
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the structural transformations of the PW11Co@ZIF-67 nanocomposite induced by thermal treatment†
Víctor K.
Abdelkader-Fernández
 *a,
Diana M.
Fernandes
*b,
Salete S.
Balula
b,
Luís
Cunha-Silva
*b,
Manuel J.
Pérez-Mendoza
a and
Cristina
Freire
b
*a,
Diana M.
Fernandes
*b,
Salete S.
Balula
b,
Luís
Cunha-Silva
*b,
Manuel J.
Pérez-Mendoza
a and
Cristina
Freire
b
aDepartamento de Química Inorgánica, Facultad de Ciencias, Universidad de Granada, Granada 18071, Spain. E-mail: victorkarim@ugr.es
bREQUIMTE/LAQV, Departamento de Química e Bioquímica, Faculdade de Ciências da Universidade de Porto, Rua do Campo Alegre, 4169-007 Porto, Portugal. E-mail: diana.fernandes@fc.up.pt; l.cunha.silva@fc.up.pt
First published on 2nd November 2022
Abstract
A guest@host POM@ZIF nanocomposite—PW11Co@ZIF-67—has been synthesized using an in situ strategy. This new nanocomposite exhibits (i) individually ZIF-67-cage-confined POM units, (ii) structural defects in the ZIF-67 host induced by the POM, and (iii) charge transfer from the ZIF-67 to the confined POM. In addition, it has served as a template to produce a set of derived samples by applying thermal treatment at various temperatures (200, 400, 500, 600, and 950 °C) under a N2 flow. We have used multiple characterization techniques, ICP-OES, CHNS analysis, XPS, ATR-IR, PXRD, Raman spectroscopy, N2/CO2 adsorption analysis, CV, and TEM/EDS, to fully assess the thermally-induced variation tendencies. The first two derivatives—D200 and D400—show the same nanoarrangement as the PW11Co@ZIF-67 precursor, although with incipient signs of both POM and ZIF-67 structural decompositions. The following samples—D500, D600, and D950—exhibit a carbonaceous nature consisting of C-embedded compositionally complex nanoparticles that involve Co and W present as diverse species, metallic/oxide/phosphate/phosphide. D500 presents the best intrinsic electrochemistry, probably due to the high proportion of pyridinic N moieties doping its C matrix combined with small-sized and highly dispersed Co-enriched nanoparticles. This study focuses on the need for a thorough physicochemical characterization of this class of highly nanostructured materials with a view to exploring their application in electrocatalysis.
1. Introduction
The complementary sizes of Keggin-type polyoxometalates (POMs), ∼1 nm, and zeolitic imidazole framework (ZIF) cages, 1.16 nm,1 make possible the individual encapsulation of the POM units inside the ZIF cavities. This cage-confinement/smart assembly approach, accomplished by in situ ZIF growth surrounding the previously solvated POM units, allows the preparation of highly nanostructured POM@ZIF (guest@host) nanocomposites with potential application in electrocatalysis and energy storage.1–5 In two earlier publications, we reported the cage-confinement of two cobalt-substituted silicotungstic POMs—SiW9Co3 and SiW11Co—in two different hosts: ZIF-8 ([Zn(2-MeIm)2]) and ZIF-67 ([Co(2-MeIm)2]).6,7 We found that the electrocatalytic performances shown by the resulting nanocomposites do not depend on the number of Co atoms in the POM cluster, and more importantly, that a significant synergy between ZIF-67 and the confined silicotungstic POMs is established. On one hand, an electronic charge transfer occurs from the ZIF-67 (host) to the POM units (guest), and, on the other hand, the POM generates structural defects in the ZIF-67 skeleton by partially disrupting the Co2+ node–organic ligand coordinative bonds. This bidirectional POM ↔ ZIF-67 synergistic effect resulted in a remarkable enhancement of the oxygen evolution reaction (OER) performances shown by the SiW(12−x)Cox@ZIF-67 nanocomposites, in comparison with “pristine” ZIF-67. Regardless of the reported electrocatalytic improvement, this class of nanoarchitectures also attract attention due to their role as a template for derived nanocarbons8,9 or nanoporous oxides.10 For instance, when a carbonization treatment is applied, the elevated dispersion at the nanometric scale of the POM units inside ZIF cages maximizes the homogenous distribution of the POM-derived species throughout the resulting C-matrix-embedded metal-containing nanoparticles/nanoclusters.Considering the two positive properties of POM@ZIF-67 materials, i.e., enhanced electroactivity and a high templating effect, we have developed another nanocomposite by encapsulating a cobalt-mono-substituted phosphotungstic POM, PW11Co, into ZIF-67. This Keggin-type POM has two main advantages in relation to silicotungstates: (i) a much simpler synthesis,11–15 and (ii) the possibility of generating electroactive metal phosphides by thermal decomposition.9 For these reasons, we have obtained a set of nanocomposites from the newly prepared PW11Co@ZIF-67 sample by applying thermal treatments at different temperatures (from 200 to 950 °C) under an inert gas flow. Therefore, with a view to explore its potential electrocatalytic applications, we present in this work a detailed characterization of the thermally-induced changes in the original composition, structure, texture, intrinsic electrochemistry, and morphology of PW11Co@ZIF-67.
2. Experimental
2.1. Preparation of PW11Co@ZIF-67 and its derivatives
See technical data regarding materials and reagents in the experimental details section of the ESI.†2.2. Physico(electro)chemical characterization of PW11Co@ZIF-67 and its derivatives
See technical data regarding the characterization equipment, instrumental setups, and the modification of the working electrode with sample inks in the experimental details section of the ESI.†The changes produced by the thermal treatments on the PW11Co@ZIF-67 composition/structure were assessed by inductively coupled plasma optical emission spectrometry (ICP-OES), CHNS analysis, X-ray photoelectron spectroscopy (XPS), attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR), powder X-ray diffraction (PXRD), and Raman spectroscopy. The modification of the porous texture has been evaluated by N2 and CO2 adsorption/desorption measurements. The intrinsic electrochemical behavior of the samples was characterized in 0.1 M KOH(aq) electrolyte (pH = 13.0) by (i) estimating their electroactive surface areas (ECSA) from the capacitance data obtained by charge–discharge tests consisting of successive cyclic voltammetry (CV) registered between 0.46 and 1.06 V vs. RHE at increasingly scan rates, and (ii) studying their intrinsic redox processes revealed in the corresponding CV curves registered between 0.27 and 1.41 V vs. RHE, avoiding lower and higher potentials to suppress water electrolysis. Finally, the morphological variations were studied by transmission electron microscopy (TEM) with energy-dispersive X-ray spectroscopy (EDS).
3. Results and discussion
3.1. Characterization of the guest@host PW11Co@ZIF-67 nanocomposite
PW11Co@ZIF-67 has been prepared using an in situ approach, following the protocol included in our previous study but replacing the SiW11Co POM with PW11Co.6 Analogously to the SiW11Co-ZIF-67 combination, this novel nanocomposite shows a well-defined guest@host nanoarrangement in which PW11Co units (guests) are individually confined inside the ZIF-67 cages (host). The incorporation of PW11Co into ZIF-67 is demonstrated by the detection of P and W, elements that unambiguously belong to PW11Co, in both surface and bulk PW11Co@ZIF-67 compositions (Table 1), obtained by XPS and ICP-OES analysis, respectively. This fact is further corroborated by the presence of characteristic IR vibrational bands from PW11Co in the nanocomposite spectrum (Fig. 1a). Regarding the POM content, approximately one in ten ZIF-67 cages is occupied by a PW11Co unit (see occupancy in Table 1). | ||
| Fig. 1 (a) FTIR spectra (the pink spots signalize IR bands produced by PW11Co) and (b) PXRD diffractograms (numeric values correspond to the average crystallite size, La, for each sample) of the “pristine” materials and PW11Co@ZIF-67. (c) N2 and CO2 adsorption isotherms (desorption branches are omitted for clarity) and (d) TEM images of “pristine” ZIF-67 and PW11Co@ZIF-67. (e) High-resolution XPS W 4f peak profiles of “pristine” PW11Co and PW11Co@ZIF-67 (see the deconvolutions of W 4f and the rest of core-level regions in Fig. S1 in the ESI†), and (f) cyclic voltammograms for “pristine” ZIF-67 and PW11Co@ZIF-67. | ||
| Sample | N2 adsorption | CO2 adsorption | ECSAj | ||||
|---|---|---|---|---|---|---|---|
| S BET (m2 g−1) | V pore (cm3 g−1) | Median pore size (nm) | V micropore (cm3 g−1) | Micropore surface aream (m2 g−1) | Median pore size (nm) | (m2 g−1) | |
| a XPS surface atomic percentages derived from high-resolution C 1s, O 1s, N 1s, Co 2p, P 2p, W 4f, and K 2p core-level peak areas. b The theoretical W/P atomic ratio for PW11Co is 11. c N1 stands for nitrogen atoms coordinated to Co ions, calculated from the N1 component area in N 1s peak deconvolutions (see Fig. S1 in the ESI†). d The theoretical N1/Co atomic ratio of ZIF-67 is 4. e Calculated from the ICP-OES bulk W content. f CHNS analysis bulk mass and atomic percentages. g Calculated by difference: O content = 100 − (C + N + H + Co + P + W). h ICP-OES bulk mass and atomic percentages. i Expressed in POM units per 100 ZIF cages (see occupancy calculation section in the ESI†). j Electrochemically active surface area (see ECSA estimation section in the ESI†). k Surface area calculated from the N2 isotherm data using the Brunauer–Emmett–Teller (BET) equation. l Pore volume calculated from N2 adsorbed at P/P0 = 0.99. m Calculated from the CO2 isotherm data using the Dubinin–Reduskevich method. | |||||||
| ZIF-67 | 1491 | 0.69 | 1.3 | 0.19 | 462 | 0.35 | 490 |
| PW11Co@ZIF-67 | 1422 | 0.65 | 1.3 | 0.15 | 369 | 0.40 | 555 |
A variety of characterization results supports the individual encapsulation of PW11Co inside the ZIF-67 cages. For instance, the nanocomposite W/P atomic ratio (4.0) is far from the ratio of 10.7 of “pristine” PW11Co. This “distortion” is due to the surface character of the XPS technique in combination with a complex 3D guest@host architecture in which the PW11Co units are intimately incorporated into the ZIF-67 skeleton.6,7 See a graphic explanation of this phenomenon in section 3 of the ESI.† In addition, note that POMs merely deposited on surfaces show element atomic ratios closer to the stoichiometric values.17,18 Besides, the absence of K+ ions in PW11Co@ZIF-67 strongly suggests that the POM salt was completely solvated during the ZIF-67 growth, this being an essential requirement for the individual confinement of the PW11Co anion units, probably balanced by H+ ions in the resulting nanocomposite.1 In addition, the absence of PW11Co salt crystallites in the nanocomposite is definitively demonstrated by the nonappearance of diffraction peaks attributable to the POM salt in its PXRD pattern (Fig. 1b). As verified in the case of the previously reported SiW11Co@ZIF-67,6 PW11Co encapsulation generates a structurally defective ZIF-67 skeleton. Thus, the low atomic ratio between the nitrogen atoms from Co2+-coordinated 2-methylimidazole ligands and the Co2+ ions (N1/Co) of PW11Co@ZIF-67 in comparison with that of “pristine” ZIF-67 implies a deficit of coordinated ligands, i.e. the formation of undercoordinated Co nodes (see Table 1, and high-resolution N 1s peak deconvolutions in Fig. S1 in the ESI†).19 This vacancy-type defect results in a more open framework which, along with the occupation of the cages by POM units, causes a loss of microporosity and surface area, as well as an increment of the mean CO2-accessible pore size (see the comparison with ZIF-67 textural parameters, the corresponding isotherm plots, and the pore size distributions in Table 1, Fig. 1c, and Fig. S2,† respectively). Nevertheless, this limited loss of microporosity does not affect its electrochemically active surface area, ECSA, (see Table 1, ECSA estimation section in the ESI,† CV plots in Fig. S3,† and intensity vs. scan rate plots in Fig. S4†), since PW11Co@ZIF-67 exhibits a slightly larger one ≈555 m2 g−1 than ZIF-67, probably due to the elevated intrinsic electroactivity of the incorporated PW11Co units.
The ZIF-67 host defects also partially disrupt the crystallinity of the sample, decreasing its average crystallite size in relation to “pristine” ZIF-67 (crystallite size estimated from the FWHM values of diffraction peaks in the PXRD patterns; see numeric values in Fig. 1b). Additionally, also as a consequence of the PW11Co-induced structural defects, the perfectly faceted polyhedral morphology typical of ZIF-67 samples is moderately modified (Fig. 1d, see other TEM micrographs in Fig. S5†).
In the opposite direction, ZIF-67 influences the confined PW11Co units by establishing a charge transfer from the ZIF-67 skeleton to the POM, more specifically, to its low-lying W-centered LUMOs.1 This electronic interaction is detected by the binding energy shift (ΔBE ≈ −0.5 eV) that the high-resolution XPS W 4f peak of PW11Co@ZIF-67 undergoes compared with the position of this peak in the “pristine” PW11Co (Fig. 1e). This phenomenon is presumably reflected in the PW11Co@ZIF-67 electrochemical behavior: the CVs plotted in Fig. 1f show that the anodic peak of ZIF-67—ascribed to the oxidation process of Co2+ to Co3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20—is shifted toward a lower potential, 1.18 V vs. RHE, in the case of the nanocomposite.
20—is shifted toward a lower potential, 1.18 V vs. RHE, in the case of the nanocomposite.
All these data show that PW11Co@ZIF-67—prepared with a POM of facile synthesis—reveals physico- and electro-chemical features analogous to those of the previously obtained SiW11Co@ZIF-67 nanocomposite.6,7 This demonstrates that the in situ approach followed to encapsulate silicotungstic POMs inside ZIF-67 cages6,7 can be easily extended to other Keggin-type POMs, producing similar guest@host nanoarrangements with POM ↔ ZIF-67 bidirectional synergy.
3.2. Effect of thermal treatments on PW11Co@ZIF-67 features
Several thermal treatments under a nitrogen flow were applied to PW11Co@ZIF-67 (see 2.1.2. Thermal treatments in the experimental section) to obtain a set of nanostructured derivatives by modulating its properties in terms of composition, structure, morphology, and electrochemistry. Temperatures were selected based on the PW11Co@ZIF-67 thermogravimetric (TGA) curve shown in Fig. S6.† The mass loss profile of this material shows a pronounced drop in the range of 500–600 °C, corresponding to 60% of the total mass loss, originated by the collapse of the ordered framework along with a concomitant carbonization process. Thereby, to explore the complete range of thermally induced changes, we have selected two pre-collapse temperatures (200 and 400 °C), 500 °C—when the collapse starts—and two post-collapse temperatures (600 and 950 °C).| a CHNS analysis of bulk mass and atomic percentages. b Calculated by difference: O content = 100 − (C + N + H + Co + P + W). c ICP-OES bulk mass and atomic percentages. d Sum of Co and W elements. e Sum of C, O, N, P, and H. f XPS surface mass and atomic percentages derived from high-resolution C 1s, O 1s, N 1s, Co 2p, P 2p, and W 4f core-level peak areas. g Element surface/bulk mass ratiosbetween brackets. Orange, black, and blue colors indicate high (>1.2), medium (1.2–0.8), and low (<0.8) ratios, correspondingly. |
|---|
|
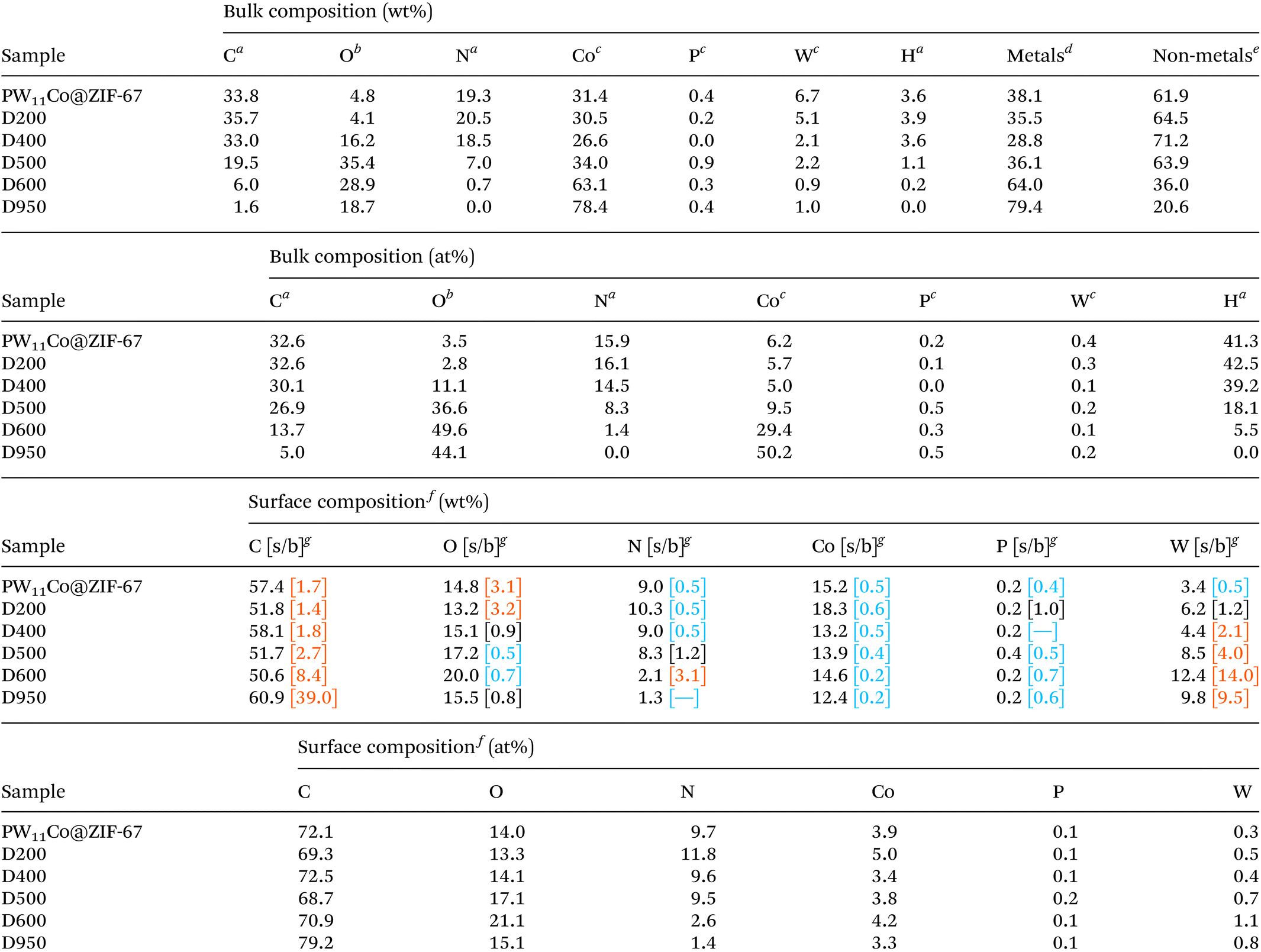
Focusing on carbon atoms, their partial gasification originated by the consecutive treatments with increasing temperatures is accompanied by a drastic migration to surface regions, reaching an extremely large surface/bulk ratio (39.0) in D950. N and W exhibit similar tendencies, i.e., accumulation in the outer regions with the increasing temperatures, with the exception that the maximum ratios are reached in D600. On the other hand, the remaining elements (Co, P, and O) reveal much lower ratios, generally below unity, showing a marked tendency to accumulate in the innermost regions regardless of the thermal treatment. The modification of the shape and position of the high-resolution XPS core-level region spectra provides useful information on the influence of thermal treatments on the PW11Co@ZIF-67 structure, see Fig. S7† and its related full description in the ESI.†
Compositional/structural changes were studied in-depth by deconvoluting the high-resolution spectra (see deconvolution plots in Fig. S1,† component assignations, component positions (BE) and FWHM, and component relative abundances in Tables S1, S2, and S3,† respectively). In Fig. 2a, the carbonization process is revealed by a new component associated with graphenic carbon (C sp2), located at ≈284.2 eV,24,25 detectable from D500 onwards. This abundance increases with the treatment, along with the shake-up (ShU) component, a satellite signal located around 290.5 eV originated by the π → π* (HOMO–LUMO) transition typical of ordered aromatic C sp2 domains.26 In parallel, the C sp3 abundance decreases, indicating that carbonization generates a partially graphitized matrix. This gain in C sp2-based structural order is also revealed by the decrease in the component widths (Fig. 2b). In Fig. 2c it is observed that the relative abundance of the lowest BE O 1s component (see O 1s deconvolution plots in Fig. S1 and abundances in Table S3†) increases with the increasing treatment T. Besides, its position is progressively shifted from ≈530.5 eV (in PW11Co@ZIF-67) to ≈529.9 eV (in D950). This shift suggests that the chemical environment that causes this component is modified with the thermal treatments. In PW11Co@ZIF-67 and D200, this component is fully originated by the W–O bonds of the POM, whilst in the case of D600 and D950, it can be attributed to CoO and WOx (2 ≤ x ≤ 3),21 produced by the high temperatures of the corresponding treatments. In D400 and D500, whose lowest BE components show an intermediate position (≈530.2 eV), the two different environments must coexist, hence an intermediate degradation state of PW11Co is found in that temperature range.
 | ||
| Fig. 2 Variations with the treatment temperature of (a) the relative abundances for C 1s deconvolution components; (b) the FWHM (full width at half maximum) values for all C 1s deconvolution components, except for ShU and plasmon; (c) the relative abundance and position (BE) of the O 1s deconvolution component associated with (i) O–W bonds in the PW11Co cluster—in PW11Co@ZIF-67, and D200—, (ii) O–W (POM)/metal oxide mixture—in D400 and D500—, and (iii) metal oxides—in D600 and D950—; (d) the relative abundance and position (BE) of the N 1s deconvolution component associated with (i) C–N–C bonds in the ZIF-67 host—in PW11Co@ZIF-67, D200, and D400—and (ii) pyridinic groups in the C matrix—in D500, D600, and D950—; and (e) the relative abundances for Co 2p deconvolution components. Note that 0 °C values—in the x-axis of (a), (b), (c), and (e)—correspond to the non-treated precursor PW11Co@ZIF-67. | ||
Additionally, the comparison of N 1s deconvolutions (Fig. S1†) demonstrates the conversion of the N atoms belonging to the pristine ZIF-67 organic ligands19,27 to pyridinic, pyrrolic, and graphitic quaternary N groups doping the carbon matrix in D500, D600, and D950.22 Even, the proportion of N from “intact” organic ligands in D400 decreases from ≈88% in PW11Co@ZIF-67 to ≈77% (Fig. 2d and Table S4†), denoting an incipient ZIF-67 decomposition. Fig. 2d also shows that the abundance of the pyridinic moiety diminishes from D500 onwards. Focusing on Co 2p deconvolutions, Fig. 2e shows that the abundance of the component originated by Co0, and possibly by low amounts of CoxPy, increases from ≈3% in D500 to ≈17% in D950, proving a limited reducing effect of the thermal treatments on cobalt. This increment of Co0 is accompanied by a concomitant diminution of the CoO proportion, also revealed by the decreasing satellite abundances associated with oxidized cobalt.23
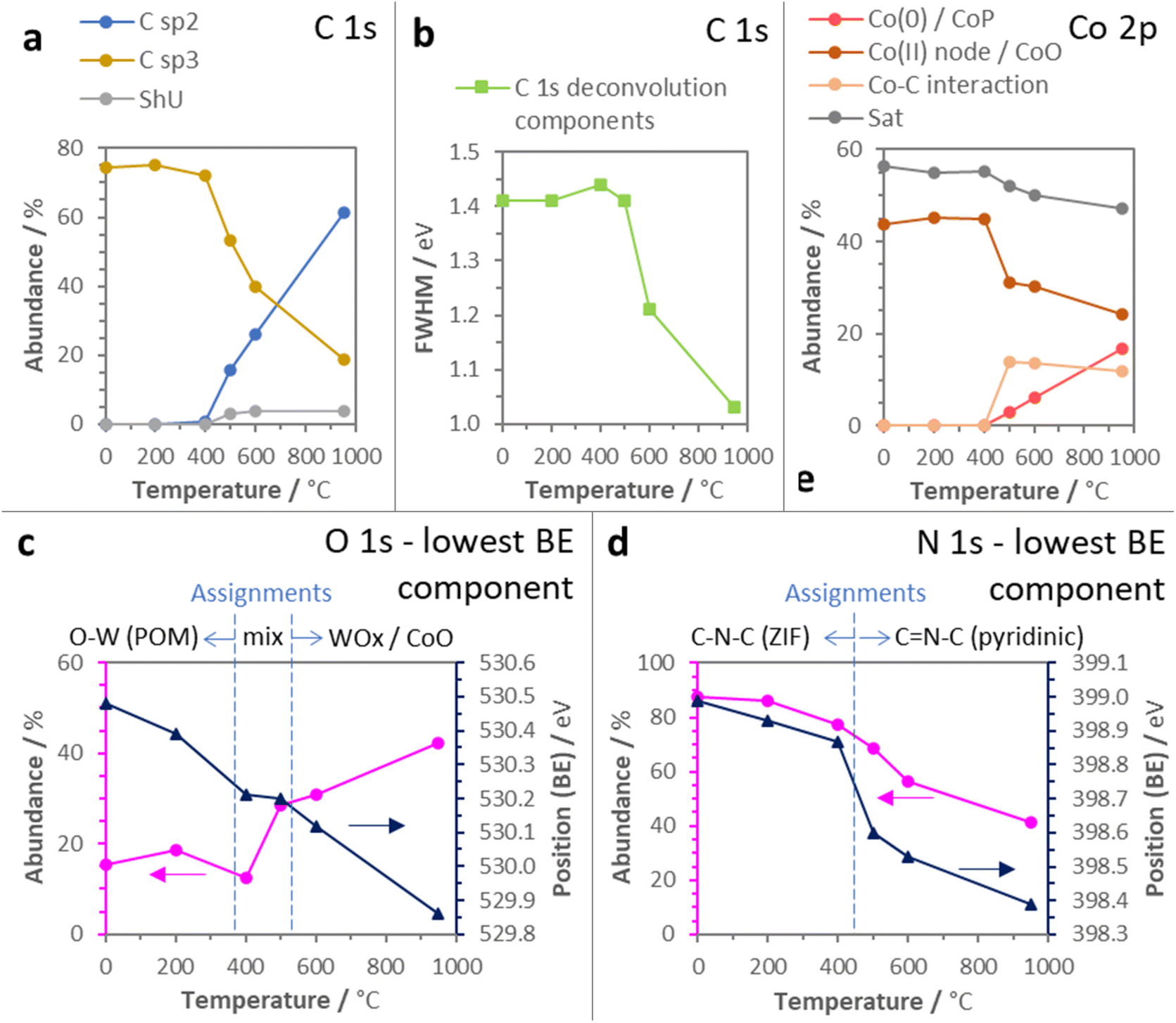
IR and Raman spectroscopies and PXRD further support the detailed assessment of the thermally induced structural modification of PW11Co@ZIF-67. Fig. 3a includes the IR spectra for each sample. ZIF-67 vibrational peaks are visible until D400, and from D500 onwards they are vanished, being substituted by indistinctive broad bands characteristic of carbon materials. Concentrating on PW11Co vibrational features (marked with pink spots in Fig. 1a), they are still completely detectable in D200, but in the material obtained at 400 °C some of them are lost, pointing out the starting point for POM structural degradation. This is in full agreement with the XPS O 1s deconvolution results exposed above (see commentaries concerning Fig. 2c). Furthermore, the powder XRD patterns indicate that the mild thermal treatments (200 and 400 °C) preserve the pristine crystallinity of the ZIF-67 skeleton in PW11Co@ZIF-67 since all the ZIF-67 diffraction peaks remain being detectable in D200 and D400 patterns, and their average crystallite sizes are not decreased (Fig. 3b). On the other hand, D500, D600, and D950 patterns reveal a total absence of the ZIF-67 features and very low intensities (note that they are 10×-scaled in Fig. 3b), showing up the total thermally-induced structural collapse of the ZIF skeleton. Moreover, they do not exhibit the graphitic (002) peak at 24–26°, revealing that no parallel stacked graphenic layers are formed in the C matrixes.
 | ||
| Fig. 3 (a) ATR-IR spectra, (b) PXRD patterns, and (c) Raman spectra (λ = 532 nm) of PW11Co@ZIF-67 and its derivatives. Note that (i) pink spots in (a) indicate IR vibrational bands originated by PW11Co (see Fig. 1a), (ii) numeric values in (b) correspond to the average crystallite sizes (Lc) calculated using the Scherrer formula, (iii) in (c), cyan spots indicate ZIF-67 vibrational features and numeric values in D500, D600, and D950 Raman spectra correspond to ID/IG ratios. | ||
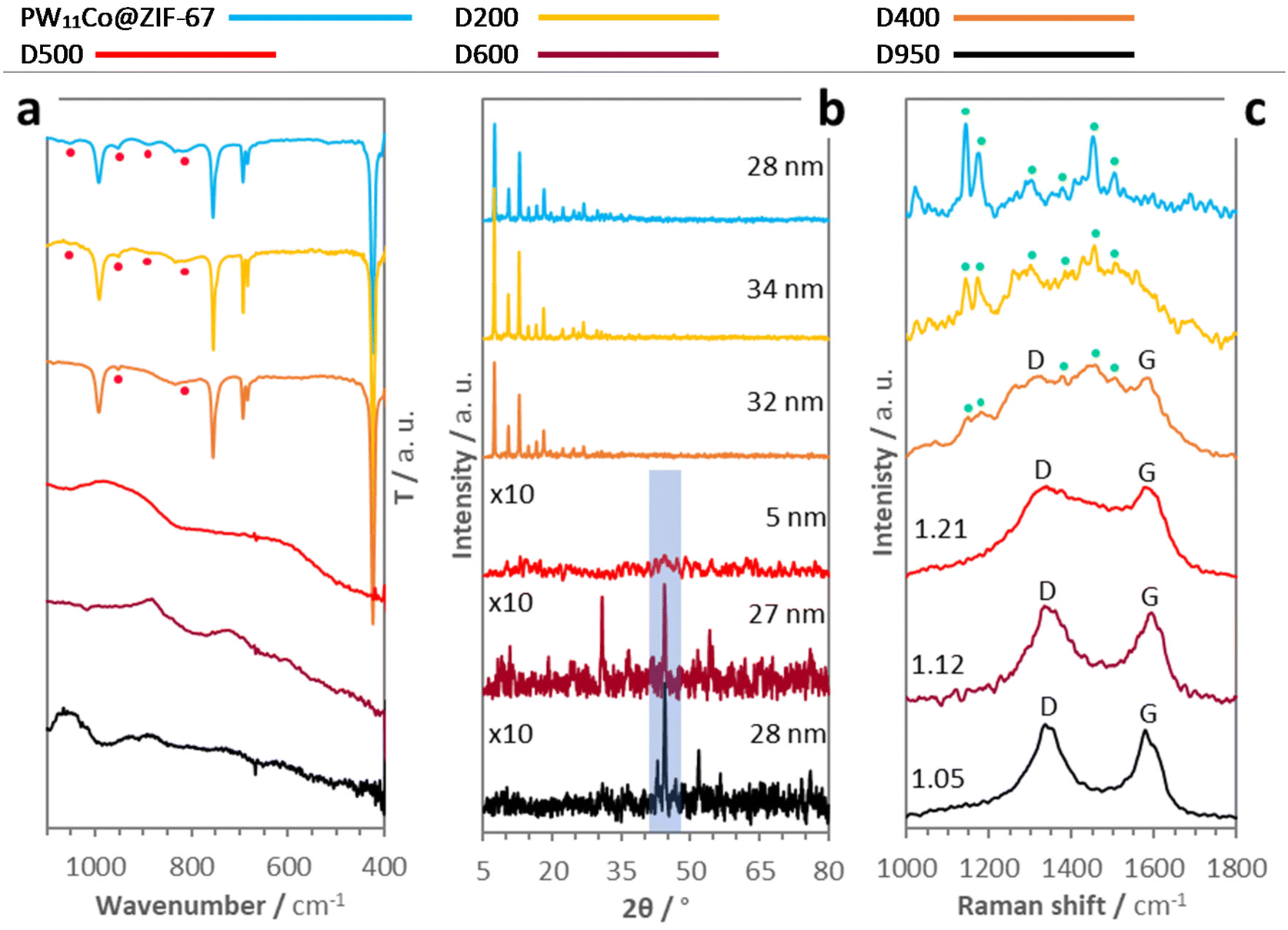
Regarding the diffraction peaks, D500 only shows a barely detectable incipient peak at a 2θ of ≈44.4°, which becomes much more intense in the D600 and D950 patterns (note the translucid rectangle in Fig. 3b and see detailed XRD patterns in Fig. S8†). Thereby, D500 represents an early stage for the nucleation of metal-containing nanoparticles (M-NPs). Specifically, the position of this feature is compatible with the formation of a Co0 (111) metallic phase28 exhibiting growing crystallite sizes (Lc) with the treatment temperature, a result that agrees with the XPS Co 2p deconvolution data. Besides, there are other diffraction peaks in the D600 and D950 diffractograms, although their elevated noise levels make difficult the unambiguous identification of all features. However, no existence of these peaks in the characteristic XRD pattern of ZIF-67-derived nanocarbons—obtained under similar carbonization conditions—29 along with their positions suggest the formation of other nanocrystalline phases directly related to the presence of PW11Co in the precursor, presumably cobalt/tungsten oxides (CoxOy/WOx), and phosphides (CoxPy/WxPy) (see Fig. S8†). Focusing on Raman analysis (see Fig. S9†), selected spectra obtained with an excitation wavelength of 532 nm are obtained in Fig. 3c. While D200 and D400 Raman spectra still present multiple features of PW11Co@ZIF-67 (signaled by cyan spots), D500, D600, and D950 show the D and G bands characteristic of carbon materials. In fact, these bands are barely detectable in the D400 spectrum as well, denoting an early and limited C matrix formation at 400 °C. The increment of the carbon structural order with the temperature is revealed by the diminishing ID/IG ratios,30,31 from 1.21 in D500 to 1.05 in D950 (these ratios were calculated from the deconvolution of Raman spectra in two components, D and G; see 2-component deconvolution plots in Fig. S10† and the intensity and FWHM parameters in Table S4†). This gain of order in the C matrixes is also unveiled by the sharpening of both D and G peaks from D500 onwards: carbon crystallite sizes (La) were calculated from D band FWHM values using the spatial confinement model equation for disordered graphene-based carbons (La = 580/FWHMD).32 These average sizes are 2.6, 4.4, and 5.1 nm for D500, D600, and D950, respectively, reflecting the graphenic domain extensions. This trend is in accordance with XPS metrics since the La values establish an inverse linear relationship with the C sp3/C sp2 abundance ratios (purple circle data series in Fig. 4a) calculated from the areas of C 1s C sp2 and C sp3 components in the C 1s deconvolutions (see Fig. 2a, S1, and Table S4†).
 | ||
| Fig. 4 (a) Correlations of carbon crystallite sizes (La)—from Raman 2-component deconvolutions—and D3/G area ratios—from Raman 5-component deconvolutions—with C sp3/C sp2 area ratios—from XPS C 1s deconvolutions—in D500, D600, and D950. And (b) 5-component deconvolution plot of a selected Raman spectrum of D600. | ||
Deconvoluting Raman spectra into five components (D4, D, D3, G, and D′) is a useful tool to delve into carbon matrix structural order. Table 3 shows the consensual carbon structures, whose vibrational modes generate each component, and their approximated Raman shifts (position).33–36Fig. 4b show the 5-component deconvolution of a selected spectrum for D600 (see the rest of the deconvolution plots in Fig. S11†). The average metrics derived from these deconvolutions, namely the component intensities and abundances, are included in Table 3, while the positions and FWHM values are collected in Table S5.† As the D3 component is associated with C sp3 amorphous carbon, ID3/Itotal ratios proportionate a measure of the carbon structural order analogous to the ID/IG ratios obtained from 2-component Raman spectra deconvolution. Similarly, ID3/Itotal ratios decrease from 0.30 in D500 to 0.07 in D950. Paying attention to component abundances, the increment of carbon structural order with the rising treatment temperatures is based on the loss of amorphous carbon: the D500 D3 component abundance is ≈45%, whilst in D950 it drops to ≈9%, and not to an extensive graphitization process. This explains the no detection of the graphitic peak (002) in the PXRD patterns of the three carbonaceous samples (see Fig. 3b and S8†).
| Description of components33–36 | ||
|---|---|---|
| Component | Assignation | Position (cm−1) |
| D4 | sp2-based carbon chains at graphenic layer edges | 1200–1310 |
| D | Defects in sp2 (point defects -in plane sp2 non-hexagonal rings, edges, curvature) | ≈1350 |
| D3 | Amorphous carbon (i.e., non-crystalline sp3-dominated carbon) | ≈1500 |
| G | Extension of sp2 ordered domains | ≈1580 |
| D′ | Defects in sp2 (similar to D) | ≈1620 |
| Raman deconvolution metrics | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Intensity | I D3/Itotala | Abundanceb (%) | |||||||||
| Sample | D4 | D | D3 | G | D′ | D4 | D | D3 | G | D′ | |
| a I total stands for the sum of the five components: ID4 + ID + ID3 + IG + ID′. b Abundances correspond to the component relative areas. | |||||||||||
| D500 | 6.81 | 46.06 | 47.79 | 45.14 | 12.24 | 0.30 | 3.6 | 31.2 | 45.5 | 17.5 | 2.2 |
| D600 | 3.05 | 16.31 | 4.06 | 15.48 | 3.56 | 0.10 | 10.6 | 45.8 | 13.9 | 28.3 | 1.5 |
| D950 | 5.91 | 48.11 | 8.20 | 44.77 | 11.96 | 0.07 | 11.3 | 48.0 | 9.5 | 28.3 | 2.9 |
Finally, we compared the D3/G area ratios with the above-mentioned XPS C sp3/C sp2 area ratios, finding an intense exponential correlation, R2 = 0.99 (blue diamond data series in Fig. 4a). This fitting supports the Raman–XPS complementarity in the study of MOF-derived nanocarbon matrixes, and the intrinsic association of the Raman D3 and G components with the XPS C sp3 and C sp2 components, respectively.
 | ||
| Fig. 5 (a) N2 sorption isotherms of PW11Co@ZIF-67 and its derivatives. (b) Regions of P/P0 < 0.025 for PW11Co@ZIF-67, D200 and D400. (c) CO2 sorption isotherms of PW11Co@ZIF-67 and its derivatives. Note that in (a) and (c) desorption branches have been omitted for clarity, and in (b) the two translucid rectangles highlight abrupt adsorption steps. | ||
PW11Co@ZIF-67, D200, and D400 N2 profiles are typical of highly microporous solids (type I isotherms), whilst the isotherms from D500 onwards show a drastic loss of microporosity due to the ZIF-67 skeleton thermal decomposition. Mild treatments (200 and 400 °C) do not have a noticeable effect on microporosity; however, especially the D400 N2 isotherm shows the two-step N2 sorption region at P/P0 < 0.025 more soften than that for pristine PW11Co@ZIF-67 (see highlighted zones in Fig. 5b). The 1st step is normally ascribed to the reorganization of the N2 molecules inside cages and the 2nd step to the imidazolate linker rotation provoked by the gas pressure, i.e. the gate-opening effect.37 Thus, mild treatments seem to partially limit the framework flexibility, likely due to the disruption of a certain fraction of Co–ligand bonds. The more pronounced slopes of the PW11Co@ZIF-67, D200, and D400 isotherms in Fig. 5c indicate that the microporosity accessible to the CO2 molecules is also more developed in these samples. Table 4 shows the textural parameters derived from both N2 and CO2 sorption measurements. BET surface areas reflect the loss of microporosity envisaged in the N2 isotherms: the two pre-collapse samples exhibit areas above 1400 m2 g−1, decreasing below 130 m2 g−1 from D500 onwards. The pore volume, Horvath–Kawazoe pore size distributions (Fig. S2†), and mean pore sizes reflect the microporosity loss as well. On the other hand, the DR micropore volume and surface areas also follow this tendency, except that they slightly increase in D950. This suggests that at 950 °C an incipient recuperation of the ultramicroporosity (<0.5 nm) occurs, non-accessible to the N2 molecules, previously lost during the ZIF degradation/carbonization process.
| Sample | N2 adsorption | CO2 adsorption | ECSAd | ||||
|---|---|---|---|---|---|---|---|
| S BET (m2 g−1) | V pore (cm3 g−1) | Mean pore size (nm) | V micropore (cm3 g−1) | Micropore surface areac (m2 g−1) | Median pore size (nm) | (m2 g−1) | |
| a Surface area calculated from the N2 isotherm data using the Brunauer–Emmett–Teller (BET) equation. b Pore volume calculated from N2 adsorbed at P/P0 = 0.99. c Calculated from the CO2 isotherm data using the Dubinin–Redushkevich (DR) equation. d Electrochemically active surface area (see ECSA estimation section in the ESI†). | |||||||
| PW11Co@ZIF-67 | 1422 | 0.65 | 1.3 | 0.15 | 369 | 0.40 | 555 |
| D200 | 1507 | 0.67 | 1.3 | 0.16 | 403 | 0.40 | 233 |
| D400 | 1422 | 0.68 | 1.4 | 0.14 | 359 | 0.40 | 490 |
| D500 | 83 | 0.17 | 21.2 | 0.06 | 141 | 0.35 | — |
| D600 | 30 | 0.15 | 13.3 | 0.04 | 106 | 0.39 | — |
| D950 | 16 | 0.04 | 21.8 | 0.08 | 189 | 0.41 | 6 |
The electrochemically active surface areas (ECSA) included in Table 4 were estimated using the following equation: ECSA = Cdl/Cref, where Cdl and Cref stand for the double-layer and reference capacitance values per unit area, respectively (see calculation details in ECSA estimation section in the ESI, CV plots in Fig. S3, current intensity vs. scan rate graphs in Fig. S4, and Cdl and Cref values in Table S6†). The mild temperature treatment (200 and 400 °C) generated ECSA decreases in comparison with PW11Co@ZIF-67, not imputable to the drop of the corresponding BET and micropore surface areas since they are similar for the three samples. Thus, the mild temperature-induced limited degradation of the POM units and the ZIF-67 skeleton is the most probable cause. The ECSA for D500 and D600 cannot be calculated due to their Cdl being in negative values (see Table S6†). This phenomenon can be produced by the low BET and micropore surface areas of these samples, accompanied by a high uncertainty in the Cdl estimation provoked by the large noise-to-signal ratios of the CV curves, exhibiting very low current intensities (see Fig. S3 and S4†). In addition, the presence of metal oxide NPs in these samples can contribute to the drop of their electroactive areas, since metal oxides typically exhibit low ECSA values associated with low BET surface areas.38 Similarly, D950—with an elevated proportion of metal oxide NPs as well—presents an extremely low ECSA, 6 m2 g−1. Probably, and unlike samples D500 and D600, in this case, the above-mentioned limited recovery of ultramicroporosity makes the ECSA calculable.
CV tests have been performed to study the intrinsic electrochemical activity of the samples (Fig. 6). Noteworthily, the D500 CV plot in Fig. 6d shows much higher current densities (j) than the CV plots of PW11Co@ZIF-67 and the other derivatives, envisaging remarkable intrinsic activity in electrocatalysis. These notable current levels can be related to the numerous and homogenously dispersed N atoms doping the D500 carbon matrix, NXPS = 9.5 at%, see Table 2, and Fig. S1†—. Besides, the six materials show faradaic processes (peaks) originated by cobalt redox reactions. All the samples clearly show a cathodic peak at ≈1.1 V vs. RHE—associated with Co3+ → Co2+ reduction—,20 however the anodic peaks change. While the PW11Co@ZIF-67 CV curve (Fig. 6a) exhibits a Co2+ → Co3+ oxidation peak at 1.18 V vs. RHE, the D200 and D400 CV plots (Fig. 6b and c, correspondingly) show a peak shifted to higher potentials—almost coincident with the anodic peak of “pure” ZIF-67 CV (see Fig. 1f)—and absence of it, respectively. These modifications are presumably originated by the progressive degradation of both ZIF-67 and PW11Co, disrupting their host → guest electronic interaction (see Fig. 1e and its commentaries). Then, CV plots of D500, D600, and D950 include another anodic feature located at lower potentials—1.06, 0.99, and 1.12 V vs. RHE, respectively—than the Co2+ → Co3+ oxidation peak. This new peak can be related to the in situ formation of cobalt hydroxides, Co(OH)2, on metallic Co surfaces during the application of a voltage in the CV tests in alkaline electrolytes.39 In fact, the CV plot of D950—the sample that contains the largest Co0 proportion (see XPS Co 2p deconvolution in Fig. 2e)—presents an expanded range of possible redox processes, involving additional peaks related to the formation of other cobalt oxide/hydroxide species with diverse oxidation states: Co3O4, Co2O3, Co(OH)3 and CoO2.39–41
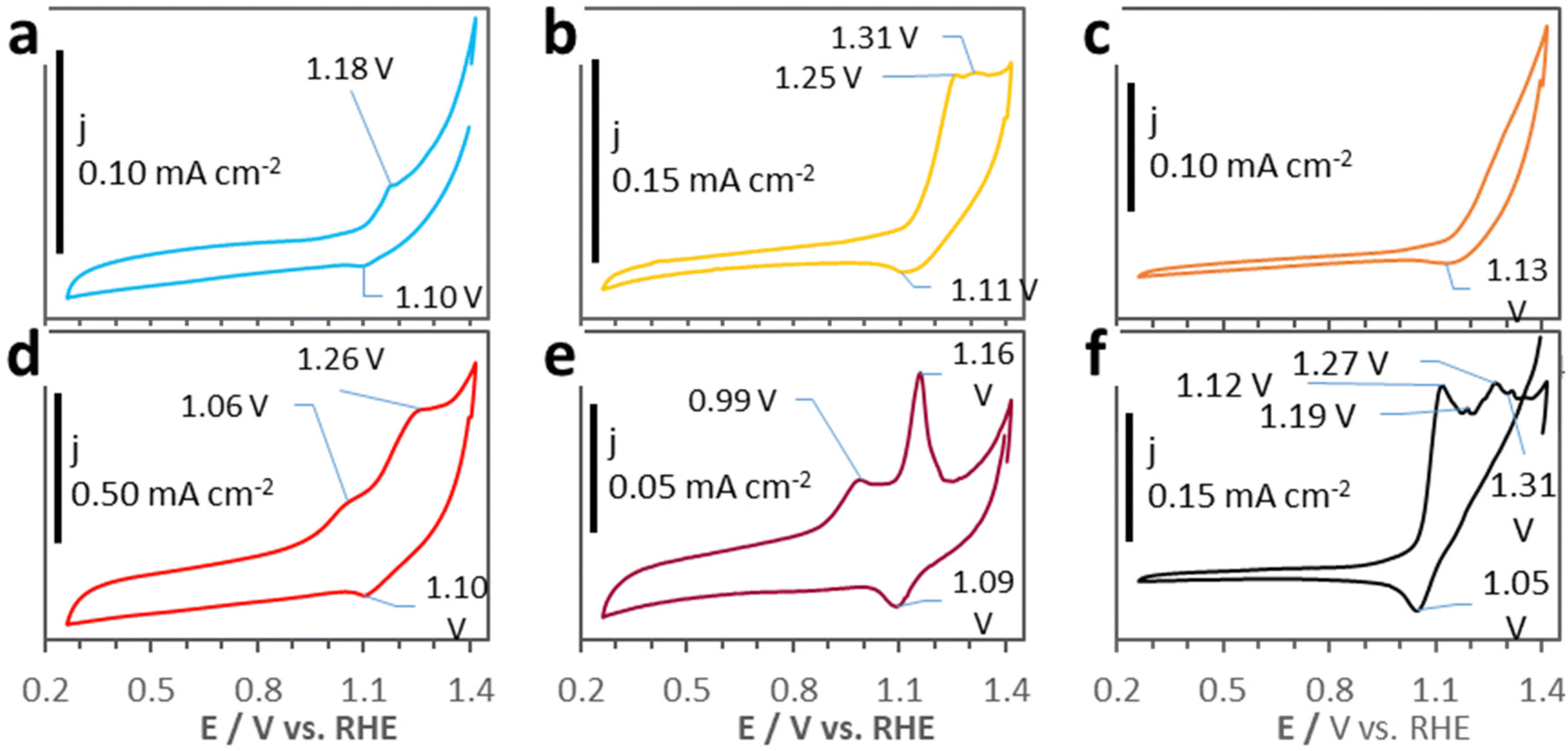 | ||
| Fig. 6 CV plots of (a) PW11Co@ZIF-67, (b) D200, (c) D400, (d) D500, (e) D600, and (f) D950. | ||
TEM images included in Fig. 7a–f (see additional micrographs in Fig. S5†) show significant morphological modification of the precursor with the thermal treatments. PW11Co@ZIF-67 exhibits ZIF characteristic polyhedral-shaped particles involving wrinkled regions originated by the POM encapsulation on ZIF-67 cages. The two following samples, D200 and D400, maintain this aspect, although D400 presents emerging granular irregularities. Since W 4f and O 1s deconvolution data, IR, Raman, and N2 adsorption analyses indicate that the 400 °C treatment leads to a limited structural decomposition of the PW11Co units and the ZIF-67 skeleton (see Fig. S7f† and Fig. 2c, 3a, c, 5b, as well as their respective commentaries), it is plausibly to relate these changes to the observed D400 morphological modifications. Then, from D500 onwards (Fig. 7d–f) carbonaceous aggregates embedding highly contrasted nanoparticles are detected. The complete loss of the characteristic ZIF-67 polyhedral shape in these samples can be ascribed to a certain destabilization effect produced on the ZIF-67 framework by the encapsulated PW11Co units, since previously reported ZIF-67-derived nanocarbons do preserve—at least partially—that geometry.29,42 The elevated contrast of the carbon-embedded NPs allows us to infer their high metal contents. This point is confirmed by the element distribution maps obtained for W and Co atoms in Fig. 7g–l and the remaining elements in Fig. S14.† Paying attention to the NP morphologies, their average sizes increase from a few nanometers to hundreds with the heat treatment, this trend being in agreement with the increasing average crystallite sizes of D500, D600, and D950 (see the PXRD data in Fig. 3b). W distribution maps in Fig. 7g–l proportionate the information on both the POM locations and NP formation.
 | ||
| Fig. 7 TEM micrographs with two amplifications of (a) PW11Co@ZIF-67, (b) D200, (c) D400, (d) D500, (e) D600, and (f) D950. Tungsten and cobalt distribution maps of (g) PW11Co@ZIF-67, (h) D200, (i) D400, (j) D500, (k) D600, and (l) D950. | ||
PW11Co@ZIF-67, D200, and D400 show fairly homogeneous W distributions, barely concentrated in the wrinkled zones, suggesting a good dispersion of the PW11Co units. D500 exhibits small-sized NPs in the TEM micrographs; however, its W map does not show a significant accumulation of tungsten in them. In contrast, its Co distribution map does present cobalt concentration in those contrasted regions, demonstrating that the formation of Co-containing NPs precedes the formation of the W-containing NPs. Regarding D600 and D950, no complete overlapping of the W- and Co-enriched regions reveals that mixed bimetallic W–Co NPs with a homogeneous distribution of these two metals are not formed in a large extension. Lastly, P atoms show distributions (Fig. S14†) similar to W and Co: high homogeneity in the precursor, D200 and D400—suggesting an elevated dispersion of the encapsulated PW11Co units—and concentration in the NPs from D500 onwards, due to the formation of metal phosphates and phosphides.
4. Conclusions
We prepared a novel guest@host nanocomposite, PW11Co@ZIF-67, exhibiting physicochemical parameters compatible with the individual cage-confinement of the POM units, POM-induced structural defects in the ZIF-67 skeleton, and ZIF-67 → POM charge transfer, properties analogous to those shown by the previously reported SiW11Co@ZIF-67, but achieved using a POM with an easier synthesis that can produce electroactive metal phosphides when heating treatments are performed.Structural modifications in PW11Co@ZIF-67 were induced via thermal treatments at different temperatures (200, 400, 500, 600, and 950 °C). D200 and D400 undergo very limited modifications in both the ZIF-67 skeleton and confined PW11Co units. These concurrent POM/ZIF partial decompositions probably compromise their electronic interaction, deteriorating their electrochemical features. Noteworthily, D500 exhibits finely dispersed few nm-sized Co-enriched NPs embedded in a highly disordered C matrix. In contrast, in D600 and D950, the higher temperatures generate two types of large-sized NPs, one enriched in Co and one enriched in W, with low amounts of phosphates and phosphides derived from PW11Co. As a consequence of the increasing T, these two samples show more ordered C matrixes. Although the three carbonaceous derivatives undergo drastic ECSA drops, likely caused by the collapse of the pristine ZIF-67 microporosity, their CV plots involve new anodic peaks indicating an enhancement of their intrinsic electrochemistry. Apart from the new peaks, the CV of D500 presents an elevated current density, probably originated from an effective interaction between its abundant doping N moieties—specially pyridinic groups—and the very small-sized Co-containing NPs.
We described the thermally induced migration of the elements by combining XPS, ICP-OES, and CHNS analysis data. Minor alterations in XPS N 1s and O 1s deconvolutions, IR and Raman spectra profiles, and low-P/P0 N2 isotherm regions have been crucial for detecting slight structural modifications in D200 and D400. Additionally, Raman-XPS C 1s deconvolution correlations provided a good estimation of carbon structural order in D500, D600, and D950. All these results highlight the importance of the in-depth physico(electro)chemical characterization of these POM@MOF derivatives.
Finally, the next step of this research will be focused on the complete characterization of PW11Co@ZIF-67 and its derivatives as electrocatalysts for the strategic oxygen-related processes—oxygen reduction (ORR) and evolution reactions (OER)—with special emphasis on structure-performance correlations.
Author contributions
Víctor K. Abdelkader-Fernández: investigation, formal analysis, and writing – original draft. Diana M. Fernandes: conceptualization, writing – review & editing, and supervision. Salete S. Balula: conceptualization and writing – review & editing. Luís Cunha-Silva: conceptualization, writing – review & editing, and supervision. Manuel J. Pérez-Mendoza: supervision, writing – review & editing, and resources. Cristina Freire: supervision, project administration, and funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work received financial support from the project POCI-01-0145-FEDER-016422 – UniRCell, financed by national funds through the FCT/MCTES (Fundaçao para a Ciência e a Tecnologia/Ministério da Ciência, Tecnologia e Ensino Superior) and when appropriate co-financed by FEDER (Fundo Europeu de Desenvolvimento Regional). This work also received support from PT national funds (FCT/MCTES) through the projects UIDB/50006/2020 and UIDP/50006/2020. VKAF thanks the project UniRCell for his previous work contracts and the Junta de Andalucía (Spanish regional government) for his current Postdoc contract (PAIDI 2020, Young Doctors Program). DMF, SSB and LCS thank FCT/MCTES for funding through the Individual Call to Scientific Employment Stimulus (2021.00771.CEECIND/CP1662/CT0007, CEECIND/03877/2018, and CEECIND/00793/2018, respectively).References
- S. Mukhopadhyay, J. Debgupta, C. Singh, A. Kar and S. K. Das, Angew. Chem., Int. Ed., 2018, 57, 1918–1923 CrossRef CAS PubMed.
- S. Mulkapuri, A. Ravi and S. K. Das, Chem. Mater., 2022, 34(8), 3624–3636 CrossRef CAS.
- D. Kumar, A. Joshi, G. Singh and R. K. Sharma, Chem. Eng. J., 2022, 431, 134085 CrossRef CAS.
- P. Mialane, C. Mellot-Draznieks, P. Gairola, M. Duguet, Y. Benseghir, O. Oms and A. Dolbecq, Chem. Soc. Rev., 2021, 50, 6152–6220 RSC.
- S. Mukhopadhyay, O. Basu, A. Kar and S. K. Das, Inorg. Chem., 2020, 59, 472–483 CrossRef CAS PubMed.
- V. K. Abdelkader-Fernandez, D. M. Fernandes, S. S. Balula, L. Cunha-Silva and C. Freire, J. Mater. Chem. A, 2020, 8, 13509–13521 RSC.
- V. K. Abdelkader Fernández, D. Mesquita Fernandes, S. Silva Balula, L. Cunha-Silva and C. Freire, ACS Appl. Energy Mater., 2020, 3, 2925–2934 CrossRef.
- S. Q. Wang, Y. L. Cao, W. Jia, Z. J. Lu and D. Z. Jia, Appl. Catal., B, 2021, 298, 120579 CrossRef CAS.
- Y. J. Tang, H. J. Zhu, L. Z. Dong, A. M. Zhang, S. L. Li, J. Liu and Y. Q. Lan, Appl. Catal., B, 2019, 245, 528–535 CrossRef CAS.
- Q. Lan, Z. M. Zhang, C. Qin, X. L. Wang, Y. G. Li, H. Q. Tan and E. B. Wang, Chem. – Eur. J., 2016, 22, 15513–15520 CrossRef CAS PubMed.
- S. S. Balula, L. Cunha-Silva, I. Santos, A. C. Estrada, A. C. Fernandes, J. A. S. Cavaleiro, J. Pires, C. Freire and A. M. V. Cavaleiro, New J. Chem., 2013, 37, 2341–2350 RSC.
- M. S. Balula, J. A. Gamelas, H. M. Carapuca, A. M. V. Cavaleiro and W. Schlindwein, Eur. J. Inorg. Chem., 2004, 619–628 CrossRef CAS.
- M. M. Q. Simoes, C. M. M. Conceicao, J. A. F. Gamelas, P. Domingues, A. M. V. Cavaleiro, J. A. S. Cavaleiro, A. J. V. Ferrer-Correia and R. A. W. Johnstone, J. Mol. Catal. A: Chem., 1999, 144, 461–468 CrossRef CAS.
- J. G. Liu, F. Ortega, P. Sethuraman, D. E. Katsoulis, C. E. Costello and M. T. Pope, J. Chem. Soc., Dalton Trans., 1992, 1901–1906 RSC.
- G. Herve and A. Teze, Inorg. Chem., 1977, 16, 2115–2117 CrossRef CAS.
- C. M. Tourne, G. F. Tourne, S. A. Malik and T. J. R. Weakley, J. Inorg. Nucl. Chem., 1970, 32, 3875–3890 CrossRef CAS.
- V. K. Abdelkader-Fernandez, D. M. Fernandes, L. Cunha-Silva, A. J. S. Fernandes and C. Freire, Electrochim. Acta, 2021, 389, 138719 CrossRef CAS.
- D. M. Fernandes, M. P. Araujo, A. Haider, A. S. Mougharbel, A. J. S. Fernandes, U. Kortz and C. Freire, ChemElectroChem, 2018, 5, 273–283 CrossRef CAS.
- F. Y. Tian, A. M. Cerro, A. M. Mosier, H. K. Wayment-Steele, R. S. Shine, A. Park, E. R. Webster, L. E. Johnson, M. S. Johal and L. Benz, J. Phys. Chem. C, 2014, 118, 14449–14456 CrossRef CAS.
- W. R. Zheng, M. J. Liu and L. Y. S. Lee, ACS Catal., 2020, 10, 81–92 CrossRef CAS.
- W. Grunert, E. S. Shpiro, R. Feldhaus, K. Anders, G. V. Antoshin and K. M. Minachev, J. Catal., 1987, 107, 522–534 CrossRef.
- M. Ayiania, M. Smith, A. J. R. Hensley, L. Scudiero, J. S. McEwen and M. Garcia-Perez, Carbon, 2020, 162, 528–544 CrossRef CAS.
- D. R. Sun, L. Ye, F. X. Sun, H. Garcia and Z. H. Li, Inorg. Chem., 2017, 56, 5203–5209 CrossRef CAS PubMed.
- S. Hanelt, J. F. Friedrich, G. Orts-Gil and A. Meyer-Plath, Carbon, 2012, 50, 1373–1385 CrossRef CAS.
- N. Lachman, X. Sui, T. Bendikov, H. Cohen and H. D. Wagner, Carbon, 2012, 50, 1734–1739 CrossRef CAS.
- B. Brena, Y. Luo, M. Nyberg, S. Carniato, K. Nilson, Y. Alfredsson, J. Ahlund, N. Martensson, H. Siegbahn and C. Puglia, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 195214 CrossRef.
- A. Awadallah-F, F. Hillman, S. A. Al-Muhtaseb and H. K. Jeong, J. Mater. Sci., 2019, 54, 5513–5527 CrossRef CAS.
- V. K. Abdelkader-Fernandez, D. M. Fernandes, S. S. Balula, L. Cunha-Silva, M. J. Perez-Mendoza, F. J. Lopez-Garzon, M. F. Pereira and C. Freire, ACS Appl. Energy Mater., 2019, 2, 1854–1867 CrossRef CAS.
- W. H. Li, A. F. Zhang, X. Jiang, C. Chen, Z. M. Liu, C. S. Song and X. W. Guo, ACS Sustainable Chem. Eng., 2017, 5, 7824–7831 CrossRef CAS.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cancado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276–1291 RSC.
- P. Puech, J.-M. Plewa, P. Mallet-Ladeira and M. Monthioux, Carbon, 2016, 105, 275–281 CrossRef CAS.
- M. Couzi, J. L. Bruneel, D. Talaga and L. Bokobza, Carbon, 2016, 107, 388–394 CrossRef CAS.
- M. Asadullah, S. Zhang, Z. H. Min, P. Yimsiri and C. Z. Li, Bioresour. Technol., 2010, 101, 7935–7943 CrossRef CAS PubMed.
- X. J. Li, J. Hayashi and C. Z. Li, Fuel, 2006, 85, 1700–1707 CrossRef CAS.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Poschl, Carbon, 2005, 43, 1731–1742 CrossRef CAS.
- D. Fairen-Jimenez, S. A. Moggach, M. T. Wharmby, P. A. Wright, S. Parsons and T. Duren, J. Am. Chem. Soc., 2011, 133, 8900–8902 CrossRef CAS PubMed.
- S. Jung, C. C. L. McCrory, I. M. Ferrer, J. C. Peters and T. F. Jaramillo, J. Mater. Chem. A, 2016, 4, 3068–3076 RSC.
- M. Mirzaeian, N. Akhanova, M. Gabdullin, Z. Kalkozova, A. Tulegenova, S. Nurbolat and K. Abdullin, Energies, 2020, 13(19), 5228 CrossRef CAS.
- H. G. Meier, J. R. Vilche and A. J. Arvia, J. Electroanal. Chem., 1982, 134, 251–272 CrossRef.
- H. G. Meier, J. R. Vilche and A. J. Arvia, J. Electroanal. Chem., 1982, 138, 367–379 CrossRef.
- N. L. Torad, R. R. Salunkhe, Y. Q. Li, H. Hamoudi, M. Imura, Y. Sakka, C. C. Hu and Y. Yamauchi, Chem. – Eur. J., 2014, 20, 7895–7900 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables. Component assignations, component positions (BE) and FWHM, and component relative abundances (XPS deconvolutions). Component intensities and FWHM (Raman 2-component deconvolutions). Component positions and FWHM (Raman 5-component deconvolutions). Double-layer capacitances, reference capacitances per unit area, and ECSA. Figures. High-resolution core-level spectra deconvolutions (XPS). Pores size distributions (PSD) (N2 adsorption). Double-layer discharging tests (CV) and the corresponding linear fittings. Additional micrographs (TEM). The thermogravimetric curve of PW11Co@ZIF-67 (TGA). Diffraction patterns (PXRD). Raman spectra. 2-Component and 5-component deconvolutions (Raman). N2 and CO2 adsorption/desorption isotherms. Element distribution maps (TEM/EDS). See DOI: https://doi.org/10.1039/d2dt02976f |
| This journal is © The Royal Society of Chemistry 2022 |