
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Secondary aerosol formation during the dark oxidation of residential biomass burning emissions†
John K.
Kodros
 a,
Christos
Kaltsonoudis
a,
Marco
Paglione
b,
Kalliopi
Florou
a,
Spiro
Jorga
c,
Christina
Vasilakopoulou
ad,
Manuela
Cirtog
e,
Mathieu
Cazaunau
e,
Bénédicte
Picquet-Varrault
e,
Athanasios
Nenes
*af and
Spyros N.
Pandis
*ad
a,
Christos
Kaltsonoudis
a,
Marco
Paglione
b,
Kalliopi
Florou
a,
Spiro
Jorga
c,
Christina
Vasilakopoulou
ad,
Manuela
Cirtog
e,
Mathieu
Cazaunau
e,
Bénédicte
Picquet-Varrault
e,
Athanasios
Nenes
*af and
Spyros N.
Pandis
*ad
aInstitute of Chemical Engineering Sciences, ICE-HT, Patras, 26504, Greece. E-mail: athanasios.nenes@epfl.ch; spyros@chemeng.upatras.gr
bInstitute of Atmospheric Sciences and Climate, Italian National Research Council, Bologna 40129, Italy
cDepartment of Chemical Engineering, Carnegie Mellon University, Pittsburgh, 15213, USA
dUniv Paris Est Creteil and Université Paris Cité, CNRS, LISA, F-94010 Créteil, France
eLISA, UMR CNRS 7583, Université Paris-Est Créteil, Université de Paris, Institut Pierre Simon Laplace (IPSL), Créteil, France
fSchool of Architecture, Civil and Environmental Engineering, Swiss Federal Institute of Technology Lausanne, Lausanne 1015, Switzerland
First published on 23rd August 2022
Abstract
Particulate matter from biomass burning emissions affects air quality, ecosystems and climate; however, quantifying these effects requires that the connection between primary emissions and secondary aerosol production is firmly established. We performed atmospheric simulation chamber experiments on the chemical oxidation of residential biomass burning emissions under dark conditions. Biomass burning organic aerosol was found to age under dark conditions, with its oxygen-to-carbon ratio increasing by 7–34% and producing 1–38 μg m−3 of secondary organic aerosol (5–80% increase over the fresh organic aerosol) after 30 min of exposure to NO3 radicals in the chamber (corresponding to 1–3 h of exposure to typical nighttime NO3 radical concentrations in an urban environment). The average mass concentration of SOA formed under dark-oxidation conditions was comparable to the mass concentration formed after 3 h (equivalent to 7–10 h of ambient exposure) under ultraviolet lights (6 μg m−3 or a 47% increase over the emitted organic aerosol concentration). The dark-aging experiments showed a substantial increase in secondary nitrate aerosol (0.12–3.8 μg m−3), 46–100% of which is in the form of organic nitrates. The biomass burning aerosol pH remained practically constant at 2.8 throughout the experiment. This value promotes inorganic nitrate partitioning to the particulate phase, potentially contributing to the buildup of nitrate aerosol in the boundary layer and enhancing long-range transport. These results suggest that oxidation through reactions with the NO3 radical is an additional secondary aerosol formation pathway in biomass burning emission plumes that should be accounted for in atmospheric chemical-transport models.
Environmental significanceUnderstanding the pathways for chemical aging of biomass burning emissions is essential to identifying and quantifying the impact of this source on air quality and climate; however, the role of chemical oxidation occurring in the absence of sunlight is not well understood. We present evidence of rapid dark chemical oxidation of biomass burning emissions and secondary aerosol production under laboratory conditions. This overnight production of secondary aerosol may contribute to aerosol burdens in winter urban environments. Thermodynamic modeling suggests that the pH of biomass burning remains at a level that can further promote inorganic aerosol production. This dark oxidation is not included in chemical transport models thus under-representing the link between biomass burning and its impact on health and climate. |
1 Introduction
Biomass burning (BB), both in the form of open fires (including wildfires) and residential burning, is an increasingly important source of ambient particulate matter (PM) air pollution, affecting both climate and human health.1–5 BB emits substantial amounts of primary organic aerosol (POA), black carbon (BC), and brown carbon (BrC) directly into the particle phase.2,6–9 These primary emissions are a major contributor to global organic aerosol (OA) and absorbing aerosol burdens.2 BB is also an important contributor to the oxidative potential of PM, which is currently thought to be one of the causes of adverse health outcomes from PM inhalation.10,11BB emits semi (SVOCs), intermediate (IVOCs), and volatile organic compounds (VOCs) directly into the atmosphere.12 Moreover, as the plume dilutes, POA can evaporate and further contribute to SVOC and IVOC availability.13,14 These organic vapors may react with oxidants in the atmosphere (e.g., OH, O3, and NO3) to form lower-volatility compounds, which may then condense to the particle phase forming secondary organic aerosol (SOA). These chemical and physical transformations within the BB plume may form SOA on a time scale of hours to days further impacting climate and air quality.14–17 This SOA is often chemically different to the POA emitted directly from the BB source,18,19 complicating the link between BB emissions and net PM concentrations in the days following emission. In order to fully quantify the impacts of BB plumes on human health and climate, it is necessary to account for both the corresponding primary and secondary PM.
Previous research has mainly focused on the oxidation of BB plumes in the presence of sunlight (i.e., via the OH radical).18,20–22 While the conditions that lead to net OA formation (the increase in SOA outweighs the decrease in POA due to dilution and evaporation) are not entirely understood, it is clear that BB plumes can undergo extensive chemical processing resulting in SOA production through oxidation with the OH radical. Hennigan et al.23 performed laboratory smog chamber experiments on BB emissions from a variety of fuels and reported a wide range of outcomes ranging from a net OA increase to a net decrease across all experiments. Hodshire et al.22 reported that laboratory experiments often showed a larger net increase in OA compared to field studies; however, both laboratory and field studies show extensive production of chemically aged aerosol, suggesting substantial SOA formation even if net OA remains roughly constant.
The established understanding of the daytime formation mechanism for SOA production has led to increasing complexity in atmospheric models, which are better able to reproduce observations under conditions of adequate sunlight.24–27 Fountoukis et al.27 found that oxidized OA (OOA) is fairly well represented in Paris in the summer; however, the model under-predicted OOA concentrations in the winter, suggesting a possible additional formation pathway for SOA under conditions of low-photochemical activity. One such proposed secondary mechanism for SOA production in BB plumes is via reactions with the NO3 radical.28
The NO3 radical is formed primarily through the reaction of NO2 and O3, and is rapidly photolyzed during the day.29,30 Thus, oxidation through reactions with the NO3 radical may only take place during periods of low photochemical activity (such as at night or in winter). Measurements of nighttime NO3 radical concentrations display a large degree of spatiotemporal variability. Two-year measurements of NO3 in an urban site in Jerusalem indicated an average nighttime concentration of 27 ± 43 ppt, with measured levels during 21 nights exceeding 220 ppt.31,32 The NO3 radical concentration has also been shown to exhibit a strong vertical gradient.33 Brown et al.34 found that on average, NO3 levels in a New England area increase with altitude reaching a maximum within the first 500 meters above the surface (with concentrations ranging from 0–90 ppt) and decrease at higher elevations. Urban and rural surface-layer measurements show average nighttime NO3 radical concentrations in the range of 10–40 ppt: Wang et al.35 reported 16 ± 9 ppt in Shanghai, China; Li et al.36 measured 22 ± 2 ppt in Guangzhou, China; McLaren et al.37 found median overnight concentrations of 10 ppt near Vancouver, Canada. Conversely, concentrations in the range of 3–10 ppt have been reported in remote sites.38–40 The chemical transport simulations of Khan et al.29 suggest that annual-mean nighttime NO3 radical concentrations ranged between 10–25 ppt across the northeastern US.
Studies to date have established that the NO3 radical is a very efficient oxidant of many biogenic VOC emissions, particularly during periods of transport and mixing with air with high NOx concentrations.30,41–45 In particular, the NO3 radical has been shown to be very reactive toward unsaturated VOCs. Recently, there has been increasing interest in exploring the extent of nighttime chemical processing of BB emission plumes. Tiitta et al.46 and Hartikainen et al.47 reported substantial SOA production (factor of 2 increases over the emitted POA concentration) in laboratory experiments under dark conditions, with dominant gas-phase reactions taking place between the NO3 radical and phenolic and furanoic compounds. Decker et al.28 initialized a chemical box model with aircraft observations and predicted substantial overnight oxidation of BB VOCs largely through reactions with NO3. Li et al.36 found substantial absorption enhancement of brown carbon after NO3 radicals were allowed to react with tar aerosols from BB in a flow reactor. Kodros et al.16 argued that the rapid and extensive chemical processing of BB plumes in the dark observed in laboratory experiments produces SOA that is chemically similar to ambient observations of oxidized and secondary OA factors, thus suggesting dark oxidation of BB plumes may be an additional formation pathway of OOA not included in most models.
Dark oxidation of BB plumes via the NO3 radical may also lead to secondary inorganic and organic nitrate aerosol formation.16,28,46 Kiendler-Scharr et al.48 found substantial organic nitrate mass concentrations in PM in urban and rural areas and estimated that the dominant formation pathway involves reactions with the NO3 radical. Similarly, Rollins et al.49 reported organic nitrate aerosol production as a result of nocturnal NO3 radical chemistry in Bakersfield, California. The contribution of residential BB specifically to organic and inorganic nitrate aerosol through dark oxidation in urban areas is not well understood.
Biomass burning aerosol exhibits substantial hygroscopicity.50 Biomass burning emits considerable amounts of NH3 and NOx, that in turn can transform into inorganic nitrate aerosol. The paucity of sulfate and the presence of potassium and other non-volatile cations also imply that the aerosol generated during biomass burning is much less acidic than other types of combustion aerosol (e.g., from fossil fuels). Observations51,52 and model studies to date53 suggest that ambient aerosol dominated by biomass burning emissions tends to exhibit elevated levels of pH compared to other aerosol types. This in turn carries important implications for the susceptibility of the aerosol to NH3 and HNO3 levels, as well as the tendency of inorganic nitrate aerosol to accumulate in the boundary layer during BB events and the resulting nitrogen being able to transport over long distances before deposition.54,55 It is unclear if the elevated pH levels are an “inherent” property of BB aerosol, and if it is driven primarily by the semi-volatile inorganic components (NH3, HNO3) or the non-volatile cations that may be present (primarily K+). Finally, the importance of the organic water in controlling aerosol pH levels also needs to be determined.
In this work we investigate if BB emissions can age rapidly in the dark and form substantial concentrations of secondary organic and inorganic aerosol. This dark oxidation is not included in most atmospheric chemical-transport models, suggesting an under-representation of the link between BB emissions and secondary aerosol. In Section 2, we detail the environmental smog chamber experimental procedure and instrumentation. In Section 3, we discuss secondary aerosol formation and mass spectra evolution for a typical experiment (Section 3.1), discuss the range of results across all experiments (Section 3.2), calculate aerosol acidity (Section 3.3), and finally estimate the atmospheric relevant timescales of the major processes (Section 3.4). In Section 4, we discuss our conclusions and study limitations.
2 Methods
2.1 Experimental facility and instrumentation
Experiments on the dark chemical processing of BB emissions took place at the Foundation for Research and Technology-Hellas (FORTH) atmospheric simulation chamber and combustion chamber facilities at the Center for the Study of Air Quality and Climate Change. This facility consists of a 30 m3 temperature and light-controlled room capable of sustaining variable sized reactors. In this study, all experiments took place in a 10 m3 Teflon reactor. Combustion of biomass took place in the combustion facility located in a separate room directly beneath the chamber. BB emissions were transferred into the chamber through a dilution system.In the chamber, a suite of online instrumentation measured the concentrations of particle- and gas-phase species. Non-refractory PM1 aerosol was monitored by a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS, Aerodyne Research Inc.) working in V mode. A scanning mobility particle sizer (SMPS; classifier model 3080, DMA model 3081, CPC model 3787, TSI) measured the aerosol number size distribution. In these experiments, the sheath flow rate was 3 L min−1 and the aerosol sample flow rate was 0.6 L min−1, allowing for a diameter size range of 14 to 790 nm. Black carbon was monitored using a multiple-angle absorption photometer (MAAP, Thermo Scientific Inc.) and a single particle soot photometer (SP2, Droplet Measurement Technology). A proton-transfer-reaction mass spectrometer (PTR-MS, Ionicon Analytik) measured VOCs. Inorganic gas-phase species were measured using a series of gas monitors: nitrogen oxides (NO and NO2, Teledyne model T201), ozone (O3, Teledyne model 400E), carbon monoxide (CO, Teledyne model 300E), and carbon dioxide (CO2, Teledyne model T360). In a subset of experiments, NO3 radical concentrations were measured by incoherent broad-band cavity-enhanced absorption spectroscopy (IBB-CEAS) as described by Fouqueau et al.56 Gas-phase concentrations of NH3 were measured using a photoacoustic monitor (LSE, model NH3-1700).
2.2 Experimental procedure
All eight dark-aging experiments (experiments 1–8; Table 1) followed the same general procedure. First, the BB emissions were injected into the chamber through the dilution system (with a dilution rate of approximately 10-to-1). In all experiments, a small amount (approximately 40–70 ppb) of d9-butanol was also injected to determine the OH concentrations. The fresh emissions remained in the chamber under dark conditions for approximately 2 hours to allow sufficient time for mixing and characterization. During this period, some chemical processing may occur through reactions among the primary pollutants and other compounds already present in the mixture (e.g. ozone). To initiate oxidation by NO3 radicals, which we defined as time zero in all experiments, we injected a variable concentration of NO2 and O3 (precursors to the NO3 radical). We then allowed at least 3 hours for chemical processing and characterization of the aged aerosol and vapors. In a subset of experiments, we injected ammonium sulfate immediately following the chemical processing period in order to quantify the size-resolved loss rates of particles to the chamber walls. We also performed 2 reference experiments (experiments 9–10). In experiment 9, we initiated oxidation (at time zero) through turning on ultraviolet (UV) lights to simulate daytime (OH radical) oxidation. In experiment 10, we left the BB emissions in the chamber under dark conditions for several hours without injection of NO2 and O3.| Exp. | Biomass burning emissions | Experimental conditions | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| OA [μg m−3] | Inorganic nitrate [μg m−3] | Organic nitrate [μg m−3] | BC [μg m−3] | O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C :C |
Lights | RH [%] | T [K] | O3 [ppb] | NO2 [ppb] | |
| a In experiment 10, no additional NO2 or O3 was injected. The levels reported here represent emissions directly from combustion as a comparison. | ||||||||||
| 1 | 14 | 0.2 | 0.2 | 7.9 | 0.3 | Dark | 9 | 27 | 123 | 93 |
| 2 | 14 | 0.1 | 0.1 | 3.8 | 0.4 | Dark | 9 | 21 | 44 | 43 |
| 3 | 83 | 0.7 | 0.8 | 109 | 0.3 | Dark | 10 | 25 | 228 | 99 |
| 4 | 39 | 0.4 | 0.2 | 0.8 | 0.4 | Dark | 58 | 27 | 91 | 57 |
| 5 | 33 | 0.7 | 0.4 | 7.0 | 0.4 | Dark | 58 | 28 | 52 | 36 |
| 6 | 9 | 0.7 | 0.4 | 36 | 0.4 | Dark | 48 | 27 | 143 | 70 |
| 7 | 47 | 0.8 | 0.5 | 4.5 | 0.4 | Dark | 45 | 27 | 173 | 97 |
| 8 | 30 | 0.8 | 0.7 | 0.1 | 0.4 | Dark | 60 | 26 | 118 | 103 |
| 9 | 13 | 0.2 | 0.2 | 6 | 0.4 | UV | 15 | 19 | — | — |
| 10 | 45 | 0.5 | 0.4 | 8.4 | 0.4 | Dark | 8 | 21 | (23)a | (7)a |
Initial concentrations of biomass burning aerosol, NO2 and O3, as well as relative humidity are presented for all experiments in Table 1. Initial OA concentrations measured by the AMS ranged from 9–83 μg m−3 across all experiments. To simulate oxidation in dark conditions in urban areas, we injected between 36–103 ppb of NO2 and 44–228 ppb of O3 for each experiment. In all experiments, NO2 was injected prior to the injection of O3 to prevent reaction of ozone with organic compounds. The relatively high concentrations of NO2 and O3 are intended to accelerate oxidation that may take place over night. Section 3.4 discusses the corresponding atmospheric timescales which are longer than those of our experiments. Finally, to test the sensitivity of dark oxidation to relative humidity and possible contribution of aqueous-phase oxidation to chemical processing, we varied the relative humidity in the chamber ranging from dry conditions (about 10% relative humidity) to moderate levels of around 60%. The temperature of the experiments ranged from 19–28 °C, values that are in general higher than those typically expected for winter nighttime conditions. We expect these higher temperatures to favor NO3 radical production. This issue will also be addressed in Section 3.4.
The combustion of olive tree wood in a residential wood stove was the BB source of our experiments. Both the wood and stove were purchased locally and represent typical residential heating devices and fuels used in Greece. Previous studies have suggested that combustion conditions such as flaming phase or combustion temperature may play a role in explaining the variability of the extent of oxidation by altering the emission profile of particle- and gas-phase organic compounds. In this study, we do not systematically test the sensitivity of dark oxidation to combustion conditions. In all experiments, we sampled under flaming conditions, roughly representative of expected conditions in a residential wood stove, approximately 15–30 minutes after ignition. We note that differences in combustion conditions may play some role in the extent of oxidation through differences in emission profiles.
2.3 Data analysis
The HR-ToF-AMS measurements were analyzed using the AMS software toolkits SeQUential Igor data RetRiEvaL (SQUIRREL) v1.57 and Peak Integration by Key Analysis (PIKA) v1.16. Elemental ratios (e.g., O:C) were calculated following the method of Canagaratna et al.57 The AMS collection efficiency was calculated following the method of Kostenidou et al.58 The total nitrate aerosol measured by the AMS was apportioned into organic and inorganic nitrate following the methods discussed in Farmer et al.59 and Kiendler-Scharr et al.48 While we recognize the uncertainty in this method, we feel its use is acceptable for the purposes of this study.
The measured size-dependent particle wall-loss rates measured during the characterization period after an experiment were used to correct the AMS mass concentrations following Wang et al.60 The relative standard deviation of the measured wall loss rate constants across experiments was 35%. In experiments where a wall-loss characterization period did not take place directly following oxidation, the average size-resolved particle wall loss rate profile was used to correct for particle wall loss.
The secondary aerosol mass concentration at time t is defined as the wall-loss corrected mass concentration minus the average wall-loss corrected mass concentration during the hour before the start of oxidation. The enhancement ratio (ER) of a parameter is defined as the parameter measured at time t divided by the averaged parameter before the start of oxidation. It has been calculated for the wall-loss corrected mass concentration and for the O:C ratio.
To separate the total BB OA (bbOA) AMS spectrum into a primary (bbPOA) and a produced secondary OA (bbSOA) spectrum, we use a simplified mass-balance approach outlined in Jorga et al.61 In this method, we estimate the initial (i.e., fresh) bbPOA assuming that its mass concentration is only affected by particle wall loss with a first-order loss rate with the wall loss rate calculated as discussed above, while the normalized bbPOA spectrum is assumed to be constant in time. The produced bbSOA spectrum is thus the difference between the measured OA spectrum and the estimated fresh bbPOA spectrum at time t. We note that this method assumes that particle wall loss and SOA formation are the dominant processes taking place in the chamber, and does not account for semi-volatile vapor wall loss or heterogeneous reactions.
We use the theta angle to quantify the differences between bbOA mass spectra.62 The theta angle treats two mass spectra as n-dimensional vectors (where n is the number of m/z values in the spectrum) and calculates the inner product between them. The theta angle is calculated by eqn (1) (following the discussion in Kostenidou et al.62):
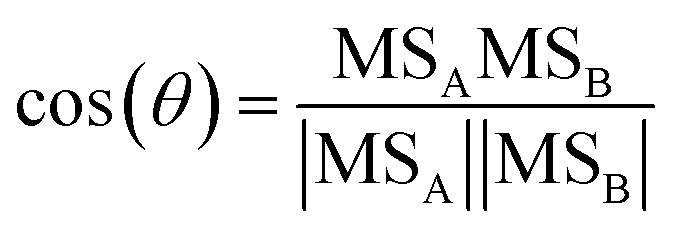 | (1) |
In all experiments, OH concentrations are inferred through the consumption of d9-butanol injected at the start of the experiment.64 Here, we define one photochemical day as continued 24 h OH exposure at a concentration of 1.5 × 106 molecule per cm3.65,66
2.4 Calculation of aerosol pH
To estimate the acidity of the BB emissions and the associated change after dark chemical aging, we simulate aerosol thermodynamic properties using ISORROPIA-Lite,67 which is based on the metastable routines of ISORROPIA II, version 2.3 (ref. 68) but expanded to include the effects of aerosol water uptake associated with the bbOA on the semi-volatile partitioning of the inorganics. Inputs to ISORROPIA-Lite include temperature, relative humidity, and the total (gas-plus particle-phase) concentrations of NH3, H2SO4, Na+, Ca2+, K+, Mg2+, HCl, and HNO3. In addition, water uptake from OA is calculated based on the OA mass concentration, density, and the hygroscopicity parameter, κ.69 Measurements from the AMS are used as inputs for the particle-phase species with the exception of the crustal elements and measurements of gas-phase NH3. Due to lack of measurements, we assume that the total HNO3 is equal to the measured particulate inorganic nitrate concentration (which is a reasonable assumption if the pH is high enough for most of the nitrate to partition to the particulate phase). We further assume that the concentrations of HCl, Na+, Ca2+, and Mg2+ are negligible. The lack of direct measurements of these species is a limitation of this analysis.In our base set of calculations, we assume that concentrations of potassium (K+) are 0.15 times the concentration of particulate nitrate following Ryu et al.70 We set the OA hygroscopicity parameter equal to 0.15 and OA density to 1.4 g cm−3. The sensitivity of our results to the above assumptions has been quantified and is described in a subsequent section.
3 Results
3.1 Results of a typical dark oxidation experiment
In a typical experiment, such as experiment 1 (see Table 1), the BB emissions were transferred into the chamber at approximately −2.5 h (Fig. 1). The particles were composed of bbOA (14 μg m−3), 8 μg m−3 of BC and less than 1 μg m−3 each of nitrate, sulfate, ammonium, and chloride (Fig. 1a). The BB emissions also included 15 ppb of O3 and 11 ppb of NO2 (Fig. 1b). During the 2 h characterization period of the fresh emissions, the mass concentration of these particle- and gas-phase species shows only slight changes, with a decreasing trend of O3 (possibly due to reactions with NO and NO2 and losses to the chamber walls).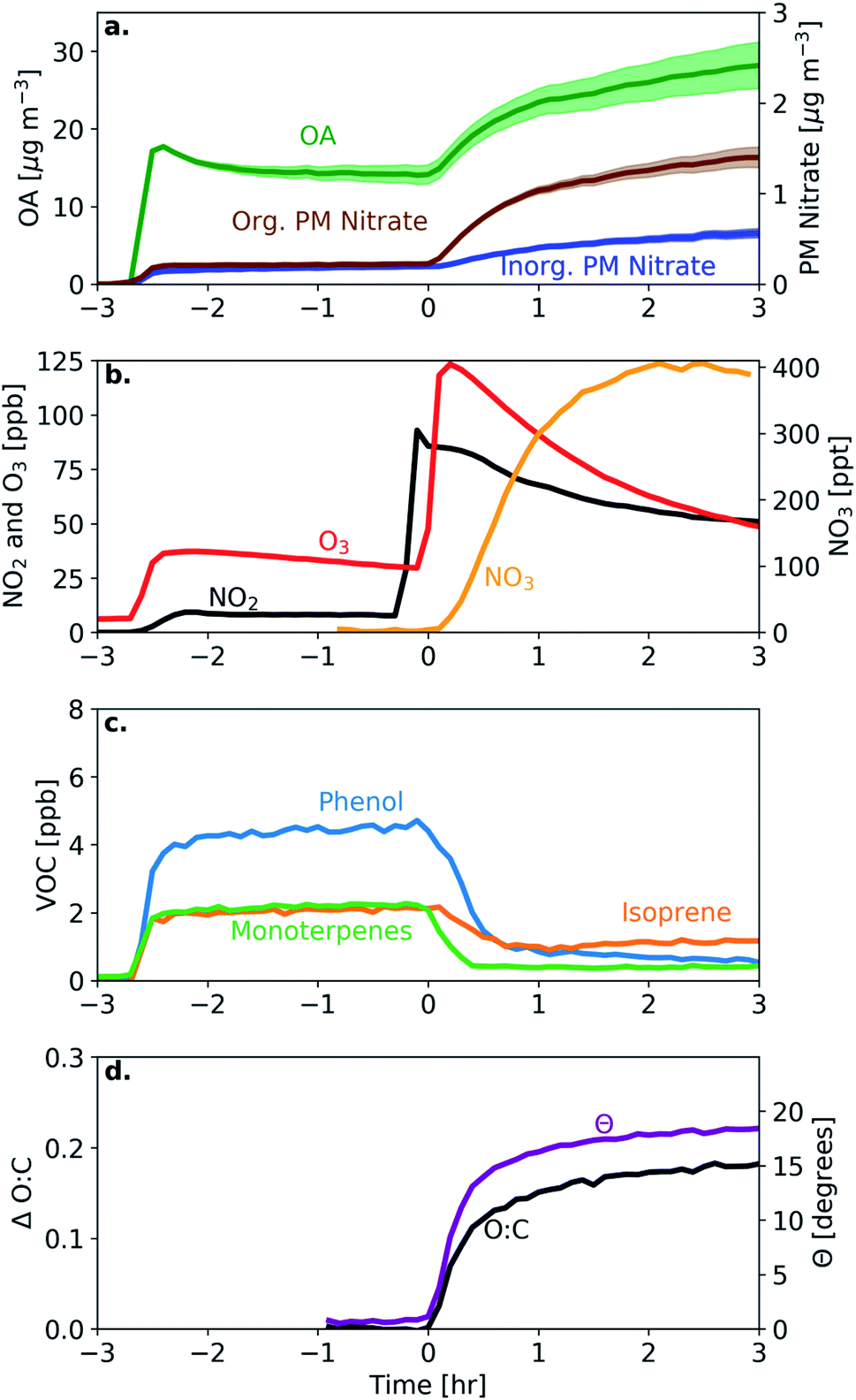 | ||
| Fig. 1 Measurements for a typical experiment (experiment 1 in Table 1) showing (a) wall-loss corrected bbOA, PM inorganic nitrate, and PM organic nitrate (shaded region represents one standard deviation around the wall-loss rate constant), (b) inorganic gas-phase species NO2, O3, and NO3 radical, (c) representative VOCs showing the largest decrease (phenol, isoprene, and monoterpenes), and (d) the change in the O:C ratio and theta angle. | ||
At time zero, dark oxidation was initiated through the injection of 93 ppb of NO2 and 123 ppb of O3, resulting in increasing NO3 radical concentrations (125 ppt after 30 min, Fig. 1b). Following this injection, rapid formation of bbSOA and PM organic nitrate is observed. In the first 30 min following the injection of O3, the bbOA mass increased by 5 μg m−3 (an enhancement of 37%), while the PM organic nitrate increased by 0.5 μg m−3 (and enhancement of almost 250%). About 40% of the secondary aerosol mass was formed in these first 30 min after the injection of O3. Also following injection, there was a corresponding decrease in VOC levels, the largest of which were phenol, isoprene, and monoterpenes (Fig. 1c).
To measure the extent of chemical processing of bbOA, we consider two metrics commonly associated with quantifying the extent of OA oxidation: the change in the O:C ratio and the evolution of the theta angle (defined in Section 2.3). In the hour prior to the initiation of oxidation both metrics remained roughly constant (the O:C ratio varied by less than 0.01 and the theta angle varied by less than 1°), suggesting little chemical changes of the fresh bbOA emissions (Fig. 1d). This suggests that on a broad level, BB emission plumes may not age rapidly in the dark without being exposed to sufficiently high concentrations of NO2 and O3 (that react to form the highly oxidizing NO3 radical). In the 30 min following the injection of NO2 and O3, the O:C ratio increases by 0.12 (34% increase), and the theta angle by 14°, indicating notable evolution of the bbOA spectrum while still maintaining some similarity to the fresh bbOA aerosol. The majority of the total increase in these metrics over the course of the experiment takes place in the first 30 min (65% and 74% of the total increase over the experiment in O:C and the theta angle, respectively).
We separate the measured OA mass spectra into a fresh bbPOA and produced bbSOA spectrum (Fig. 2). Similar to previous studies on BB oxidation, the normalized produced bbSOA spectrum in these experiments shows enhancements at m/z 28 (CO+), 29 (CHO+), and 44 (CO2+), and decreases in m/z 60 (C2H4O2+), 57 (C4H9+), and 73 (C3H5O2+), relative to the normalized fresh bbPOA spectrum (Fig. 2). Both the fresh bbPOA and the produced bbSOA display prominent peaks commonly associated with combustion at m/z 69 (C4H5O+) and 91 (C7H7+). The theta angle between the fresh and aged factor is 30°, indicating substantial dissimilarity between the two spectra.
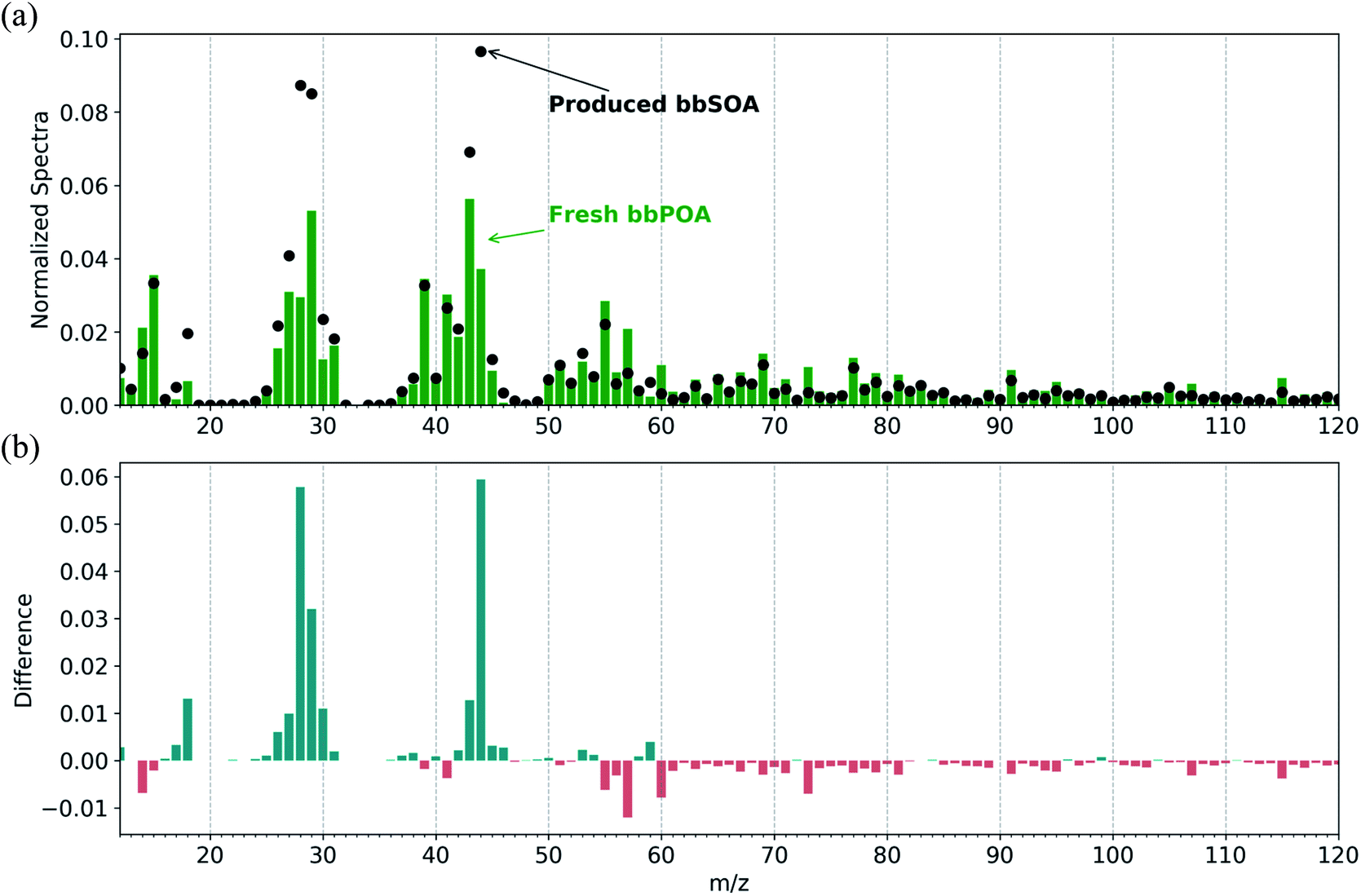 | ||
| Fig. 2 (a) The normalized fresh bbPOA (green bars) and produced bbSOA (black circles) factors and (b) the difference between them for the typical experiment (experiment 1). The blue bars indicate an increasing contribution while the red bars represent a decreasing contribution in OA at the given m/z for the produced bbSOA relative to the fresh bbPOA. | ||
To estimate the relative contribution of reactions with NO3, O3, and OH to the oxidation of BB emissions, we calculate the average lifetime of the VOCs with the largest decreasing trends: phenol, isoprene, and monoterpenes (assumed to be α-pinene for this calculation). As a simplification we use the concentrations of these VOCs in the fresh BB emissions and the average concentration of the oxidants in the first hour after the initiation of oxidation. For all three VOCs, the average lifetime for reactions with OH are greater than 9 hours (three times the length of the oxidation portion of the experiment), suggesting OH concentrations are too low (approximately 9.5 × 105 molecule per cm3) to play an important role in the oxidation reactions in our experiments (Tables S2 and S3†). In contrast, the average lifetime against reactions with NO3 are all less than 1 h (0.9 h for phenol, 0.5 h for isoprene, and 0.08–0.16 h for α-pinene), indicating NO3 likely plays a dominant role in the oxidation of all three VOCs. Finally, reactions with O3 are likely only relevant for α-pinene (with an average lifetime of 1.9–2.9 h). While the reactions of phenol are most likely dominated by the NO3 radical, it is possible that O3 may react with products of this initial reaction. For instance, the reaction of phenol with NO3 produces a variety of products, one of which is the organic molecule, C6H5O. The average lifetime of C6H5O against reactions with O3 is less than 1 min (Table S3†). Additionally, O3 may react heterogeneously with organic compounds in the aerosol phase.
3.2 Results across all experiments
:C ratio ranged from 0.3–0.4. This ratio remained roughly constant over the 1–2 h prior to oxidation. Following the injection of NO2 and O3, all experiments show an increase in O:C ratio ranging from 0.03–0.12 (enhancement ratio of 1.07–1.34) after 30 min and 0.09–0.23 (enhancement of 1.2–1.58) after 3 h (Table 2). The experiments with the lowest concentration of injected NO2 and O3 (experiments 2 and 5) had the lowest increase in O:C ratio (0.03–0.04) after the first 30 min. Similarly, the theta angle increased in all dark-aging experiments by 3–16° in the first 30 min following the initiation of oxidation and by 8–27° after 3 h (Fig. 3). While experiments 2 and 5 had the lowest exposure to NO2 and O3 and corresponding lowest increase in O:C ratio, only experiment 2 had a similarly limited increase in theta angle (3° after 30 minutes) while the theta angle in experiment 5 increased by a modest 8° after the first 30 min. The reason for this discrepancy may be the effect of relative humidity. Experiment 2 was performed under dry (less than 10% RH) conditions, while experiment 5 was performed at an RH of 60%.
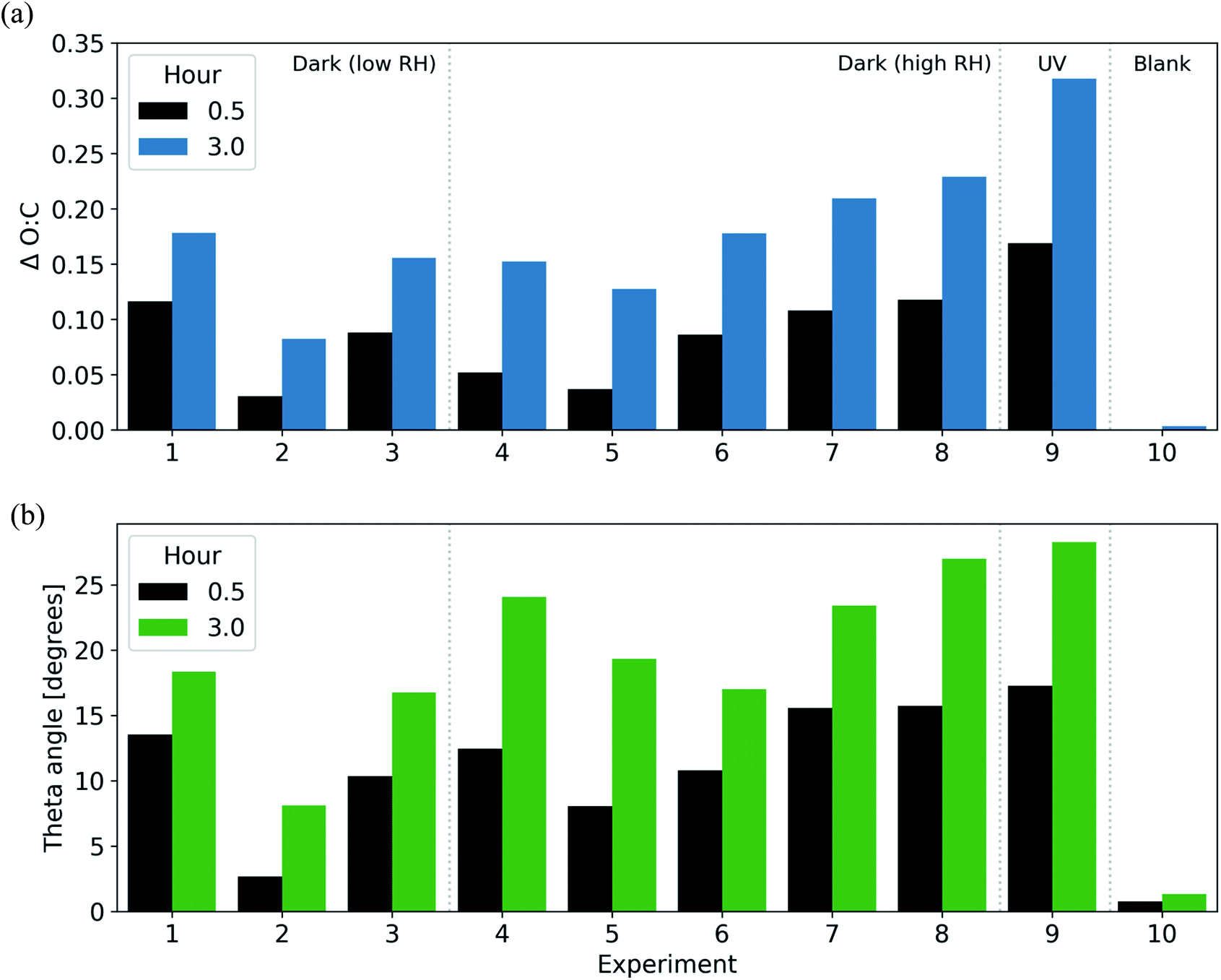 | ||
| Fig. 3 The change in (a) O:C ratio and (b) theta angle across all BB experiments. Experiments 1–3 were performed in dark and dry conditions, exp. 4–8 in dark and humid conditions, and exp. 9 and 10 are the reference experiments. The black bars represent the change in the first 0.5 h following the initiation of oxidation, while the green/black bars denote the change following 3 hours after the initiation of oxidation. | ||
:C ratio and the theta angle
| Experiment | 0.5 hours | 3 hours | ||||
|---|---|---|---|---|---|---|
| ΔO:C |
O:CER |
Theta angle | ΔO:C |
O:CER |
Theta angle | |
| 1 | 0.12 | 1.34 | 13.5 | 0.18 | 1.52 | 18.3 |
| 2 | 0.03 | 1.07 | 2.7 | 0.09 | 1.20 | 8.1 |
| 3 | 0.09 | 1.26 | 10.3 | 0.16 | 1.46 | 16.7 |
| 4 | 0.05 | 1.12 | 12.4 | 0.14 | 1.33 | 24.1 |
| 5 | 0.04 | 1.09 | 8.0 | 0.12 | 1.32 | 19.3 |
| 6 | 0.09 | 1.19 | 10.8 | 0.18 | 1.38 | 17.0 |
| 7 | 0.10 | 1.28 | 15.6 | 0.20 | 1.53 | 23.4 |
| 8 | 0.11 | 1.30 | 15.7 | 0.23 | 1.58 | 27.0 |
| 9 | 0.17 | 1.43 | 17.3 | 0.32 | 1.81 | 28.3 |
| 10 | 0.00 | 1.00 | 0.8 | 0.0 | 1.00 | 1.3 |
All dark oxidation experiments showed production of SOA following injection of NO2 and O3 (Table 3 and Fig. 4). Wall-loss corrected SOA ranged from 0.7–38 μg m−3 (an OA ER of 1.05–1.81) in the 30 min following the initiation of oxidation. Between 11–53% of the total SOA was formed within these first 30 min; however, all experiments except for experiment 5 show a continued increasing trend in wall-loss corrected OA throughout the 3 h oxidation period. Similar to the O:C ratio, experiments with lower levels of NO2 and O3 (experiments 2 and 5) tended to form lower concentrations of SOA with OA enhancements in the range of 4–5% as compared to similar experiments with higher levels of NO2 and O3 with enhancements ranging from 24–81%.
| Experiment | 0.5 hours | 3 hours | ||||||
|---|---|---|---|---|---|---|---|---|
| bbSOA [μg m−3] | OAER | PM org. nitrate [μg m−3] | PM inorg. nitrate [μg m−3] | bbSOA [μg m−3] | OAER | PM org. nitrate [μg m−3] | PM inorg. nitrate [μg m−3] | |
| 1 | 5.3 | 1.37 | 0.47 | 0.11 | 13.6 | 1.95 | 1.18 | 0.37 |
| 2 | 0.68 | 1.05 | 0.09 | 0.04 | 5.6 | 1.41 | 0.39 | 0.19 |
| 3 | 38.2 | 1.46 | 1.85 | 2.03 | 74 | 1.89 | 3.9 | 3.7 |
| 4 | 4.9 | 1.24 | 0.48 | 0.04 | 4.2 | 1.11 | 0.88 | 0.05 |
| 5 | 1.4 | 1.04 | 0.12 | 0.00 | 0.1 | 1.00 | 0.32 | −0.1 |
| 6 | 7.5 | 1.81 | 0.30 | 0.47 | 26.6 | 3.9 | 1.3 | 1.5 |
| 7 | 16.5 | 1.35 | 1.49 | 0.41 | 21 | 1.44 | 2.17 | 0.53 |
| 8 | 19.4 | 1.64 | 1.17 | 0.45 | 21 | 1.70 | 1.70 | 0.26 |
| 9 | 1.16 | 1.09 | 0.05 | 0.02 | 6.1 | 1.47 | 0.08 | 0.03 |
| 10 | 0.00 | 1.00 | 0.08 | 0.06 | 3.9 | 1.1 | 0.20 | 0.20 |
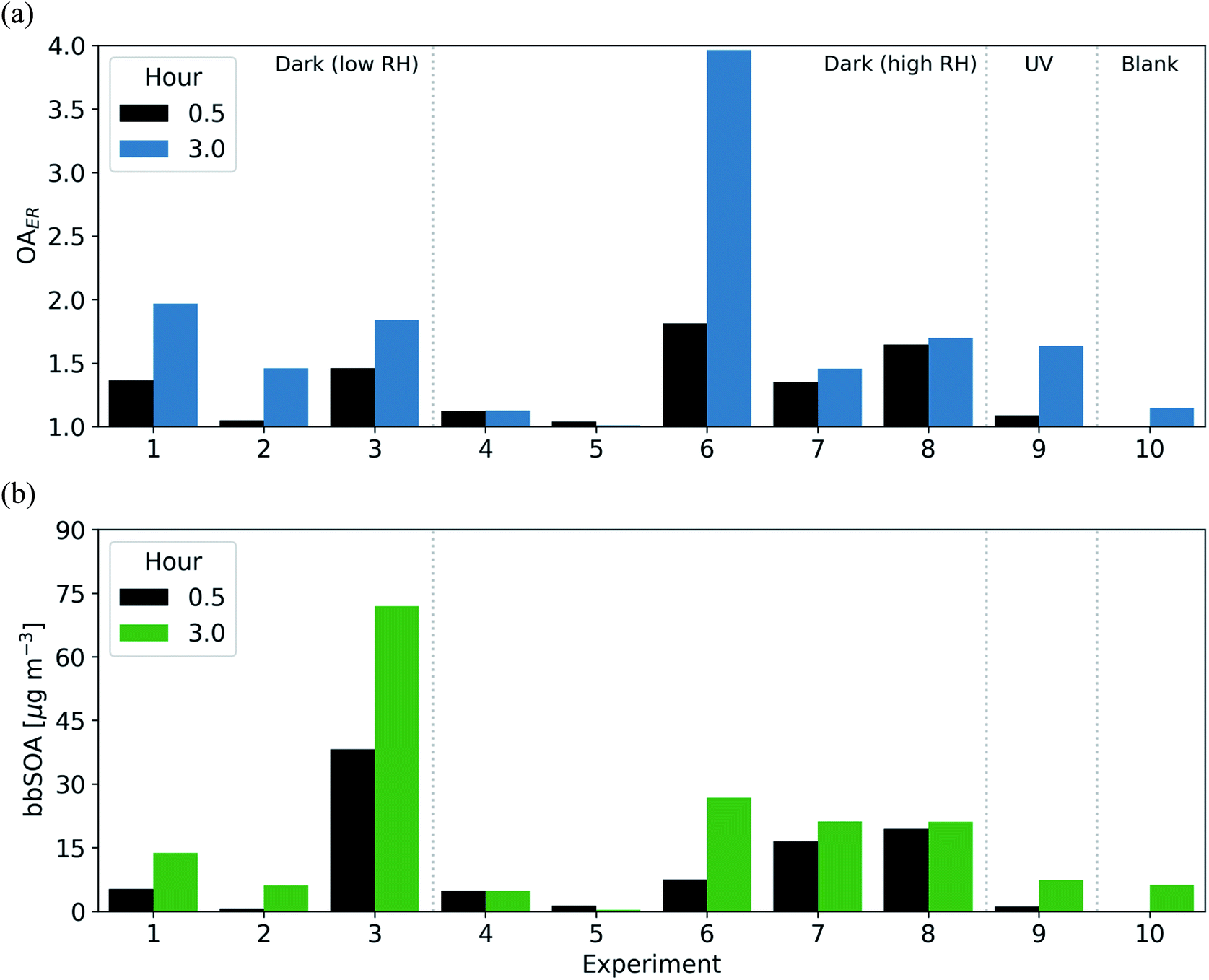 | ||
| Fig. 4 The (a) OAER and (b) bbSOA mass concentration after 30 minutes (black) and 3 hours (blue/green) for all dark oxidation and reference experiments. | ||
We compare these dark-oxidation experiments to a reference experiment in which the BB emissions were aged under UV lights (via the OH radical). The BB emissions in this experiment (experiment 9) were exposed to UV lights (JNO2 of 0.59 min−1) for 3 h. The experiment under UV conditions had a larger increase in O:C ratio (0.32 or an 80% increase) than the experiments under dark conditions (0.09–0.23 or a 20–60% increase) after 3 h. Similarly, the UV experiment showed the largest change in theta angle (28° as opposed to 8–27° in the dark-aging experiments). Conversely, the SOA formation in the UV experiment tended to be similar or slightly lower than in experiments under dark conditions. In the UV experiment, the OA enhancement ratio was 1.47 (6 μg m−3), while in the dark oxidation experiments it ranged between 1.00–1.95 (0.1–74 μg m−3) with one experiment reaching an OA enhancement of 3.9 (experiment 6).
In experiment 10, where no external oxidation was initiated, the O:C ratio remained constant throughout the experiment and the theta angle between the bbOA at the end of the experiment compared to the emitted spectrum was only 1.3°, indicating near-identical spectra.
The fresh bbOA AMS mass spectrum exhibited moderate variability across the various experiments with an average theta angle between pairs of experiments of 17° (standard deviation of 7°, Fig. S1†). Among the dark oxidation experiments, the produced bbSOA spectrum tended to be more similar with (i.e., a lower theta angle) between two experiments than the fresh bbOA factor for the same pair of experiments (an average theta angle of 12.5° with a standard deviation of 6°). This increase in similarity may be due to the increasing prominence of OA mass at m/z 28 (CO+), 29 (CHO+), and 44 (CO2+) in all aged bbOA spectra.
After the injection of NO2 and O3, both organic and inorganic nitrate increased rapidly. In 7 of 8 experiments aged under dark conditions, the majority of the secondary nitrate aerosol was organic, resulting in 46–100% of the post-oxidation (after 3 h) nitrate aerosol being organic (Fig. 5). In the 30 min after the initiation of oxidation, the organic nitrate mass concentration increased by 0.1–1.9 μg m−3. For comparison, under UV conditions, the organic nitrate mass concentration increased by only 0.08 μg m−3 in the three hours after the initiation of oxidation (Fig. 5). The large enhancements of inorganic and organic nitrate aerosol are likely characteristic of dark oxidation due to the presence of NO2 and the NO3 radical.
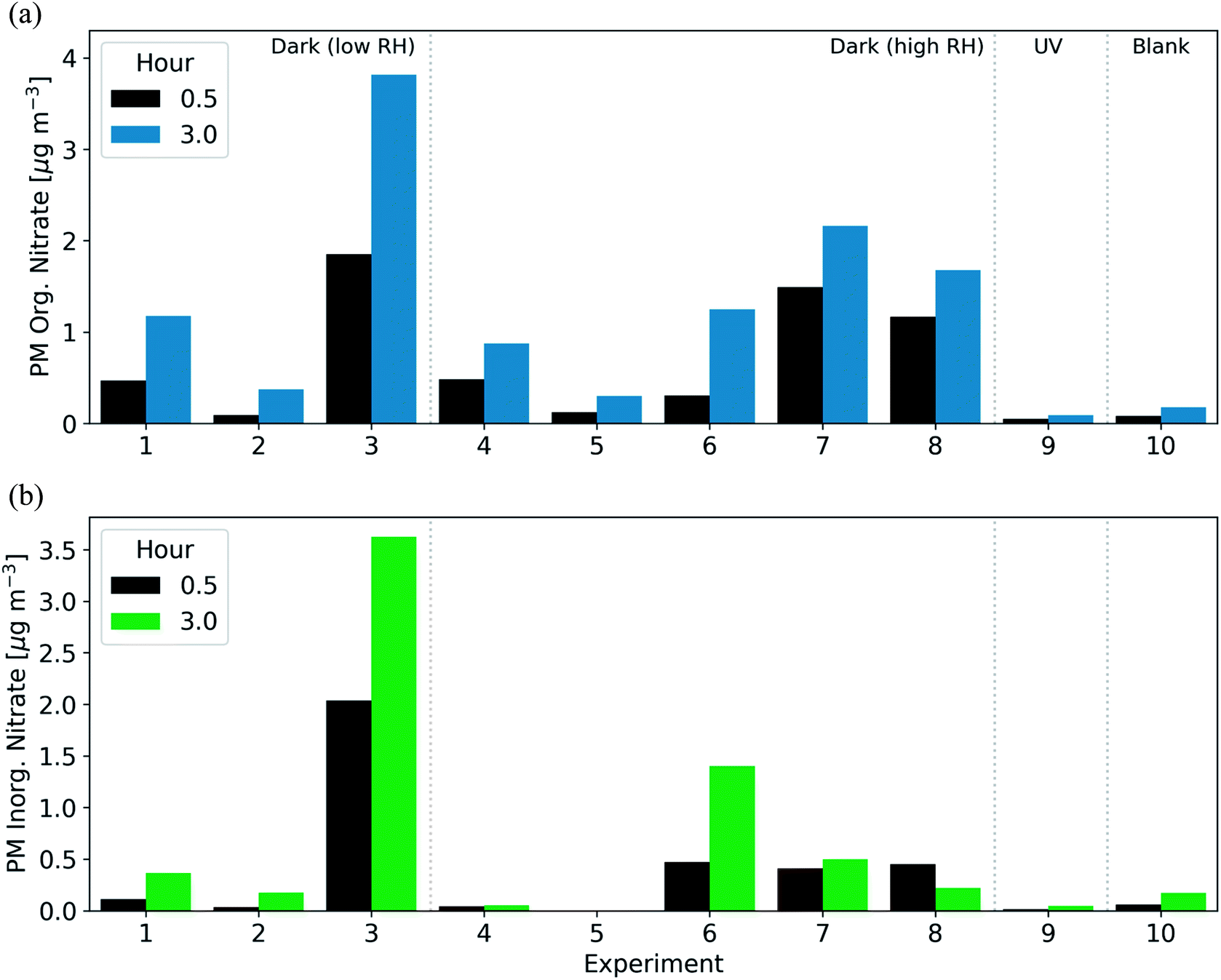 | ||
| Fig. 5 Production of secondary (a) PM organic nitrate and (b) PM inorganic nitrate aerosol after 0.5 h (black) and 3 h (blue/green) following the initiation of oxidation across all experiments. | ||
3.3 Aerosol acidity
We simulated the thermodynamic properties of the bbOA, in order to estimate the aerosol acidity before and after oxidation. Under the base assumptions (outlined in Section 2.4), we calculate an average pH for the fresh emissions of 2.8 (with standard deviation of 0.25) across all experiments (Table S1†). The pH of the aerosol did not change appreciably following dark oxidation, with an average pH of the aged aerosol of 2.8 (standard deviation of 0.12) (Fig. 6). The largest change in estimated pH occurred in experiment 6 with a 0.3 unit increase between the fresh (pH of 2.5) and aged (pH of 2.8) aerosol.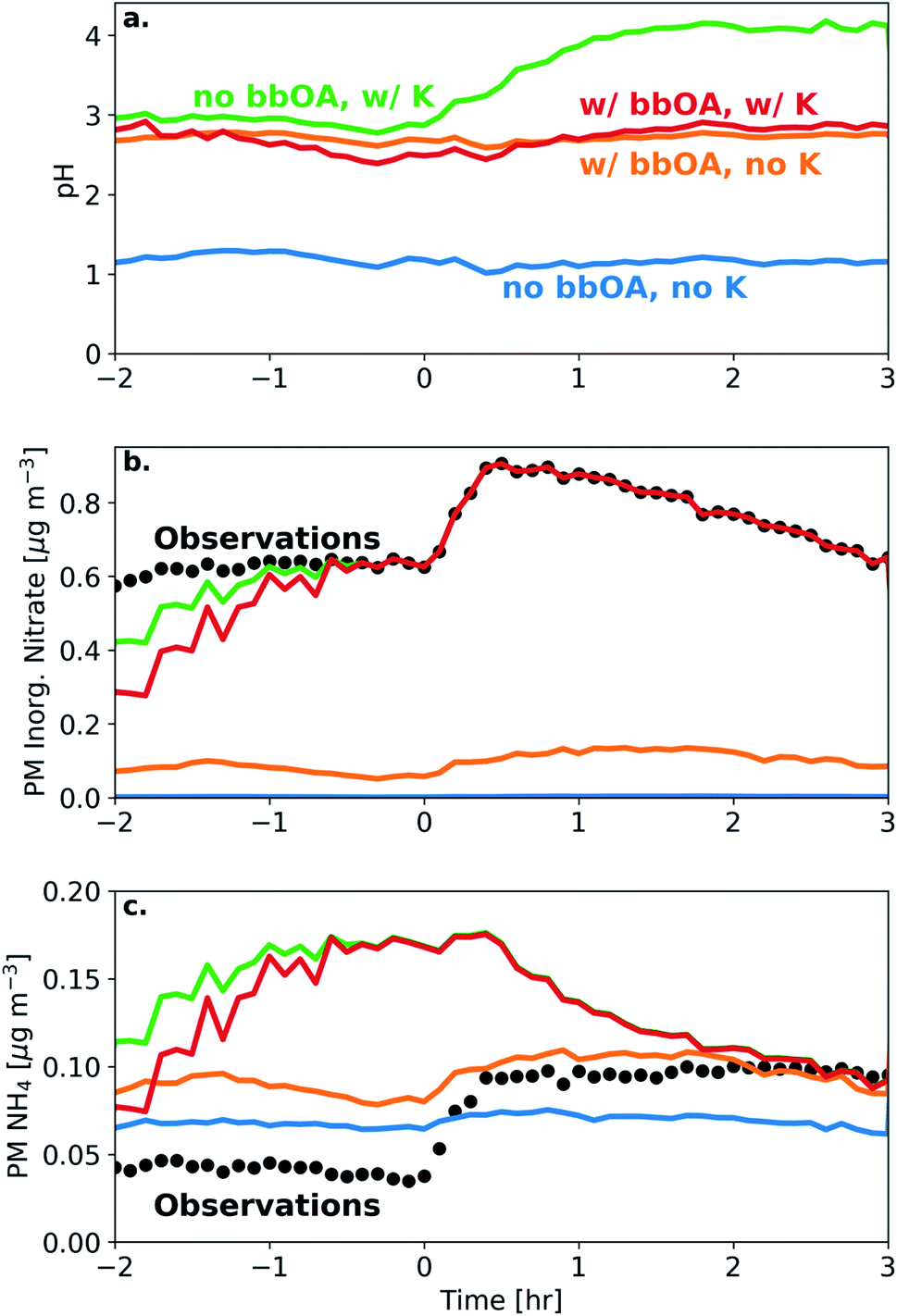 | ||
| Fig. 6 Model simulations and corresponding measurements for experiment 8 showing (a) aerosol pH, (b) PM inorganic nitrate, and (c) PM NH4. Included are sensitivity simulations with neither bbOA water uptake nor K included in the model (blue), only bbOA water uptake included (orange), only K included (green), and the base assumptions with bbOA water uptake and K included (red). Measurements from the HR-ToF-AMS are included as black dots. | ||
To test the sensitivity of including bbOA water uptake into the model and the assumed K+ levels, we perform additional simulations: (1) a simulation with no bbOA water uptake and K+ concentrations set to zero; (2) K+ concentrations equal to 15% of the PM nitrate (but without bbOA water uptake); (3) bbOA water uptake but with K+ concentrations set to zero; (4) and with both bbOA water uptake and potassium concentrations included in the model. When including neither bbOA water uptake nor K+ in the model, the pH of the BB aerosol stays roughly constant at a value of 1.2 throughout the experiment (Fig. 6). When bbOA water uptake alone is included, the pH of the bbOA increases to 2.7. Similarly, when an estimate of K+ emissions from BB is included (but without bbOA water uptake), the pH of the freshly emitted BB aerosol increases to 2.9. Interestingly, when K+ is included without bbOA water uptake, the pH of the BB aerosol increases from 2.9 to 4.1 following oxidation; however, when both bbOA water uptake and K+ are included the pH ranges from only 2.5–2.8 throughout the experiment.
These differences are due to the change in simulated PM inorganic nitrate and ammonium when bbOA water uptake and K+ are included in the model. When neither bbOA water uptake nor K+ are included in the simulation, nitrate is entirely in the gas phase (in the form of HNO3). Including bbOA water uptake leads to a slight increase in simulated particle-phase inorganic nitrate (0.1 μg m−3). However, when K+ is included, the simulated PM inorganic nitrate matches the observations after one hour. On the other hand, the inclusion of K+ leads to an over prediction of PM ammonium relative to the observations. This highlights the importance of both bbOA water uptake and K+ concentration in determining aerosol pH. This also means that while pH in biomass burning aerosol is buffered largely by the NH3–NH4+ partitioning,71–73 the amount of organic water uptake plays a pivotal role in the case of biomass burning to ensure that bbOA pH is virtually constant throughout its lifetime.
In additional sensitivity simulations, we tested a range of hygroscopicity parameters from 0.1 to 0.2, and found little change in estimated pH.
3.4 Equivalent atmospheric time scales
In a subset of experiments (experiments 1, 4, 5, 7, and 8), we measured the NO3 radical concentrations during the BB experiments. Prior to the initiation of oxidation, NO3 radical concentrations were below the detection limit (approximately 3 ppt for 10 s of acquisition time) of the instrument, suggesting that the NO2 and O3 emitted from this source were not sufficient to form substantial concentrations of NO3. In the 30 min following the injection of NO2 and O3, NO3 concentrations increased to 100–400 ppt for experiments 1, 7, and 8 (with relatively higher injections of NO2 and O3) and 0–50 ppt for experiments 4 and 5 (with relatively lower injections of NO2 and O3). In experiment 5, NO3 stayed near the detection limit of the instrument for the duration of the experiment. In the remaining four experiments (experiments 1, 4, 7 and 8) there were significant concentrations (80–400 ppt) of NO3 throughout the 3 h oxidation period, suggesting that our dark-aging experiments were likely limited by VOC levels as opposed to oxidants.To relate the experimental timescale and injected concentrations of oxidant precursor species (NO2 and O3), we assume a typical nighttime ambient urban NO3 radical concentration of 20 ppt, in line with previous measurements at urban sites.29,31,34,35 In the 30 min following the injection of NO2 and O3, the NO3 exposure across the experiments with measured NO3 radical concentrations above the limit of detection (experiments 1, 4, 7, and 8) ranges from 0.4–2.3 equivalent hours. After 3 h, this range increases to 11–43 h. Assuming 10 h of darkness in an ambient environment, the integrated NO3 radical exposure after 0.9–2.8 h is roughly equivalent to one night of NO3 exposure in a typical urban environment (Fig. 7). As a comparison, we estimate OH exposure in the UV-aging experiment (experiment 9) and calculate that 3 h of experimental time is roughly equivalent to approximately 7–10 photochemical equivalent hours (assuming [OH] = 1.5 × 106 molecule per cm3 throughout a 24 h period).
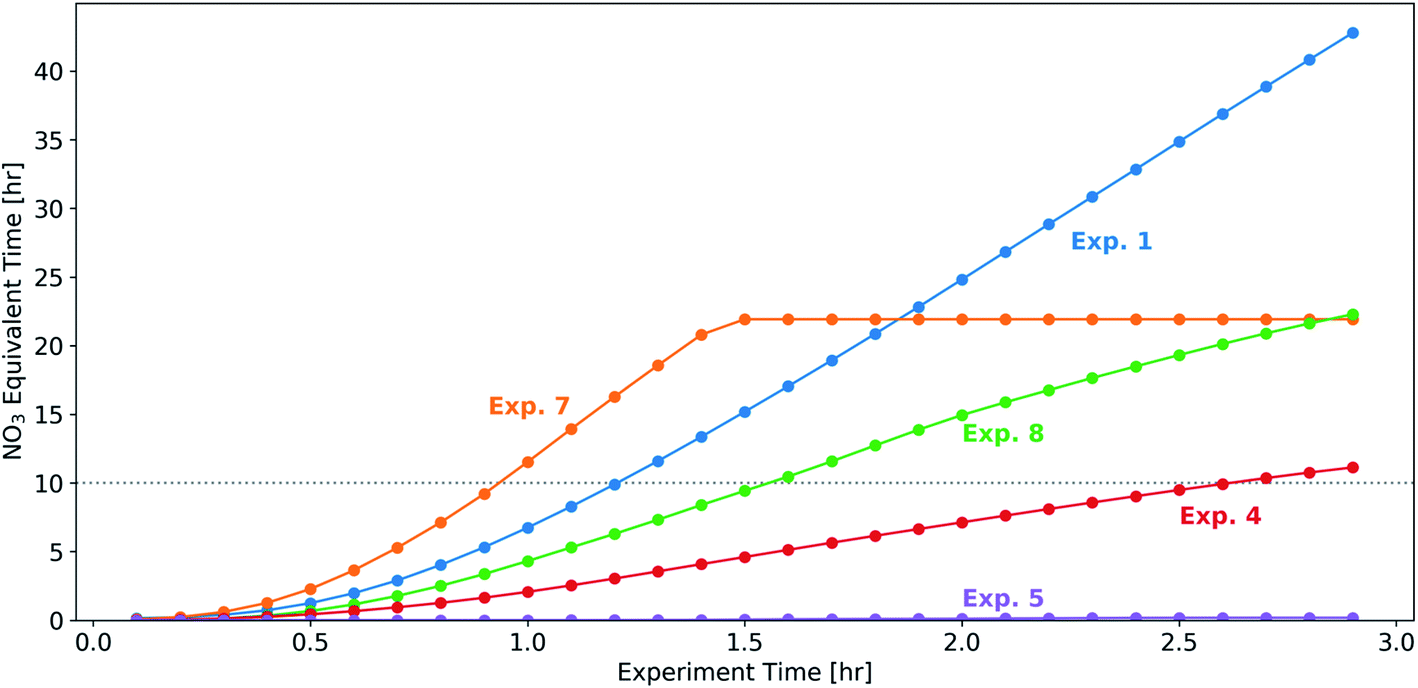 | ||
| Fig. 7 The equivalent number of hours in an ambient urban environment (assuming an NO3 radical concentration 20 ppt throughout the night) for the subset of experiments with NO3 radical measurements. | ||
One important limitation in this analysis is the possibility of O3 or OH contributing to the oxidation in the dark-aging experiments. Following the discussion in Section 3.1, our estimates of the average lifetime of phenol, isoprene, and α-pinene suggest an important role of NO3 in the fate of all three VOCs (with average lifetimes one hour or less), while OH likely does not contribute to the oxidation of these VOCs (with the shortest estimated lifetime of six hours, or twice the length of the oxidation portion of the experiment; Table S4†). As discussed previously, O3 may contribute to reactions with α-pinene or to reactions with the products of the reaction of phenol with NO3. In this case, the equivalent time scale in an ambient urban environment for our experiments would be longer than discussed here, as the O3 concentration in our experiments (44–228 ppb) is higher than is typical in nighttime ambient environments. Further, this analysis does not take into account heterogeneous reactions which may affect the O:C ratio.
4 Conclusions and implications
In this study, we performed a suite of smog chamber experiments to explore the sensitivity of the chemical processing and formation of secondary aerosol of biomass burning emissions to aging under dark (dominated by the NO3 radical) conditions. The biomass combustion was designed to be representative of residential heating in an urban environment. We find that biomass burning organic aerosol (bbOA) does age rapidly under dark conditions following exposure to NO2 and O3. The oxygen-to-carbon ratio increased by 7–34% in the first 30 min following the injection of NO2 and O3. The experiments with the lowest increase in the oxygen-to-carbon ratio were experiments with the lowest levels of NO2 and O3 (suggesting low NO3 radical concentrations). Production of secondary organic aerosol across these experiments ranged from 0.7–38 μg m−3 (corresponding to a 4–81% increase over the initial emitted organic aerosol concentration). In addition, the dark-aging experiments produced 0.1–3.8 μg m−3 of secondary nitrate aerosol, much of which (46–100%) is in the form of organic nitrate. The results presented here are in broad agreement with the results of Tiitta et al.46 and Hartikainen et al.47 which reported large enhancements of secondary organic aerosol and consumption of phenolic compounds after exposure of biomass burning emissions to the NO3 radical.In a subset of experiments, the NO3 radical concentration was measured using incoherent broad-band cavity-enhanced absorption spectroscopy (IBB-CEAS). In the 30 min following the injection of additional NO2 and O3, the NO3 radical concentration increased to as much as 400 ppt across these experiments. Assuming an average nighttime concentration of 20 ppt (typical of an urban environment), roughly 1–3 h of NO3 exposure at these experimental concentrations would correspond to one night of continued 20 ppt exposure (defined as 10 hours of darkness). While this comparison of NO3 radical exposure to ambient urban conditions is a rough estimate, it does suggest that freshly emitted bbOA can undergo substantial chemical processing, including the formation of secondary organic and inorganic aerosol, overnight.
We compare these dark-aging experiments to 2 reference experiments: one experiment with no external initiation of oxidation and one experiment with oxidation initiated with UV lights. While there were low levels of NO2 and O3 (7 and 23 ppb, respectively) emitted with the biomass burning OA and VOCs, the experiment with no external initiation of oxidation showed no increase in the oxygen-to-carbon ratio and only a slight increase in aerosol concentration (within the uncertainty range of the wall loss correction) after 3 hours of the experiment. During an experiment in which the BB emissions were aged under UV lights, the O:C increased more than in the dark-aging experiments (43% after 0.5 h and 81% after 3 h). Conversely the production of secondary organic aerosol was comparable to the dark-aging experiments; however, we note that we did not test the same range of initial conditions in the UV experiments as in the eight dark-aging experiments.
The enhancement in secondary inorganic and organic aerosol did not appear to substantially affect the calculated pH of the biomass burning aerosol if the water uptake from the organics is considered in the calculations. Using the ISORROPIA-Lite thermodynamical model, we estimate the pH of the BB aerosol is roughly constant at 2.8 before and after the dark oxidation. The modeled partitioning of ammonium is consistent with the observed values. This result does exhibit moderate sensitivity to the amount of potassium in the aerosol, which is largely mitigated from the presence of organic water. These levels of pH promote most of the total nitrate to be in the form of aerosol nitrate, which implies that its dry deposition rates in the ambient atmosphere would significantly decrease compared to if it were residing in the gas phase in the form of HNO3.55 This observation carries two significant implications: inorganic nitrate would tend to accumulate much more in the boundary layer before it deposits, increasing the PM levels considerably and its sensitivity to NH3 and/or HNO3,54,55 while the relatively slow deposition of nitrate means that the corresponding nitrogen can be transported over long distances before it is deposited.54 This in itself increases the range of influence of BB emissions, their impact on air quality and ecosystem productivity over large regions of the globe.53
There are a number of limitations regarding this study. First, we did not systematically test the sensitivity of the production of secondary aerosol under dark conditions to differences in combustion conditions. Different combustion conditions may alter the profile of gas- and particle-phase emission, and hence alter the degree to which the fresh emission may be aged by the NO3 radical. While our laboratory emissions are representative of residential heating, we note that open fires may behave differently. In addition, we note the possible loss of semi-volatile organic species during the transfer of the emission plume into the experimental chamber. Second, while the NO3 radical appears to dominate the oxidation process (the estimated average lifetime of phenol, isoprene, and α-pinene against reaction with NO3 are much less than the length of the oxidation portion of the experiment), it is possible that O3 may react with monoterpenes or with later-generation VOC products to enhance chemical aging. In this case, the equivalent time scales based on NO3 exposure would be an underestimate of nighttime oxidation in an ambient environment. Finally, when separating fresh from produced organic aerosol spectra, we assume only homogenous gas-phase oxidation takes place without heterogeneous reactions on the particle surface. Recent work by Yazdani et al. (in review) suggest that both aerosol- and gas-phase aging of primary BBOA may occur to a significant extent, perhaps more than AMS-based analysis alone may imply. Despite these limitations, our results suggest that biomass burning emissions are able to age substantially under dark conditions on a time scale of roughly one night under polluted urban conditions, and the inherent properties of the aerosol (acidity, hygroscopicity) favor the condensation of significant amounts of nitrate to the aerosol phase. This in turn reduces the deposition rate of nitrates, promotes the accumulation of nitrate aerosol in the boundary layer and its transport over long distances before deposition to the surface. Not including this dark aging mechanism and acidity effect in chemical-transport models underestimates the connection between biomass burning emissions and nocturnal air pollution.
Data availability
Experimental results are available through the EUROCHAMP Data Centre (https://data.eurochamp.org/). The EUROCHAMP Data Centre is maintained by the French national center for Atmospheric data and services AERIS.Author contributions
J. K. K., A. N., and S. N. P. contributed to conceptualization; J. K. K., C. K., M. P., K. F., A. N., and C. V. contributed to investigation; S. J., M. C., M. C., A. N. and B. P. V., contributed to novel methods and instrumentation; J. K. K., C. K., M. P., K. F., S. J., A. N. and C. S. contributed to formal analysis; J. K. K. wrote the original draft; all authors contributed to review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the European Research Council (ERC-2016-COG), Project Pyrogenic TRansformations Affecting Climate and Health (PyroTRACH – 726165), and the Horizon-2020 Project REMEDIA of the European Union under grant agreement no. 874753.References
- A. L. Westerling, H. G. Hidalgo, D. R. Cayan and T. W. Swetnam, Warming and Earlier Spring Increase Western U.S. Forest Wildfire Activity, Science, 2006, 313, 940–943 CrossRef CAS PubMed.
- T. C. Bond, S. J. Doherty, D. W. Fahey, P. M. Forster, T. Berntsen, B. J. DeAngelo, M. G. Flanner, S. Ghan, B. Kärcher, D. Koch, S. Kinne, Y. Kondo, P. K. Quinn, M. C. Sarofim, M. G. Schultz, M. Schulz, C. Venkataraman, H. Zhang, S. Zhang, N. Bellouin, S. K. Guttikunda, P. K. Hopke, M. Z. Jacobson, J. W. Kaiser, Z. Klimont, U. Lohmann, J. P. Schwarz, D. Shindell, T. Storelvmo, S. G. Warren and C. S. Zender, Bounding the Role of Black Carbon in the Climate System: A Scientific Assessment, J. Geophys. Res.: Atmos., 2013, 118, 5380–5552 CrossRef CAS.
- S. N. Pandis, K. Skyllakou, K. Florou, E. Kostenidou, C. Kaltsonoudis, E. Hasa and A. A. Presto, Urban Particulate Matter Pollution: A Tale of Five Cities, Faraday Discuss., 2016, 189, 277–290 RSC.
- B. Ford, M. Val Martin, S. E. Zelasky, E. V. Fischer, S. C. Anenberg, C. L. Heald and J. R. Pierce, Future Fire Impacts on Smoke Concentrations, Visibility, and Health in the Contiguous United States, GeoHealth, 2018, 2, 229–247 CrossRef CAS PubMed.
- I. Tsiodra, G. Grivas, K. Tavernaraki, A. Bougiatioti, M. Apostolaki, D. Paraskevopoulou, A. Gogou, C. Parinos, K. Oikonomou, M. Tsagkaraki, P. Zarmpas, A. Nenes and N. Mihalopoulos, Annual Exposure to Polycyclic Aromatic Hydrocarbons in Urban Environments Linked to Wintertime Wood-Burning Episodes, Atmos. Chem. Phys., 2021, 21, 17865–17883 CrossRef CAS.
- G. R. Van Der Werf, J. T. Randerson, L. Giglio, G. J. Collatz, M. Mu, P. S. Kasibhatla, D. C. Morton, R. S. Defries, Y. Jin and T. T. Van Leeuwen, Global Fire Emissions and the Contribution of Deforestation, Savanna, Forest, Agricultural, and Peat Fires (1997-2009), Atmos. Chem. Phys., 2010, 10, 11707–11735 CrossRef CAS.
- C. Wiedinmyer, S. K. Akagi, R. J. Yokelson, L. K. Emmons, J. A. Al-Saadi, J. J. Orlando and A. J. Soja, The Fire INventory from NCAR (FINN): A High Resolution Global Model to Estimate the Emissions from Open Burning, Geosci. Model Dev., 2011, 4, 625–641 CrossRef.
- H. Forrister, J. Liu, E. Scheuer, J. Dibb, L. Ziemba, K. L. Thornhill, B. Anderson, G. Diskin, A. E. Perring, J. P. Schwarz, P. Campuzano-Jost, D. A. Day, B. B. Palm, J. L. Jimenez, A. Nenes and R. J. Weber, Evolution of Brown Carbon in Wildfire Plumes, Geophys. Res. Lett., 2015, 42, 4623–4630 CrossRef CAS.
- Y. Zhang, H. Forrister, J. Liu, J. Dibb, B. Anderson, J. P. Schwarz, A. E. Perring, J. L. Jimenez, P. Campuzano-Jost, Y. Wang, A. Nenes and R. J. Weber, Top-Of-Atmosphere Radiative Forcing Affected by Brown Carbon in the Upper Troposphere, Nat. Geosci., 2017, 10, 486 CrossRef CAS.
- V. Verma, T. Fang, L. Xu, R. E. Peltier, A. G. Russell, N. L. Ng and R. J. Weber, Organic Aerosols Associated with the Generation of Reactive Oxygen Species (ROS) by Water-Soluble PM2.5, Environ. Sci. Technol., 2015, 49, 4646–4656 CrossRef CAS PubMed.
- D. Paraskevopoulou, A. Bougiatioti, I. Stavroulas, T. Fang, M. Lianou, E. Liakakou, E. Gerasopoulos, R. Weber, A. Nenes and N. Mihalopoulos, Yearlong Variability of Oxidative Potential of Particulate Matter in an Urban Mediterranean Environment, Atmos. Environ., 2019, 206, 183–196 CrossRef CAS.
- T. G. Karl, T. J. Christian, R. J. Yokelson, P. Artaxo, W. M. Hao and A. Guenther, The Tropical Forest and Fire Emissions Experiment: Method Evaluation of Volatile Organic Compound Emissions Measured by PTR-MS, FTIR, and GC from Tropical Biomass Burning, Atmos. Chem. Phys., 2007, 7, 5883–5897 CrossRef CAS.
- A. L. Robinson, N. M. Donahue, M. K. Shrivastava, E. A. Weitkamp, A. M. Sage, A. P. Grieshop, T. E. Lane, J. R. Pierce and S. N. Pandis, Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging, Science, 2007, 315, 1259–1262 CrossRef CAS.
- Q. Bian, S. H. Jathar, J. K. Kodros, K. C. Barsanti, L. E. Hatch, A. A. May, S. M. Kreidenweis and J. R. Pierce, Secondary Organic Aerosol Formation in Biomass-Burning Plumes: Theoretical Analysis of Lab Studies and Ambient Plumes, Atmos. Chem. Phys., 2017, 17, 5459–5475 CrossRef CAS.
- A. Akherati, Y. He, M. M. Coggon, A. R. Koss, A. L. Hodshire, K. Sekimoto, C. Warneke, J. de Gouw, L. Yee, J. H. Seinfeld, T. B. Onasch, S. C. Herndon, W. B. Knighton, C. D. Cappa, M. J. Kleeman, C. Y. Lim, J. H. Kroll, J. R. Pierce and S. H. Jathar, Oxygenated Aromatic Compounds are Important Precursors of Secondary Organic Aerosol in Biomass-Burning Emissions, Environ. Sci. Technol., 2020, 54, 8568–8579 CrossRef CAS PubMed.
- J. K. Kodros, D. K. Papanastasiou, M. Paglione, M. Masiol, S. Squizzato, K. Florou, K. Skyllakou, C. Kaltsonoudis, A. Nenes and S. N. Pandis, Rapid Dark Aging of Biomass Burning as an Overlooked Source of Oxidized Organic Aerosol, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(52), 33028–33033 CrossRef CAS.
- K. O'Dell, R. S. Hornbrook, W. Permar, E. J. T. Levin, L. A. Garofalo, E. C. Apel, N. J. Blake, A. Jarnot, M. A. Pothier, D. K. Farmer, L. Hu, T. Campos, B. Ford, J. R. Pierce and E. V. Fischer, Hazardous Air Pollutants in Fresh and Aged Western US Wildfire Smoke and Implications for Long-Term Exposure, Environ. Sci. Technol., 2020, 54, 11838–11847 CrossRef.
- A. M. Ortega, D. A. Day, M. J. Cubison, W. H. Brune, D. Bon, J. A. de Gouw and J. L. Jimenez, Secondary Organic Aerosol Formation and Primary Organic Aerosol Oxidation from Biomass-Burning Smoke in a Flow Reactor During FLAME-3, Atmos. Chem. Phys., 2013, 13, 11551–11571 CrossRef.
- M. Paglione, S. Gilardoni, M. Rinaldi, S. Decesari, N. Zanca, S. Sandrini, L. Giulianelli, D. Bacco, S. Ferrari, V. Poluzzi, F. Scotto, A. Trentini, L. Poulain, H. Herrmann, A. Wiedensohler, F. Canonaco, A. S. H. Prévôt, P. Massoli, C. Carbone, M. C. Facchini and S. Fuzzi, The Impact of Biomass Burning and Aqueous-phase Processing on Air Quality: A Multi-Year Source Apportionment Study in the Po Valley, Italy, Atmos. Chem. Phys., 2020, 20, 1233–1254 CrossRef CAS.
- A. P. Grieshop, N. M. Donahue and A. L. Robinson, Laboratory Investigation of Photochemical Oxidation of Organic Aerosol from Wood Fires 2: Analysis of Aerosol Mass Spectrometer Data, Atmos. Chem. Phys., 2009, 9, 2227–2240 CrossRef CAS.
- C. J. Hennigan, A. P. Sullivan, J. L. Collett Jr and A. L. Robinson, Levoglucosan Stability in Biomass Burning Particles Exposed to Hydroxyl Radicals, Geophys. Res. Lett., 2010, 37, L09806 CrossRef.
- A. L. Hodshire, A. Akherati, M. J. Alvarado, B. Brown-Steiner, S. H. Jathar, J. L. Jimenez, S. M. Kreidenweis, C. R. Lonsdale, T. B. Onasch, A. M. Ortega and J. R. Pierce, Aging Effects on Biomass Burning Aerosol Mass and Composition: A Critical Review of Field and Laboratory Studies, Environ. Sci. Technol., 2019, 53, 10007–10022 CrossRef CAS PubMed.
- C. J. Hennigan, M. A. Miracolo, G. J. Engelhart, A. A. May, A. A. Presto, T. Lee, A. P. Sullivan, G. R. McMeeking, H. Coe, C. E. Wold, W. M. Hao, J. B. Gilman, W. C. Kuster, J. De Gouw, B. A. Schichtel, J. L. Collett, S. M. Kreidenweis and A. L. Robinson, Chemical and Physical Transformations of Organic Aerosol from the Photo-Oxidation of Open Biomass Burning Emissions in an Environmental Chamber, Atmos. Chem. Phys., 2011, 11, 7669–7686 CrossRef CAS.
- G. N. Theodoritsi and S. N. Pandis, Simulation of the Chemical Evolution of Biomass Burning Organic Aerosol, Atmos. Chem. Phys., 2019, 19, 5403–5415 CrossRef CAS.
- A. Hodzic, P. Campuzano-Jost, H. Bian, M. Chin, P. R. Colarco, D. A. Day, K. D. Froyd, B. Heinold, D. S. Jo, J. M. Katich, J. K. Kodros, B. A. Nault, J. R. Pierce, E. Ray, J. Schacht, G. P. Schill, J. C. Schroder, J. P. Schwarz, D. T. Sueper, I. Tegen, S. Tilmes, K. Tsigaridis, P. Yu and J. L. Jimenez, Characterization of Organic Aerosol Across the Global Remote Troposphere: A Comparison of ATom Measurements and Global Chemistry Models, Atmos. Chem. Phys., 2020, 20, 4607–4635 CrossRef CAS.
- S. J. Pai, C. L. Heald, J. R. Pierce, S. C. Farina, E. A. Marais, J. L. Jimenez, P. Campuzano-Jost, B. A. Nault, A. M. Middlebrook, H. Coe, J. E. Shilling, R. Bahreini, J. H. Dingle and K. Vu, An Evaluation of Global Organic Aerosol Schemes Using Airborne Observations, Atmos. Chem. Phys., 2020, 20, 2637–2665 CrossRef CAS.
- C. Fountoukis, A. G. Megaritis, K. Skyllakou, P. E. Charalampidis, H. A. C. Denier van der Gon, M. Crippa, A. S. H. Prévôt, F. Fachinger, A. Wiedensohler, C. Pilinis and S. N. Pandis, Simulating the Formation of Carbonaceous Aerosol in a European Megacity (Paris) During the MEGAPOLI Summer and Winter Campaigns, Atmos. Chem. Phys., 2016, 16, 3727–3741 CrossRef CAS.
- Z. C. J. Decker, K. J. Zarzana, M. Coggon, K.-E. Min, I. Pollack, T. B. Ryerson, J. Peischl, P. Edwards, W. P. Dubé, M. Z. Markovic, J. M. Roberts, P. R. Veres, M. Graus, C. Warneke, J. de Gouw, L. E. Hatch, K. C. Barsanti and S. S. Brown, Nighttime Chemical Transformation in Biomass Burning Plumes: A Box Model Analysis Initialized with Aircraft Observations, Environ. Sci. Technol., 2019, 53, 2529–2538 CrossRef CAS PubMed.
- M. A. H. Khan, M. C. Cooke, S. R. Utembe, A. T. Archibald, R. G. Derwent, P. Xiao, C. J. Percival, M. E. Jenkin, W. C. Morris and D. E. Shallcross, Global Modeling of the Nitrate Radical (NO3) for Present and Pre-Industrial Scenarios, Atmos. Res., 2015, 164–165, 347–357 CrossRef CAS.
- N. L. Ng, S. S. Brown, A. T. Archibald, E. Atlas, R. C. Cohen, J. N. Crowley, D. A. Day, N. M. Donahue, J. L. Fry, H. Fuchs, R. J. Griffin, M. I. Guzman, H. Herrmann, A. Hodzic, Y. Iinuma, J. L. Jimenez, A. Kiendler-Scharr, B. H. Lee, D. J. Luecken, J. Mao, R. McLaren, A. Mutzel, H. D. Osthoff, B. Ouyang, B. Picquet-Varrault, U. Platt, H. O. T. Pye, Y. Rudich, R. H. Schwantes, M. Shiraiwa, J. Stutz, J. A. Thornton, A. Tilgner, B. J. Williams and R. A. Zaveri, Nitrate Radicals and Biogenic Volatile Organic Compounds: Oxidation, Mechanisms, and Organic Aerosol, Atmos. Chem. Phys., 2017, 17, 2103–2162 CrossRef CAS.
- D. Asaf, D. Pedersen, V. Matveev, M. Peleg, C. Kern, J. Zingler, U. Platt and M. Luria, Long-Term Measurements of NO3 Radical at a Semiarid Urban Site: 1. Extreme Concentration Events and Their Oxidation Capacity, Environ. Sci. Technol., 2009, 43, 9117–9123 CrossRef CAS PubMed.
- D. Asaf, E. Tas, D. Pedersen, M. Peleg and M. Luria, Long-Term Measurements of NO3 Radical at a Semiarid Urban Site: 2. Seasonal Trends and Loss Mechanisms, Environ. Sci. Technol., 2010, 44, 5901–5907 CrossRef CAS PubMed.
- J. Stutz, K. W. Wong, L. Lawrence, L. Ziemba, J. H. Flynn, B. Rappenglück and B. Lefer, Nocturnal NO3 Radical Chemistry in Houston, TX, Atmos. Environ., 2010, 44, 4099–4106 CrossRef CAS.
- S. S. Brown, W. P. Dubé, H. D. Osthoff, J. Stutz, T. B. Ryerson, A. G. Wollny, C. A. Brock, C. Warneke, J. A. de Gouw, E. Atlas, J. A. Neuman, J. S. Holloway, B. M. Lerner, E. J. Williams, W. C. Kuster, P. D. Goldan, W. M. Angevine, M. Trainer, F. C. Fehsenfeld and A. R. Ravishankara, Vertical Profiles in NO3 and N2O5 Measured from an Aircraft: Results from the NOAA P-3 and Surface Platforms During the New England Air Quality Study, J. Geophys. Res.: Atmos., 2007, 112, D22304 CrossRef.
- S. Wang, C. Shi, B. Zhou, H. Zhao, Z. Wang, S. Yang and L. Chen, Observation of NO3 radicals over Shanghai, China, Atmos. Environ., 2013, 70, 401–409 CrossRef CAS.
- C. Li, Q. He, A. P. S. Hettiyadura, U. Käfer, G. Shmul, D. Meidan, R. Zimmermann, S. S. Brown, C. George, A. Laskin and Y. Rudich, Formation of Secondary Brown Carbon in Biomass Burning Aerosol Proxies through NO3 Radical Reactions, Environ. Sci. Technol., 2020, 54, 1395–1405 CrossRef CAS PubMed.
- R. McLaren, P. Wojtal, D. Majonis, J. McCourt, J. D. Halla and J. Brook, NO3 Radical Measurements in a Polluted Marine Environment: Links to Ozone Formation, Atmos. Chem. Phys., 2010, 10, 4187–4206 CrossRef CAS.
- B. J. Allan, G. McFiggans, J. M. C. Plane, H. Coe and G. G. McFadyen, The Nitrate Radical in the Remote Marine Boundary Layer, J. Geophys. Res.: Atmos., 2000, 105, 24191–24204 CrossRef.
- J. Matsumoto, K. Imagawa, H. Imai, N. Kosugi, M. Ideguchi, S. Kato and Y. Kajii, Nocturnal Sink of NOx via NO3 and N2O5 in the Outflow from a Source Area in Japan, Atmos. Environ., 2006, 40, 6294–6302 CrossRef CAS.
- M. Vrekoussis, N. Mihalopoulos, E. Gerasopoulos, M. Kanakidou, P. J. Crutzen and J. Lelieveld, Two-Years of NO3 Radical Observations in the Boundary Layer Over the Eastern Mediterranean, Atmos. Chem. Phys., 2007, 7, 315–327 CrossRef CAS.
- C. Warneke, J. A. de Gouw, P. D. Goldan, W. C. Kuster, E. J. Williams, B. M. Lerner, R. Jakoubek, S. S. Brown, H. Stark, M. Aldener, A. R. Ravishankara, J. M. Roberts, M. Marchewka, S. Bertman, D. T. Sueper, S. A. McKeen, J. F. Meagher and F. C. Fehsenfeld, Comparison of Daytime and Nighttime Oxidation of Biogenic and Anthropogenic VOCs Along the New England Coast in Summer During New England Air Quality Study 2002, J. Geophys. Res.: Atmos., 2004, 109, D10309 CrossRef.
- N. L. Ng, A. J. Kwan, J. D. Surratt, A. W. H. Chan, P. S. Chhabra, A. Sorooshian, H. O. T. Pye, J. D. Crounse, P. O. Wennberg, R. C. Flagan and J. H. Seinfeld, Secondary Organic Aerosol (SOA) Formation from Reaction of Isoprene with Nitrate Radicals (NO3), Atmos. Chem. Phys., 2008, 8, 4117–4140 CrossRef CAS.
- S. S. Brown, J. A. deGouw, C. Warneke, T. B. Ryerson, W. P. Dubé, E. Atlas, R. J. Weber, R. E. Peltier, J. A. Neuman, J. M. Roberts, A. Swanson, F. Flocke, S. A. McKeen, J. Brioude, R. Sommariva, M. Trainer, F. C. Fehsenfeld and A. R. Ravishankara, Nocturnal Isoprene Oxidation Over the Northeast United States in Summer and Its Impact on Reactive Nitrogen Partitioning and Secondary Organic Aerosol, Atmos. Chem. Phys., 2009, 9, 3027–3042 CrossRef CAS.
- S. S. Brown, W. P. Dubé, R. Bahreini, A. M. Middlebrook, C. A. Brock, C. Warneke, J. A. de Gouw, R. A. Washenfelder, E. Atlas, J. Peischl, T. B. Ryerson, J. S. Holloway, J. P. Schwarz, R. Spackman, M. Trainer, D. D. Parrish, F. C. Fehshenfeld and A. R. Ravishankara, Biogenic VOC Oxidation and Organic Aerosol Formation in an Urban Nocturnal Boundary Layer: Aircraft Vertical Profiles in Houston, TX, Atmos. Chem. Phys., 2013, 13, 11317–11337 CrossRef CAS.
- J. L. Fry, D. C. Draper, K. C. Barsanti, J. N. Smith, J. Ortega, P. M. Winkler, M. J. Lawler, S. S. Brown, P. M. Edwards, R. C. Cohen and L. Lee, Secondary Organic Aerosol Formation and Organic Nitrate Yield from NO3 Oxidation of Biogenic Hydrocarbons, Environ. Sci. Technol., 2014, 48, 11944–11953 CrossRef CAS PubMed.
- P. Tiitta, A. Leskinen, L. Hao, P. Yli-Pirilä, M. Kortelainen, J. Grigonyte, J. Tissari, H. Lamberg, A. Hartikainen, K. Kuuspalo, A.-M. Kortelainen, A. Virtanen, K. E. J. Lehtinen, M. Komppula, S. Pieber, A. S. H. Prévôt, T. B. Onasch, D. R. Worsnop, H. Czech, R. Zimmermann, J. Jokiniemi and O. Sippula, Transformation of Logwood Combustion Emissions in a Smog Chamber: Formation of Secondary Organic Aerosol and Changes in the Primary Organic Aerosol Upon Daytime and Nighttime Aging, Atmos. Chem. Phys., 2016, 16, 13251–13269 CrossRef CAS.
- A. Hartikainen, P. Yli-Pirilä, P. Tiitta, A. Leskinen, M. Kortelainen, J. Orasche, J. Schnelle-Kreis, K. E. J. Lehtinen, R. Zimmermann, J. Jokiniemi and O. Sippula, Volatile Organic Compounds from Logwood Combustion: Emissions and Transformation under Dark and Photochemical Aging Conditions in a Smog Chamber, Environ. Sci. Technol., 2018, 52, 4979–4988 CrossRef CAS PubMed.
- A. Kiendler-Scharr, A. A. Mensah, E. Friese, D. Topping, E. Nemitz, A. S. H. Prevot, M. Äijälä, J. Allan, F. Canonaco, M. Canagaratna, S. Carbone, M. Crippa, M. Dall Osto, D. A. Day, P. De Carlo, C. F. Di Marco, H. Elbern, A. Eriksson, E. Freney, L. Hao, H. Herrmann, L. Hildebrandt, R. Hillamo, J. L. Jimenez, A. Laaksonen, G. McFiggans, C. Mohr, C. O'Dowd, R. Otjes, J. Ovadnevaite, S. N. Pandis, L. Poulain, P. Schlag, K. Sellegri, E. Swietlicki, P. Tiitta, A. Vermeulen, A. Wahner, D. Worsnop and H.-C. Wu, Ubiquity of Organic Nitrates from Nighttime Chemistry in the European Submicron Aerosol, Geophys. Res. Lett., 2016, 43, 7735–7744 CrossRef CAS.
- A. W. Rollins, E. C. Browne, K.-E. Min, S. E. Pusede, P. J. Wooldridge, D. R. Gentner, A. H. Goldstein, S. Liu, D. A. Day, L. M. Russell and R. C. Cohen, Evidence for NOx Control over Nighttime SOA Formation, Science, 2012, 337, 1210–1212 CrossRef CAS PubMed.
- A. Bougiatioti, S. Bezantakos, I. Stavroulas, N. Kalivitis, P. Kokkalis, G. Biskos, N. Mihalopoulos, A. Papayannis and A. Nenes, Biomass-Burning Impact on CCN Number, Hygroscopicity and Cloud Formation During Summertime in the Eastern Mediterranean, Atmos. Chem. Phys., 2016, 16, 7389–7409 CrossRef CAS.
- A. Bougiatioti, P. Nikolaou, I. Stavroulas, G. Kouvarakis, R. Weber, A. Nenes, M. Kanakidou and N. Mihalopoulos, Particle Water and pH in the Eastern Mediterranean: Source Variability and Implications for Nutrient Availability, Atmos. Chem. Phys., 2016, 16, 4579–4591 CrossRef CAS.
- H. O. T. Pye, A. Nenes, B. Alexander, A. P. Ault, M. C. Barth, S. L. Clegg, J. L. Collett Jr, K. M. Fahey, C. J. Hennigan, H. Herrmann, M. Kanakidou, J. T. Kelly, I.-T. Ku, V. F. McNeill, N. Riemer, T. Schaefer, G. Shi, A. Tilgner, J. T. Walker, T. Wang, R. Weber, J. Xing, R. A. Zaveri and A. Zuend, The Acidity of Atmospheric Particles and Clouds, Atmos. Chem. Phys., 2020, 20, 4809–4888 CrossRef CAS PubMed.
- A. R. Baker, M. Kanakidou, A. Nenes, S. Myriokefalitakis, P. L. Croot, R. A. Duce, Y. Gao, C. Guieu, A. Ito, T. D. Jickells, N. M. Mahowald, R. Middag, M. M. G. Perron, M. M. Sarin, R. Shelley and D. R. Turner, Changing Atmospheric Acidity as a Modulator of Nutrient Deposition and Ocean Biogeochemistry, Sci. Adv., 2021, 21, 13067–13076 CAS.
- A. Nenes, S. N. Pandis, R. J. Weber and A. Russell, Aerosol pH and Liquid Water Content Determine When Particulate Matter is Sensitive to Ammonia and Nitrate aAvailability, Atmos. Chem. Phys., 2020, 20, 3249–3258 CrossRef CAS.
- A. Nenes, S. N. Pandis, M. Kanakidou, A. G. Russell, S. Song, P. Vasilakos and R. J. Weber, Aerosol Acidity and Liquid Water Content Regulate the Dry Deposition of Inorganic Reactive Nitrogen, Atmos. Chem. Phys., 2021, 21, 6023–6033 CrossRef CAS.
- A. Fouqueau, M. Cirtog, M. Cazaunau, E. Pangui, P. Zapf, G. Siour, X. Landsheere, G. Méjean, D. Romanini and B. Picquet-Varrault, Implementation of an Incoherent Broadband Cavity-Enhanced Absorption Spectroscopy Technique in an Atmospheric Simulation Chamber for In Situ NO3 Monitoring: Characterization and Validation for Kinetic Studies, Atmos. Meas. Tech., 2020, 13, 6311–6323 CrossRef CAS.
- M. R. Canagaratna, J. L. Jimenez, J. H. Kroll, Q. Chen, S. H. Kessler, P. Massoli, L. Hildebrandt Ruiz, E. Fortner, L. R. Williams, K. R. Wilson, J. D. Surratt, N. M. Donahue, J. T. Jayne and D. R. Worsnop, Elemental Ratio Measurements of Organic Compounds Using Aerosol Mass Spectrometry: Characterization, Improved Calibration, and Implications, Atmos. Chem. Phys., 2015, 15, 253–272 CrossRef.
- E. Kostenidou, R. K. Pathak and S. N. Pandis, An Algorithm for the Calculation of Secondary Organic Aerosol Density Combining AMS and SMPS Data, Aerosol Sci. Technol., 2007, 41, 1002–1010 CrossRef CAS.
- D. K. Farmer, A. Matsunaga, K. S. Docherty, J. D. Surratt, J. H. Seinfeld, P. J. Ziemann and J. L. Jimenez, Response of an Aerosol Mass Spectrometer to Organonitrates and Organosulfates and Implications for Atmospheric Chemistry, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6670–6675 CrossRef CAS PubMed.
- N. Wang, S. D. Jorga, J. R. Pierce, N. M. Donahue and S. N. Pandis, Particle Wall-Loss Correction Methods in Smog Chamber Experiments, Atmos. Meas. Tech., 2018, 11, 6577–6588 CrossRef CAS.
- S. D. Jorga, C. Kaltsonoudis, A. Liangou and S. N. Pandis, Measurement of Formation Rates of Secondary Aerosol in the Ambient Urban Atmosphere Using a Dual Smog Chamber System, Environ. Sci. Technol., 2020, 54, 1336–1343 CrossRef CAS PubMed.
- E. Kostenidou, B.-H. Lee, G. J. Engelhart, J. R. Pierce and S. N. Pandis, Mass Spectra Deconvolution of Low, Medium, and High Volatility Biogenic Secondary Organic Aerosol, Environ. Sci. Technol., 2009, 43, 4884–4889 CrossRef CAS PubMed.
- C. Kaltsonoudis, E. Kostenidou, E. Louvaris, M. Psichoudaki, E. Tsiligiannis, K. Florou, A. Liangou and S. N. Pandis, Characterization of Fresh and Aged Organic Aerosol Emissions from Meat Charbroiling, Atmos. Chem. Phys., 2017, 17, 7143–7155 CrossRef CAS.
- P. Barmet, J. Dommen, P. F. DeCarlo, T. Tritscher, A. P. Praplan, S. M. Platt, A. S. H. Prévôt, N. M. Donahue and U. Baltensperger, OH Clock Determination by Proton Transfer Reaction Mass Spectrometry at an Environmental Chamber, Atmos. Meas. Tech., 2012, 5, 647–656 CrossRef CAS.
- P. F. DeCarlo, I. M. Ulbrich, J. Crounse, B. de Foy, E. J. Dunlea, A. C. Aiken, D. Knapp, A. J. Weinheimer, T. Campos, P. O. Wennberg and J. L. Jimenez, Investigation of the Sources and Processing of Organic Aerosol Over the Central Mexican Plateau from Aircraft Measurements During MILAGRO, Atmos. Chem. Phys., 2010, 10, 5257–5280 CrossRef CAS.
- B. A. Nault, P. Campuzano-Jost, D. A. Day, J. C. Schroder, B. Anderson, A. J. Beyersdorf, D. R. Blake, W. H. Brune, Y. Choi, C. A. Corr, J. A. de Gouw, J. Dibb, J. P. DiGangi, G. S. Diskin, A. Fried, L. G. Huey, M. J. Kim, C. J. Knote, K. D. Lamb, T. Lee, T. Park, S. E. Pusede, E. Scheuer, K. L. Thornhill, J.-H. Woo and J. L. Jimenez, Secondary Organic Aerosol Production from Local Emissions Dominates the Organic Aerosol Budget Over Seoul, South Korea, During KORUS-AQ, Atmos. Chem. Phys., 2018, 18, 17769–17800 CrossRef CAS.
- S. Kakavas, S. N. Pandis and A. Nenes, ISORROPIA-lite: A Comprehensive Atmospheric Aerosol Thermodynamics Module for Earth System Models, Tellus B, 2022, 74(1), 1–23 CrossRef.
- C. Fountoukis and A. Nenes, ISORROPIA II: A Computationally Efficient Thermodynamic Equilibrium Model for K+, Ca2+, Mg2+, NH4+, Na+, SO42−, NO3, Atmos. Chem. Phys., 2007, 7, 4639–4659 CrossRef CAS.
- M. D. Petters and S. M. Kreidenweis, A Single Parameter Representation of Hygroscopic Growth and Cloud Condensation Nucleus Activity, Atmos. Chem. Phys., 2007, 7, 1961–1971 CrossRef CAS.
- S. Y. Ryu, J. E. Kim, H. Zhuanshi, Y. J. Kim and G. U. Kang, Chemical Composition of Post-Harvest Biomass Burning Aerosols in Gwangju, Korea, J. Air Waste Manage. Assoc., 2004, 54, 1124–1137 CrossRef CAS PubMed.
- R. J. Weber, H. Guo, A. G. Russell and A. Nenes, High Aerosol Acidity Despite Declining Atmospheric Sulfate Concentrations Over the Past 15 years, Nat. Geosci., 2016, 9, 282–285 CrossRef CAS.
- H. Guo, R. J. Weber and A. Nenes, High Levels of Ammonia Do Not Raise Fine Particle pH Sufficiently to Yield Nitrogen Oxide-Dominated Sulfate Production, Sci. Rep., 2017, 7, 12109 CrossRef PubMed.
- G. Zheng, H. Su, S. Wang, M. O. Andreae, U. Pöschl and Y. Cheng, Multiphase Buffer Theory Explains Contrasts in Atmospheric Aerosol Acidity, Science, 2020, 369, 1374–1377 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ea00031h |
| This journal is © The Royal Society of Chemistry 2022 |