
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mapping boron catalysis onto a phosphorus cluster platform†
Benjamin L. L.
Réant
 a,
Bono
van IJzendoorn
a,
George F. S.
Whitehead
b and
Meera
Mehta
*a
a,
Bono
van IJzendoorn
a,
George F. S.
Whitehead
b and
Meera
Mehta
*a
aDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: meera.mehta@manchester.ac.uk
bX-Ray Diffraction Facility, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 17th November 2022
Abstract
Clusters of main group elements, such as phosphorus, arsenic, germanium, and tin – called Zintl clusters – have been known for more than a century. However, their application in main group catalysis is largely unknown. Here, we tether boranes to a seven-atom phosphorus cluster ({C8H14}BCH2CH2SiMe2)3P7 (2) and we demonstrate Lewis acid catalysis as proof-of-principle that boron chemistry can be mapped onto clusters using this method. Catalyst 2 was employed to mediate key organic transformations, including the hydroboration of carbodiimides, isocyanates, ketones, alkenes, alkynes, and nitriles. To the best of our knowledge, this is the first application of Zintl-based clusters as an innocent platform in metal-free catalysis. By chaining boron, its treasure chest of chemistry can be unlocked at these clusters. Hence, beyond catalysis this method could find applications for main group clusters in neutron capture therapy, stimuli responsive materials, and cross-coupling, and frustrated Lewis pair and functional polymer chemistries.
Introduction
Organoboranes, compounds that feature B–C bonds, are highly important in synthetic science, functional polymers, and neutron capture therapy.1–6 Within synthesis, boranes are crucial as stoichiometric reagents, for example in the reduction of carbonyls to alcohols and in cross-coupling reactions, and also as catalysts.7–10 Boron centered catalysts are particularly exciting because they are sustainable environment-friendly alternatives to expensive transition metal catalysts, based on their higher crustal abundance and lower costs.11–15Zintl clusters, clusters of main group elements, can be understood as molecular prototypes of heterogenous materials.16 Heptaphosphane ([P7]) clusters are particularly interesting because they are structural fragments of red phosphorus.17–21 Red phosphorous is an inexpensive, highly available, and shelf-stable material, but it is also very insoluble in common laboratory solvents and thus can be difficult to study. In contrast, the heptaphosphane cluster, especially when functionalized, is soluble in many common laboratory solvents such as dimethylformamide, pyridine, tetrahydrofuran, toluene, ether, and pentane. This solubility enables solution-state studies into its chemistry and may inform future applications of red phosphorus-based materials.
Although known for decades, the chemistry of hepta pnictogen ([Pn7]; Pn = P or As) clusters towards small molecules is not well known. Historically, hepta pnictogen clusters have been protonated, functionalized with group 14 electrophiles, and coordinated to d- and f-block metals.17,18,22 In 2012 and 2014, the protonated heptapnictide cages [HPn7]2− (Pn = P, As) were reacted with carbodiimides and isocyanates in hydropnictination reactions by the Goicoechea group.23–25 Goicoechea and co-workers also reacted the trianionic clusters [Pn7]3− (Pn = P, As) with alkynes to yield 1,2,3-tripnictolides by transfer of a [Pn3]− unit,26,27 while reaction of [P7]3− with carbon monoxide gave the [PCO]− anion by P− transfer.28 In 2021, we found that tris-silyl functionalized (R3Si)3P7 clusters captured and exchanged heteroallenes between all three P–Si bonds on the cluster.29
In 2021 Fässler prepared a boron functionalized [Ge9] cluster.30 Although the reactivity of the boron-[Ge9] cluster itself has not yet been reported, the [Ge9] cluster and chloroborane precursors were found to stoichiometrically ring open ethers and capture an acetonitrile.31 Further, group 14 clusters have been coordinated to transition metals and applied as spectator ligands in transition metal catalysis (Fig. 1).16,32,33 Very recently, we have reported on the first transition-metal free Zintl catalysis with a boron-functionalized [P7] cluster, which was also found to be noninnocent in the transformation.34
 | ||
| Fig. 1 Previous Zintl-based catalysts and this work. | ||
Here we tether boranes to a heptaphosphane cluster and use them in Lewis acid catalysis (Fig. 1). We mediate key organic transformations, namely the hydroboration of heteroallenes (carbodiimides and isocyanates), ketones, alkenes, alkynes, and nitriles. To the best of our knowledge, this is the first application of Zintl-based clusters as an innocent platform in main group catalysis. Boron tethers on clusters offer the exciting possibility to translate the wealth of boron chemistry to less developed main group clusters.
Results and discussion
Synthesis of cluster catalysts
First, [Na(DME)x]3[P7] (DME = dimethoxyethane) was prepared using a literature method.35 Next, [Na(DME)x]3[P7] was reacted with chlorodimethylvinylsilane in toluene. Nuclear magnetic resonance (NMR) spectroscopy investigations revealed 3 new multiplet resonances in the 31P NMR spectrum and complete consumption of starting material. The 29Si NMR spectrum showed a doublet at −2.36 ppm with 1JPSi = 42.5 Hz consistent with installation of the silyl groups on the [P7] cluster. Further, three doublets of doublets at 5.63 (3JHH = 20.1 Hz, 2JHH = 3.2 Hz), 5.83 (3JHH = 14.4 Hz, 2JHH = 3.2 Hz), and 6.18 (3JHH = 20.1 Hz, 14.4 Hz) were observed in the 1H NMR spectrum, confirming that the vinyl moiety is conserved. These NMR data are in line with the formation of (CH2CHSiMe2)3P7 (1) (Scheme 1). The solid-state structure of 1 was elucidated by single crystal X-ray diffraction (XRD) studies (Fig. 2). The XRD data of 1 further confirmed that the silyl groups are tethered to the bridging phosphorus atoms of the cluster (average P–Si bond lengths of 2.288 Å), and the preservation of the vinyl moiety (average C–C bond lengths of 1.304 Å, consistent with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds).36,37 Compound 1 was isolated in 86% yield.
C double bonds).36,37 Compound 1 was isolated in 86% yield.
 | ||
| Scheme 1 Synthesis of boron tethered [P7] cluster 2. | ||
 | ||
| Fig. 2 Molecular structure of 1. Anisotropic displacement ellipsoids pictured at 50% probability. Hydrogen atoms omitted for clarity. Phosphorus: orange; silicon: pink; carbon: black. | ||
Next, 1 was reacted with 9-borabicyclo(3.3.1)nonane (9-BBN; R2BH) dimer in toluene (Scheme 1, route A). 1H NMR spectroscopy showed the disappearance of the vinyl moieties, consistent with hydroboration of all three vinyl groups to yield the boron tethered [P7] cluster (R2BCH2CH2SiMe2)3P7 (2). The 11B NMR spectrum revealed a broad signal at 86.5 ppm, consistent with three-coordinate boron. Despite multiple efforts, single crystals only of sufficient quality to verify the connectivity of 2 by XRD could be obtained (Fig. 3). Analysis of the crystal packing together with the downfield 11B NMR resonance confirmed no self-assembly in the solution- or solid-state. Interestingly, it was found that cluster 2 could also be prepared by first hydroborating the chlorosilane to give ClSiMe2CH2CH2BR2 (3), then installing the silyl groups onto the cluster through salt metathesis (Scheme 1, route B). Regardless of the route taken, compound 2 was isolated in high yield (76–95%).
 | ||
| Fig. 3 Molecular structure of 2. Anisotropic displacement ellipsoids pictured at 50% probability. Hydrogen atoms omitted for clarity. Phosphorus: orange; silicon: pink; carbon: black. | ||
Lewis acidity tests
In order to quantify the Lewis acidity of the boron centres on 2, the adapted Gutmann–Beckett test was performed and fluoride ion affinities (FIAs) computed (see ESI, Section 3†).38–42 The Gutmann–Beckett acceptor number was determined to be 46.7. Computationally, the FIA was found to be 350 kJ mol−1 upon addition of one fluoride, 220 kJ mol−1 for the second fluoride, and 113 kJ mol−1 for the third fluoride. The FIA values indicate that when one boron on 2 coordinates an anion, reactivity at the others decreases. This is thought to be because FIA computes anion coordination and not neutral bases, hence after the addition of the first fluoride the overall complex is anionic thus columbic repulsion influences the 2nd and 3rd values. The Gutmann–Beckett acceptor number and first FIA for 2 are consistent with those of the untethered chlorosilane 3 (acceptor number = 46.6; FIA = 340 kJ mol−1). These studies confirm that the Lewis acidity at the boron centres of 2 is not perturbed by tethering to the cluster, and thus their reactivity should be preserved.Hydroboration catalysis
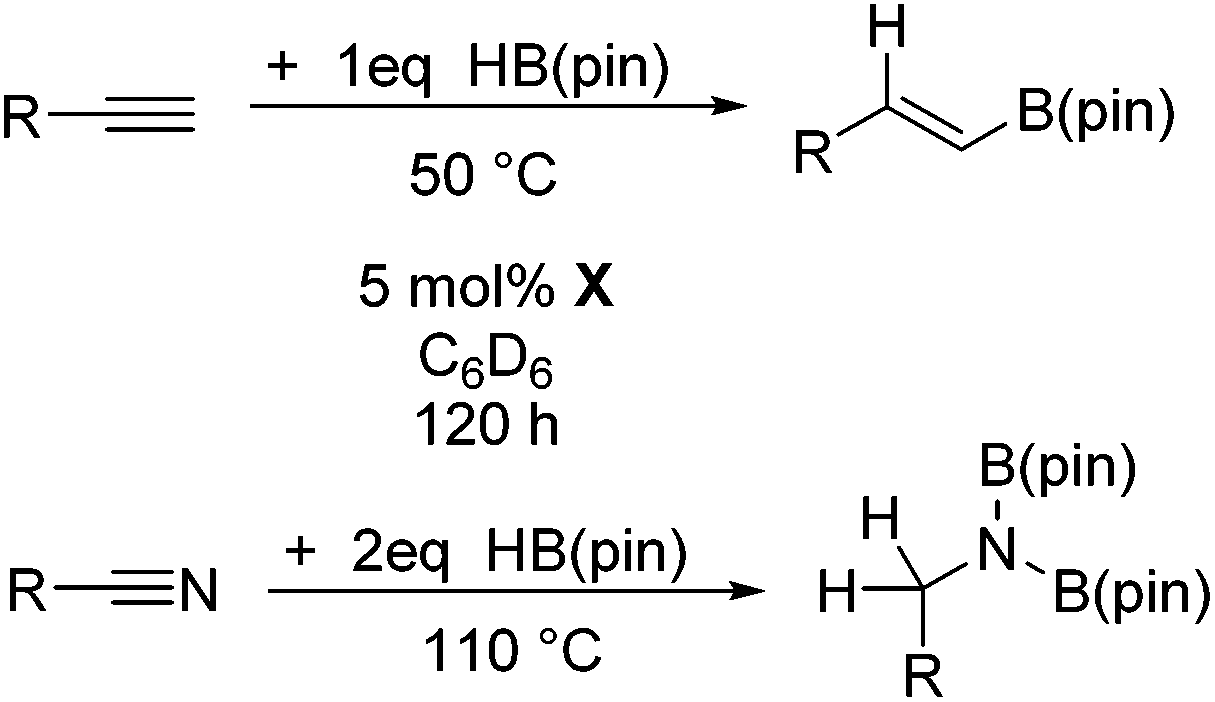
The Lewis acidity of boron has been widely exploited; one such area is in Lewis acid hydroboration catalysis.1,43 Previously, the 9-BBN dimer has been used as a catalyst to hydroborate carbodiimides at 25–60 °C.44 To show that boron chemistry can be mapped onto Zintl-derived clusters via the silyl tethers, compound 2 was studied as a catalyst for the hydroboration of carbodiimides and isocyanates.First, the stability of 2 in C6D6 was tested. No decomposition was observed by NMR spectroscopy after 72 hours at 25 °C, 120 hours at 50 °C, and 168 hours at 110 °C. Next, di-isopropylcarbodiimide (4a) and phenyl isocyanate (5a) were reacted with pinacol borane, HB(pin), in the presence of 5 mol% 2 (Table 1). HB(pin) was selected as the boron reductant because the organic product is expected to display greater stability towards protodeboronation compared to the 9-BBN analogue.9 Boronic esters are also widely employed moieties for important organic transformations, such as Suzuki–Miyaura couplings. In C6D6 at 50 °C after 2 hours complete hydroboration of 4a to 4b was observed by NMR spectroscopy. In contrast, 5a required 72 hours under similar conditions to give 43% of the mono-hydroborated product 5b. In the case of 5a, increasing the equivalents of HB(pin) from 1 to 3 and increasing the reaction temperature from 50 to 110 °C gave mixtures of the mono-hydroborated (5b), bis-hydroborated (5c), and deoxygenated (5d) products, with higher temperatures favouring the deoxygenated product 5d. For 4a, even upon increasing the equivalents of HB(pin) and temperature no bis-hydroborated product was observed. Additionally, changing the solvent from C6D6 to tetrahydrofuran gave lower conversions, presumably because donor solvents will coordinate to the boron active site and quench some of its reactivity.
|
|
||||||
|---|---|---|---|---|---|---|
| Solvent | Temp. (°C) | HB(pin) eq. | Time (h) | 4b, % conv.a | Time (h) |
5b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5c:5d, % conv.a :5c:5d, % conv.a |
| a Determined by 1H NMR spectroscopy. Isolated yields are given in parenthesis. | ||||||
| C6D6 | 25 | 1 | 12 | 92 | 18 | 0 |
| C6D6 | 50 | 1 | 2 | >99 (91) | 72 | 43:0:0 |
| C6D6 | 50 | 2 | 24 | >99 | 48 | 0:50:2 |
| C6D6 | 50 | 3 | — | — | 48 | 0:54:2 |
| C6D6 | 110 | 1 | — | — | 48 | 18:0:9 |
| C6D6 | 110 | 2 | 2 | >99 | 48 | 0:0:21 |
| C6D6 | 110 | 3 | — | — | 48 | 0:0:53 |
| THF | 25 | 1 | 12 | 70 | 18 | 0 |
| THF | 50 | 1 | 2 | 95 | 72 | 18 |

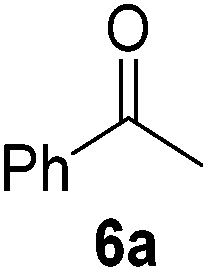
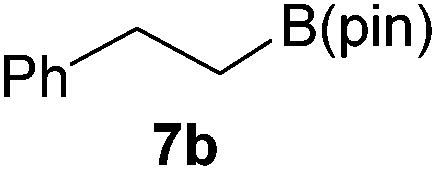
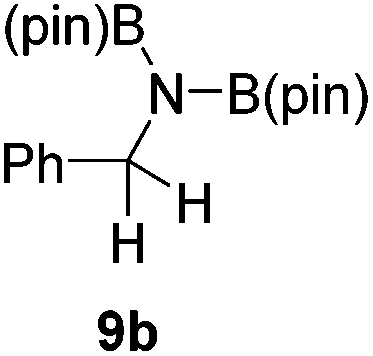
It is noteworthy that other s- and p-block catalysts have previously mediated these types of hydroborations.1,12,43,45–47 In fact, 9-BBN itself has also been employed to mediate the hydroboration of terminal alkynes and nitriles.48,49 To probe the generality of 2 as a hydroboration catalyst, we expanded our scope to other carbon-element multiple bonds, including a ketone, alkene, alkyne, and nitrile (Table 2). Acetophenone (6a) and styrene (7a) were converted to their hydroborated products 6b and 7b with 1 equivalent of HB(pin) and 5 mol% 2 in C6D6 at 110 °C after 15 h and 72 h respectively. In line with literature precedence,49 only anti-Markovnikov hydroboration of 7a was observed. Similarly, hydroboration of phenylacetylene (8a) exclusively gave the syn-addition to the trans-isomer 8b in 79% conversion after 120 h at 50 °C. In contrast, when benzonitrile (9a) was reacted with 1 equivalent HB(pin) and 5 mol% 2 in C6D6 at 110 °C, 44% conversion to the bis-hydroborated product 9b was observed after 120 h with no evidence of mono-hydroboration, thus 2 equivalent of HB(pin) was added to achieve conversion of 9a to 9b in 84% yield. Control reactions further confirmed that in the absence of 2 no hydroboration of 4a–9a was observed. Further, no deoxygenation of acetophenone (6a) or bis-hydroborations of phenylacetylene (8a) was observed with an excess of HB(pin) and 5 mol% 2.
| Substrate | Catalyst | Temp. (°C) | Time (h) | % conv.a | Product |
|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy. b Additional 8% 5c and 3% 5d detected. Isolated yields are given in parenthesis. | |||||

|
— | 50 | 24 | 0 |

|
| 2 | 50 | 2 | >99 (91) | ||
| 3 | 50 | 2 | 89 | ||
| (Me3Si)3P7 | 50 | 2 | 0 | ||

|
— | 50 | 72 | 0 |

|
| 2 | 50 | 72 | 43 | ||
| 3 | 50 | 72 | 31b | ||
| (Me3Si)3P7 | 50 | 72 | 0 | ||
|
— | 110 | 48 | 0 |

|
| 2 | 110 | 15 | 91 | ||
| 3 | 110 | 15 | 96 | ||
| (Me3Si)3P7 | 110 | 15 | 0 | ||

|
— | 110 | 48 | 0 |
|
| 2 | 110 | 72 | 80 | ||
| 3 | 110 | 72 | 68 | ||
| (Me3Si)3P7 | 110 | 72 | 0 | ||

|
— | 50 | 120 | 0 |

|
| 2 | 50 | 120 | 79 | ||
| 3 | 50 | 120 | 68 | ||
| (Me3Si)3P7 | 50 | 120 | 0 | ||

|
— | 110 | 120 | 0 |
|
| 2 | 110 | 120 | 84 | ||
| 3 | 110 | 120 | 70 | ||
| (Me3Si)3P7 | 110 | 120 | 0 | ||
In order to probe the role of the cluster in catalysis, 15 mol% of the untethered chlorosilane 3 was investigated as a hydroboration catalyst (Table 2, see ESI, Section 5.3†). It was found that under the established reaction conditions for each substrate, when 15 mol% 3 was employed in lieu of 5 mol% 2 conversions to the respective hydroborated products 4b–9b were nearly the same. With a minor increase in conversion between 10–14% observed for catalyst 2, except for in the case of acetophenone (6a) hydroboration. It is also noteworthy that no degradation of the Cl–Si bond could be observed in any of the reaction mixtures by 29Si NMR spectroscopy. Further, in the case of phenyl isocyanate (5b) hydroboration with 3 minor amounts of 5c and 5d were also observed by 1H NMR spectroscopy, showing that the transformation is less selective than when the boranes are tethered. Next, (Me3Si)3P7 was prepared, using a literature method,35 and tested in place of catalyst 2 at 5 mol% catalyst loading where it showed no catalytic activity. These findings demonstrate that the [P7] cluster acts as a mostly innocent platform and the catalysis originates at the B-tethered arms.
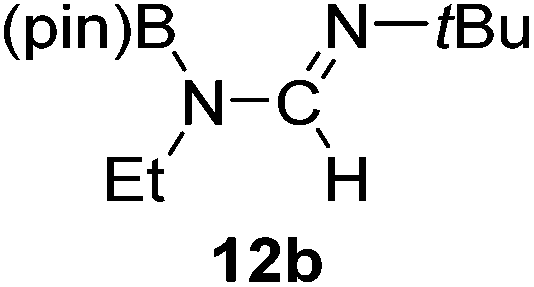
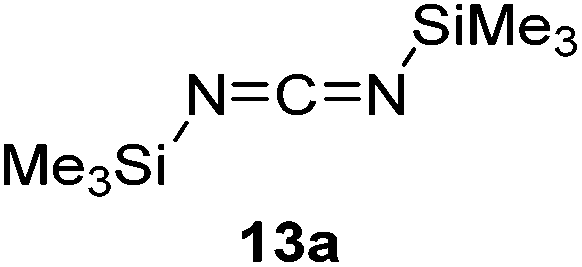
Having demonstrated the generality of 2 as a hydroboration catalyst, the substrate scope was expanded (Tables 3–5). First, carbodiimides and isocyanates were investigated with 1 equivalent of HB(pin) and 5 mol% 2 in C6D6 at 50 °C (Table 3). Similar to the reactivity of di-isopropylcarbodiimide (4a), dicyclohexylcarbodiimide (10a) gave the hydroborated product 10b after 6 h in high yield. The bulkier carbodiimides di-tert-butylcarbodiimide (11a) and 1-tert-butyl-3-ethylcarbodiimide (12a) required longer reaction times to give the hydroborated products 11b and 12b, respectively. Carbodiimide 13a, which features the electron-withdrawing trimethylsilyl group only gave 19% 13b after 72 h, whereas the aromatic functionalized carbodiimide bis(2,6-di-isopropylphenyl)carbodiimide (14a) afforded product 14b in 88% conversion after 36 h. Similar to the reactivity of phenyl isocyanate (5a), para-phenyl functionalized isocyanates 15a–18a all gave moderate conversions, between 32–42%, to the mono-hydroborated products 15b–18b. Those with the electron withdrawing trifluoromethyl- (16a) and fluoro- (17a) groups required longer reaction times. Meanwhile, cyclohexyl isocyanate (19a) yielded 19b in 60% conversion after 72 h. The bulkier tert-butyl isocyanate (20a) required 90 h to afford 49% 20b, and benzyl isocyanate (21a) required 120 h to give 41% of the hydroborated product 21b.











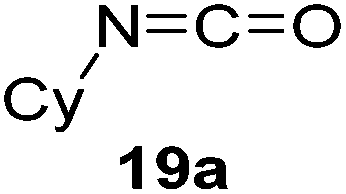

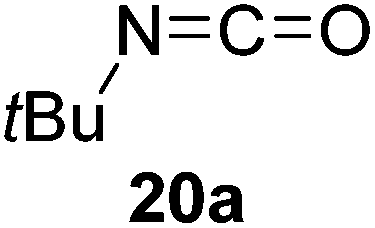

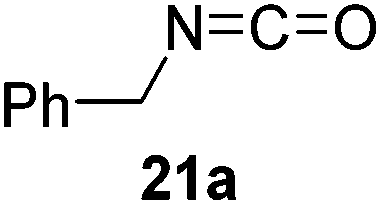



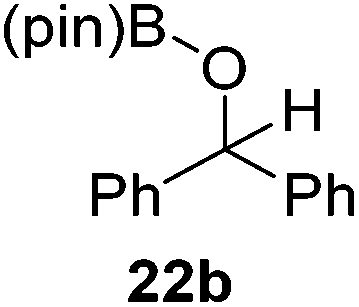
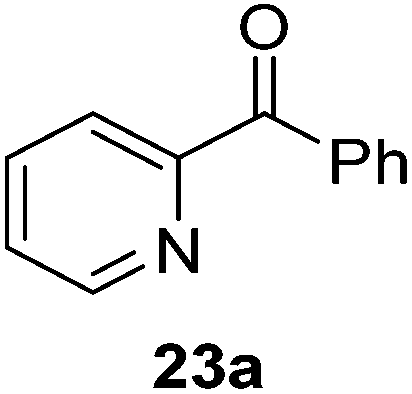



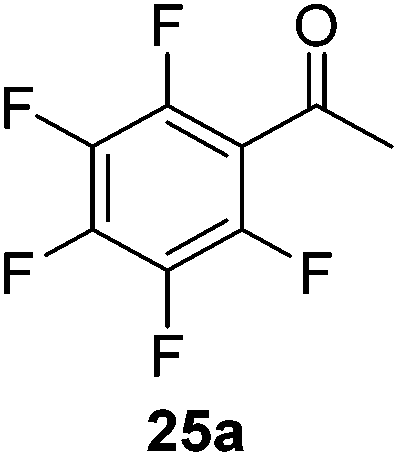


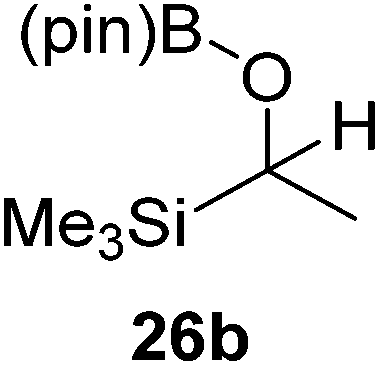

















Then, the scope of carbonyls and alkenes was expanded at 110 °C with 5 mol% 2 (Table 4). Benzophenone (22a) was hydroborated to 22b after 72 h in 42% conversion. Pyridine (23a) and thiophene (24a) functionalized ketones were hydroborated to 23b and 24b, correspondingly, in high yield but required 36 h, presumably because the pyridine and thiophene groups compete with substrate coordination. The pentafluorophenyl functionalized ketone 25a required longer reaction times, 72 hours, to give moderate conversion to 25b. While the silyl functionalized ketone 26a was converted to 26b in quantitative yield in 19 hours. For alkenes, 4-bromostyrene (27a), 4-vinylanisole (28a), and 2-vinylnaphthalene (29a) hydroborated to give 27b, 28b, and 29b, respectively, in similar conversions (between 76–78%). 4-Vinylpyridine (30a) and 9-vinyl-9H-carbazole (31a) were hydroborated to 30b and 31b, respectively, in moderate conversions.
Finally, the scope of terminal alkynes and nitriles was expanded (Table 5). Phenylacetylene derivatives were investigated with 1 equivalent of HB(pin) and 5 mol% 2 in C6D6 at 50 °C. 4-Fluorophenylacetylene (32a), 4-chlorophenylacetylene (33a), 4-bromophenylacetylene (34a), 4-trifluoromethylphenylacetylene (35a), 4-methoxyphenylacetyelene (36a), 4-methylphenylacetylene (37a), and 3-methylphenylacetylene (38a) all gave the corresponding hydroborated products 32b–38b with similarly high conversions (between 76–90%). Next, nitriles were investigated with 2 equivalent of HB(pin) in a similar fashion. 4-Bromobenzonitrile (39a), 4-methoxybenzonitrile (40a), cyclohexanecarbonitrile (41a), and butyronitrile (42a) were all bis-hydroborated to the 39b–42b products in moderate to high yield. While, similar to the ketone and alkene hydroborations, the pyridine functionalized nitriles 43a and 44a could only be bis-hydroborated to 43b and 44b, respectively, in low conversions.
Mechanistically, compound 2 is expected to operate in line with previously well-established p- and s-block Lewis acid mediated hydroboration and hydrosilylation reactions.13,50–52 Catalyst 2 is expected to activate the H–B bond of HB(pin) which then can undergo attack by the substrate to transfer the B(pin) followed by hydride transfer from the catalyst. However, when compound 2 was reacted with 3 equivalents of HB(pin) no coordination could be observed by NMR spectroscopy, at room temperature and −70 °C. It must be noted that even undetected small amounts of adduct formation between 2 and HB(pin) could still promote catalysis. And in fact, would be consistent with the mild catalyst performance of 2. Additionally, 1:3 reactions of 2 with phenyl isocyanate (5a), acetophenone (18a), and phenylacetylene (30a) also showed no evidence of substrate coordination by NMR spectroscopy. In contrast, 1:3 reactions of 2 with di-isopropylphenylcarbodiimide (4a) and benzonitrile (38a) both showed a small upfield shift in the 11B NMR spectra, consistent with substrate coordination. However, no noticeable changes were observed in the 31P NMR spectra, consistent with the cluster being innocent towards substrate activation (see ESI, Section 8†). Because no decomposition or retro-hydroboration of 2 has been observed, secondary borane catalyst mechanisms that invoke initial catalyst hydroboration at the substrate are not expected to be possible.44,53,54 Further, Thomas and co-workers have previously reported the ‘hidden role’ that BH3 and borohydrides can play in hydroboration catalysis.43 In order to test for hidden BH3 and borohydride generation, 5 mol% of 2 was heated with HBpin and benzonitrile (9a) for 5 days at 110 °C, the most pushing conditions employed for catalyst 2. Next an excess of tetramethylethylenediamine (TMEDA) was added to capture borohydrides and allow for their detection by NMR spectroscopy. No evidence of TMEDA captured BH3 or borohydride could be detected by 11B and 1H NMR spectroscopy (see ESI, Section 9†).55
It is also noteworthy that no catalyst decomposition was observed after any of the catalytic transformations. This stability opens the door to catalyst recycling. In the case of di-isopropylcarbodiimde (4a) hydroboration, after complete conversion the reaction was reloaded with substrates and the catalyst recycled 8 additional times with no loss in catalytic performance. We believe that the catalyst could have been reused >8 times, but detection of this living catalyst was not possible by 31P NMR spectroscopy after cycle 9 (see ESI, Section 10†).
Conclusions
In conclusion we find that boron catalysts can be tethered onto a heptaphosphane cluster (2). We show that once tethered, the reactivity of the borane is fully retained. As proof-of-principle, compound 2 was applied as a catalyst for the hydroboration of carbon-element multiple bonds, including carbodiimides, isocyanates, ketones, alkenes, alkynes, and nitriles. This work shows the diverse chemistry possible with boron can be chained onto main group clusters. This work represents the first application of Zintl-based clusters as an innocent platform in main group catalysis. At its heart this is not a study of highly active boron catalysis, but in fact a method that could be applied to unlock the vault of boron chemistry at main group clusters. Applications with Zintl-derived clusters is in its infancy. Beyond catalysis, boron tethers on clusters could be used to find applications for main group clusters in neutron capture therapy, cross-coupling reactions to build polymers, and frustrated Lewis pair (FLP) chemistry. This work reminds us of boron functionalized polymers, interest in which has since exploded with the development of them as components in stimuli-responsive, optical, self-healing, and sensor materials.56,57 Thus, a similar trajectory could be imagined.Author contributions
B. L. L. R. and B. v. I. performed the synthesis of compounds 1–3. B. L. L. R. performed the catalysis. G. F. S. W. collected and solved the XRD data. B. v. I. performed the DFT calculations. B. L. L. R. constructed the ESI.† M. M. wrote and edited the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC (for EP/V012061/1 & a DPT student B. v. I.) and the Royal Society (RGS/R1/211101) for funding. We thank Gareth Smith and the Leigh group for mass spectrometric analyses. We also thank Anne Davies and Martin Jennings for elemental analyses.Notes and references
-
M. Mehta, in Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O'Hare, Elsevier, Kidlington, UK, 2022, pp. 122–195 Search PubMed
.
- G. Berionni, Chem. Synth., 2021, 1, 10 Search PubMed
-
H. DeFrancesco, J. Dudley and A. Coca, Boron Reagents in Synthesis, Am. Chem. Soc., 2016, ch. 1, vol. 1236, pp. 1–25 Search PubMed
- S.-Y. Liu and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3434–3435 RSC
- L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC
- S. M. Berger, M. Ferger and T. B. Marder, Chem. – Eur. J., 2021, 27, 7043–7058 CrossRef CAS PubMed
-
M. Hatano and K. Ishihara, Boron Reagents in Synthesis, Am. Chem. Soc., 2016, ch. 2, vol. 1236, pp. 27–66 Search PubMed
- K. Smith, Chem. Soc. Rev., 1974, 3, 443–465 RSC
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC
- E. R. Burkhardt and K. Matos, Chem. Rev., 2006, 106, 2617–2650 CrossRef CAS PubMed
- V. Nori, F. Pesciaioli, A. Sinibaldi, G. Giorgianni and A. Carlone, Catalysts, 2022, 12, 5 CrossRef CAS
-
J. R. Lawson and R. L. Melen, Organometallic Chemistry: Volume 41, RSC, 2017, vol. 41, pp. 1–27 Search PubMed
-
M. Mehta and C. B. Caputo, Synthetic Inorganic Chemistry, ed. E. J. M. Hamilton, Elsevier, 2021, pp. 169–220 Search PubMed
- W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345–354 RSC
- W. E. Piers, Adv. Organomet. Chem., 2004, 52, 1–76 CrossRef
- O. P. E. Townrow, C. Chung, S. A. Macgregor, A. S. Weller and J. M. Goicoechea, J. Am. Chem. Soc., 2020, 142, 18330–18335 CrossRef CAS PubMed
- B. van IJzendoorn and M. Mehta, Dalton Trans., 2020, 49, 14758–14765 RSC
- R. S. P. Turbervill and J. M. Goicoechea, Chem. Rev., 2014, 114, 10807–10828 CrossRef CAS PubMed
- A. Dragulescu-Andrasi, L. Z. Miller, B. Chen, D. T. McQuade and M. Shatruk, Angew. Chem., Int. Ed., 2016, 55, 3904–3908 CrossRef CAS PubMed
- M. Jo, A. Dragulescu-Andrasi, L. Z. Miller, C. Pak and M. Shatruk, Inorg. Chem., 2020, 59, 5483–5489 CrossRef CAS PubMed
- M. Ruck, D. Hoppe, B. Wahl, P. Simon, Y. Wang and G. Seifert, Angew. Chem., Int. Ed., 2005, 44, 7616–7619 CrossRef CAS PubMed
- R. J. Wilson, B. Weinert and S. Dehnen, Dalton Trans., 2018, 47, 14861–14869 RSC
- R. S. P. Turbervill and J. M. Goicoechea, Organometallics, 2012, 31, 2452–2462 CrossRef CAS
- R. S. P. Turbervill and J. M. Goicoechea, Chem. Commun., 2012, 48, 1470–1472 RSC
- R. S. P. Turbervill and J. M. Goicoechea, Eur. J. Inorg. Chem., 2014, 1660–1668 CrossRef CAS
- R. S. P. Turbervill and J. M. Goicoechea, Inorg. Chem., 2013, 52, 5527–5534 CrossRef CAS PubMed
- R. S. P. Turbervill, A. R. Jupp, P. S. B. McCullough, D. Ergöçmen and J. M. Goicoechea, Organometallics, 2013, 32, 2234–2244 CrossRef CAS
- A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064–10067 CrossRef CAS PubMed
- B. van IJzendoorn, I. Vitorica-Yrezabal, G. Whitehead and M. Mehta, Chem. – Eur. J., 2021, 28, e202103737 Search PubMed
- C. Wallach, F. S. Geitner, A. J. Karttunen and T. F. Fässler, Angew. Chem., Int. Ed., 2021, 60, 2648–2653 CrossRef CAS PubMed
- C. Wallach, F. S. Geitner and T. F. Fässler, Chem. Sci., 2021, 12, 6969–6976 RSC
- N. E. Poitiers, L. Giarrana, V. Huch, M. Zimmer and D. Scheschkewitz, Chem. Sci., 2020, 11, 7782–7788 RSC
- O. P. E. Townrow, S. B. Duckett, A. S. Weller and J. M. Goicoechea, Chem. Sci., 2022, 13, 7626–7633 RSC
- B. van IJzendoorn, S. F. Albawardi, I. J. Vitorica-Yrezabal, G. F. S. Whitehead, J. E. McGrady and M. Mehta, J. Am. Chem. Soc., 2022 DOI:10.1021/jacs.2c08559
- M. Cicač-Hudi, J. Bender, S. H. Schlindwein, M. Bispinghoff, M. Nieger, H. Grützmacher and D. Gudat, Eur. J. Inorg. Chem., 2016, 649–658 CrossRef
- L. Pauling and L. O. Brockway, J. Am. Chem. Soc., 1937, 59, 1223–1236 CrossRef CAS
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed
- K. Christe, D. Dixon, D. McLemore, W. Wilson, J. Sheehy and J. Boatz, J. Fluor. Chem., 2000, 101, 151–153 CrossRef CAS
- P. Erdmann, J. Leitner, J. Schwarz and L. Greb, ChemPhysChem, 2020, 21, 987–994 CrossRef CAS PubMed
- I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75–88 CrossRef CAS
- U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS
- M. A. Beckett, G. C. Strickland, J. R. Holland and K. Sukumar Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS
- A. D. Bage, K. Nicholson, T. A. Hunt, T. Langer and S. P. Thomas, ACS Catal., 2020, 10, 13479–13486 CrossRef CAS
- A. Ramos, A. Antiñolo, F. Carrillo-Hermosilla, R. Fernández-Galán and D. García-Vivó, Chem. Commun., 2019, 55, 3073–3076 RSC
- M. Magre, M. Szewczyk and M. Rueping, Chem. Rev., 2022, 122, 8261–8312 CrossRef CAS PubMed
- K. Kuciński and G. Hreczycho, Green Chem., 2020, 22, 5210–5224 RSC
- A. Das, S. Rej and T. K. Panda, Dalton Trans., 2022, 51, 3027–3040 RSC
- S. Pradhan, R. V. Sankar and C. Gunanathan, J. Org. Chem., 2022, 87, 12386–12398 CrossRef CAS PubMed
- S. L. R. Garett, L. Ruesch, M. C. Mifflin and N. S. Werner, Am. J. Undergrad. Res., 2020, 17, 3–12 Search PubMed
- W. E. Piers, A. J. V. Marwitz and L. G. Mercier, Inorg. Chem., 2011, 50, 12252–12262 CrossRef CAS PubMed
- B. Rao, C. C. Chong and R. Kinjo, J. Am. Chem. Soc., 2018, 140, 652–656 CrossRef CAS PubMed
- M. Mewald and M. Oestreich, Chem. – Eur. J., 2012, 18, 14079–14084 CrossRef CAS PubMed
- Q. Yin, S. Kemper, H. F. T. Klare and M. Oestreich, Chem. – Eur. J., 2016, 22(39), 13840–13844 CrossRef CAS PubMed
- M. Fleige, J. Möbus, T. vom Stein, F. Glorius and D. W. Stephan, Chem. Commun., 2016, 52, 10830–10833 RSC
- A. R. Gatti and T. Wartik, Inorg. Chem., 1966, 5, 329–330 CrossRef CAS
- F. Cheng and F. Jäkle, Polym. Chem., 2011, 2, 2122–2132 RSC
- F. Jäkle, J. Inorg. Organomet. Polym. Mater., 2005, 15, 293–307 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: The general information, experimental procedures, characterization data, and computational details. CCDC 2192816 (1) and 2192817 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03657f |
| This journal is © The Royal Society of Chemistry 2022 |