
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDilution and photooxidation driven processes explain the evolution of organic aerosol in wildfire plumes†
Ali
Akherati‡
 a,
Yicong
He
a,
Lauren A.
Garofalo
b,
Anna L.
Hodshire
b,
Delphine K.
Farmer
b,
Sonia M.
Kreidenweis
c,
Wade
Permar
d,
Lu
Hu
d,
Emily V.
Fischer
c,
Coty N.
Jen
e,
Allen H.
Goldstein
f,
Ezra J. T.
Levin
c,
Paul J.
DeMott
c,
Teresa L.
Campos
g,
Frank
Flocke
g,
John M.
Reeves
g,
Darin W.
Toohey
h,
Jeffrey R.
Pierce
*c and
Shantanu H.
Jathar
*a
a,
Yicong
He
a,
Lauren A.
Garofalo
b,
Anna L.
Hodshire
b,
Delphine K.
Farmer
b,
Sonia M.
Kreidenweis
c,
Wade
Permar
d,
Lu
Hu
d,
Emily V.
Fischer
c,
Coty N.
Jen
e,
Allen H.
Goldstein
f,
Ezra J. T.
Levin
c,
Paul J.
DeMott
c,
Teresa L.
Campos
g,
Frank
Flocke
g,
John M.
Reeves
g,
Darin W.
Toohey
h,
Jeffrey R.
Pierce
*c and
Shantanu H.
Jathar
*a
aDepartment of Mechanical Engineering, Colorado State University, Fort Collins, CO, USA. E-mail: shantanu.jathar@colostate.edu
bDepartment of Chemistry, Colorado State University, Fort Collins, CO, USA
cDepartment of Atmospheric Science, Colorado State University, Fort Collins, CO, USA. E-mail: jeffrey.pierce@colostate.edu
dDepartment of Chemistry and Biochemistry, University of Montana, Missoula, MT, USA
eDepartment of Chemical Engineering, Carnegie Mellon University, Pittsburgh, PA, USA
fDepartment of of Environmental Science, Policy & Management, University of California Berkeley, Berkeley, CA, USA
gEarth Observing Laboratory, National Center for Atmospheric Research, Boulder, CO, USA
hDepartment of Atmospheric and Oceanic Science, University of Colorado Boulder, Boulder, CO, USA
First published on 15th June 2022
Abstract
Wildfires are an important atmospheric source of primary organic aerosol (POA) and precursors for secondary organic aerosol (SOA) at regional and global scales. However, there are large uncertainties surrounding the emissions and physicochemical processes that control the transformation, evolution, and properties of POA and SOA in large wildfire plumes. We develop a plume version of a kinetic model to simulate the dilution, oxidation chemistry, thermodynamic properties, and microphysics of organic aerosol (OA) in wildfire smoke. The model is applied to study the in-plume OA in four large wildfire smoke plumes intercepted during an aircraft-based field campaign in summer 2018 in the western United States. Based on estimates of dilution and oxidant concentrations before the aircraft first intercepted the plumes, we simulate the OA evolution from very close to the fire to several hours downwind. Our model results and sensitivity simulations suggest that dilution-driven evaporation of POA and simultaneous photochemical production of SOA are likely to explain the observed evolution in OA mass with physical age. The model, however, substantially underestimates the change in the oxygen-to-carbon ratio of the OA compared to measurements. In addition, we show that the rapid chemical transformation within the first hour after emission is driven by higher-than-ambient OH concentrations (3 × 106–107 molecules per cm3) and the slower evolution over the next several hours is a result of lower-than-ambient OH concentrations (<106 molecules per cm3) and depleted SOA precursors. Model predictions indicate that the OA measured several hours downwind of the fire is still dominated by POA but with an SOA fraction that varies between 30% and 56% of the total OA. Semivolatile, heterocyclic, and oxygenated aromatic compounds, in that order, were found to contribute substantially (>90%) to SOA formation. Future work needs to focus on better understanding the dynamic evolution closer to the fire and resolving the rapid change in the oxidation state of OA with physical age.
Environmental significanceWildfires are an important atmospheric source of primary organic aerosol (POA) and precursors for secondary organic aerosol (SOA) at regional and global scales. However, there are large uncertainties surrounding the emissions and physicochemical processes that control the transformation, evolution, and properties of POA and SOA in large wildfire plumes. In this work, we develop a plume version of a kinetic model to simulate the dilution, oxidation chemistry, thermodynamic properties, and microphysics of organic aerosol (OA) in wildfire smoke. We find that dilution-driven evaporation of POA and simultaneous photochemical production of SOA are likely to explain the observed evolution of OA in wildfire plumes. Further, we show that there is rapid chemical transformation of wildfire smoke aerosol within the first hour after emission, driven by high concentrations of the hydroxyl radical. |
1. Introduction
Wildfires are an important source of organic aerosol (OA) and OA precursors to the atmosphere.1–6 Wildfire OA, as a major component of the submicron atmospheric aerosol mass,7,8 has been estimated to exert a strong influence on the Earth's radiative budget,9,10 and adversely affect regional and global air quality,8,11–13 human health,14,15 and visibility.14,16 Yet, there are large uncertainties surrounding the emissions and processes that control the abundance, distribution, and properties of wildfire OA in the atmosphere. For example, primary emissions of biomass burning OA, which includes OA from wildfires, agricultural fires, and biofuel combustion, vary by a factor of 2 in published inventories17 while atmospheric production rates of biomass burning OA, have been shown to vary over two orders of magnitude in several recent global modeling studies (1–100 Tg per year).18 These uncertainties have made it extremely challenging to represent wildfire OA in large-scale atmospheric models, which, in part, have limited the ability of these models to predict the atmospheric and environmental impacts of wildfire emissions.Wildfires directly emit particles that are dominated by primary organic aerosol (POA) with smaller amounts of black carbon (BC) and inorganic species (e.g., sulfate, nitrate, potassium).4,19,20 Wildfire POA has been shown to be semivolatile and evaporates with dilution.21–23 Furthermore, wildfire POA and the vapors in equilibrium with the POA (or semivolatile organic compounds, SVOC) are also known to be reactive.23–25 Wildfires emit OA precursors that include intermediate-volatility and volatile organic compounds (IVOC and VOC)26–28 and these oxidize in the atmosphere to form secondary organic aerosol (SOA).29 In addition to emissions of reduced hydrocarbons such as alkanes, aromatic, and biogenic VOCs,4 wildfires also emit oxygenated IVOCs and VOCs.23,27,28,30–34 These include oxygenated aromatic and heterocyclic organic compounds that have recently been shown to be important precursors for SOA formation.35–39 Although a lot has been understood about the composition, oxidation chemistry, and properties of OA and OA precursors from biomass burning emissions, especially in laboratory experiments, a detailed understanding of the physicochemical evolution of OA and OA precursors in real wildfire plumes is less well understood.
Recently, Hodshire et al.40 undertook a comprehensive review of wildfire OA data gathered in four laboratory campaigns and thirteen field campaigns performed over the past two decades. An analysis of these data suggested that while photooxidation of biomass burning emissions resulted in an enhancement in OA mass in laboratory experiments (mean = 1.25, median = 1.44, and interquartile range or IQR = 1.1–1.54),37–39,41–43 a similar enhancement was missing for the OA tracked in real wildfire plumes (mean = 1.1, median = 1.0, and IQR = 0.77–1.0).20,44–60 In addition, the OA measured within wildfire plumes closest to the fire (<1 h) was more oxidized (i.e., a higher oxygen-to-carbon (O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C) ratio for the OA) (mean = 0.36, median = 0.38, and IQR = 0.18–0.54) than the initial OA measured in laboratory experiments (mean = 0.23, median = 0.23, and IQR = 0.17–0.29), and the OA from wildfires exhibited a stronger O:C enhancement with photochemical age (mean = 1.82, median = 1.85, and IQR = 1.5–2.0) than the OA from laboratory fires (mean = 1.6, median = 1.5, and IQR = 1.3–1.8). Similar to the trends in OA O:C, a different enhancement was also observed for the ratio of two mass fragments (m44/m60) measured by the aerosol mass spectrometer between the field (mean = 4.5, median = 3.8, and IQR = 2.3–6.7) and the laboratory (mean = 3.5, median = 3.0 and IQR = 1.8–3.6). m44 refers to the mass fragment that is associated with the low-volatility organic compounds found in SOA61 while m60 refers to the mass fragment arising from fragmentation of primary emissions of anhydrous sugars such as levoglucosan.54
:C) ratio for the OA) (mean = 0.36, median = 0.38, and IQR = 0.18–0.54) than the initial OA measured in laboratory experiments (mean = 0.23, median = 0.23, and IQR = 0.17–0.29), and the OA from wildfires exhibited a stronger O:C enhancement with photochemical age (mean = 1.82, median = 1.85, and IQR = 1.5–2.0) than the OA from laboratory fires (mean = 1.6, median = 1.5, and IQR = 1.3–1.8). Similar to the trends in OA O:C, a different enhancement was also observed for the ratio of two mass fragments (m44/m60) measured by the aerosol mass spectrometer between the field (mean = 4.5, median = 3.8, and IQR = 2.3–6.7) and the laboratory (mean = 3.5, median = 3.0 and IQR = 1.8–3.6). m44 refers to the mass fragment that is associated with the low-volatility organic compounds found in SOA61 while m60 refers to the mass fragment arising from fragmentation of primary emissions of anhydrous sugars such as levoglucosan.54
Based on this analysis, Hodshire et al.40 put forth four hypotheses to explain this field versus laboratory difference. First, they argued that the dilution of the wildfire plume could result in evaporation of POA and this evaporation could be approximately balanced by SOA production to result in a small change in the OA mass with photochemical aging. Since the condensing SOA is likely to have a higher O:C ratio than the evaporating POA, this POA-SOA swap would not affect the OA mass but instead produce an increase in the O:C ratio with aging. This hypothesis has been supported by theoretical calculations.62,63 Second, the authors hypothesized that because wildfire plumes are typically only sampled at least 15 minutes after emission (often longer), the plume measurements could have missed the OA evolution that happened prior to the first measurement. Early sampling poses safety issues as well as the prospect of measuring poorly mixed plumes. If rapid chemistry does indeed occur early on, this may explain both the higher O:C ratio measured in the field and the reduced propensity to form SOA after the aircraft first intercepts a plume. Third, they postulated that the field and laboratory differences could arise from differences in the emissions and chemistry of OA and OA precursors, attributed to differences in the fuel mixtures, burn conditions, combustion efficiency, and environmental conditions (e.g., temperature, relative humidity) and regimes (e.g., photolytic rates, NOX). Finally, they noted several experimental artifacts linked to laboratory experiments, including losses of OA and OA precursors to transfer ducts38 and walls of the environmental chamber,64–66 that could drive differences between laboratory and field measurements. For the most part, these hypotheses remain untested and will serve as the basis for this work.
Several studies have tested the first hypothesis above, i.e. that dilution-driven evaporation of POA is roughly balanced by SOA production in wildfire plumes.33,67 In one of these studies based on wildfire plumes sampled in the western United States (US), Palm et al.33 quantified the evolution of OA mass as a function of dilution and photochemical age. They found that plumes that had diluted but not yet undergone photooxidation showed evidence for POA evaporation, while plumes that had both diluted and undergone photooxidation showed evidence for replacement of the lost POA mass with SOA. However, Palm et al.33 did not account for the photochemical evolution prior to the first sample (i.e. <30 minutes since emission), nor did they account for the oxidation of all potential precursors (i.e., SVOCs) leading to SOA formation. Based on closure calculations, they argued that nearly 90% of the SOA came from oxidation of evaporated POA vapors with marginal contributions from other VOCs. To the best of our knowledge, no bottom-up, detailed models have been used to simulate and evaluate the OA mass and composition evolution in wildfire plumes.
In this study, we simulated the physicochemical evolution of OA in a subset of wildfire plumes sampled during a recent airborne field campaign based in the western US. The OA evolution was simulated using a kinetic model that accounts for the dilution, oxidation chemistry, thermodynamic properties, and microphysics of OA. A novel contribution of this work is that we incorporate estimates of dilution and photochemical age before the first airborne plume transect, to simulate the OA evolution from very close to the fire to several hours downwind. Model results of mass and composition were compared to plume measurements to study the contribution of precursors and processes to OA evolution.
2. Materials and methods
2.1 Aircraft measurements
The analysis in this work is centered around measurements made during the Western Wildfire Experiment for Cloud Chemistry, Aerosol Absorption, and Nitrogen (WE-CAN) field campaign. An extensive description of the field campaign and instrumentation used can be found in Lindaas et al.68 and Juncosa Calahorrano et al.69 Briefly, during WE-CAN, the NSF/NCAR C-130 research aircraft was deployed between July and September 2018 to sample the evolution of trace gases and particles in wildfire smoke plumes over the western US. Smoke was sampled from 23 distinct wildfires over this period and for 12 of these wildfires, the aircraft was flown along horizontal transects orthogonal to the wind direction to perform pseudo-Lagrangian sampling of plumes. Multiple transects (4 to 14 per fire) were executed along the length of the wildfire plume over multiple hours (2 to 6 hours of physical age) and several hundred kilometers from the source of the fire (10 to 220 km). We should note that near-Lagrangian sampling was accomplished for only a few of the wildfire plumes (e.g., Taylor Creek, later transects for Sharps) and, in most cases, the sampling was pseudo-Lagrangian. As shown in Fig. S1,† the aircraft sampled much faster (twice as fast, on average) than the physical age of the plume for four out of the five transect sets studied in this work. The modeling and analysis undertaken in this work assumes that the fuel and burn conditions and, therefore, the emissions remained constant during the measurement time period. In other words, we assumed that the measurements represent a true Lagrangian dataset and this assumption should be considered while interpreting the results. In addition to performing transects through the center/core of the wildfire plume, for some wildfire plumes the aircraft also performed transects at different altitudes to probe vertical variability in composition and environmental conditions. These data were excluded from the analysis presented in this work.Here, we focused on the evolution of the smoke emitted from the following four wildfires: Taylor Creek Fire (southwest Oregon), Sharps Fire (southern Idaho), Bear Trap Fire (eastern Utah), and Silver Creek Fire (northwest Colorado). The aircraft completed two distinct pseudo-Lagrangian sampling efforts for the Bear Trap Fire and each of these transect sets were analyzed separately. In total, the analysis focused on five transect sets. The location and dominant fuel(s) for each wildfire can be found in Table 1 of Lindaas et al.68 These wildfires were chosen because of their ideal sampling strategy, clear trends in the evolution of OA mass and O:C with time, and because all the required measurements were available to perform the proposed OA modeling.
| Fire | OH concentration (molecules per cm3) | OH exposure (molecules h per cm3) | ||
|---|---|---|---|---|
| Before 1st transect | After 1st transect | At 1st transect | At final transect | |
| Taylor Creek | 8.9 × 106 | 0.97 × 106 | 3.1 × 106 | 5.7 × 106 |
| Sharps | 4.0 × 106 | 0.47 × 106 | 3.0 × 106 | 6.5 × 106 |
| Bear Trap 1 | 3.0 × 106 | 0.57 × 106 | 2.7 × 106 | 4.6 × 106 |
| Bear Trap 2 | 2.8 × 106 | 0.65 × 106 | 2.6 × 106 | 4.7 × 106 |
| Silver Creek | 3.5 × 106 | 1.0 × 106 | 1.9 × 106 | 5.5 × 106 |
A suite of gas, particle, meteorological, and remote sensing instrumentation was deployed on the NSF/NCAR C-130 to comprehensively characterize the smoke emissions and its environment. Below, we briefly discuss the measurements and data products that relate to the scope of this work. Pressure and temperature were measured using base instrumentation available onboard the aircraft.68,69 CO was quantified using a Quantum-Cascade Tunable Infrared Laser Direct Absorption Spectrometer (QC-TILDAS, Aerodyne Research) and reported at 1 Hz.70 CO was assumed to be an inert tracer and used to model dilution with the background air and to develop convenient metrics to characterize the OA evolution with physical age. VOC mixing ratios were quantified using a Proton-Transfer-Reaction Time-of-Flight Mass Spectrometer (PTR-ToF-MS 4000, Ionicon Analytik) at 1 Hz.34 The PTR-ToF-MS quantified concentrations for 122 VOCs, including the most important SOA precursors that have been identified for biomass burning in previous work.37,38 A High-Resolution Aerosol Mass Spectrometer (HR-AMS; Aerodyne Research) measured the mass concentrations and composition of the sub-micron, non-refractory aerosol every 5 seconds.20 Measurements of aerosol mass concentrations and OA elemental ratios of H:C and O:C from the HR-AMS measurements were used here. A Single Particle Soot Photometer (SP2; Droplet Measurement Technologies) quantified BC mass concentrations every 10 seconds.20 Finally, aerosol size distributions were measured using a Passive Cavity Aerosol Spectrometer Probe (PCASP) at 1 Hz (ref. 71) and these were used to inform the shape of the initial aerosol size distribution (median and geometric standard deviation). We used the 10 seconds merge data for all the measurements mentioned above and conversions were performed when necessary to calculate concentrations at ambient conditions (instead of at standard temperature and pressure conditions). Links to the data are provided in the ‘Data Availability’ section.
We determined transect-average values for OA, non-OA (sulfate + nitrate + ammonium + chloride + BC), CO, and VOC concentrations as well as the OA O:C and the number size distributions. Following earlier published work with the WE-CAN data,20,33,68,69 we identified the plume-sampling periods by visually examining the CO time series for increases and decreases in mixing ratios. These averages were then corrected for background conditions by computing a universal background value for each measured species (e.g., CO, OA, benzene) using all data from outside the plume while the aircraft performed a transect set. This method assumed that the composition of the gas and aerosol species in the background air remained constant in time and space. We also calculated a normalized excess mixing ratio (NEMR) for OA and VOCs by ratioing its background-corrected concentration (in μg m−3 for OA and ppbv for VOCs) with the background-corrected CO (in ppbv). The background-corrected OA O:C was calculated as a ratio of the background-corrected molar concentrations for O and C; equations can also be found in Hodshire et al.72 For this calculation, we assumed the OA to be only composed of C, H, and O such that the molar concentrations of C, H, and O were determined from the available OA, O:C, and H:C data. While wildfire OA is likely to be composed of nitrogen-containing organic compounds, the low N:C values measured during WE-CAN (∼0.02)20 meant that accounting for nitrogen had a negligible impact on the reported OA mass concentrations and O:C and H:C values. Background corrections were performed separately for each wildfire plume transect set. We studied the sensitivity of the findings from this work to other ways in which the background values can be calculated and these are described later (Section 3.2).
2.2 OH concentration and exposure estimates
The hydroxyl radical (OH) exposure in the wildfire plumes was determined by examining the laboratory-normalized and reactivity-differentiated VOC decay. For each transect in a wildfire plume, this calculation was done by first taking the natural logarithm of the ratio of the VOC NEMRs at that transect to the average values of the VOC NEMRs measured in laboratory experiments. The VOC NEMRs from laboratory experiments were based on the work of Koss et al.28 who measured emissions from 58 separate burns performed on 18 different fuels. We decided to use the average values of the VOC NEMR across the 58 burns instead of using fuel-specific VOC NEMRs because the wildfire emissions sampled during WE-CAN arose from the combustion of a mixture of fuel types. Furthermore, we also did not consider differences in the burn conditions (e.g., differences in the modified combustion efficiency) between the laboratory experiments and wildfire plumes. As this calculation could only be performed when the corresponding VOC NEMRs were also available from the laboratory experiments, we performed this calculation on 106 out of the 122 VOCs measured by the PTR-ToF-MS. We assumed that the reactive oxygenated VOC concentrations in the wildfire plume were largely from direct emissions from the fire rather than produced through in-plume chemical reactions. This field-to-laboratory ratio of VOC NEMRs was then plotted against the reaction rate constant for those VOCs with OH (kOH). The kOH values for the VOCs are from those reported in Koss et al.28 noting that they reflect kOH values for either the most dominant isomer, weighted average of the potential isomers, or a VOC that is structurally similar. Here, we used kOH values reported at 300 K to perform this analysis. As kOH values for well-studied VOCs such as alkanes, alkenes, and aromatics are only ±20% off at the cooler temperatures in the wildfire plume compared to those at 300 K, accounting for the temperature-dependent kOH is unlikely to change the OH estimates presented here. Isoprene, monoterpenes, and catechol can also react with O3 in addition to OH and were excluded from the analysis, although including these VOCs did not appear to change the estimated OH exposure (not shown). This result was partly because the O3 mixing ratios in the wildfire plume were low enough (45–90 ppbv) that these VOCs still preferentially reacted with OH rather than O3.Results for the first transect from the five transect sets analyzed in this work are shown in Fig. 1, as an example. The ratio of VOC NEMRs exhibited an inverse relationship with kOH. The likely explanation for this inverse trend was that the oxidation chemistry prior to this transect resulted in stronger depletion of VOCs with a higher kOH and weaker depletion of VOCs with a lower kOH. An alternate explanation is that the ratio of VOC NEMRs was biased lower for the higher kOH species compared to the lower kOH species on account of differences in fuel and burn conditions as well as the timing of the emissions, but we are not aware of why there might be systematic changes in the emissions with the species kOH. Assuming that chemistry prior to the first transect explains the trends in Fig. 1, the slope in the ratio of VOC NEMRs with kOH was fit to determine an effective OH exposure for the time period before this transect. We should note that we did not assume that the field and laboratory VOC NEMRs at the time of emission were the same, which would require setting the intercept for the fit to 0, which we did not do. Rather, we assumed that there were no systematic kOH-dependent differences in emissions between the field and laboratory cases. This exercise was repeated for all transects to calculate a time-varying OH exposure estimate for all transect sets, results for which are shown in Fig. 2. A line was fit through the OH exposure data to calculate an average OH concentration for the wildfire plume after the first transect. We assumed that the OH exposure changed linearly from zero, close to the fire, to the OH exposure estimated at the first transect, which resulted in a separate, time-invariant OH concentration estimate for the time period before the first transect.
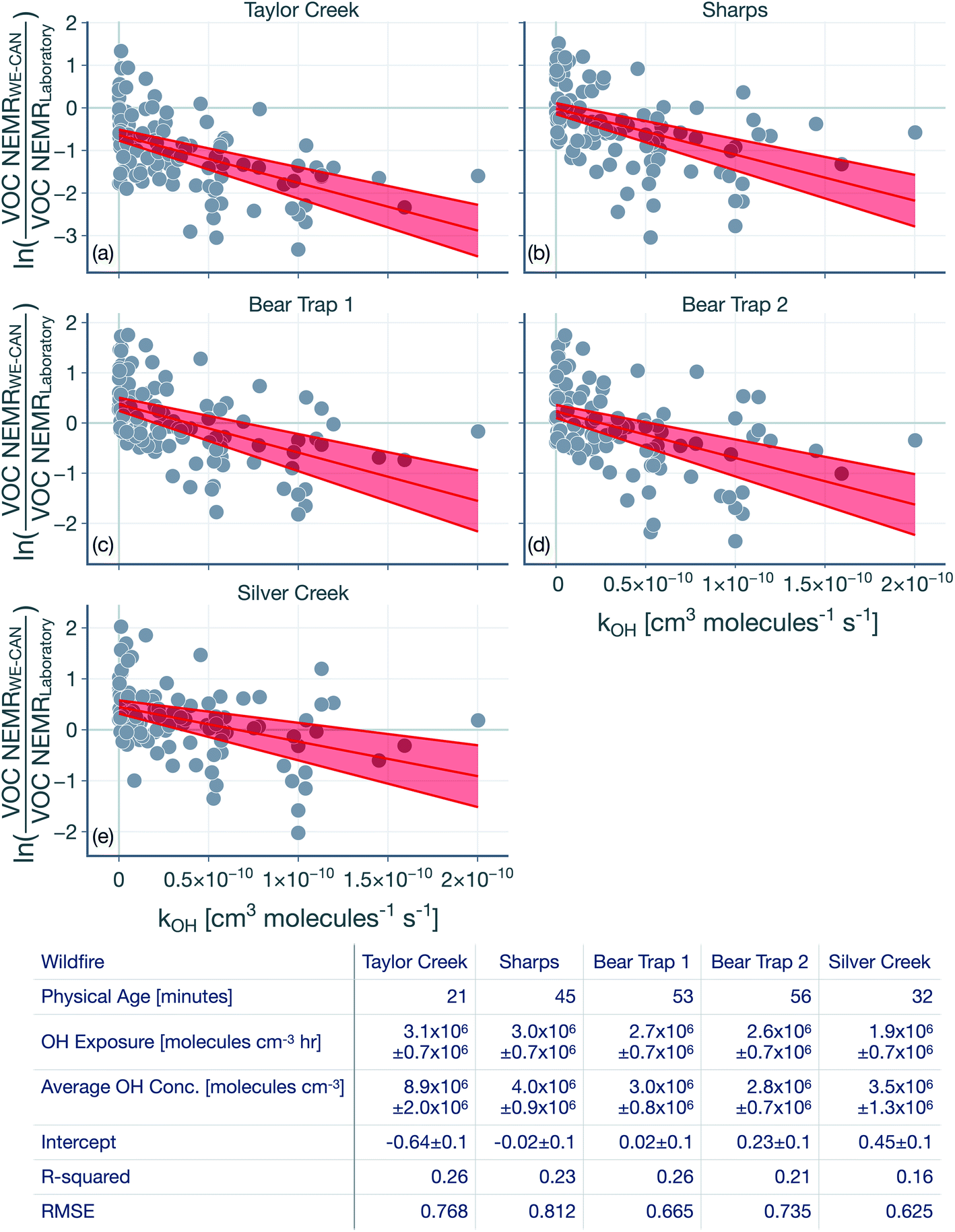 | ||
| Fig. 1 Logarithm of the ratios of the VOC NEMR in the first transect of the wildfire plume to the average value of the VOC NEMR measured in laboratory experiments plotted against kOH. Results are shown for the (a) Taylor Creek, (b) Sharps, (c) Bear Trap 1, (d) Bear Trap 2, and (e) Silver Creek Fire transect sets. Solid red lines represent the linear fit to the data and the red bands capture the standard error. The table lists statistics based on the linear fit including the OH concentration and exposure. The time-varying OH exposures for all five transect sets are shown in Fig. 2 and the OH concentrations are listed again in Table 1. | ||
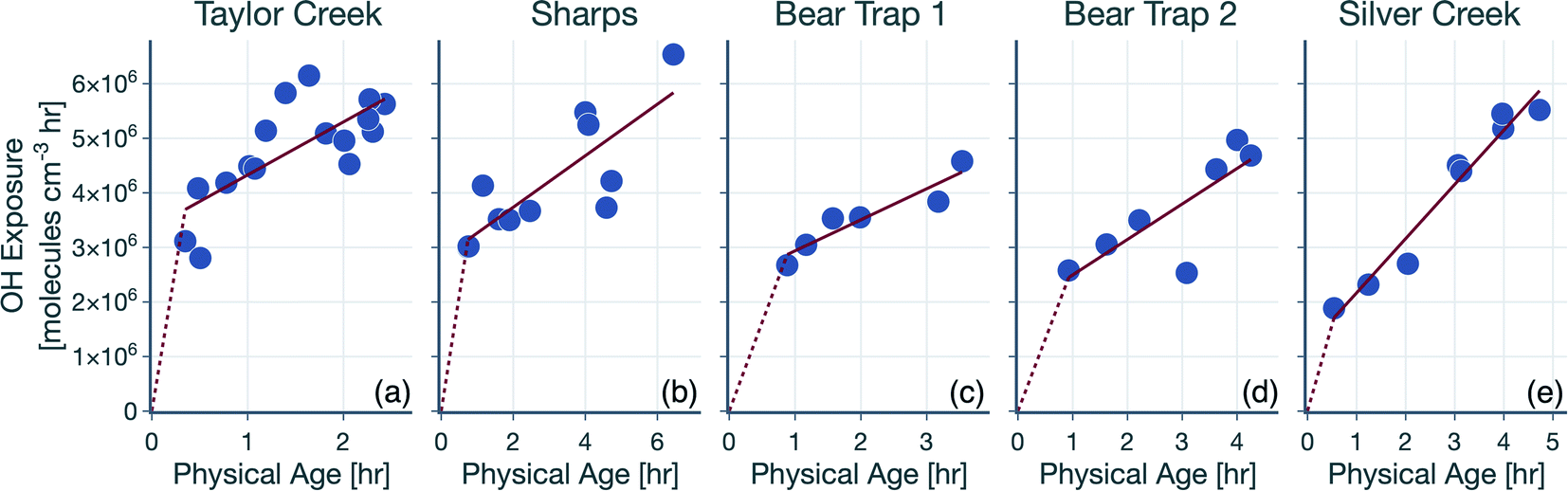 | ||
| Fig. 2 Time-varying OH exposures estimated for the five transects sets: (a) Taylor Creek, (b) Sharps, (c) Bear Trap 1, (d) Bear Trap 2, and (e) Silver Creek. The solid red lines are linear fits to time periods after the first transect while the dotted red lines represent the estimated OH exposure prior to the first transect. | ||
The OH concentrations before and after the first transect are tabulated for the five transect sets in Table 1. For the five transect sets and four fires analyzed, the total mean OH exposure varied between 4.6 × 106 (Bear Trap 2) and 6.5 × 106 (Sharps) molecules h per cm3 to produce 3.1 to 4.4 hours of photochemical aging at an average OH concentration of 1.5 × 106 molecules per cm3. The OH concentrations varied between 2.8 × 106 (Bear Trap 2) and 8.9 × 106 (Taylor Creek) before the first transect and between 0.47 × 106 (Sharps) and 1.0 × 106 (Silver Creek) beyond the first transect. This analysis approach suggests that OH concentrations, on average, were a factor of six larger before the first transect compared to after the first transect suggesting that photochemical oxidation was as relevant to the wildfire plume in the near field (between the point of emission and the first transect) as it was for the far field (beyond the first transect) after accounting for differences in the physical age before and after the first transect; the first transect was estimated between 21 and 56 minutes of the physical age of the wildfire plume.
We assessed our estimates of OH concentrations in the wildfire plumes after the first transect and found them to be consistently lower than those estimated in previous work. For instance, our estimates for OH concentration in the Taylor Creek and Bear Trap wildfire plumes after the first transect (0.97 × 106 and ∼0.6 × 106 molecules per cm3, respectively) were a factor of eight and five smaller than previous OH estimates for the same wildfire plumes calculated from the observed decay for a handful of VOCs in the core of the plume (∼8 × 106 and ∼3 × 106 molecules per cm3, respectively).69 Similarly, our estimates of OH concentration after the first transect, across all five transect sets, were a factor of five to ten smaller than those estimated in Mexican and African biomass burning plumes44,48,53 where OH concentrations were calculated using techniques similar to those outlined in Juncosa Calahorrano et al.69 We tentatively argue that the historical estimates of OH concentrations in wildfire plumes are larger and different than those measured here because earlier work has only used a few species (<5) to calculate OH concentrations. We hypothesize that our approach, which uses a much larger number of species (106), provides a more robust estimate for mean OH concentrations in the plume. However, this discrepancy in OH concentrations needs to be investigated in detail in future work. Finally, our estimated OH concentrations were found to be lower than the average OH concentrations typically found in urban environments and regional backgrounds (∼1.5 × 106 molecules per cm3).73 In this specific urban and regional comparison, the low OH concentrations in the wildfire plume might be a result of the large OH sinks present therein as well as the low photochemical activity expected in optically dense plumes.74
We note that the OH estimates used in this work are uncertain. The OH estimates are likely to be unbiased if the field and laboratory emissions ratios have no systematic relationship with kOH. In case there is a relationship and that this relationship is different between the field and laboratory, our OH estimates could be biased lower or higher. Hence, there is no clear way in which we could argue that our OH estimates bound the range of expected values in a wildfire plume. Since the OH estimates were developed based on the observed decay of all species including reactive oxygenated VOCs, any chemical production of oxygenated species should bias our OH estimate lower. Hence, based on the inclusion of the reactive oxygenated VOCs alone, our OH estimates potentially present a lower bound estimate. Although there is limited evidence, Coggon et al.75 have found in laboratory experiments performed on biomass burning smoke that there was little to no production of reactive oxygenated VOCs, such as phenol and furan, relative to these species' primary emissions. When OH was calculated from hydrocarbon VOCs measured by the PTR-ToF-MS to eliminate the influence from including oxygenated VOCs, the inverse relationship was weakened and produced OH concentrations that were at least a factor of 2 lower than those listed in Table 1, both before and after the first transect. We also calculated OH concentrations beyond the first transect by using the VOC NEMRs at the first transect as the reference instead of using the VOC NEMRs from the laboratory. The concentrations so calculated and limited to the time period beyond the first transect were found to be only slightly higher to those listed in Table 1 (0.67 × 106–1.2 × 106 molecules per cm3) but still lower than those in historical studies mentioned earlier. The VOC NEMRs at the first transect, by definition, cannot be used to determine the OH concentrations prior to the first transect. Given the uncertainty, we studied the sensitivity of the findings from this work to the OH concentrations before and after the first transect and these are described later (Section 3.2).
2.3 Organic aerosol model
We developed a plume version of the SOM-TOMAS model to simulate the formation, composition, and evolution of OA in wildfire plumes. The statistical oxidation model (SOM), uses a statistical approach to track the multigenerational oxidation chemistry and thermodynamic properties of OA precursors and its oxidation products.76,77 The TwO Moment Aerosol Sectional model (TOMAS), uses a sectional approach to track the number and mass moments of the aerosol size distribution to simulate the microphysical processes of nucleation, coagulation, and condensation/evaporation.78,79 The SOM has been extensively used to study the influence of multi-generational aging,80 IVOCs,81 NOX,82 and vapor wall losses in chambers65,83 on OA evolution. More recently, the SOM-TOMAS model was used to study the SOA formation from biogenic VOCs,84 phenols,85 and evaporated petro- and bio-fuels.86 Pertinent to this work, the SOM-TOMAS model was used to study the SOA formation in chamber experiments performed on wildfire emissions.38 The SOM-TOMAS model configuration described in detail in Akherati et al.38 was used in this work with modifications to account for dilution, different environmental conditions (e.g., pressure, temperature), and an updated treatment of POA and SVOCs.The SOM tracks the chemical evolution of OA and its precursors using a two-dimensional, carbon (NC) and oxygen (NO) number grid. The properties of each model species (e.g. reactivity (kOH), volatility (c*)) are parameterized based on their NC and NO. The SOM has six adjustable parameters that govern the oxidation chemistry and thermodynamic properties of the model species: (i–iv) pf,1–pf,4, the yields of four functionalized products that add one, two, three, and four oxygen atoms to the carbon backbone, respectively; (v) mfrag, the parameter that characterizes the fragmentation probability (Pfrag); and (vi) ΔLVP, the decrease in the c* of the model species per addition of an oxygen atom. The particle-phase species in SOM are tracked in 36 TOMAS size sections that span dry diameters between 3 to 10000 nm. TOMAS simulates coagulation between size sections and the kinetic condensation/evaporation of mass between the particle and vapor phases for all SOM model species. The SOM-TOMAS model also accounts for formation of highly oxygenated organic molecules (HOMs) and formation/dissociation of oligomers.84 The SOM-TOMAS model was also updated recently with the diffusive-reactive framework described in Zaveri et al.87 to model the influence of phase state on the kinetic gas/particle partitioning of OA.84 Although SOA precursors found in biomass burning emissions (e.g., monoterpenes) are known to form HOMs88,89 and oligomers90,91 and certain biomass burning particles can be viscous, neither HOM nor oligomer formation was modeled and the OA was assumed to be liquid-like with a diffusion coefficient of 10−10 m2 s−1. These assumptions surrounding HOMs, oligomers, and phase state will need to be examined in future work.
For each wildfire plume, the SOM-TOMAS model was used to simulate the OA evolution from the time just after emission up to the last measured transect. While this should include both vertical plume rise and horizontal plume transport after reaching the equilibrium height, we do not explicitly model the change in the pressure and temperature of the air parcel during vertical plume rise. The background-corrected CO concentrations past the first transect were first fit to an exponential function and the CO values including those extrapolated to t = 0 were then used to determine dilution of the wildfire plume with background air starting at t = 0. Since we only used background-corrected values in this work, we assumed the background air to be free of any trace gases and particles. This assumption affects non-linear processes, such as equilibrium partitioning and coagulation; however, because background concentrations are generally much lower than in-plume concentrations, these are expected to have a minimal effect.63 While the pressure and temperature values changed modestly between wildfire plumes, they were found to be in a relatively narrow range within each individual wildfire plume (Fig. S2†). Hence, an average pressure and temperature value was used for the entire wildfire plume. Model predictions and measurements of concentrations and mixing ratios in this work were expressed at the plume pressure and temperature.
2.4 SOA formation from VOCs
SOA formation from VOCs was modeled similarly to the treatment presented in Akherati et al.38 Briefly, we considered five broad classes of SOA precursors, with the surrogates informing the SOA formation listed in parentheses: (i) alkanes (n-dodecane; Loza et al.92), (ii) aromatic hydrocarbons (benzene, toluene, m-xylene; Ng et al.93 and Zhang et al.65), (iii) oxygenated aromatics (phenol, guaiacol, syringol; Yee et al.94), (iv) heterocyclics (2-methylfuran + dimethylfuran; He et al.86), and (v) biogenics (isoprene, α-pinene; Chhabra et al.95). We did not model the SOA formation from partially speciated VOCs as we did not find them to be important SOA precursors in our previous work.38 Akherati et al.38 found that the five organic classes mentioned above, on average, were able to explain most of the SOA measured in chamber experiments performed on emissions from laboratory fires, with oxygenated aromatics and heterocyclic compounds accounting for 80% of the SOA produced. The SOM-TOMAS parameters, specific to high NOX conditions, to model SOA formation from these precursors are listed in Table S1.† Each of the 67 SOA precursors in the model were assigned an SOA surrogate, mentioned in parentheses above, and this surrogate assignment along with the molecular weight and kOH value for the precursor is listed in Table S2.† We also modeled SOA formation from SVOCs and that treatment is described in Section 2.5.The VOC NEMRs at the first transect were extended to t = 0 by correcting for the OH exposure before the first transect (see Table 1 for transect-set-specific OH exposures). These field VOC NEMRs at t = 0 and the first transect were aggregated by precursor class and compared to the VOC NEMRs measured by Koss et al.28 during a recent laboratory campaign. Results of this comparison, combined over all five transect sets and four wildfires, are shown in Fig. 3. Results when separated by transect set were similar to those presented in Fig. 3 and, hence, are not shown or discussed. The VOC NEMRs for the five SOA precursor classes at the first transect were substantially lower than those measured in the laboratory. The median values at the first transect were 74%, 58%, 28%, 15%, and 75% lower for the biogenic, oxygenated aromatic, heterocyclic, aromatic hydrocarbons, and alkane classes, respectively, compared to the laboratory values. The largest differences were observed for the most reactive SOA precursors with respect to OH (e.g., oxygenated aromatics) and the smallest differences were observed for the least reactive SOA precursors with respect to OH (e.g., aromatic hydrocarbons), a feature that was leveraged to determine the OH concentrations and exposure in Section 2.2. Even after correcting for OH exposure prior to the first transect, the VOC NEMRs for three of the organic classes (i.e., biogenic, oxygenated aromatic, and alkane) were still modestly lower compared to those measured in the laboratory. The median values at t = 0 were 18%, 38%, and 61% lower for the biogenic, oxygenated aromatic, and alkane classes, respectively, compared to the laboratory values. The median values were 32% and 15% higher for the heterocyclic and aromatic hydrocarbon classes, respectively, compared to the laboratory values.
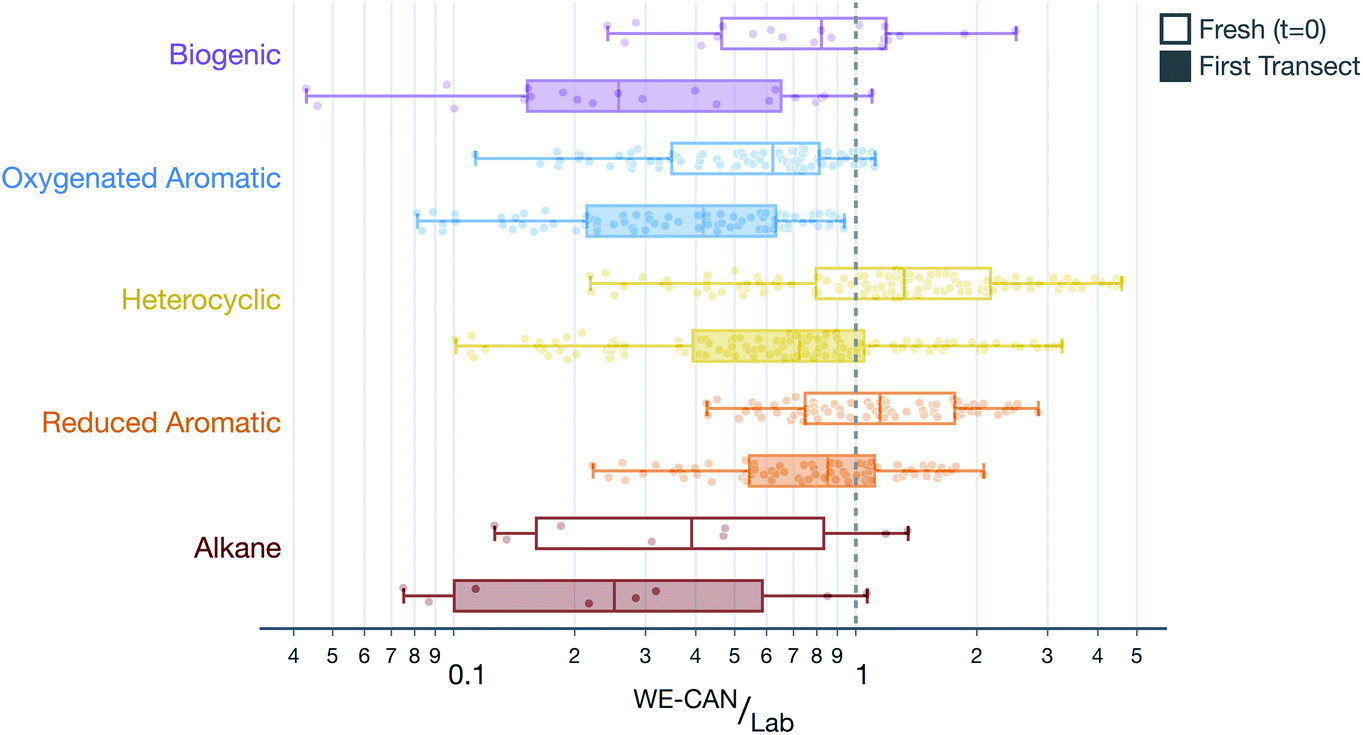 | ||
| Fig. 3 Ratio of VOC NEMRs at t = 0 and the first transect in the wildfire plume to the average values of VOC NEMRs measured in laboratory experiments. Data are aggregated by the following SOA precursor class: biogenics, oxygenated aromatics, heterocyclics, aromatic hydrocarbons, and alkanes. Within each SOA precursor class, data are presented together for all five transect sets such that each point represents the ratio for a given VOC NEMR in a specific transect. | ||
There are two important implications of the differences in the ratios of VOC NEMRs shown in Fig. 3 and described in the previous paragraph. First, field emissions of SOA precursors (those at t = 0) are lower than those measured in laboratory experiments, for at least a few of the important organic classes. It is expected then that the SOA precursor mixture would be relatively less potent in forming SOA in wildfire plumes than what has been observed in laboratory experiments.37–39,41–43 Second, a significant fraction of the SOA precursor emissions are depleted between t = 0 and the first transect (between 15% and 74%). This implies that not only is SOA being actively produced in the near field after emission (<1 hour) but also that there is significantly reduced potential for continued production of SOA beyond the first transect. These comparisons partly explain the field versus laboratory differences outlined by Hodshire et al.,40 for the OA evolution after the first transect.
2.5 Treatment of POA and SVOCs
POA was treated as semivolatile and reactive, following a history of observations from laboratory experiments.21,22,96 Fresh emissions of POA close to the fire were determined in an iterative manner. Using an initial guess for the POA mass concentration at t = 0, we fit a mass distribution for eight model species in the SOM grid that was able to reproduce the average volatility behavior observed by May et al.22 for POA emissions (Fig. S3(a)†). May et al.22 measured this average volatility behavior by studying the response of fresh POA emissions from 19 separate fires and 12 different fuels to dilution and thermal denuding in laboratory experiments. The model species were placed in a SOM grid for multi-ring aromatics (ΔLVP = 1.4922), the reason for which is discussed later in this section. The number of model species (i.e., 8) used to represent the POA and SVOC mass in the SOM grid was arbitrary and the use of a larger number of model species (e.g., 10, 15) did not seem to affect our results (not discussed further). The following species were used to represent the POA/SVOC mass in the SOM grid: C5O7, C9O2, C9O5, C11O2, C12O2, C12O3, C14O5, and C15O6 (Fig. S3(c)†). An explanation for why this precise set of species was used is presented later when describing results from the sensitivity (Monte-Carlo) simulations (Section 3.2). In addition to constraining the mass distribution in the SOM grid to match observations of POA volatility, the model species were deliberately chosen to produce a specific response in the aerosol O:C with POA mass concentration, following observations of biomass burning OA in laboratory21 and field97 environments (Fig. S3(b)†): an increasing OA O:C from with a decrease in OA mass concentration from 1000 to 1 μg m−3. The SOM-TOMAS model was used to simulate the time period between t = 0 and the first transect using the POA value assumed at t = 0, and this process was iterated until the OA mass concentration at the first transect was the same as the measured value. This method necessarily assumes that the OA mass and composition at t = 0 is different from that at the first transect. As the assumptions about POA volatility and oxidation chemistry were altered across different simulations, this iterative approach to determining the POA mass concentration at t = 0 was done separately for each simulation and transect set.
We used the detailed speciation data for POA and SVOCs from Jen et al.25 to create a substitute volatility distribution, which was then used to study the sensitivity in model results to the volatility distribution assumed for POA and SVOCs. Jen et al.25 used two-dimensional gas chromatography with time-of-flight mass spectrometry (GC × GC-ToF-MS) on derivatized samples to measure the detailed chemical composition of POA and SVOC emissions from 29 laboratory-generated fires performed on 24 different fuels. They measured ∼150 unique species in the sample collected on quartz filters and these amounted to between 10 and 65% of the total POA/SVOC mass. The averaged and normalized volatility distribution and mass distribution in carbon-oxygen space for the POA and SVOC emissions is shown in Fig. S4(a) and (b),† respectively. The volatility distribution was constructed by binning the ∼150 species by c*, which was estimated for each species using EPISuite 4.11,98 a numerical model that estimates physical and chemical properties of pure substances.
Jen et al.25 found that the SVOC emissions were dominated by sugars, phenols, and other complex organic compounds (e.g., terpenoids, heterocyclics), organic compound classes also measured by the PTR-ToF-MS. In theory, there should be little if any overlap between the SOA precursors quantified by the PTR-ToF-MS and GC × GC-ToF-MS since the PTR-ToF-MS exclusively sampled gas-phase compounds with a c* higher than 105 μg m−328 and the GC × GC-ToF-MS measurement was performed on quartz filters that generally trap gas- and particle-phase compounds lower than a c* of 105 μg m−3.22 In this work, we assumed that the SVOCs and VOCs represent a mutually exclusive set of SOA precursors despite similarities in the compound classes within these two categories. SVOCs have been hypothesized to be important precursors of SOA formation from biomass burning emissions,20,33 but there are few laboratory datasets that can be called upon to inform the oxidation chemistry of SVOCs in our model. In addition to Jen et al.,25 several studies have measured semivolatile multi-ring aromatics in biomass burning emissions and they are expected to serve as SOA precursors.23,35,99 Hence, in the absence of any model SOA precursors for which laboratory data are available, we simulated the oxidation chemistry of SVOCs in the base configuration of the SOM-TOMAS model assuming that they have a similar potential to form SOA as multi-ring aromatics (i.e., naphthalene). However, given the uncertainty in this assumption, we performed sensitivity simulations where we modeled the oxidation chemistry of SVOCs as oxygenated aromatics (i.e., phenol), heterocyclics (i.e., 2-methylfuran + dimethylfuran), linear alkanes (i.e., n-dodecane), or biogenics (i.e., α-pinene) with the species in parentheses used as the surrogate to model SOA formation (Section 3.2).
2.6 Model configuration and simulations
The base simulations were performed with the following model configuration. We assumed POA to be semivolatile and reactive. The representation of POA/SVOC mass in the SOM grid was based on the volatility distribution of May et al.22 and the oxidation chemistry for SVOCs was modeled based on SOM parameters for multi-ring aromatics (naphthalene). We assumed a liquid-like phase state to model the kinetics of OA gas/particle partitioning. All SOA parameters, including those for SVOCs and VOCs, were corrected for the influence of vapor losses to the walls of the Teflon chamber. Background corrections were performed on the raw observations using a background value that was specific to each wildfire transect set, but not varying in time or space for that transect set. The OH concentrations were based on an analysis of the ratios of VOC NEMRs, with separate but constant values for the time periods before and after the first transect. The first transect for each transect set was hand chosen based on the transect closest to the fire that had the highest observed VOC NEMR. Simulations performed with this base model configuration are also referred to as the ‘SV-POA + FullChem’ simulations, representative of the most updated treatment of OA and OA precursors in wildfire plumes. Nearly all of the assumptions in the base model were rigorously tested by performing sensitivity simulations, as described below:(1) In addition to the base simulations (SV-POA + FullChem), we performed a systematic sequence of simulations where we tested assumptions about POA volatility and SVOC and VOC chemistry: (i) non-volatile POA and no chemistry (NV-POA + NoChem), (ii) non-volatile POA and VOC chemistry (NV-POA + Chem), (iii) semivolatile POA and no chemistry (SV-POA + NoChem), and (iv) semivolatile POA and SVOC chemistry (SV-POA + NoVOCChem). Results from these simulations are presented in Fig. 4 and 5.
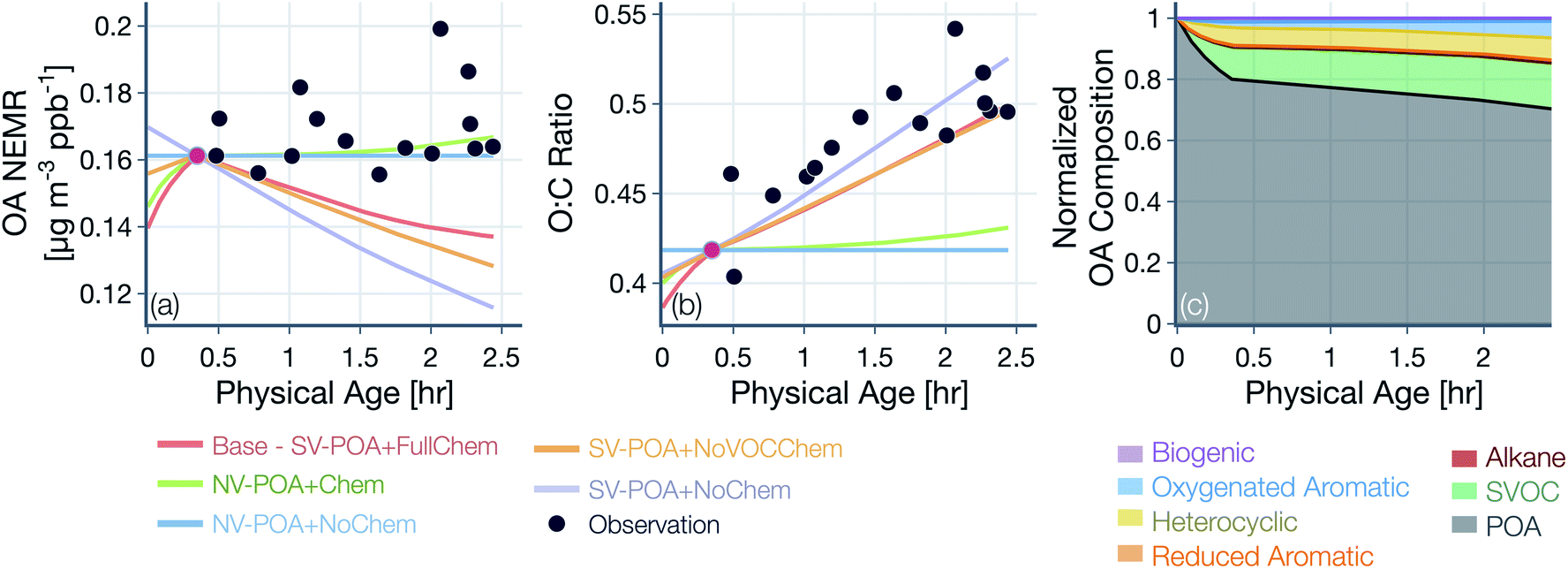 | ||
| Fig. 4 Predictions of (a) OA NEMR and (b) OA O:C from the SOM-TOMAS model (solid colored lines) compared against measurements (solid black circles) from the Taylor Creek Fire. Model predictions are shown for five different simulations that vary in their assumptions about POA volatility and SVOC and VOC oxidation chemistry. Model predictions were always constrained to the measurements of OA NEMR and OA O:C at the 1st transect, marked by the red solid circle. Predictions of the normalized OA composition from the base simulations are shown in panel (c). | ||
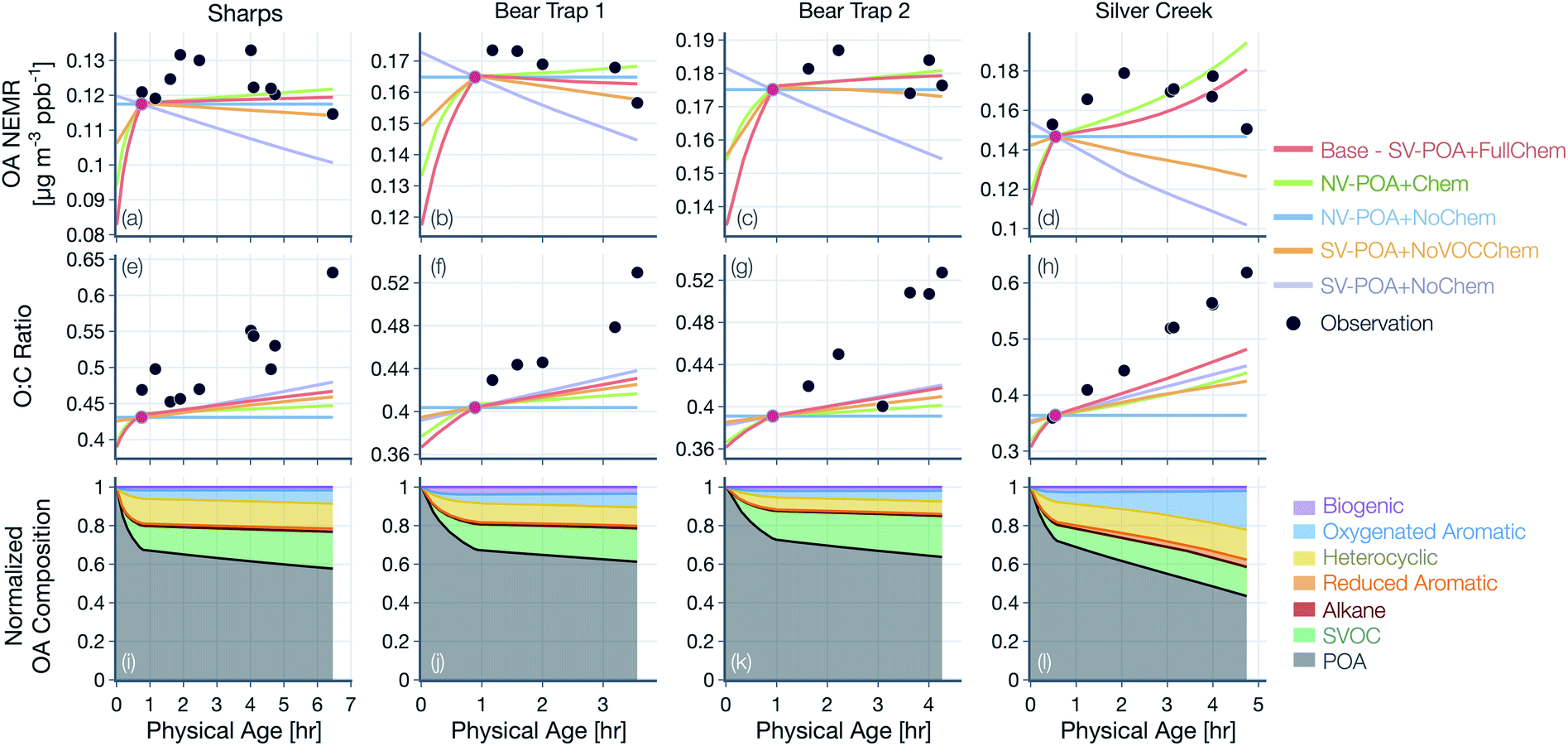 | ||
| Fig. 5 Predictions of (a–d) OA NEMR and (e–h) OA O:C from the SOM-TOMAS model compared against measurements (solid black circles) from four different wildfire transect sets (Sharps, Bear Trap × 2, Silver Creek). Model predictions are shown for five different simulations that vary in their assumptions about POA volatility and oxidation chemistry. (i–l) Model predictions of the normalized OA composition from the base simulations (SV-POA + FullChem). | ||
(2) Simulations were performed to assess the sensitivity in model results to the treatment of POA and SVOCs: (i) we simulated the oxidation chemistry for SVOCs using the SOM parameters for oxygenated aromatics (i.e., phenol), heterocyclics (i.e., 2-methylfuran + dimethylfuran), linear alkanes (i.e., n-dodecane), or biogenics (i.e., α-pinene), with the species in parentheses used as the surrogate to model SOA formation, (ii) we used the POA volatility distribution determined from the work of Jen et al.25 in combination with the five surrogate species to model the oxidation chemistry of SVOCs, and (iii) we performed a thousand Monte-Carlo simulations where we randomly chose eight species in the SOM grid to represent the POA and SVOC mass and fit a mass distribution with these eight species that reproduced the POA volatility behavior observed by May et al.22 Results from these simulations are presented in Fig. 6, S9 and S10.†
(3) Simulations were performed to assess the sensitivity in model results to the assumed OH concentrations, ‘first’ transect, and background correction: (i) we assumed a low or high OH concentration over the entire wildfire plume with the low value based on the OH estimate after the first transect and the high value based on the OH estimate before the first transect (Table 1), (ii) we assumed a time-varying OH concentration that was determined by fitting a power function through the OH exposure data presented in Fig. 2, (iii) we assumed a constant OH concentration of 1.5 × 106 molecules per cm3 that is commonly used to reflect average OH concentrations in polluted environments, (iv) instead of using the transect closest to the fire as the first transect, we assumed the second or third closest transects to be the ‘first’ transect to perform the simulations, and (v) measurements and model inputs were calculated by performing background corrections by using the transect-specific background concentrations or assuming the concentrations upwind of the fire to be representative of the true background. Results from these simulations are presented in Fig. 7, S11 and S12.†
3. Results
3.1 OA mass and composition evolution
Results from simulations performed with the base configuration and with assumptions about POA volatility and oxidation chemistry are shown in Fig. 4 for the Taylor Creek Fire. We present results for Taylor Creek first because the sampling strategy was the most Lagrangian in our dataset (Fig. S1†). Model predictions and measurements of OA mass are presented using the NEMR metric (Fig. 4(a)) and those for OA composition as the background-corrected OA O:C (Fig. 4(b)). Model predictions and measurements of the background-corrected OA and CO concentrations at ambient volume are shown in Fig. S5.† The measurements show mildly increasing OA NEMR (0.16 μg m−3 ppbv−1) with an increase in the OA O:C (from ∼0.41 to ∼0.52) and these trends were used to evaluate predictions from the five different model simulations. Uncertainties in the observed OA NEMR and O:C were deliberately left out since those were found to be much larger than the overall trend, which made it harder to evaluate the model predictions. A version of Fig. 4 that includes the standard error in the mean for the observed OA NEMR and O:C is presented as Fig. S6.†
For the simulations with non-volatile POA and no oxidation chemistry (NV-POA + NoChem), the model reproduced observations of OA NEMR (Mean Bias Error (MBE) = -0.009, Mean Absolute Error (MAE) = 0.011; μg m−3 ppbv−1) but, by definition, produced no change in the OA O:C (MBE = −0.060, MAE = 0.062). With the oxidation chemistry turned on to produce SOA from VOCs (NV-POA + Chem), the model only produced a marginal increase in the OA NEMR compared to the NV-POA + NoChem simulation and, hence, reproduced observations of OA NEMR (MBE = −0.007, MAE = 0.011; μg m−3 ppbv−1). The NV-POA + Chem simulation produced a slight increase in OA O:C with physical age compared to the NV-POA + NoChem model but still significantly lower than the observed increase in OA O:C (MBE = −0.055, MAE = 0.057). The relatively small increase in OA NEMR and OA O:C can be explained by the marginal amounts of SOA formed from the VOC mixture beyond the first transect. As noted in Section 2.4, this VOC mixture was substantially depleted in important SOA precursors by the first transect. For the simulation with semivolatile POA with no oxidation chemistry (SV-POA + NoChem) and the simulation with semivolatile POA with oxidation chemistry for SVOCs alone (SV-POA + NoVOCChem), the model appeared to underestimate the OA NEMR (MBE = −0.035 and −0.028, MAE = 0.035 and 0.028, respectively; μg m−3 ppbv−1) with a substantial increase in OA O:C. The predicted decrease in OA NEMR compared to the NV-POA + NoChem model stemmed from the evaporation of POA with dilution that was only partly recovered through SOA formation in the SV-POA + NoVOCChem model. Results from the SV-POA + NoChem simulation suggested that POA evaporation alone could explain the increase in the OA O:C with physical age as the lower-volatility material left in the particle phase after evaporation had a relatively higher O:C than the semivolatile material that had evaporated (Fig. S3(b)†).
The base simulation that assumed a semivolatile POA and oxidation chemistry for both SVOCs and VOCs (SV-POA + FullChem) underestimated the OA NEMR by 15% compared to observations (MBE = −0.024, MAE = 0.024; μg m−3 ppbv−1) but produced a large increase in OA O:C consistent with the observations (MBE = −0.019, MAE = 0.032; μg m−3 ppbv−1). The base simulation predicted a lower increase in O:C compared to the SV-POA + NoChem simulation because the SOA being formed in the base simulation had a lower O:C than the remaining POA. Overall, two of the simulations appeared to be the most consistent with observations of OA NEMR but significantly underestimated the observations of OA O:C (NV-POA + NoChem and NV-POA + Chem), and two of the simulations came close to reproducing the increase in OA O:C with physical age but underestimated the observations of OA NEMR (SV-POA + NoChem and SV-POA + NoVOCChem). The base or SV-POA + FullChem simulation seemed to offer a balanced comparison with observations where both the OA NEMR and O:C were only slightly underestimated.
Results from simulations for the remaining fires and transect sets (Sharps, Bear Trap, and Silver Creek) are shown in Fig. 5(a–h), where the relative trends across the five different simulations were similar to those presented for Taylor Creek in Fig. 4. Therefore, the differences in these simulations are not described further. However, as discussed below, the absolute performance of the base simulation for these other transect sets was mixed. The base simulation could not reproduce the initial increase and later decrease in observed OA NEMR for the Sharps and Silver Creek Fires. Both of these fires were sampled by the aircraft much faster than the physical age (Fig. S1†), suggesting that the measurements may partly reflect changes in emissions rather than those from their physicochemical evolution. The base simulation produced a mildly increasing OA NEMR for the Sharps Fire (MBE = −0.004, MAE = 0.006; μg m−3 ppbv−1) and a sharply increasing OA NEMR for the Silver Creek Fire (MBE = −0.004, MAE = 0.012; μg m−3 ppbv−1). Unlike the comparison for the Taylor Creek Fire, the base simulation appeared to reproduce the relatively constant observations of OA NEMR for the Bear Trap Fire transects. The base simulations produced a different trend in the modeled OA NEMR with physical age across the five different transect sets because presumably, in each of these transect sets, there were differences in the dilution rate, environmental conditions, and absolute concentrations of the OA, OA precursors, and oxidants. The base simulation consistently underestimated the change in OA O:C with physical age, with the strongest comparison for the Bear Trap 1 Fire (MBE = −0.044, MAE = 0.044) and the weakest comparison for the Silver Creek Fire (MBE = −0.087, MAE = 0.087).
Taken together, we draw the following conclusions from the model-measurement comparisons presented in Fig. 4 and 5. First, a non-volatile and non-reactive treatment of POA, regardless of the inclusion of SOA produced from VOC oxidation, is unlikely to explain the combined observations of OA NEMR and O:C. Second, POA evaporation with dilution alone can potentially explain the change in the OA O:C with time but results in loss of OA mass (decrease in OA NEMR) that is not consistent with observations. Third, we argue that POA evaporation with dilution and SOA formation from both SVOCs and VOCs, as captured by the base model, best explains the trends in both the OA NEMR and OA O:C across the five transect sets and four wildfires: average MBE = −0.007 and MAE = 0.011 (μg m−3 ppbv−1) for OA NEMR and average MBE = −0.046 and MAE = 0.055 for OA O:C across all five transect sets. We note that the model treatment in the base simulations closely reflects our updated understanding of the POA and SOA system from wildfire emissions.
Model predictions of the normalized OA composition with physical age from the base simulations are shown in Fig. 4(c) for the Taylor Creek Fire and in Fig. 5(i–l) for all other transect sets while model predictions of the POA-SOA split with photochemical age for all transect sets are shown in Fig. S7.† There are several interesting features to note. The OA composition changed rapidly starting at t = 0 from the dilution-driven evaporation of directly emitted POA and the SOA produced from the oxidation of SVOCs and VOCs. By the first transect, the average OA across all five transect sets, 21 to 56 minutes after emission, was 73% POA and 27% SOA. As the base-simulation-predicted OA NEMR was found to universally increase during the period before the first transect for all five transect sets, the changes in OA composition were largely driven by SOA condensation rather than POA evaporation. The rapid SOA production was facilitated by the higher OH concentrations experienced before the first transect (2.8–8.9 × 106 molecules per cm3; see Table 1). Generally speaking, the observed and base-simulation-predicted OA NEMRs did not vary much past the first transect for any of the transect sets. Despite that fact, the base simulations predicted a modest change in the OA composition with physical age after the first transect, suggesting a roughly equal replacement of POA with SOA. Over all five transect sets, POA continued to dominate the total OA mass beyond the first transect (>45%) but there was continued production of SOA over this time period. By the last transect, the SOA contribution to the total OA varied between 30% for the Taylor Creek Fire and 56% for the Silver Creek Fire.
The base simulations predicted that the majority of the SOA was formed from the oxidation of SVOCs, heterocyclics, and oxygenated aromatics, in that order. The contribution of the different VOC classes to SOA formation was similar between the different transect sets, although oxygenated aromatics contributed much more to SOA formation in the Silver Creek Fire than in the other Fires. On average, these three precursor classes accounted for 45, 25, and 21% of total SOA and 18, 11, and 9% of the total OA by the last transect. Heterocyclics and oxygenated aromatics have already been implicated as important SOA precursors in laboratory experiments performed on biomass burning emissions37–39,100 and our results here confirm their relevance for wildfire plumes as well. Biogenic VOCs were found to be significantly less influential compared to the precursor classes just discussed where they accounted for less than 5% of total SOA and 2% of total OA by the last transect.
These simulations provide model-based evidence for dilution-driven evaporation of POA mass being replaced by SOA mass formed from the oxidation of SVOCs and VOCs in wildfire plumes during WE-CAN. This conclusion is consistent with the theoretical findings of Bian et al.62 and Hodshire et al.,63 who showed that POA evaporation can be approximately replaced with SOA condensation under certain conditions pertaining to the fire size, background concentrations, and atmospheric stability. The base simulations predicted a mean POA-SOA split of 59–41% by the last transect over all five transect sets. These model-predicted POA-SOA splits agreed well with the theoretical findings of Hodshire et al.,63 who predicted a POA-SOA split of ∼50–50% for 1 km2 fires and ∼75–25% for 100 km2 fires, and the findings of Palm et al.,33 who analytically determined a maximum POA-SOA split of 66–33% for OA measured over several wildfire plumes during WE-CAN. Note that Palm et al.33 estimated their maximum POA-SOA split assuming that the OA at the first transect was exclusively POA. In contrast to the precursor-resolved findings discussed above, Palm et al.33 proposed that SVOCs were responsible for nearly 90% of the SOA formed within the plume. However, their approach would overestimate the SVOC contribution to SOA because the underlying closure calculation subtracted the SOA estimated from non-SVOC precursors from the total SOA formed.
3.2 Sensitivity in model predictions
Results from simulations performed to assess the sensitivity to the model treatment of POA and SVOCs are shown in Fig. 6. Here, we compare model predictions of OA NEMR (top row) and OA O:C (middle row) against measurements for the Taylor Creek Fire and plot model predictions of the fractional contributions of POA and SOA to OA (bottom row). We also include predictions from the base simulation shown in Fig. 4. Results from sensitivity simulations performed for the other transect sets are shown in Fig. S9 and S10.† In the base simulation, SVOCs accounted for <20% of the total OA by the last transect. Despite the relatively small contribution of SVOCs to total OA, the use of different surrogate species to simulate the oxidation chemistry of SVOCs resulted in a moderate spread in the OA NEMR predictions, with all predictions biased lower than the measurements. The use of a heterocyclic surrogate (i.e., 2-methylfuran + dimethylfuran) seemed to agree the best, and the use of an oxygenated aromatic surrogate (i.e., phenol) seemed to agree the least with the OA NEMR observations. The differences in the model predictions were understandable since the potential to form SOA is known to vary substantially across the five surrogate species considered: naphthalene, n-dodecane, 2-methylfuran + dimethylfuran, phenol/guaiacol, and α-pinene. There was a similar spread in the model predictions of OA O:C but, in contrast to the OA NEMR comparisons, all model predictions compared reasonably with the observed increase in the OA O:C. The spread in the model predictions of OA O:C was between 0.5 and 0.53 at the last transect. The average OA split was 75% POA and 25% SOA by the last transect.
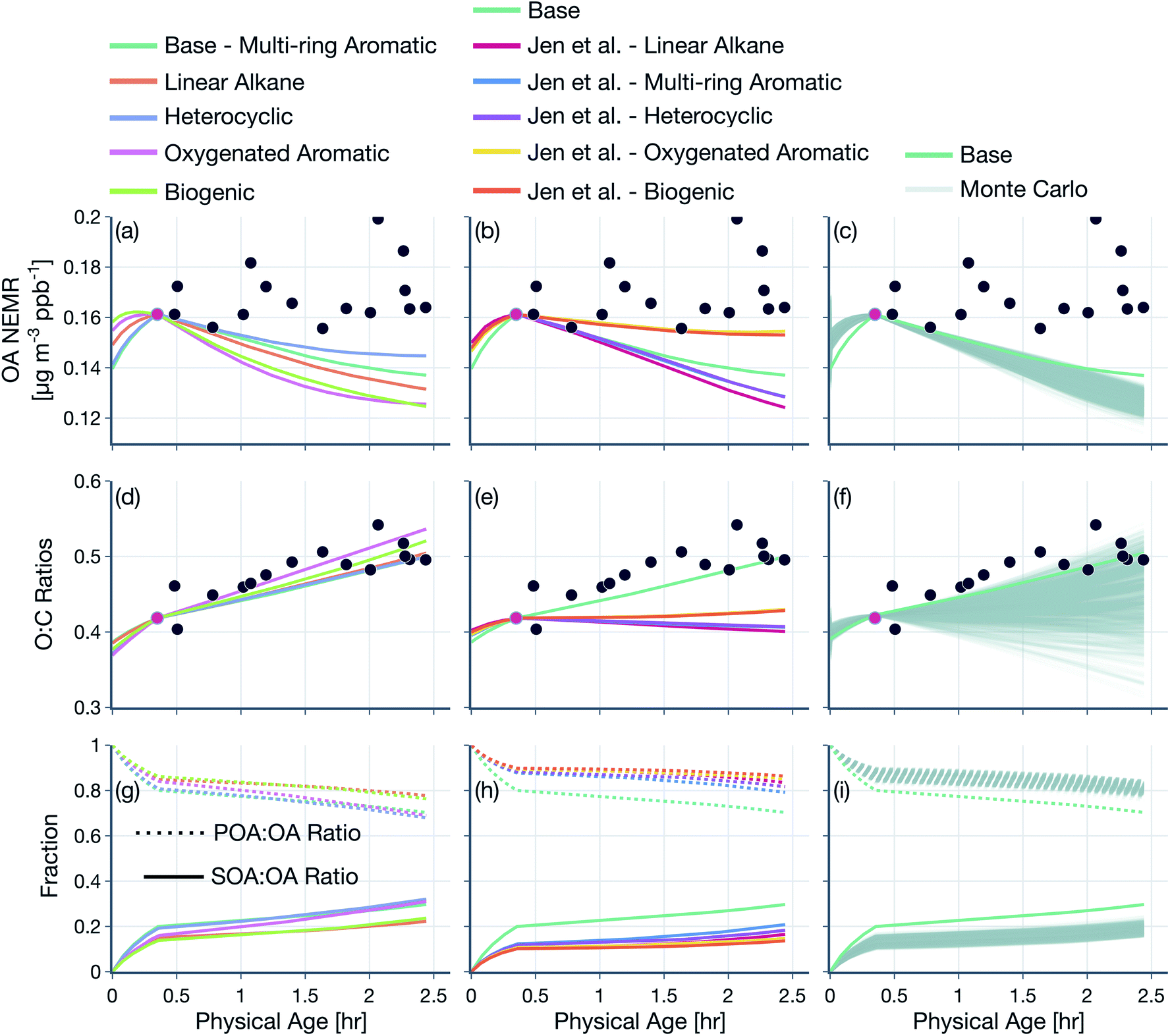 | ||
| Fig. 6 Predictions of (a–c) OA NEMR and (d–f) OA O:C from the SOM-TOMAS model (solid colored lines) compared against measurements (solid black circles) from the Taylor Creek Fire. (g–i) Predictions of the fractional contributions of POA and SOA to OA. Model predictions are shown for sensitivity simulations performed with varying assumptions for the (a, d and g) SVOC oxidation chemistry, (b, e and h) POA volatility and SVOC oxidation chemistry, and (c, f and i) POA + SVOC mass distribution in the SOM grid (Monte-Carlo). | ||
On using the substitute volatility distribution based on the speciation data of Jen et al.,25 the model produced a relatively larger spread in the OA NEMR predictions compared to the sensitivity result discussed above. The use of an oxygenated aromatic (i.e., phenol/guaiacol) and biogenic (i.e., α-pinene) surrogate seemed to agree the best and the use of a heterocyclic (i.e., 2-methylfuran + dimethylfuran), multi-ring aromatic (i.e., naphthalene), and linear alkane (i.e., n-dodecane) surrogate seemed to agree the least with the OA NEMR observations. All simulations produced a flat response in the OA O:C with physical age despite a gradual change in the POA-SOA split and dilution-driven evaporation of the semivolatile material. This flat response, which was inconsistent with the observed change in the OA O:C, was primarily from the lower-volatility material that was left in the particle phase being less oxidized than the higher volatility material that had evaporated (Fig. S4(c)†). The substitute volatility distribution resulted in a larger POA-SOA split (average of 83–17%) compared to the first set of sensitivity results (average of 75–25%) likely because a smaller fraction of the fresh POA mass was lost to evaporation from the use of a less volatile volatility distribution (Fig. S4(a) and (d)†). The mixed comparisons for OA NEMR and O:C suggest that model inputs determined from speciation data hold promise but might be limited because, in this specific case, the speciated compounds only represented a fraction (10–65%) of the total POA + SVOC mass.
Finally, we performed a thousand Monte-Carlo simulations where we randomly specified the mass distribution of the POA + SVOC mass in the SOM grid while ensuring that this mass distribution reproduced the POA volatility behavior observed by May et al.22 (Fig. S8†). The iterations produced a relatively narrower spread in the OA NEMR predictions compared to the previous sensitivity simulations but they all seemed to underestimate the observed trends. By the last transect, the predicted OA NEMR was 17 to 29% lower than the average observed OA NEMR. Compared to the OA NEMR, there was a much larger spread in the predicted OA O:C with iterations predicting a decrease in O:C with physical age at the one end (from 0.41 to 0.31) to reproducing the observed increase at the other end (0.38 to 0.54). It appears that the mass distribution of POA + SVOC in the SOM grid had a significant, non-linear influence on POA evaporation, SOA production, and subsequently on the OA O:C evolution with photochemical age. We note that the mass distribution of POA + SVOC in the SOM grid across the thousand simulations was always constrained to observations of POA volatility but had enough degrees of freedom to produce a substantial spread in model predictions. The iteration that resulted in the largest absolute OA NEMR at the end of the simulation and then the largest increase in OA O:C was chosen to represent the base simulation results presented in Fig. 4 and 5. In other words, the distribution of POA + SVOC mass in the SOM grid for the base simulation was chosen from amongst those used in the Monte-Carlo simulations that produced the most optimum comparison against measurements of OA NEMR and O:C for the Taylor Creek Fire. There is some indirect evidence for this POA + SVOC mass distribution in that the O:C dependence with OA mass loading seemed to agree qualitatively with a subset of laboratory and field observations of biomass burning OA (not shown).21,97
Results from simulations performed to assess the sensitivity to the OH estimates are presented in Fig. 7(a, d and g). The use of a power function fitted to the OH exposure data to determine OH concentrations (OH-power fit) produced results that were slightly higher compared to those from the base simulation. Similarly, if we assumed that the lower OH concentration after the first transect was also relevant to the time period before the first transect (OH-low; 0.97 × 106 molecules per cm3), the model predictions of OA NEMR and O:C were slightly higher than those from the base simulation. When using a constant OH concentration of 1.5 × 106 molecules per cm3 (OH-ambient) or the higher OH concentration from before the first transect (OH-high; 8.9 × 106 molecules per cm3) for the entire evolution, the model predicted a higher OA NEMR compared to predictions from the base, OH-power fit, and OH-low simulations. This was because the OH concentrations after the first transect in both of these instances were larger than those used in the base, OH-power fit, and OH-low simulations and these higher OH concentrations, which were ∼50% larger in OH-ambient and a factor of ∼10 larger in OH-high, promoted SOA formation.
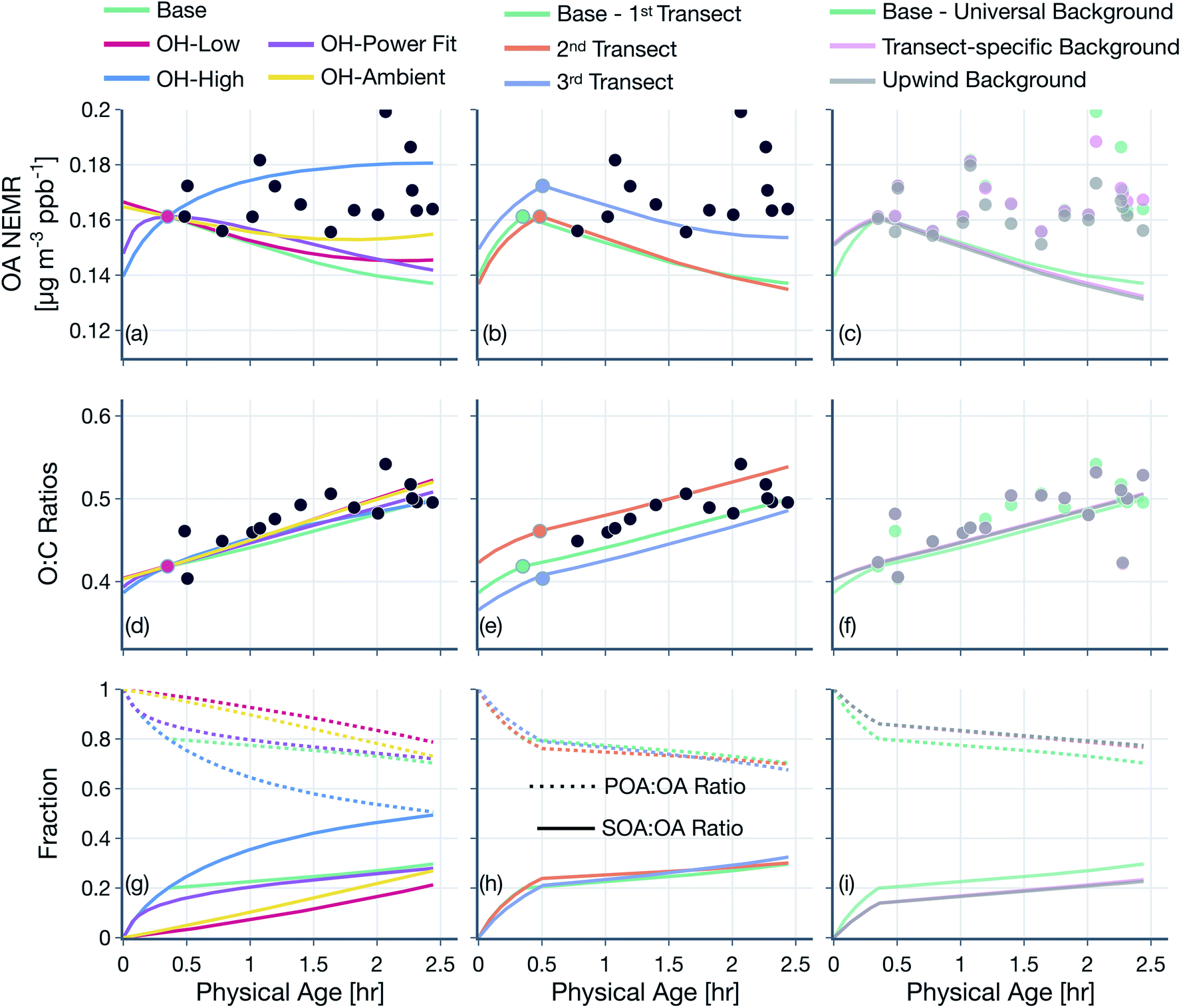 | ||
| Fig. 7 Predictions of (a–c) OA NEMR and (d–f) OA O:C from the SOM-TOMAS model (solid colored lines) compared against measurements (solid black circles) from the Taylor Creek Fire. (g–i) Predictions of the fractional contributions of POA and SOA to OA. Model predictions are shown for sensitivity simulations performed with varying assumptions for OH (a, d and g), reference transect (b, e and h), and (c, f and i) approach to performing background corrections. | ||
For the OH-high simulation, the increase in OA NEMR was found to be relatively consistent with the evolution in the observations indicating that the OH concentrations may continue to be elevated even after the first transect. However, the OH-high simulations overestimated the OA NEMR compared to the observations for the other Fires (Fig. S11†), where the base, OH-power fit, and OH-low simulations produced results that were more in line with the observations. As the Taylor Creek Fire dataset is the most Lagrangian amongst all Fires, the OH sensitivity simulation results presented here provide some evidence that our OH concentration estimates after the first transect (Table 1) may be biased low and would need to be revised in future work to be consistent with the higher OH concentrations estimated in earlier work (see Section 2.2 for a longer discussion). Interestingly, a higher OA NEMR in the OH-high simulation did not change predictions for OA O:C presumably because the additional SOA formed had an O:C similar to the existing OA's O:C. Overall, the model predictions appeared to be somewhat sensitive to the OH concentration inputs that produced a significant spread in the OA NEMR and POA-SOA splits but not so much in the OA O:C.
Qualitatively, the modeled trends in the OA NEMR and OA O:C were not very different when we assigned different transect sets (i.e., ‘2nd Transect’, ‘3rd Transect’) to be the ‘1st transect’ where model predictions of OA NEMR and OA O:C were anchored to observations; results are presented in Fig. 7(b, e and h). There appeared to be some tradeoff in the model-measurement comparisons for OA NEMR and OA O:C with the transect chosen. For instance, anchoring the model predictions to the information at the third transect seemed to produce better agreement with observations of OA NEMR but underestimated observations of OA O:C. The opposite was found to be true when anchoring the model predictions to the information at the second transect. Regardless of the variability in the OA NEMR and O:C, the POA-SOA splits were nearly identical between the three simulations. The use of ‘Transect-Specific’ or ‘Upwind’ data to calculate values to perform the background corrections did not seem to have any significant effect on model predictions; results are presented in Fig. 7(c, f and i).
4. Implications of the base simulation results
A summary of the model predictions from the base simulations for POA, SOA, and the SOA precursor NEMRs at t = 0 and the first and last transects, for all five transect sets is presented in Fig. 8. Some of these results have been presented in Fig. 4 and 5 earlier but this specific presentation of the results provides an opportunity to summarize the modeling effort and draw broader implications.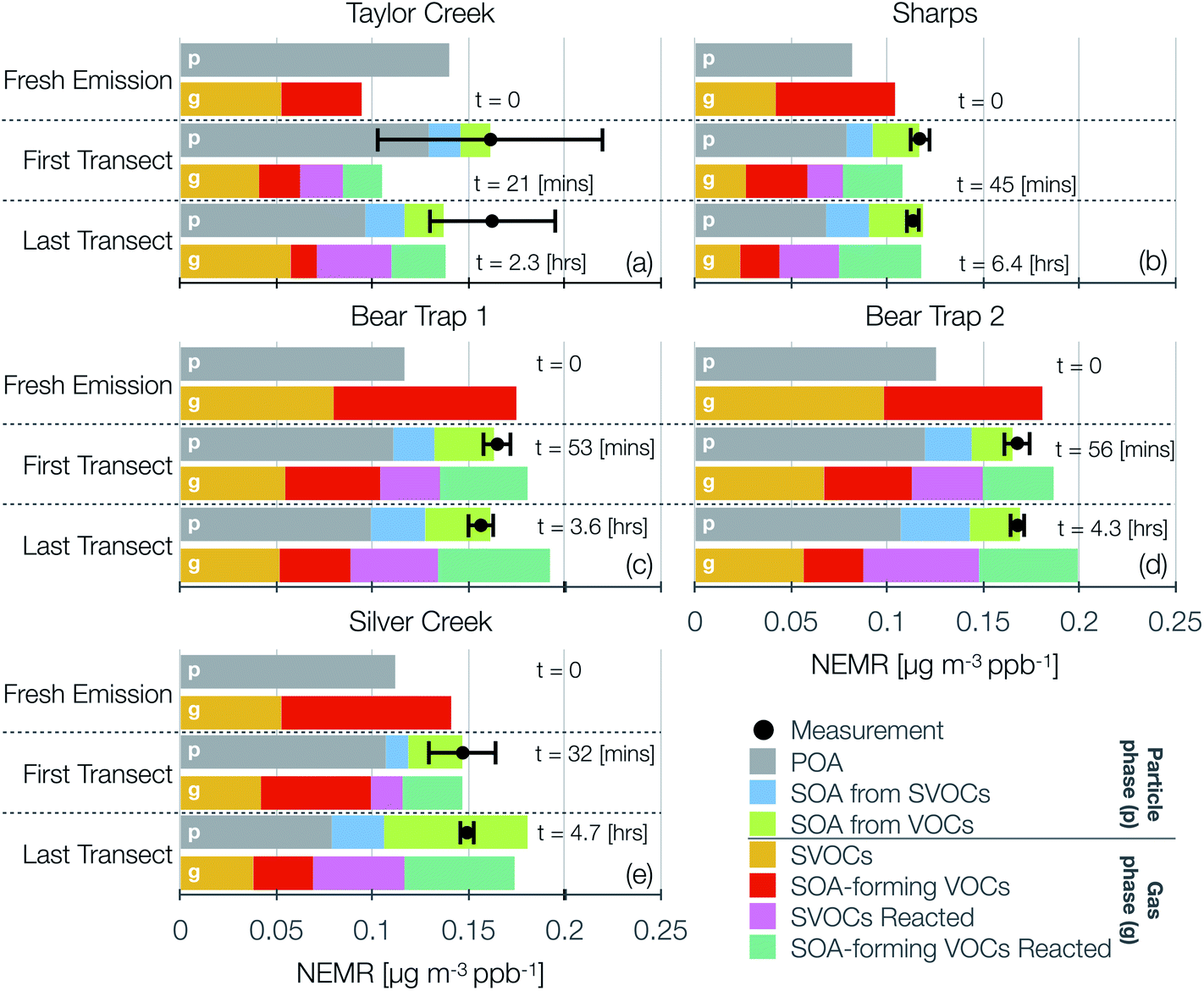 | ||
| Fig. 8 Model predictions of OA and SOA precursor NEMRs at t = 0 (‘Fresh Emission’) and the first and last transects for all five transect sets; (a) Taylor Creek, (b) Sharps, (c) Bear Trap 1, (d) Bear Trap 2, and (e) Silver Creek. Observations of OA NEMR are also presented for the first and last transects in black circles with error bars. | ||
First, fresh emissions of POA were found to be similar in magnitude to the sum of SOA-forming SVOCs and VOCs. For reference, for mobile sources, SOA precursor emissions are easily an order of magnitude larger than those for POA.29,101 This means that direct emissions of POA are likely to be an important constituent of smoke aerosol downwind of the fire, even as some fraction of it is lost to evaporation and surface reactions (not modeled in this work) and SVOCs and VOCs oxidize to form SOA and add to OA mass.
Second, up to half of the SOA precursor mass was rapidly oxidized to form SOA early on, which primarily resulted in an increase in the OA NEMR and O:C prior to the first transect. This result implies that a substantial fraction of the total SOA is actually formed very close to the fire and that the OA measured on the first aircraft transect is likely to be enhanced compared to the fresh OA emissions measured in laboratory experiments, after accounting for differences in gas/particle partitioning at different OA mass loadings. Furthermore, the rapid evolution of the OA system close to the fire is bound to alter the chemical, microphysical, and optical properties of smoke aerosols early on and confound comparisons of aerosol measurements at the first transect with similar measurements made on fresh emissions in laboratory environments.
Third, for most of the modeled and measured transect sets, except for the model prediction for the Silver Creek Fire, the OA NEMR did not vary much between the first and last transects, but there was a gradual change in the modeled OA composition with POA evaporation and SOA formation from SVOCs and VOCs. This is consistent with both theoretical and analytical findings in Bian et al.,62 Hodshire et al.,63 Palm et al.,33 and Liang et al.67 The change in composition implies that while the OA mass may remain constant, its atmospheric properties will continue to evolve with physical age. Furthermore, as the SOA precursors were heavily depleted and the OA mass concentrations at the last transect (5–30 μg m−3) were only marginally larger than the background concentrations (1–20 μg m−3), we postulate that the OA NEMR and O:C are unlikely to change dramatically with additional photochemical aging, any different than the changes experienced by background aerosol.
Fourth, by the last transect, SVOCs and VOCs contributed about equally to SOA formation in our wildfire plumes. The dominant SVOCs and VOCs contributing to SOA formation are very likely to be oxygenated organic compounds (e.g., sugars, heterocyclics, oxygenated aromatics), classes that are not explicitly included or are represented too coarsely in emissions inventories and chemical mechanisms, part of atmospheric models. Hence, the representation of SOA formation, as studied here, needs to be reflected for biomass burning sources in atmospheric models.
5. Conclusions, uncertainties, and directions for future work
In this study, we used a plume version of a kinetic model to simulate the dilution and physicochemical evolution of OA in wildfire plumes measured during the WE-CAN field campaign. The model was built on parameterizations developed from laboratory data, initialized using field measurements, and evaluated against the OA mass and composition measurements gathered from pseudo-Lagrangian transect sets. Our work suggests that it is very likely that dilution-driven evaporation of semivolatile POA and simultaneous photochemical production of SOA from SVOCs and VOCs explains the relative invariability in OA enhancements with photochemical age in observations of ambient wildfire plumes. These findings around OA evolution are consistent with the theoretical analyses presented by Bian et al.62 and Hodshire et al.63 as well as the analytical findings of Palm et al.33 and Liang et al.67 In addition, our model predictions indicate an important role for oxidation chemistry and rapid SOA formation before the first aircraft measurements, which is likely to be driven by higher-than-ambient OH concentrations in the wildfire plume (3 × 106 to 107 molecules per cm3). Notionally, for the fires studied here, we expect the OA measured within an hour after emission to be 80% POA and 20% SOA and 60% POA and 40% SOA after several additional hours of evolution. SOA precursor emissions for a few important organic classes (i.e., oxygenated aromatics, biogenics) appear to be systematically lower than those measured in laboratory experiments, and these lower emissions might partly explain the reduced propensity to form SOA in wildfire plumes. Finally, oxygenated compound classes such as sugars, heterocyclics, and oxygenated aromatics within SVOCs and VOCs are likely to serve as important precursors for SOA formation in wildfire plumes.Model results were found to be moderately sensitive to the treatment for POA and SVOCs. Hence, continued work to fully speciate the POA and SVOC mass to inform the volatility properties of POA and to identify surrogate species to model the oxidation chemistry of SVOCs will likely lead to improvements in model predictions. A point of contention for SVOCs is that they have not been explicitly considered when studying the SOA formation from biomass burning emissions in laboratory experiments.37,38 Akherati et al.38 observed that lower-volatility SOA precursors, especially in the SVOC range, were susceptible to loss in transfer ducts used to direct smoke emissions into environmental chambers. If this is indeed true, this might be one reason why SVOCs remain highly relevant for wildfire plumes but may not have been for laboratory experiments. Regardless, the chemical composition and oxidation chemistry of SVOCs relevant to SOA formation needs to be studied in the future.
We acknowledged a significant discrepancy in OH concentrations in the wildfire plume based on techniques used in this work and OH concentrations estimated in earlier work. In addition, model predictions were found to be somewhat sensitive to the OH concentrations assumed in the wildfire plume. Hence, ongoing and future work needs to focus on developing and applying analytical and modeling techniques to better estimate and evaluate OH concentrations in wildfire plumes. For instance, recently, Peng et al.74 calculated HOX (OH + HO2) production rates in wildfire plumes sampled during WE-CAN from the photolysis of nitrous acid (HONO), O3, and other smaller aldehydes (e.g., formaldehyde) and ozonolysis of alkenes. These HOX production rates could be used to inform OH concentrations. Similarly, OH concentrations could be constrained by applying explicit gas-phase chemical mechanisms to reproduce the time-dependent evolution of VOCs and their oxidation products in wildfire plumes.
In addition to the uncertainties alluded to in this work, there are several additional aspects to consider while modeling the OA evolution in wildfire plumes. First, the model parameterizations in this work (e.g., POA volatility, SOA parameters) were based on simpler model systems studied in laboratory environments and these parameters may not accurately represent the processes in real wildfire plumes. Most obviously, differences in the fuel complex, burn conditions, combustion efficiency, and environmental conditions (e.g., temperature, relative humidity) and regimes (e.g., photolytic rates, NOX) could produce differences in the emissions, chemistry, and properties of OA and OA precursors between the laboratory and the field.34,68 A more specific example is that the parameters that we used to model the SOA formation from SVOCs, heterocyclics, and oxygenated aromatics (precursor classes that contributed the most to SOA production) came from environmental chamber experiments performed on a handful of surrogate species (phenol, guaiacol, and syringol, 2-methylfuran + dimethylfuran, and naphthalene, respectively) under relatively dry (relative humidity < 20%) and high NOX conditions (200–800 ppbv).86,94,102 While this extrapolation is typical for how laboratory data are translated into parameters for use in atmospheric models, these laboratory versus field differences need to be considered when evaluating model predictions against measurements.
Second, the physicochemical evolution modeled prior to the first transect remains extremely uncertain as there are no observations to evaluate those model predictions. Aircraft campaigns in the future should aim to characterize the near-field evolution in the hour after emission by performing transects closer to the fire when conditions allow. Moreover, campaigns should also accommodate repeated sampling of the near-field to assess changes in emissions over the same timescales used to perform the transect set. Any emissions changes would then need to be considered in interpreting the plume evolution inferred from the transect dataset.
Third, the current version of the SOM-TOMAS model does not simulate the photolysis or aqueous chemistry of OA or OA precursors. Photolysis has been shown to be an important loss pathway for SOA formed from monoterpenes.103,104 Oxygenated aromatics that include phenols, methoxyphenols, and phenolic carbonyls, after uptake into aerosol water, can participate in aqueous reactions to form low-volatility and light absorbing SOA.26,105 Both of these chemical processes are likely occurring in wildfire plumes and, hence, need to be included in future modeling efforts.
Fourth, the model initialization and evaluation in this work only relied on a subset of measurements made during WE-CAN. Future work could certainly benefit from leveraging an extended set of measurements gathered during WE-CAN and similar field campaigns focused on studying biomass burning emissions (e.g., BBOP,106 LASIC,107 FIREX-AQ (https://csl.noaa.gov/projects/firex-aq/)). For example, model predictions could be compared against measurements of the evolving composition (e.g., oligomers), size distribution, and thermodynamic (e.g., volatility), optical (e.g., scattering, extinction), and climate (e.g., cloud condensation nuclei) properties.
Fifth, Peng et al.74 and Hodshire et al.72 were recently able to study the distinct evolution of trace species resolved over the width of the wildfire plume. Both found evidence for increased photochemical activity near the edges and wings of the plume since these regions diluted much faster and were less optically dense compared to the core of the plume. Modeling in the future could use the information inherent in gradients within the transect to constrain the OA evolution under varying dilution and photochemical conditions.
And finally, the modeling in this work focused on simulating the plume evolution in a subset of large, daytime fires in the western US. In the future, the model will need to be applied to study a diversity of fires in terms of size, fuels, and geography (e.g., agricultural fires in the southeast US) to assess the broader applicability of the findings presented in this work.
Data availability
Field campaign data from WE-CAN can be found at the permanent archival link: https://data.eol.ucar.edu/master_lists/generated/we-can/. The latest version of the SOM-TOMAS plume model along with the simulation data will be shared upon request.Author contributions
AA, ALH, JRP, and SHJ designed the study. AA and YH developed the model and AA performed the simulations and analyzed the data. LAG, DKF, SMK, WP, LH, EVF, CNJ, AHG, TLC, FF, JMR, and DWT facilitated access to the field data and its appropriate use in the model. AA, JRP, and SHJ wrote the paper with contributions from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Oceanic and Atmospheric Administration (NA17OAR4310003, NA17OAR4310001, NA17OAR4310010) and the U.S. Department of Energy (DOE), Office of Science (DE-SC0017975). The WE-CAN field campaign was supported by the National Science Foundation through grants AGS-1650786 and AGS-1650275. This publication was partly developed under Assistance Agreement No. R840008 awarded by the U.S. Environmental Protection Agency to SHJ. It has not been formally reviewed by EPA. The views expressed in this document are solely those of the authors and do not necessarily reflect those of the Agency. EPA does not endorse any products or commercial services mentioned in this publication.References
- M. O. Andreae and P. Merlet, Emission of Trace Gases and Aerosols from Biomass Burning. Global Biogeochem, Cycles, 2001, 15(4), 955–966, DOI:10.1029/2000GB001382.
- J. S. Reid, R. Koppmann, T. F. Eck and D. P. Eleuterio, A Review of Biomass Burning Emissions Part II: Intensive Physical Properties of Biomass Burning Particles, Atmos. Chem. Phys., 2005, 5(3), 799–825, DOI:10.5194/acp-5-799-2005.
- G. R. van der Werf, J. T. Randerson, L. Giglio, G. J. Collatz, M. Mu, P. S. Kasibhatla, D. C. Morton, R. S. DeFries, Y. Jin and T. T. van Leeuwen, Global Fire Emissions and the Contribution of Deforestation, Savanna, Forest, Agricultural, and Peat Fires (1997–2009), Atmos. Chem. Phys., 2010, 10(23), 11707–11735, DOI:10.5194/acp-10-11707-2010.
- S. K. Akagi, R. J. Yokelson, C. Wiedinmyer, M. J. Alvarado, J. S. Reid, T. Karl, J. D. Crounse and P. O. Wennberg, Emission Factors for Open and Domestic Biomass Burning for Use in Atmospheric Models, Atmos. Chem. Phys., 2011, 11(9), 4039–4072, DOI:10.5194/acp-11-4039-2011.
- R. J. Yokelson, I. R. Burling, J. B. Gilman, C. Warneke, C. E. Stockwell, J. de Gouw, S. K. Akagi, S. P. Urbanski, P. Veres, J. M. Roberts, W. C. Kuster, J. Reardon, D. W. T. Griffith, T. J. Johnson, S. Hosseini, J. W. Miller, D. R. Cocker III, H. Jung and D. R. Weise, Coupling Field and Laboratory Measurements to Estimate the Emission Factors of Identified and Unidentified Trace Gases for Prescribed Fires, Atmos. Chem. Phys., 2013, 13(1), 89–116, DOI:10.5194/acp-13-89-2013.
- M. O. Andreae, Emission of Trace Gases and Aerosols from Biomass Burning – an Updated Assessment, Atmos. Chem. Phys., 2019, 19(13), 8523–8546, DOI:10.5194/acp-19-8523-2019.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Evolution of Organic Aerosols in the Atmosphere, Science, 2009, 326(5959), 1525–1529, DOI:10.1126/science.1180353.
- G. P. Schill, K. D. Froyd, H. Bian, A. Kupc, C. Williamson, C. A. Brock, E. Ray, R. S. Hornbrook, A. J. Hills, E. C. Apel, M. Chin, P. R. Colarco and D. M. Murphy, Widespread Biomass Burning Smoke throughout the Remote Troposphere. Nat, Geosci, 2020, 13(6), 422–427, DOI:10.1038/s41561-020-0586-1.
- E. Ramnarine, J. K. Kodros, A. L. Hodshire, C. R. Lonsdale, M. J. Alvarado and J. R. Pierce, Effects of near-Source Coagulation of Biomass Burning Aerosols on Global Predictions of Aerosol Size Distributions and Implications for Aerosol Radiative Effects, Atmos. Chem. Phys., 2019, 19(9), 6561–6577, DOI:10.5194/acp-19-6561-2019.
- P. V. Hobbs, J. S. Reid, R. A. Kotchenruther, R. J. Ferek and R. Weiss, Direct Radiative Forcing by Smoke from Biomass Burning, Science, 1997, 275(5307), 1776–1778, DOI:10.1126/science.275.5307.1777.
- X. Yue, L. J. Mickley, J. A. Logan and J. O. Kaplan, Ensemble Projections of Wildfire Activity and Carbonaceous Aerosol Concentrations over the Western United States in the Mid-21st Century. Atmos, Environ, 2013, 77, 767–780, DOI:10.1016/j.atmosenv.2013.06.003.
- K. O'Dell, B. Ford, E. V. Fischer and J. R. Pierce, Contribution of Wildland-Fire Smoke to US PM2.5 and Its Influence on Recent Trends, Environ. Sci. Technol., 2019, 53(4), 1797–1804, DOI:10.1021/acs.est.8b05430.
- C. D. McClure and D. A. Jaffe, US Particulate Matter Air Quality Improves except in Wildfire-Prone Areas, Proc. Natl. Acad. Sci. U. S. A, 2018, 115(31), 7901–7906, DOI:10.1073/pnas.1804353115.
- B. Ford, M. Val Martin, S. E. Zelasky, E. V. Fischer, S. C. Anenberg, C. L. Heald and J. R. Pierce, Future Fire Impacts on Smoke Concentrations, Visibility, and Health in the Contiguous United States, GeoHealth, 2018, 2(8), 229–247, DOI:10.1029/2018GH000144.
- J. Chen, C. Li, Z. Ristovski, A. Milic, Y. Gu, M. S. Islam, S. Wang, J. Hao, H. Zhang, C. He, H. Guo, H. Fu, B. Miljevic, L. Morawska, P. Thai, Y. F. Lam, G. Pereira, A. Ding, X. Huang and U. C. Dumka, A Review of Biomass Burning: Emissions and Impacts on Air Quality, Health and Climate in China, Sci. Total Environ, 2017, 579, 1000–1034, DOI:10.1016/j.scitotenv.2016.11.025.
- D. V. Spracklen, L. J. Mickley, J. A. Logan, R. C. Hudman, R. Yevich, M. D. Flannigan and A. L. Westerling, Impacts of Climate Change from 2000 to 2050 on Wildfire Activity and Carbonaceous Aerosol Concentrations in the Western United States, J. Geophys. Res., 2009, 114(D20) DOI:10.1029/2008jd010966.
- T. S. Carter, C. L. Heald, J. L. Jimenez, P. Campuzano-Jost, Y. Kondo, N. Moteki, J. P. Schwarz, C. Wiedinmyer, A. S. Darmenov, A. M. da Silva and J. W. Kaiser, How Emissions Uncertainty Influences the Distribution and Radiative Impacts of Smoke from Fires in North America, Atmos. Chem. Phys., 2020, 20(4), 2073–2097, DOI:10.5194/acp-20-2073-2020.
- M. Shrivastava, C. D. Cappa, J. Fan, A. H. Goldstein, A. B. Guenther, J. L. Jimenez, C. Kuang, A. Laskin, S. T. Martin, N. L. Ng, T. Petaja, J. R. Pierce, P. J. Rasch, P. Roldin, J. H. Seinfeld, J. Shilling, J. N. Smith, J. A. Thornton, R. Volkamer, J. Wang, D. R. Worsnop, R. A. Zaveri, A. Zelenyuk and Q. Zhang, Recent Advances in Understanding Secondary Organic Aerosol: Implications for Global Climate Forcing: Advances in Secondary Organic Aerosol, Rev. Geophys., 2017, 55(2), 509–559, DOI:10.1002/2016RG000540.
- K. A. Lewis, W. P. Arnott, H. Moosmüller, R. K. Chakrabarty, C. M. Carrico, S. M. Kreidenweis, D. E. Day, W. C. Malm, A. Laskin, J. L. Jimenez, I. M. Ulbrich, J. A. Huffman, T. B. Onasch, A. Trimborn, L. Liu and M. I. Mishchenko, Reduction in Biomass Burning Aerosol Light Absorption upon Humidification: Roles of Inorganically-Induced Hygroscopicity, Particle Collapse, and Photoacoustic Heat and Mass Transfer, Atmos. Chem. Phys., 2009, 9(22), 8949–8966, DOI:10.5194/acp-9-8949-2009.
- L. A. Garofalo, M. A. Pothier, E. J. T. Levin, T. Campos, S. M. Kreidenweis and D. K. Farmer, Emission and Evolution of Submicron Organic Aerosol in Smoke from Wildfires in the Western United States, ACS Earth Space Chem., 2019, 3(7), 1237–1247, DOI:10.1021/acsearthspacechem.9b00125.
- J. A. Huffman, K. S. Docherty, C. Mohr, M. J. Cubison, I. M. Ulbrich, P. J. Ziemann, T. B. Onasch and J. L. Jimenez, Chemically-Resolved Volatility Measurements of Organic Aerosol from Different Sources, Environ. Sci. Technol., 2009, 43(14), 5351–5357, DOI:10.1021/es803539d.
- A. A. May, E. J. T. Levin, C. J. Hennigan, I. Riipinen, T. Lee, J. L. Collett Jr, J. L. Jimenez, S. M. Kreidenweis and A. L. Robinson, Gas-Particle Partitioning of Primary Organic Aerosol Emissions: 3. Biomass Burning, ACS Earth Space Chem., 2013, 118(19), 327–338, DOI:10.1002/jgrd.50828.
- L. E. Hatch, A. Rivas-Ubach, C. N. Jen, M. Lipton, A. H. Goldstein and K. C. Barsanti, Measurements of I/SVOCs in Biomass-Burning Smoke Using Solid-Phase Extraction Disks and Two-Dimensional Gas Chromatography, Atmos. Chem. Phys., 2018, 18(24), 17801–17817, DOI:10.5194/acp-18-17801-2018.
- C. J. Hennigan, A. P. Sullivan, J. L. Collett Jr and A. L. Robinson, Levoglucosan Stability in Biomass Burning Particles Exposed to Hydroxyl Radicals: Levoglucosan Stability, Aerosol. Geophys. Res. Lett., 2010, 37(9) DOI:10.1029/2010gl043088.
- C. N. Jen, L. E. Hatch, V. Selimovic, R. J. Yokelson, R. Weber, A. E. Fernandez, N. M. Kreisberg, K. C. Barsanti and A. H. Goldstein, Speciated and Total Emission Factors of Particulate Organics from Burning Western US Wildland Fuels and Their Dependence on Combustion Efficiency, Atmos. Chem. Phys., 2019, 19(2), 1013–1026, DOI:10.5194/acp-19-1013-2019.
- J. D. Smith, V. Sio, L. Yu, Q. Zhang and C. Anastasio, Secondary Organic Aerosol Production from Aqueous Reactions of Atmospheric Phenols with an Organic Triplet Excited State, Environ. Sci. Technol., 2014, 48(2), 1049–1057, DOI:10.1021/es4045715.
- L. E. Hatch, R. J. Yokelson, C. E. Stockwell, P. R. Veres, I. J. Simpson, D. R. Blake, J. J. Orlando and K. C. Barsanti, Multi-Instrument Comparison and Compilation of Non-Methane Organic Gas Emissions from Biomass Burning and Implications for Smoke-Derived Secondary Organic Aerosol Precursors, Atmos. Chem. Phys., 2017, 17(2), 1471–1489, DOI:10.5194/acp-17-1471-2017.
- A. R. Koss, K. Sekimoto, J. B. Gilman, V. Selimovic, M. M. Coggon, K. J. Zarzana, B. Yuan, B. M. Lerner, S. S. Brown, J. L. Jimenez, J. Krechmer, J. M. Roberts, C. Warneke, R. J. Yokelson and J. Gouw, de. Non-Methane Organic Gas Emissions from Biomass Burning: Identification, Quantification, and Emission Factors from PTR-ToF during the FIREX 2016 Laboratory Experiment, Atmos. Chem. Phys., 2018, 18(5), 3299–3319, DOI:10.5194/acp-18-3299-2018.
- S. H. Jathar, T. D. Gordon, C. J. Hennigan, H. O. T. Pye, G. Pouliot, P. J. Adams, N. M. Donahue and A. L. Robinson, Unspeciated Organic Emissions from Combustion Sources and Their Influence on the Secondary Organic Aerosol Budget in the United States, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(29), 10473–10478, DOI:10.1073/pnas.1323740111.
- C. E. Stockwell, P. R. Veres, J. Williams and R. J. Yokelson, Characterization of Biomass Burning Emissions from Cooking Fires, Peat, Crop Residue, and Other Fuels with High-Resolution Proton-Transfer-Reaction Time-of-Flight Mass Spectrometry, Atmos. Chem. Phys., 2015, 15(2), 845–865, DOI:10.5194/acp-15-845-2015.
- L. E. Hatch, W. Luo, J. F. Pankow, R. J. Yokelson, C. E. Stockwell and K. C. Barsanti, Identification and Quantification of Gaseous Organic Compounds Emitted from Biomass Burning Using Two-Dimensional Gas Chromatography–time-of-Flight Mass Spectrometry, Atmos. Chem. Phys., 2015, 15(4), 1865–1899, DOI:10.5194/acp-15-1865-2015.
- K. Sekimoto, A. R. Koss, J. B. Gilman, V. Selimovic, M. M. Coggon, K. J. Zarzana, B. Yuan, B. M. Lerner, S. S. Brown, C. Warneke, R. J. Yokelson, J. M. Roberts and J. A. de Gouw, High- and Low-Temperature Pyrolysis Profiles Describe Volatile Organic Compound Emissions from Western US Wildfire Fuels, Atmos. Chem. Phys., 2018, 18(13), 9263–9281, DOI:10.5194/acp-18-9263-2018.
- B. B. Palm, Q. Peng, C. D. Fredrickson, B. H. Lee, L. A. Garofalo, M. A. Pothier, S. M. Kreidenweis, D. K. Farmer, R. P. Pokhrel, Y. Shen, S. M. Murphy, W. Permar, L. Hu, T. L. Campos, S. R. Hall, K. Ullmann, X. Zhang, F. Flocke, E. V. Fischer and J. A. Thornton, Quantification of Organic Aerosol and Brown Carbon Evolution in Fresh Wildfire Plumes, Proc. Natl. Acad. Sci. U. S. A, 2020, 117(47), 29469–29477, DOI:10.1073/pnas.2012218117.
- W. Permar, Q. Wang, V. Selimovic, C. Wielgasz, R. J. Yokelson, R. S. Hornbrook, A. J. Hills, E. C. Apel, I.-T. Ku, Y. Zhou, B. C. Sive, A. P. Sullivan, J. L. Collett Jr, T. L. Campos, B. B. Palm, Q. Peng, J. A. Thornton, L. A. Garofalo, D. K. Farmer, S. M. Kreidenweis, E. J. T. Levin, P. J. DeMott, F. Flocke, E. V. Fischer and L. Hu, Emissions of Trace Organic Gases from Western U.S. Wildfires Based on WE-CAN Aircraft Measurements, J. Geophys. Res., 2021, 126(11), e2020JD033838, DOI:10.1029/2020jd033838.
- E. A. Bruns, J. G. Slowik, I. El Haddad, D. Kilic, F. Klein, J. Dommen, B. Temime-Roussel, N. Marchand, U. Baltensperger and A. S. H. Prévôt, Characterization of Gas-Phase Organics Using Proton Transfer Reaction Time-of-Flight Mass Spectrometry: Fresh and Aged Residential Wood Combustion Emissions, Atmos. Chem. Phys., 2017, 17(1), 705–720, DOI:10.5194/acp-17-705-2017.
- G. Stefenelli, J. Jiang, A. Bertrand, E. A. Bruns, S. M. Pieber, U. Baltensperger, N. Marchand, S. Aksoyoglu, A. S. H. Prévôt, J. G. Slowik and I. E. Haddad, Secondary Organic Aerosol Formation from Smoldering and Flaming Combustion of Biomass: A Box Model Parametrization Based on Volatility Basis Set, Atmos. Chem. Phys., 2019, 19(17), 11461–11484, DOI:10.5194/acp-19-11461-2019.
- A. T. Ahern, E. S. Robinson, D. S. Tkacik, R. Saleh, L. E. Hatch, K. C. Barsanti, C. E. Stockwell, R. J. Yokelson, A. A. Presto, A. L. Robinson, R. C. Sullivan and N. M. Donahue, Production of Secondary Organic Aerosol During Aging of Biomass Burning Smoke From Fresh Fuels and Its Relationship to VOC Precursors, J. Geophys. Res. D: Atmos, 2019, 124(6), 3583–3606, DOI:10.1029/2018JD029068.
- A. Akherati, Y. He, M. M. Coggon, A. R. Koss, A. L. Hodshire, K. Sekimoto, C. Warneke, J. de Gouw, L. Yee, J. H. Seinfeld, T. B. Onasch, S. C. Herndon, W. B. Knighton, C. D. Cappa, M. J. Kleeman, C. Y. Lim, J. H. Kroll, J. R. Pierce and S. H. Jathar, Oxygenated Aromatic Compounds Are Important Precursors of Secondary Organic Aerosol in Biomass-Burning Emissions, Environ. Sci. Technol., 2020 DOI:10.1021/acs.est.0c01345.
- C. Y. Lim, D. H. Hagan, M. M. Coggon, A. R. Koss, K. Sekimoto, J. de Gouw, C. Warneke, C. D. Cappa and J. H. Kroll, Secondary Organic Aerosol Formation from the Laboratory Oxidation of Biomass Burning Emissions, Atmos. Chem. Phys., 2019, 19(19), 12797–12809, DOI:10.5194/acp-19-12797-2019.
- A. L. Hodshire, A. Akherati, M. J. Alvarado, B. Brown-Steiner, S. H. Jathar, J. L. Jimenez, S. M. Kreidenweis, C. R. Lonsdale, T. B. Onasch, A. M. Ortega and J. R. Pierce, Aging Effects on Biomass Burning Aerosol Mass and Composition: A Critical Review of Field and Laboratory Studies, Environ. Sci. Technol., 2019, 53(17), 10007–10022, DOI:10.1021/acs.est.9b02588.
- C. J. Hennigan, M. A. Miracolo, G. J. Engelhart, A. A. May, A. A. Presto, T. Lee, A. P. Sullivan, G. R. McMeeking, H. Coe, C. E. Wold, W.-M. Hao, J. B. Gilman, W. C. Kuster, J. Gouw, B. A. de Schichtel, J. L. Collett Jr, S. M. Kreidenweis and A. L. Robinson, Chemical and Physical Transformations of Organic Aerosol from the Photo-Oxidation of Open Biomass Burning Emissions in an Environmental Chamber, Atmos. Chem. Phys., 2011, 11(15), 7669–7686, DOI:10.5194/acp-11-7669-2011.
- A. M. Ortega, D. A. Day, M. J. Cubison, W. H. Brune, D. Bon, J. A. de Gouw and J. L. Jimenez, Secondary Organic Aerosol Formation and Primary Organic Aerosol Oxidation from Biomass-Burning Smoke in a Flow Reactor during FLAME-3, Atmos. Chem. Phys., 2013, 13(22), 11551–11571, DOI:10.5194/acp-13-11551-2013.
- D. S. Tkacik, E. S. Robinson, A. Ahern, R. Saleh, C. Stockwell, P. Veres, I. J. Simpson, S. Meinardi, D. R. Blake, R. J. Yokelson, A. A. Presto, R. C. Sullivan, N. M. Donahue and A. L. Robinson, A Dual-Chamber Method for Quantifying the Effects of Atmospheric Perturbations on Secondary Organic Aerosol Formation from Biomass Burning Emissions: Investigation of Biomass Burning SOA, J. Geophys. Res. D: Atmos, 2017, 122(11), 6043–6058, DOI:10.1002/2016JD025784.
- P. V. Hobbs, P. Sinha, R. J. Yokelson, T. J. Christian, D. R. Blake, S. Gao, T. W. Kirchstetter, T. Novakov and P. Pilewskie, Evolution of Gases and Particles from a Savanna Fire in South Africa, J. Geophys. Res. D: Atmos, 2003, 108(D13) DOI:10.1029/2002JD002352.
- M. J. Alvarado and R. G. Prinn, Formation of Ozone and Growth of Aerosols in Young Smoke Plumes from Biomass Burning: 1. Lagrangian Parcel Studies, J. Geophys. Res, 2009, 114(D9), D09307, DOI:10.1029/2008JD011144.
- J. C. Liu, L. J. Mickley, M. P. Sulprizio, F. Dominici, X. Yue, K. Ebisu, G. B. Anderson, R. F. A. Khan, M. A. Bravo and M. L. Bell, Particulate Air Pollution from Wildfires in the Western US under Climate Change. Clim, Change, 2016, 138(3), 655–666, DOI:10.1007/s10584-016-1762-6.
- P. F. DeCarlo, E. J. Dunlea, J. R. Kimmel, A. C. Aiken, D. Sueper, J. Crounse, P. O. Wennberg, L. Emmons, Y. Shinozuka, A. Clarke, J. Zhou, J. Tomlinson, D. R. Collins, D. Knapp, A. J. Weinheimer, D. D. Montzka, T. Campos and J. L. Jimenez, Fast Airborne Aerosol Size and Chemistry Measurements above Mexico City and Central Mexico during the MILAGRO Campaign, Atmos. Chem. Phys., 2008, 8(14), 4027–4048, DOI:10.5194/acp-8-4027-2008.
- R. J. Yokelson, J. D. Crounse, P. F. DeCarlo, T. Karl, S. Urbanski, E. Atlas, T. Campos, Y. Shinozuka, V. Kapustin, A. D. Clarke, A. Weinheimer, D. J. Knapp, D. D. Montzka, J. Holloway, P. Weibring, F. Flocke, W. Zheng, D. Toohey, P. O. Wennberg, C. Wiedinmyer, L. Mauldin, A. Fried, D. Richter, J. Walega, J. L. Jimenez, K. Adachi, P. R. Buseck, S. R. Hall and R. Shetter, Emissions from Biomass Burning in the Yucatan, Atmos. Chem. Phys., 2009, 9(15), 5785–5812, DOI:10.5194/acp-9-5785-2009.
- M. D. Jolleys, H. Coe, G. McFiggans, G. Capes, J. D. Allan, J. Crosier, P. I. Williams, G. Allen, K. N. Bower, J. L. Jimenez, L. M. Russell, M. Grutter and D. Baumgardner, Characterizing the Aging of Biomass Burning Organic Aerosol by Use of Mixing Ratios: A Meta-Analysis of Four Regions, Environ. Sci. Technol., 2012, 46(24), 13093–13102, DOI:10.1021/es302386v.
- V. Vakkari, V.-M. Kerminen, J. P. Beukes, P. Tiitta, P. G. van Zyl, M. Josipovic, A. D. Venter, K. Jaars, D. R. Worsnop, M. Kulmala and L. Laakso, Rapid Changes in Biomass Burning Aerosols by Atmospheric Oxidation, Geophys. Res. Lett., 2014, 41(7), 2644–2651, DOI:10.1002/2014GL059396.
- V. Vakkari, J. P. Beukes, M. Dal Maso, M. Aurela, M. Josipovic and P. G. van Zyl, Major Secondary Aerosol Formation in Southern African Open Biomass Burning Plumes. Nat, Geosci, 2018, 11(8), 580–583, DOI:10.1038/s41561-018-0170-0.
- A. A. May, T. Lee, G. R. McMeeking, S. Akagi, A. P. Sullivan, S. Urbanski, R. J. Yokelson and S. M. Kreidenweis, Observations and Analysis of Organic Aerosol Evolution in Some Prescribed Fire Smoke Plumes, Atmos. Chem. Phys., 2015, 15(11), 6323–6335, DOI:10.5194/acp-15-6323-2015.
- S. K. Akagi, J. S. Craven, J. W. Taylor, G. R. McMeeking, R. J. Yokelson, I. R. Burling, S. P. Urbanski, C. E. Wold, J. H. Seinfeld, H. Coe, M. J. Alvarado and D. R. Weise, Evolution of Trace Gases and Particles Emitted by a Chaparral Fire in California, Atmos. Chem. Phys., 2012, 12(3), 1397–1421, DOI:10.5194/acp-12-1397-2012.
- M. J. Cubison, A. M. Ortega, P. L. Hayes, D. K. Farmer, D. Day, M. J. Lechner, W. H. Brune, E. Apel, G. S. Diskin, J. A. Fisher, H. E. Fuelberg, A. Hecobian, D. J. Knapp, T. Mikoviny, D. Riemer, G. W. Sachse, W. Sessions, R. J. Weber, A. J. Weinheimer, A. Wisthaler and J. L. Jimenez, Effects of Aging on Organic Aerosol from Open Biomass Burning Smoke in Aircraft and Laboratory Studies, Atmos. Chem. Phys., 2011, 11(23), 12049–12064, DOI:10.5194/acp-11-12049-2011.
- J. Brito, L. V. Rizzo, W. T. Morgan, H. Coe, B. Johnson, J. Haywood, K. Longo, S. Freitas, M. O. Andreae and P. Artaxo, Ground-Based Aerosol Characterization during the South American Biomass Burning Analysis (SAMBBA) Field Experiment, Atmos. Chem. Phys., 2014, 14(22), 12069–12083, DOI:10.5194/acp-14-12069-2014.
- W. T. Morgan, J. D. Allan, S. Bauguitte, E. Darbyshire, M. J. Flynn, J. Lee, D. Liu, B. Johnson, J. Haywood, K. M. Longo, P. E. Artaxo and H. Coe, Transformation and Ageing of Biomass Burning Carbonaceous Aerosol over Tropical South America from Aircraft in Situ Measurements during SAMBBA, Atmos. Chem. Phys., 2020, 20(9), 5309–5326, DOI:10.5194/acp-20-5309-2020.
- H. Forrister, J. Liu, E. Scheuer, J. Dibb, L. Ziemba, K. L. Thornhill, B. Anderson, G. Diskin, A. E. Perring, J. P. Schwarz, P. Campuzano-Jost, D. A. Day, B. B. Palm, J. L. Jimenez, A. Nenes and R. J. Weber, Evolution of Brown Carbon in Wildfire Plumes, Geophys. Res. Lett., 2015, 42(11), 4623–4630, DOI:10.1002/2015GL063897.
- S. Collier, S. Zhou, T. B. Onasch, D. A. Jaffe, L. Kleinman, A. J. Sedlacek, N. L. Briggs, J. Hee, E. Fortner, J. E. Shilling, D. Worsnop, R. J. Yokelson, C. Parworth, X. Ge, J. Xu, Z. Butterfield, D. Chand, M. K. Dubey, M. S. Pekour, S. Springston and Q. Zhang, Regional Influence of Aerosol Emissions from Wildfires Driven by Combustion Efficiency: Insights from the BBOP Campaign, Environ. Sci. Technol., 2016, 50(16), 8613–8622, DOI:10.1021/acs.est.6b01617.
- G. Capes, B. Johnson, G. McFiggans, P. I. Williams, J. Haywood and H. Coe, Aging of Biomass Burning Aerosols over West Africa: Aircraft Measurements of Chemical Composition, Microphysical Properties, and Emission Ratios, J. Geophys. Res. D: Atmos, 2008, 113(D23) DOI:10.1029/2008JD009845.
- M. D. Jolleys, H. Coe, G. McFiggans, J. W. Taylor, S. J. O'Shea, M. Le Breton, S. J.-B. Bauguitte, S. Moller, P. Di Carlo, E. Aruffo, P. I. Palmer, J. D. Lee, C. J. Percival and M. W. Gallagher, Properties and Evolution of Biomass Burning Organic Aerosol from Canadian Boreal Forest Fires, Atmos. Chem. Phys., 2015, 15(6), 3077–3095, DOI:10.5194/acp-15-3077-2015.
- N. L. Ng, M. R. Canagaratna, J. L. Jimenez, P. S. Chhabra, J. H. Seinfeld and D. R. Worsnop, Changes in Organic Aerosol Composition with Aging Inferred from Aerosol Mass Spectra, Atmos. Chem. Phys., 2011, 11(13), 6465–6474, DOI:10.5194/acp-11-6465-2011.
- Q. Bian, S. H. Jathar, J. K. Kodros, K. C. Barsanti, L. E. Hatch, A. A. May, S. M. Kreidenweis and J. R. Pierce, Secondary Organic Aerosol Formation in Biomass-Burning Plumes: Theoretical Analysis of Lab Studies and Ambient Plumes, Atmos. Chem. Phys., 2017, 17(8), 5459–5475, DOI:10.5194/acp-17-5459-2017.
- A. L. Hodshire, Q. Bian, E. Ramnarine, C. R. Lonsdale, M. J. Alvarado, S. M. Kreidenweis, S. H. Jathar and J. R. Pierce, More Than Emissions and Chemistry: Fire Size, Dilution, and Background Aerosol Also Greatly Influence Near-Field Biomass Burning Aerosol Aging, J. Geophys. Res. D: Atmos, 2019, 124(10), 5589–5611, DOI:10.1029/2018JD029674.
- A. Matsunaga and P. J. Ziemann, Gas-Wall Partitioning of Organic Compounds in a Teflon Film Chamber and Potential Effects on Reaction Product and Aerosol Yield Measurements, Aerosol Sci. Technol., 2010, 44(10), 881–892, DOI:10.1080/02786826.2010.501044.
- X. Zhang, C. D. Cappa, S. H. Jathar, R. C. McVay, J. J. Ensberg, M. J. Kleeman and J. H. Seinfeld, Influence of Vapor Wall Loss in Laboratory Chambers on Yields of Secondary Organic Aerosol, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(16), 5802–5807, DOI:10.1073/pnas.1404727111.
- J. E. Krechmer, D. Pagonis, P. J. Ziemann and J. L. Jimenez, Quantification of Gas-Wall Partitioning in Teflon Environmental Chambers Using Rapid Bursts of Low-Volatility Oxidized Species Generated in Situ, Environ. Sci. Technol., 2016, 50(11), 5757–5765, DOI:10.1021/acs.est.6b00606.
- Y. Liang, C. N. Jen, R. J. Weber, P. K. Misztal and A. H. Goldstein, Chemical composition of PM2.5 in October 2017 Northern California wildfire plumes, Atmos. Chem. Phys., 2021, 21, 5719–5737, DOI:10.5194/acp-21-5719-2021.
- J. Lindaas, I. B. Pollack, L. A. Garofalo, M. A. Pothier, D. K. Farmer, S. M. Kreidenweis, T. L. Campos, F. Flocke, A. J. Weinheimer, D. D. Montzka, G. S. Tyndall, B. B. Palm, Q. Peng, J. A. Thornton, W. Permar, C. Wielgasz, L. Hu, R. D. Ottmar, J. C. Restaino, A. T. Hudak, I.-T. Ku, Y. Zhou, B. C. Sive, A. Sullivan, J. L. Collett Jr and E. V. Fischer, Emissions of Reactive Nitrogen from Western U.S. Wildfires during Summer 2018, J. Geophys. Res., 2021, 126(2), e2020JD032657, DOI:10.1029/2020jd032657.
- J. Calahorrano, J. F. Lindaas, J. O'Dell, K. Palm, B. B. Peng, Q. Flocke, F. Pollack, I. B. Garofalo, L. A. Farmer, D. K. Pierce, J. R. Collett, J. L. Weinheimer Jr., A. Campos, T. Hornbrook, R. S. Hall, S. R. Ullmann, K. Pothier, M. A. Apel, E. C. Permar, W. Hu, L. Hills, A. J. Montzka, D. Tyndall, G. Thornton and J. A. Fischer, E. V. Daytime Oxidized Reactive Nitrogen Partitioning in Western U.s. Wildfire Smoke Plumes, J. Geophys. Res., 2021, 126(4), e2020JD033484, DOI:10.1029/2020jd033484.
- B. Lebegue, M. Schmidt, M. Ramonet, B. Wastine, C. Yver Kwok, O. Laurent, S. Belviso, A. Guemri, C. Philippon, J. Smith and S. Conil, Comparison of Nitrous Oxide (N2O) Analyzers for High-Precision Measurements of Atmospheric Mole Fractions, Atmos. Meas. Tech., 2016, 9(3), 1221–1238, DOI:10.5194/amt-9-1221-2016.
- A. Kupc, C. Williamson, N. L. Wagner, M. Richardson and C. A. Brock, Modification, Calibration, and Performance of the Ultra-High Sensitivity Aerosol Spectrometer for Particle Size Distribution and Volatility Measurements during the Atmospheric Tomography Mission (ATom) Airborne Campaign, Atmos. Meas. Tech, 2018, 11(1), 369–383, DOI:10.5194/amt-11-369-2018.
- A. L. Hodshire, E. Ramnarine, A. Akherati, M. L. Alvarado, D. K. Farmer, S. H. Jathar, S. M. Kreidenweis, C. R. Lonsdale, T. B. Onasch, S. R. Springston, J. Wang, Y. Wang, L. I. Kleinman, A. J. Sedlacek III and J. R. Pierce, Dilution Impacts on Smoke Aging: Evidence in Biomass Burning Observation Project (BBOP) Data, Atmos. Chem. Phys., 2021, 21(9), 6839–6855, DOI:10.5194/acp-21-6839-2021.
- S. M. Griffith, R. F. Hansen, S. Dusanter, V. Michoud, J. B. Gilman, W. C. Kuster, P. R. Veres, M. Graus, J. A. de Gouw, J. Roberts, C. Young, R. Washenfelder, S. S. Brown, R. Thalman, E. Waxman, R. Volkamer, C. Tsai, J. Stutz, J. H. Flynn, N. Grossberg, B. Lefer, S. L. Alvarez, B. Rappenglueck, L. H. Mielke, H. D. Osthoff and P. S. Stevens, Measurements of Hydroxyl and Hydroperoxy Radicals during CalNex-LA: Model Comparisons and Radical Budgets, J. Geophys. Res. D: Atmos, 2016, 121(8), 4211–4232, DOI:10.1002/2015JD024358.
- Q. Peng, B. B. Palm, K. E. Melander, B. H. Lee, S. R. Hall, K. Ullmann, T. Campos, A. J. Weinheimer, E. C. Apel, R. S. Hornbrook, A. J. Hills, D. D. Montzka, F. Flocke, L. Hu, W. Permar, C. Wielgasz, J. Lindaas, I. B. Pollack, E. V. Fischer, T. H. Bertram and J. A. Thornton, HONO Emissions from Western U.S. Wildfires Provide Dominant Radical Source in Fresh Wildfire Smoke, Environ. Sci. Technol., 2020, 54(10), 5954–5963, DOI:10.1021/acs.est.0c00126.
- M. M. Coggon, C. Y. Lim, A. R. Koss, K. Sekimoto, B. Yuan, J. B. Gilman, D. H. Hagan, V. Selimovic, K. J. Zarzana, S. S. Brown, J. M. Roberts, M. Müller, R. Yokelson, A. Wisthaler, J. E. Krechmer, J. L. Jimenez, C. Cappa, J. H. Kroll, J. de Gouw and C. Warneke, Atmos. Chem. Phys., 2019, 19, 14875–14899 CrossRef CAS.
- C. D. Cappa and K. R. Wilson, Multi-Generation Gas-Phase Oxidation, Equilibrium Partitioning, and the Formation and Evolution of Secondary Organic Aerosol, Atmos. Chem. Phys., 2012, 12(20), 9505–9528, DOI:10.5194/acp-12-9505-2012.
- S. H. Jathar, C. D. Cappa, A. S. Wexler, J. H. Seinfeld and M. J. Kleeman, Multi-Generational Oxidation Model to Simulate Secondary Organic Aerosol in a 3-D Air Quality Model, Geosci. Model Dev., 2015, 8(8), 2553–2567, DOI:10.5194/gmd-8-2553-2015.
- P. J. Adams and J. H. Seinfeld, Predicting Global Aerosol Size Distributions in General Circulation Models, J. Geophys. Res. D: Atmos, 2002, 107(D19), AAC 4-1–AAC 4-23, DOI:10.1029/2001JD001010.
- J. R. Pierce, I. Riipinen, M. Kulmala, M. Ehn, T. Petäjä, H. Junninen, D. R. Worsnop and N. M. Donahue, Quantification of the Volatility of Secondary Organic Compounds in Ultrafine Particles during Nucleation Events, Atmos. Chem. Phys., 2011, 11(17), 9019–9036, DOI:10.5194/acp-11-9019-2011.
- S. H. Jathar, C. D. Cappa, A. S. Wexler, J. H. Seinfeld and M. J. Kleeman, Simulating Secondary Organic Aerosol in a Regional Air Quality Model Using the Statistical Oxidation Model – Part 1: Assessing the Influence of Constrained Multi-Generational Ageing, Atmos. Chem. Phys., 2016, 16(4), 2309–2322, DOI:10.5194/acp-16-2309-2016.
- S. Eluri, C. D. Cappa, B. Friedman, D. K. Farmer and S. H. Jathar, Modeling the Formation and Composition of Secondary Organic Aerosol from Diesel Exhaust Using Parameterized and Semi-Explicit Chemistry and Thermodynamic Models, Atmos. Chem. Phys., 2018, 18(19), 13813–13838, DOI:10.5194/acp-18-13813-2018.
- A. Akherati, C. D. Cappa, M. J. Kleeman, K. S. Docherty, J. L. Jimenez, S. M. Griffith, S. Dusanter, P. S. Stevens and S. H. Jathar, Simulating Secondary Organic Aerosol in a Regional Air Quality Model Using the Statistical Oxidation Model – Part 3: Assessing the Influence of Semi-Volatile and Intermediate-Volatility Organic Compounds and NOx, Atmos. Chem. Phys., 2019, 19(7), 4561–4594, DOI:10.5194/acp-19-4561-2019.
- C. D. Cappa, S. H. Jathar, M. J. Kleeman, K. S. Docherty, J. L. Jimenez, J. H. Seinfeld and A. S. Wexler, Simulating Secondary Organic Aerosol in a Regional Air Quality Model Using the Statistical Oxidation Model–Part 2: Assessing the Influence of Vapor Wall Losses, Atmos. Chem. Phys., 2016, 16(5), 3041–3059 CrossRef CAS.
- Y. He, A. Akherati, T. Nah, N. L. Ng, L. A. Garofalo, D. K. Farmer, M. Shiraiwa, R. A. Zaveri, C. D. Cappa, J. R. Pierce and S. H. Jathar, Particle Size Distribution Dynamics Can Help Constrain the Phase State of Secondary Organic Aerosol, Environ. Sci. Technol., 2021, 55(3), 1466–1476, DOI:10.1021/acs.est.0c05796.
- L. A. Garofalo, Y. He, S. H. Jathar, J. R. Pierce, C. D. Fredrickson, B. B. Palm, J. A. Thornton, F. Mahrt, G. V. Crescenzo, A. K. Bertram, D. C. Draper, J. L. Fry, J. Orlando, X. Zhang and D. K. Farmer, Heterogeneous Nucleation Drives Particle Size Segregation in Sequential Ozone and Nitrate Radical Oxidation of Catechol, Environ. Sci. Technol., 2021 DOI:10.1021/acs.est.1c02984.
- Y. He, B. King, M. Pothier, L. Lewane, A. Akherati, J. Mattila, D. K. Farmer, R. L. McCormick, M. Thornton, J. R. Pierce, J. Volckens and S. H. Jathar, Secondary Organic Aerosol Formation from Evaporated Biofuels: Comparison to Gasoline and Correction for Vapor Wall Losses, Environ. Sci. Process. Impacts, 2020, 22(7), 1461–1474, 10.1039/d0em00103a.
- R. A. Zaveri, R. C. Easter, J. E. Shilling and J. H. Seinfeld, Modeling Kinetic Partitioning of Secondary Organic Aerosol and Size Distribution Dynamics: Representing Effects of Volatility, Phase State, and Particle-Phase Reaction, Atmos. Chem. Phys., 2014, 14(10), 5153–5181, DOI:10.5194/acp-14-5153-2014.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipilä, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann, B. Lee, F. Lopez-Hilfiker, S. Andres, I.-H. Acir, M. Rissanen, T. Jokinen, S. Schobesberger, J. Kangasluoma, J. Kontkanen, T. Nieminen, T. Kurtén, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard, M. Canagaratna, M. Dal Maso, T. Berndt, T. Petäjä, A. Wahner, V.-M. Kerminen, M. Kulmala, D. R. Worsnop, J. Wildt and T. F. Mentel, Nature, 2014, 506, 476–479 CrossRef CAS PubMed.
- D. Stolzenburg, L. Fischer, A. L. Vogel, M. Heinritzi, M. Schervish, M. Simon, A. C. Wagner, L. Dada, L. R. Ahonen, A. Amorim, A. Baccarini, P. S. Bauer, B. Baumgartner, A. Bergen, F. Bianchi, M. Breitenlechner, S. Brilke, S. Buenrostro Mazon, D. Chen, A. Dias, D. C. Draper, J. Duplissy, I. El Haddad, H. Finkenzeller, C. Frege, C. Fuchs, O. Garmash, H. Gordon, X. He, J. Helm, V. Hofbauer, C. R. Hoyle, C. Kim, J. Kirkby, J. Kontkanen, A. Kürten, J. Lampilahti, M. Lawler, K. Lehtipalo, M. Leiminger, H. Mai, S. Mathot, B. Mentler, U. Molteni, W. Nie, T. Nieminen, J. B. Nowak, A. Ojdanic, A. Onnela, M. Passananti, T. Petäjä, L. L. J. Quéléver, M. P. Rissanen, N. Sarnela, S. Schallhart, C. Tauber, A. Tomé, R. Wagner, M. Wang, L. Weitz, D. Wimmer, M. Xiao, C. Yan, P. Ye, Q. Zha, U. Baltensperger, J. Curtius, J. Dommen, R. C. Flagan, M. Kulmala, J. N. Smith, D. R. Worsnop, A. Hansel, N. M. Donahue and P. M. Winkler, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9122–9227 CrossRef CAS PubMed.
- E. L. D’Ambro, S. Schobesberger, R. A. Zaveri, J. E. Shilling, B. Hwan Lee, F. D. Lopez-Hilfiker, C. Mohr and J. A. Thornton, ACS Earth Space Chem., 2018, 2, 1058–1067 CrossRef.
- R. A. Zaveri, J. E. Shilling, A. Zelenyuk, M. A. Zawadowicz, K. Suski, S. China, D. M. Bell, D. Veghte and A. Laskin, Environ. Sci. Technol., 2020, 54, 2595–2605 CrossRef CAS PubMed.
- C. L. Loza, J. S. Craven, L. D. Yee, M. M. Coggon, R. H. Schwantes, M. Shiraiwa, X. Zhang, K. A. Schilling, N. L. Ng, M. R. Canagaratna, P. J. Ziemann, R. C. Flagan and J. H. Seinfeld, Secondary Organic Aerosol Yields of 12-Carbon Alkanes, Atmos. Chem. Phys., 2014, 14(3), 1423–1439, DOI:10.5194/acp-14-1423-2014.
- N. L. Ng, J. H. Kroll, A. W. H. Chan, P. S. Chhabra, R. C. Flagan and J. H. Seinfeld, Secondary Organic Aerosol Formation from m-Xylene, Toluene, and Benzene, Atmos. Chem. Phys., 2007, 7(14), 3909–3922, DOI:10.5194/acp-7-3909-2007.
- L. D. Yee, K. E. Kautzman, C. L. Loza, K. A. Schilling, M. M. Coggon, P. S. Chhabra, M. N. Chan, A. W. H. Chan, S. P. Hersey, J. D. Crounse, P. O. Wennberg, R. C. Flagan and J. H. Seinfeld, Secondary Organic Aerosol Formation from Biomass Burning Intermediates: Phenol and Methoxyphenols, Atmos. Chem. Phys., 2013, 13(16), 8019–8043, DOI:10.5194/acp-13-8019-2013.
- P. S. Chhabra, N. L. Ng, M. R. Canagaratna, A. L. Corrigan, L. M. Russell, D. R. Worsnop, R. C. Flagan and J. H. Seinfeld, Elemental Composition and Oxidation of Chamber Organic Aerosol, Atmos. Chem. Phys., 2011, 11(17), 8827–8845, DOI:10.5194/acp-11-8827-2011.
- A. P. Grieshop, M. A. Miracolo, N. M. Donahue and A. L. Robinson, Constraining the Volatility Distribution and Gas-Particle Partitioning of Combustion Aerosols Using Isothermal Dilution and Thermodenuder Measurements, Environ. Sci. Technol., 2009, 43(13), 4750–4756, DOI:10.1021/es8032378.
- J. A. Huffman, K. S. Docherty, A. C. Aiken, M. J. Cubison, I. M. Ulbrich, P. F. DeCarlo, D. Sueper, J. T. Jayne, D. R. Worsnop, P. J. Ziemann and J. L. Jimenez, Chemically-Resolved Aerosol Volatility Measurements from Two Megacity Field Studies, Atmos. Chem. Phys., 2009, 9(18), 7161–7182, DOI:10.5194/acp-9-7161-2009.
- EPA, EPI SuiteTM-estimation program interface, https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface, accessed Mar 20, 2021 Search PubMed.
- G. J. Stewart, B. S. Nelson, W. J. F. Acton, A. R. Vaughan, N. J. Farren, J. R. Hopkins, M. W. Ward, S. J. Swift, R. Arya, A. Mondal, R. Jangirh, S. Ahlawat, L. Yadav, S. K. Sharma, S. S. M. Yunus, C. N. Hewitt, E. Nemitz, N. Mullinger, R. Gadi, L. K. Sahu, N. Tripathi, A. R. Rickard, J. D. Lee, T. K. Mandal and J. F. Hamilton, Emissions of Intermediate-Volatility and Semi-Volatile Organic Compounds from Domestic Fuels Used in Delhi, India, Atmos. Chem. Phys., 2021, 21(4), 2407–2426, DOI:10.5194/acp-21-2407-2021.
- T. Joo, J. C. Rivera-Rios, M. Takeuchi, M. J. Alvarado and N. L. Ng, Secondary Organic Aerosol Formation from Reaction of 3-Methylfuran with Nitrate Radicals, ACS Earth Space Chem., 2019, 3(6), 922–934, DOI:10.1021/acsearthspacechem.9b00068.
- Q. Lu, Y. Zhao and A. L. Robinson, Comprehensive Organic Emission Profiles for Gasoline, Diesel, and Gas-Turbine Engines Including Intermediate and Semi-Volatile Organic Compound Emissions, Atmos. Chem. Phys., 2018, 18(23), 17637–17654, DOI:10.5194/acp-18-17637-2018.
- A. W. H. Chan, M. N. Chan, J. D. Surratt, P. S. Chhabra, C. L. Loza, J. D. Crounse, L. D. Yee, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Role of Aldehyde Chemistry and NOx Concentrations in Secondary Organic Aerosol Formation, Atmos. Chem. Phys., 2010, 10(15), 7169–7188, DOI:10.5194/acp-10-7169-2010.
- K. M. Henry and N. M. Donahue, Photochemical Aging of α-Pinene Secondary Organic Aerosol: Effects of OH Radical Sources and Photolysis, J. Phys. Chem. A, 2012, 116(24), 5932–5940, DOI:10.1021/jp210288s.
- M. A. Zawadowicz, B. H. Lee, M. Shrivastava, A. Zelenyuk, R. A. Zaveri, C. Flynn, J. A. Thornton and J. E. Shilling, Photolysis Controls Atmospheric Budgets of Biogenic Secondary Organic Aerosol, Environ. Sci. Technol., 2020, 54(7), 3861–3870, DOI:10.1021/acs.est.9b07051.
- D. D. Huang, Q. Zhang, H. H. Y. Cheung, L. Yu, S. Zhou, C. Anastasio, J. D. Smith and C. K. Chan, Formation and Evolution of aqSOA from Aqueous-Phase Reactions of Phenolic Carbonyls: Comparison between Ammonium Sulfate and Ammonium Nitrate Solutions, Environ. Sci. Technol., 2018, 52(16), 9215–9224, DOI:10.1021/acs.est.8b03441.
- L. I. Kleinman and A. J. Sedlacek, Biomass Burning Observation Project (BBOP) Final Campaign Report; DOE/SC-ARM-15–083; DOE ARM, Climate Research Facility, Washington, DC (United States), 2016 Search PubMed.
- P. Zuidema, M. Alvarado, C. Chiu, S. de Szoeke, C. Fairall, G. Feingold, A. Freedman, S. Ghan, J. Haywood, P. Kollias, E. Lewis, G. McFarquhar, A. McComiskey, D. Mechem, T. Onasch, J. Redemann, D. Romps, D. Turner, H. Wang, R. Wood, S. Yuter and P. Zhu, Layered Atlantic Smoke Interactions with Clouds (LASIC) Field Campaign Report; DOE/SC-ARM-18–018; DOE Office of Science Atmospheric Radiation Measurement (ARM) Program (United States), 2018.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d1ea00082a |
| ‡ Now at the Department of Civil and Environmental Engineering, University of California Davis, Davis, CA, USA. |
| This journal is © The Royal Society of Chemistry 2022 |