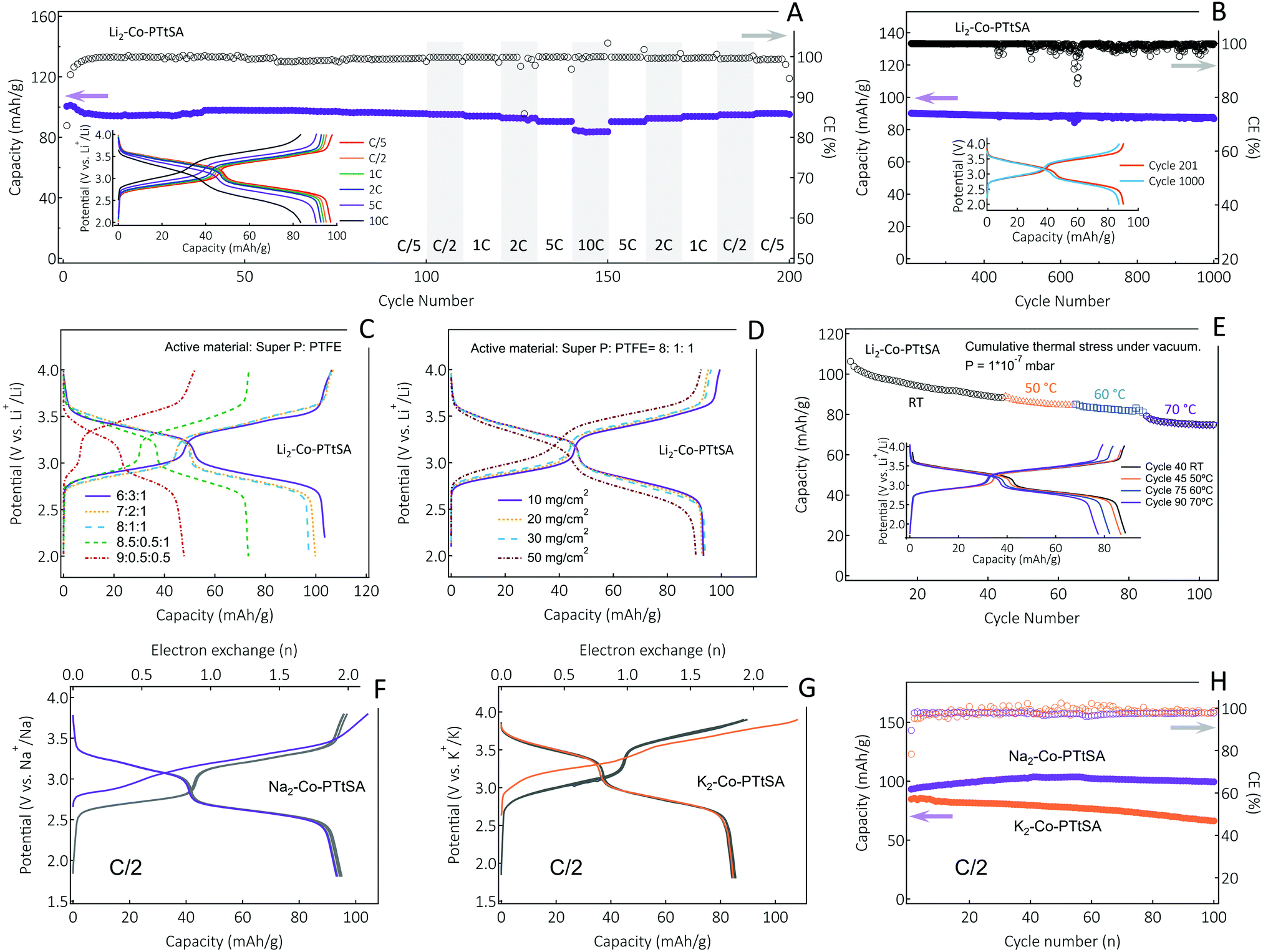 | ||
| Fig. 4 Charge storage performances of the selected A2-Co-PTtSA CP electrodes tested in half-cell configuration with the Li, Na, or K metal counter- and pseudo-reference electrodes. (A) Cycling stability of Li2-Co-PTtSA measured at different cycling rates ranging from C/5 to 10C, with the corresponding charge-discharge profiles shown in the inset, and (B) long-term cycling stability plot at a cycling rate of 5C. Charge-discharge profiles of Li2-Co-PTtSA electrodes with (C) different carbon content, and (D) high mass loading. (E) Cycling stability of Li2-Co-PTtSA measured on combined thermal-vacuum stress testing at current 2C, with charge–discharge profiles recorded at fixed temperature shown in the inset. The first three cycle potential-capacity curves for (F) Na2-Co-PTtSA and (G) K2-Co-PTtSA, and (H) the corresponding cycling stability plots at a rate of C/2. | ||
Electrodes with variable carbon content and mass loading have been also assembled and analyzed (Fig. 4C and D). The electrodes with 30, 20 and 10 wt% carbon content show negligible differences with low polarization and high material utilization (Fig. 4C). This is indicative of fast electrode kinetics, assigned to intrinsic electronic conductivity and also high density of these materials and thus low carbon fractions needed for continuous charge conduction percolation. Only by further decreasing the carbon content to 5%, the electrode shows lower material utilization of ∼75%, with nevertheless low charge-discharge polarization, which is due to under-optimized electrode formulation, inhomogeneous mixing of conductive carbon and electrode material, as well as particle size control (Fig. S13, ESI†). This phenomenon is also evidenced by further decreasing the content of PTFE binder to 5%, where electrode-carbon contact becomes worse, leading to further decrease of active material utilization (Fig. 4C).
Different mass loading tests were performed with 10 wt% Super P formulation, showing excellent cell performance up to 30 mg cm−2 loading of active material, with no obvious capacity decrease and electrode polarization noticed (Fig. 4D). Only for very high mass loading of 50 mg cm−2, slightly lower material utilization (< 5% drop) and higher polarization (∼100 mV) were observed. The sodium and potassium CP cathodes show similar electrochemical behavior to the lithium version, with also two distinguishable plateaus as well as high efficiency and stability attained with cycling (Fig. 4F–H). The K2-Co-PTtSA cells show excellent cycling performance with over 78% of capacity retaining after 100 cycles, whereas Na2-Co-PTtSA shows negligible capacity loss per cycle and a coulombic efficiency (CE) in average above 99% (Fig. 4H).
Besides high active material content and mass loading, energy storage devices that withstand extreme operation conditions like temperature, radiative (cosmic rays), and mechanical (vibration, shock, vacuum) stresses are also of interest, particularly, for the deployment of autonomous self-powered devices (sensors, actuators) in harsh environments. Testing of innovative battery materials should be performed in an environment that emulates as close as possible the stresses and strains associated to the real conditions. Thus, we also tested A2-TM-PTtSA cells under the low earth orbit space environment conditions of high vacuum (down to 10−8 pascals), and stepped temperature ramping (steps of 2 hours at 50, 60 and 70 °C). Samples showed excellent cycling stability with more than 85% of the initial capacity retained over the entire duration of the thermal stress experiment (Fig. 4E), highlighting the resilience of these materials to harsh environmental conditions.
As revealed by data shown in Fig. 3, 4 and Fig. S8, S11 (ESI†), all A2-TM-PTtSA CPs display a two-plateau reversible electrochemical behavior, irrespective of the nature of stored alkali (Li+, Na+, K+) or spectator TM cations (Fe2+, Mn2+, Co2+), pointing towards a similar redox mechanism. This was subsequently thoroughly investigated with the use of various ex situ techniques, over the course of one full charge–discharge cycle (using Li2-Co-PTtSA as model electrode material, Fig. 5). First, FTIR survey shows similar vibration fingerprints between the pristine and cycled electrode (position I and V, respectively; Fig. 5A and B), corroborating the full chemical reversibility of the electrochemical charge and discharge process. The FTIR analysis of the 2-electron (fully) oxidized, as well as for the 1-electron redox intermediate states reveals the loss of aromatic signature and the formation of electron delocalized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN bands, corroborating the reversible ligand-based 2-electron redox.
C and CN bands, corroborating the reversible ligand-based 2-electron redox.
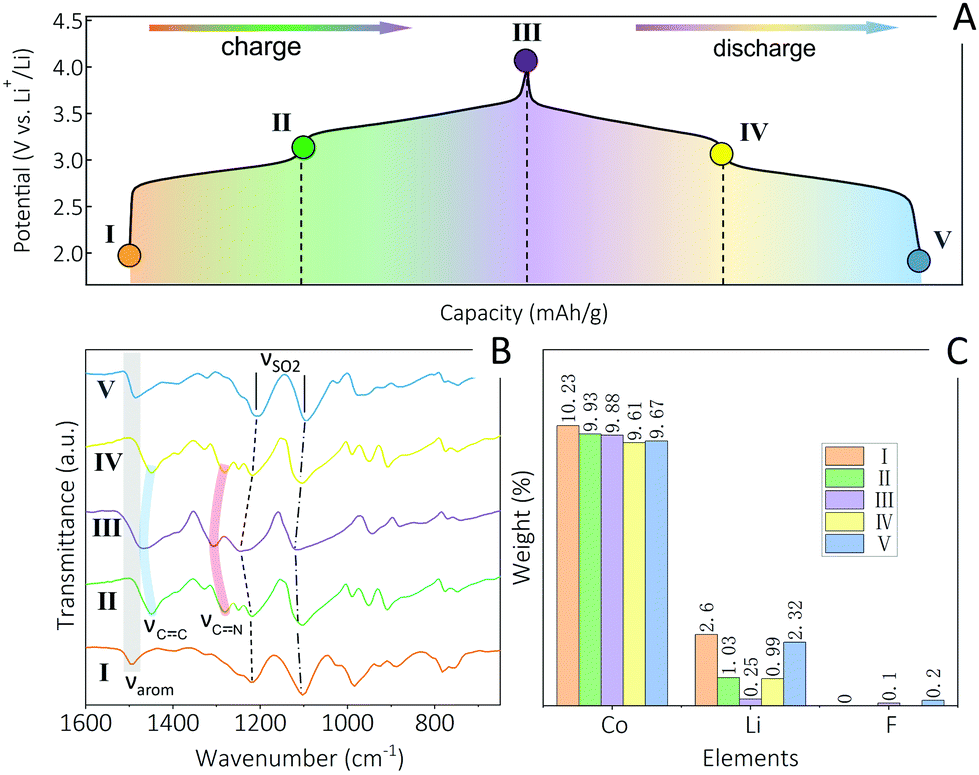 | ||
| Fig. 5 Ex situ analysis of redox in Li2-Co-PTtSA. (A) The galvanostatic charge–discharge curve of with selected different dilithiation/lithiation states, and the five points marked represent various charge/discharge states of Li2-Co-PTtSA electrode: pristine (I), one electron half-charged (II), two electron full-charged (III), one electron half-discharged (IV) and two electron full-discharged (V). The corresponding ex situ FTIR spectra (B) and ex situ elemental analysis results (C). | ||
The redox was also followed by elemental analysis, with a particular focus on the nature of the exchanged cations, and possible counter-anions (Fig. 5C and Fig. S14, ESI†). The weight content of Co to Li in the pristine Li2-Co-PTtSA sample are 10.23% and 2.60% (Co/Li = 1/2.15 atom ratio, position I). Upon half-charging to the intermediate II, the Co content remains constant, whereas lithium content drops to 1.03% (Co/Li = 1/0.87). With further oxidation to the fully charged state III, lithium content drops to 0.25%, corresponding to atomic ratio of Co/Li = 1/0.21 (the remaining lithium is probably from the residual lithium electrolyte salt). Upon discharge, lithium content regains the initial value of 0.99% (Co/Li = 1/0.86) and 2.32% (Co/Li = 1/2.04) at the intermediate position IV and full-discharged state V, respectively. This analysis implies that 2 Li+ are being exchanged during the redox, excluding any significative cation exchange, dissolution, and shuttle of Co2+. Furthermore, low F amount was detected (assigned to residual electrolyte), implying also no TFSI− anion uptake for charge compensation during redox.
The significant changes in redox behavior of A2-TM-PTtSA CP vs. A4-PTtSA equivalents (i.e., two plateau, higher voltage) could potentially indicate a mixed one-electron ligand and one electron TM2+/TM3+ redox. To further clarify this, ex situ XPS analysis was done (Fig. S15, ESI†), showing that the oxidation state of Co2+ is retained throughout the entire charge–discharge process. Given nevertheless the possible redox activity of the applied TMs, we also investigated the reversibility of the 3rd electron exchange (TMs redox) in the disclosed CPs, thus potentially increasing the charge storage capacity. To enable this, the electrodes were cycled up to 4.5 V (vs. Li+/Li) for all Li2-(Fe,Co,Mn)-PTtSA compositions (Fig. S16, ESI†). All materials were found to display an additional high-voltage plateau at above >4V (vs. Li+/Li), with however capacities occasionally exceeding the theoretical expected 1-electron redox of TM's, for example for Co(II) and Mn(II). The redox was also found irreversible, accompanied by rapid degradations, all these assigned to combined effects of electrolyte decomposition, structural instability of the coordination structure upon accessing the high redox states (3+) of TM centers,43 as well as upon the insertion of the bulky TFSI− anions required for charge compensation.
The ex situ elemental analysis, XPS, FTIR and extended voltage window cycling data, combined altogether provide strong indication of the two-electron reversible redox centered on the organic ligand and not on TM(II/III) redox couple. The redox mechanism of A2-TM-PTtSA is schematized in Scheme 2, implying a 2-electron stepwise reversible process. During the charge process, one coordination unit of A2-TM-PTtSA (position I) extracts one A+ to A1-TM-PTtSA (position II) followed by the second A+ to A0-TM-PTtSA (position III) with the ligand oxidation state change of PTtSA4- → PTtSA3− → PTtSA2−, accounting for the two distinct charge plateaus, respectively. During the reverse discharge process, one coordination unit of A0-TM-PTtSA accepts consecutively two A+ and two electrons at different discharge potentials, reforming the pristine A2-TM-PTtSA phase (position IV and V).
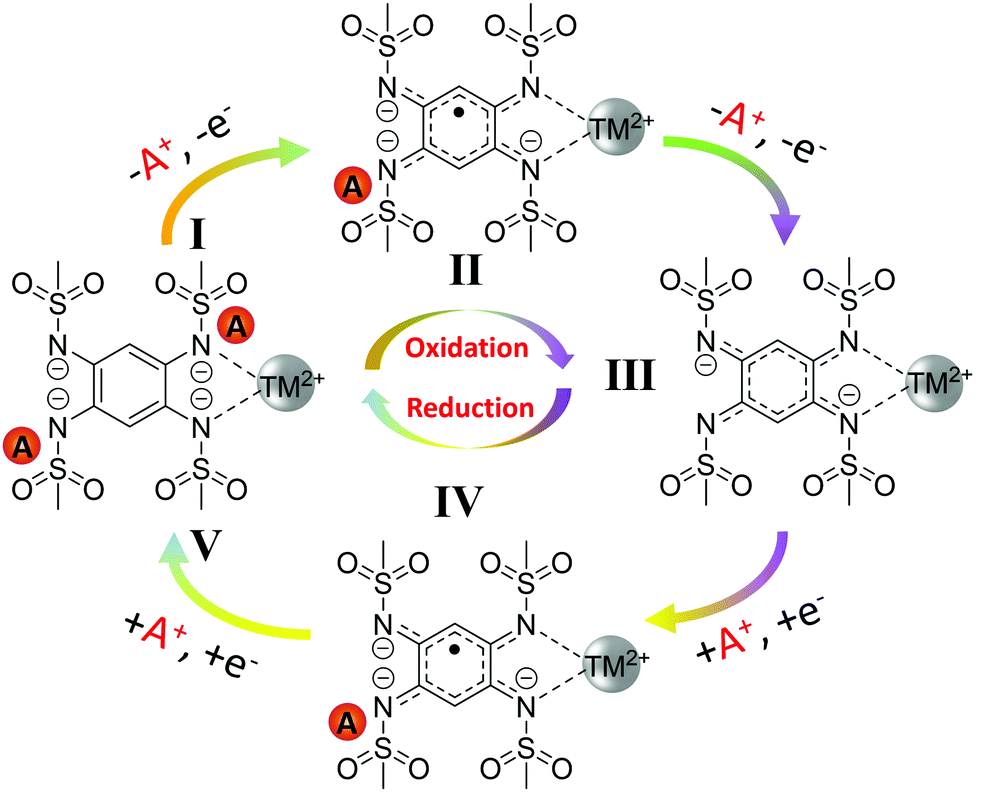 | ||
| Scheme 2 Redox reaction pathway of A2-TM-PTtSA. | ||
Finally, a key feature of A2-TM-PTtSA materials developed here is their alkali-ion containing nature, making them compatible with conventional Li-ion, or other metal-ion cell assembly approaches. Thus, we paired a Li2-Co-PTtSA cathode with a graphite anode in a full-cell configuration (Fig. 6). A commercial electrolyte (LP30, 1 M LiPF6 in EC/DMC 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol%) was used, further emphasizing the compatibility of the developed cathode materials for practical applications. Given the high redox potential difference between Li2-Co-PTtSA cathode and graphite anode (Fig. 6A), the respective cell displays a high average output voltage of ∼3V (Fig. 6B). This is amongst the highest achieved so far for an organic redox Li-ion cell. After the first cycle formation, a reversible capacity of 103 mA h g−1 (based on the active mass of the Li2-Co-PTtSA) is attained. The CE stabilizes above 98% after the first 5 cycles, with a retained capacity of 86% over 200 cycles (Fig. 6C).
:1 vol%) was used, further emphasizing the compatibility of the developed cathode materials for practical applications. Given the high redox potential difference between Li2-Co-PTtSA cathode and graphite anode (Fig. 6A), the respective cell displays a high average output voltage of ∼3V (Fig. 6B). This is amongst the highest achieved so far for an organic redox Li-ion cell. After the first cycle formation, a reversible capacity of 103 mA h g−1 (based on the active mass of the Li2-Co-PTtSA) is attained. The CE stabilizes above 98% after the first 5 cycles, with a retained capacity of 86% over 200 cycles (Fig. 6C).
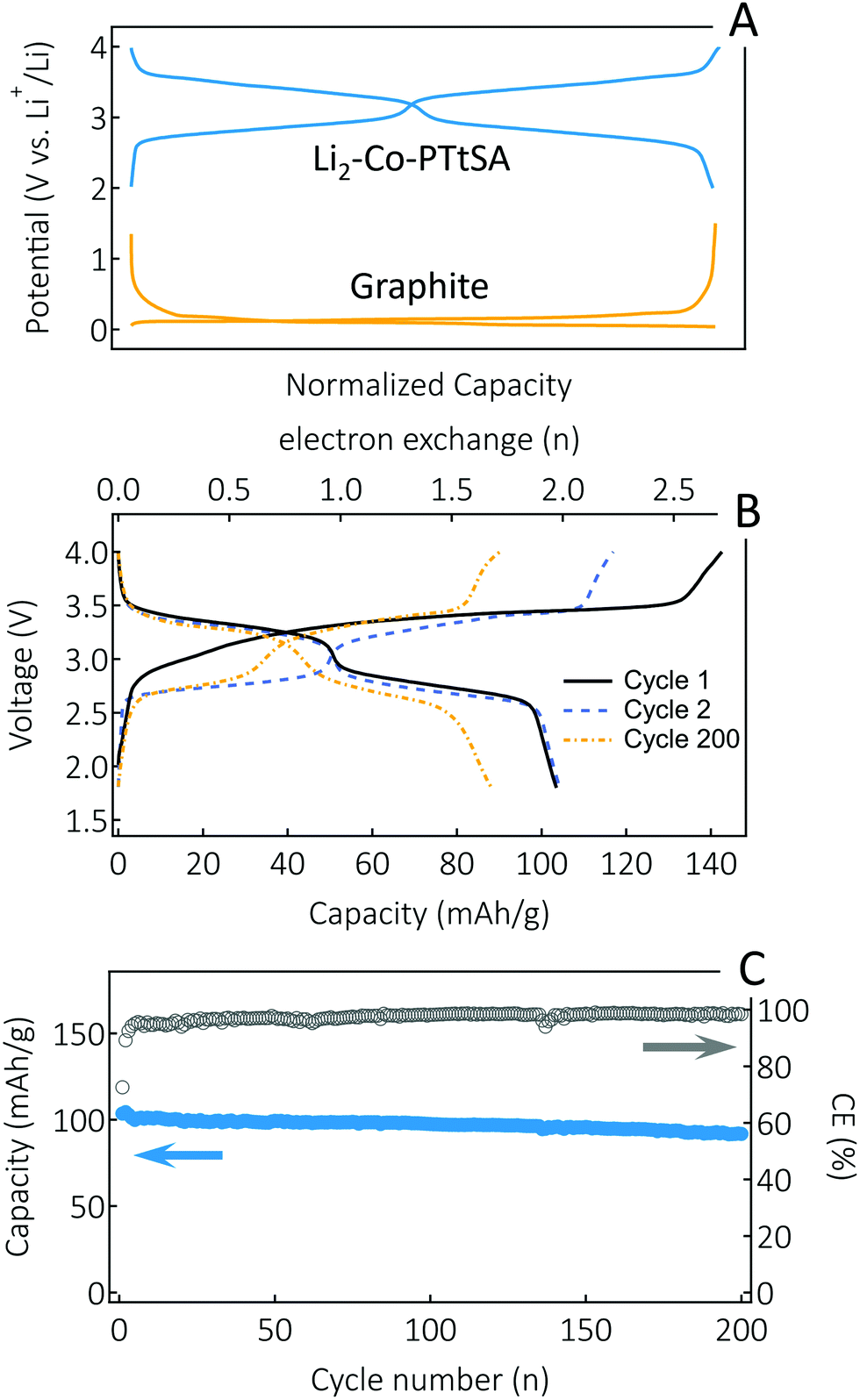 | ||
| Fig. 6 Full cell assembly and performances. (A) Galvanostatic charge–discharge profiles for Li2-Co-PTtSA and graphite as measured in half cells vs. Li metal pseudo-reference/counter electrode. (B) Capacity-voltage profiles of the full cell cycled at a rate of 0.5C. (C) Specific capacity and coulombic efficiency vs. cycle number at a cycling rate of 0.5C. | ||
Conclusions
A new series of alkali-ion containing CPs based on the benzene-1,2,4,5-tetrayltetrakis methylsulfonylamide tetra-anionic ligand has been designed and synthesized, and their physicochemical properties combined with electrochemical analysis are disclosed. The A2-TM-PTtSA phases show high electrical conductivity (10−7 to 10−6 S cm−1 at room temperature), display a high redox potential above 3V (vs. Li, Na, or K), good electrochemical reversibility, as well as excellent rate and cycling performances, making them potential candidates for future battery applications (Table S1, ESI†). The practical feasibility is further corroborated by the fabrication of Li-ion full cells using graphite as anode material, where a high cell voltage and competitive cycling stability is achieved. Batteries based on PTtSA coordination polymer cathodes may be less attractive than inorganic cathodes for portable electronic due to the slight lower material density, thus lower volumetric energy density. However, these can be cheaper and more environmentally sustainable, as well as more suitable for large-scale energy storage with higher sustainability requirements. With this work, we further extend the possibility and knowledge of using alkali-cation containing conductive coordination polymers as alkali-ion reservoir cathode materials for electrochemical energy storage applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. W., X. L., and X. G. acknowledge China Scholarship Council for the financial support. A. V. acknowledges the European Research Council under the European Union's Horizon 2020 research and innovation program (grant agreement no. 770870, MOOiRE, ERC-CoG), as well as support from F. R. S.-FNRS through F.4552.21-P grant and WBI DG Wallonie-Bruxelles in Chili. L. G. acknowledges support from the Project Grant ANID PIA Anillo ACT192023. C. M. acknowledge UEFISCDI for financial support, grant PN-III-P4-ID-PCE-2020-0824, Contract nr PCE-22/2021.Notes and references
- D. M. D'Alessandro, Chem. Commun., 2016, 52, 8957–8971 RSC.
- H. H. Lee, J. B. Lee, Y. Park, K. H. Park, M. S. Okyay, D.-S. Shin, S. Kim, J. Park, N. Park, B.-K. An, Y. S. Jung, H.-W. Lee, K. T. Lee and S. Y. Hong, ACS Appl. Mater. Interfaces, 2018, 10, 22110–22118 CrossRef CAS PubMed.
- D. Sheberla, J. C. Bachman, J. S. Elias, C.-J. Sun, Y. Shao-Horn and M. Dincă, Nat. Mater., 2017, 16, 220–224 CrossRef CAS PubMed.
- H. Wang, Q.-L. Zhu, R. Zou and Q. Xu, Chem, 2017, 2, 52–80 CAS.
- R. Zhao, Z. Liang, R. Zou and Q. Xu, Joule, 2018, 2, 2235–2259 CrossRef CAS.
- A. E. Lakraychi, S. De Kreijger, D. Gupta, B. Elias and A. Vlad, ChemSusChem, 2020, 13, 2225–2231 CrossRef CAS PubMed.
- J. Wang, A. E. Lakraychi, X. Liu, L. Sieuw, C. Morari, P. Poizot and A. Vlad, Nat. Mater., 2021, 20, 665–673 CrossRef CAS PubMed.
- B. Esser, F. Dolhem, M. Becuwe, P. Poizot, A. Vlad and D. Brandell, J. Power Sources, 2021, 482, 228814 CrossRef CAS.
- L. Sieuw, A. Jouhara, E. Quarez, C. Auger, J. F. Gohy, P. Poizot and A. Vlad, Chem. Sci., 2019, 10, 418–426 RSC.
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- H. Banda, J.-H. Dou, T. Chen, N. J. Libretto, M. Chaudhary, G. M. Bernard, J. T. Miller, V. K. Michaelis and M. Dincă, J. Am. Chem. Soc., 2021, 143, 2285–2292 CrossRef CAS PubMed.
- D. Rambabu, A. E. Lakraychi, J. Wang, L. Sieuw, D. Gupta, P. Apostol, G. Chanteux, T. Goossens, K. Robeyns and A. Vlad, J. Am. Chem. Soc., 2021, 143, 11641–11650 CrossRef CAS PubMed.
- G. Férey, F. Millange, M. Morcrette, C. Serre, M.-L. Doublet, J.-M. Grenèche and J.-M. Tarascon, Angew. Chem., Int. Ed., 2007, 46, 3259–3263 CrossRef PubMed.
- G. de Combarieu, M. Morcrette, F. Millange, N. Guillou, J. Cabana, C. P. Grey, I. Margiolaki, G. Férey and J. M. Tarascon, Chem. Mater., 2009, 21, 1602–1611 CrossRef CAS.
- A. E. Lakraychi, F. Dolhem, A. Vlad and M. Becuwe, Adv. Energy Mater., 2021, 2101562 CrossRef CAS.
- Q. Jiang, P. Xiong, J. Liu, Z. Xie, Q. Wang, X.-Q. Yang, E. Hu, Y. Cao, J. Sun, Y. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 5273–5277 CrossRef CAS PubMed.
- B. Tian, G.-H. Ning, Q. Gao, L.-M. Tan, W. Tang, Z. Chen, C. Su and K. P. Loh, ACS Appl. Mater. Interfaces, 2016, 8, 31067–31075 CrossRef CAS PubMed.
- Z. Zhang, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2014, 136, 16112–16115 CrossRef CAS PubMed.
- Z. Zhang, H. Yoshikawa and K. Awaga, Chem. Mater., 2016, 28, 1298–1303 CrossRef CAS.
- M. E. Ziebel, C. A. Gaggioli, A. B. Turkiewicz, W. Ryu, L. Gagliardi and J. R. Long, J. Am. Chem. Soc., 2020, 142, 2653–2664 CrossRef CAS PubMed.
- X. Lou, F. Geng, B. Hu, C. Li, M. Shen and B. Hu, ACS Appl. Energy Mater., 2019, 2, 413–419 CrossRef CAS.
- Z. Peng, X. Yi, Z. Liu, J. Shang and D. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 14578–14585 CrossRef CAS PubMed.
- L. Sieuw, A. E. Lakraychi, D. Rambabu, K. Robeyns, A. Jouhara, G. Borodi, C. Morari, P. Poizot and A. Vlad, Chem. Mater., 2020, 32, 9996–10006 CrossRef CAS.
- D.-Y. Wang, R. Liu, W. Guo, G. Li and Y. Fu, Coord. Chem. Rev., 2021, 429, 213650 CrossRef CAS.
- C. Zhang, K. Fan, Y. Chen, Y. Wu and C. Wang, ACS Appl. Electron. Mater., 2021, 3, 1947–1958 CrossRef CAS.
- J. Cepeda, S. Pérez-Yáñez, G. Beobide, O. Castillo, E. Goikolea, F. Aguesse, L. Garrido, A. Luque and P. A. Wright, Chem. Mater., 2016, 28, 2519–2528 CrossRef CAS.
- E. M. Miner, S. S. Park and M. Dincă, J. Am. Chem. Soc., 2019, 141, 4422–4427 CrossRef CAS PubMed.
- S. S. Park, Y. Tulchinsky and M. Dincă, J. Am. Chem. Soc., 2017, 139, 13260–13263 CrossRef CAS PubMed.
- B. M. Wiers, M.-L. Foo, N. P. Balsara and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14522–14525 CrossRef CAS PubMed.
- Z. Xie, Z. Wu, X. An, X. Yue, J. Wang, A. Abudula and G. Guan, Energy Storage Mater., 2020, 32, 386–401 CrossRef.
- D. H. Hong, H. S. Shim, J. Ha and H. R. Moon, Bull. Korean Chem. Soc., 2021, 42, 956–969 CrossRef CAS.
- B. Xu, H. Zhang, H. Mei and D. Sun, Coord. Chem. Rev., 2020, 420, 213438 CrossRef CAS.
- Y. Chen, M. Tang, Y. Wu, X. Su, X. Li, S. Xu, S. Zhuo, J. Ma, D. Yuan, C. Wang and W. Hu, Angew. Chem., Int. Ed., 2019, 58, 14731–14739 CrossRef CAS PubMed.
- Y. Wu, Y. Chen, M. Tang, S. Zhu, C. Jiang, S. Zhuo and C. Wang, Chem. Commun., 2019, 55, 10856–10859 RSC.
- H. J. Kim, Y. Kim, J. Shim, K. H. Jung, M. S. Jung, H. Kim, J. C. Lee and K. T. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 3479–3486 CrossRef CAS PubMed.
- R. Weber, M. Genovese, A. J. Louli, S. Hames, C. Martin, I. G. Hill and J. R. Dahn, Nat. Energy, 2019, 4, 683–689 CrossRef CAS.
- X. Xue and Y. Xu, ACS Appl. Mater. Interfaces, 2019, 11, 38849–38858 CrossRef CAS PubMed.
- T. Suga, Y.-J. Pu, K. Oyaizu and H. Nishide, Bull. Chem. Soc. Jpn., 2004, 77, 2203–2204 CrossRef CAS.
- C. Wang and J. Hong, Electrochem. Solid-State Lett., 2007, 10, A65 CrossRef CAS.
- H. Tukamoto and A. R. West, J. Electrochem. Soc., 1997, 144, 3164–3168 CrossRef CAS.
- S. Yang, D. O. Schmidt, A. Khetan, F. Schrader, S. Jakobi, M. Homberger, M. Noyong, A. Paulus, H. Kungl, R.-A. Eichel, H. Pitsch and U. Simon, Materials, 2018, 11 Search PubMed.
- J. Marzec, K. Świerczek, J. Przewoźnik, J. Molenda, D. R. Simon, E. M. Kelder and J. Schoonman, Solid State Ionics, 2002, 146, 225–237 CrossRef CAS.
- H. Bamberger, U. Albold, J. Dubnická Midlíková, C.-Y. Su, N. Deibel, D. Hunger, P. P. Hallmen, P. Neugebauer, J. Beerhues, S. Demeshko, F. Meyer, B. Sarkar and J. van Slageren, Inorg. Chem., 2021, 60, 2953–2963 CrossRef CAS PubMed.
- M. Tang, S. Zhu, Z. Liu, C. Jiang, Y. Wu, H. Li, B. Wang, E. Wang, J. Ma and C. Wang, Chem, 2018, 4, 2600–2614 CAS.
- J. Wang, X. Liu, H. Jia, P. Apostol, X. Guo, F. Lucaccioni, X. Zhang, Q. Zhu, C. Morari, J.-F. Gohy and A. Vlad, ACS Energy Lett., 2022, 668–674, DOI:10.1021/acsenergylett.1c02571.
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4, 127–142 CrossRef CAS.
- A. Jouhara, N. Dupré, A.-C. Gaillot, D. Guyomard, F. Dolhem and P. Poizot, Nat. Commun., 2018, 9, 4401 CrossRef PubMed.
- L. H. Ahrens, Nature, 1952, 169, 463 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee00566b |
| This journal is © The Royal Society of Chemistry 2022 |