
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolybdenum precursor delivery approaches in atomic layer deposition of α-MoO3†
Daniela
Lorenzo
,
David Maria
Tobaldi
 *,
Vittorianna
Tasco
*,
Marco
Esposito
,
Adriana
Passaseo
and
Massimo
Cuscunà
*,
Vittorianna
Tasco
*,
Marco
Esposito
,
Adriana
Passaseo
and
Massimo
Cuscunà
CNR NANOTEC Institute of Nanotechnology, Via Monteroni, Lecce 73100, Italy. E-mail: david.tobaldi@nanotec.cnr.it; vittorianna.tasco@nanotec.cnr.it
First published on 5th December 2022
Abstract
In this research work, we present a study on time-sequenced plasma-enhanced atomic layer deposition (PE-ALD) processes towards the achievement of high-quality α-MoO3 thin films which are suitable for exfoliation. In particular, a conventional precursor injection method along with a boosted precursor delivery approach are discussed and analysed. In the latter, the proposed gas supply mechanism ensures a large number of deposited Mo atoms per unit of time, which, along with a proper thermal energy, leads to high-quality and oriented orthorhombic α-MoO3 films. The proposed boosted approach is also compared with post growth annealing steps, resulting in more effective achievement of a highly oriented orthorhombic α-MoO3 phase and less time consumption.
1. Introduction
In the emerging field of transition metal oxides, thin films of molybdenum oxide have received great interest.1,2 Because of its high transparency and high work function, MoOx can be used as a hole extraction layer in silicon heterojunction solar cells,3–6 as the anode buffer layer in organic photovoltaics,7 or as the anode interface on indium tin oxide to improve the stability of organic light-emitting diodes.8 Other applications include, but are not limited to, solid-state lithium ion batteries,9 plasmonics,10 as an alternative material to the metallic counterpart,11–14 photochromic and thermochromic devices,15 and pseudocapacitors,16 since thin films of molybdenum oxide exhibit high capacity, low electrical resistivity, and high thermal and chemical stability. Molybdenum oxide has also demonstrated good catalytic properties for applications in nanostructured gas sensors.17While molybdenum oxide can be found in several stoichiometries – ranging from the fully stoichiometric MoO3 to the semimetallic MoO22 – the orthorhombic MoO3 polymorph (α-MoO3) is particularly interesting because of its layered crystal structure, which can be exfoliated, giving rise to a two-dimensional (2D) morphology. Thus, obtaining layered α-MoO3 films, over other MoO3 polymorphs, is highly desirable for exploiting electronic/optoelectronic devices with novel capabilities.18 Various chemical and physical methods are available for the preparation of molybdenum oxide thin films on different substrates, such as the sol–gel process,19,20 pulsed laser deposition,21,22 thermal23 or electron beam evaporation,10,24 chemical vapour deposition,25,26 and dc reactive magnetron sputtering.27 Plasma-enhanced atomic layer deposition (PE-ALD) technique is an alternative method, exhibiting precise control of the thickness, very low contamination content, and excellent uniformity along with conformal coverage on structured surfaces with complex shapes.28 Such benefits result from the time-sequenced introduction of precursors inside the deposition zone, where selective and self-limiting half-reactions occur on the surface. Hence, thin-film growth is determined by the surface kinetics, allowing parasitic gas-phase reactions to be avoided.29,30 Time-sequenced (temporal) ALD processes are conventionally characterised by the slow deposition rates, related to the need to purge reactants during each growth half-cycle. To tackle the conventional pulse/purge sequence towards industrial-scale thin-film ALD processes, the spatial ALD (SALD) approach has been developed.31–34 In this approach, the substrate moves itself toward stationary nozzles of the precursors and inert gases. Nevertheless, in the case of metalorganic precursors with low vapour pressure, a long precursor dosing time is still needed during each ALD cycle,35 further extending the deposition time. In addition, the SALD approach is based on a different and expensive reactor compared to those used for conventional ALD, and therefore a like-for-like comparison is unfit.
In this study, we describe an alternative PE-ALD approach, suitable for conventional time-sequenced ALD systems, which allows for the deposition of high-quality α-MoO3 thin films using (NtBu)2(NMe2)2Mo as the metalorganic precursor.35 While a “boosted” technique for precursor delivery has been assessed by Bertuch et al.,36 this process, with a full ALD cycle of around 20 s, was tested up to 250 °C, solely giving amorphous MoO3 films. In the present work, the growth characteristics are examined and a detailed film characterisation is provided. Not only has the full ALD cycle been reduced to 9 s (versus 20 s in ref. 36), but also the high quality of the film is assured, giving rise to oriented α-MoO3. This is assumed to be due to the large number of deposited Mo atoms per unit of time, together with the proper thermal energy supplied.
A comparison of results from the traditional precursor delivery was also performed, showing that the combination of the boosted precursor delivery with proper thermal energy gave a more α-oriented MoO3 film with respect to a traditional precursor injection. The boosted precursor delivery approach is also discussed in comparison with the effect on the film microstructure of time-consuming post-growth annealing steps.
2. Experimental section
2.1. Film deposition
Different MoOx films were deposited on a PE-ALD system (SI ALD LL, SENTECH Instruments). It is composed of a reactor unit connected to a remote capacitively coupled plasma (CCP) source, a load-lock for ergonomic wafer loading, a precursor cabinet, and a pumping unit consisting of a foreline dry pump and a turbomolecular pump reaching a base pressure of 10−6 mbar. It is worth noting that the remote CCP source, driven by a radio frequency (RF) generator (13.56 MHz), is attached to the upper flange of the reactor. During the deposition process, the substrate is placed outside of the plasma generation region, and the coated surface is not bombarded with destructive ions. The gas carrier flow for precursor pulsing was 40 standard cubic centimeters per minute (sccm) of ultra-high purity grade N2 (99.999%). As a metalorganic precursor, we used (NtBu)2(NMe2)2Mo (Strem Chemicals Inc.), given its stability at high temperature, which enables good oxide quality,35 and O2 (150 sccm) as the reactant. The reactor wall temperature was kept uniform at 100 °C during the whole film growth process, and for all the depositions at substrate temperatures higher than RT. The Mo cylinder was heated at 55 °C. All the delivery lines were heated at 120 °C to prevent condensation of the precursor.37 Two different precursor delivery approaches (boosted and traditional) were used for the deposition of the films. This is discussed in detail in the ESI (Fig. S1–†). The base pressure used for both the approaches was set in the range of 20.5–21 Pa (about 150 mTorr) using a throttle valve. The RF power applied to the capacitive plasma source was set at 200 W, with reflected power in the range of 2–3 W.The films were grown (i) on p-type Si (100) wafers with a resistivity of 1–30 Ω cm (low-doped substrates), a thickness of ∼650–700 μm, and a native oxide thickness of 2.8 nm, as measured by spectroscopic ellipsometry (SE) using the SiO2 fit,38 and (ii) on glass substrates to study the optical transmittance. All the substrates were pre-cleaned with acetone and isopropyl alcohol. Different samples were grown by the repetition of 300 cycles with substrate temperature tuned between RT and 400 °C.
2.2. Post-deposition annealing
The MoOx films deposited at 100 °C were selected for annealing experiments, performed ex situ. The as-deposited MoOx samples were heated in a furnace (Nabertherm) under a N2 atmosphere at 250, 400 and 500 °C, respectively. The heating ramp rate was 4 °C min−1, followed by 3 hours of soaking time at the maximum temperature.2.3. Film analysis
SE measurements were recorded in the visible/NIR range (400–900 nm) with a J.A. Woollam M-2000 system at three angles of incidence (50°, 55°, and 60°). This was used to determine the optical constants of the MoOx films on the silicon substrate with native oxide on top and their thicknesses. Scanning electron microscopy (SEM Zeiss Merlin, operating at 5 kV) cross-section investigations were combined with SE for comparison purposes. The model used to fit the SE data consisted of three layers: silicon for substrate,39 native silicon oxide,40,41 and MoOx as the top layer. In a simplified picture, a Cauchy model42,43 was used for fitting the MoOx layer.44,45 The thickness was determined as the averaged value retrieved on a grid of 5 points on every sample. Transmittance spectra of the films grown on the glass substrates were recorded by SE in the transmission mode in the wavelength range of 300–900 nm (UV-visible spectroscopy). Moreover, the film morphology was studied by atomic force microscopy (AFM) (Nanosurf EasyScan 2) in the non-contact mode using silicon tips with a nominal tip radius being less than 10 nm and WS × M was used for the image analysis.46X-ray analysis was used to detect the structural and morphological features of the MoOx films, as well as the sample mineralogy, using an X-ray diffractometer (XRD, Malvern PANalytical X'Pert Pro MRD) equipped with a fast PiXcel detector – CuKα radiation. XRD patterns were recorded in grazing incidence (GI) geometry, at an incident angle of 0.5°, a step size of 0.01°2θ, and a counting time of 0.5 s, over the interval 5–85°2θ. In addition, specular X-ray reflectivity (XRR) was also used to retrieve the information about the density of the films. XRR measurements were recorded on the same instrument as that used for GIXRD, in parallel beam geometry, with an incidence angle from 0 to 4°2θ. The XRR patterns were fitted using the X'Pert Reflectivity software package; the sample model consisted of a MoOx/native SiO2/Si stack with surface and interface roughness. In the fittings, the native SiO2 thickness was constrained to the value determined via SE measurements, with the density and thickness of the MoOx layer being the fitting parameters left free.
3. Results and discussion
Fig. 1a shows the growth per cycle (GPC), calculated from the film thicknesses determined by SE, as a function of the deposition temperatures used in the boosted and traditional precursor delivery approaches. SE-retrieved values were compared with those from the cross-sectional SEM investigation (see Fig. S4†). It should be noted that the layers exhibit good uniformity, and the indicated values retrieved by ellipsometry investigation (table of Fig. 1a) are in good agreement with those from the SEM analysis, as discussed in the ESI.†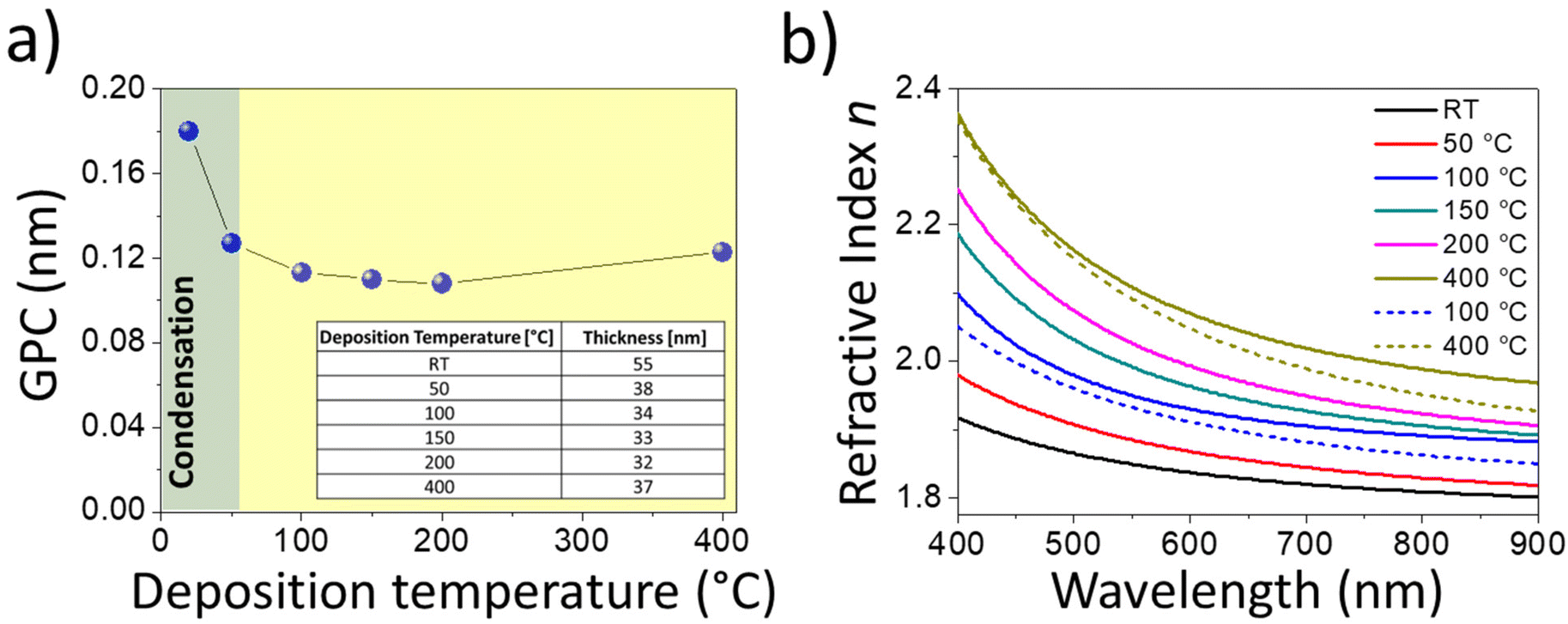 | ||
| Fig. 1 (a) Growth per cycle (GPC) for the boosted approach, determined by SE, as a function of the deposition temperature. GPC is calculated by considering the ratio between film thicknesses (reported in the table) and the number of cycles, which is 300. The green area represents the temperature range in which the metalorganic precursor undergoes condensation, whilst the yellow zone highlights the temperature where proper atomic layer deposition occurs. (b) Refractive indices of the MoOx films (300 cycles) deposited by the boosted approach at different temperatures, obtained by SE (solid lines). The refractive indices of MoOx films deposited by ALD, through the traditional bubbling approach, are also reported for comparison, at two deposition temperatures of 100 °C and 400 °C (dashed lines). | ||
As can be seen in Fig. 1a, condensation of the Mo precursor is likely to occur in the green area – where the substrate temperature is lower than that of the bubbler (55 °C) – thus leading to the largest GPC (0.18 nm at RT). Conversely, the yellow area marks the temperature at which proper atomic layer deposition occurs.47
By increasing the temperature of the substrate, condensation is expected to be suppressed. The GPC decreases to values of about 0.11 nm for the intermediate temperatures (100, 150 and 200 °C), to increase again at 400 °C (about 0.12 nm). Actually, at higher temperatures, the ALD deposition could be affected by the presence of a thermal chemical vapour deposition (CVD) component. However, it was verified that MoOx does not grow when using O2 gas instead of O2 plasma as the reactant. Therefore, such an increase in the GPC is likely due to the transition from amorphous to polycrystalline growth, leading to the formation of nanograins (as detailed below) that enhances the reactive surface area.35
SE investigation, accompanied by the Cauchy fitting model,44,45 permitted us to assess the optical properties of the MoOx thin films. It is worth noting that the validity of the simplified Cauchy modelling was supported by SEM cross-sectional data, as discussed in the supplementary file (section S2†). This was also supported by the presence of an absorption edge lower than 400 nm in the deposited MoOx films, as observed in the transmission spectrum of Fig. S5.† The refractive index (n), shown in Fig. 1b, was found to increase with the deposition temperature, and to overcome the values obtained by the traditional precursor delivery approach, as demonstrated for two ALD deposition temperatures of 100 °C and 400 °C. For such temperatures, a bubbling valve opening time of 10 s was chosen as the standard, to work with a GPC in a saturation regime (see Fig. S3†). It should be pointed out that the extinction coefficient (k) in the visible/NIR (400–900 nm) is null for the investigated temperature range.
The morphology of the film surface for all the temperatures employed with the boosted approach was examined by SEM and AFM. At 100 °C, a flat surface appears (Fig. 2a, top panel), with a root mean square (RMS) roughness of 0.2 nm (Fig. 2a, bottom panel). Conversely, at a maximum temperature (400 °C), the roughness of the film increases to 1.5 nm (Fig. 2b, bottom panel). The morphology evolves to nanograins with lateral size in the range of 20–30 nm (Fig. 2b, top panel), and the film appears to be very compact, as observed by the SEM cross-sectional image (see Fig. S4†). Fig. 2c also reports the morphology of the MoOx films deposited using the traditional Mo precursor delivery mode (thickness of about 41 nm, as retrieved by ellipsometry measurements). Both SEM and AFM images report the formation of nanograins, slightly larger than those observed for the boosted approach at the same temperature. This is likely due to the slightly larger GPC, as already observed in Fig. S3† for a lower temperature, 100 °C, that leads to a greater thickness with a consequential nanograin enlargement.
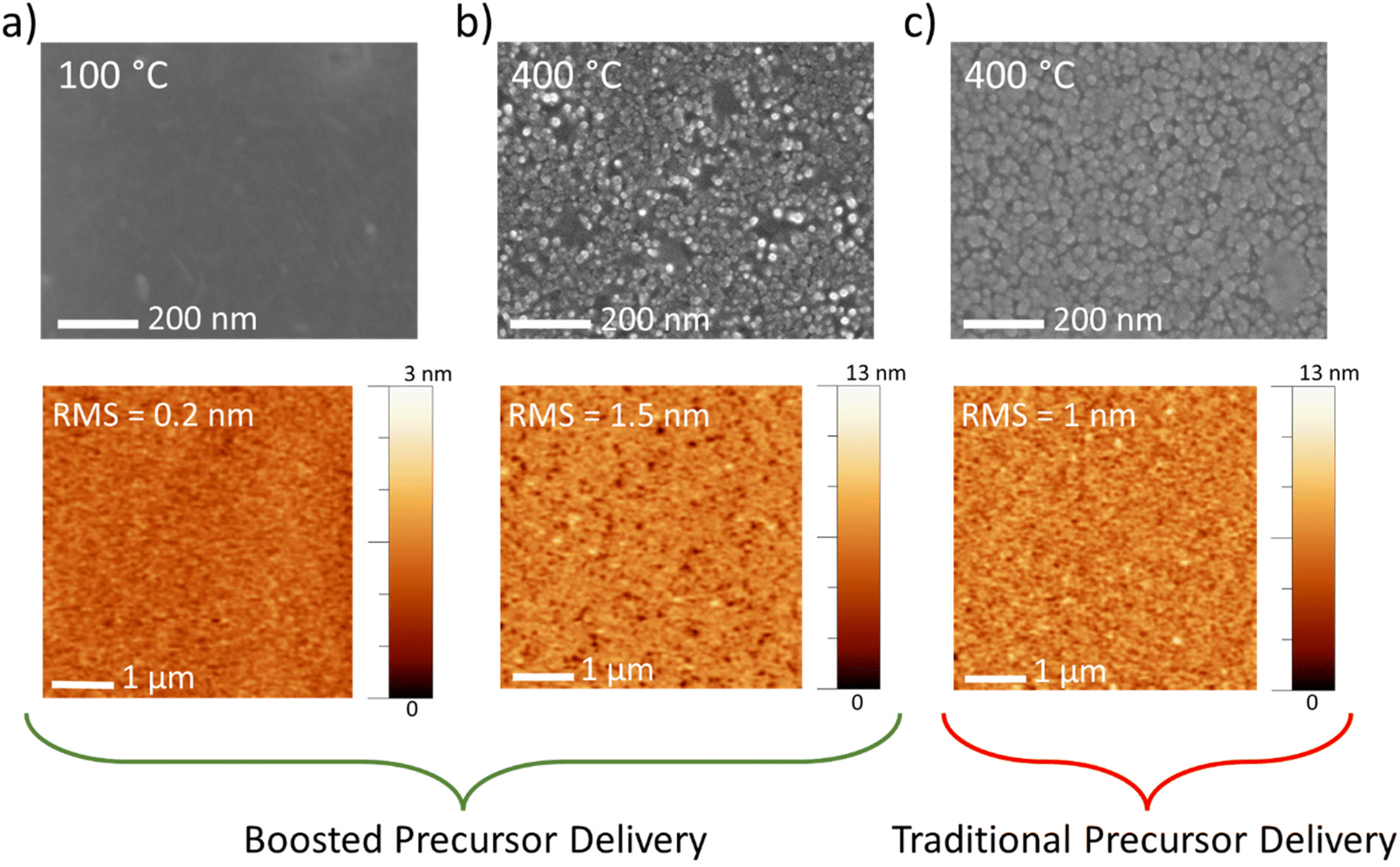 | ||
| Fig. 2 SEM (top panels) and AFM (bottom panels) images of MoOx thin films deposited by means of the boosted precursor injection (300 ALD cycles) on the silicon substrate with native oxide on top at different temperatures: (a) 100 °C and (b) 400 °C. The panel (c) reports the SEM and AFM images of the films deposited at 400 °C using a traditional Mo precursor delivery method (300 ALD cycles). | ||
The effect of growth temperature on the molybdenum oxide structure was investigated in detail through an X-ray analysis for boosted and traditional Mo precursor delivery methods. The diffractograms, shown in Fig. 3a, are related to MoOx thin films deposited at 100 and 400 °C (300 ALD cycles) by the boosted approach. While the film deposited at 100 °C exhibits amorphous layer features, for deposition at 400 °C, three distinct peaks qualitatively reveal the prevalence of orthorhombic α-MoO3, the thermodynamically stable phase observed for crystalline MoO3,35,44 over the monoclinic and metastable β-MoO3 polymorph. Indeed, only a weak (011) reflection of the β-MoO3 polymorph was detected. Moreover, it is worth noting that such an α-MoO3 film exhibits preferential orientation along the [0k0] crystallographic direction. We can argue that during the growth of the whole film, a β-MoO3 phase is initially formed, and then a topotactic transition, consistent with the findings of Carcia and McCarron,48 occurred from β- to α-MoO3. This is usually expected to happen upon thermal treatment above 350 °C.2
 | ||
| Fig. 3 (a) GIXRD and (b) XRR patterns of MoOx thin films deposited by means of boosted (100 °C and 400 °C) and traditional (400 °C) precursor delivery methods. Vertical red lines in (a) represent the [0k0] reflection of α-MoO3, whilst the green line represents the (011) reflection of the β-MoO3 polymorph. The diffractogram of the Si (100) substrate was also reported for comparison. The orange continuous lines in (b) represent the XRR fits to extrapolate the thickness and density of the films. Inset in (b): Yoneda wings around the second Kiessig fringe for the α-MoO3 film deposited at 400 °C by means of the boosted precursor injection method. | ||
As a comparison, a GIXRD pattern of MoOx film grown at 400 °C by means of a traditional precursor injection method is displayed in Fig. 3a. As it can be noted, weakly crystalline β- and α-MoO3 polymorphs coexist together with an amorphous phase. It is worth noting that the structural difference cannot be ascribed to an unintentional CVD component of the traditional approach, and indeed it was verified that no growth occurs when using O2 gas instead of O2 plasma as the reactant. It is clear that the boosted precursor delivery method favours the formation of crystalline MoO3, with the dominance of α-MoO3 over the β-MoO3 polymorph. This means that the presented ALD deposition not only is effective in reducing the process time-scale but, in the case of a growth temperature of 400 °C, also enables the attainment of oriented α-MoO3 films, which can be exfoliated into 2D sheets, as shown by Kalantar-zadeh et al.49 All of the GIXRD patterns in Fig. 3a only showed reflections belonging to MoO3 polymorphs, thus displaying the absence of legacies from the metalorganic precursor. The XRR measurements of such films, and related fitting, are displayed in Fig. 3b. As a general trend, deposition by means of the boosted precursor delivery method provides films with higher densities by increasing the deposition temperature. The extracted density evolves from 3.5 g cm−3 in the amorphous film deposited at 100 °C, to 4.6 g cm−3 for the α-MoO3 film deposited at 400 °C, respectively. In the latter case, XRR off-specular scans exhibit Yoneda wings (the inset of Fig. 3b) at the sides of the second specular Kiessig fringe.50 The Yoneda wings were modelled assuming a fractal interface.51 In this way, we obtained the lateral correlation length of the α-MoO3 layer to be 40 nm. The latter is in good agreement with the nanograin size observed through the SEM analysis of Fig. 2b (film deposited at 400 °C), which is in the range of 20–30 nm. This further confirms that optimised growth with the proper thermal energy favours the formation of crystalline and more compact films, with a density close to the value of bulk α-MoO3, i.e. 4.69 g cm−3.52 Such an increase in the film density with the temperature can also be directly correlated with the increase in the refractive index of the deposited films, as discussed in Fig. 1b.
As a comparison, XRR measurements and related fitting of the film deposited at 400 °C using the traditional Mo precursor delivery approach provided a density of 4.3 g cm−3, lower than that reported for the developed boosted precursor injection method. We can assume that the lower density and the dominance of β-MoO3 over the α-MoO3 polymorph result in a lower refractive index as reported in Fig. 1b.
Therefore, our data about the boosted precursor delivery approach support the assumption that the extra injection step provides a high density of metalorganic precursor molecules in the unit of time. If the surface of the substrate has a suitable thermal energy, this results in a high nucleation density. This yields a uniform and oriented α-MoO3 film deposited in a shorter time, and with improved quality compared to the traditional delivery approach.
A further insight into the proposed growth process was sought by decoupling it from thermally induced effects. In particular, we selected the MoOx film deposited at 100 °C (300 cycles using the boosted deposition method), consisting of amorphous MoOx on the silicon substrates (Fig. 3a), and exposed it to post deposition annealing performed at three temperatures: 250, 400 and 500 °C. The effects of annealing were investigated by the X-ray analysis and SEM. For the sample heated at 500 °C under N2, the film sublimed, exposing the silicon surface underneath the film, with the formation of MoOx islands (not shown), as already observed in ref. 44.
Upon annealing at 250 °C, XRD shows that the amorphous suboxide phase dominates the structural composition (Fig. S6†). After annealing at 400 °C, the initial suboxide phase also shows the onset of a transformation into crystalline α-MoO3 (Fig. S6†). However, despite the films showing XRD [0k0] reflections related to the orthorhombic α-polymorph of molybdenum oxide, they are less intense (by a factor of 3) as compared to those detected in the film directly grown at 400 °C by means of the boosted precursor delivery method (see Fig. S6†). Moreover, from the XRR analysis of the annealed film at 400 °C, the film density is 4.1 g cm−3, which is lower than that obtained with the deposition at 400 °C, and the thickness is about 25 nm. The latter is also confirmed by the SEM cross-section (Fig. S7,† bottom panel), reporting a MoO3 layer with a thickness of about 25 nm after annealing at 400 °C, lower than the as-deposited one, as observed in Fig. S4† (film deposited at 100 °C). Therefore, time and energy consuming post-growth annealing processes are shown to produce a limited effect in comparison with an optimised boosted growth at 400 °C.
4. Conclusions
The orthorhombic MoO3 polymorph (α-MoO3) is particularly interesting because it possesses a layered crystal structure, giving rise to a two-dimensional (2D) morphology to be leveraged in electronic/optoelectronic devices with novel capabilities.While time-sequenced atomic layer deposition represents an ideal candidate for the deposition of α-MoO3 with precise control of the thickness, very low contamination content and excellent uniformity, this technique suffers from slow deposition rates.
In this work, we have developed a time-saving alternative plasma-enhanced atomic layer deposition process for the α-MoO3 thin films. It has been hypothesised that an increase in the number of deposited Mo atoms per unit of time is granted by pushing the vapour pressure in the headspace of the metalorganic precursor cylinder. This led not only to a decrease in the deposition time, but also to a higher quality of the films, compared to a traditional precursor injection method, as proved by the XRD measurements. Moreover, the resulting α-MoO3 film preferentially orients itself along the [0k0] crystallographic direction, thus being particularly promising for exfoliation into 2D sheets. While good-quality films are obtained via deposition at 400 °C, this was not achieved through post-deposition annealing at 400 °C of the amorphous films deposited at lower temperatures, demonstrating the scarce effect of thermal energy alone. Thus, the post-deposition annealing step can be avoided, which allows for a time- and energy-saving process.
Conflicts of interest
The authors declare no competing financial interest.Data availability
The raw/processed data required to reproduce these findings cannot be shared at this time as the data also form part of an ongoing study.Acknowledgements
This work has been partially funded by the National project “HYSPID” funded by the National Research Council (CUP: B86C18000430006), the Regional projects Innonetwork “IN-AIR” (CUP: B37H17004840007) and “nmSensors TransfoClean” (CUP: B37H17005010007) funded by InnovaPuglia and the National project Electronics on GaN-based Technologies “EleGaNTe” funded by MUR – Italian Ministry of Research (CUP: B91G18000200005). David M. Tobaldi thanks the EleGaNTe project for funding. We are very much obliged to Ms Iolena Tarantini (University of Salento) and Mr Gianmichele Epifani (CNR NANOTEC) for technical support. We would like to thank Sentech Instruments GmbH for technical support. Dr Francesco Gabellone (CNR NANOTEC) is kindly acknowledged for his help in the graphical editing.References
- M. Diskus, O. Nilsen, H. Fjellvåg, S. Diplas, P. Beato, C. Harvey, E. van Schrojenstein Lantman and B. M. Weckhuysen, J. Vac. Sci. Technol., A, 2012, 30, 01A107 CrossRef.
- I. A. de Castro, R. S. Datta, J. Z. Ou, A. Castellanos-Gomez, S. Sriram, T. Daeneke and K. Kalantar-zadeh, Adv. Mater., 2017, 29, 1701619 CrossRef PubMed.
- C. Battaglia, X. Yin, M. Zheng, I. D. Sharp, T. Chen, S. McDonnell, A. Azcatl, C. Carraro, B. Ma, R. Maboudian, R. M. Wallace and A. Javey, Nano Lett., 2014, 14, 967–971 CrossRef CAS.
- C. Battaglia, S. M. de Nicolás, S. De Wolf, X. Yin, M. Zheng, C. Ballif and A. Javey, Appl. Phys. Lett., 2014, 104, 113902 CrossRef.
- J. Bullock, A. Cuevas, T. Allen and C. Battaglia, Appl. Phys. Lett., 2014, 105, 232109 CrossRef.
- B. Macco, M. F. J. Vos, N. F. W. Thissen, A. A. Bol and W. M. M. Kessels, Phys. Status Solidi RRL, 2015, 9, 393–396 CrossRef CAS.
- Y.-C. Tseng, A. U. Mane, J. W. Elam and S. B. Darling, Sol. Energy Mater. Sol. Cells, 2012, 99, 235–239 CrossRef CAS.
- F. Wang, X. Qiao, T. Xiong and D. Ma, Org. Electron., 2008, 9, 985–993 CrossRef CAS.
- W. Chen, H. Zhang, Y. Wang, Z. Ma and Z. Li, Electrochim. Acta, 2014, 144, 369–375 CrossRef CAS.
- S. Kasani, P. Zheng, J. Bright and N. Wu, ACS Appl. Electron. Mater., 2019, 1, 2389–2395 CrossRef CAS.
- D. Simeone, V. Tasco, M. Esposito, M. Manoccio, D. Lorenzo, M. Scuderi, A. D. Luca, S. Cabrini, A. Passaseo and M. Cuscunà, Nanotechnology, 2021, 32, 025305 CrossRef CAS.
- D. Simeone, M. Esposito, M. Scuderi, G. Calafiore, G. Palermo, A. De Luca, F. Todisco, D. Sanvitto, G. Nicotra, S. Cabrini, V. Tasco, A. Passaseo and M. Cuscunà, ACS Photonics, 2018, 5, 3399–3407 CrossRef CAS.
- M. Esposito, F. Todisco, S. Bakhti, A. Passaseo, I. Tarantini, M. Cuscunà, N. Destouches and V. Tasco, Nano Lett., 2019, 19, 1922–1930 CrossRef CAS.
- M. Scuderi, M. Esposito, F. Todisco, D. Simeone, I. Tarantini, L. De Marco, M. De Giorgi, G. Nicotra, L. Carbone, D. Sanvitto, A. Passaseo, G. Gigli and M. Cuscunà, J. Phys. Chem. C, 2016, 120, 24314–24323 CrossRef.
- J. N. Yao, K. Hashimoto and A. Fujishima, Nature, 1992, 355, 624–626 CrossRef.
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater, 2010, 9, 146–151 CrossRef PubMed.
- F. Rahman, A. Zavabeti, Md. A. Rahman, A. Arash, A. Mazumder, S. Walia, S. Sriram, M. Bhaskaran and S. Balendhran, ACS Appl. Mater. Interfaces, 2019, 11, 40189–40195 CrossRef PubMed.
- A. Arash, T. Ahmed, A. G. Rajan, S. Walia, F. Rahman, A. Mazumder, R. Ramanathan, S. Sriram, M. Bhaskaran, E. Mayes, M. S. Strano and S. Balendhran, 2D Mater., 2019, 6, 035031 CrossRef.
- C. Girotto, E. Voroshazi, D. Cheyns, P. Heremans and B. P. Rand, ACS Appl. Mater. Interfaces, 2011, 3, 3244–3247 CrossRef.
- G. R. Mutta, S. R. Popuri, J. I. B. Wilson and N. S. Bennett, Solid State Sci., 2016, 61, 84–88 CrossRef.
- M. F. Al-Kuhaili, S. M. A. Durrani and I. A. Bakhtiari, Appl. Phys. A, 2010, 98, 609–615 CrossRef.
- S. S. Sunu, E. Prabhu, V. Jayaraman, K. I. Gnanasekar and T. Gnanasekaran, Sens. Actuators, B, 2003, 94, 189–196 CrossRef.
- T. S. Sian and G. B. Reddy, Sol. Energy Mater. Sol. Cells, 2004, 82, 375–386 CrossRef.
- R. Sivakumar, R. Gopalakrishnan, M. Jayachandran and C. Sanjeeviraja, Curr. Appl. Phys., 2007, 7, 51–59 CrossRef.
- A. Abdellaoui, L. Martin and A. Donnadieu, Phys. Status Solidi A, 1988, 109, 455–462 CrossRef.
- A. Szekeres, T. Ivanova and K. Gesheva, J. Solid State Electrochem., 2002, 7, 17–20 CrossRef.
- V. Nirupama, K. R. Gunasekhar, B. Sreedhar and S. Uthanna, Curr. Appl. Phys., 2010, 10, 272–278 CrossRef.
- J. Cai, X. Han, X. Wang and X. Meng, Matter, 2020, 2, 587–630 CrossRef.
- R. W. Johnson, A. Hultqvist and S. F. Bent, Mater. Today, 2014, 17, 236–246 CrossRef.
- R. L. Puurunen, J. Appl. Phys., 2005, 97, 121301 CrossRef.
- A. Illiberi, F. Roozeboom and P. Poodt, ACS Appl. Mater. Interfaces, 2012, 4, 268–272 CrossRef PubMed.
- D. B. Fullager, G. D. Boreman, C. D. Ellinger and T. Hofmann, Thin Solid Films, 2018, 653, 267–273 CrossRef.
- G. Gregory, C. Luderer, H. Ali, T. S. Sakthivel, T. Jurca, M. Bivour, S. Seal and K. O. Davis, Adv. Mater. Interfaces, 2020, 7, 2000895 CrossRef.
- D. Muñoz-Rojas, T. Maindron, A. Esteve, F. Piallat, J. C. S. Kools and J.-M. Decams, Mater. Today Chem., 2019, 12, 96–120 CrossRef.
- M. F. J. Vos, B. Macco, N. F. W. Thissen, A. A. Bol and W. M. M. (Erwin) Kessels, J. Vac. Sci. Technol., A, 2016, 34, 01A103 CrossRef.
- A. Bertuch, G. Sundaram, M. Saly, D. Moser and R. Kanjolia, J. Vac. Sci. Technol., A, 2014, 32, 01A119 CrossRef.
- T. Muneshwar and K. Cadien, J. Vac. Sci. Technol., A, 2015, 33, 060603 CrossRef.
- C. M. Herzinger, B. Johs, W. A. McGahan, J. A. Woollam and W. Paulson, J. Appl. Phys., 1998, 83, 3323–3336 CrossRef.
- D. E. Aspnes and A. A. Studna, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 27, 985–1009 CrossRef.
- I. H. Malitson, J. Opt. Soc. Am., 1965, 55, 1205 CrossRef.
- C. Z. Tan, J. Non-Cryst. Solids, 1998, 223, 158–163 CrossRef.
- E. Langereis, S. B. S. Heil, H. C. M. Knoops, W. Keuning, M. C. M. van de Sanden and W. M. M. Kessels, J. Phys. D: Appl. Phys., 2009, 42, 073001 CrossRef.
- B. Johs, J. A. Woollam, C. M. Herzinger, J. N. Hilfiker, R. A. Synowicki and C. L. Bungay, Overview of variable-angle spectroscopic ellipsometry (VASE): II. Advanced applications, Bellingham, United States, 1999, p. 1029404, DOI:10.1117/12.351667.
- M. Mattinen, P. J. King, L. Khriachtchev, M. J. Heikkilä, B. Fleming, S. Rushworth, K. Mizohata, K. Meinander, J. Räisänen, M. Ritala and M. Leskelä, Mater. Today Chem., 2018, 9, 17–27 CrossRef.
- M. Mattinen, J.-L. Wree, N. Stegmann, E. Ciftyurek, M. E. Achhab, P. J. King, K. Mizohata, J. Räisänen, K. D. Schierbaum, A. Devi, M. Ritala and M. Leskelä, Chem. Mater., 2018, 30, 8690–8701 CrossRef.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef PubMed.
- S. E. Potts, W. Keuning, E. Langereis, G. Dingemans, M. C. M. van de Sanden and W. M. M. Kessels, J. Electrochem. Soc., 2010, 157, P66 CrossRef.
- P. F. Carcia and E. M. McCarron, Thin Solid Films, 1987, 155, 53–63 CrossRef.
- K. Kalantar-zadeh, J. Tang, M. Wang, K. L. Wang, A. Shailos, K. Galatsis, R. Kojima, V. Strong, A. Lech, W. Wlodarski and R. B. Kaner, Nanoscale, 2010, 2, 429–433 RSC.
- B. K. Tanner, in Handbook of Advanced Nondestructive Evaluation, ed. N. Ida and N. Meyendorf, Springer International Publishing, Cham, 2019, pp. 1181–1214 Search PubMed.
- S. K. Sinha, E. B. Sirota, S. Garoff and H. B. Stanley, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 2297–2311 CrossRef.
- CRC handbook of chemistry and physics, ed. J. R. Rumble, CRC Press, Taylor & Francis Group, Boca Raton, London, New York, 98th edition 2017–2018, 2017 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt03702e |
| This journal is © The Royal Society of Chemistry 2023 |