
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toward low-cost biological and hybrid biological/catalytic conversion of cellulosic biomass to fuels†
Lee R.
Lynd‡
 *ab,
Gregg T.
Beckham‡
*ac,
Adam M.
Guss
ad,
Lahiru N.
Jayakody
e,
Eric M.
Karp
c,
Costas
Maranas
af,
Robert L.
McCormick
g,
Daniel
Amador-Noguez
ah,
Yannick J.
Bomble
ai,
Brian H.
Davison
ad,
Charles
Foster
af,
Michael E.
Himmel
ai,
Evert K.
Holwerda
ab,
Mark S.
Laser
ab,
Chiam Yu
Ng
af,
Daniel G.
Olson
ab,
Yuriy
Román-Leshkov
aj,
Cong T.
Trinh
ak,
Gerald A.
Tuskan
ad,
Vikas
Upadhayay
af,
Derek R.
Vardon
l,
Lin
Wang
af and
Charles E.
Wyman
am
*ab,
Gregg T.
Beckham‡
*ac,
Adam M.
Guss
ad,
Lahiru N.
Jayakody
e,
Eric M.
Karp
c,
Costas
Maranas
af,
Robert L.
McCormick
g,
Daniel
Amador-Noguez
ah,
Yannick J.
Bomble
ai,
Brian H.
Davison
ad,
Charles
Foster
af,
Michael E.
Himmel
ai,
Evert K.
Holwerda
ab,
Mark S.
Laser
ab,
Chiam Yu
Ng
af,
Daniel G.
Olson
ab,
Yuriy
Román-Leshkov
aj,
Cong T.
Trinh
ak,
Gerald A.
Tuskan
ad,
Vikas
Upadhayay
af,
Derek R.
Vardon
l,
Lin
Wang
af and
Charles E.
Wyman
am
aCenter for Bioenergy Innovation, Oak Ridge National Laboratory, Oak Ridge, TN, USA. E-mail: lee.r.lynd@dartmouth.edu
bThayer School of Engineering, Dartmouth College, Hanover, NH, USA
cRenewable Resources and Enabling Sciences Center, National Renewable Energy Laboratory, Golden, CO, USA
dBiosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN, USA
eSchool of Biological Sciences, Southern Illinois University, IL, USA
fDepartment of Chemical Engineering, The Pennsylvania State University, University Park, PA, USA
gCenter for Integrated Mobility Science, National Renewable Energy Laboratory, Golden, CO, USA
hDepartment of Bacteriology, University of Wisconsin, Madison, USA
iBiosciences Center, National Renewable Energy, Golden, CO, USA
jDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA
kDepartment of Chemical and Biomolecular Engineering, University of Tennessee, Knoxville, USA
lCatalytic Carbon Transformation Center, National Renewable Energy, Golden, CO, USA
mDepartment of Chemical and Environmental Engineering, University of California Riverside, Riverside, CA, USA
First published on 9th February 2022
Abstract
Developing economically viable, scalable, and sustainable technologies for the conversion of lignocellulosic polysaccharides to liquid fuels is widely seen as a centerpiece of the global bioeconomy, and a key part of a multi-pronged approach to achieve carbon neutrality. Here we identify technology challenges and opportunities to achieve this promise. An overview of feedstocks, processes and products indicates that (1) biorefining at a scale sufficient to meaningfully impact climate change will likely involve fuels as the primary products, chemicals and biomaterials as co-products, and lignocellulose as the preferred feedstock; (2) microbial processing of cellulosic biomass will likely occur in the presence of solids, rather than involving solids-free sugar syrups, giving rise to challenges and constraints distinctive to lignocellulose; (3) anaerobic processing involves much lower costs than aerobic processing, making it more promising for fuel production; and (4) anaerobic production at high yields and broth titers has to date been reported only for molecules with ≤4 carbons. Some anaerobic bacteria are substantially more effective at polysaccharide deconstruction than aerobic fungi. Processes based on these microbes have great potential for cost reduction but require substantial research-driven advances. A mechanistic, functional group approach to product tolerance and inhibition is presented, separation technologies applicable to different product classes are surveyed, and perspectives are offered on opportunities to decrease product inhibition and the cost of product recovery. Pathways and research opportunities are considered for chemo-catalytic conversion of anaerobic fermentation products to larger fuel molecules. Fuel properties are considered for a broad range of biologically-derived products in relation to their suitability for various transport applications. Strategic perspectives are presented drawing on these diverse topics and insights. For multiple compounding reasons, features of small molecules make it less expensive to produce them biologically compared to large molecules, and this is particularly true for production from lignocellulose. Yet the fuels the world would most value producing from lignocellulosic biomass to address climate stabilization are large molecules compatible with heavy-duty, difficult-to-electrify transport applications. Hybrid processes wherein lignocellulose is converted biologically to small molecule intermediates and then converted chemo-catalytically to larger fuel molecules are a promising approach to reconciling this discrepancy.
Broader contextThe production of liquid transport fuels from lignocellulose has long been pursued via biochemical conversion, yet production at costs competitive with conventional alternatives has yet to be achieved. Here, we offer an overview of research advances in the field as well as two key points: (1) there are alternative processing paradigms that have the potential to produce low molecular weight fuels or fuel precursors at much lower cost than the conventional paradigm based on thermochemical pretreatment and added cellulase, and (2) catalytic conversion of low molecular weight intermediates, notably ethanol, to hydrocarbon fuels is a promising approach to bridge the current gap between the fuel molecules that are most readily biologically produced and the fuel molecules that the world would most value producing from biomass. |
I. Introduction
Producing transportation fuels from biomass is a substantial endeavor in the world today, and has long been a focus of research and development. Factors motivating the development and deployment of biofuels include rural economic development and employment, greenhouse gas emission reductions, improved urban air quality, enhanced energy security, and improved balance of trade. Cellulosic feedstocks have purchase costs lower than petroleum at prices seen over the last decade,1 are potentially available on larger scales than other biomass feedstocks,2,3 and have the potential to improve the sustainability of agricultural landscapes.4,5 Such feedstocks are either derived from activities undertaken for purposes other than fuel production – e.g. agricultural and forestry residues, municipal waste – or herbaceous or woody crops grown primarily for solar energy capture.6Roughly two thirds of the mass, and typically over half of the energy of cellulosic feedstocks, is composed of carbohydrate, with lignin the next most prominent component. Conversion technologies can be either biological or non-biological, with many processes containing elements of each type. This Review addresses processes that include biological conversion of the carbohydrate fraction of cellulosic biomass and includes catalytic conversion of small biomolecules into larger fuel molecules. Prior reviews have addressed thermochemical processing routes for production of fuels and chemicals,7,8 synthesis gas fermentation,9–12 and lignin valorization.13–16 As reviewed elsewhere,17,18 biological conversion of lignocellulose involves lower temperatures and pressures but longer reaction times compared to thermochemical processing. Biological processing has the potential to benefit from emergent advances in the life sciences, but is in many cases less technologically mature than thermochemical processing. Both approaches have merits, and the preferred approach will depend on particular features of the application considered. There is also considerable potential for the two approaches to be beneficially combined,19 as explored in the concluding section of this review.
To be converted to fuels by microorganisms, insoluble lignocellulose carbohydrates must be transformed into a soluble form that can be taken up by cells and then transformed intracellularly into one or more desired compounds. Research challenges and opportunities arise with respect to each of these transformations, referred to here as solubilization and fermentation. For solubilization, key research foci include understanding and increasing the effectiveness of biocatalysts that mediate solubilization, pretreatment or other methods to augment biological solubilization, and issues associated with achieving effective solubilization under industrial conditions including high solids loading. For fermentation, key research foci involve diagnosis and alleviation of limitations to achieving high product yields and titers and again doing so under industrial conditions. The rate of microbially-mediated product formation can be important in some process configurations, but in many configurations this rate is constrained by the rate of solubilization, which is usually slower than fermentation. Challenges and opportunities associated with fermentation can be and often are considered independently of solubilization; there are cogent arguments for doing so from both fundamental and applied perspectives, considered further in Section II. However, if and how solubilization is integrated is an important aspect of process and biocatalyst development that gives rise to challenges, opportunities, and key constraints.
Six precommercial pioneer cellulosic ethanol facilities have been brought online over the last decade, all based on thermochemical pretreatment, added cellulase enzymes produced by aerobic fungi in a dedicated set of unit operations, and – in all cases but one – yeast fermentation. This important development provides opportunities for validation, as well as cost reductions informed by experience. The technology embodied in pioneer cellulosic ethanol facilities is, however, still far from being cost-competitive for stand-alone fuel production on an unsubsidized cost/energy basis.1 Liquid fuels other than ethanol produced via fermentation of the carbohydrate component of cellulosic biomass have been proposed but remain at earlier stages of development. The investment in cellulosic biofuels has decreased markedly since its peak in 2011 and is now far outpaced by the investment in solar and wind energy, and cellulosic biofuel deployment over the last decade has fallen far short of expectations.20,21 Evidence that cellulosic biofuels are needed to realize a low-carbon future has strengthened over the same period,22 but the world increasingly looks to biofuels for carbon-neutral aviation and other heavy duty transport modes and to batteries and hydrogen to decarbonize light duty vehicles.23–25 Given these trends, a strategic reset and increased consideration of alternatives to conventional processing paradigms appear to be warranted.
The overall objective of this Review is to inform consideration of some promising emergent approaches for producing cellulosic biofuels. Based on the rationale presented below, we emphasize production of small molecules that can be produced biologically under anaerobic conditions at high titers in the presence of solids, either anticipating that they will be used as fuels directly or that they will be catalytically converted into targeted fuel molecules after biological processing. We focus on molecules and processes with potential to be used for bulk fuel applications, rather than for low-level fuel additives.
Section II considers distinctive features of cellulosic biomass conversion, with respect to feedstocks, products, and processing. Section III considers microbial deconstruction of cellulosic biomass. Cellularly-mediated transformations are addressed in Section IV with respect to stoichiometry, host organism choice, and product tolerance and inhibition. Product recovery is discussed in Section V, post-biological catalytic processing in Section VI, and fuel utility in Section VII. We close by offering perspectives on paths to producing cellulosic biofuels at scale in Section VIII.
II. Feedstocks, products, and processes
II.a. Feedstocks
Lignocellulosic feedstocks include herbaceous and woody plants, as well as plant residues arising from agricultural, industrial, and municipal processes. The carbohydrate component of unmodified flowering plants (angiosperms), including both herbaceous and woody species, is composed primarily of cellulose (homopolymer of β-D-cellobiose) and arabinoxylan-rich hemicellulose. Softwood (gymnosperm) hemicellulose is rich in glucomannan. For most (but not all) cellulosic feedstocks, carbohydrate represents about two thirds of the total dry weight, with lignin comprising about a quarter of the dry weight followed by smaller quantities of extractives and ash. The composition and structure of cellulosic feedstocks are reviewed in detail elsewhere.26–28Lignocellulose has evolved to be resistant to deconstruction, or ‘to be recalcitrant’,29,30 with respect to both biological and physical attack over a period of at least months and often decades or centuries. The property of lignocellulosic recalcitrance reflects its biological function as the structural support for leaves, the solar collector of the plant. Seeds, by contrast, have evolved to be easily deconstructed, consistent with their biological function of fostering the emergence of the next generation when growing conditions are favorable. As might be expected, given these different functions, technology for solubilization of the main carbohydrate components is mature and low cost for seeds, whereas such technology is costly, not yet mature, and a key economic bottleneck for lignocellulosic polysaccharides. Operationally, lignocellulose-derived carbohydrates have generally been regarded as no longer recalcitrant once they are solubilized. Moreover, among microorganisms considered for industrial application, the ability to metabolize hexoses is typically more widespread than the ability to metabolize pentoses and the ability to metabolize monomers is more widespread than the ability to metabolize soluble oligomers.
Comprehensive studies have projected that there is potential to produce over a billion tons of cellulosic biomass annually in both the United States6 and European Union.31 The “Billion Tons” study, updated in 2014, estimated that most of this potential could be accessed for $60 to $80 per delivered dry ton in the US.6 Prices near the upper end of this cost range have been projected for corn stover, including dedicated harvest and transport.32 Feedstocks produced at a processing facility can have lower costs, for example in the case of sugarcane bagasse, which is estimated to be available in Brazil at an average of $35 (range $14 to $55) per dry ton.33 Feedstocks that currently incur a disposal cost, for example municipal waste or sludge produced at a paper mill, are likely to have not only the lowest prices but also the lowest volumes.
Cellulosic biomass purchased at a representative price of $75 per oven dry metric ton costs approximately $4.4 per GJ on an energy basis, equivalent to oil at $27 per barrel,1 and about $0.115 per kg carbohydrate assuming a carbohydrate mass fraction of 0.65. By comparison, the purchase price of carbohydrate from corn at $3.75 per bushel, typical of recent years in the U.S.,34 is about $0.23 per kg carbohydrate. Dividing the carbohydrate purchase cost of corn or lignocellulose (PC, $ per kg carbohydrate) by the maximum stoichiometric product yield (YMaxP/S, kg product per kg carbohydrate), the minimum feedstock purchase cost (MFPC) can be calculated for both lignocellulose and corn in units of $ per kg product. That is:
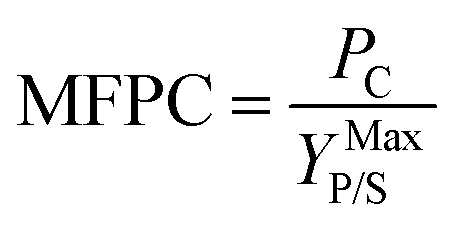 | (1) |
The MFPC represents the cost of feedstock expressed on a per unit product basis assuming conversion of the feedstock to the product at theoretical yields and exclusive of conversion. By assigning the entire cost of feedstock to carbohydrate, the analysis presented in Fig. 1 implicitly assumes that non-carbohydrate feedstock fractions have no value. If net values from such fractions were realized – e.g., oil or protein-rich residues from corn, or lignin-derived coproducts from lignocellulose – the purchase cost of feedstock would be lower than the MFPC calculated using eqn (1).
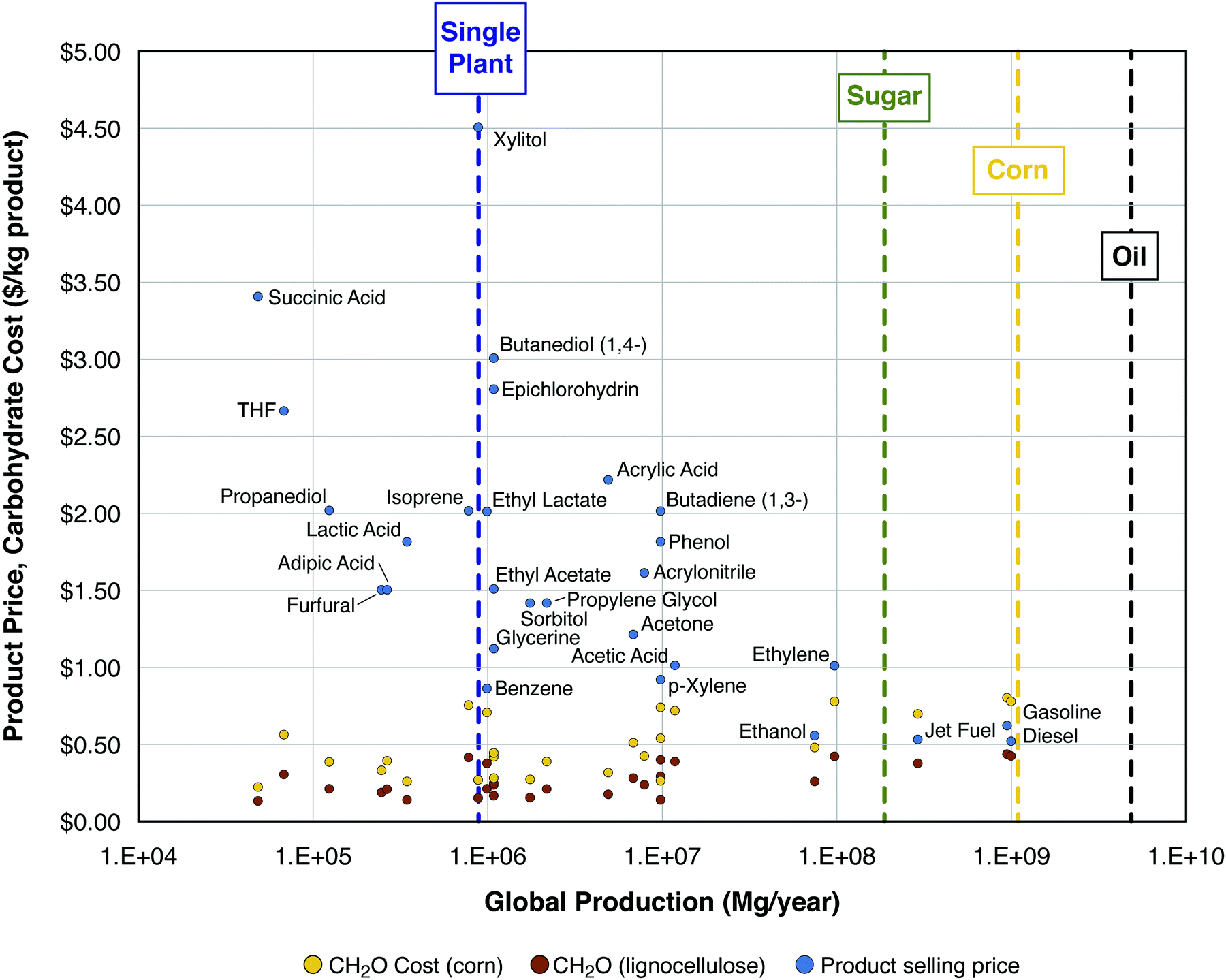 | ||
| Fig. 1 Product selling price ($ per kg, blue dots) and minimum feedstock purchase cost ($ per kg; yellow dots for corn, brown dots for cellulosic biomass, see text for definitions and assumptions). Selling price and global consumption data for all product points marked in blue come from Biddy et al. (2016), except for diesel, gasoline, jet fuel, and ethanol – data for these products come from the EIA: (1) wholesale price (https://www.eia.gov/dnav/pet/pet_pri_refoth_dcu_nus_a.htm) and (2) global consumption (https://www.eia.gov/outlooks/aeo/data/browser/#/?id=49-IEO2017&cases=Reference). Scales of production are shown (see vertical dashed lines) for a typical first-generation ethanol plant,36 global sugar production (https://apps.fas.usda.gov/psdonline/circulars/sugar.pdf), global corn production (https://apps.fas.usda.gov/psdonline/circulars/grain-corn-coarsegrains.pdf), and global petroleum production (https://www.eia.gov/beta/international/data/browser). | ||
Fig. 1 compares the selling prices and MFPCs, both in $ per kg product, as functions of annual global consumption for 25 potential bioproducts, as well as gasoline, diesel, and jet fuel. It can be observed that many chemicals sell for ≥$1.50 per kg and nearly all sell for >$1 per kg. For the 28 chemical products in the figure, the average MFPC values are $0.46 per kg product from corn and $0.25 per kg product from cellulosic biomass, with the difference between these values being $0.21 per kg chemical product. Gasoline, jet fuel, and diesel all sell for on the order of $0.5 per kg, depending on the price of oil. Because these products are more reduced than most chemicals, the calculated MFPC values are higher for production of fuels from carbohydrates. In particular, the average MPFC value for gasoline, jet fuel, and diesel is $0.74 kg−1 for production from corn and $0.40 kg−1 for production from cellulosic biomass, with a difference of $0.34 per kg fuel product. If the difference between the purchase cost of corn and that of lignocellulose carbohydrate is $0.34 kg−1 product for fuels, which sell at $0.5 kg−1 (Δ = 68% of the selling price), there is a strong incentive to overcome processing challenges associated with cellulosic biomass. On the other hand, if this difference is $0.21 kg−1 for chemicals which sell at $1.50 kg−1, corresponding to 14% of the selling price, then the incentive for overcoming the processing challenges is a great deal less.
Scales of production are also shown in Fig. 1 for a single biorefinery at the scale typical of a modern first-generation ethanol plant (processing corn or sugarcane, about 100 million gallons of ethanol per year36), global sugar production, global corn production, and global petroleum production. For roughly half of the chemical products listed, the current global production is commensurate with the scale of carbohydrate utilization of ≤2 modern ethanol plants; for all the chemical products listed except for ethylene, the current global production is commensurate with the carbohydrate utilization of ≤20 ethanol plants. By contrast, the world's over 500 ethanol plants produce <2% of the global consumption of liquid transport fuels.37 The scale of global production of sugar, most of which is currently used for purposes other than as a bioprocessing feedstock, is larger than that of chemicals, but smaller than that of fuels. The scale of global production of corn kernels is almost equal to that of fuels on a mass basis (shown in Fig. 1), but about 11% on an energy basis (not shown). Compared to the current global petroleum consumption, the potential cellulosic biomass resource is of comparable magnitude according to low-end estimates on which there is good agreement, and much larger according to high-end estimates on which there is low agreement.38
Comparative features of chemicals and fuels produced from lignocellulose are presented in Table 1. Because of the higher profit margins of chemicals compared to fuels, there is less economic motivation to produce chemicals from cellulosic biomass than there is to produce fuels, as elaborated above. The higher margins of chemicals can potentially lower the price of fuels in an integrated biorefinery,39,40 as occurs today in the case of established processes for refining petroleum and coal. Chemicals have higher profit margins than fuels, which means that the cost of processing is a smaller fraction of the selling price. Thus, chemicals are more readily economically feasible at smaller scales than fuels. Chemicals also have smaller markets than fuels, by 1 to 4 orders of magnitude (Fig. 1). Taking these factors together, a lignocellulose-to-chemical plant will be economically feasible at a smaller scale than a lignocellulose-to-fuel plant. A small-scale lignocellulose-to-chemical plant requires a large investment to achieve the economies of scale required for fuel production. By contrast, a large-scale fuel plant provides infrastructure and scale to a bolt-on chemical process at lower cost than the chemical process could realize on its own. As stated in Table 1, the incremental investment for chemicals once fuel production is established is smaller, whereas the incremental investment for fuels once chemical production is established is larger. A fuel-producing host plant is enabling for a chemical bolt-on, whereas a chemical-producing host plant is less enabling for a fuel bolt-on.
In consideration of the factors considered in this section, if biorefining is to be carried out at a scale sufficiently large to meaningfully impact fuel production and climate change, it is reasonable to anticipate that fuels will be the primary products, chemicals and biomaterials will be coproducts, and lignocellulose will be the preferred feedstock. Put differently, fuels are the biorefining product with the highest volume and lowest unit value and are most compatible with the biorefining feedstock with the highest volume and the lowest unit value, lignocellulosic biomass.
II.b. Products
Many dozens of biologically-derived molecules have been suggested in the literature as potential fuels or chemicals. Fig. 2 and 3 present data for a substantial collection of such molecules in order of increasing number of carbon atoms, and – for each number of carbons – increasing degree of reductance (available electrons per carbon).41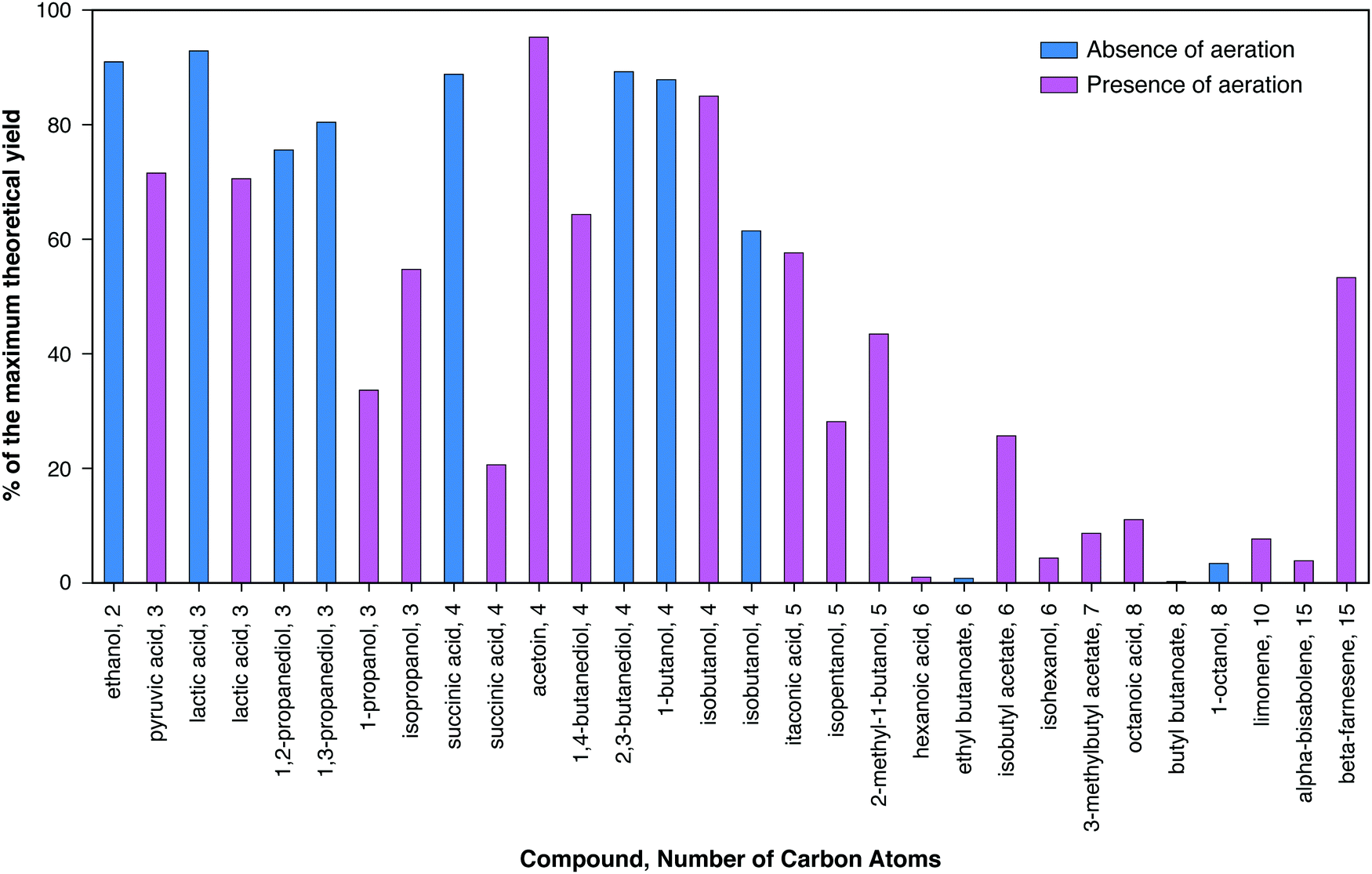 | ||
| Fig. 2 Comparison of the current best yields represented as the percentages of the maximum theoretical carbon yields obtained from the publications we surveyed. The maximum theoretical carbon yield of each molecule is calculated as the ratio of the degrees of reductance per carbon between the substrate and the product. For example, the degrees of reductance per carbon for glucose and ethanol are 4 and 6, respectively. Thus, the maximum theoretical carbon yield for ethanol equals 4/6 = 0.67. Blue and magenta bars represent experiments that were conducted in the absence and presence of aeration (including microaerophilic conditions), respectively. | ||
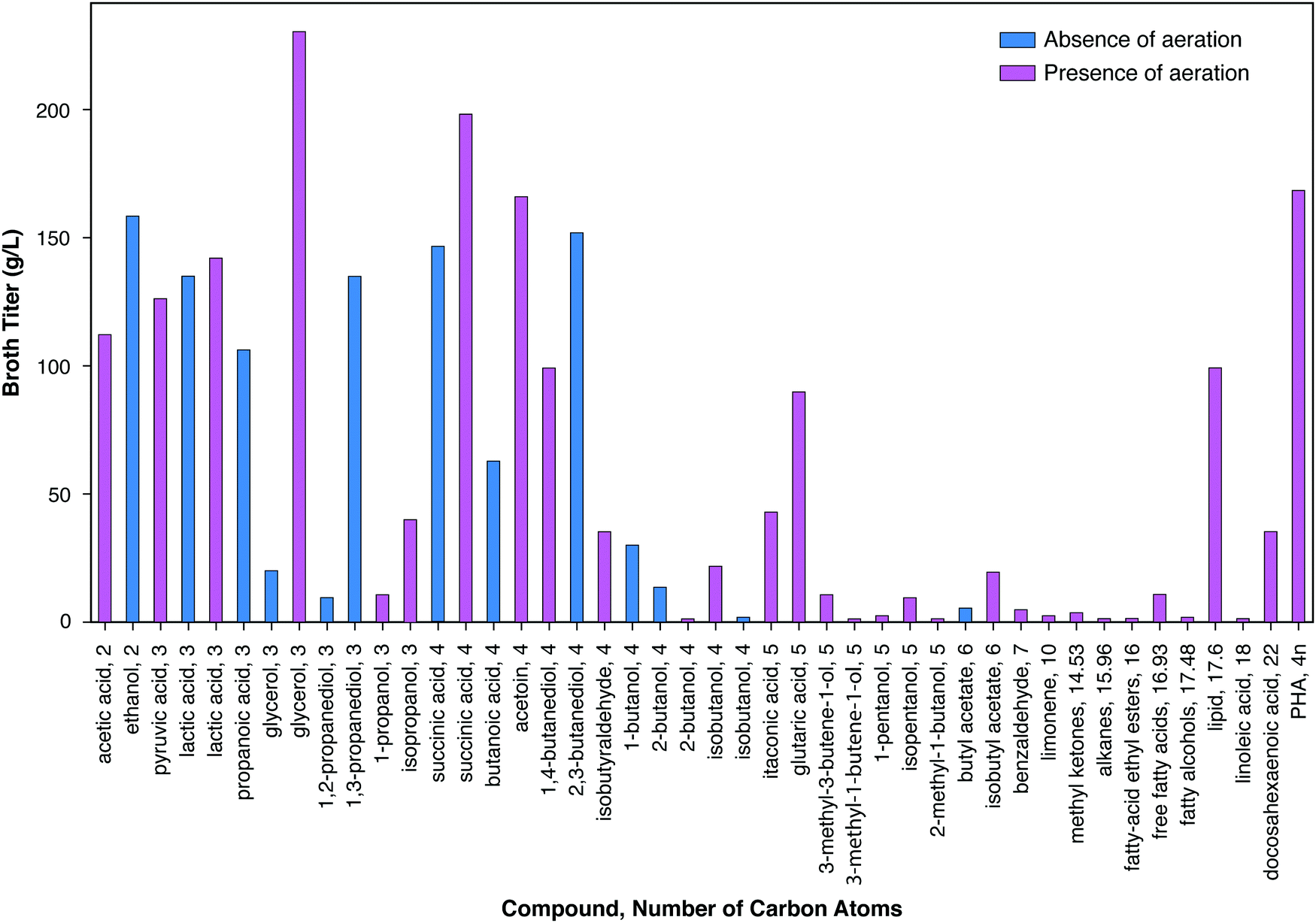 | ||
| Fig. 3 Highest experimental titer (g L−1) extracted from our literature survey for each molecule. Blue and magenta colors represent experiments carried out in the absence and presence of aeration (including microaerophilic conditions), respectively. | ||
Fig. 2 presents the maximum yields reported in the literature expressed as percents of the maximum theoretical yields based on the conservation of available electrons in the substrate, as explained in the caption. Maximum theoretical yields based on this definition are sometimes higher than values based on the maximum possible by known or envisioned pathways,42 although values for the two definitions are in most cases similar and for many cases the same. It can be observed that the highest product yields realized have been obtained in the absence of aeration, as can be seen for lactic and succinic acids. The achieved yields decrease substantially for carbon numbers above five.
Fig. 3 presents the maximum reported titer in the bioreactor in which microbial transformation occurs for a similar range of bioproducts. A pronounced trend of decreasing titer with increasing number of carbon atoms may be observed, with the notable exception of lipids (mostly triacyl glycerides) and polyhydroxybutyrate. Of the eleven compounds with maximum titers ≥100 g L−1, ten have ≤4 carbons. Of the fifteen additional products with maximum titers between 10 g L−1 and 100 g L−1, two have three carbons, six have four carbons, and three have five carbons. The reasons for this trend generally include the higher toxicity of products with higher numbers of carbons (Section IV.c), the lower solubility of such products, and the differences in the amount of R&D effort expended. Maximum titers under both aerobic and anaerobic conditions are presented for lactic acid, glycerol, and succinic acid. In all cases, higher titers have been achieved under aerobic conditions as compared to anaerobic conditions. The high titers achieved in the case of lipid and PHB production were also obtained in the presence of aeration. However, aerobic production entails substantial costs compared to products from anaerobic metabolism, as considered in the following section, and in particular Table 2. Fully anaerobic production of lipids or polyhydroxyalkanoates would be a significant advance.
| Aerobic | Anaerobic | |
|---|---|---|
| Capital | ||
| Bioreactor vessel | $52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 460771 460771 |
$5589903 |
| Bioreactor air compressor | $1631302 |
$0 |
| Bioreactor agitator | $0 | $347713 |
| Bioreactor cooler | $932368 |
$351837 |
| Seed train equipment | $2663748 |
$3511609 |
| Recirculation/transfer pump | $389057 |
$103570 |
| Total |
$58077246 |
$9904632 |
| Operating | ||
| Bioreactor system including agitator | $1916323 |
$191674 |
| Chiller system | $2909972 |
$1220396 |
| Total |
$4826295 |
$1412070 |
II.c. Processes
An overview of process options for conversion of cellulosic biomass to fuels and chemicals is presented in Fig. 4. We fully acknowledge that additional configurations may be imagined; however, those listed in Fig. 4 are representative of the range of possibilities. | ||
| Fig. 4 Processing landscape for conversion of lignocellulose to fuels. See text for explanation. | ||
After washing and initial milling, most processes involve pretreatment using some combination of added chemicals and/or heat prior to biological processing to increase the accessibility of lignocellulose to saccharolytic enzymes and thus increase the fractional solubilization of the polysaccharides. One pretreatment process configuration, Fig. 4(I.a), uses water or steam at elevated temperature with no added chemicals. Examples include steam43 and liquid hot water pretreatments.44 A second configuration, Fig. 4(I.b), involves added chemicals that are not recovered,45,46 with dilute acid hydrolysis being a prominent example. The pretreatment process configuration in Fig. 4(I.c) involves added chemicals that are recovered and reused, with examples being ammonia,47 ionic liquids,48 and organic solvents.49
Biological processing configurations II.a, II.b and II.c in Fig. 4 involve added enzymes, but differ with respect to whether solids are removed prior to fermentation (configuration II.a50–52) or after fermentation (configuration II.b53), and whether solubilization and fermentation occur sequentially (configurations II.a and II.b54–56) or simultaneously (configuration II.c57,58). Configuration II.d does not involve added enzymes and achieves hydrolysis and fermentation simultaneously.59–61 Configuration II.e integrates not only fermentation and hydrolysis, but also milling during fermentation to augment biologically-mediated solubilization in lieu of thermochemical pretreatment.62,63
The biological processing configurations listed in Fig. 4 are given in approximate order of not only increasing consolidation of process steps, but also increasing organism and process development challenges. Some of these configurations have also received a great deal more study than others.
Recovery and purification of bioproducts may be accomplished based on differences between the desired product and the fermentation medium such as volatility, charge, size, and solubility as considered in Section V. Fig. 4 distinguishes between product purification following removal of solids (III.a) and in the presence of solids (III.b).
Most of the twelve combinations of pretreatment and biological processing configurations presented in Fig. 4 have been proposed in the literature. Considering that each process configuration has many variations within it (e.g., with respect to the chemical(s) added during pretreatment, the biocatalyst(s) used for biological conversion, batch or continuous processing, and the product made), the resulting combinatoric space is very large.
Of the four biological conversion configurations listed in Fig. 4, only one (II.a) involves fermentation in the absence of solids. Use of this configuration has the advantage that it allows fermentation and product recovery to proceed without complications associated with the presence of solids. However, this configuration entails intrinsic challenges. These challenges include feedback inhibition of saccharolytic enzymes at high sugar concentrations, the difficulty of achieving high recovery of sugars from solids without excessive dilution, and the need to either do solids–liquid separation aseptically by sterilizing an extra time after solids removal or by running the entire process non-aseptically. Based on these considerations and the availability of alternatives, we would be surprised to see widespread industrial deployment of processes that deliver a concentrated, solids-free stream of lignocellulose-derived sugars to biological conversion – at least for bulk fuel production.
The physical characteristics of lignocellulose give rise to considerations and constraints that do not arise for processing of soluble substrates. Unreacted lignocellulose–water mixtures are highly hygroscopic and typically have free water up to a solids loading of about 15 wt%,64 corresponding to a carbohydrate concentration of about 10 wt%. Pumping, mixing, heat transfer, and mass transfer become progressively more challenging at solids loadings approaching this threshold.64 Strategies to ameliorate these challenges have been investigated, including partial hydrolysis in advance of introducing solids into the vessel in which fermentation occurs,65 and fed-batch operation.64,66,67 Both of these strategies take advantage of the dramatic liquefaction that accompanies biologically-mediated solubilization of lignocellulose (e.g., viscosity reduction by about 30-fold for pretreated lignocellulose35,36 and as much as 2000-fold for unpretreated lignocellulose),70 with most of this reduction occurring during the first 10% of the reaction. Even with such strategies, solids handling constraints limit product concentrations for lignocellulosic feedstocks to lower values than can be achieved with starch-rich feedstocks. Cellulosic ethanol titers ≤60 g L−1 feature in most technoeconomic analyses as well as most laboratory or pilot studies reported in the literature, with the highest titer reported to date being 86 g L−1.71 Titers of 150 g L−1 are achieved in ethanol production from corn,72 and about half of this for production from sugarcane.73
Mass transfer of any kind is more difficult in the presence of lignocellulose slurries compared to solids-free fermentation broth, including aeration, steam or gas stripping, membrane separation, and liquid–liquid extraction. For laminar flow, relevant for high-solids lignocellulose slurries, the mass transfer coefficient is proportional to the viscosity to the −0.5 power. Because the viscosity of biomass slurries at high solids is in general markedly higher than that of water,70 the mass transfer coefficients for such slurries are typically much lower than that for water.
Aeration provides an illustrative example of the impact of slurries on processing costs. Gunukula et al.74 concluded that the minimum selling price of commodity chemical production is increased by 30% for aerobic processing compared to anaerobic processing due to higher capital costs. Weusthuis et al.75 observed that new microbial conversion processes for bulk chemical production should be anaerobic, because aerobic processes have lower product yields and productivity limited by oxygen transfer. For biotransformation of lignocellulose slurries, not considered by either Gunukula et al. or Weusthuis et al., the cost advantage for anaerobic processing relative to aerobic processing will in general be even larger than that for transformation of soluble feedstocks. For example, Table 2 compares an aerobic process for lipid production from cellulosic biomass to an otherwise similar anaerobic process based on models developed by Biddy et al.40 Relative to the anaerobic process, aerobic processing entails over 9-fold higher cost for the bioreactor vessel, over 5-fold higher total capital cost, and an operating cost over 3-fold higher. Unless subsequent work finds some factor specific to lipid production that does not apply to other processes mediated by aerobic microorganisms, which is possible, but we believe unlikely, the data in Table 2 support the working hypothesis that the cost of aerobic processing of cellulosic biomass is impractically high in the context of fuel production.
The lower product concentrations and longer reaction times for lignocellulose processing as compared to processing soluble substrates have a multiplicative impact on volumetric productivity (g product per L day). As an illustration, consider the case of cellulosic ethanol, the lignocellulose-derived defined culture fermentation product with by far the largest technoeconomic analysis literature and the closest to commercialization. Publicly-available process designs for cellulosic ethanol production feature reaction times on the order of 5 days and ethanol titers on the order of 50 g L−1.76,77 Thus, the volumetric productivity is on the order of 10 g ethanol per L day. This productivity is about 7-fold lower than that achieved for batch production of ethanol from corn via the dry milling process,72 and 20-fold lower than that achieved in production of ethanol from sugarcane with cell recycle.73
Although the cost of fermentation vessels at a productivity of 10 g product per L day is not prohibitive, it rapidly becomes so at values below this value, particularly when compounded by increased separation costs and low-priced fuel products. As an illustration, Fig. 5 presents capital and operating costs for distillation and fermentation of ethanol as functions of ethanol titer. A reaction time of 5 days is assumed for production from corn stover, with costs based on the model of Humbird et al.,77 as detailed in the figure caption. Compared to production at 70 g L−1, the cumulative cost of distillation and fermentation at a titer of 40 g L−1 increases by 12 cents per gallon (32%). However, reducing the titer to 20 g L−1 increases the cost by 60 cents per gallon (2.6-fold) compared to 70 g L−1, and 48 cents per gallon (2.1-fold) compared to 40 g L−1. The two largest cost components, distillation operating costs and fermentation capital costs. Both are inversely proportional to the product concentration, as does the cumulative cost.
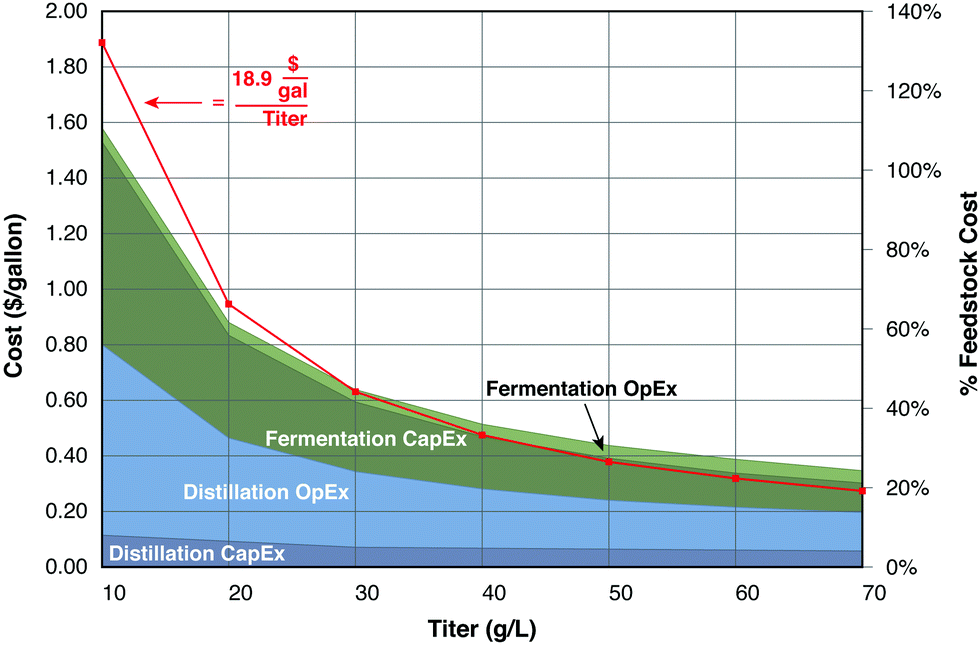 | ||
| Fig. 5 Capital (CapEx) and operating (OpEx, exclusive of feedstock) costs for cellulosic ethanol fermentation and distillation as functions of ethanol titer and percentages of feedstock cost. The red curve corresponds to a constant value (18.9 $ per gallon) divided by titer, a relationship that closely follows the sum of these costs. This illustrative scenario is based on data from the design of Humbird et al. (2011), and assumes a scale of 50 million gallons of ethanol per year, a yield of 65 gallons per dry ton feedstock, a fermentation residence time of 5 days, and a feedstock cost of $84.5 per dry ton. | ||
In the absence of continuous product removal, achieving a productivity of 10 g L−1 day at reaction times typically assumed for lignocellulose processing implies a product concentration of ≥40 g L−1. Yet there are many products for which such titers have not been achieved (Fig. 3) and are not likely to be achieved due to either toxicity or solubility constraints. Productivity and product concentration can in principle be uncoupled by continuous product removal, accomplished either in situ within the bioreactor or via a recirculated side stream as illustrated in Fig. 4 and discussed further in Section VI. Most and likely all candidate separation processes for continuous product removal are based on mass transfer and are thus more difficult to implement in the presence of lignocellulose slurries. Continuous removal of a biologically derived product from a concentrated lignocellulose slurry has not been reported to our knowledge and represents a challenging research frontier as discussed in Section VI. Without such removal, the range of products that can feasibly be produced from lignocellulose is constrained to those that can be produced at moderate to high concentrations – that is, several tens of grams per liter.
000 digesters worldwide, including over 7000 on large scales, and the global capacity doubling about every 7 years,78 biogas (methane and CO2) production is by far the largest application of biological conversion of cellulosic biomass practiced in the world today. Of the roughly six demonstration-scale cellulosic ethanol plants built around the world during the last decade, most are not in production today and none have been replicated.79 Processes for biological conversion of lignocellulose to fuels other than biogas have yet to become established on an industrial scale.
III. Microbial deconstruction of cellulosic biomass
Extensive research has been devoted over the last half-century to the enzymatically-mediated solubilization of the carbohydrate component of cellulosic biomass, including substrate features that contribute to the recalcitrance of cellulosic biomass,80,81 functional and structural categorization of carbohydrate active enzymes (http://www.cazy.org), the mechanisms of enzymatic hydrolysis,80,82–85 cellulase production,86 kinetic models,87,88 pretreatment,81,89 and genetic modification of plants to be more easily deconstructed.90,91 Caution is appropriate in making generalizations in light of the diversity of biocatalysts and their mode of action;80,92 as well as differences between plants and plant tissues.93 With this in mind, and realizing that understanding is most developed for the cellulase systems of aerobic fungi, we posit that the following observations are robust:• Catalytically-active carbohydrate-active enzymes (CAZymes) are known to exist in three primary enzyme architecture categories: mono-functional enzymes, multi-functional enzymes, and complexed (cellulosomal) enzymes.80 For model substrates, the specific activity of cellulosomal enzymes operating in vitro appears to be higher than that of unfractionated mixtures of mono-functional enzymes by about an order of magnitude,59 and to be further enhanced by a factor of 3 to 5 in the presence of metabolically-active cells.94 Comparing the specific activity of unfractionated secretomes containing CAZymes with different architectures on lignocellulosic substrates is of interest but is not possible based on available data.
• Rates of in vitro lignocellulose deconstruction have repeatedly been observed to slow markedly as the reaction progresses.59,82 For example, the rate per cellulase adsorbed to cellulose was observed by South et al. to decrease as the 5th power of the fraction of the substrate remaining.95
• Lytic polysaccharide monooxgenases, first described in 2010,96 cleave cellulose and other biomass polysaccharides in the presence of molecular oxygen or hydrogen peroxide and an electron donor and show potential to reduce enzyme loading, but require rethinking of industrial processing procedures considering that they may, in some cases, also degrade the saccharolytic enzymes.96,97
• Given the recalcitrance of plant cell wall polysaccharides and the fact that cellulose digestion in aqueous media is an interfacial process, the performance of saccharolytic enzymes is approximately 100-fold slower than starch hydrolysis.82
• Due to the solid nature of cellulosic biomass, only a small fraction (e.g., 1 in 10000) of the bonds joining sugar moieties are accessible to saccharolytic enzymes at any given time.59 This partially explains the slow rate of enzymatically-mediated plant cell solubilization. A further result is that rate saturation with respect to biocatalyst concentration as well as substrate concentration is commonly encountered, and thus Michaelis–Menten kinetics (for which rate is proportional to enzyme) are generally not valid.
• Solubilization of the carbohydrate component of plant cell walls generally only progresses to completion on industrially relevant timescales if some form of non-biological augmentation is employed. A wide range of thermochemical pretreatment processes has been proposed pursuant to this objective, and biological pretreatment has also received some attention.98,99 Although pretreatments differ with respect to their mode of action and impact on substrate properties, they have in common increasing the accessibility of feedstocks to attack by enzymes.81,84,89
Compared to the study of enzymatically mediated hydrolysis of cellulosic biomass, a smaller although still substantial body of work has been devoted to plant cell wall deconstruction mediated by microorganisms. Understanding lignocellulose solubilization as a microbial phenomenon requires consideration of several factors in addition to those associated with the action of enzymes.59,80 These factors include: (1) the biophysical and kinetic impacts of microbial cells as part of enzyme–substrate–microbe complexes, (2) the capture and transformation of solubilization products to support microbial growth, (3) bioenergetics and metabolic control of cellulase synthesis, (4) the relative capacity of different microbes to deconstruct plant cell walls and the mechanistic basis for observed differences, (5) the effectiveness of various approaches to enhance microbially-mediated solubilization, (6) the impact of high solids concentrations and other features of industrial processing environments on the physiology and activity of microorganisms, and (7) the form and function of plant cell wall-fermenting microbial consortia. These factors are key determinants of performance for biomass processing via the concept of consolidated bioprocessing (CBP), which has outstanding potential for low-cost processing, but is less fully developed than processes based on added enzymes.1
As reviewed in detail elsewhere,59 the ability to deconstruct plant cell walls is widely distributed among many genera in the domain Bacteria and in the fungal groups within the domain Eukarya. No species of the domain Archaea have been shown to grow on crystalline cellulose, although aerobic enrichments of group 8 Bathyarchaeota appear to be capable of utilizing recalcitrant organic matter.100 Within the eubacteria, there is considerable concentration of cellulolytic anaerobes in the anaerobic order Clostridiales (phylum Firmicutes). Fungal cellulose utilization is distributed across the entire kingdom, from the primitive, protist-like Chytridiomycetes to the advanced Basidiomycetes. Described anaerobic fungi are from the phylum Neocallimastigomycota, including representatives of the Orpinomyces and Neocallimastix genera among others,101 possess a large array of enzymes involved in biomass deconstruction102 but are not yet easy to cultivate.103 The discussion below considers anaerobic microorganisms able to deconstruct cellulosic biomass in light of their potential utility for CBP and the cost advantages of anaerobic processing in general (Section II.c). Acknowledging other cellulolytic anaerobes, including those yet-to-be-discovered, we focus on Clostridium thermocellum (recently renamed Acetivibio thermocellus) in light of its distinctively effectiveness at lignocellulose solubilization and because it has received the most study among cellulolytic anaerobes to date.
Adhesion of microbes to lignocellulose fibers and expression of saccharolytic enzymes on the cell surface are common among anaerobic microbes.59 Consistent with these observations, ternary enzyme–substrate–microbe complexes are thought to be the primary agents of lignocellulose solubilization in anaerobic, lignocellulose-solubilizing environments.59 The microbe appears to play an active role in such complexes. Metabolically-active C. thermocellum cultures were observed by Lu et al. to enhance the rate of cellulose solubilization by several fold compared to controls with the same amount of cellulase present.104 Bomble et al.80 observed that cellulase enzymes operating in the space between the surface of the cell and the substrate are in a distinctive biophysical environment, that the properties of this space are impacted by the cell surface, and that both the enzymes and the cell surface have likely been subject to selective pressure to maximize substrate solubilization and capture. Cellulolytic anaerobes form a monolayer biofilm on cellulose particles in studies to date.105,106 Pronounced differences in gene expression between adhered and planktonic populations of cellulose-fermenting C. thermocellum cultures have been observed, indicating that these populations exist in different metabolic states, and consistent with most of the products of biomass solubilization being consumed by adhered microorganisms without entering the bulk solution.107
Mechanistic kinetic models encompassing all known complexities do not yet exist for enzymatically mediated plant cell wall solubilization and are even less developed for the yet more complex case of microbially-mediated solubilization. Phenomenological microbial models, however, do exist and offer some useful insights. In the case of Avicel (microcrystalline cellulose) fermentation by C. thermocellum in batch culture, the solubilization rates are initially first-order in the concentration of biocatalyst, inferred based on nitrogen measurements, and are first-order in substrate thereafter. The overall kinetics are described well by a model that is first order in biocatalyst, first order in substrate, and second order overall.108 In continuous cultures involving both pure and mixed cultures and both model cellulosic substrates and lignocellulose, kinetics are described well by a model that is first order in remaining accessible carbohydrate.59,109,110 This observation is remarkable in light of the complexity of the reactions involved, and further underscores the importance of substrate accessibility as a determinant of the rate of lignocellulose solubilization.
When fermenting cellulose, available evidence indicates that C. thermocellum transports cellodextrins containing on average four glucose moieties (G4) into the cell via an adenosine binding cassette (ABC) system.111 Cellodextrins of length Gn are cleaved intracellularly to Gn−1 + glucose-1-phosphate (G1P) by cellodextrin and cellobiose phosphorylases. As a result of these events, approximately three quarters of the β-glucosidic bonds originally present in cellulose are cleaved intracellularly rather than extracellularly by a reaction that is phosphorolytic rather than hydrolytic. Glucose and G1P resulting from phosphorylase enzymes are metabolized via a non-standard Embden–Meyerhof pathway with distinctive features including a pyrophosphate-linked rather than ATP-linked phosphofructokinase, GTP- as well as ATP-linked hexokinase and phosphoglycerate kinase,112 and conversion of phosphoenolpyruvate (PEP) via a ‘‘malate shunt’’ rather than pyruvate kinase.113 Although most thoroughly described in C. thermocellum, most and perhaps all of these features appear to be shared by other cellulolytic anaerobic bacteria, including both mesophiles and thermophiles.114,115
The net result of these many deviations from canonical glycolysis is a greater yield of ATP and smaller thermodynamic driving force for C. thermocellum – and likely other cellulolytic anaerobes – compared to most anaerobes that ferment soluble substrates. In particular, fermentation of cellulose to equimolar ethanol and acetate is thought to yield about five ATP units per glucose moiety.116 This may be compared to about three ATP per glucose for Escherichia coli,117 two for Saccharomyces cerevisae,118 and one for Zymomonas mobilis.119 Consistent with this high ATP yield, glycolysis in C. thermocellum is distinctively reversible compared to several other microbes and appears to operate near thermodynamic equilibrium.120 Microbes utilizing soluble substrates can maximize either the specific substrate utilization rate (e.g., g substrate g cells−1 h−1) or the cell yield (g cells g substrate−1), the mathematical product of which is the specific growth rate, μ (h−1). For microbes growing on cellulosic biomass, however, the specific substrate consumption rate is highly constrained by low kcat values and limited substrate accessibility. It may be speculated that this situation leads to strong selective pressure for C. thermocellum to maximize the cell yield, enabled by high ATP yields.
Plant cell wall solubilization in Nature is mediated by communities of microorganisms rather than pure cultures. Individual microbial species have co-evolved in the presence of other species, and are widely thought to provide complementary metabolic functions resulting in interspecies synergy such that metabolic functions in general,121–125 and plant cell wall solubilization in particular,80,126–128 are more effective when mediated by microbial communities than by pure cultures. Taha et al.129 observed that defined cocultures are more effective at plant cell wall solubilization than their component microbes evaluated in pure cultures. Considerable effort has been devoted to characterization of lignocellulose-solubilizing microbial communities using next-generation sequencing, proteomics, and bioinformatics,130–133 and it has been posited that by understanding such communities, we can hope to develop engineered biomass solubilization biocatalysts and systems that approach the performance of those operative in Nature.134,135
Paye et al. reported controlled comparison of the effectiveness of various biocatalysts for plant cell wall deconstruction.136 Under the conditions tested, the ranked order of 5 day carbohydrate solubilization yields on mid-season switchgrass was C. thermocellum > a thermophilic horse manure enrichment > Clostridium clariflavum > Clostridium cellulolyticum > Caldicellulosiruptor bescii. Izquierdo et al. found C. clariflavum to be somewhat more effective than C. thermocellum.137 For unpretreated plant cell walls from several sources under a broad range of controlled conditions – including various feedstocks, fungal cellulase loadings and incubation temperatures, enzyme and feedstock loadings, particle sizes, and feedstock maturities – C. thermocellum cultures achieve 2- to 6-fold higher carbohydrate solubilization than commercial fungal cellulase (Ctec2/Htec2).63,116,136,137 In the case of unpretreated corn stover, hydrolysis mediated by commercial cellulase preparations with supplemental β-glucosidase, in some cases at impractically high enzyme loadings, results in 14 to 25% carbohydrate solubilization116,138,139 By contrast, C. thermocellum cultures achieve 66% solubilization of corn stover with no added enzymes and no pretreatment other than autoclaving.116 Factors underlying the effectiveness of C. thermocellum at plant cell wall deconstruction are thought to include the ability of the large C. thermocellum cellulosome to unravel the ends of plant cell wall fibers,140,141 complementary action of cellulosomal complexes and non-cellulosomal enzymes,142 and the impact of microbial cells as components of ternary enzyme–substrate–microbe complexes.59,80 The relative importance of these factors remains to be elucidated.
Considering various factors, or “recalcitrance levers”, Holwerda et al. found the relative impact on carbohydrate solubilization to be as follows: non-biological augmentation (cosolvent enhanced lignocellulose fractionation or milling during fermentation) > biocatalyst choice (C. thermocellum, fungal cellulase, or C. bescii) > feedstock choice (switchgrass or Populus), feedstock modification (three switchgrass lines chosen from a large population) > natural variants (two Populus lines chosen from a large population).63 These trends are evident from Fig. 6.
 | ||
| Fig. 6 Comparative solubilization by cultures of Clostridium thermocellum (green bars) and simultaneous saccharification and fermentation (SSF) featuring commercial cellulase (Ctec2 with supplemental β-glucosidase) in the presence of Saccharomyces cerevisiae (brown bars) for various lignocellulosic substrates. Dark blue indicates the difference of solubilization as the result of transgenic feedstock engineered for lower recalcitrance. Light blue indicates the difference in solubilization per application of ball-milling mediated cotreatment. All substrates were evaluated at 5 g L−1 glucose equivalents as duplicate bioreactor runs. More information on transgenic and natural variants feedstocks, culturing conditions and methodology can be found in Holwerda et al.63 | ||
Whereas differences in the deconstruction effectiveness of various biocatalysts and impact of recalcitrance levers are notable and potentially important, it should be realized that data for C. thermocellum and other thermophiles under industrial conditions – and particularly solids loading at the high levels likely required for industrial processes – have not yet been reported. Verbeke et al.143 found that switchgrass solubilization by C. thermocellum cultures decreases with increasing solids loading. Beyond feedstock solubilization, engineering the conversion of desired products into thermophilic, lignocellulose-fermenting anaerobes is a work in progress as reviewed elsewhere.116,144
A notable feature of plant cell wall deconstruction in Nature is augmentation of biological attack by mechanical comminution, employed by, for example, both ruminants and insects. In the case of termites, ingested lignocellulose particles are reduced to a particle size of 10 to 20 μm by the cutting action of the mandibles and mechanical grinding in the foregut gizzard, increasing the efficiency of biological attack.145 In the case of ruminants, exemplified by the cow, fiber is alternately exposed to biological attack in the rumen and mechanical comminution as the cow “chews its cud”. This results in an increase in forage particle surface area by as much as 104, at a cost of 1 to 2% of the metabolizable energy ingested.146
Inspired by these examples, together with the hypothesis that anaerobic cellulolytic bacteria such as C. thermocellum are likely to be mechanically tough because of their small size and thick cell wall, investigation of milling during fermentation as an alternative to thermochemical pretreatment has recently been initiated. Beginning with Paye et al., milling during fermentation has been referred to as “cotreatment136”. Weimer and Hall147 observed that close examination of the rumination process and the animal behavior that underlies it suggests design configurations that could be incorporated into a bioreactor to achieve an effective co-treatment that could obviate the need for more expensive and environmentally unfriendly chemical pretreatments. Milling as a stand-alone pretreatment prior to biological conversion is generally thought to be impractical due to excessively high energy requirements.148–151 However, the profound physical changes that accompany biologically-mediated plant cell wall polysaccharide deconstruction may enable much-reduced energy requirements for milling at an intensity sufficient to enhance solubilization.68–70 In experiments involving fermentation without milling and milling without fermentation followed by a second fermentation, Paye et al.136 found that ball milling for five minutes nearly doubled solubilization of senescent switchgrass by C. thermocellum compared to two fermentations without milling. Balch et al.62 found a similar enhancement of senescent switchgrass solubilization for C. thermocellum fermentation with continuous ball milling as compared to an unmilled control. Microbes capable of vigorous growth under anaerobic conditions, including E. coli and T. saccharolyticum, as well as C. thermocellum, were found to readily withstand ball milling at an intensity sufficient to allow near-complete carbohydrate solubilization. This was not the case for bacteria with less vigorous anaerobic growth, such as Z. mobilis or B. subtilis, or with yeast.152 It is interesting to note that the carbohydrate solubilization achieved by C. thermocellum on Populus in the presence of ball milling is comparable to that achieved by termites.145
The realization of near total carbohydrate solubilization via microbial fermentation in the presence of aggressive milling/grinding suggests that physical inaccessibility is a more important factor limiting microbially-mediated lignocellulose deconstruction than recalcitrant chemical linkages. This observation also indicates that the presence of lignin does not prevent high carbohydrate solubilization by microbial fermentation, although lignin is known to inhibit cellulase enzymes.153 Lignin is not significantly modified by C. thermocellum fermentation in the presence of cotreatment, which may enhance its value for subsequent conversion to value-added products compared to the modified lignin resulting from processes featuring thermochemical pretreatment.62 We know of no convincing evidence that the aromatic backbone of lignin undergoes significant reaction under anaerobic conditions as envisioned for consolidated bioprocessing.
The potential impact of cotreatment combined with consolidated bioprocessing was explored for corn stover conversion to ethanol in a paper based on a process design and equipment cost database developed at the National Renewable Energy Laboratory (NREL1). In this analysis, a Base Case scenario featuring dilute acid pretreatment and added fungal cellulase defined by NREL is compared to an Advanced Scenario featuring consolidated bioprocessing with cotreatment (C-CBP) with projected R&D-driven advances and coproduction of fuel pellets. Fig. 7 contrasts the conventional lignocellulosic fuel production process, featuring thermochemical pretreatment and added enzymes, to C-CBP. The authors acknowledged, as do we, that the technology in the Base Case is more mature than C-CBP, which is more speculative and more uncertain. Substantial further research advances will be required, proof of concept will have to be demonstrated, and current performance limitations will have to be overcome before C-CBP can be implemented industrially.
 | ||
| Fig. 7 Flow sheets comparing the Base Case (A) and Advanced Scenario (B) from Lynd et al. (2017). “DSP” denotes downstream processing and separation. Carbohydrate-active enzymes are produced aerobically in a dedicated unit operation for (A), but are produced during fermentation by an anaerobic, biofuel-producing culture for (B). | ||
Compared to the Base Case, the Advanced Scenario with C-CBP was found to have an 8-fold shorter payback period (25 versus 3 years) and economic feasibility at a 10-fold smaller scale. Life cycle analysis indicated that the greenhouse gas mitigation per ton biomass was 25% greater for the Advanced Scenario compared to the Base Case scenario. The performance parameters for the Advanced Scenario include a solids loading of 20 wt%, an ethanol titer of 59 g L−1, a volumetric productivity of 10 g ethanol per L per day, and an ethanol yield of 330 L per dry metric ton corn stover. All these parameter values are comparable to those of the Base Case, although they exceed what have been obtained thus far for the conversion process based on thermophilic CBP and cotreatment assumed in the Advanced Scenario. Rather than improved titer, productivity and yield values, the projected cost savings in the Advanced Scenario arise primarily from configurational changes, and in particular from eliminating added enzymes and increasing substrate accessibility via cotreatment in lieu of thermochemical pretreatment.
Status. Cellulase enzymes are produced at an industrial scale with applications in the pulp and paper, textile, and food processing industries. Cellulase preparations produced by aerobic fungi for use in lignocellulose conversion to fuels have been an intensive focus of both academic and industrial research for several decades. Comparative studies over the last few years indicate that cellulolytic anaerobic bacteria have superior lignocellulose deconstruction capability compared to aerobic fungi under controlled – but to date not industrial – conditions. Progress has been made in the development and application of genetic tools for engineering cellulose- and hemicellulose-fermenting cellulolytic anaerobes potentially capable of one-step consolidated bioprocessing of cellulosic biomass without added enzymes. However, commercial implementation will require considerable further understanding and development. Milling during fermentation, cotreatment, is a potential alternative to thermochemical pretreatment. Although some key features have been demonstrated, cotreatment is still a nascent technology and has yet to be widely studied.
Challenges and opportunities
The realization that Nature offers biocatalysts with biomass deconstruction capability superior to commercial cellulases produced from aerobic fungi has profound implications for the development of biofuel production processes. Anaerobic production of cellulase enzymes as reagents to be added to cultures of non-cellulolytic microbes appears unlikely to be practical because of the high ATP demand for protein synthesis together with the low ATP supply of non-oxidative anaerobic metabolism. Transfer of the complex cellulolytic machinery of thermophilic anaerobes such as C. thermocellum to proven industrial microorganisms such as S. cerevisiae and E. coli is interesting for the purpose of gaining fundamental insights. However, full functional replication of native cellulase and cellulosome systems via heterologous expression is considerably more complex than any biotechnological achievement thus far, and in our view is not promising in the context of industrial process development and deployment for the foreseeable future. Most promising in our estimation is the development of industrially-desired traits in non-model cellulolytic microbes that embody Nature's best biomass deconstruction capability. Such development is a grand challenge in the field of cellulosic biofuels, and is as yet far from realized. Success in this endeavor will require improving product-related properties such as titers and yields as well as industrial robustness, drawing from both biotechnological and process development tools.IV. Cellularly-mediated transformations
Stoichiometry, host organism choice, and product tolerance and inhibition are considered in this section. Biochemical pathways for cellularly-mediated transformations are comprehensively reviewed elsewhere.154–156IV.a. Stoichiometry
Cost-effective processing will in general be facilitated by maximizing the product value per unit raw material, and this is particularly important for commercial production of fuels and commodity chemicals for which the cost of raw material is generally a substantial contributor to selling price. For fuels, which are sold based on their energy content, and in the future potentially on the basis of displaced greenhouse gas emissions from fossil fuel combustion, high energy yields (product energy per unit energy in the feedstock component converted) are the main concern. For chemicals, which are sold on a mass basis, high mass yields (product mass per unit mass in the feedstock component converted) are the priority. As presented in Table 3 the maximum stoichiometrically possible yield on an energy basis is similar and over 90% for most products derived by non-oxidative pathways, and thus not a great differentiator for fuel applications. By contrast, the maximum possible yield on a mass basis varies by more than 4-fold across the spectrum of products considered, and is inversely related to the degree of reductance and energy density. As shown in the right-most column, the enthalpy per available electron is very nearly constant for most biologically derived products. Compounds with degrees of reductance ≤4 do not concentrate energy relative to carbohydrates and are generally of interest for chemical rather than fuel applications.| Compound | Formula | Degree of reductance (available electrons/C)a | Maximum yield | Enthalpy of combustion (higher heating value) | ||
|---|---|---|---|---|---|---|
| (Energy basis) | (Mass basis) | (MJ kg−1) | (MJ per available e−) | |||
| a Higher heating values (HHVs) for all compounds except for cellulose were calculated using standard enthalpy of formation values obtained from NIST Standard Reference Database 103b; https://www.nist.gov/mml/acmd/trc/thermodata-engine/srd-nist-tde-103b. The cellulose HHV value was obtained from ref. 157. | ||||||
| Methane | CH4 | 8 | 0.948 | 0.266 | 55.5 | 0.110 |
| Ethanol | C2H6O | 6 | 0.948 | 0.512 | 28.9 | 0.111 |
| Butanol | C4H10O | 6 | 0.950 | 0.411 | 36.0 | 0.111 |
| Farnesene | C15H24 | 5.6 | 0.972 | 0.325 | 46.8 | 0.114 |
| Ethyl butyrate | C6H12O2 | 5.33 | 0.946 | 0.484 | 30.5 | 0.111 |
| Lactic acid | C3H6O3 | 4 | 0.973 | 1.0 | 15.2 | 0.114 |
| Succinic acid | C4H6O4 | 3.5 | 0.911 | 1.12 | 12.7 | 0.107 |
| Cellulose | (C6H10O5)n | 4 | — | — | 17.0 | |
IV.b Host organism choice
The choice of production host is often critical to the success of bioprocess development efforts. The important host properties include (1) the ability to utilize the feedstock of interest, (2) the extent to which a well-characterized product formation pathway exists, (3) the ability to withstand high product titers (considered in detail below), (4) knowledge of metabolic features, (5) the availability of genetic tools, (6) the relative ease of obtaining regulatory approval, and (7) a collection of features often referred to as “robustness”. Generally defined as the ability to function well under industrial conditions, the elements of robustness include good performance on a low-cost fermentation medium, competing successfully against contaminants, tolerance to inhibitors, and vigorous fermentation performance if a substrate is available.Prior to the advent of genetic engineering, microbial hosts were chosen based on an organism's native ability to produce a desired product from a desired feedstock. Prominent examples include ethanol production by S. cerevisiae, citric acid production by Aspergillus niger, acetone/butanol/ethanol (ABE) production by Clostridium acetobutylicum, glutamate production by Corynebacterium glutamicum, and lactic acid production by Lactobacillus strains and other lactic acid bacteria (Table 4). The tools were lacking to make targeted changes to an organism's genome, but random mutagenesis (e.g., UV irradiation, chemical mutagenesis) combined with screening or selection allowed for identification of strains with enhanced performance under desired conditions.
| Product or substrate | Host organism | Major deciding factors | |
|---|---|---|---|
| Prior to genetic engineering | Ethanol | Saccharomyces cerevisiae | Native pathway, robustness, high titer, rate and yield |
| Citric acid | Aspergillus niger 174 | Native high yield from cheap feedstocks, tolerance to low pH | |
| Glutamate | Corynebacterium glutamicum | Natively high yield | |
| Lactic acid | Lactobacillus sp. | Native producers, GRAS status | |
| Acetone–butanol–ethanol (ABE) | Clostridium acetobutylicum, Clostridium beijerinckii | Native producers, high titer | |
| Genetic engineering applied to initial model microbes | 1,3-Propanediol | Escherichia coli | Available genetic tools and understanding of physiology |
| 1,4-Butanoediol | Escherichia coli | Available genetic tools and understanding of physiology | |
| Isobutanol | Escherichia coli, S. cerevisiae | Available genetic tools and understanding of physiology | |
| Farnesene | S. cerevisiae | Available genetic tools and understanding of physiology | |
| Succinate | Escherichia coli, S. cerevisiae | Available genetic tools and understanding of physiology | |
| Commercial processes enabled by engineering a broadened range of hosts | Lysine | Corynebacterium glutamicum | Native producer of amino acids |
| Lactic acid | Low pH yeasts such as Kluyveromyces marxianus and Issachenkia orientalis | Acid tolerance | |
| n-Butanol | Clostridium acetobutylicum | Native producer of n-butanol | |
| Syngas conversion | Clostridium autoethanogenum | Native syngas utilizer | |
| Emergent processes involving yet more host diversity | Lignocellulose conversion via CBP | C. thermocellum | Native ability to rapidly deconstruct lignocellulose |
| Methane conversion | Methylomicrobium buryatense | Native methane utilizer | |
| Formate | Cupriavidus necator | Native formate utilizer | |
Enabled by the molecular biology revolution, rational metabolic engineering became possible in a handful of model microorganisms, especially S. cerevisiae and E. coli. The most prominent commercial success story from this era is 1,3-propanediol (PDO) production by DuPont,158 where E. coli was engineered to produce PDO from sugars at a substantially lower cost than was possible using petroleum. Other notable examples include production of 1,4-butanediol,159 isobutanol,160,161 farnesene,162 and succinic acid.163 Indeed, the vast majority of metabolic engineering and synthetic biology research continues to be performed in model organisms such as E. coli and S. cerevisiae due to the availability of well-developed genetic tools and the deep fundamental understanding of the physiology of these model organisms. However, engineering complex physiological traits, including the use of lignocellulosic biomass as a feedstock, remains a challenge for model organisms.
The scope of industrial biotechnology was further expanded as researchers realized that some industrially important phenotypes are too complex to engineer into model organisms. Therefore, non-model microbes offer advantages over traditional hosts, and accessing these phenotypes is often worth the effort needed to develop genetic tools and physiological understanding. Several of these organisms were prominent in period one, including Z. mobilis, C. acetobutylicum and C. glutamicum. This effort led to a much deeper understanding of non-model host organisms, and examples of successful engineering include production of lysine and other amino acids in C. glutamicum164 and isopropanol/butanol/ethanol (IBE) production by C. acetobutylicum.165 The development of Clostridium autoethanogenum,166 an acetogen that is natively capable of growing on CO and H2 in syngas, has opened new processing paradigms for bioconversion of various feedstocks. Recently, acid-tolerant yeasts such as Kluyveromyces marxianus and Issatchenkia orientalis have been developed for production of organic acids such as lactic acid.167 For CBP, the most effort has focused on C. thermocellum because it has one of the fastest known growth rates on crystalline cellulose, and its cellulosome allows for efficient deconstruction of lignocellulose.142,168 The development of genetic tools for this organism has enabled the development of strains with ethanol yields exceeding 75% of the theoretical maximum and titers over 25 g L−1 from crystalline cellulose.159 Thermostable heterologous pathways can be now engineered in this organism to produce a broad range of fuel molecules such as isobutanol,169n-butanol170 and a wide range of ester molecules including isobutyl acetate, n-butyl acetate, n-pentyl acetate, isoamyl acetate, geranyl acetate, and isobutyl isobutyrate.171–173
Table 4 lists some representative commodity products, the host organisms used for commercial production, and deciding factors underlying the choice of host organism.
Recent advances in genetic tool development for non-model microbes have simplified and accelerated the development of transformation for new organisms,175 which is starting to allow researchers to choose host organisms based on desired traits rather than initial genetic tractability. In particular, Restriction–Modification systems are prokaryotic host defense systems that degrade foreign DNA that is methylated differently than the host.176–178 Methylome analysis reveals the sites that are targeted for degradation, allowing for rational approaches to developing genetic transformation systems.175 This, combined with the development of modular collections of genetic parts (e.g., selectable markers, origins of replication),179 allows rapid evaluation of genetic constructs in target hosts. For microbes that are already genetically tractable, more advanced tools such as recombineering180–183 and site-specific recombination are being developed,184 which will further accelerate strain engineering in these organisms.
The understanding of cellular physiology needed to rationally engineer non-model organisms can also be rapidly developed via systems biology. Genomics,185 transcriptomics,186 proteomics,187 and metabolomics188 can all be readily applied to desired hosts, revealing details of central and secondary metabolism, gene regulation, and metabolic flux that are needed for engineering efforts. Metabolic modeling is also rapidly advancing for non-model microbes,189 which can serve as a platform for generating hypotheses to test experimentally.
The ability to deconstruct lignocellulose at high rates and yields is a phenotype that has proven to be difficult to import into model microbes.116 Because of this, cellulolytic anaerobes with the ability to rapidly ferment cellulosic biomass are under active consideration for processing cellulosic biomass. When weighing the use of model or non-model host microbes for industrial fermentation of lignocellulose, one must evaluate the relative difficulty of adding the desired features that each class of microbes does not have, including strong lignocellulose fermentation capability in the case of model hosts, and high product yields, titers, and robustness in the case of non-model hosts. Experience and recent opinion appear to favor non-model hosts for this application, but time will tell.
Early determination of key complex traits for a given process is essential for choosing the best host. Particular focus on hosts that enable robust feedstock utilization and integration with downstream processing will likely be critical to developing an economical process.
Once a host is selected, substantial early effort on genetic tool development can pay large dividends by accelerating strain engineering. Basic tools and information such as genetic transformation, gene insertion/deletion methods, characterized promoters and ribosome binding sites for tuning gene expression, and -omics datasets need to be rapidly developed and serve as the foundation for rational metabolic engineering.
As new genetic tools are developed, such as the current expanding application of CRISPR-Cas genome editing to diverse microorganisms, they have the potential to dramatically increase the rate and efficiency of genetic engineering. However, adapting these tools to new organisms can be challenging and time consuming, often forcing a choice between proceeding with strain engineering using the tools that exist and waiting until more advanced tools are available.
Thermophiles and other extremophiles offer potential complex phenotypes that could open new bioprocessing opportunities, but extra effort is often required for genetic tool development. As more synthetic biology “parts” are developed for these organisms, the rate of tool development will likely increase, making these organisms more attractive for bioengineering.
IV.c. Product tolerance and inhibition
Mechanisms by which biologically-produced fuels and fuel precursors can inhibit cellular function include impaired function of biological macromolecules (DNA, RNA protein, and lipids), loss of cell structural integrity (membranes), and disrupted metabolic circuits (enzyme inhibition, cofactor leakage, uncoupling or creating redox cofactor imbalance, and futile ATP or reducing equivalent consumption). The extent and mechanism of inhibition observed are commonly associated with the functional group(s), the inherent reactivity, and the molecular weight of the inhibitor.190–193 Here we review the salient structure–activity relationships of potentially toxic intermediates and target products using a functional group classification, which has been widely employed to assess the toxicity of inhibitors found in biomass-derived intermediate streams.194–196Several physicochemical parameters have been identified that influence microbial toxicity. These include the following:
(i) The electrophilicity index (ω)197 – many inhibitors inactivate biological molecules in the cell through direct addition reactions, and the electrophilicity index provides an indication of the potential reactivity to undergo an adduct reaction with a biomolecule of importance for cellular function.192,198
(ii) The steric hindrance of the reactive group – when the reactive group of an inhibitor is sterically hindered, this will naturally reduce the propensity to target a biomolecule within the cell.190 Accordingly, steric factors are another critical physicochemical parameter of toxicity.
(iii) The octanol–water partition coefficient (logP) – membrane damage is another common component of microbial toxicity, and logP is a measure of relative hydrophobicity and thus the potential of an inhibitor for membrane partitioning. Molecules that exhibit logP values between 1 and 5 are often found to be toxic to microbes.199,200 Although nuances can lead to deviations from ideal behavior, the more similar a molecule is to a membrane the more it impairs the membrane, and hence biological, function.201
Based on these parameters, we can generally rank the toxicity threshold of identical chain length fermentation-derived intermediates and products, as shown in Fig. 8: aldehydes > ketones > alcohols > esters > organic acids > alkanes. Given the diversity and complexity of microbial micro-environments, metabolic circuits, and cell physiology, it should be noted that the chemical structural indicators of toxicity are not always perfectly correlated with the actual outcome of biological toxicity measurements, but provide useful guidelines on their potential toxicity. Moreover, different functional groups pose distinct molecular toxicity mechanisms on host microbes, and as mentioned, the “R-group” attached to a functional group also can contribute substantially to the toxicity of a particular compound. To that end, below we briefly review toxicity mechanisms for each functional group separately and provide salient examples from common anaerobic fermentation intermediates and target products.
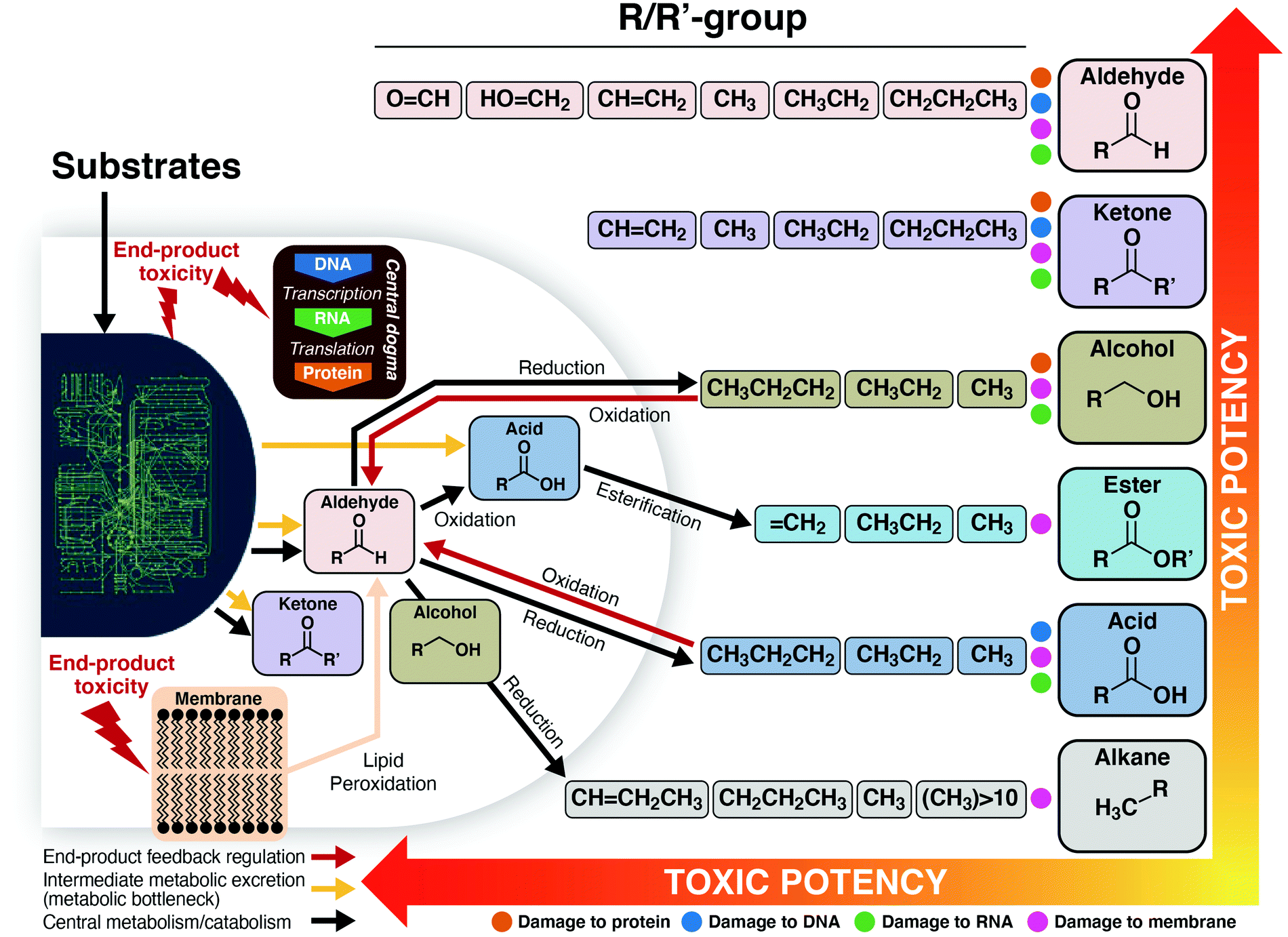 | ||
| Fig. 8 Illustration of the functional group-dependent toxic potency of the products on microbial hosts. Substrates, intermediates, and products may damage biological macromolecules and disrupt metabolism through multiple mechanisms. During the conversion of substrates to products, toxic intermediates such as aldehydes, acids, and ketones are often produced. In addition, some target products may induce feedback regulation and increase the accumulation of toxic intermediates in the cells. The toxic potency was ranked for the compounds at the same concentration (soluble form), carbon-chain length, and pH. | ||
Protein damage is a critical component of aldehyde toxicity, often through adduct reactions between aldehydes and proteins.191,192 The extent of damage is closely related to the electrophilicity (ω) and chemical structure of the aldehyde (sterics).204 “Hard” aldehydes (which are highly charged, small, and weakly polarizable aldehydes, e.g., acetaldehyde and formaldehyde) target hard-neutrophilic lysine residues on proteins to form carboxymethyl lysine, leading to deleterious crosslinking events.204 Alternatively, “soft” α,β-unsaturated aldehydes (which are weakly charged, larger, and more polarizable than hard aldehydes, e.g., acrolein) target hard-neutrophilic lysine residues and the β-carbon of the aldehyde then reacts with soft-neutrophilic cysteine residues through Michael addition, again leading to cross-linking events.204 However, the most toxic aldehydes are oxoaldehydes (e.g., glycolaldehyde), which exhibit a distinct mechanism to crosslink proteins by targeting hard-neutrophilic lysine residues and/or soft-neutrophilic cysteine residues via the formation of Schiff-base and concurrent Amadori rearrangement. This reaction in turn leads to regeneration of a hard-aldehyde carbonyl group after the first attack on a protein, which can then efficiently form a second covalent bond with a different protein.203,205
Beyond attacking proteins, short aliphatic aldehydes that exhibit low steric hindrance also damage DNA by inducing DNA single-strand breaks and DNA–protein crosslinking.206,207 Aldehydes can also react with lipids in multiple ways, such as via the amino residues of membrane lipids (e.g., with phosphatidylethanolamine) to form glycation end products or through glycoxidation of lipids.208 The aldehyde can also generate reactive oxygen species, and cause cell damage. Taken together, the toxicity of aldehydes is highly correlated with the reactivity properties of the “R-group”. For small aldehydes likely to be formed in anaerobic fermentation processes to produce C2–C6 fuels or fuel precursors, the toxicity trend of sub-groups of aldehydes could be described as follows: α-oxoaldehydes (e.g., glycolaldehyde) > soft α,β-unsaturated non-hindered aldehydes (e.g., acrolein) > hard, non-hindered aldehydes (e.g., acetaldehyde) > hard aldehydes with short or long-chain alkanals (e.g., pentaldehyde).
P value.201,214 Thus, the logP value is an effective indicator of alcohol toxicity; for example, the order of toxicity of selected short-chain alcohols is as follows: n-hexanol > n-pentanol ≥ n-butanol > n-propanol > ethanol > methanol.
Haft and coworkers showed that membrane damage only explains one component of alcohol-mediated damage.215 In addition, it was revealed that ethanol toxicity is related to an uncoupling of transcription and translation in E. coli through ribosomal stalling, intragenic transcriptional termination, and inhibition of an RNA polymerase.215 In addition, it has been reported that short-chain alcohols are able to damage proteins directly, thus inducing unfolded protein response in bacteria,200,216,217 and these findings have enabled new metabolic engineering strategies for strain development towards improved alcohol toxicity.
P), pKa, and chain length.225 Toxicodynamic studies have demonstrated that increasing molecular weight generally leads to decreases in the toxicity of organic acids on bacteria, but this observation is microbe specific.226 Going forward, a deeper understanding of the mechanisms of anion-specific toxicity is vital to further develop anaerobic acid-producing strains. As with aldehydes and ketones, in many cases, the toxicity of organic acids is highly associated with the molecular properties of the “R-group”.
P value and solubility of the compound.201 Notably, alkenes are more toxic than alkanes with similar molecular lengths.227 Also, the toxicity of alkanes or alkenes on microbes is often highly correlated with intermediate toxic compound accumulation. For instance, in isobutene production, the intermediates isobutyraldehyde and isobutanol are hypothesized to cause toxicity response.228,229
P, and decreasing solubility. For the same carbon numbers and metabolite concentrations, branched chain metabolites tend to be less toxic than linear chain ones.221 Up to the point that solubility becomes limiting, both theoretical considerations and empirical data indicate that inhibition of microbial metabolism increases with increasing hydrophobicity and hence chain length. This has been shown for organic acids, alcohols, solvents, esters, and cyclic hydrocarbons, and is also consistent with the data shown in Fig. 3.221,230–233 Given that membrane partitioning is one of the vital mechanisms of inhibition by alkanes, alcohols, ethers, and esters, the toxic potency is positively correlated with the chain length of these functional groups. However, increasing the chain length enhances steric hindrance and impairs the electrophilicity of aldehydes and ketones, and reduces the toxic potency to attack neutrophilic macromolecules.192 Hence, the toxicity trends with respect to chain length depend on the functional group of the product. With long chain lengths and high hydrophobicity, the solubility becomes very low and can indeed be lower than the toxic threshold.
Product insolubility can provide spontaneous separation from an aqueous milieu, e.g., into an organic phase in extractive fermentation, and thus may be regarded as an advantageous property. Indeed, this detoxification strategy has been successfully exploited for microbial biosynthesis of shorter chain esters at high levels exceeding their solubility limit and inhibition threshold.171 On the other hand, continuous product removal appears to be challenging to implement in the presence of solid cellulosic feedstocks (see Section II.c).
V. Product recovery
In this section, we review product recovery for prominent classes of organic compounds, provide strategic perspectives on product recovery for bulk fuel applications, and conclude with consideration of in situ product recovery. It may be noted that much of the literature and experience associated with purifying commodity bioproducts is for chemicals rather than fuels, and that nearly all such literature and experience is for feedstocks other than lignocellulose.Separation of ethanol
In the corn ethanol industry, fermentation broth containing about 16 wt% ethanol,72 residual solids, and yeast is typically fed to a beer column with internals designed to function in the presence of solids. Rectification to the 95 wt% azeotrope is achieved in a second column, and the azeotrope is then broken with molecular sieves, which are a regenerable desiccant, or through the use of additional pressure swing columns.253,254Process designs for cellulosic ethanol also feature distillation in the presence of solid fermentation residues.76,77 However, solids handling constraints limit ethanol titers from lignocellulose to about half of those realized industrially from starch (Section II.c). In the absence of heat integration, estimates for the heat required to produce pure ethanol from fermentation broth at 6 wt% ethanol via distillation and molecular sieve dehydration are in the range of 6.5 to 9.4 MJ kg−1, corresponding to 20 to 30% of the ethanol low heating value.254,255 However, since the economics of ethanol recovery are dominated by the cost of energy (steam) rather than the cost of capital (Fig. 5), there is an economic incentive to include heat integration. For example, NREL cellulosic ethanol designs reuse heat available from high temperature pretreatment for distillation at lower temperatures.77 Reducing the energy requirements of distillation can also be approached by multi-effect (multiple pressure) and vapor compression strategies. For example, Diaz and Tost256 estimated a fuel requirement of 2.5 MJ kg−1 for a recovery of 10 wt% ethanol via double-effect distillation and dehydration (about 9% of the low heating value). At a titer of 6 wt% ethanol, more realistic for lignocellulosic feedstocks, a likely conservative extrapolation based on an inverse relationship between distillation energy requirements and titers (Fig. 5) yields a heat requirement of about 4.2 MJ kg−1 (about 15% of the low heating value). Yet lower distillation energy requirements are possible by combining strategies if there were economic incentives to do so. Combustion of solid, lignin-rich process residues is sufficient to provide all heat and electricity requirements for cellulosic ethanol production from both corn stover77 and sugarcane bagasse.76 Considering a scenario where lignin is not burned and electricity is not cogenerated, Pourhashem et al.257 found that biogas produced from organics present in liquid effluents is sufficient to meet the process heat demand.
Separation of mixed alcohols and ketones
Like ethanol, acetone–butanol–ethanol (ABE) fermentation is a well-established fermentation process, with the recovery of ABE also centering on distillation. In the ABE process, the classical design is batch fermentation with ABE titers reaching approximately 2 to 3 wt% of the broth. The unfiltered broth is fed to a beer column where ABE is separated through steam stripping. The overhead of the column contains the quaternary azeotrope of water–acetone–butanol–ethanol, which is ∼30 wt% ABE and 70 wt% water. Acetone, butanol, and ethanol are then separated as individual components through a series of four distillation columns operated at below atmospheric pressures. A process flow diagram of the operation can be found in Fig. 1 of Roffler et al. (1987).258 Minimum energy requirements for industrial production of ABE were reported by Diaz and Tost259 to be about 30 MJ kg−1 solvents, which is close to the low heating value of the solvents. However, these authors projected heat requirements of about 6 MJ kg−1 for ABE recovery via distillation with heat integration.Separation of higher boiling alcohols
Examples of higher boiling alcohols produced biologically at an industrial scale include butanol260/isobutanol,261 and the short chain diols propanediol and butanediol.262 In general, the standard industrial practice for the post-fermentation recovery of these higher boiling alcohols involves distillation.Both n-butanol (B.P. 117 °C) and isobutanol (B.P. 108 °C) are separated from fermentation broth using two distillation columns.254 In this process, cells and debris are first removed and the cell free broth is sent to the first column that concentrates butanol up to its azeotropic composition (55.5 wt% for n-butanol and 70 wt% for isobutanol)1 in the vapor. The vapor is condensed and the butanol–water mixture phase separates into upper and lower layers in a decanter. The upper layer is enriched in butanol at a composition above the azeotrope concentration, which is decanted off and sent to the second distillation column, where it is distilled to a dry butanol product.
The lower layer is recycled back into the first column as reflux (Fig. 9). This process is commonly referred to as continuous heteroazeotropic distillation. Estimates for the heat required to recover n-butanol by distillation without heat integration range from 17 MJ kg−1 to 24.2 MJ kg−1,255,263,264 corresponding to 51 to 73% of the low heating value, and about 19 MJ kg−1 for isobutanol (57% of the low heating value).255 Diaz and Tost256 estimated that the heat requirement for isobutanol separation can be reduced to 5.7 MJ kg−1 (17% of the low heating value) via heat integrated distillation, with yet lower values available if vapor compression is employed. Although distillation is used commercially to recover butanol-containing fermentation broths, alternatives to distillation are an active area of research. These alternatives include supercritical extraction, membrane extraction, solvent extraction, and salting out.265
 | ||
| Fig. 9 Illustration of heteroazeotropic distillation for the separation of butnanols from fermentation broth. A detailed set of operation parameters for these columns can be found in Luyben, 2008.210 | ||
Diols are a more recent entry into the fermentation derived chemicals market, with industrial scale processes operated by DuPont for 1,3-propanediol (PDO) and a newer facility operated by Novamont and Genomatica for 1,4-butanediol (BDO) production.266 While these diols could conceivably be used as fuel precursors using the dehydration and oligomerization chemistry described below, their primary target use today is for bioplastic and biofiber precursors. Polymer precursors have stringent purity requirements that are generally >99%.267 As a result, additional unit operations are needed in combination with distillation to achieve these high purities. In the case of PDO and BDO, the high boiling point of these compounds, 211 °C and 235 °C, respectively, requires a clarification step before distillation operations are performed. After cell and protein removal, the broth is subjected to nanofiltration and then “polishing” by passing the broth through a series of cation and anion exchange resins to remove trace impurities that yield off-color specifications in the final product before distillation. These yellowing compounds are common to fermentation processes but are undesirable in precursor products due to their interference in the dyeability of the final polymer product. Thus, the removal of yellowing compounds is the focus of many patents.268 After the cation and anion exchange step PDO, or BDO, is separated with a series of four distillation columns operated at reduced pressure. In the first column, water is removed in the distillate, while the higher boiling bottom components including the diol are sent to the second column. In the second column, the diol is removed in the distillate and sent to a hydrogenation reactor where it is mixed with hydrogen and passed over a solid acid catalyst to convert impurities to ‘more reduced products such as alcohols. The third column removes these hydrogenated light impurities in the distillate, and the fourth column recovers the diol in the distillate at polymer grade purity.269 This industrial DSP process for diols is designed to meet the purity requirements for polymer applications. If diols are used as fuel precursors the purity requirements would be much less stringent and an alternative DSP approach driven by minimizing energy consumption would need to be developed.
Separation of carboxylic acids
Carboxylates are emerging fuel intermediates and have garnered much interest as platforms for renewable chemicals and fuels (including diesel and aviation fuels)270,271 because they can be produced at industrially relevant titers, rates and yields by anaerobic fermentation.272,273 Currently, several industrial operations exist for the production of carboxylic acids including succinic acid,274 citric acid, lactic acid, acetic acid, itaconic acid, and pyruvic acid.275Carboxylates have boiling points higher than those of water and unfavorable azeotropes, and as such, their separation cannot be achieved through distillation. Instead, separation of these products first begins with a cell and debris removal step. After clarification, salt breaking via addition of an acid (e.g. sulfuric acid) is often performed.276 Salt breaking is necessary in the common case where the pKa of the produced acid(s) is lower than the pH tolerated by the fermenting organism(s), and when employed is the most costly operation in the separation of carboxylates.40 In the literature, calcium precipitation is commonly referred to as the industrially standard method for salt breaking.277 In this approach, calcium hydroxide is added to the fermenter as a neutralizing agent to produce the calcium salt of the acid. The calcium salt is insoluble and precipitates out of solution. The solid calcium-carboxylate salt is recovered via filtration, and then aqueous sulfuric acid is added to break the salt forming the free acid in solution and a precipitate of calcium sulfate (gypsum).277 This method is challenging from an economic and waste management perspective due to the large amount of gypsum formed. Currently, the preferred industrial approach is to use cation exchange (IX) as a means to break the salt in a continuous ion exchange system.269 Another approach used is neutralization of the fermentation with ammonia and salt breaking with the addition of aqueous sulfuric acid to produce the free acid and ammonium sulfate. Ammonia is recovered through thermal cracking of the ammonium sulfate and recycled back to the fermenter to be used again as a neutralizing agent.278 These approaches have proven to be more economical than the calcium precipitation method; however, they still add substantial production costs to the final acid product. In one case an additional 1$ was added to the minimum fuel selling price due to the salt breaking step.279 As a result, it is not uncommon for separation costs to approach 60 to 70% of the production costs if a salt breaking step is employed, which is far above the typical 20 to 40% range of DSP.280 Ideally, fermentation would occur at low pH, below the pKa of the produced carboxylic acid, yielding the free acid directly in the broth, and the salt-breaking step would not be needed. However, host selection is critical in this case, as maintaining metabolic activity at low pH requires specialized adaptations to survive, and it is not likely to be practical to replicate these adaptations in model microbial hosts. To date, citric acid,281 succinic,282 and lactic acid283 are produced at pHs of ∼2 to 4. Cargill engineered yeast capable of producing lactic acid at low pH74 and this advance has been key to the large scale deployment of renewable lactic and polylactic acid products, because of the economic benefits of avoiding the salt breaking operation.
After salt breaking, an optional decolorization step is employed using either non-functional resins or activated carbon.284 The spent carbon is regenerated through heating in air, resulting in some attrition74 of the carbon, and makeup carbon must be added over time. This step is usually employed only for polymer applications.
Finally, the free acid is recovered from the dilute solution with a method suitable for the physiochemical properties of acid. For moderately soluble acids like succinic278 and itaconic acids,285 evaporative crystallization allows recovery of products at very high purities that are suitable for fuel and polymer applications.286,287 For more soluble acids like lactic, citric, and pyruvic acids, product recovery and concentration are usually achieved chromatographically using weak anion exchange resins in a simulated moving bed (SMB) system.286 Here the broth is first thermally dewatered using mechanical vapor recompression to produce a liquor that is then fed to an SMB to chromatographically separate the target acid.
Ester derivatives of acids can also be separated if required. Ester recovery entails dewatering the dilute acid through an SMB system using weakly basic solid adsorbents like polyvinylpyridine (PVP) and eluting the acid from the adsorbent with an alcohol.288 The acid/alcohol solution is then reactively distilled, catalyzed with a mineral acid in a continuous stirred tank reactor (CSTR). The CSTR effluent feeds the middle of a distillation column to recover the alcohol in the overhead of the first column. The ester and mineral acid from the bottoms of the first column are sent to the second column where the ester is recovered in the overhead and the mineral acid in the bottoms is recycled back to the CSTR.289
If esters are produced in the bioreactor218 that have boiling points lower than that of water (e.g., ethyl acetate) separation can occur via gas stripping.290 Here gas is bubbled through the fermentation broth and carries the ester into the overhead where the ester is condensed to recover an equilibrium mixture of the ester and water which can be subsequently purified. This approach could in principle be operated in the presence of slurries. However, this approach requires a significant amount of energy both for evaporation and to cool the large volumetric flowrate of gas utilized, and, instead, Hybrid Extraction Distillation–In Situ Product Recovery (HED-ISPR) systems are often more energetically favorable (discussed below), but are difficult to operate in slurry environments.291 If the produced ester has a boiling point greater than that of water, direct phase separation approaches are the most attractive option.291 However, this requires production at levels such that phase separation occurs, which to our knowledge has not yet been achieved.
Strategic perspective on product recovery for bulk fuel applications
Distillation is in general the least-cost separation method for products and concentrations thereof for which the volatility of the biomolecule is less than that of water.292 Distillation is particularly advantageous for lignocellulose processing because it can readily be applied in the presence of solids. The boiling points of pure compounds at 1 atm provide an initial indication of the feasibility of distillation, although the situation is complicated by the formation of azeotropes as discussed below.Evaporating water from a less volatile biomolecule present at low titers is generally not practical for bulk fuel applications. For example, consider a hypothetical product with volatility much less than water for all mole fractions produced in an aqueous broth at a titer of 20 g L−1 – higher than those that have been achieved for all but a few bioproducts to date (Fig. 3). Evaporation of over 980 grams of water per liter would be required to perform separation by distillation with a non-zero reflux ratio. Multiplying >0.98 kg L−1 broth by the latent heat of water (2.45 MJ kg−1) and dividing by 0.02 kg product per L broth, we obtain a heat requirement of >120 MJ kg−1 product, which is about three times the heating value of jet fuel. Heat requirements on this order are very likely prohibitive for bulk fuel production even with heat integration. They may, however, be acceptable for chemicals, which typically have processing cost margins (product price minus feedstock cost) many-fold higher than fuels (Fig. 1). The situation changes significantly for low-volatility components produced at higher broth titers. As an illustration, for C4 diols produced at 140 g L−1, the same calculation above gives a lower limit at zero reflux of 0.86 kg water × 2.45 MJ kg−1 water/(0.14 kg product) = 15 MJ kg−1, which is not prohibitive for bulk fuel applications. Regardless of titer, a challenge associated with the recovery of low-volatility products via distillation is that growth medium components tend to be concentrated with the product in the distillation column bottoms.
As discussed in Section II.c, fermentation capital costs rapidly become prohibitive at volumetric productivities <10 g L−1 per day, corresponding to titers of about 40 to 50 g L−1 for the four to five-day reaction times typical of reported designs for biological conversion of lignocellulose. Thus, for the many contemplated bioproducts with broth titers less than about 40 g L−1, In Situ Product Removal (ISPR) is likely to be required for low-cost production of bulk transport fuels.
In Fig. 10, boiling points are plotted vs. their maximum reported broth titers for bioproducts presented in Fig. 3 that are produced anaerobically with degrees of reductance greater than that of carbohydrate, recalling that compounds with lower degrees of reductance are generally not considered for use as fuels. See Section IV.a. Of the eight compounds meeting these criteria, the number of carbon atoms ranges from two (ethanol) to four (1-butanol, 2,3-butanediol, butanoic acid), and the degree of reductance ranges from 4.7 (glycerol, propanoic acid) to 6.0 (ethanol, butanol).
 | ||
| Fig. 10 Boiling points and maximum reported broth titers for the eight anaerobically derived bioproducts from Fig. 3 with degrees of reductance >4. The reason that the boundary between quadrants II and IV is 125 °C is that some azeotrope-forming compounds have greater volatility than water at dilution concentrations and thus can be removed from fermentation broth via evaporation. | ||
Four classes of products may be defined based on whether separation by distillation is possible and whether ISPR is likely required (Fig. 10). Separation is least complex for Class I products that are recoverable by distillation and are produced at titers sufficiently high that ISPR is not required, with ethanol being a prime example. The next level of complexity, at least in the presence of solids, is products produced at sufficiently low broth titers that ISPR is likely required but can be recovered with distillation, for example butanol. Class III products are less volatile than water but are produced at sufficiently high titers (generally >40 g L−1) to warrant post fermentation recovery schemes that are non-thermal in nature. Some DSP approaches for Class III are crystallization, adsorption, LLE, etc., and some exemplary products are succinic acid, lactic acid, etc. Class IV products are the most complex to separate. These products are less volatile than water and are produced at low titers (generally <30 g L−1). Here ISPR is required. Some examples include volatile fatty acids (VFAs) from arrested anaerobic digestion and glycerol. In general, an increasing complexity of separation processes is expected to be accompanied by increasing cost and reduced operational robustness, although verification of these trends for a particular compound requires product-specific analysis. For products other than those shown in Fig. 10, the most immediate challenge for fuel applications would appear to be increasing broth titer.
The product recovery landscapes presented in Fig. 10 have a bearing on where in the process flow sheet solids–liquid (S/L) separation occurs. For Classes I and II, solids removal can likely be accomplished after both fermentation and product recovery. For Class III, S/L separation can occur after fermentation, but solids will need to be removed prior to product recovery for most separation technologies. Washing of the solid cake will also likely be required to avoid significant yield losses due to entrainment. For Class IV, it will likely be necessary to either remove solids prior to fermentation or separate solids multiple times within an ISPR side stream loop. Both are challenging for bulk fuel production from cellulosic feedstocks and are likely impractical in the absence of innovative approaches.
For S/L separation after product recovery, a filter press can be used to produce a filter cake with a solids concentration of ca. 50 wt% – depending on the characteristics of the solids – without concern over the unrecovered product. For S/L separation after fermentation but before product recovery, a filter press can also be employed. However, the product must be removed from the solids to prevent yield loss. The extent of the yield loss depends on the solids loading in the fermenter and the solids content of the pressed cake. For example, given a fermenter operating with 15 wt% solids and the resulting cake after S/L separation with a solids content of 55 wt%, approximately 16% of the product will remain in the cake. Thus, washing is required to recover the product and that wash water will enter the DSP train, increasing energy consumption and cost. Studies are needed to minimize excess wash water carried through the DSP operations. If S/L separation occurs before fermentation to produce clarified hydrolysate as the fermentation feed, solids removal would likely involve a screw press or filter press. However, in this configuration the solids must be washed to avoid loss of entrained sugars.
In situ product recovery (ISPR)
ISPR is an important process intensification strategy that has the potential to dramatically lower DSP costs, reduce DSP energy footprints, decrease process water footprints, and increase volumetric productivity.293 This is especially true for biomolecules with lower volatilities that cannot be separated directly from slurries in a beer column. Briefly, ISPR systems operate by continuously separating the target product from the broth while the fermentation is ongoing. In the absence of solids, ISPR can involve the target product being recovered by adsorption to a resin, adsorption into a liquid organic overlayer, or selective chemical precipitation, or stripped from the broth with a gas.294 Two main process approaches exist, with the first being an internal separation configuration wherein the product is recovered from the broth in the bioreactor and the second being an external loop design where the broth is pumped out of the fermenter to recover the product and water is recycled back to the process. External loop ISPR systems are more common, but often require the use of a cell and debris retention device to confine the cells to the fermenter and avoid their direct contact with the DSP operations. By continuously removing the product, ISPR alleviates end product toxicity, allowing greater volumetric productivity and longer fermentation uptimes than batch operations. However, we note that for ISPR to be advantageous, it must be practical to provide fermentable substrates at concentrations higher than the maximum product concentration tolerated divided by the stoichiometric product yield on the substrate. This condition would not appear to be met for ethanol production from cellulosic biomass (discussed above), but likely is met for many other products.Distillation can be used for ISPR, not only for compounds such as ethanol with lower boiling points than water,295 but also for compounds that have higher boiling points than water but form azeotropes such that the bioproduct is more volatile at low concentrations. For example, the relative volatility of n-butanol and i-butanol – which boil at 118 °C and 108 °C, respectively – is higher than that of water by about 20- and 30-fold, respectively, at the concentrations produced by fermentation. GEVO uses this property to advantage in their commercial process for isobutanol production from corn mash, which features ISPR using distillation in the presence of solids.296 Amyris exploits phase separation to produce >130 g farnesene per liter broth processed from sugar syrup containing glucose at 800 g L−1.297 The enhancement in bioreactor volumetric productivity (e.g., g product per L broth per h) achieved via ISPR can be quite substantial. For example, Ezeji et al.298 achieved a 4-fold increase in n-butanol production by gas stripping, and Daugulis et al. achieved more than doubled ethanol productivity by liquid–liquid extraction299 – both using concentrated sugar syrups.
It is important to realize that there have been very few reports of ISPR for lignocellulose-derived bioproducts. In particular, we know of no reports of liquid–liquid extraction, distillation, gas stripping, membrane separation, and adsorption which implement ISPR in the presence of lignin-rich slurries that arise from lignocellulose processing.
ISPR for the production of organic acids appears to be especially beneficial, because it not only increases the microorganism productivity and yield, but also autoregulates the fermenter pH85,86 and can eliminate the need for the expensive salt breaking step described above. In situ separation of carboxylic acids has typically been studied with LLE using organic phase extractants like trioctylamine (TOA) and trioctylphosphine oxide (TOPO) or Cyanex (a commercially available liquid mixture of various chain length hydrocarbons attached to a phosphine oxide group). These extractants are often dissolved in a hydrophobic diluent (e.g., mineral oil).300,301 These extractants selectively remove carboxylic acids through either an acid–base interaction as with TOA or hydrogen bonding to the carboxylic acid as with TOPO.300,301 However, due to the toxicity of these extractants, fermentation systems are designed to mitigate leakage of the organic phase into the aqueous medium of the fermentation vessel. These fermentation systems can be either free cell cultures or immobilized cultures where the broth is circulated in an external loop and contacts the organic phase through a membrane contacting unit and extraction of the carboxylic acids is achieved through perstraction.302 The broth side of the membrane is often held at a slightly higher pressure (∼Δ10 psi) than the organic side, mitigating crossover of the extractant into the fermentation vessel. The free acid is then separated from the organic phase through a stripping operation. For lower boiling acids (e.g., acetic acid, propionic acid, and butyric acid) distillation is used to directly recover the free acid and regenerate the organic extractant303 in the so called Hybrid Extractive Distillation–In Situ Product Recovery (HED-ISPR) system (Fig. 11). The energy footprint of HED-ISRP systems has been shown to be <20% of the higher heating value for volatile fatty acids, provided more than ∼4 wt% of the target product is present in the organic phase.301,304
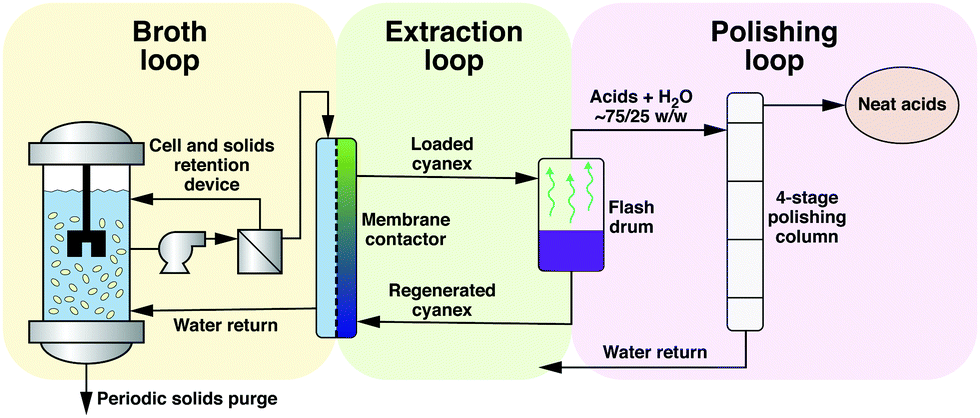 | ||
| Fig. 11 HED-ISPR system diagram with an external loop for recovery of light carboxylic acids. Broth is continuously pumped from the bioreactor into a cell and debris retention device. Cell-free permeate is then passed through a hollow fiber membrane contactor that provides high surface area contact between the aqueous phase and the phosphine oxide extractant (Cyanex). Acids are removed from the Cyanex phase by flashing and the overhead vapor is sent to a 4-stage polishing column to recover neat acids. | ||
Biologically produced esters can be separated in a similar way but using extractants, such as oleyl alcohol and mineral oil. Work has shown that HED-ISPR is especially effective for direct ester separation requiring ∼30 fold less energy than conventional gas stripping ISPR and 3.6× less energy than the HED-ISPR of the carboxylic acid counterparts. Implementation of the HED-ISPR concept would very likely require S/L separation in advance of fermentation.
For non-volatile acids (e.g., succinic acid, itaconic acid, fumaric acid) the free acids are extracted into a TOA organic phase that contains ∼20 wt% water. The acids are then recovered through evaporative dehydration of the organic phase, resulting in precipitation of the crystalized acid.305,306 Water is then recombined with TOA to regenerate the organic extractant.
As described above, ISPR allows decoupling of aqueous product concentration from DSP, allowing greater volumetric productivity and alleviating end product toxicity limitations. However, ISPR involves a tradeoff between lowered fermenter costs on the one hand and increased process complexity on the other hand. This approach has promise and will likely be necessary for some conversions to be practical. However, there are few examples from industry, and constraints imposed by process considerations will be important in determining what gets deployed.
Status
Currently, ethanol is the only commodity scale fuel produced by fermentation. This is largely enabled by its ease of separation from fermentation broth. Specifically, ethanol's high volatility and titers enable energy efficient recovery by distillation without solids removal. Emerging biomolecules such as butanediol and carboxylic acids that could be used as diesel and aviation fuel precursors are being produced at high titers, near that of ethanol. However, these intermediates are used today in higher value applications such as renewable polymers and fibers. Industrial separation processes employed for these applications are focused on meeting high purity requirements.Challenges and opportunities
For C2 to C4 alcohols and other compounds with similar relative volatility differences compared to water, distillation is a satisfactory separation method with generally practical costs and energy requirements. However, opportunities for alternative separation processes exist that outperform or complement distillation. At titers realized thus far, bioproducts other than ethanol, diols, and carboxylic acids likely require ISPR to avoid bioreactor costs that are prohibitively high for bulk fuel applications. ISPR is applied commercially in the presence of solids for volatile compounds produced from corn. ISPR for processes based on lignocellulose represents an important frontier with few studies reported thus far. Technologies and process configurations that enable ISPR for compounds that are not practical to recover by distillation or volatility are particularly challenging, especially in the presence of solids, but could substantially expand the range of biomolecules useful for fuel applications if advances were made. Using ISPR to realize benefits beyond enhancement of volumetric productivity, for example in the case of pH regulation and avoiding of salt breaking in organic acid recovery, is also an emergent area.VI. Post-biological catalytic processing
Substitutes and blend stocks for gasoline, diesel, jet, and marine fuels will be required for renewable biofuels to make a meaningful impact across the entire transportation sector. Current fuels exhibit a wide range of physical, chemical, and thermal properties (vide supra), and replacement biofuels for existing infrastructure must exhibit characteristics concomitant with current fuel and engine property requirements, although they need not necessarily be exact molecular replicates of today's petroleum-derived transportation fuels.307 Given that anaerobic fermentation products that can be obtained at high titers, rates, and yields via atom-efficient metabolic pathways typically range from C2 to C6 in carbon number, many of these compounds do not directly exhibit the necessary fuel properties, especially for jet, diesel, and maritime fuels or blend stocks. Accordingly, chemo-catalytic transformation of fermentation-derived products or product mixtures often merits consideration to expand the range of fuel molecules that can be produced from fermentation-derived intermediates. In nearly all cases, the primary reactions that must be catalyzed involve some form of deoxygenation (e.g., dehydration, decarboxylation) and carbon–carbon bond coupling (e.g., oligomerization, condensation, alkylation, ketonization) to form higher molecular weight compounds or fuel additives and to enhance energy density.308Fig. 12 provides a summary of these transformations, each of which is discussed in turn in this section.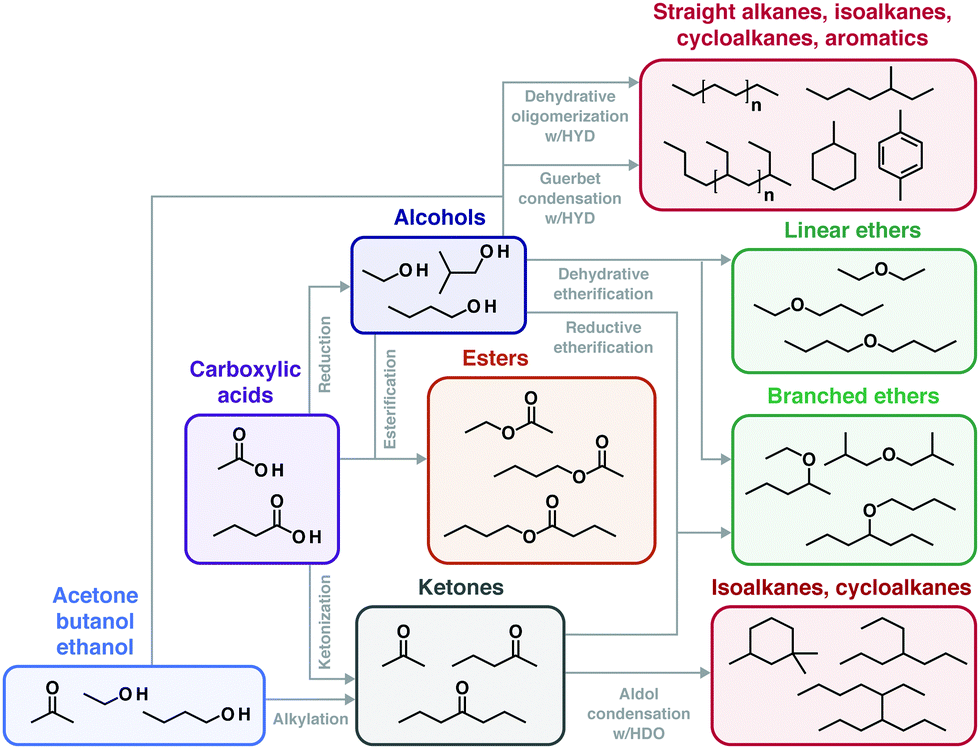 | ||
| Fig. 12 Chemo-catalytic pathways to upgrade anaerobic fermentation products into oxygenated and hydrocarbon fuels. | ||
Alcohol upgrading
Perhaps the most prevalent chemo-catalytic approach that has been studied for decades is alcohol dehydration and oligomerization to hydrocarbons, most often with ethanol, n-butanol, and isobutanol, all of which can be derived from anaerobic fermentation. Several examples of catalytic upgrading of ethanol and butanol to hydrocarbon fuels, especially to jet fuel and gasoline, are being scaled now.309–311 Dehydration of ethanol to ethylene is an industrial process that utilizes γ-Al2O3 at temperatures >300 °C and atmospheric pressure. Other classes of solid acids are also highly active for alcohol dehydration, including zeolites, metal oxides, and heteropolyacids. Near quantitative yields of ethylene have been demonstrated over modified HZSM-5 at 260 °C,312 and alcohol dehydration can occur in the presence of water at high yields, albeit at elevated temperatures to avoid inhibition.313,314Following dehydration, olefins can undergo oligomerization to increase their chain length to be suitable for blending into gasoline, diesel, or jet fuel. Oligomerization of C2–C3 olefins is an industrial process that employs both homogenous and heterogeneous catalysts to steer product distributions.315,316 A large body of work has also been conducted to examine the oligomerization of C4–C6 olefins derived from biomass conversion processes.317–319 For fuels, conversion conditions and catalyst formulations must be tailored to achieve the target chain length, branching, and degree of aromaticity. Highly linear oligomers are desirable for diesel fuels due to their high cetane numbers, while branched oligomers are desirable for gasoline and aviation fuels due to their low-temperature performance. Following oligomerization, a final hydrogenation step is often necessary to remove olefins in the finished fuel (Scheme 1).
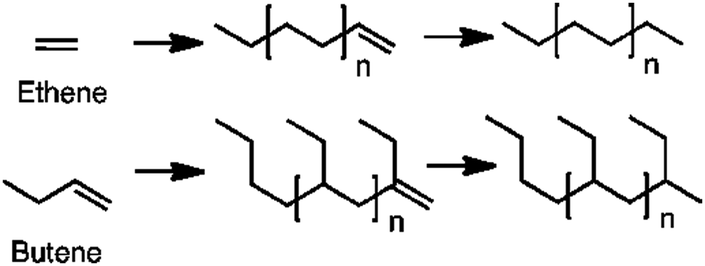 | ||
| Scheme 1 Olefin oligomerization of ethene and butene with subsequent hydrogenation. | ||
To intensify the conversion of alcohols to hydrocarbon fuels, efforts have focused on integrating dehydration and oligomerization reactions over a common catalyst (Scheme 2).320 Direct alcohol-to-hydrocarbon conversions can be challenging to control due to the greater range of potential products, undesirable formation of light hydrocarbons, and catalyst coke formation. Additionally, reduction power may be needed to maximize aliphatic hydrocarbon yields (over aromatic compounds and coke), and catalysts must be active and durable in the presence of water and impurities. The cost of catalytic conversion of wet ethanol vapor to hydrocarbon blendstocks has been estimated to be comparable to that for purification to anhydrous ethanol.321
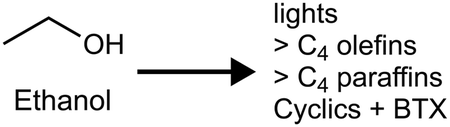 | ||
| Scheme 2 Direct catalytic conversion of ethanol to hydrocarbons. | ||
With regard to oxygenated fuel products, short chain alcohols can be catalytically converted to long chain alcohols to improve their blendability and energy density. The Guerbet reaction has received increased interest for converting ethanol into n-butanol via one-pot dehydrogenation, aldol condensation, and re-hydrogenation (Scheme 3).322–324 The Guerbet reaction is facilitated by acid–base redox catalysts at moderate temperatures (200 to 300 °C), but control is required to minimize further coupling reactions of reactive intermediates that form long chain branched alcohols. Although ethanol has been the primary alcohol of interest, the Guerbet reaction also works well for coupling longer chain primary alcohols.325
 | ||
| Scheme 3 Guerbet reaction to convert ethanol to n-butanol. | ||
Esters and ethers are another class of alcohol-derived products that can be added to gasoline and diesel fuel to improve combustion performance. Ethyl acetate is an exemplary ester that can be produced industrially from ethanol dehydrogenation over copper chromite catalysts (Scheme 4). The reaction takes place at 220–240 °C and 20 bar with high selectivity (99% at 65% conversion) and produces hydrogen that be reclaimed.245 Ethyl acetate is hydrophobic, which improves its blending properties, and it has a higher RON than ethanol.
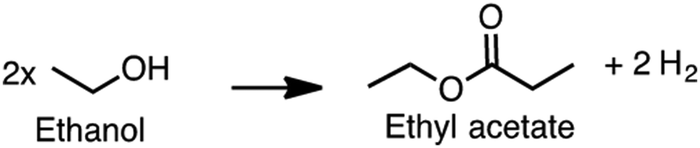 | ||
| Scheme 4 Ethanol dehydrogenation to ethyl acetate. | ||
Ethers are advantageous oxygenated additives for diesel fuel that have high cetane numbers (DEE 85–96, DBE ∼100) and enhance low temperature ignition and emission profiles.106 Etherification of alcohols can occur in the gas or liquid phase at moderate temperatures (180 to 220 °C) and result in high yields by tuning the acidity and reaction conditions of alcohol dehydration catalysts (Scheme 5).326–329
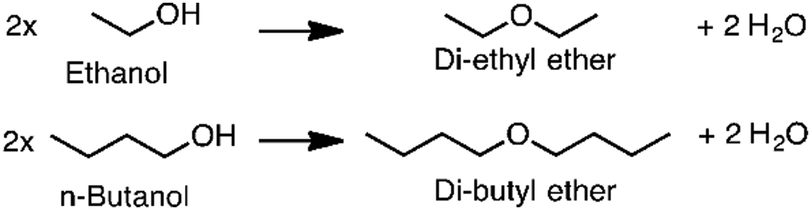 | ||
| Scheme 5 Alcohol partial dehydration to ethers. | ||
Mixed alcohol and ketone upgrading
In addition to mono-alcohols, mixed acetone, butanol, and ethanol (ABE) streams can afford additional chemocatalytic pathways for producing fuel precursors. As shown in Scheme 3, mixed alcohols can undergo the Guebert reaction to extend their carbon chain lengths as well as produce branched alcohols through aldehyde condensation side reactions.330 When acetone is added to alcohols, alkylation can occur to form longer chain methyl ketones with no carbon loss or external hydrogen addition (Scheme 6). Alkylations are promoted by heterogeneous basic oxide catalysts at moderate temperature (250 °C) in near quantitative yield.330–332 This reaction is sensitive to water over basic oxide catalysts,331 although tailored homogenous catalysts have been successfully applied.333 Alkylation can also be performed with mixtures of isopropanol and acetone, which are produced by strains during the biological production of isopropanol.334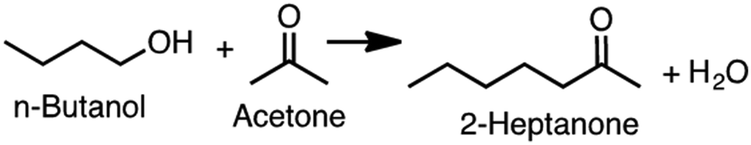 | ||
| Scheme 6 Butanol and acetone alkylation to form 2-heptanone. | ||
Methyl ketones, in the form of either acetone or longer chain ketones derived from alkylation, are powerful coupling agents that readily undergo aldol condensation over basic oxide catalysts. Condensation couples molecules with no loss of carbon and removes oxygen as water. Furthermore, sequential dimer and trimer reactions can produce ring-closed products that are well suited for hydrodeoxygenation to saturated cyclic hydrocarbons for aviation fuels (Scheme 7).332 Similar to alkylation, condensation is sensitive to water over heterogeneous oxides.335 In addition, care must be taken to ensure that coupling retains the terminal ketone position, as central ketones are far less reactive for condensation coupling.
 | ||
| Scheme 7 Acetone condensation to form dimers and trimers, followed by HDO to produce naphthene suitable for jet fuel blending. | ||
Carboxylic acid upgrading
Carboxylates are another class of anaerobic fermentation products that often range in chain length from C2 to C6. Although challenging to separate by conventional distillation, carboxylates can enable multiple chemo-catalytic transformations to produce either partially oxygenated fuel or fully deoxygenated fuel blendstocks. Direct reduction of aliphatic acids to alcohols can be performed in the gas or condensed phase, which typically requires high hydrogen pressures (>500 psi) and moderate temperatures (<200 °C) over precious metal catalysts;313,336,337 however, non-precious metals have proven to be effective as well. Of note, the reduction of longer chain acids in water, such as hexanoic acid (Scheme 8), can potentially enable the formation of phase-separating alcohols to integrate catalytic conversion and separation. | ||
| Scheme 8 Hexanoic acid reduction to hexanol. | ||
The resulting alcohols are amenable to the same catalytic conversion pathways shown above (Schemes 1–5), but they may also react with unconverted carboxylic acids to form esters (Scheme 9). As noted with ethyl acetate (Scheme 4), short chain esters can display high RONs and serve as oxygenated gasoline fuel additives. Multiple studies have examined carboxylate esterification with butanol due to the biphasic reaction system that results with water. Ester reactions can occur at mild temperatures (<120 °C) in near quantitative yields over acid resin catalysts. Reactions with butanol have been studied with acetic acid, propionic acid,338 and butyric acid.339
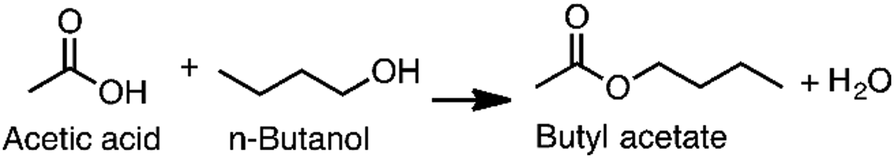 | ||
| Scheme 9 Acetic acid esterification with butanol. | ||
To extend the chain length of carboxylic acids, catalytic ketonization can couple two carboxylic acids to produce a ketone with one carbon less than the acid pair, while forming one mole of CO2 as a byproduct (Scheme 10). Although not atom efficient for coupling shorter chain acids, medium chain acids can be converted to longer chain hydrophobic ketones with high selectivity and yields greater than 95%.340–342 Ketonization commonly takes place over amphoteric oxides at high temperatures (350–425 °C) and atmospheric pressure in the vapor phase. Alternatively, recent studies have investigated ketonization in the condensed aqueous phase, although water significantly inhibits the reaction yield.343,344
 | ||
| Scheme 10 Hexanoic acid ketonization to form 6-undecanone. | ||
Following ketonization, multiple routes are accessible for converting ketones to hydrocarbon and oxygenated fuels. Ketones can be selectively reduced to alcohols in near quantitative yields at moderate temperatures and high hydrogen pressures over Ru/C catalysts (>500 psig).345 Ketones can also react with alcohols to form branched ethers that display similarly advantageous oxygenated fuel properties to linear ethers.270 Alternatively, full deoxygenation of ketones can occur over precious metal and non-precious metal catalysts to produce linear hydrocarbons suitable as heavy duty ground transportation and aviation fuel blendstocks, depending on the ketone chain length.341,346,347 Lastly, methyl ketones can readily undergo condensation dimer and trimer reactions to further extend their chain length,332 similar to the exemplary acetone pathway shown in Scheme 7.
Status
Both ethanol dehydration to ethylene and ethylene oligomerization to long chain hydrocarbons are processes that have been implemented at the industrial scale separately. Several green ethylene facilities using ethanol from first generation biorefineries are currently under operation at the hundreds of ktons per year scale. For example, Braskem in Brazil is set to expand its current ethylene production capacity from 200 to 260 ktons per year by the end of 2022. Although no large-scale facilities currently produce mid-distillate fuels directly from ethanol, several demonstration projects are underway, including LanzaJet's efforts to convert ethanol into sustainable aviation fuel in an alcohol-to-jet facility at the Freedom Pines Fuels biorefinery at a capacity of 10 million gallons per year.Challenges and opportunities
To facilitate industrial-scale catalytic transformations, robust and tailored catalyst materials are needed to withstand the unique process challenges of anaerobically derived intermediates. For gas phase conversions, bifunctional dehydration and oligomerization catalysts are needed that can selectively produce target fuel classes, while withstanding volatile impurities, water, and coking.310,348 For condensed phase reactions, the water tolerance of heterogeneous metal oxides remains a challenge,335,349 as well as transitioning from model to complex fermentation substrates that contain residual organics and salts that can foul and deactivate catalysts.273,350,351 Supported catalysts used in condensed phase media are particularly susceptible to active site leaching, which represents an irreversible and costly mode of deactivation further exacerbated by the chelating abilities of oxygenates.352,353 Ultimately, hybrid biological and chemo-catalytic process scenarios will rely fundamentally on cost-effective separations and robust catalysts for subsequent upgrading, typically in the condensed phase – which represents a substantial departure from most petroleum-based chemical catalyses.363 However, many opportunities for creative catalyst and process development in this area remain. These opportunities include, and indeed necessitate, the design and testing of realistic catalyst systems that must be able to operate for extended time on-stream (thousands of hours of operation).With regard to active site design, the molecular tunability of homogenous catalysts offers promising directions for further development to improve activity and selectivity, although cost effective recovery strategies must be integrated into design.354,355 Likewise, the increased tunability of heterogeneous catalyst materials to facilitate tailored pore geometries, specified metal–support interactions, cascade chemistries, and robust active site anchoring strategies holds promise for advancing the conversion of anaerobic intermediates.356–358 Finally, unresolved questions remain regarding the mechanism of key transformations359–361 that require insight to improve rational catalyst design to use novel materials beyond the laboratory.362 New strategies for catalyst design must be hand in hand with process intensification approaches, including the use of membrane reactors, reactive distillation, and continuous manufacturing, to maximize efficiency.
As the cost of renewable electricity continues to decrease with the sustained growth of the solar and wind sectors, there will be opportunities for “green” electrons to play increasingly central roles in the manufacture of chemicals and fuels at the biorefinery. The past two decades have seen an explosion of research activity that has generated several important fundamental advances in photoelectrochemical water splitting for the production of CO2-free H2. While renewable hydrogen is still 3–4 times more expensive than that obtained from traditional methane steam reforming, competitive prices could materialize in the short- to mid-term horizons. This opens promising future opportunities for the development of hybrid bioelectrocatalytic reactors to couple processes that generally would require separate vessels. A standout example is the use of bioelectrochemical reactors as an alternative to sparging H2 and O2 gases for growing chemolithoautotrophic microorganisms that produce oxygenates from CO2 metabolism. By coupling electrochemical water splitting (for H2 production) with CO2 fixation, hybrid inorganic-biological systems can feature growth rates, carbon yields, and energy efficiencies with potential to drastically exceed those of the state-of-the-art.364,365
Unlike most thermochemical upgrading schemes, electrochemistry and photocatalysis offer unique opportunities to carry out these reactions under low temperature and pressure conditions. However, while our understanding of how to use solar photons to generate energetic electron/hole carriers to drive redox-based catalysis is advanced, the science of efficient direct activation of oxygenated intermediates for chemoselective C–C bond formation remains nascent. This challenge is intertwined with the related challenges of selective electron–proton and oxide transfer toward C–O and C–H bond activation with control over key kinetic branchpoints across complex reaction cascade networks. The formation of liquid fuels from bioderived intermediates requires the coupling of large numbers of protons and electrons with bond forming and bond breaking reactions. This myriad of conditions presents the imperative for developing reaction cascades wherein each individual step is performed under ideal operating environments and presents the basic science challenge of how to integrate photo-state excited states, redox carriers, hydrogen activation, phase transfer catalysts, hydrides, and carbanions, between disparate reaction microenvironments in ways that provide for flux balance, kinetic coupling, rapid equilibration, and selective transport. Once direct photo-electrochemical activation is better understood, one can envision an expanded role of solar photons in their coupling to lower the activation barriers for forming reactive intermediates along liquid fuel reaction cascades.
VII. Fuel utility
On-vehicle energy storage, vehicles, and infrastructure have had a dynamic relationship in the past, with each changing in response to developments in the other two. This can confidently be expected to continue in the future, for example to accommodate the expanded use of batteries and hydrogen in transport applications. Changes in vehicles and infrastructure in response to emergent biofuels are conceivable, and in general far smaller than the changes required for batteries or hydrogen. The extent to which such changes are considered will, however, depend on the extent to which biofuels are able to offer scalable benefits compared to the status quo in terms of price, performance, and social and environmental externalities. Other things being equal, fuels that require less change in vehicles and infrastructure will gain market acceptance more rapidly.To make an impact on transport energy supply, biofuels must be suitable for use in one or more transport modes and the engines that power them. These include light duty transport powered by spark-ignited (SI) or compression-ignited (CI) engines, heavy-duty transport and equipment, freight rail, and ocean shipping – all of which are powered by CI engines, and aviation, powered primarily by jet fuel burned in gas turbines. Responsive to the different requirements of these applications, current fuels exhibit a wide range of physical, chemical, and thermal properties. Biofuels must exhibit properties suitable for use in these applications, although they need not necessarily be exact molecular replicates of today's petroleum-derived transportation fuels.307,366–368 Fuel standards, for example ASTM and CEN standards, provide a basis to determine such suitability. Today, biofuels are most used as low-level blends in petroleum refinery fuels. Gasoline containing from zero to 15 volume percent (vol%) ethanol is described in terms of fuel properties by ASTM standard D4814 and diesel containing up to 5 vol% biodiesel is described by the properties in ASTM standard D975. Oxygenates are not allowed in jet fuel and alternative jet fuel blendstocks – which in some cases are biofuels – are referred to as synthesized hydrocarbons. The properties of currently allowed materials that can be blended at up to 50 vol% in some cases are described in ASTM standard D7566; however, the final blend with petroleum-derived jet fuel must meet the jet fuel standard, ASTM D1655.
A new fuel would be most easily introduced into the market if it meets the same property requirements applied to these conventional fuels – whether used as a blend or in its pure form. Blends containing higher levels of the new fuel or oxygenate such as ethanol contents of 51 vol% or higher, or biodiesel over 5 vol% to 20 vol%, exhibit different properties than their petroleum-derived counterparts and therefore have their own ASTM standards (D5798 and D7467, respectively). Fuel properties arise from the molecular structure of the molecule or molecules that make up the fuel. However, many of the most important fuel properties are measured using performance tests developed many years ago and for which detailed structure–property relationships are only qualitatively understood.
Property requirements
Table 5 lists some of the most important properties from the ASTM standards for gasoline, diesel, and jet fuels. These properties were chosen because they are embodied in the molecular structure of the fuel. Properties not included in Table 5 include those that limit impurities, such as sulfur and chloride, as these are primarily functions of the production process. Certain other property requirements are commonly addressed using fuel additives – compounds added at typically 10 to 100 ppm levels. Examples include lubricity and electrical conductivity, which are also not included in Table 5.| Gasoline | Diesel | Jet | |
|---|---|---|---|
| ASTM standard | D4814a (D02 Committee, n.d.) | D975b (D02 Committee, n.d.) | D1655c and D4054 (D02 Committee, n.d.) |
| Boiling point | 60–185 °C | 150–338 °C | 150–300 °C |
| Vapor pressure or flashpoint | Approximately 40–100 kPa at 37.8 °C | >38 °C winter | >38 °C |
| >52 °C summer | |||
| Freezing point/fluidity | <−10 °C or soluble in hydrocarbons | <−10 °C or soluble in hydrocarbons | Freezing point <−40 °C/viscosity at −20 °C ≤ 8 mm2 s−1 |
| Combustion | Research octane number ≥91 | Cetane number ≥40 | Smoke point ≥25 mm |
| 35 ≤ cetane number ≤ 60 | |||
| Stability | 240 min D525 | — | D3241 |
| Composition | Benzene <0.62 vol% | — | Aromatics <25 vol% |
| Density/heat of combustion | — | — | 775–840 kg m−3 |
| >42.8 MJ kg−1 |
Boiling point
A proposed fuel's boiling point must be in the designated range for proper engine operation. For wide boiling point mixtures such as petroleum-derived fuels, the boiling point distribution (known as the distillation curve) is described in terms of the temperature for a certain volume percent of the fuel to evaporate (for example, T50 is the temperature where 50 volume percent of a fuel has evaporated). Boiling points defining the low end of the acceptable range in Table 5 are approximate as the most highly volatile components in fuels are limited by vapor pressure (for gasoline) or the flashpoint (for diesel or jet fuel) rather than the boiling point. The upper boiling point limits are hard limits, and the most desirable biofuels would likely boil well below these upper limits to allow blending at high levels, or must meet these limits for neat biofuels.For SI engines fueled with gasoline, the T90 and final boiling point are limited to prevent introduction of fuel components that do not evaporate, mix with air, and burn, and instead are swept into the engine lubricant causing degraded lubricant performance or emitted into the air. T90 is limited to 185 °C and the final boiling point to 225 °C for this purpose, and a single component blendstock should boil well below the T90 limit if it is to be blended at significant percentages. A recent study367 that screened many biofuel candidates for suitability as gasoline components applied a maximum boiling point of 165 °C in an effort to accommodate blends up to 30 vol%. Many bio-derived components meeting this T90 limit were identified, including C1 to C5 alcohols, a broad range of esters and ketones, diisobutylene, and a mixture of 2-methyl and 2,5-dimethyl furans. There are several examples of materials proposed for use in gasoline that were ultimately found unworkable because of their high boiling point. For example, 2-phenylethanol has been discussed as a biofuel,243,369,370 and appears to have a high octane number, suggesting that it be used in gasoline.368 However, it boils at 218 °C – well above the T90 limit for gasoline – and was found to not completely evaporate and burn, accumulating in the engine lubricant in a recent study.371 A similar example372 is ethyl levulinate, also predicted to have a very high octane number, but boiling at 206 °C. High boiling gasoline components can also contribute to particulate matter emissions.288
Diesel fuel is limited to a T90 of 338 °C to prevent introduction of difficult to vaporize components that could lead to engine operational issues such as lubricant dilution or in-cylinder carbon deposits. As for gasoline, single component blendstocks should boil well below these T90 and end point limits. An interesting example is fatty acid methyl ester biodiesel, a mixture of several esters produced from fats and oils that boil between 330 °C and 357 °C. When blended into diesel fuel, the high boiling range of biodiesel can cause fuels to fail the 338 °C T90 limit, which is one of the reasons that conventional diesel fuels are limited to containing no more than 5 vol% biodiesel. Blends of 6 vol% to 20 vol% biodiesel are considered alternative fuels and have a separate ASTM standard (D7467) that limits T90 to 343 °C. Experience has shown that for biodiesel blends – but not necessarily for any other fuels – this higher allowable T90 does not cause operational problems. While 100% biodiesel is sometimes used, it is not approved by any diesel engine manufacturers today and one potential issue is its high boiling point. Changes to engine designs to accommodate higher boiling point fuels are possible and could be considered in the future if fuel benefits were seen to outweigh costs.
Jet fuel has a T10 limit of 205 °C, which ensures the fuel is volatile enough for ease of starting, and a final boiling point upper limit of 300 °C to exclude difficult to vaporize materials. The distillation curve is also correlated with fuel performance at critical jet engine operating points (lean blowout, cold start, and altitude relight373). Therefore, for synthesized hydrocarbons there are typically additional requirements to ensure that the fuel is not a narrow boiling point range material that would cause a dramatic shift in the distillation curve if blended at high levels. For example, synthetic blending components made from the Fischer–Tropsch process or via hydroprocessing of fats and oils have the additional requirement that the difference between T90 and T10 be 22 °C or higher. These components are allowed at up to 50 vol%. In contrast, farnesane (referred to as Synthesized Iso-Paraffins from Hydroprocessed Fermented Sugars) is only allowed at 10 vol% because it is a pure compound or a mixture of isomers that boil over a very narrow range.
Vapor pressure and flashpoint
Gasoline must have adequate vapor pressure for starting the engine.374 An adequately high vapor pressure is also necessary from a safety perspective to ensure that the vapor space in a tank of fuel is too fuel rich to be ignited. However, if the vapor pressure is too high, vapor lock issues can occur in warm weather,374 and fuel evaporative emissions can exceed regulatory limits.375 ASTM and CEN standards vary the allowable maximum vapor pressure seasonally and geographically to allow higher vapor pressures in colder weather, but limit vapor pressures in warmer weather to meet vapor lock and environmental requirements. The example of ethanol blending into gasoline is instructive. Ethanol forms highly non-ideal solutions with gasoline (does not follow Raoult's law).375 The vapor pressure of pure ethanol is approximately 10 kPa at 38 °C (the temperature used by the ASTM for gasoline vapor pressure measurements), while gasoline for use in the summer months is limited to a maximum vapor pressure of 62 kPa in the United States. If ethanol blended ideally, blending would lower the final blend vapor pressure, but in fact the opposite happens and blending of 5 vol% to 30 vol% ethanol will typically increase the finished gasoline vapor pressure by 10 kPa. Only at higher blend levels, above 50 vol%, does ethanol cause the vapor pressure to be reduced. In contrast, use of 100% ethanol in place of gasoline is not workable because of its very low vapor pressure in its pure form, making it difficult to start the engine. This is the reason that the fuel known as E85 was developed. Originally, this fuel consisted of 85 vol% fuel grade ethanol (containing 5 vol% hydrocarbon denaturant) to which was added 15 vol% of a high vapor pressure gasoline. Today, so-called E85 can contain 51 vol% to 83 vol% chemical ethanol, with gasoline added at the volume needed to meet the minimum vapor pressure requirements of the ASTM D5798 standard. Note that E85 requires that vehicles be specifically designed to tolerate the higher ethanol content. These are known as flex-fuel vehicles and can run on any blend of ethanol from zero to 83 vol%. Other potential fuels have been shown to blend non-ideally to a greater or lesser extent than ethanol, including methanol, 1-propanol, 2-propanol, and methyl acetate.367 Isobutanol has been widely discussed as a more suitable gasoline blend component than ethanol because it blends much more ideally, causing a reduction in the finished fuel vapor pressure. Other C4 and C5 alcohols, various esters and ketones, alkyl furans, and diisobutylene are also shown to blend relatively ideally with gasoline. A list of potentially bio-derived molecules and mixtures that could function as gasoline blendstocks is shown in Table 6.| Fuel application | Candidate biologically-derived molecules |
|---|---|
| a The example composition can vary with biomass feedstock. b Approved for use in jet fuel at up to 10 vol%. c Approved for use in jet fuel at up to 50 vol%. Fischer–Tropsch derived kerosine does not involve biological process steps but can be produced from biomass. | |
| Spark-ignition engine fuels (gasoline) | Alcohols |
| Methanol | |
| Ethanol | |
| n-Propanol | |
| 2-Propanol | |
| 1-Butanol | |
| 2-Butanol | |
| 2-Methylpropan-1-ol (isobutanol) | |
| 2-Methyl-1-butanol | |
| 2-Pentanol | |
| Prenol (3-methyl-2-buten-1-ol) | |
| Fusel alcohol mixture (15 vol% ethanol, 55 vol% isobutanol, 5 vol% 2-methylbutan-1-ol, 10 vol% 3-methylbutan-1-ol, 15 vol% 2-phenylethanol)a | |
| Esters | |
| Methylacetate | |
| Methylbutanoate | |
| Methylisobutanoate (2-methylpropanate) | |
| Methylpentanoate | |
| Methyl-2-methylbutanoate | |
| Ethylacetate | |
| Ethylbutanoate | |
| Ethylisobutyrate (2-methylpropanoate) | |
| 1-Methylethylacetate (isopropylacetate) | |
| Butylacetate | |
| Isobutylacetate (2-methylpropylacetate) | |
| Isoamylacetate (3-methylbutylacetate) | |
| Ethers | |
| Methoxybenzene (anisole) | |
| Ketones | |
| 2-Butanone (methyl ethyl ketone) | |
| 2-Pentanone | |
| 3-Pentanone | |
| 3-Methyl-2-butanone | |
| Cyclopentanone | |
| 3-Hexanone | |
| 4-Methyl-2-pentanone (methylisobutyl ketone) | |
| 2,4-Dimethyl-3-pentanone | |
| Ketone mixture (42.5 wt% 2-pentanone, 11.4 wt% methyl–isobutyl ketone, 30.3 wt% 4-heptanone, 15.8 wt% 2-heptanone)a | |
| Compression ignition engine fuels (diesel) | 2-Nonanol |
| Butylcyclohexane | |
| Farnesane (2,6,10-trimethyl dodecane) | |
| n-Undecane | |
| 5-Ethyl-4-propylnonane (80%) | |
| Hexyl hexanoate | |
| Methyl decanoate | |
| Dibutoxymethane (DBM) | |
| 4-Butoxy heptane (95%) | |
| Dipentyl ether | |
| Di-isoamyl ether | |
| Aviation turbine fuels (Jet) | Fischer–Tropsch hydroprocessed synthesized paraffinic kerosineb |
| Synthesized paraffinic kerosine from hydroprocessed esters and fatty acidsb | |
| Farnesane (2,6,10-trimethyl-dodecane)c | |
| Synthesized kerosine with aromatics derived by alkylation of light aromatics from nonpetroleum sourcesb | |
| Alcohol-to-jet synthetic paraffinic keroseneb | |
| Synthesized kerosine from hydrothermal conversion of fatty acid esters and fatty acidsb | |
| Synthesized paraffinic kerosine from hydroprocessed hydrocarbons, esters and fatty acidsc | |
| Menthane (1-methyl-4-propan-2-ylcyclohexane) | |
| Butylcyclohexane | |
For safe handling of diesel and jet fuel the flashpoint must be above the limits in Table 5, which limits the amount of highly volatile components and ensures that the vapor space in a tank of fuel is too fuel lean to be ignited. Ethanol–diesel blends are an example of a poor match between a biofuel's vapor pressure or flashpoint and the required fuel properties. In its pure form, the flashpoint of ethanol is 13 °C. Because ethanol is not very soluble in diesel fuel, surfactants are used to form an emulsion (other additives are required to increase the cetane number and improve lubricity). An ethanol–diesel emulsion has a flashpoint in the 13 to 20 °C range for nominally 10% blends.376 At this level of flashpoint, the vapor space above the fuel in a vehicle tank is an ignitable mixture under normal ambient conditions, creating an extreme safety hazard.
Fluidity
It is important for liquid fuels and fuel components to remain liquid under ambient conditions where they are handled and blended, as well as under conditions of use whether in high altitude aviation applications or cold wintertime temperatures. This places a maximum value on the freezing point and the temperature where a fuel component can crystallize out of a blend (cloud point). For gasoline and diesel there are no specific standard requirements for low temperature fluidity other than that the finished fuel be fit for use in the local environment – and for gasoline boiling range hydrocarbons’ freezing points are so low that this is not an issue. However, certain oxygenates may come out of the gasoline solution at low temperatures and form a separate liquid phase,368 and this phase separation temperature should be below the temperature anticipated for fuel use. Cold temperature fluidity and phase stability are more important for diesel fuels where the cloud point of market fuels can vary seasonally from 5 °C to −40 °C. Oxygenate components can also exhibit the formation of a separate liquid phase at cold temperatures, as has been reported for diesel blends with ethyl levulinate.372 The value of −10 °C maximum listed in Table 5 is meant to ensure that a bioblendstock is liquid in a terminal handling environment and would have a reasonably large wintertime market; however, much lower freezing point levels may be needed for blending of diesel fuels to be used in the coldest wintertime areas.For hydrocarbon fuels, n-alkanes crystallize at the highest temperatures. For example, the enthalpy of fusion for n-alkanes is 36 kJ mol−1 for dodecane and increases with chain length. In comparison, the enthalpy of fusion for C10 isoalkanes, cycloalkanes, and aromatics is below 20 kJ mol−1.377 However, crystallization also depends on the composition of the non-crystallizing components of the solution in complex ways.378 The presence of polar oxygen functional groups on an alkyl chain increases the freezing point and reduces solubility (note the following melting points, dodecane: −9.6 °C, 2-dodecanone: 20 °C, dodecanol: 24 °C, dodecanoic acid: 43.2 °C), while the less polar ester group can have little effect (methyl undecanoate: −10 °C). In some cases impurities rather than the primary biofuel component can cause cold weather operational issues.379 This has been the case for biodiesel – of which over 6 billion liters are consumed annually in the United States. Saturated monoglycerides are a common impurity in biodiesel, and have been implicated as the cause of widely reported cold weather issues that occurred early in biodiesel's introduction.380,381 These issues have largely been eliminated today by changes to biodiesel ASTM standard requirements and production processes.
Fluidity at cold temperatures is also critical for jet fuel, which is required to meet a maximum freezing point limit of −40 °C (Jet A grade). The cloud point is measured by cooling the sample from room temperature and measuring the temperature where crystals first appear. The freezing point is measured with the opposite approach; the sample is frozen and then heated until all crystals disappear. Freezing points are typically a few degrees higher than cloud points for hydrocarbon fuels, and so more conservative from the standpoint of low-temperature operability of the fuel. The Federal Aviation Administration and ASTM have approved seven synthesized hydrocarbon blendstocks for use in jet fuel. All must meet the −40 °C freezing point requirement, except for farnesane, which has a −60 °C freezing point requirement. Additionally, for jet fuels it is critical that the viscosity over the operational temperature range not exceed a critical value to ensure pumpability and a consistent fuel spray pattern. The maximum limit of 8 mm2 s−1 at −20 °C is used to ensure adequate performance. Approved alternative jet fuel blend components as well as potential future bio-derived jet components are listed in Table 6.
Combustion
For gasoline and diesel fuel the combustion requirements are about resistance to autoignition, while for jet fuel both resistance to autoignition and the soot formation tendency are important. Avoidance of autoignition is critical for SI engines. In these engines, the fuel–air mixture is ignited by a spark. If the fuel has inadequate resistance to autoignition, the unburned mixture (referred to as the end-gas) will autoignite at some time after the spark but before being consumed in the spark-initiated flame. This uncontrolled ignition is commonly referred to as knock and can damage the engine. A minimum resistance to autoignition is ensured by requiring a minimum octane number,382 although other fuel properties can also influence knock resistance.383 In the United States, knock resistance is ensured by using the average of the research octane number (RON) and motor octane number (MON) – both derived from engine tests conducted under slightly different conditions. Recent studies have shown that the MON is much less relevant for modern engines298 and a minimum RON of 91 may be considered to be the effective octane number lower limit. In Europe standard EN 228 requires a minimum RON of 95 and a minimum MON of 85. Future engines may have a significantly higher RON requirement because of regulations demanding higher efficiency, which will be achieved by increasing the compression ratio, turbocharging, engine downsizing, and operation at lower engine speeds.384 These technologies produce higher end-gas temperatures and pressures or allow more time for the end-gas to autoignite. Additionally, a RON of 91 for a bioblendstock may not be adequate for blending with petroleum refinery gasoline blendstock which typically has a RON of 85 to 87 in the United States because the refinery blendstock is currently optimized for blending with 10 vol% ethanol (RON of 109). Many proposed bioblendstock molecules with RONs above 91 do not adequately increase the RON when blended into a conventional sub-octane gasoline blendstock.367Gasoline bioblendstocks with the highest potential to enable more efficient engine designs exhibit non-linear octane number blending when combined with petroleum refinery blendstocks.385 This non-linear blending is synergistic in that the final blend RON is significantly higher than that would be predicted based on the volume or molar weighted average of the RON values for the components being blended. A molar blending RON can be defined for a bioblendstock as shown in eqn (2). This becomes a normalized molar blending RON if divided by the bioblendstock's pure component RON – eqn (3). A normalized molar blending RON is equal to one for linear blending, is greater than one for synergistic blending, and is less than one for antagonistic blending. These behaviors are illustrated in Fig. 13 for several potential bioblendstocks. Synergistic blending is observed for alcohols, alkyl furans, and olefins, and weakly for ketones. Esters can show antagonistic blending (methyl and ethyl acetate) or near linear blending. Synergistic blending is, hypothetically, caused by the bioblendstock functioning as a radical scavenger at relatively low temperatures, shutting down the early autoignition chemistry of the most reactive refinery gasoline components through chain terminating reactions.386 Given that products of biomass conversion are frequently oxygenates, it is notable that several classes of oxygenates are shown to blend synergistically with refinery hydrocarbons for RONs. The details of non-linear blending are currently a very active area of research in combustion kinetics.
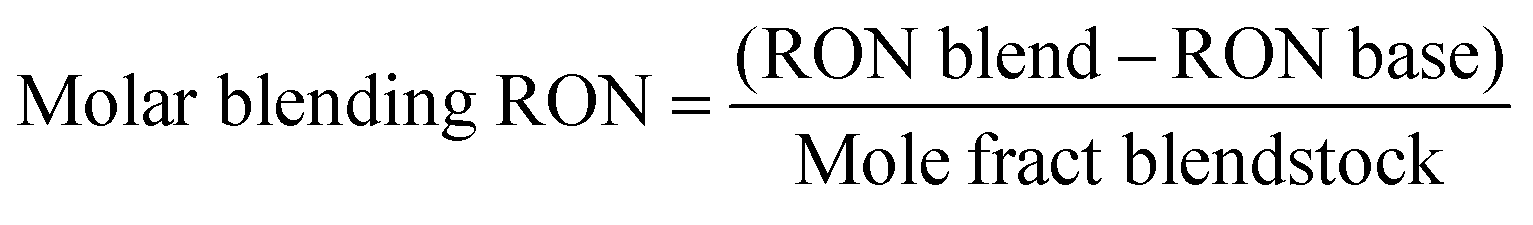 | (2) |
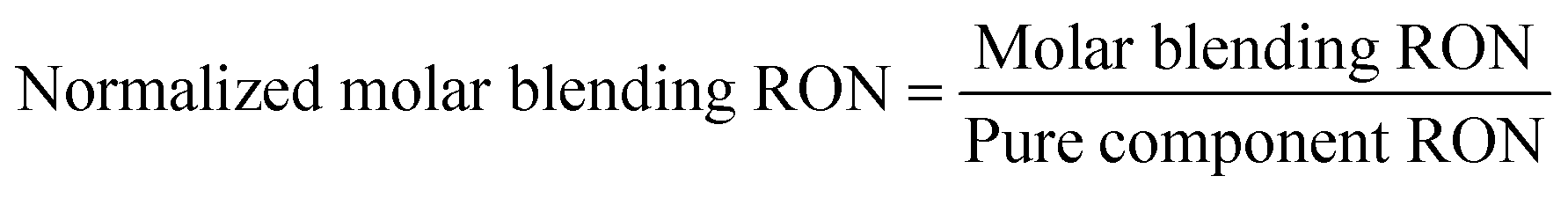 | (3) |
 | ||
| Fig. 13 Normalized molar blending RONs for a range of potential bioblendstocks blended into a 4-component gasoline surrogate (McCormick et al. (2017); Monroe et al. (2019)). | ||
 | ||
| Scheme 11 Key reactions in low-temperature autoignition. | ||
The larger radical species formed in Scheme 11 reaction 4 abstracts a second hydrogen internally, leading to the formation of a ketohydroperoxide and an OH radical, as shown in Scheme 12 for n-heptane. As the temperatures increases, the O–O bond in the ketohydroperoxide breaks, producing two radical species. The overall reaction therefore produces three radical species (two OH radicals and an oxidized hydrocarbon radical) and is the primary chain branching process in low-temperature autoignition – which dominates autoignition in high CN materials.387
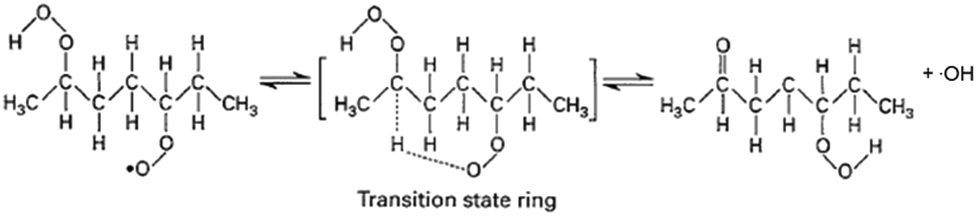 | ||
| Scheme 12 The second isomerization step in the low-temperature autoignition mechanism.387 | ||
Isoalkanes have less opportunity to undergo this low-temperature reaction sequence, especially if highly branched, because there are fewer methylene groups. For example, 2,2,4-trimethylpentane (isooctane) has very low reactivity and defines RON = 100 on the octane number scale. Small alkanes such as methane and ethane are also unable to undergo this reaction sequence. Ethane which is estimated to have a RON of 115 yet is a gas under ambient conditions, making it impossible to use in gasoline. Addition of an OH group to form ethanol slightly reduces the RON to 109 but creates a liquid that can be blended with conventional gasoline or used in its neat form. However, other small molecules can be highly reactive, such as dimethyl ether with a CN of over 55. In this molecule weak C–H388 bonds lead to peroxyradical formation and a similar reaction sequence to that shown in reactions R1–R4.389 Aromatics cannot undergo this low-temperature reaction sequence and so have low reactivity (high RON, low CN).
Note that reactions R2 and R4 in Scheme 11 are reversible with the forward reaction favored by higher pressure and the reverse reaction favored at higher temperature. As the temperature increases further (above 850 K), the reversibility of these reactions shuts down low-temperature autoignition. At higher temperatures fuel molecules are consumed by sequential hydrogen abstractions from OH and HO2 radicals. Above 1000 K, hydrogen peroxide decomposes to produce a large number of hydroxyl radicals which rapidly begin to consume the fuel, regardless of molecular structure.387
A wide range of molecular structures can exhibit high CNs, and in many cases these molecules will have a significant chain of uninterrupted methylene groups. In a recent study examining a wide range of potentially biomass-derived structures for their properties as diesel blendstocks, Fioroni and coworkers390 found examples of ethers, esters, alcohols, cycloalkanes, isoalkanes and branched alkanes with adequately high CNs while also meeting other property requirements. The very high CNs available from certain ethers are notable. For example, we can compare undecane with a CN of 71 with dipentyl ether (essentially replacing the central methylene in undecane with oxygen) and the CN increases to over 100. The presence of oxygen weakens the adjacent C–H bonds and, combined with the favorable geometry of ethers for forming a 6-membered ring transition state, leads to increased reactivity. The case of long chain alcohols is interesting, as longer chains provide not only higher CNs (the CNs for 1-octanol, 1-nonanol, and 1-decanol are 39, 46, and 50, respectively) but also higher freezing points (−16 °C, −6 °C, and 6 °C, respectively) such that none of these alcohols could simultaneously meet the Table 5 requirements for the freezing point and CN. Moving of the alcohol group to the number 2 carbon slightly reduces the CN but dramatically reduces the melting point, perhaps providing a better pathway to long-chain alcohol utilization in diesel (a 2-nonanol CN of 40 and a melting point of −36 °C).
A primary negative environmental impact of diesel combustion is the formation of fine particles or soot.391,392 On-road engines in the United States and many other markets are required to employ a ceramic particle filter to prevent soot emissions,393 and the use of particle filters for many off-road and stationary diesel engine applications is increasing. While not a property requirement for use as a diesel fuel, oxygenates in general have been shown to reduce soot formation in diesel combustion.394 Given that oxygenates are in many respects more easily produced from biomass than are hydrocarbons, this benefit of biomass-derived fuels should not be overlooked. Soot reduction of over 50% has been shown for 100% biodiesel.395 Highly oxygenated polyoxymethylene ethers can be burned in diesel engines with almost no soot formation.396 For engines equipped with a particle filter, reduced soot formation in the engine improves filter performance and reduces pressure drop and the frequency of filter regeneration, consequently reducing the fuel economy penalty for filter operation.
Avoidance of soot formation is critical for jet engine operation because soot increases engine maintenance costs, has negative effects on engine efficiency by increasing radiant heat transfer397 and has significant environmental and health impacts.398 Soot is limited by requiring a minimum smoke point (a higher smoke point indicates a lower sooting fuel). Olefins and aromatics are higher soot formers than n-alkanes, isoalkanes, and cycloalkanes,399 and the aromatic content also is limited in jet fuels to control soot formation. With one exception, the forms of synthesized hydrocarbons currently allowed to be blended into jet fuels are all isoalkanes, or mixtures of normal and isoalkanes. Note that alkyl substituted cycloalkanes are considered to be desirable jet fuel components because of their high specific energy (MJ kg−1) relative to the minimum requirement shown in Table 5, and their high energy density (MJ L−1) which provides economic benefit to the user. As noted, oxygenates typically reduce the sooting tendency;399 however, oxygenates are not currently allowed in aviation fuels. The cetane number of alternative jet fuel blendstocks must be in the 35 to 60 range. This ensures adequate reactivity for cold start and high altitude relight, while also preventing premature ignition under high temperature, high pressure conditions.
Stability
Fuels should not polymerize or undergo oxidation or thermal degradation in storage or during handling. Generally, this eliminates aldehydes because of their tendency to undergo condensation reactions. Molecules with conjugated double bonds can undergo Diels–Alder type reactions characteristic of thermal instability, and molecules with bis-allylic double bonds have a low energy barrier pathway for hydrogen abstraction from the central carbon and the formation of a radical that can react with oxygen, leading to poor oxidation stability.400 Gasoline401 and jet fuel402 have well-established thermal oxidation stability tests. There are examples of proposed bioblendstocks that exhibit poor stability in the gasoline stability test, including 2-methyl furan and dimethyl furan, as well as cyclopentanone.367,403 All of the currently approved alternative jet fuels meet a higher stability requirement than conventional jet fuel. While stability is also important for diesel fuels, no accepted standard test for petroleum diesel has been developed, primarily because petroleum refinery diesel is widely regarded as stable. Because biodiesel can contain significant amounts of linoleic and linolenic fatty acid chains, both of which exhibit bis-allylic double bonds, a stability test was developed for the biodiesel blendstock and blends.404 Biodiesel is amenable to the use of antioxidant additives to ensure adequate stability,405,406 and this may also be true of other proposed biofuels with potential stability issues.Other requirements
Jet fuel has additional requirements for density and heat of combustion to achieve the desired aircraft range. Because cycloalkanes are denser than normal or branched alkanes, they are highly desirable components in jet fuel. Recently, a new pre-screening procedure for potential sustainable aviation fuels has been published, and identifies surface tension, viscosity, density, heat of combustion, the flashpoint, the cetane number, and the freezing point as important for predicting the behavior of jet fuels under critical jet engine operating conditions.373 Benzene is limited in gasoline because it is a known human carcinogen. In general, new fuels must have health effects no greater than conventional fuels and there have been occasional examples of proposed biofuels with toxicity or other health issues. For example, γ-valerolactone can be made by cellulose hydrolysis and hydrogenation of the resulting levulinic acid.407 This compound has been proposed as a biofuel and is estimated to have a RON of about 100.408 However, it is listed by the US Department of Justice as an addictive drug subject to abuse by ingestion.409 It is a central nervous system depressant similar to gamma-hydroxybutyrate (gHB). Given these properties, it may be difficult to commercialize γ-valerolactone as a biofuel.Neat fuels
Most of the discussion of fuel use provided above has described fuels for the scenario of blending into a petroleum refinery blendstock. However, to achieve large carbon emission reductions from the transportation sector – that is, near zero or even negative net carbon emissions – the blending strategy will likely have to give way to a strategy of using 100% biofuel or other low-net carbon fuels. In the market today, there are only a few examples of the commercial use of 100% (or near 100%) biofuel. The use of E85 was described above. The vehicles that can use E85 are referred to as ethanol-tolerant – they are not optimized to take advantage of the high RON and other properties of the E85 fuel. Volumes of E85 have remained small in the United States; however, in Brazil flex-fuel or ethanol tolerant vehicles dominate the car market and run on fuel containing 20% ethanol, hydrous (95%) ethanol, or any blend of the two, with ethanol capturing roughly 50% of the gasoline market. One can imagine a future where low-net carbon ethanol produced from cellulosic feedstocks is blended with a high vapor pressure bio-derived material to meet minimum vapor pressure requirements and used in vehicles with SI engines optimized for performance and efficiency on the high ethanol blend fuel. Another approach is cars powered with direct ethanol fuel cells that could run on neat ethanol or hydrous (95%) ethanol.410 In Brazil, where hydrous ethanol is already widely distributed, this may be a lower cost path to decarbonization than electrification – and could potentially also be applied to heavy-duty vehicles.Ethanol has also been used in its neat or near-neat form in compression ignition (diesel) engines. The use of ethanol as a diesel engine fuel is not an obvious choice because of ethanol's very low cetane number (and consequently very high RON). This is overcome using organo-nitrate fuel additives that increase fuel reactivity and specially designed engines with very high compression ratios. Since 1985 the Swedish engine maker Scania has produced heavy-duty engines designed to run on a fuel it refers to as ED95.411 This fuel is 95 vol% ethanol with 5 wt% ignition improver additive plus other additives for lubricity and to increase the flashpoint. In 2015 Scania launched the fourth generation of their ED95 engine, optimized with ethanol tolerant materials, fuel injection and combustion chamber geometry specifically designed for the fuel, and a compression ratio of 28:1 (compared to a typical diesel compression ratio of 17:1). Hundreds of vehicles employing this technology are currently in use, generally by centrally refueled fleets such as transit buses and industry delivery fleets. A second approach to use of ethanol in diesel engines is dual fuel combustion.412 This also requires an optimized engine design that uses port-fuel injection for ethanol that is pre-mixed with air during the intake stroke, and direct injection for a small quantity of diesel fuel that serves as a pilot to ignite the ethanol–air mixture. Results show high thermal efficiency similar to diesel combustion with large reductions in soot and NOx emissions. Additional research is required to fully optimize engine design for this approach and to maximize the ethanol energy fraction.
Neat biodiesel (B100) has been used in some applications; however, because of the relatively high cloud point and boiling point of B100, this is not common. Engine manufacturers typically approve the use of blends up to B20, and ongoing research is examining the properties and performance of higher blends. A hydrocarbon form of biomass-based diesel, commonly referred to as renewable diesel (RD), is produced by hydroprocessing of esters and fatty acids, the same feedstocks used to make biodiesel. This fuel consists entirely of normal and iso-paraffins413 and can meet all requirements of the ASTM D975 standard for diesel fuels. Many engine makers approve the use of this fuel at 100% (RD100) if it meets this ASTM standard.
None of the synthetic hydrocarbon fuels approved for use in jet fuel is allowed at more than 50 vol%. However, turbine engine and air frame makers are committed to moving to 100% sustainable fuels in the coming decades.414
Experience using neat biofuels (or other forms of low-carbon fuels) suggests that if these fuels can meet existing ASTM standards then they could be used in existing engines without modification – as is the case for RD. Fuels that cannot meet existing fuel standards may require the development of optimized engines to be used in neat form. This argues for targeting products from biomass, in the near-term, that meet current ASTM standard requirements and will not require a new engine technology. In the longer term, engines can be developed to tolerate or even exploit unique properties of biofuels that are significantly different from the fuels of today. The development of engine technology specific for a neat (or nearly neat) biofuel has only been pursued for the case of ethanol, where commercial production is highly developed, and projections of future production volumes are large. If a new biofuel with advantageous properties in neat form could achieve a similar technology readiness level to ethanol, the development of dedicated engines to take advantage of it might become a possibility.
Survey of potential biologically-derived fuel molecules
Careful evaluation of possible production routes of hydrocarbon and oxygenated products led to a list of candidate fuel molecules (those listed in Table 6).Status
Ethanol is the only widely available commercial biofuel for use in SI engines and is widely used globally at low to moderate blend levels in petroleum gasoline. A dramatic expansion in ethanol production is possible using lignocellulosic biomass, and vehicles that are at least tolerant of much higher levels of ethanol have been produced in the United States and currently dominate the market in Brazil. Two biofuels produced from fats and oils (RD and biodiesel) are also widely used at low blend levels in petroleum diesel. But feedstock resources for expanded production are relatively limited, and fuels produced from more widely available lignocellulosic biomass will have different properties, benefits, and limitations. Seven alternative jet fuel blendstocks have been approved, many of which could be produced via sustainable pathways. Yet the production volumes remain relatively small.Challenges and opportunities
Decarbonization goals require the replacement of high carbon emitting fuels with low carbon alternatives – even if engines are not fully optimized for their use. For light duty automobiles a switch to fully electric powertrains is likely over the next few decades and can address decarbonization as the electric grid also expands and decarbonizes. In the interim, expanded use of low-net carbon ethanol from lignocellulosic biomass could reduce carbon emissions from existing vehicles and near future vehicles designed to tolerate high level ethanol blends. As the fleet electrifies ethanol production facilities could refit to produce other fuels from their feedstocks, or convert ethanol to jet and diesel hydrocarbons, for example.415,416A critical challenge is the development of viable technologies for decarbonization of larger vehicles such as long-haul trucks, freight rail, large marine vessels, and aircraft. New biomass derived diesel fuels will likely exhibit significantly different properties from the RD and biodiesel that supply these markets today. The development of new or modified fuel quality standards is likely to be required. Similarly for aviation fuels, new production pathways need to be developed and moved through the relatively arduous alternative jet fuel blendstock qualification process, and the yet to be defined neat alternative jet fuel qualification process.
A wide variety of molecular structures is available from microbial metabolism and subsequent catalytic upgrading. This opens the opportunity to produce both hydrocarbons and oxygenated compounds with unique and advantageous properties not accessible by conventional routes. Advantageous properties include synergistic octane blending, soot reduction, and higher volumetric energy content (for cycloalkanes versus branched alkanes in jet fuel): for example, the extremely high blending octane numbers of alkyl furans, and polyoxymethylene ethers that eliminate soot formation in diesel combustion.367,396 Some researchers have begun to carefully target molecular structures exhibiting desirable properties for specific fuel applications. Vardon and colleagues270,346,347 have described a ‘fuel property first’ design approach to production of diesel blendstocks with advantageous properties from short chain carboxylic acids. It is also possible to imagine engine modifications being made in response to the fuel molecules that offer economic or other benefits. However, this would likely require a clearer indication of what these molecules and benefits are than we have today, together with strong motivation to adopt new fuel molecules. The main candidate motivation we see is climate stabilization. Aggressive expansion of biofuels in a climate-constrained scenario would benefit from and likely require that GHG emission reductions be reflected in their prices.
VIII. Paths to large scale cellulosic biofuels
Based on the product surveys presented in Section II (Fig. 2 and 3), there are seven molecules that are more reduced than carbohydrate, produced anaerobically, and have at least one of the following desirable features: a maximum reported yield of ≥75% stoichiometric, a maximum reported broth titer of ≥40 g L−1, and a boiling point lower than that of water. As shown in Fig. 14, three of these molecules are monohydroxy alcohols, three are dihydroxy alcohols, one is an organic acid, and all seven of these molecules have 4 carbons or less. As the only product that has all three features, ethanol occupies a singular space in the bioproduct landscape, is the lowest cost liquid fuel or fuel intermediate produced biologically today, and is in our view likely to remain so. Isobutanol is actively being investigated for fuel applications,296 but is not included in Fig. 14 because there are no reports in the literature to date of it being produced in the absence of aeration with yields above 75%. | ||
| Fig. 14 Potential fuel molecules/precursors with desirable properties (see text for explanation). | ||
Other things being equal, aeration increases the cost of biological conversion (Section II), and in situ product recovery increases the cost and complexity of separation (Section V). Although biological processing in the presence of residual lignocellulose solids is expected to be less expensive than removing solids in advance of biological processing (Section II), both aeration and ISPR are more challenging to implement in the presence of solids as discussed in Sections II and V respectively. Nearly all biologically produced molecules with ≥4 carbons produced at more than very low yields and titers are produced aerobically (Fig. 2 and 3). Increasing the chain length of candidate fuel molecules with a given R-group is generally correlated with decreased titers due to either toxicity or solubility constraints (Section IV).
For multiple compounding reasons, features of small molecules make it less expensive to produce them biologically than large molecules, and this is particularly true for production from lignocellulose.
Approaching cellulosic biofuel production from the point of view of fuel utility rather than biology, the need for biofuels to achieve climate stabilization can be ranked roughly as follows based on the unavailability of alternatives: aviation > ocean shipping > long-haul trucking > light duty vehicles. This ranking is, however, inversely related to the number of candidate biologically-produced fuel molecules (Table 6). Moreover, large fuel molecules are used today, and generally preferred over small fuel molecules, for the fuel applications for which biofuels are most likely needed to mitigate climate change.
In consideration of these observations together with the emergence of battery-powered light duty vehicles over the past decade, there is a mismatch between the fuel molecules biological conversion most readily provides, at least today, and the fuels humanity would most value obtaining from cellulosic biomass. Reconciling this gap will be a key determinant of whether and how biofuels play a large role in enabling a carbon-neutral transport sector.22
Fig. 15 maps research and development strategies for cellulosic biofuel production featuring biological transformations onto a two-dimensional landscape defined in terms of feedstock (from easily fermented feedstocks to lignocellulose) and product (small fuel molecules to large fuel molecules) properties. Starting with the lower left corner, commercial production of small fuel molecules from easily fermented feedstocks (e.g. corn kernels and juice from sugarcane or sugar beets) with costs that are competitive with fossil fuels on an energy basis in some markets has been realized in the case of ethanol. These objectives have in general not yet been achieved for other small fuel molecules but are being actively pursued.
 | ||
| Fig. 15 Alternative research and commercialization paths. Green denotes biological conversion; magenta denotes non-biological (catalytic or thermochemical) conversion. | ||
From this departure point, there are multiple research and commercialization paths by which to approach the goal of producing large fuel molecules from cellulosic biomass. Three such paths rely entirely on biological conversion. Path 1 involves activities aimed at producing large molecules from lignocellulose as an immediate objective rather than proceeding in a stepwise fashion. Path 2 features first resolving the technical and economic challenges to enable industrially feasible production of small molecules from lignocellulose, and then addressing the distinctive set of technical and economic challenges associated with production of large molecules from lignocellulose. Path 3 approaches these two objectives in the opposite order – that is, first large molecules from easily-fermented feedstocks and then large molecules from lignocellulose. Two paths are also shown that include non-biological processing. In path 4, production of small fuel molecules from lignocellulose is approached via improved biological conversion, and conversion of lignocellulose-derived small molecules to large fuel molecules is approached by non-biological catalysis. Path 5 features conversion of easily fermented feedstocks to large fuel molecules via improved catalytic or thermochemical processes.
Stepwise-approaches – that is, paths other than path 1 in Fig. 15 – avoid trying to do too many new things at once, and are more practical and more promising in our view.20 In particular, it makes sense for research and commercialization efforts aimed at advancing biological conversion of lignocellulose to small fuel molecules to be approached without the substantial added complexities, challenges and risks associated with production of high molecular weight fuels. Conversely, it makes sense for efforts aimed at advancing biological conversion of easily fermented feedstocks to large molecules to be approached without the substantial complexities, challenges, and risks associated with small fuel molecule production from lignocellulose. Consistent with these observations, we know of few efforts in either the research or commercialization domain involving biologically-mediated production of large fuel molecules based on path 1.
While biological production of large molecules has revolutionized the pharmaceutical industry, industrially feasible production of large molecules for fuel applications is challenging and has yet to be demonstrated. Such production is, however, required in order for paths 2 and 3 to be commercially feasible.
Path 4 becomes attractive to the extent that (a) biological production of large fuel molecules from lignocellulose remains more costly than biological production of small molecules and (b) catalytic conversion of small biologically derived intermediates can be accomplished at low cost. The former outcome seems likely to us based on fundamental considerations that would appear unlikely to change, as considered above. Catalytic conversion of small biologically derived intermediates can be accomplished by a variety of mechanisms (see Section VI) and involves chemical catalysis in reactors that are relatively small, do not incur large costs in other applications, and can likely operate under progressively more mild conditions as they receive more attention. Ethanol conversion to hydrocarbons without adding substantially increasing cost per GJ appears to be within sight today, although further advances will be necessary to better align the distribution of hydrocarbon products with requirements for aviation and (especially) diesel engines.311,321,417 Conversion of ethanol to hydrocarbons has the added advantage that it can build on the large existing production capacity and thus conceivably expand quite rapidly once technological obstacles have been overcome.
Path 5 is equivalent to path 4 except for starting with small fuel molecules from easily-fermented feedstocks rather than lignocellulose. This path represents an important step and could be implemented commercially based on existing ethanol production capacity independent of advances in biological conversion of lignocellulose or biological production of large fuel molecules.
Cost-competitive biological production of liquid fuels or liquid fuel precursors from lignocellulose has proved to be challenging thus far, even for small molecules. These challenges have been encountered in the context of a processing paradigm featuring thermochemical pretreatment and added fungal cellulase, which has been the focus of intensive research and development for over the past three decades. As noted herein (Sections II and III), alternative process concepts have been proposed that avoid both thermochemical pretreatment and added enzymes. Moreover, it has recently become clear that Nature offers cellulase enzyme systems that are more effective than fungal cellulase at lignocellulose deconstruction, albeit thus far under laboratory conditions. The possibility of utilizing superior biocatalysts, the existence of alternative biological processing paradigms, and combining biological production of small molecules with catalytic production of larger molecules together provide important optionality for continuing efforts to realize the widely anticipated, but thus-far elusive potential of cellulosic biofuels.
Author contributions
LRL and GTB: conceptualization, lead responsibility for writing and editing the overall manuscript, and lead responsibility for writing some sections. AMG, LJ, EMK, and RLM: lead responsibility for writing some sections, review, and editing the overall manuscript. DA-N, YJB, BHD, MEH, EKH, MSL, DGO, YRL, CTT, GAT, DRV, and CEW: review and editing the overall manuscript. CF, CYN, VU, and LW: data curation. Funding acquisition: led by GAT and BHD, with contributions by LRL, GTB, AMG, DAN, YJB, MEH, EKH, and DGO.Conflicts of interest
LRL is involved in a cellulosic biofuel start-up company. CEW and BHD are involved in a catalytic upgrading start-up company.Acknowledgements
We acknowledge funding from the Center for Bioenergy Innovation, which is a U.S. Department of Energy Bioenergy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science. LJ, EMK, DRV, and GTB also acknowledge support from the U.S. DOE Bioenergy Technologies Office (BETO), and RLM acknowledges support from BETO and the U.S. DOE Vehicle Technologies Office under the Co-Optimization of Fuels and Engines Consortium, all via Contract No. DE-AC36-08GO28308 with the National Renewable Energy Laboratory. The authors thank Brett Hopwood (ORNL) for expert assistance with figure preparation.References
- L. R. Lynd, X. Liang, M. J. Biddy, A. Allee, H. Cai, T. Foust, M. E. Himmel, M. S. Laser, M. Wang and C. E. Wyman, Cellulosic ethanol: Status and innovation, Curr. Opin. Biotechnol, 2017, 45, 202–211 CrossRef CAS PubMed.
- B. E. Dale, J. E. Anderson, R. C. Brown, S. Csonka, V. H. Dale, G. Herwick, R. D. Jackson, N. Jordan, S. Kaffka and K. L. Kline, Take a closer look: Biofuels can support environmental, economic and social goals, ACS Publications, 2014 Search PubMed.
- G. W. Huber, S. Iborra and A. Corma, Synthesis of transportation fuels from biomass: chemistry, catalysts, and engineering, Chem. Rev., 2006, 106(9), 4044–4098 CrossRef CAS PubMed.
- L. R. Lynd, M. S. Laser, D. Bransby, B. E. Dale, B. Davison, R. Hamilton, M. Himmel, M. Keller, J. D. McMillan and J. Sheehan, et al., How biotech can transform biofuels, Nat. Biotechnol., 2008, 26(2), 169–172, DOI:10.1038/nbt0208-169 , [accessed 2019 Jul 2].
- N. Jordan, G. Boody, W. Broussard, J. D. Glover, D. Keeney, B. H. McCown, G. McIsaac, M. Muller, H. Murray and J. Neal, Sustainable development of the agricultural bio-economy, Science, 2007, 316(5831), 1570–1571 CrossRef CAS PubMed.
- A. Turhollow, R. Perlack, L. Eaton, M. Langholtz, C. Brandt, M. Downing, L. Wright, K. Skog, C. Hellwinckel and B. Stokes, et al., The updated billion-ton resource assessment, Biomass Bioenergy, 2014, 70, 149–164, DOI:10.1016/j.biombioe.2014.09.007 , [accessed 2019 Oct 8].
- J. Q. Bond, A. A. Upadhye, H. Olcay, G. A. Tompsett, J. Jae, R. Xing, D. M. Alonso, D. Wang, T. Zhang and R. Kumar, Production of renewable jet fuel range alkanes and commodity chemicals from integrated catalytic processing of biomass, Energy Environ. Sci., 2014, 7(4), 1500–1523 RSC.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Catalytic conversion of biomass to biofuels, Green Chem., 2010, 12(9), 1493, 10.1039/c004654j , [accessed 2021 Jun 30].
- K. Asimakopoulos, H. N. Gavala and I. V. Skiadas, Reactor systems for syngas fermentation processes: A review, Chem. Eng. J., 2018, 348, 732–744 CrossRef CAS.
- P. Dürre and B. J. Eikmanns, C1-carbon sources for chemical and fuel production by microbial gas fermentation, Curr. Opin. Biotechnol, 2015, 35, 63–72 CrossRef PubMed.
- F. Liew, M. E. Martin, R. C. Tappel, B. D. Heijstra, C. Mihalcea and M. Köpke, Gas fermentation—A flexible platform for commercial scale production of low-carbon-fuels and chemicals from waste and renewable feedstocks, Front. Microbiol., 2016, 7, 694 Search PubMed.
- D. Verma, A. Singla, B. Lal and P. M. Sarma, Conversion of biomass-generated syngas into next-generation liquid transport fuels through microbial intervention: Potential and current status, Curr. Sci., 2016, 329–336 CrossRef CAS.
- Z.-H. Liu, R. K. Le, M. Kosa, B. Yang, J. Yuan and A. J. Ragauskas, Identifying and creating pathways to improve biological lignin valorization, Renewable Sustainable Energy Rev., 2019, 105, 349–362 CrossRef CAS.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. Bruijnincx and B. M. Weckhuysen, Paving the way for lignin valorisation: Recent advances in bioengineering, biorefining and catalysis, Angew. Chem., Int. Ed., 2016, 55(29), 8164–8215 CrossRef CAS PubMed.
- W. Schutyser, T. Renders, S. Van den Bosch, S.-F. Koelewijn, G. T. Beckham and B. F. Sels, Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading, Chem. Soc. Rev., 2018, 47(3), 852–908 RSC.
- J. Zakzeski, P. C. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, The catalytic valorization of lignin for the production of renewable chemicals, Chem. Rev., 2010, 110(6), 3552–3599 CrossRef CAS PubMed.
- M. M. Wright and R. C. Brown, Comparative economics of biorefineries based on the biochemical and thermochemical platforms, Biofuels, Bioprod. Biorefining, 2007, 1(1), 49–56, DOI:10.1002/bbb.8 , [accessed 2021 Oct 5].
- T. D. Foust, A. Aden, A. Dutta and S. Phillips, An economic and environmental comparison of a biochemical and a thermochemical lignocellulosic ethanol conversion processes, Cellulose, 2009, 16(4), 547–565, DOI:10.1007/s10570-009-9317-x , [accessed 2021 Oct 5].
- M. Laser, H. Jin, K. Jayawardhana, B. E. Dale and L. R. Lynd, Projected mature technology scenarios for conversion of cellulosic biomass to ethanol with coproduction thermochemical fuels, power, and/or animal feed protein, Biofuels, Bioproducts and Biorefining, 2009, 3(2), 231–246, DOI:10.1002/bbb.131 , [accessed 2021 Oct 5].
- L. R. Lynd, The grand challenge of cellulosic biofuels, Nat. Biotechnol., 2017, 35(10), 912 CrossRef CAS PubMed.
- Advanced biofuels: What holds them back?/publications/2019/Nov/Advanced-biofuels-What-holds-them-back, https://irena.org/publications/2019/Nov/Advanced-biofuels-What-holds-them-back, [accessed 2021 Aug 13].
- A. Brown, ‘Technology Roadmap: Delivering Sustainable Bioenergy’, International Energy Agency, Paris, 2017, https://www.iea.org/reports/technology-roadmap-delivering-sustainable-bioenergy.
- L. M. Fulton, L. R. Lynd, A. Körner, N. Greene and L. R. Tonachel, The need for biofuels as part of a low carbon energy future, Biofuels, Bioproducts Biorefining, 2015, 9(5), 476–483 CrossRef CAS.
- D. J. Gaspar, C. J. Mueller, R. L. McCormick, J. Martin, S. Som, G. M. Magnotti, J. Burton, D. Vardon, V. Dagle, T. L. Alleman, et al., Top 13 Blendstocks Derived from Biomass for Mixing-Controlled Compression-Ignition (Diesel) Engines: Bioblendstocks with Potential for Decreased Emissions and Improved Operability. Pacific Northwest National Lab. (PNNL), Richland, WA (United States); 2021. Report No.: PNNL-31421 DOI:10.2172/1806564.
- https://www.osti.gov/biblio/1660415-sustainable-aviation-fuel-review-technical-pathways – Google Search, [accessed 2021 Aug 13]. https://www.google.com/search?q=.+https%3A%2F%2Fwww.osti.gov%2Fbiblio%2F1660415-sustainable-aviation-fuel-review-technical-pathways&rlz=1C1GCEA_enUS890US890&oq=.++https%3A%2F%2Fwww.osti.gov%2Fbiblio%2F1660415-sustainable-aviation-fuel-review-technical-pathways&aqs=chrome..69i57.1243j0j15&sourceid=chrome&ie=UTF-8.
- S. P. Chundawat, G. T. Beckham, M. E. Himmel and B. E. Dale, Deconstruction of lignocellulosic biomass to fuels and chemicals, Ann. Rev. Chem. Biomol. Eng., 2011, 2, 121–145 CrossRef CAS PubMed.
- A. Wiselogel, S. Tyson and D. Johnson, Biomass feedstock resources and composition, in Handbook on Bioethanol, Routledge, 2018, pp. 105–118 Search PubMed.
- X. Zhao, L. Zhang and D. Liu, Biomass recalcitrance. Part I: the chemical compositions and physical structures affecting the enzymatic hydrolysis of lignocellulose, Biofuels, Bioproducts and Biorefining, 2012, 6(4), 465–482, DOI:10.1002/bbb.1331 , [accessed 2019 Oct 16].
- M. E. Himmel, S.-Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Biomass recalcitrance: Engineering plants and enzymes for biofuels production, Science, 2007, 315(5813), 804–807 CrossRef CAS PubMed.
- L. R. Lynd, C. E. Wyman and T. U. Gerngross, Biocommodity engineering, Biotechnol. Prog., 1999, 15(5), 777–793 CrossRef CAS PubMed.
- D8.2_S2Biom_Vision_for_1_billion_tonnes_biomass_2030.pdf, [accessed 2021 Nov 15], https://www.s2biom.eu/images/Publications/D8.2_S2Biom_Vision_for_1_billion_tonnes_biomass_2030.pdf.
- J. Thompson and W. E. Tyner, Corn Stover for Bioenergy Production: Cost Estimates and Farmer Supply Response, Purdue University, Purdue Extension, 2020, https://nam12.safelinks.protection.outlook.com/?url=https%3A%2F%2Fwww.extension.purdue.edu%2Fextmedia%2FEC%2FRE-3-W.pdf&data=02%7C01%7Cmarge.heggison%40dartmouth.edu%7Ca318264de5c84ea72e0b08d8607fdc40%7C995b093648d640e5a31ebf689ec9446f%7C0%7C1%7C637365450328914337&sdata=qUpXKLejKMfzb8bFpnr8k4y%2FGShNxsOTNisw%2BVvZTuE%3D&reserved=0 Search PubMed.
- A. Brown, Advanced Biofuels – Potential for Cost Reduction, https://www.ieabioenergy.com/wp-content/uploads/2020/02/T41_CostReductionBiofuels-11_02_19-final.pdf.
- National Agricultural Statistics Service, United States Department of Agriculture, 2020, https://www.nass.usda.gov/Charts_and_Maps/Agricultural_Prices/pricecn.php.
- L. Lynd, Application of material and energy balances to biological systems, Chem. Eng. Probl. Biotechnol., 1989, 29–49 Search PubMed.
- Global ethanol as a percent of liquid transport fuels: Global ethanol production, 2015: 25.7 BGY; 1.95 quad Btu. U.S. Department of Energy, Alternative Fuels Data Center, https://afdc.energy.gov/data/categories/biofuels-production.
- EIA projects energy consumption in air transportation to increase through 2050 – Today in Energy – U.S. Energy Information Administration (EIA), [accessed 2020 Sep 2] https://www.eia.gov/todayinenergy/detail.php?id=41913.
- F. Creutzig, N. H. Ravindranath, G. Berndes, S. Bolwig, R. Bright, F. Cherubini, H. Chum, E. Corbera, M. Delucchi and A. Faaij, Bioenergy and climate change mitigation: an assessment, GCB Bioenergy, 2015, 7(5), 916–944 CrossRef CAS.
- L. R. Lynd, Converting plant biomass to fuels and commodity chemicals in South Africa: A third chapter?, S. Afr. J. Sci., 2003, 11 Search PubMed.
- M. J. Biddy, R. Davis, D. Humbird, L. Tao, N. Dowe, M. T. Guarnieri, J. G. Linger, E. M. Karp, D. Salvachúa and D. R. Vardon, The techno-economic basis for coproduct manufacturing to enable hydrocarbon fuel production from lignocellulosic biomass, ACS Sustainable Chem. Eng., 2016, 4(6), 3196–3211 CrossRef CAS.
- L. E. Erickson, S. E. Selga and U. E. Viesturs, Application of mass and energy balance regularities to product formation, Biotechnol. Bioeng., 1978, 20(10), 1623–1638 CrossRef CAS.
- D. Dugar and G. Stephanopoulos, Relative potential of biosynthetic pathways for biofuels and bio-based products, Nat. Biotechnol., 2011, 29(12), 1074 CrossRef CAS PubMed.
- M. Heitz, E. Capek-Menard, P. G. Koeberle, J. Gagne, E. Chornet, R. P. Overend, J. D. Taylor and E. Yu, Fractionation of Populus tremuloides at the pilot plant scale: Optimization of steam pretreatment conditions using the STAKE II technology, Bioresour. Technol., 1991, 35(1), 23–32 CrossRef CAS.
- N. Mosier, R. Hendrickson, N. Ho, M. Sedlak and M. R. Ladisch, Optimization of pH controlled liquid hot water pretreatment of corn stover, Bioresour. Technol., 2005, 96(18), 1986–1993 CrossRef CAS PubMed.
- T.-C. Hsu, G.-L. Guo, W.-H. Chen and W.-S. Hwang, Effect of dilute acid pretreatment of rice straw on structural properties and enzymatic hydrolysis, Bioresour. Technol., 2010, 101(13), 4907–4913 CrossRef CAS PubMed.
- B. C. Saha, L. B. Iten, M. A. Cotta and Y. V. Wu, Dilute acid pretreatment, enzymatic saccharification and fermentation of wheat straw to ethanol, Process Biochem., 2005, 40(12), 3693–3700 CrossRef CAS.
- V. Balan, B. Bals, S. P. Chundawat, D. Marshall and B. E. Dale, Lignocellulosic biomass pretreatment using AFEX, Biofuels, 2009, 61–77 CAS.
- S. Singh, B. A. Simmons and K. P. Vogel, Visualization of biomass solubilization and cellulose regeneration during ionic liquid pretreatment of switchgrass, Biotechnol. Bioeng., 2009, 104(1), 68–75 CrossRef CAS PubMed.
- T. Y. Nguyen, C. M. Cai, R. Kumar and C. E. Wyman, Co-solvent pretreatment reduces costly enzyme requirements for high sugar and ethanol yields from lignocellulosic biomass, ChemSusChem, 2015, 8(10), 1716–1725 CrossRef CAS PubMed.
- M. W. Lau and B. E. Dale, Cellulosic ethanol production from AFEX-treated corn stover using Saccharomyces cerevisiae 424A(LNH-ST), Proc. Natl. Acad. Sci. U. S. A., 2009, 106(5), 1368–1373, DOI:10.1073/pnas.0812364106 , [accessed 2021 Feb 4].
- A. Mohagheghi, N. Dowe, D. Schell, Y.-C. Chou, C. Eddy and M. Zhang, Performance of a newly developed integrant of Zymomonas mobilis for ethanol production on corn stover hydrolysate, Biotechnol. Lett., 2004, 26(4), 321–325 CrossRef CAS PubMed.
- B. C. Saha, N. N. Nichols, N. Qureshi and M. A. Cotta, Comparison of separate hydrolysis and fermentation and simultaneous saccharification and fermentation processes for ethanol production from wheat straw by recombinant Escherichia coli strain FBR5, Appl. Microbiol. Biotechnol., 2011, 92(4), 865–874 CrossRef CAS PubMed.
- K. Öhgren, R. Bura, G. Lesnicki, J. Saddler and G. Zacchi, A comparison between simultaneous saccharification and fermentation and separate hydrolysis and fermentation using steam-pretreated corn stover, Process Biochem., 2007, 42(5), 834–839 CrossRef.
- J. Méndez, D. de França Passos, D. Wischral, L. F. Modesto and N. Pereira Jr, Second-generation ethanol production by separate hydrolysis and fermentation from sugarcane bagasse with cellulose hydrolysis using a customized enzyme cocktail, Biofuels, 2019, 1225–1231 Search PubMed.
- R. Gupta, K. K. Sharma and R. C. Kuhad, Separate hydrolysis and fermentation (SHF) of Prosopis juliflora, a woody substrate, for the production of cellulosic ethanol by Saccharomyces cerevisiae and Pichia stipitis-NCIM 3498, Bioresour. Technol., 2009, 100(3), 1214–1220 CrossRef CAS PubMed.
- F. Xu, J. Sun, N. M. Konda, J. Shi, T. Dutta, C. D. Scown, B. A. Simmons and S. Singh, Transforming biomass conversion with ionic liquids: process intensification and the development of a high-gravity, one-pot process for the production of cellulosic ethanol, Energy Environ. Sci., 2016, 9(3), 1042–1049 RSC.
- B. C. Saha, N. N. Nichols, N. Qureshi, G. J. Kennedy, L. B. Iten and M. A. Cotta, Pilot scale conversion of wheat straw to ethanol via simultaneous saccharification and fermentation, Bioresour. Technol., 2015, 175, 17–22 CrossRef CAS PubMed.
- D. Cannella, P. V. Sveding and H. Jørgensen, PEI detoxification of pretreated spruce for high solids ethanol fermentation, Appl. Energy, 2014, 132, 394–403 CrossRef CAS.
- L. R. Lynd, P. J. Weimer, W. H. Van Zyl and I. S. Pretorius, Microbial cellulose utilization: Fundamentals and biotechnology, Microbiol. Mol. Biol. Rev., 2002, 66(3), 506–577 CrossRef CAS PubMed.
- D. G. Olson, J. E. McBride, A. J. Shaw and L. R. Lynd, Recent progress in consolidated bioprocessing, Curr. Opin. Biotechnol, 2012, 23(3), 396–405 CrossRef CAS PubMed.
- G. Salehi Jouzani and M. J. Taherzadeh, Advances in consolidated bioprocessing systems for bioethanol and butanol production from biomass: A comprehensive review, Biofuel Res. J., 2015, 2(1), 152–195 CrossRef.
- M. L. Balch, E. K. Holwerda, M. F. Davis, R. W. Sykes, R. M. Happs, R. Kumar, C. E. Wyman and L. R. Lynd, Lignocellulose fermentation and residual solids characterization for senescent switchgrass fermentation by Clostridium thermocellum in the presence and absence of continuous in situ ball-milling, Energy Environ. Sci., 2017, 10(5), 1252–1261 RSC.
- E. K. Holwerda, R. S. Worthen, N. Kothari, R. C. Lasky, B. H. Davison, C. Fu, Z.-Y. Wang, R. A. Dixon, A. K. Biswal and D. Mohnen, Multiple levers for overcoming the recalcitrance of lignocellulosic biomass, Biotechnol. Biofuels, 2019, 12(1), 15 CrossRef PubMed.
- D. B. Hodge, M. N. Karim, D. J. Schell and J. D. McMillan, Model-based fed-batch for high-solids enzymatic cellulose hydrolysis, Appl. Biochem. Biotechnol., 2008, 152(1), 88, DOI:10.1007/s12010-008-8217-0 , [accessed 2020 Oct 22].
- H. Jørgensen, J. Vibe-Pedersen, J. Larsen and C. Felby, Liquefaction of lignocellulose at high-solids concentrations, Biotechnol. Bioeng., 2007, 96(5), 862–870, DOI:10.1002/bit.21115 , [accessed 2020 Oct 22].
- A. A. Modenbach and S. E. Nokes, Enzymatic hydrolysis of biomass at high-solids loadings – A review, Biomass Bioenergy, 2013, 56, 526–544, DOI:10.1016/j.biombioe.2013.05.031 , http://www.sciencedirect.com/science/article/pii/S0961953413002948, [accessed 2020 Oct 22].
- M. J. Cardona, E. J. Tozzi, N. Karuna, T. Jeoh, R. L. Powell and M. J. McCarthy, A process for energy-efficient high-solids fed-batch enzymatic liquefaction of cellulosic biomass, Bioresour. Technol., 2015, 198, 488–496, DOI:10.1016/j.biortech.2015.09.042 , http://www.sciencedirect.com/science/article/pii/S0960852415013139, [accessed 2020 Oct 22].
- D. M. Lavenson, E. J. Tozzi, N. Karuna, T. Jeoh, R. L. Powell and M. J. McCarthy, The effect of mixing on the liquefaction and saccharification of cellulosic fibers, Bioresour. Technol., 2012, 111, 240–247 CrossRef CAS PubMed.
- P. A. Skovgaard, L. G. Thygesen, H. Jørgensen, M. Cardona, E. Tozzi, M. McCarthy, M. Siika-Aho and T. Jeoh, The role of endoglucanase and endoxylanase in liquefaction of hydrothermally pretreated wheat straw, Biotechnol. Prog., 2014, 30(4), 923–931 CrossRef CAS PubMed.
- S. Ghosh, E. K. Holwerda, R. S. Worthen, L. R. Lynd and B. P. Epps, Rheological properties of corn stover slurries during fermentation by Clostridium thermocellum, Biotechnol. Biofuels, 2018, 11(1), 246 CrossRef PubMed.
- X. Chen, E. Kuhn, E. W. Jennings, R. Nelson, L. Tao, M. Zhang and M. P. Tucker, DMR (deacetylation and mechanical refining) processing of corn stover achieves high monomeric sugar concentrations (230 g L−1) during enzymatic hydrolysis and high ethanol concentrations (>10% v/v) during fermentation without hydrolysate purification or concentration, Energy Environ. Sci., 2016, 9(4), 1237–1245 RSC.
- M. D. Stowers The US ethanol industry. Regional Economic Development, 2009, pp. 3–11 Search PubMed.
- L. Carlos, T. Olitta and S. Nitsche, Ethanol production in Brazil: The industrial process and its impact on yeast fermentation, in Dos Santos Bernardes M. A., ed. Biofuel Production-Recent Developments and Prospects, InTech, 2011, http://www.intechopen.com/books/biofuel-production-recent-developments-and-prospects/ethanol-production-in-brazil-the-industrial-process-and-its-impact-on-yeast-fermentation DOI:10.5772/17047.
- S. Gunukula, T. Runge and R. Anex, Assessment of biocatalytic production parameters to determine economic and environmental viability, ACS Sustainable Chem. Eng., 2017, 5(9), 8119–8126 CrossRef CAS.
- R. A. Weusthuis, I. Lamot, J. van der Oost and J. P. Sanders, Microbial production of bulk chemicals: Development of anaerobic processes, Trends Biotechnol., 2011, 29(4), 153–158 CrossRef CAS PubMed.
- T. L. Junqueira, M. F. Chagas, V. L. Gouveia, M. C. Rezende, M. D. Watanabe, C. D. Jesus, O. Cavalett, A. Y. Milanez and A. Bonomi, Techno-economic analysis and climate change impacts of sugarcane biorefineries considering different time horizons, Biotechnol. Biofuels, 2017, 10(1), 50 CrossRef PubMed.
- D. Humbird, R. Davis, L. Tao, C. Kinchin, D. Hsu, A. Aden, P. Schoen, J. Lukas, B. Olthof and M. Worley, Process design and economics for biochemical conversion of lignocellulosic biomass to ethanol: Dilute-acid pretreatment and enzymatic hydrolysis of corn stover, National Renewable Energy Lab.(NREL), Golden, CO, 2011 Search PubMed.
- WBA-globalreport-56ppa4_digital.pdf, [accessed 2021 Nov 15] https://www.worldbiogasassociation.org/wp-content/uploads/2019/07/WBA-globalreport-56ppa4_digital.pdf.
- IAEE Online Conference Proceedings, http://www.iaee.org/proceedings/article/17363, [accessed 2021 Nov 15].
- Y. J. Bomble, C.-Y. Lin, A. Amore, H. Wei, E. K. Holwerda, P. N. Ciesielski, B. S. Donohoe, S. R. Decker, L. R. Lynd and M. E. Himmel, Lignocellulose deconstruction in the biosphere, Curr. Opin. Chem. Biol., 2017, 41, 61–70, DOI:10.1016/j.cbpa.2017.10.013 (Mechanistic Biology Energy), http://www.sciencedirect.com/science/article/pii/S1367593117300960, [accessed 2019 Jul 1].
- X. Meng and A. J. Ragauskas, Recent advances in understanding the role of cellulose accessibility in enzymatic hydrolysis of lignocellulosic substrates, Curr. Opin. Biotechnol., 2014, 27, 150–158 CrossRef CAS PubMed.
- Y.-H. P. Zhang and L. R. Lynd, Toward an aggregated understanding of enzymatic hydrolysis of cellulose: Noncomplexed cellulase systems, Biotechnol. Bioeng., 2004, 88(7), 797–824 CrossRef CAS PubMed.
- S. M. Cragg, G. T. Beckham, N. C. Bruce, T. D. Bugg, D. L. Distel, P. Dupree, A. G. Etxabe, B. S. Goodell, J. Jellison and J. E. McGeehan, et al., Lignocellulose degradation mechanisms across the tree of life, Curr. Opin. Chem. Biol., 2015, 29, 108–119, DOI:10.1016/j.cbpa.2015.10.018 (Energy Mechanistic Biology), http://www.sciencedirect.com/science/article/pii/S1367593115001246, [accessed 2020 Jul 16].
- S.-Y. Leu and J. Y. Zhu, Substrate-related factors affecting enzymatic saccharification of lignocelluloses: Our recent understanding, Bioenergy Res., 2013, 6(2), 405–415 CrossRef CAS.
- C. M. Payne, B. C. Knott, H. B. Mayes, H. Hansson, M. E. Himmel, M. Sandgren, J. Stahlberg and G. T. Beckham, Fungal cellulases, Chem. Rev., 2015, 115(3), 1308–1448 CrossRef CAS PubMed.
- D. de França Passos, N. Pereira Jr and A. M. de Castro, A comparative review of recent advances in cellulases production by Aspergillus, Penicillium and Trichoderma strains and their use for lignocellulose deconstruction, Curr. Opin. Green Sustainable Chem., 2018, 14, 60–66 CrossRef.
- T. Ezaki, K. Nishinari, M. Samejima and K. Igarashi, Bridging the micro–macro gap between single-molecular behavior and bulk hydrolysis properties of cellulase, Phys. Rev. Lett., 2019, 122(9), 098102 CrossRef CAS PubMed.
- S. E. Levine, J. M. Fox, H. W. Blanch and D. S. Clark, A mechanistic model of the enzymatic hydrolysis of cellulose, Biotechnol. Bioeng., 2010, 107(1), 37–51 CrossRef CAS PubMed.
- R. Kumar, M. Tabatabaei, K. Karimi and I. Sárvári Horváth, Recent updates on lignocellulosic biomass derived ethanol – A review, Biofuel Res. J., 2016, 3(1), 347–356, DOI:10.18331/BRJ2016.3.1.4 , http://www.biofueljournal.com/article_13498.html, [accessed 2019 Jul 18].
- U. C. Kalluri, H. Yin, X. Yang and B. H. Davison, Systems and synthetic biology approaches to alter plant cell walls and reduce biomass recalcitrance, Plant Biotechnol. J., 2014, 12(9), 1207–1216 CrossRef CAS PubMed.
- M. Yadav, K. Paritosh, A. Chawade, N. Pareek and V. Vivekanand, Genetic engineering of energy crops to reduce recalcitrance and enhance biomass digestibility, Agriculture, 2018, 8(6), 76 CrossRef.
- S. D. Mansfield, C. Mooney and J. N. Saddler, Substrate and enzyme characteristics that limit cellulose hydrolysis, Biotechnol. Prog., 1999, 15(5), 804–816 CrossRef CAS PubMed.
- M. Pauly and K. Keegstra, Plant cell wall polymers as precursors for biofuels, Curr. Opin. Plant Biol., 2010, 13(3), 304–311 CrossRef PubMed.
- Full Text, [accessed 2019 Jul 2], https://www.pnas.org/content/pnas/103/44/16165.full.pdf.
- C. R. South, D. A. L. Hogsett and L. R. Lynd, Modeling simultaneous saccharification and fermentation of lignocellulose to ethanol in batch and continuous reactors, Enzyme Microb. Technol., 1995, 17(9), 797–803 CrossRef CAS.
- G. Vaaje-Kolstad, B. Westereng, S. J. Horn, Z. Liu, H. Zhai, M. Sørlie and V. G. H. Eijsink, An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides, Science, 2010, 330(6001), 219–222, DOI:10.1126/science.1192231 , https://science.sciencemag.org/content/330/6001/219, [accessed 2020 Nov 24.
- S. J. Horn, G. Vaaje-Kolstad, B. Westereng and V. Eijsink, Novel enzymes for the degradation of cellulose, Biotechnol. Biofuels, 2012, 5(1), 45, DOI:10.1186/1754-6834-5-45 , [accessed 2020 Nov 24].
- D. Salvachúa, A. Prieto, M. López-Abelairas, T. Lu-Chau, Á. T. Martínez and M. J. Martínez, Fungal pretreatment: An alternative in second-generation ethanol from wheat straw, Bioresour. Technol., 2011, 102(16), 7500–7506 CrossRef PubMed.
- R. Sindhu, P. Binod and A. Pandey, Biological pretreatment of lignocellulosic biomass – An overview, Bioresour. Technol., 2016, 199, 76–82, DOI:10.1016/j.biortech.2015.08.030 (Pretreatment of Biomass), http://www.sciencedirect.com/science/article/pii/S0960852415011347, [accessed 2019 Oct 8].
- B. J. Baker, V. De Anda, K. W. Seitz, N. Dombrowski, A. E. Santoro and K. G. Lloyd, Diversity, ecology and evolution of Archaea, Nat. Microbiol., 2020, 1–14 Search PubMed.
- S. S. Paul, D. Bu, J. Xu, K. D. Hyde and Z. Yu, A phylogenetic census of global diversity of gut anaerobic fungi and a new taxonomic framework, Fungal Divers., 2018, 89(1), 253–266 CrossRef.
- K. V. Solomon, C. H. Haitjema, J. K. Henske, S. P. Gilmore, D. Borges-Rivera, A. Lipzen, H. M. Brewer, S. O. Purvine, A. T. Wright and M. K. Theodorou, Early-branching gut fungi possess a large, comprehensive array of biomass-degrading enzymes, Science, 2016, 351(6278), 1192–1195 CrossRef CAS PubMed.
- I. A. Podolsky, S. Seppälä, T. S. Lankiewicz, J. L. Brown, C. L. Swift and M. A. O’Malley, Harnessing Nature's Anaerobes for Biotechnology and Bioprocessing, 2019 Search PubMed.
- Y. Lu, Y.-H. P. Zhang and L. R. Lynd, Enzyme–microbe synergy during cellulose hydrolysis by Clostridium thermocellum, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(44), 16165–16169 CrossRef CAS PubMed.
- A. Dumitrache, G. Wolfaardt, G. Allen, S. N. Liss and L. R. Lynd, Form and function of Clostridium thermocellum biofilms, Appl. Environ. Microbiol., 2013, 79(1), 231–239 CrossRef CAS PubMed.
- Z.-W. Wang, S.-H. Lee, J. G. Elkins and J. L. Morrell-Falvey, Spatial and temporal dynamics of cellulose degradation and biofilm formation by Caldicellulosiruptor obsidiansis and Clostridium thermocellum, AMB Express, 2011, 1(1), 30 CrossRef PubMed.
- A. Dumitrache, D. M. Klingeman, J. Natzke, M. Rodriguez Jr, R. J. Giannone, R. L. Hettich, B. H. Davison and S. D. Brown, Specialized activities and expression differences for Clostridium thermocellum biofilm and planktonic cells, Sci. Rep., 2017, 7, 43583 CrossRef PubMed.
- E. K. Holwerda and L. R. Lynd, Testing alternative kinetic models for utilization of crystalline cellulose (Avicel) by batch cultures of Clostridium thermocellum, Biotechnol. Bioeng., 2013, 110(9), 2389–2394 CrossRef CAS PubMed.
- X. Liang, J. M. Whitham, E. K. Holwerda, X. Shao, L. Tian, Y.-W. Wu, V. Lombard, B. Henrissat, D. M. Klingeman and Z. K. Yang, et al., Development and characterization of stable anaerobic thermophilic methanogenic microbiomes fermenting switchgrass at decreasing residence times, Biotechnol. Biofuels, 2018, 11(1), 243, DOI:10.1186/s13068-018-1238-1 , [accessed 2019 Oct 8].
- P. S. Van, The uniformity and nutritive availability of cellulose, in Federation proceedings, vol. 32, 1973, pp. 1804–1808 Search PubMed.
- Y.-H. P. Zhang and L. R. Lynd, Cellulose utilization by Clostridium thermocellum: bioenergetics and hydrolysis product assimilation, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(20), 7321–7325 CrossRef CAS PubMed.
- J. Zhou, D. Olson, D. A. Argyros, Y. Deng, W. M. van Gulik, J. P. van Dijken and L. R. Lynd, Atypical glycolysis in Clostridium thermocellum, Appl. Environ. Microbiol., 2013, 3000–3008 CrossRef CAS PubMed , https://aem.asm.org/content/79/9/3000.short, [accessed 2020 Dec 1].
- R. Lamed and J. G. Zeikus, Ethanol production by thermophilic bacteria: Relationship between fermentation product yields of and catabolic enzyme activities in Clostridium thermocellum and Thermoanaerobium brockii, J. Bacteriol., 1980, 144(2), 569–578 CrossRef CAS PubMed , https://jb.asm.org/content/144/2/569, [accessed 2020 Dec 1].
- C. R. Carere, T. Rydzak, T. J. Verbeke, N. Cicek, D. B. Levin and R. Sparling, Linking genome content to biofuel production yields: A meta-analysis of major catabolic pathways among select H2 and ethanol-producing bacteria, BMC Microbiol., 2012, 12(1), 295, DOI:10.1186/1471-2180-12-295 , [accessed 2020 Dec 1].
- J. O. Park, L. B. Tanner, M. H. Wei, D. B. Khana, T. B. Jacobson, Z. Zhang, S. A. Rubin, S. H.-J. Li, M. B. Higgins and D. M. Stevenson, Near-equilibrium glycolysis supports metabolic homeostasis and energy yield, Nat. Chem. Biol., 2019, 15(10), 1001–1008 CrossRef CAS PubMed.
- L. R. Lynd, A. M. Guss, M. E. Himmel, D. Beri, C. Herring, E. K. Holwerda, S. J. Murphy, D. G. Olson, J. Paye and T. Rydzak, Advances in consolidated bioprocessing using Clostridium thermocellum and Thermoanaerobacter saccharolyticum, Ind. Biotechnol. Microorg., 2016, 10, 365–394 Search PubMed.
- A. Hasona, Y. Kim, F. G. Healy, L. O. Ingram and K. T. Shanmugam, Pyruvate formate lyase and acetate kinase are essential for anaerobic growth of Escherichia coli on xylose, J. Bacteriol., 2004, 186(22), 7593–7600, DOI:10.1128/JB.186.22.7593-7600.2004 , [accessed 2021 Jun 30].
- S. Kok, B. U. Kozak, J. T. Pronk and A. J. A. Maris, Energy coupling in Saccharomyces cerevisiae: Selected opportunities for metabolic engineering, FEMS Yeast Res., 2012, 12(4), 387–397, DOI:10.1111/j.1567-1364.2012.00799.x , [accessed 2021 Jun 30].
- T. Conway, The Entner–Doudoroff pathway: History, physiology and molecular biology, FEMS Microbiol. Lett., 1992, 103(1), 1–28, DOI:10.1111/j.1574-6968.1992.tb05822.x , [accessed 2021 Jun 30].
- T. B. Jacobson, T. K. Korosh, D. M. Stevenson, C. Foster, C. Maranas, D. G. Olson, L. R. Lynd and D. Amador-Noguez, In vivo thermodynamic analysis of glycolysis in Clostridium thermocellum and Thermoanaerobacterium saccharolyticum using 13C and 2H tracers, Msystems, 2020, 5(2), E00736-19 CrossRef PubMed.
- J. K. Fredrickson, Ecological communities by design, Science, 2015, 348(6242), 1425–1427 CrossRef CAS PubMed.
- N. I. Johns, T. Blazejewski, A. L. Gomes and H. H. Wang, Principles for designing synthetic microbial communities, Curr. Opin. Microbiol., 2016, 31, 146–153 CrossRef PubMed.
- M. T. Mee, J. J. Collins, G. M. Church and H. H. Wang, Syntrophic exchange in synthetic microbial communities, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(20), E2149–E2156 CrossRef CAS PubMed.
- R. Tsoi, F. Wu, C. Zhang, S. Bewick, D. Karig and L. You, Metabolic division of labor in microbial systems, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(10), 2526–2531 CrossRef CAS PubMed.
- K. Zhou, K. Qiao, S. Edgar and G. Stephanopoulos, Distributing a metabolic pathway among a microbial consortium enhances production of natural products, Nat. Biotechnol., 2015, 33(4), 377 CrossRef CAS PubMed.
- L. Cortes-Tolalpa, D. J. Jiménez, M. J. de Lima Brossi, J. F. Salles and J. D. van Elsas, Different inocula produce distinctive microbial consortia with similar lignocellulose degradation capacity, Appl. Microbiol. Biotechnol., 2016, 100(17), 7713–7725 CrossRef CAS PubMed.
- M. J. de Lima Brossi, D. J. Jiménez, L. Cortes-Tolalpa and J. D. van Elsas, Soil-derived microbial consortia enriched with different plant biomass reveal distinct players acting in lignocellulose degradation, Microb. Ecol., 2016, 71(3), 616–627 CrossRef PubMed.
- Z. Wen, Q. Li, J. Liu, M. Jin and S. Yang, Consolidated bioprocessing for butanol production of cellulolytic Clostridia: Development and optimization, Microbial Biotechnol., 2019, 410–422 Search PubMed.
- M. Taha, E. Shahsavari, K. Al-Hothaly, A. Mouradov, A. T. Smith, A. S. Ball and E. M. Adetutu, Enhanced biological straw saccharification through coculturing of lignocellulose-degrading microorganisms, Appl. Biochem. Biotechnol., 2015, 175(8), 3709–3728 CrossRef CAS PubMed.
- D. J. Jiménez, D. Chaves-Moreno and J. D. Van Elsas, Unveiling the metabolic potential of two soil-derived microbial consortia selected on wheat straw, Sci. Rep., 2015, 5, 13845 CrossRef PubMed.
- D. J. Jiménez, M. J. de Lima Brossi, J. Schückel, S. K. Kračun, W. G. T. Willats and J. D. van Elsas, Characterization of three plant biomass-degrading microbial consortia by metagenomics-and metasecretomics-based approaches, Appl. Microbiol. Biotechnol., 2016, 100(24), 10463–10477 CrossRef PubMed.
- S. P. Lillington, P. A. Leggieri, K. A. Heom and M. A. O’Malley, Nature's recyclers: Anaerobic microbial communities drive crude biomass deconstruction, Curr. Opin. Biotechnol., 2020, 62, 38–47 CrossRef CAS PubMed.
- J. J. Rosnow, L. N. Anderson, R. N. Nair, E. S. Baker and A. T. Wright, Profiling microbial lignocellulose degradation and utilization by emergent omics technologies, Crit. Rev. Biotechnol., 2017, 37(5), 626–640 CrossRef CAS PubMed.
- S. P. Gilmore and M. A. O’Malley, Microbial communities for bioprocessing: Lessons learned from nature, Curr. Opin. Chem. Eng., 2016, 14, 103–109 CrossRef.
- K. V. Solomon, C. H. Haitjema, D. A. Thompson and M. A. O’Malley, Extracting data from the muck: Deriving biological insight from complex microbial communities and non-model organisms with next generation sequencing, Curr. Opin. Biotechnol., 2014, 28, 103–110 CrossRef CAS PubMed.
- J. M. Paye, A. Guseva, S. K. Hammer, E. Gjersing, M. F. Davis, B. H. Davison, J. Olstad, B. S. Donohoe, T. Y. Nguyen and C. E. Wyman, Biological lignocellulose solubilization: Comparative evaluation of biocatalysts and enhancement via cotreatment, Biotechnol. Biofuels, 2016, 9(1), 8 CrossRef PubMed.
- J. A. Izquierdo, S. Pattathil, A. Guseva, M. G. Hahn and L. R. Lynd, Comparative analysis of the ability of Clostridium clariflavum strains and Clostridium thermocellum to utilize hemicellulose and unpretreated plant material, Biotechnol. Biofuels, 2014, 7(1), 136 CrossRef PubMed.
- T. H. Kim and Y. Y. Lee, Pretreatment and fractionation of corn stover by ammonia recycle percolation process, Bioresour. Technol., 2005, 96(18), 2007–2013 CrossRef CAS PubMed.
- F. Teymouri, L. Laureano-Perez, H. Alizadeh and B. E. Dale, Optimization of the ammonia fiber explosion (AFEX) treatment parameters for enzymatic hydrolysis of corn stover, Bioresour. Technol., 2005, 96(18), 2014–2018 CrossRef CAS PubMed.
- G. Resch, S. Donohoe, O. Baker, R. Decker, A. Bayer, T. Beckham and E. Himmel, Fungal cellulases and complexed cellulosomal enzymes exhibit synergistic mechanisms in cellulose deconstruction, Energy Environ. Sci., 2013, 6(6), 1858–1867, 10.1039/C3EE00019B , [accessed 2020 Nov 24].
- B. S. Donohoe and M. G. Resch, Mechanisms employed by cellulase systems to gain access through the complex architecture of lignocellulosic substrates, Curr. Opin. Chem. Biol., 2015, 29, 100–107, DOI:10.1016/j.cbpa.2015.08.014 (Energy Mechanistic biology).
- Q. Xu, M. G. Resch, K. Podkaminer, S. Yang, J. O. Baker, B. S. Donohoe, C. Wilson, D. M. Klingeman, D. G. Olson and S. R. Decker, Dramatic performance of Clostridium thermocellum explained by its wide range of cellulase modalities, Sci. Adv., 2016, 2(2), e1501254 CrossRef PubMed.
- T. J. Verbeke, G. M. Garcia and J. G. Elkins, The effect of switchgrass loadings on feedstock solubilization and biofuel production by Clostridium thermocellum, Biotechnol. Biofuels, 2017, 10(1), 233 CrossRef PubMed.
- D. G. Olson, R. Sparling and L. R. Lynd, Ethanol production by engineered thermophiles, Curr. Opin. Biotechnol., 2015, 33, 130–141 CrossRef CAS PubMed.
- A. Brune, Symbiotic digestion of lignocellulose in termite guts, Nat. Rev. Microbiol., 2014, 12(3), 168 CrossRef CAS PubMed.
- P. J. Weimer, J. B. Russell and R. E. Muck, Lessons from the cow: What the ruminant animal can teach us about consolidated bioprocessing of cellulosic biomass, Bioresour. Technol., 2009, 100(21), 5323–5331 CrossRef CAS PubMed.
- P. J. Weimer and M. B. Hall, The potential for biomimetic application of rumination to bioreactor design, Biomass Bioenergy, 2020, 143, 105822 CrossRef CAS.
- A. Barakat, H. de Vries and X. Rouau, Dry fractionation process as an important step in current and future lignocellulose biorefineries: A review, Bioresour. Technol., 2013, 134, 362–373 CrossRef CAS PubMed.
- A. Hendriks and G. Zeeman, Pretreatments to enhance the digestibility of lignocellulosic biomass, Bioresour. Technol., 2009, 100(1), 10–18 CrossRef CAS PubMed.
- P. Kumar, D. M. Barrett, M. J. Delwiche and P. Stroeve, Methods for pretreatment of lignocellulosic biomass for efficient hydrolysis and biofuel production, Ind. Eng. Chem. Res., 2009, 48(8), 3713–3729 CrossRef CAS.
- B. C. Vidal, B. S. Dien, K. C. Ting and V. Singh, Influence of feedstock particle size on lignocellulose conversion—A review, Appl. Biochem. Biotechnol., 2011, 164(8), 1405–1421 CrossRef CAS PubMed.
- M. L. Balch, M. B. Chamberlain, R. S. Worthen, E. K. Holwerda and L. R. Lynd, Fermentation with continuous ball milling: Effectiveness at enhancing solubilization for several cellulosic feedstocks and comparative tolerance of several microorganisms, Biomass Bioenergy, 2020, 134, 105468 CrossRef CAS.
- J. K. Saini, A. K. Patel, M. Adsul and R. R. Singhania, Cellulase adsorption on lignin: A roadblock for economic hydrolysis of biomass, Renewable Energy, 2016, 98, 29–42, DOI:10.1016/j.renene.2016.03.089 , (Special Issue: New Horizons in Biofuels Production and Technologies), https://www.sciencedirect.com/science/article/pii/S0960148116302737, [accessed 2021 Oct 15].
- S. Y. Lee, H. U. Kim, T. U. Chae, J. S. Cho, J. W. Kim, J. H. Shin, D. I. Kim, Y.-S. Ko, W. D. Jang and Y.-S. Jang, A comprehensive metabolic map for production of bio-based chemicals, Nat. Catal., 2019, 2(1), 18–33 CrossRef CAS.
- J. C. Liao, L. Mi, S. Pontrelli and S. Luo, Fuelling the future: Microbial engineering for the production of sustainable biofuels, Nat. Rev. Microbiol., 2016, 14(5), 288 CrossRef CAS PubMed.
- Y. J. Zhou, E. J. Kerkhoven and J. Nielsen, Barriers and opportunities in bio-based production of hydrocarbons, Nat. Energy, 2018, 1, S41560-018-0197-x Search PubMed.
- R. J. Wooley and V. Putsche, Development of an ASPEN PLUS physical property database for biofuels components, 1996, p. NREL/TP-425-20685 DOI:10.2172/257362.
- C. E. Nakamura and G. M. Whited, Metabolic engineering for the microbial production of 1, 3-propanediol, Curr. Opin. Biotechnol, 2003, 14(5), 454–459 CrossRef CAS PubMed.
- L. Tian, B. Papanek, D. G. Olson, T. Rydzak, E. K. Holwerda, T. Zheng, J. Zhou, M. Maloney, N. Jiang and R. J. Giannone, et al., Simultaneous achievement of high ethanol yield and titer in Clostridium thermocellum, Biotechnol. Biofuels, 2016, 9(1), 116, DOI:10.1186/s13068-016-0528-8 , [accessed 2020 Jul 16].
- B. Bhadra, R. Bhalla, A. L. Kruckeberg, V. Nagarajan, R. Patnaik and W. Suh, Fermentative production of alcohols, 2018, https://patents.google.com/patent/US9909148B2/en, [accessed 2020 Jul 20].
- R. M. R. Feldman, U. Gunawardena, J. Urano, P. Meinhold, A. Aristidou, C. A. Dundon and C. Smith, Yeast organism producing isobutanol at a high yield, 2018 Search PubMed.
- K. R. Benjamin, I. R. Silva, J. P. Cherubim, D. McPhee and C. J. Paddon, Developing commercial production of semi-synthetic artemisinin, and of β-farnesene, an isoprenoid produced by fermentation of Brazilian sugar, J. Braz. Chem. Soc., 2016, 27(8), 1339–1345, DOI:10.5935/0103-5053.20160119.
- N. Nghiem, S. Kleff and S. Schwegmann, Succinic acid: Technology development and commercialization, Fermentation, 2017, 3(2), 26, DOI:10.3390/fermentation3020026 , http://www.mdpi.com/2311-5637/3/2/26, [accessed 2020 Jul 16].
- F. K. Félix, C. do, L. A. J. Letti, G. Vinícius de Melo Pereira, P. G. B. Bonfim, V. T. Soccol and C. R. Soccol, L-Lysine production improvement: A review of the state of the art and patent landscape focusing on strain development and fermentation technologies, Crit. Rev. Biotechnol., 2019, 39(8), 1031–1055, DOI:10.1080/07388551.2019.1663149 , [accessed 2020 Jul 16].
- J. Lee, Y.-S. Jang, S. J. Choi, J. A. Im, H. Song, J. H. Cho, D. Y. Seung, E. T. Papoutsakis, G. N. Bennett and S. Y. Lee, Metabolic engineering of Clostridium acetobutylicum ATCC 824 for isopropanol–butanol–ethanol fermentation, Appl. Environ. Microbiol., 2012, 78(5), 1416–1423, DOI:10.1128/AEM.06382-11 , [accessed 2020 Jul 16].
- R. O. J. Norman, T. Millat, K. Winzer, N. P. Minton and C. Hodgman, Progress towards platform chemical production using Clostridium autoethanogenum, Biochem. Soc. Trans., 2018, 46(3), 523–535, DOI:10.1042/BST20170259 , https://portlandpress.com/biochemsoctrans/article/46/3/523/67397/Progress-towards-platform-chemical-production, [accessed 2020 Jul 16].
- P. Suominen, A. Aristidou, M. Pentilla, M. Ilmen, L. Ruohonen, K. Koivuranta and K. Roberg-Perez, Genetically modified yeast of the species Issatchenkia orientalis and closely relates species, and fermentation processes using same, 2012, https://www.osti.gov/biblio/1034912.
- L. Artzi, E. A. Bayer and S. Morais, Cellulosomes: Bacterial nanomachines for dismantling plant polysaccharides, Nat. Rev. Microbiol., 2017, 15(2), 83 CrossRef CAS PubMed.
- P. P. Lin, L. Mi, A. H. Morioka, K. M. Yoshino, S. Konishi, S. C. Xu, B. A. Papanek, L. A. Riley, A. M. Guss and J. C. Liao, Consolidated bioprocessing of cellulose to isobutanol using Clostridium thermocellum, Metab. Eng., 2015, 31, 44–52, DOI:10.1016/j.ymben.2015.07.001 , https://linkinghub.elsevier.com/retrieve/pii/S1096717615000804, [accessed 2021 Jun 30].
- L. Tian, P. M. Conway, N. D. Cervenka, J. Cui, M. Maloney, D. G. Olson and L. R. Lynd, Metabolic engineering of Clostridium thermocellum for n-butanol production from cellulose, Biotechnol. Biofuels, 2019, 12(1), 186 CrossRef PubMed.
- H. Seo, J.-W. Lee, R. J. Giannone, N. J. Dunlap and C. T. Trinh, Engineering promiscuity of chloramphenicol acetyltransferase for microbial designer ester biosynthesis, Metab. Eng., 2021, 66, 179–190 CrossRef CAS PubMed.
- H. Seo, J.-W. Lee, S. Garcia and C. T. Trinh, Single mutation at a highly conserved region of chloramphenicol acetyltransferase enables isobutyl acetate production directly from cellulose by Clostridium thermocellum at elevated temperatures, Biotechnol. Biofuels, 2019, 12, 245, DOI:10.1186/s13068-019-1583-8 , [accessed 2021 Jun 30].
- H. Seo, P. N. Nicely and C. T. Trinh, Endogenous carbohydrate esterases of Clostridium thermocellum are identified and disrupted for enhanced isobutyl acetate production from cellulose, Biotechnol. Bioeng., 2020, 117(7), 2223–2236, DOI:10.1002/bit.27360 , [accessed 2021 Jun 30].
- P. L. Show, K. O. Oladele, Q. Y. Siew, F. A. Aziz Zakry, J. C.-W. Lan and T. C. Ling, Overview of citric acid production from Aspergillus niger, Front. Life Sci., 2015, 8(3), 271–283, DOI:10.1080/21553769.2015.1033653 , [accessed 2020 Jul 16].
- L. A. Riley, L. Ji, R. J. Schmitz, J. Westpheling and A. M. Guss, Rational development of transformation in Clostridium thermocellum ATCC 27405 via complete methylome analysis and evasion of native restriction–modification systems, J. Ind. Microbiol. Biotechnol., 2019, 46(9–10), 1435–1443, DOI:10.1007/s10295-019-02218-x , [accessed 2020 Jul 16].
- L. D. Mermelstein, N. E. Welker, G. N. Bennett and E. T. Papoutsakis, Expression of cloned homologous fermentative genes in Clostridium acetobutylicum ATCC 824, Bio/Technology, 1992, 10(2), 190–195, DOI:10.1038/nbt0292-190 , [accessed 2021 Jul 9].
- R. J. Roberts, T. Vincze, J. Posfai and D. Macelis, REBASE—A database for DNA restriction and modification: Enzymes, genes and genomes, Nucleic Acids Res., 2015, 43(D1), D298–D299 CrossRef CAS PubMed.
- M. R. Tock and D. T. Dryden, The biology of restriction and anti-restriction, Curr. Opin. Microbiol., 2005, 8(4), 466–472 CrossRef CAS PubMed.
- J. T. Heap, O. J. Pennington, S. T. Cartman and N. P. Minton, A modular system for Clostridium shuttle plasmids, J. Microbiol. Methods, 2009, 78(1), 79–85, DOI:10.1016/j.mimet.2009.05.004 , https://linkinghub.elsevier.com/retrieve/pii/S0167701209001298, [accessed 2020 Jul 16].
- M. E. Pyne, M. R. Bruder, M. Moo-Young, D. A. Chung and C. P. Chou, Harnessing heterologous and endogenous CRISPR-Cas machineries for efficient markerless genome editing in Clostridium, Sci. Rep., 2016, 6(1), 25666, DOI:10.1038/srep25666 , [accessed 2020 Jul 16].
- J. M. Vento, N. Crook and C. L. Beisel, Barriers to genome editing with CRISPR in bacteria, J. Ind. Microbiol. Biotechnol., 2019, 46(9–10), 1327–1341, DOI:10.1007/s10295-019-02195-1 , [accessed 2020 Jul 16].
- J. E. Walker, A. A. Lanahan, T. Zheng, C. Toruno, L. R. Lynd, J. C. Cameron, D. G. Olson and C. A. Eckert, Development of both type I–B and type II CRISPR/Cas genome editing systems in the cellulolytic bacterium Clostridium thermocellum, Metab. Eng. Commun., 2020, 10, e00116 CrossRef PubMed.
- J. Zhang, W. Zong, W. Hong, Z.-T. Zhang and Y. Wang, Exploiting endogenous CRISPR-Cas system for multiplex genome editing in Clostridium tyrobutyricum and engineer the strain for high-level butanol production, Metab. Eng., 2018, 47, 49–59, DOI:10.1016/j.ymben.2018.03.007 , https://linkinghub.elsevier.com/retrieve/pii/S1096717618300594, [accessed 2020 Jul 16].
- J. R. Elmore, A. Furches, G. N. Wolff, K. Gorday and A. M. Guss, Development of a high efficiency integration system and promoter library for rapid modification of Pseudomonas putida KT2440, Metab. Eng. Commun., 2017, 5, 1–8 CrossRef PubMed.
- H. Ellegren, Genome sequencing and population genomics in non-model organisms, Trends Ecol. Evol., 2014, 29(1), 51–63, DOI:10.1016/j.tree.2013.09.008 , https://linkinghub.elsevier.com/retrieve/pii/S0169534713002310, [accessed 2020 Jul 16].
- V. Eldem, G. Zararsiz, T. Taşçi, I. P. Duru, Y. Bakir and M. Erkan, Transcriptome analysis for non-model organism: Current status and best-practices. Applications of RNA-Seq and Omics Strategies-From Microorganisms to Human Health, 2017, pp. 55–78 Search PubMed.
- J. Armengaud, J. Trapp, O. Pible, O. Geffard, A. Chaumot and E. M. Hartmann, Non-model organisms, a species endangered by proteogenomics, J. Proteomics, 2014, 105, 5–18, DOI:10.1016/j.jprot.2014.01.007 , https://linkinghub.elsevier.com/retrieve/pii/S1874391914000190, [accessed 2020 Jul 16].
- D. Amador-Noguez, X.-J. Feng, J. Fan, N. Roquet, H. Rabitz and J. D. Rabinowitz, Systems-level metabolic flux profiling elucidates a complete, bifurcated tricarboxylic acid cycle in Clostridium acetobutylicum, J. Bacteriol., 2010, 192(17), 4452–4461, DOI:10.1128/JB.00490-10 , [accessed 2021 Jul 9].
- L. Heirendt, S. Arreckx, T. Pfau, S. N. Mendoza, A. Richelle, A. Heinken, H. S. Haraldsdóttir, S. M. Keating, V. Vlasov and J. Wachowiak, Creation and analysis of biochemical constraint-based models: The COBRA Toolbox v3. 0, 2017, arXiv preprint, arXiv:1710.04038 Search PubMed.
- K. Chan and P. J. O’Brien, Structure–activity relationships for hepatocyte toxicity and electrophilic reactivity of α,β-unsaturated esters, acrylates and methacrylates, J. Appl. Toxicol., 2008, 28(8), 1004–1015 CrossRef CAS PubMed.
- L. N. Jayakody, C. W. Johnson, J. M. Whitham, R. J. Giannone, B. A. Black, N. S. Cleveland, D. M. Klingeman, W. E. Michener, J. L. Olstad and D. R. Vardon, Thermochemical wastewater valorization via enhanced microbial toxicity tolerance, Energy Environ. Sci., 2018, 11(6), 1625–1638 RSC.
- R. M. LoPachin and T. Gavin, Molecular mechanisms of aldehyde toxicity: A chemical perspective, Chem. Res. Toxicol., 2014, 27(7), 1081–1091 Search PubMed.
- L. O. Ingram, Ethanol tolerance in bacteria, Crit. Rev. Biotechnol., 1989, 9(4), 305–319 CrossRef PubMed.
- L. J. Jönsson, B. Alriksson and N.-O. Nilvebrant, Bioconversion of lignocellulose: Inhibitors and detoxification, Biotechnol. Biofuels, 2013, 6(1), 16 CrossRef PubMed.
- H. B. Klinke, A. B. Thomsen and B. K. Ahring, Inhibition of ethanol-producing yeast and bacteria by degradation products produced during pre-treatment of biomass, Appl. Microbiol. Biotechnol., 2004, 66(1), 10–26 CrossRef CAS PubMed.
- R. Xie, M. Tu and T. Elder, Substituent effect of phenolic aldehyde inhibition on alcoholic fermentation by Saccharomyces cerevisiae, Energy Fuels, 2016, 30(4), 3078–3084 CrossRef CAS.
- R. M. LoPachin, T. Gavin, A. DeCaprio and D. S. Barber, Application of the hard and soft, acids and bases (HSAB) theory to toxicant–target interactions, Chem. Res. Toxicol., 2012, 25(2), 239–251 Search PubMed.
- K. S. Akers, G. D. Sinks and T. W. Schultz, Structure–toxicity relationships for selected halogenated aliphatic chemicals, Environ. Toxicol. Pharmacol., 1999, 7(1), 33–39 CrossRef CAS PubMed.
- S. A. Nicolaou, S. M. Gaida and E. T. Papoutsakis, A comparative view of metabolite and substrate stress and tolerance in microbial bioprocessing: From biofuels and chemicals, to biocatalysis and bioremediation, Metab. Eng., 2010, 12(4), 307–331 CrossRef CAS PubMed.
- K. A. Zingaro and E. T. Papoutsakis, GroESL overexpression imparts Escherichia coli tolerance to i-, n-, and 2-butanol,1,2,4-butanetriol and ethanol with complex and unpredictable patterns, Metab. Eng., 2013, 15, 196–205 CrossRef CAS PubMed.
- J. Sikkema, J. A. de Bont and B. Poolman, Mechanisms of membrane toxicity of hydrocarbons, Microbiol. Mol. Biol. Rev., 1995, 59(2), 201–222 CrossRef CAS PubMed.
- J. M. Pérez, F. A. Arenas, G. A. Pradenas, J. M. Sandoval and C. C. Vásquez, Escherichia coli YqhD exhibits aldehyde reductase activity and protects from the harmful effect of lipid peroxidation-derived aldehydes, J. Biol. Chem., 2008, 283(12), 7346–7353 CrossRef PubMed.
- L. N. Jayakody and Y.-S. Jin, In-depth understanding of molecular mechanisms of aldehyde toxicity to engineer robust Saccharomyces cerevisiae, Appl. Microbiol. Biotechnol., 2021, 105(7), 2675–2692, DOI:10.1007/s00253-021-11213-1 , [accessed 2021 Jun 30].
- M. L. van Iersel, J.-P. H. Ploemen, M. L. Bello, G. Federici and P. J. van Bladeren, Interactions of α, β-unsaturated aldehydes and ketones with human glutathione S-transferase P1-1, Chem.-Biol. Interact., 1997, 108(1–2), 67–78 CrossRef CAS PubMed.
- L. N. Jayakody, J. Ferdouse, N. Hayashi and H. Kitagaki, Identification and detoxification of glycolaldehyde, an unattended bioethanol fermentation inhibitor, Crit. Rev. Biotechnol., 2017, 37(2), 177–189 CrossRef CAS PubMed.
- J. R. Kuykendall and M. S. Bogdanffy, Formation and stability of acetaldehyde-induced crosslinks between poly-lysine and poly-deoxyguanosine, Mut. Res./Fundam. Mol. Mech. Mutagen., 1994, 311(1), 49–56 CrossRef CAS.
- Y. Liang, C. Li and F. Hou, Study of glycolaldehyde on DNA damage in mouse spleen lymphocytes, J. Hygiene Res., 2004, 33(2), 214–215 CAS.
- H. M. Knott, B. E. Brown, M. J. Davies and R. T. Dean, Glycation and glycoxidation of low-density lipoproteins by glucose and low-molecular mass aldehydes: Formation of modified and oxidized particles, Eur. J. Biochem., 2003, 270(17), 3572–3582 CAS.
- J. Park, M. Rodríguez-Moyá, M. Li, E. Pichersky, K.-Y. San and R. Gonzalez, Synthesis of methyl ketones by metabolically engineered Escherichia coli, J. Ind. Microbiol. Biotechnol., 2012, 39(11), 1703–1712 CrossRef CAS PubMed.
- O. E. Melkina, I. A. Khmel, V. A. Plyuta, O. A. Koksharova and G. B. Zavilgelsky, Ketones 2-heptanone, 2-nonanone, and 2-undecanone inhibit DnaK-dependent refolding of heat-inactivated bacterial luciferases in Escherichia coli cells lacking small chaperon IbpB, Appl. Microbiol. Biotechnol., 2017, 101(14), 5765–5771, DOI:10.1007/s00253-017-8350-1 , [accessed 2021 Jun 30].
- A. Ajit, A. Z. Sulaiman and Y. Chisti, Production of bioethanol by Zymomonas mobilis in high-gravity extractive fermentations, Food Bioprod. Process., 2017, 102, 123–135 CrossRef CAS.
- B. S. Dien, M. A. Cotta and T. W. Jeffries, Bacteria engineered for fuel ethanol production: Current status, Appl. Microbiol. Biotechnol., 2003, 63(3), 258–266 CrossRef CAS PubMed.
- M. D. Smith, S. V. Pingali, J. G. Elkins, D. Bolmatov, R. F. Standaert, J. D. Nickels, V. S. Urban, J. Katsaras, B. H. Davison and J. C. Smith, Solvent-induced membrane stress in biofuel production: Molecular insights from small-angle scattering and all-atom molecular dynamics simulations, Green Chem., 2020, 22(23), 8278–8288 RSC.
- L. O. Ingram, Adaptation of membrane lipids to alcohols, J. Bacteriol., 1976, 125(2), 670–678 CrossRef CAS PubMed.
- R. J. Haft, D. H. Keating, T. Schwaegler, M. S. Schwalbach, J. Vinokur, M. Tremaine, J. M. Peters, M. V. Kotlajich, E. L. Pohlmann and I. M. Ong, Correcting direct effects of ethanol on translation and transcription machinery confers ethanol tolerance in bacteria, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(25), E2576–E2585 CrossRef CAS PubMed.
- D. Benndorf, N. Loffhagen and W. Babel, Induction of heat shock proteins in response to primary alcohols in Acinetobacter calcoaceticus, Electrophoresis, 1999, 20(4–5), 781–789 CrossRef CAS PubMed.
- C. Li, R. W. Peoples and F. F. Weight, Alcohol action on a neuronal membrane receptor: Evidence for a direct interaction with the receptor protein, Proc. Natl. Acad. Sci. U. S. A., 1994, 91(17), 8200–8204 CrossRef CAS PubMed.
- D. S. Layton and C. T. Trinh, Engineering modular ester fermentative pathways in Escherichia coli, Metab. Eng., 2014, 26, 77–88 CrossRef CAS PubMed.
- G. M. Rodriguez and S. Atsumi, Toward aldehyde and alkane production by removing aldehyde reductase activity in Escherichia coli, Metab. Eng., 2014, 25, 227–237 CrossRef CAS PubMed.
- Y.-S. Tai, M. Xiong and K. Zhang, Engineered biosynthesis of medium-chain esters in Escherichia coli, Metab. Eng., 2015, 27, 20–28 CrossRef CAS PubMed.
- B. Wilbanks and C. T. Trinh, Comprehensive characterization of toxicity of fermentative metabolites on microbial growth, Biotechnol. Biofuels, 2017, 10(1), 262 CrossRef PubMed.
- S. Brul and P. Coote, Preservative agents in foods: Mode of action and microbial resistance mechanisms, Int. J. Food Microbiol., 1999, 50(1–2), 1–17 CrossRef CAS PubMed.
- T. Warnecke and R. T. Gill, Organic acid toxicity, tolerance, and production in Escherichia coli biorefining applications, Microb. Cell Fact., 2005, 4(1), 25 CrossRef PubMed.
- N. Guaragnella, L. Antonacci, S. Passarella, E. Marra and S. Giannattasio, Achievements and perspectives in yeast acetic acid-induced programmed cell death pathways, Portland Press Ltd, 2011 Search PubMed.
- J. A. Vázquez, A. Durán, I. Rodríguez-Amado, M. A. Prieto, D. Rial and M. A. Murado, Evaluation of toxic effects of several carboxylic acids on bacterial growth by toxicodynamic modelling, Microb. Cell Fact., 2011, 10(1), 100 CrossRef PubMed.
- Microbial mechanisms of tolerance to weak acids – an overview, ed. Mira N. P. and Teixeira M. C., Frontiers SA Media, 2014 (Frontiers Research Topics) DOI:10.3389/978-2-88919-305-9, http://journal.frontiersin.org/ResearchTopic/912.
- M.-H. Yu, H. Tsunoda and M. Tsunoda, Environmental Toxicology: Biological and Health Effects, 3rd edn, CRC Press, Boca Raton, 2011 Search PubMed.
- S. Atsumi, W. Higashide and J. C. Liao, Direct photosynthetic recycling of carbon dioxide to isobutyraldehyde, Nat. Biotechnol., 2009, 27(12), 1177 CrossRef CAS PubMed.
- B. N. van Leeuwen, A. M. van der Wulp, I. Duijnstee, A. J. van Maris and A. J. Straathof, Fermentative production of isobutene, Appl. Microbiol. Biotechnol., 2012, 93(4), 1377–1387 CrossRef PubMed.
- L. O. Ingram and T. M. Buttke, Effects of alcohols on micro-organisms, in Advances in microbial physiology, vol. 25, Elsevier, 1985, pp. 253–300 Search PubMed.
- S. C. McKarns, C. Hansch, W. S. Caldwell, W. T. Morgan, S. K. Moore and D. J. Doolittle, Correlation between hydrophobicity of short-chain aliphatic alcohols and their ability to alter plasma membrane integrity, Fundam. Appl. Toxicol., 1997, 36(1), 62–70 CrossRef CAS PubMed.
- J. L. Ramos, E. Duque, M.-T. Gallegos, P. Godoy, M. I. Ramos-Gonzalez, A. Rojas, W. Terán and A. Segura, Mechanisms of solvent tolerance in gram-negative bacteria, Ann. Rev. Microbiol., 2002, 56(1), 743–768 CrossRef CAS PubMed.
- J. Zaldivar and L. O. Ingram, Effect of organic acids on the growth and fermentation of ethanologenic Escherichia coli LY01, Biotechnol. Bioeng., 1999, 66(4), 203–210 CrossRef CAS PubMed.
- S. Liu and N. Qureshi, How microbes tolerate ethanol and butanol, New Biotechnol., 2009, 26(3–4), 117–121 CrossRef CAS PubMed.
- L. Benov and I. Fridovich, Induction of the soxRS regulon of Escherichia coli by glycolaldehyde, Arch. Biochem. Biophys., 2002, 407(1), 45–48 CrossRef CAS PubMed.
- L. N. Jayakody, M. Kadowaki, K. Tsuge, K. Horie, A. Suzuki, N. Hayashi and H. Kitagaki, SUMO expression shortens the lag phase of Saccharomyces cerevisiae yeast growth caused by complex interactive effects of major mixed fermentation inhibitors found in hot-compressed water-treated lignocellulosic hydrolysate, Appl. Microbiol. Biotechnol., 2015, 99(1), 501–515 CrossRef CAS PubMed.
- L. N. Jayakody, K. Horie, N. Hayashi and H. Kitagaki, Engineering redox cofactor utilization for detoxification of glycolaldehyde, a key inhibitor of bioethanol production, in yeast Saccharomyces cerevisiae, Appl. Microbiol. Biotechnol., 2013, 97(14), 6589–6600 CrossRef CAS PubMed.
- K. A. Zingaro and E. T. Papoutsakis, Toward a semisynthetic stress response system to engineer microbial solvent tolerance, mBio, 2012, 3(5), e00308, DOI:10.1128/mBio.00308-12.
- N. Sandoval and E. Papoutsakis, Engineering membrane and cell-wall programs for tolerance to toxic chemicals: Beyond solo genes, Curr. Opin. Microbiol., 2016, 33, 56–66, DOI:10.1016/j.mib.2016.06.005 , https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5069143, [accessed 2021 Nov 15].
- H. Alper, J. Moxley, E. Nevoigt, G. R. Fink and G. Stephanopoulos, Engineering Yeast Transcription Machinery for Improved Ethanol Tolerance and Production, Science, 2006, 314(5805), 1565–1568 CrossRef CAS PubMed , https://www.jstor.org/stable/20032980, [accessed 2021 Nov 15].
- J. M. Skerker, D. Leon, M. N. Price, J. S. Mar, D. R. Tarjan, K. M. Wetmore, A. M. Deutschbauer, J. K. Baumohl, S. Bauer and A. B. Ibánez, Dissecting a complex chemical stress: Chemogenomic profiling of plant hydrolysates, Mol. Syst. Biol., 2013, 9(1), 674 CrossRef PubMed.
- J. I. Martien and D. Amador-Noguez, Recent applications of metabolomics to advance microbial biofuel production, Curr. Opin. Biotechnol., 2017, 43, 118–126 CrossRef CAS PubMed.
- P. P. Peralta-Yahya, F. Zhang, S. B. Del Cardayre and J. D. Keasling, Microbial engineering for the production of advanced biofuels, Nature, 2012, 488(7411), 320–328 CrossRef CAS PubMed.
- N. R. Sandoval, J. Y. Kim, T. Y. Glebes, P. J. Reeder, H. R. Aucoin, J. R. Warner and R. T. Gill, Strategy for directing combinatorial genome engineering in Escherichia coli, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(26), 10540–10545 CrossRef CAS PubMed.
- A. S. Abdelaal, A. M. Ageez, H. A. A. El AE and N. A. Abdallah, Genetic improvement of n-butanol tolerance in Escherichia coli by heterologous overexpression of groESL operon from Clostridium acetobutylicum, 3 Biotech, 2015, 5(4), 401–410 CrossRef PubMed.
- C. A. Tomas, N. E. Welker and E. T. Papoutsakis, Overexpression of groESL in Clostridium acetobutylicum results in increased solvent production and tolerance, prolonged metabolism, and changes in the cell's transcriptional program, Appl. Environ. Microbiol., 2003, 69(8), 4951–4965 CrossRef CAS PubMed.
- M. A. Fisher, S. Boyarskiy, M. R. Yamada, N. Kong, S. Bauer and D. Tullman-Ercek, Enhancing tolerance to short-chain alcohols by engineering the Escherichia coli AcrB efflux pump to secrete the non-native substrate n-butanol, ACS Synth. Biol., 2014, 3(1), 30–40 CrossRef CAS PubMed.
- S. Atsumi, T.-Y. Wu, I. M. Machado, W.-C. Huang, P.-Y. Chen, M. Pellegrini and J. C. Liao, Evolution, genomic analysis, and reconstruction of isobutanol tolerance in Escherichia coli, Mol. Syst. Biol., 2010, 6(1), 449 CrossRef PubMed.
- L. Lindahl, S. Genheden, L. A. Eriksson, L. Olsson and M. Bettiga, Sphingolipids contribute to acetic acid resistance in Zygosaccharomyces bailii, Biotechnol. Bioeng., 2016, 113(4), 744–753 CrossRef CAS PubMed.
- L. H. Luo, P.-S. Seo, J.-W. Seo, S.-Y. Heo, D.-H. Kim and C. H. Kim, Improved ethanol tolerance in Escherichia coli by changing the cellular fatty acids composition through genetic manipulation, Biotechnol. Lett., 2009, 31(12), 1867 CrossRef CAS PubMed.
- Z. Tan, J. M. Yoon, D. R. Nielsen, J. V. Shanks and L. R. Jarboe, Membrane engineering via trans unsaturated fatty acids production improves Escherichia coli robustness and production of biorenewables, Metab. Eng., 2016, 35, 105–113 CrossRef CAS PubMed.
- G. Stephanopoulos, Challenges in engineering microbes for biofuels production, Science, 2007, 315(5813), 801–804 CrossRef CAS PubMed.
- M. Simo, C. J. Brown and V. Hlavacek, Simulation of pressure swing adsorption in fuel ethanol production process, Comput. Chem. Eng., 2008, 32(7), 1635–1649 CrossRef CAS.
- L. M. Vane, Separation technologies for the recovery and dehydration of alcohols from fermentation broths, Biofuels, Bioprod. Biorefining, 2008, 2(6), 553–588 CrossRef CAS.
- L. Tao, E. C. Tan, R. McCormick, M. Zhang, A. Aden, X. He and B. T. Zigler, Techno-economic analysis and life-cycle assessment of cellulosic isobutanol and comparison with cellulosic ethanol and n-butanol, Biofuels, Bioprod. Biorefining, 2014, 8(1), 30–48 CrossRef CAS.
- V. H. Grisales Díaz and G. Olivar Tost, Ethanol and isobutanol dehydration by heat-integrated distillation, Chem. Eng. Process.: Process Intens., 2016, 108, 117–124, DOI:10.1016/j.cep.2016.07.005 , https://www.sciencedirect.com/science/article/pii/S0255270116301829, [accessed 2021 Feb 15].
- G. Pourhashem, P. R. Adler, A. J. McAloon and S. Spatari, Cost and greenhouse gas emission tradeoffs of alternative uses of lignin for second generation ethanol, Environ. Res. Lett., 2013, 8(2), 025021 CrossRef CAS.
- S. Roffler, H. W. Blanch and C. R. Wilke, Extractive fermentation of acetone and butanol: Process design and economic evaluation, Biotechnol. Prog., 1987, 3(3), 131–140 CrossRef CAS.
- V. H. Grisales Diaz and G. Olivar Tost, Energy efficiency of acetone, butanol, and ethanol (ABE) recovery by heat-integrated distillation, Bioprocess Biosyst. Eng., 2018, 41(3), 395–405, DOI:10.1007/s00449-017-1874-z , [accessed 2021 Feb 8].
- S. Y. Lee, J. H. Park, S. H. Jang, L. K. Nielsen, J. Kim and K. S. Jung, Fermentative butanol production by Clostridia, Biotechnol. Bioeng., 2008, 101(2), 209–228 CrossRef CAS PubMed.
- E. M. Green, Fermentative production of butanol—the industrial perspective, Curr. Opin. Biotechnol., 2011, 22(3), 337–343 CrossRef CAS PubMed.
- Y. Jiang, W. Liu, H. Zou, T. Cheng, N. Tian and M. Xian, Microbial production of short chain diols, Microb. Cell Fact., 2014, 13(1), 165 CrossRef PubMed.
- A. P. Mariano and R. Maciel Filho, Improvements in biobutanol fermentation and their impacts on distillation energy consumption and wastewater generation, BioEnergy Res., 2012, 5(2), 504–514 CrossRef CAS.
- N. Qureshi, S. Hughes, I. S. Maddox and M. A. Cotta, Energy-efficient recovery of butanol from model solutions and fermentation broth by adsorption, Bioprocess Biosyst. Eng., 2005, 27(4), 215–222, DOI:10.1007/s00449-005-0402-8 , [accessed 2021 Jan 20].
- C. Fu, Z. Li, C. Jia, W. Zhang, Y. Zhang, C. Yi and S. Xie, Recent advances on bio-based isobutanol separation, Energy Convers. Manage.: X, 2020, 100059 Search PubMed.
- News, Chemistry & Industry, 2014, [accessed 2020 Nov 24], 78(5), 6–19 DOI:10.1002/cind.785_3.x.
- D. R. Vardon, N. A. Rorrer, D. Salvachúa, A. E. Settle, C. W. Johnson, M. J. Menart, N. S. Cleveland, P. N. Ciesielski, K. X. Steirer and J. R. Dorgan, cis,cis-Muconic acid: Separation and catalysis to bio-adipic acid for nylon-6,6-polymerization, Green Chem., 2016, 18(11), 3397–3413 RSC.
- O. S. Fruchey, Processes for removing color during production of runway deicer, 2011, [accessed 2020 Nov 24], https://patents.google.com/patent/EP2305766A1/pt-PT.
- D. M. Adkesson, A. W. Alsop, T. T. Ames, L. A. Chu, J. M. Disney, B. C. Dravis, P. Fitzgibbon, J. M. Gaddy, F. G. Gallagher and W. F. Lehnhardt, Purification Of Biologically-Produced 1,3-Propanediol, 2011 Search PubMed.
- N. A. Huq, X. Huo, G. R. Hafenstine, S. M. Tifft, J. Stunkel, E. D. Christensen, G. M. Fioroni, L. Fouts, R. L. McCormick and P. A. Cherry, Performance-advantaged ether diesel bioblendstock production by a priori design, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(52), 26421–26430 CrossRef CAS PubMed.
- R. Hafenstine, A. Huq, R. Conklin, R. Wiatrowski, X. Huo, Q. Guo, A. Unocic and R. Vardon, Single-phase catalysis for reductive etherification of diesel bioblendstocks, Green Chem., 2020, 22(14), 4463–4472, 10.1039/D0GC00939C , [accessed 2021 Feb 24].
- T. J. Schwartz, B. H. Shanks and J. A. Dumesic, Coupling chemical and biological catalysis: A flexible paradigm for producing biobased chemicals, Curr. Opin. Biotechnol., 2016, 38, 54–62 CrossRef CAS PubMed.
- T. J. Schwartz, B. J. O’Neill, B. H. Shanks and J. A. Dumesic, Bridging the chemical and biological catalysis gap: Challenges and outlooks for producing sustainable chemicals, ACS Catal., 2014, 4(6), 2060–2069 CrossRef CAS.
- Succinic acid, once a biobased chemical star, is barely being made, Chemical & Engineering News, [accessed 2020 Nov 30]. https://cen.acs.org/business/biobased-chemicals/Succinic-acid-once-biobased-chemical/97/i12.
- T. Werpy and G. Petersen, Top value added chemicals from biomass: Volume I – results of screening for potential candidates from sugars and synthesis gas, National Renewable Energy Lab, Golden, CO (US), 2004 Search PubMed.
- C. S. López-Garzón and A. J. Straathof, Recovery of carboxylic acids produced by fermentation, Biotechnol. Adv., 2014, 32(5), 873–904 CrossRef PubMed.
- S.-T. Yang, H. El-Ensashy and N. Thongchul, Bioprocessing technologies in biorefinery for sustainable production of fuels, chemicals, and polymers, John Wiley & Sons, 2013 Search PubMed.
- S. Yedur, K. A. Berglund and D. D. Dunuwila, Succinic acid production and purification, 2001 Search PubMed.
- Production of Caproic Acid from Mixed Organic Waste: An Environmental Life Cycle Perspective | Environmental Science & Technology, DOI: 10.1021/acs.est.6b06220, [accessed 2020 Nov 25].
- Y. S. Huh, Y.-S. Jun, Y. K. Hong, H. Song, S. Y. Lee and W. H. Hong, Effective purification of succinic acid from fermentation broth produced by Mannheimia succiniciproducens, Process Biochem., 2006, 41(6), 1461–1465 CrossRef CAS.
- B. Max, J. M. Salgado, N. Rodríguez, S. Cortés, A. Converti and J. M. Domínguez, Biotechnological production of citric acid, Brazilian J. Microbiol., 2010, 41(4), 862–875 CrossRef CAS PubMed.
- K. R. Finley, J. M. Huryta, B. M. Mastel, T. W. McMullin, G. M. Poynter, B. J. Rush, K. T. Watts, A. M. Fosmer, V. L. Mcintosh and K. M. Brady, Compositions and methods for succinate production, 2017, https://patents.google.com/patent/US9605285B2/en, [accessed 2020 Nov 24].
- T. L. E. M. P. Carlson, Jr Low PH lactic acid fermentation, 2002, https://patents.google.com/patent/US6475759B1/en, [accessed 2020 Jul 27].
- W. F. Lehnhardt, R. V. Schanefelt and L. L. Napier, Process for recovering organic acids, 1995 Search PubMed.
- F. Adams Jr, L. F. Rice and R. J. Taylor, Itaconic acid purification process using reverse osmosis, 1970 Search PubMed.
- M. Kleiber, U. Gnabs, J. Schulze, S. Ghanegaonkar, H. Gehrke and M. Gawenda, Method for purifying carboxylic acids from fermentation broths, 2016 Search PubMed.
- Y. Mori, G. Takahashi, H. Suda and S. Yoshida, Processes for producing succinic acid, 2013, https://patents.google.com/patent/US20130018206A1/en, [accessed 2020 Nov 24].
- M. Grendze and F. Verhoff, Thermally-managed separation and dewatering processes for recovering acid products, 2000 Search PubMed.
- R. Kumar, H. Nanavati, S. B. Noronha and S. M. Mahajani, A continuous process for the recovery of lactic acid by reactive distillation, J. Chem. Technol. Biotechnol.: Int. Res. Proc., Environ. Clean Technol., 2006, 81(11), 1767–1777 CrossRef CAS.
- T. Urit, C. Löser, M. Wunderlich and T. Bley, Formation of ethyl acetate by Kluyveromyces marxianus on whey: Studies of the ester stripping, Bioprocess Biosyst. Eng., 2011, 34(5), 547–559, DOI:10.1007/s00449-010-0504-9 , [accessed 2020 Jul 16].
- P. O. Saboe, H. R. Monroe, W. E. Michener, L. P. Manker, S. J. Haugen, G. T. Beckham and E. M. Karp, In situ product recovery of bio-based ethyl esters via hybrid extraction–distillation, Green Chem., 2019, 21(19), 5306–5315 RSC.
- P. Lutze, Distillation in bioprocessing, in Distillation, Elsevier, 2014, pp. 337–365 Search PubMed.
- W. Van Hecke, G. Kaur and H. De Wever, Advances in in situ product recovery (ISPR) in whole cell biotechnology during the last decade, Biotechnol. Adv., 2014, 32(7), 1245–1255 CrossRef CAS PubMed.
- A. Freeman, J. M. Woodley and M. D. Lilly, In situ product removal as a tool for bioprocessing, Bio/Technology, 1993, 11(9), 1007–1012, DOI:10.1038/nbt0993-1007 , [accessed 2020 Nov 25].
- G. R. Cysewski and C. R. Wilke, Rapid ethanol fermentations using vacuum and cell recycle, Biotechnol. Bioeng., 1977, 19(8), 1125–1143 CrossRef CAS.
- D. C. Ryan, An overview Of Gevo's Biobased Isobutanol Production Process, 2019, p. 5 Search PubMed.
- A. L. Meadows, K. M. Hawkins, Y. Tsegaye, E. Antipov, Y. Kim, L. Raetz, R. H. Dahl, A. Tai, T. Mahatdejkul-Meadows and L. Xu, Rewriting yeast central carbon metabolism for industrial isoprenoid production, Nature, 2016, 537(7622), 694–697 CrossRef CAS PubMed.
- T. C. Ezeji, N. Qureshi and H. P. Blaschek, Acetone butanol ethanol (ABE) production from concentrated substrate: Reduction in substrate inhibition by fed-batch technique and product inhibition by gas stripping, Appl. Microbiol. Biotechnol., 2004, 63(6), 653–658 CrossRef CAS PubMed.
- A. J. Daugulis, D. B. Axford, B. Ciszek and J. J. Malinowski, Continuous fermentation of high-strength glucose feeds to ethanol, Biotechnol. Lett., 1994, 16(6), 637–642 CrossRef CAS.
- A. S. Kertes and C. J. King, Extraction chemistry of fermentation product carboxylic acids, Biotechnol. Bioeng., 1986, 28(2), 269–282 CrossRef CAS PubMed.
- P. O. Saboe, L. P. Manker, W. E. Michener, D. J. Peterson, D. G. Brandner, S. P. Deutch, M. Kumar, R. M. Cywar, G. T. Beckham and E. M. Karp, In situ recovery of bio-based carboxylic acids, Green Chem., 2018, 20(8), 1791–1804 RSC.
- K. Schügerl and J. Hubbuch, Integrated bioprocesses, Curr. Opin. Microbiol., 2005, 8(3), 294–300 CrossRef PubMed.
- Z. Gu, B. A. Glatz and C. E. Glatz, Propionic acid production by extractive fermentation. I. Solvent considerations, Biotechnol. Bioeng., 1998, 57(4), 454–461 CrossRef CAS PubMed.
- A. A. Kiss, J.-P. Lange, B. Schuur, D. W. F. Brilman, A. G. J. van der Ham and S. R. A. Kersten, Separation technology – Making a difference in biorefineries, Biomass Bioenergy, 2016, 95, 296–309, DOI:10.1016/j.biombioe.2016.05.021 , http://www.sciencedirect.com/science/article/pii/S0961953416301751, [accessed 2020 Nov 25].
- C. J. King and J. Starr, Recovery of carboxylic acids from water by precipitation from organic solutions, 1992 Search PubMed.
- J. H. Lee, V. Van Brunt and C. J. King, Water-enhanced solvation of organic solutes in ketone and ester solvents, Ind. Eng. Chem. Res., 1994, 33(5), 1373–1379 CrossRef CAS.
- R. W. Jenkins, C. M. Moore, T. A. Semelsberger, C. J. Chuck, J. C. Gordon and A. D. Sutton, The effect of functional groups in bio-derived fuel candidates, ChemSusChem, 2016, 9(9), 922–931 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz, R. M. West and J. A. Dumesic, Catalytic conversion of renewable biomass resources to fuels and chemicals, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 79–100 CrossRef CAS PubMed.
- R. Mawhood, E. Gazis, S. de Jong, R. Hoefnagels and R. Slade, Production pathways for renewable jet fuel: A review of commercialization status and future prospects, Biofuels, Bioprod. Biorefining, 2016, 10(4), 462–484 CrossRef CAS.
- C. K. Narula, Z. Li, E. M. Casbeer, R. A. Geiger, M. Moses-Debusk, M. Keller, M. V. Buchanan and B. H. Davison, Heterobimetallic zeolite, InV-ZSM-5, enables efficient conversion of biomass derived ethanol to renewable hydrocarbons, Sci. Rep., 2015, 5, 16039 CrossRef CAS PubMed.
- W.-C. Wang and L. Tao, Bio-jet fuel conversion technologies, Renewable Sustainable Energy Rev., 2016, 53, 801–822 CrossRef CAS.
- N. Zhan, Y. Hu, H. Li, D. Yu, Y. Han and H. Huang, Lanthanum–phosphorous modified HZSM-5 catalysts in dehydration of ethanol to ethylene: A comparative analysis, Catal. Commun., 2010, 11(7), 633–637 CrossRef CAS.
- G. Chen, S. Li, F. Jiao and Q. Yuan, Catalytic dehydration of bioethanol to ethylene over TiO2/γ-Al2O3 catalysts in microchannel reactors, Catal. Today, 2007, 125(1–2), 111–119 CrossRef CAS.
- A. P. Kagyrmanova, V. A. Chumachenko, V. N. Korotkikh, V. N. Kashkin and A. S. Noskov, Catalytic dehydration of bioethanol to ethylene: Pilot-scale studies and process simulation, Chem. Eng. J., 2011, 176, 188–194 CrossRef.
- A. Finiels, F. Fajula and V. Hulea, Nickel-based solid catalysts for ethylene oligomerization – a review, Catal. Sci. Technol., 2014, 4(8), 2412–2426 RSC.
- C. T. O’connor and M. Kojima, Alkene oligomerization, Catal. Today, 1990, 6(3), 329–349 CrossRef.
- B. M. Antunes, A. E. Rodrigues, Z. Lin, I. Portugal and C. M. Silva, Alkenes oligomerization with resin catalysts, Fuel Process. Technol., 2015, 138, 86–99 CrossRef CAS.
- B. G. Harvey and R. L. Quintana, Synthesis of renewable jet and diesel fuels from 2-ethyl-1-hexene, Energy Environ. Sci., 2010, 3(3), 352–357 RSC.
- B. G. Harvey and H. A. Meylemans, 1-Hexene: A renewable C6 platform for full-performance jet and diesel fuels, Green Chem., 2014, 16(2), 770–776 RSC.
- A. Galadima and O. Muraza, Zeolite catalysts in upgrading of bioethanol to fuels range hydrocarbons: A review, J. Ind. Eng. Chem., 2015, 31, 1–14 CrossRef CAS.
- J. R. Hannon, L. R. Lynd, O. Andrade, P. T. Benavides, G. T. Beckham, M. J. Biddy, N. Brown, M. F. Chagas, B. H. Davison and T. Foust, Technoeconomic and life-cycle analysis of single-step catalytic conversion of wet ethanol into fungible fuel blendstocks, Proc. Natl. Acad. Sci. U. S. A., 2019, 12576–12583 Search PubMed.
- H. Aitchison, R. L. Wingad and D. F. Wass, Homogeneous ethanol to butanol catalysis- Guerbet renewed, ACS Catal., 2016, 6(10), 7125–7132 CrossRef CAS.
- C. Angelici, B. M. Weckhuysen and P. C. Bruijnincx, Chemocatalytic conversion of ethanol into butadiene and other bulk chemicals, ChemSusChem, 2013, 6(9), 1595–1614 CrossRef CAS PubMed.
- B. Ndaba, I. Chiyanzu and S. Marx, n-Butanol derived from biochemical and chemical routes: A review, Biotechnol. Rep., 2015, 8, 1–9 CrossRef CAS PubMed.
- T. Matsu-Ura, S. Sakaguchi, Y. Obora and Y. Ishii, Guerbet reaction of primary alcohols leading to β-alkylated dimer alcohols catalyzed by iridium complexes, J. Org. Chem., 2006, 71(21), 8306–8308 CrossRef CAS PubMed.
- B. Sow, S. Hamoudi, M. H. Zahedi-Niaki and S. Kaliaguine, 1-Butanol etherification over sulfonated mesostructured silica and organo-silica, Microporous Mesoporous Mater., 2005, 79(1–3), 129–136 CrossRef CAS.
- B. Bailey, J. Eberhardt, S. Goguen and J. Erwin, Diethyl ether (DEE) as a renewable diesel fuel, SAE Trans., 1997, 1578–1584 Search PubMed.
- H. Chiang and A. Bhan, Catalytic consequences of hydroxyl group location on the rate and mechanism of parallel dehydration reactions of ethanol over acidic zeolites, J. Catal., 2010, 271(2), 251–261 CrossRef CAS.
- D. Varisli, T. Dogu and G. Dogu, Ethylene and diethyl-ether production by dehydration reaction of ethanol over different heteropolyacid catalysts, Chem. Eng. Sci., 2007, 62(18–20), 5349–5352 CrossRef CAS.
- G. Onyestyák, G. Novodárszki, R. Barthos, S. Klébert, Á. F. Wellisch and A. Pilbáth, Acetone alkylation with ethanol over multifunctional catalysts by a borrowing hydrogen strategy, RSC Adv., 2015, 5(120), 99502–99509 RSC.
- P. Anbarasan, Z. C. Baer, S. Sreekumar, E. Gross, J. B. Binder, H. W. Blanch, D. S. Clark and F. D. Toste, Integration of chemical catalysis with extractive fermentation to produce fuels, Nature, 2012, 491(7423), 235–239 CrossRef CAS PubMed.
- M. Balakrishnan, E. R. Sacia, S. Sreekumar, G. Gunbas, A. A. Gokhale, C. D. Scown, F. D. Toste and A. T. Bell, Novel pathways for fuels and lubricants from biomass optimized using life-cycle greenhouse gas assessment, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(25), 7645–7649 CrossRef CAS PubMed.
- G. Xu, Q. Li, J. Feng, Q. Liu, Z. Zhang, X. Wang, X. Zhang and X. Mu, Direct α-alkylation of ketones with alcohols in water, ChemSusChem, 2014, 7(1), 105–109 CrossRef CAS PubMed.
- C. M. Moore, R. W. Jenkins, M. T. Janicke, W. L. Kubic Jr, E. Polikarpov, T. A. Semelsberger and A. D. Sutton, Synthesis of acetone-derived C6, C9, and C12 carbon scaffolds for chemical and fuel applications, ChemSusChem, 2016, 9(24), 3382–3386 CrossRef CAS PubMed.
- E. R. Sacia, M. Balakrishnan, M. H. Deaner, K. A. Goulas, F. D. Toste and A. T. Bell, Highly selective condensation of biomass-derived methyl ketones as a source of aviation fuel, ChemSusChem, 2015, 8(10), 1726–1736 CrossRef CAS PubMed.
- H. Olcay, Y. Xu and G. W. Huber, Effects of hydrogen and water on the activity and selectivity of acetic acid hydrogenation on ruthenium, Green Chem., 2014, 16(2), 911–924 RSC.
- S. Zhang, X. Duan, L. Ye, H. Lin, Z. Xie and Y. Yuan, Production of ethanol by gas phase hydrogenation of acetic acid over carbon nanotube-supported Pt–Sn nanoparticles, Catal. Today, 2013, 215, 260–266 CrossRef CAS.
- W.-T. Liu and C.-S. Tan, Liquid-phase esterification of propionic acid with n-butanol, Ind. Eng. Chem. Res., 2001, 40(15), 3281–3286 CrossRef CAS.
- I. B. Ju, H.-W. Lim, W. Jeon, D. J. Suh, M.-J. Park and Y.-W. Suh, Kinetic study of catalytic esterification of butyric acid and n-butanol over Dowex 50Wx8-400, Chem. Eng. J., 2011, 168(1), 293–302 CrossRef CAS.
- R. A. Baylon, J. Sun, K. J. Martin, P. Venkitasubramanian and Y. Wang, Beyond ketonization: Selective conversion of carboxylic acids to olefins over balanced Lewis acid–base pairs, Chem. Commun., 2016, 52(28), 4975–4978 RSC.
- A. Corma, B. Oliver-Tomas, M. Renz and I. L. Simakova, Conversion of levulinic acid derived valeric acid into a liquid transportation fuel of the kerosene type, J. Mol. Catal. A: Chem., 2014, 388, 116–122 CrossRef.
- B. Oliver-Tomas, M. Renz and A. Corma, Direct conversion of carboxylic acids (Cn) to alkenes (C2n−1) over titanium oxide in absence of noble metals, J. Mol. Catal. A: Chem., 2016, 415, 1–8 CrossRef CAS.
- M. T. Agler, C. M. Spirito, J. G. Usack, J. J. Werner and L. T. Angenent, Chain elongation with reactor microbiomes: Upgrading dilute ethanol to medium-chain carboxylates, Energy Environ. Sci., 2012, 5(8), 8189–8192 RSC.
- R. S. Nelson, D. J. Peterson, E. M. Karp, G. T. Beckham and D. Salvachúa, Mixed carboxylic acid production by Megasphaera elsdenii from glucose and lignocellulosic hydrolysate, Fermentation, 2017, 3(1), 10 CrossRef.
- D. M. Alonso, J. Q. Bond, J. C. Serrano-Ruiz and J. A. Dumesic, Production of liquid hydrocarbon transportation fuels by oligomerization of biomass-derived C 9 alkenes, Green Chem., 2010, 12(6), 992–999 RSC.
- X. Huo, N. A. Huq, J. Stunkel, N. S. Cleveland, A. K. Starace, A. E. Settle, A. M. York, R. S. Nelson, D. G. Brandner and L. Fouts, Tailoring diesel bioblendstock from integrated catalytic upgrading of carboxylic acids: A “fuel property first” approach, Green Chem., 2019, 21(21), 5813–5827 RSC.
- N. A. Huq, G. R. Hafenstine, X. Huo, H. Nguyen, S. M. Tifft, D. R. Conklin, D. Stück, J. Stunkel, Z. Yang and J. S. Heyne, et al., Toward net-zero sustainable aviation fuel with wet waste–derived volatile fatty acids, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(13), e2023008118 CrossRef CAS PubMed.
- A. T. Aguayo, A. G. Gayubo, A. Atutxa, M. Olazar and J. Bilbao, Catalyst deactivation by coke in the transformation of aqueous ethanol into hydrocarbons. Kinetic modeling and acidity deterioration of the catalyst, Ind. Eng. Chem. Res., 2002, 41(17), 4216–4224 CrossRef CAS.
- H. Xiong, H. N. Pham and A. K. Datye, Hydrothermally stable heterogeneous catalysts for conversion of biorenewables, Green Chem., 2014, 16(11), 4627–4643 RSC.
- R. Luque, C. S. Lin, C. Du, D. J. Macquarrie, A. Koutinas, R. Wang, C. Webb and J. H. Clark, Chemical transformations of succinic acid recovered from fermentation broths by a novel direct vacuum distillation–crystallisation method, Green Chem., 2009, 11(2), 193–200 RSC.
- Z. Zhang, J. E. Jackson and D. J. Miller, Effect of biogenic fermentation impurities on lactic acid hydrogenation to propylene glycol, Bioresour. Technol., 2008, 99(13), 5873–5880 CrossRef CAS PubMed.
- M. Besson and P. Gallezot, Deactivation of metal catalysts in liquid phase organic reactions, Catal. Today, 2003, 81(4), 547–559 CrossRef CAS.
- I. Sádaba, M. L. Granados, A. Riisager and E. Taarning, Deactivation of solid catalysts in liquid media: The case of leaching of active sites in biomass conversion reactions, Green Chem., 2015, 17(8), 4133–4145 RSC.
- D. J. Cole-Hamilton, Homogeneous catalysis–new approaches to catalyst separation, recovery, and recycling, Science, 2003, 299(5613), 1702–1706 CrossRef CAS PubMed.
- D. J. Cole-Hamilton and R. P. Tooze, Catalyst separation, recovery and recycling: Chemistry and process design, Springer Science & Business Media, 2006 Search PubMed.
- F. Héroguel, B. Rozmyslowicz and J. S. Luterbacher, Improving heterogeneous catalyst stability for liquid-phase biomass conversion and reforming. CHIMIA International Journal for, Chemistry, 2015, 69(10), 582–591 Search PubMed.
- B. J. O’Neill, D. H. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, Catalyst design with atomic layer deposition, ACS Catal., 2015, 5(3), 1804–1825 CrossRef.
- R. Rinaldi and F. Schüth, Design of solid catalysts for the conversion of biomass, Energy Environ. Sci., 2009, 2(6), 610–626 RSC.
- A. Chieregato, J. Velasquez Ochoa, C. Bandinelli, G. Fornasari, F. Cavani and M. Mella, On the chemistry of ethanol on basic oxides: Revising mechanisms and intermediates in the Lebedev and Guerbet reactions, ChemSusChem, 2015, 8(2), 377–388 CrossRef CAS PubMed.
- D. Gabriëls, W. Y. Hernández, B. Sels, P. Van Der Voort and A. Verberckmoes, Review of catalytic systems and thermodynamics for the Guerbet condensation reaction and challenges for biomass valorization, Catal. Sci. Technol., 2015, 5(8), 3876–3902 RSC.
- T. N. Pham, T. Sooknoi, S. P. Crossley and D. E. Resasco, Ketonization of carboxylic acids: Mechanisms, catalysts, and implications for biomass conversion, ACS Catal., 2013, 3(11), 2456–2473 CrossRef CAS.
- S. Mitchell, N.-L. Michels and J. Pérez-Ramírez, From powder to technical body: The undervalued science of catalyst scale up, Chem. Soc. Rev., 2013, 42(14), 6094–6112 RSC.
- B. H. Shanks, Conversion of biorenewable feedstocks: New challenges in heterogeneous catalysis, Ind. Eng. Chem. Res., 2010, 49(21), 10212–10217 CrossRef CAS.
- C. Liu, B. C. Colón, M. Ziesack, P. A. Silver and D. G. Nocera, Water splitting–biosynthetic system with CO2 reduction efficiencies exceeding photosynthesis, Science, 2016, 352(6290), 1210–1213, DOI:10.1126/science.aaf5039 , [accessed 2021 Jun 30].
- Y.-C. Yeh, S. W. Singer, S. R. Chhabra, H. R. Beller and J. Mueller, Hybrid organic-inorganic system for producing biofuels. Lawrence Berkeley National Lab. (LBNL), Berkeley, CA (United States), 2017, Report No.: 9777300, https://www.osti.gov/doepatents/biblio/1397261 Search PubMed.
- M. Dahmen and W. Marquardt, Model-based design of tailor-made biofuels, Energy Fuels, 2016, 30(2), 1109–1134 CrossRef CAS.
- R. L. McCormick, G. Fioroni, L. Fouts, E. Christensen, J. Yanowitz, E. Polikarpov, K. Albrecht, D. J. Gaspar, J. Gladden and A. George, Selection criteria and screening of potential biomass-derived streams as fuel blendstocks for advanced spark-ignition engines, SAE Int. J. Fuels Lubric., 2017, 10(2), 442–460 CrossRef.
- R. L. McCormick, M. A. Ratcliff, E. Christensen, L. Fouts, J. Luecke, G. M. Chupka, J. Yanowitz, M. Tian and M. Boot, Properties of oxygenates found in upgraded biomass pyrolysis oil as components of spark and compression ignition engine fuels, Energy Fuels, 2015, 29(4), 2453–2461 CrossRef CAS.
- S. Atsumi, T. Hanai and J. C. Liao, Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels, Nature, 2008, 451(7174), 86 CrossRef CAS PubMed.
- L. Shen, Y. Nishimura, F. Matsuda, J. Ishii and A. Kondo, Overexpressing enzymes of the Ehrlich pathway and deleting genes of the competing pathway in Saccharomyces cerevisiae for increasing 2-phenylethanol production from glucose, J. Biosci. Bioeng., 2016, 122(1), 34–39 CrossRef CAS PubMed.
- M. A. Ratcliff, J. Burton, P. Sindler, E. Christensen, L. Fouts, G. M. Chupka and R. L. McCormick, Knock resistance and fine particle emissions for several biomass-derived oxygenates in a direct-injection spark-ignition engine, SAE Int. J. Fuels Lubric., 2016, 9(1), 59–70 CrossRef CAS.
- E. Christensen, A. Williams, S. Paul, S. Burton and R. L. McCormick, Properties and performance of levulinate esters as diesel blend components, Energy Fuels, 2011, 25(11), 5422–5428 CrossRef CAS.
- J. Heyne, B. Rauch, P. Le Clercq and M. Colket, Sustainable aviation fuel prescreening tools and procedures, Fuel., 2021, 290, 120004, DOI:10.1016/j.fuel.2020.120004 , https://linkinghub.elsevier.com/retrieve/pii/S0016236120330003, [accessed 2021 Jun 30].
- J. Yanowitz and R. L. McCormick, Fuel volatility standards and spark-ignition vehicle driveability, SAE Int. J. Fuels Lubric., 2016, 9(2), 408–429 CrossRef CAS.
- R. French and P. Malone, Phase equilibria of ethanol fuel blends, Fluid Phase Equilib., 2005, 228, 27–40 CrossRef.
- R. L. McCormick and R. ParishMilestone report: Technical barriers to the use of ethanol in diesel Fuel 22.
- S. Zabarnick and N. Widmor, Studies of jet fuel freezing by differential scanning calorimetry, Energy Fuels, 2001, 15(6), 1447–1453 CrossRef CAS.
- S. S. Ray, N. K. Pandey and A. K. Chatterjee, Effect of aromatics and iso-alkanes on the pour point of different types of lube oils, Fuel, 2009, 88(9), 1629–1633 CrossRef CAS.
- R. O. Dunn, Effects of minor constituents on cold flow properties and performance of biodiesel, Prog. Energy Combust. Sci., 2009, 35(6), 481–489 CrossRef CAS.
- G. M. Chupka, L. Fouts, J. A. Lennon, T. L. Alleman, D. A. Daniels and R. L. McCormick, Saturated monoglyceride effects on low-temperature performance of biodiesel blends, Fuel Process. Technol., 2014, 118, 302–309 CrossRef CAS.
- G. M. Chupka, L. Fouts and R. L. McCormick, Effect of low-level impurities on low-temperature performance properties of biodiesel, Energy Environ. Sci., 2012, 5(9), 8734–8742 RSC.
- D. Splitter, A. Pawlowski and R. Wagner, A historical analysis of the co-evolution of light-duty fuels and engines, Front. Mech. Eng., 2016, 1(1), 16 Search PubMed.
- J. P. Szybist, S. Busch, R. L. McCormick, J. A. Pihl, D. A. Splitter, M. A. Ratcliff, C. P. Kolodziej, J. M. E. Storey, M. Moses-DeBusk and D. Vuilleumier, et al., What fuel properties enable higher thermal efficiency in spark-ignited engines?, Prog. Energy Combust. Sci., 2021, 82, 100876, DOI:10.1016/j.pecs.2020.100876 , http://www.sciencedirect.com/science/article/pii/S0360128520300861, [accessed 2020 Nov 25].
- T. G. Leone, J. E. Anderson, R. S. Davis, A. Iqbal, R. A. Reese, M. H. Shelby and W. M. Studzinski, The effect of compression ratio, fuel octane rating, and ethanol content on spark-ignition engine efficiency, Environ. Sci. Technol., 2015, 49(18), 10778–10789 CrossRef CAS PubMed.
- J. Farrell, J. Holladay and R. Wagner, Fuel blendstocks with the potential to optimize future gasoline engine performance: Identification of five chemical families for detailed evaluation, US Department of Energy, DOE/GO-102018-4970, 2018.
- G. M. Fioroni, M. J. Rahimi, C. K. Westbrook, S. W. Wagnon, W. J. Pitz, S. Kim and R. L. McCormick, Chemical kinetic basis of synergistic blending for research octane number, Fuel, 2022, 307, 121865, DOI:10.1016/j.fuel.2021.121865 , https://www.sciencedirect.com/science/article/pii/S0016236121017440, [accessed 2021 Nov 15].
- H. Zhao, HCCI and CAI engines for the automotive industry, Elsevier, 2007 Search PubMed.
- C. K. Westbrook, M. Mehl, W. J. Pitz and M. Sjöberg, Chemical kinetics of octane sensitivity in a spark-ignition engine, Combust. Flame, 2017, 175, 2–15 CrossRef CAS.
- H. J. Curran, S. L. Fischer and F. L. Dryer, The reaction kinetics of dimethyl ether. II: Low-temperature oxidation in flow reactors, Int. J. Chem. Kinet., 2000, 32(12), 741–759 CrossRef CAS.
- G. Fioroni, L. Fouts, J. Luecke, D. Vardon, N. Huq, E. Christensen, X. Huo, T. Alleman, R. McCormick and M. Kass, Screening of potential biomass-derived streams as fuel blendstocks for mixing controlled compression ignition combustion, SAE Int. J. Adv. Curr. Pract. Mobility, 2019, 1(2019-01–0570), 1117–1138 Search PubMed.
- Agency UEP, Integrated science assessment (ISA) for particulate matter (Final Report, Dec 2009), US Environmental Protection Agency, Washington, DC, 2009.
- V. Ramanathan and G. Carmichael, Global and regional climate changes due to black carbon, Nat. Geosci., 2008, 1(4), 221–227 CrossRef CAS.
- T. Johnson, Vehicular emissions in review, SAE Int. J. Eng., 2016, 9(2), 1258–1275 CrossRef.
- W. Park, S. Park, R. D. Reitz and E. Kurtz, The effect of oxygenated fuel properties on diesel spray combustion and soot formation, Combust. Flame, 2017, 180, 276–283 CrossRef CAS.
- R. L. McCormick and R. Parish, Advanced petroleum based fuels program and renewable diesel program, NREL/MP, 2001, pp. 540–32674 Search PubMed.
- J. Liu, H. Wang, Y. Li, Z. Zheng, Z. Xue, H. Shang and M. Yao, Effects of diesel/PODE (polyoxymethylene dimethyl ethers) blends on combustion and emission characteristics in a heavy duty diesel engine, Fuel, 2016, 177, 206–216 CrossRef CAS.
- A. H. Lefebvre, Flame radiation in gas turbine combustion chambers, Int. J. Heat Mass Transfer, 1984, 27(9), 1493–1510 CrossRef CAS.
- B. T. Brem, L. Durdina, F. Siegerist, P. Beyerle, K. Bruderer, T. Rindlisbacher, S. Rocci-Denis, M. G. Andac, J. Zelina and O. Penanhoat, Effects of fuel aromatic content on nonvolatile particulate emissions of an in-production aircraft gas turbine, Environ. Sci. Technol., 2015, 49(22), 13149–13157 CrossRef CAS PubMed.
- E. J. Barrientos, M. Lapuerta and A. L. Boehman, Group additivity in soot formation for the example of C-5 oxygenated hydrocarbon fuels, Combust. Flame, 2013, 160(8), 1484–1498 CrossRef CAS.
- H. L. Fang and R. L. McCormick, Spectroscopic study of biodiesel degradation pathways, SAE Technical Paper, 2006 Search PubMed.
- F. Pradelle, S. L. Braga, A. R. F. Martins, F. Turkovics and R. N. Pradelle, Gum formation in gasoline and its blends: A review, Energy Fuels, 2015, 29(12), 7753–7770 CrossRef CAS.
- L. Q. Maurice, H. Lander, T. Edwards and W. E. Harrison III, Advanced aviation fuels: A look ahead via a historical perspective, Fuel, 2001, 80(5), 747–756 CrossRef CAS.
- E. Christensen, G. M. Fioroni, S. Kim, L. Fouts, R. S. Paton and R. L. McCormick, Experimental and theoretical study of oxidative stability of alkylated furans used as gasoline blend components, Fuel, 2018, 212, 576–585 CrossRef CAS.
- R. L. McCormick and S. R. Westbrook, Storage stability of biodiesel and biodiesel blends, Energy Fuels, 2010, 24(1), 690–698 CrossRef CAS.
- E. Christensen, R. L. McCormick, J. Sigelko, S. Johnson, S. Zickmann, S. Lopes, R. Gault and D. Slade, Impact of a diesel high pressure common rail fuel system and onboard vehicle storage on B20 biodiesel blend stability, SAE Int. J. Fuels Lubric., 2016, 9(1), 203–214 CrossRef.
- E. Christensen and R. L. McCormick, Long-term storage stability of biodiesel and biodiesel blends, Fuel Process. Technol., 2014, 128, 339–348 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Gamma-valerolactone, a sustainable platform molecule derived from lignocellulosic biomass, Green Chem., 2013, 15(3), 584–595 RSC.
- I. T. Horváth, H. Mehdi, V. Fábos, L. Boda and L. T. Mika, γ-Valerolactone—A sustainable liquid for energy and carbon-based chemicals, Green Chem., 2008, 10(2), 238–242 RSC.
- Information Bulletin: GHB Analogs; GBL, BD, GHV, and GVL, https://www.justice.gov/archive/ndic/pubs1/1621/1621t.htm, [accessed 2020 Jul 29].
- F. de Oliveira Gonçalves, E. Savioli Lopes, M. Savioli Lopes and R. Maciel Filho, Evaluation of the feasibility of ethanol and gasoline in solid oxide fuel cell vehicles in Brazil, Int. J. Hydrogen Energy., 2021, 46(73), 36381–36397, DOI:10.1016/j.ijhydene.2021.08.165 , https://www.sciencedirect.com/science/article/pii/S0360319921034108, [accessed 2021 Nov 15].
- P. Stalhammar, SCANIA – ED95 development Alcohol CI combustion, http://www.lth.se/fileadmin/mot2030/filer/11._Stalhammar_-_Scania_ED95_development.pdf.
- V. B. Pedrozo, I. May, W. Guan and H. Zhao, High efficiency ethanol–diesel dual-fuel combustion: A comparison against conventional diesel combustion from low to full engine load, Fuel, 2018, 230, 440–451 CrossRef CAS.
- T. G. Smagala, E. Christensen, K. M. Christison, R. E. Mohler, E. Gjersing and R. L. McCormick, Hydrocarbon renewable and synthetic diesel fuel blendstocks: Composition and properties, Energy Fuels, 2013, 27(1), 237–246 CrossRef CAS.
- B. Website, Boeing Commits to Deliver Commercial Airplanes Ready to Fly on 100% Sustainable Fuels, 2021, https://boeing.mediaroom.com/2021-01-22-Boeing-Commits-to-Deliver-Commercial-Airplanes-Ready-to-Fly-on-100-Sustainable-Fuels.
- J. Han, L. Tao and M. Wang, Well-to-wake analysis of ethanol-to-jet and sugar-to-jet pathways, Biotechnol. Biofuels, 2017, 10(1), 21, DOI:10.1186/s13068-017-0698-z , [accessed 2021 Nov 15].
- L. Tao, J. N. Markham, Z. Haq and M. J. Biddy, Techno-economic analysis for upgrading the biomass-derived ethanol-to-jet blendstocks, Green Chem., 2017, 19(4), 1082–1101 RSC.
- The Alcohol-to-Jet Conversion Pathway for Drop-In Biofuels: Techno-Economic Evaluation – Geleynse – 2018 – ChemSusChem – Wiley Online Library, DOI: 10.1002/cssc.201801690, [accessed 2020 Nov 30].
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee02540f |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |