
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBulky ligands protect molecular ruby from oxygen quenching†
Laura
Stein
a,
Cui
Wang
 b,
Christoph
Förster
a,
Ute
Resch-Genger
b and
Katja
Heinze
*a
b,
Christoph
Förster
a,
Ute
Resch-Genger
b and
Katja
Heinze
*a
aDepartment of Chemistry, Johannes Gutenberg University of Mainz, Mainz, Germany. E-mail: katja.heinze@uni-mainz.de
bDivision Biophotonics, Federal Institute for Materials Research and Testing (BAM), Richard-Willstätter-Straße 11, 12489 Berlin, Germany
First published on 7th November 2022
Abstract
Chromium(III) complexes can show phosphorescence from the spin-flip excited doublet states 2E/2T1 in the near-infrared with high photoluminescence quantum yields and extremely long lifetimes in the absence of dioxygen. The prototype molecular ruby, [Cr(ddpd)2]3+ (ddpd = N,N′-dimethyl-N,N′-dipyridine-2-ylpyridine-2,6-diamine), has a photoluminescence quantum yield and a luminescence lifetime of 13.7% and 1.1 ms in deaerated acetonitrile, respectively. However, its luminescence is strongly quenched by 3O2via an efficient Dexter-type energy transfer process. To enable luminescence applications of molecular rubies in solution under aerobic conditions, we explored the potential of sterically demanding ddpd ligands to shield the chromium(III) center from O2 using steady state and time-resolved photoluminescence spectroscopy. The structures of the novel complexes with sterically demanding ligands were investigated by single crystal X-ray diffraction and quantum chemically by density functional theory calculations. The O2 sensitivity of the photoluminescence was derived from absolutely measured photoluminescence quantum yields and excited state lifetimes under inert and aerobic conditions and by Stern–Volmer analyses of these data. Optimal sterically shielded chromium(III) complexes revealed photoluminescence quantum yields of up to 5.1% and excited state lifetimes of 518 µs in air-saturated acetonitrile, underlining the large potential of this ligand design approach to broaden the applicability of highly emissive chromium(III) complexes.
Introduction
Research on luminescent first-row transition metal complexes has tremendously increased in recent years to replace and complement the dominating precious and rare earth elements in molecular inorganic photochemistry.1 Prominent examples are chromium(III) complexes with tridentate ligands and large bite angles which display a strong phosphorescence from spin-flip excited doublet states 2E/2T1 in the near-infrared (NIR) spectral region with extremely high quantum yields and lifetimes, yet typically only in the absence of molecular oxygen (3O2).2–4The prototypical complex is the so-called molecular ruby, [Cr(ddpd)2]3+ ([Cr5H]3+, ddpd = N,N′-dimethyl-N,N′-dipyridine-2-ylpyridine-2,6-diamine, Scheme 1a) with a luminescence quantum yield of ϕArPL = 13.7% and an excited state lifetime of τArPL = 1.1 ms at room temperature in deaerated acetonitrile solution (λem = 738, 775 nm).5,6 Modifying the ligand structure and substitution pattern enabled the tuning of the emission wavelength from 709 nm to 1067 nm.7,8
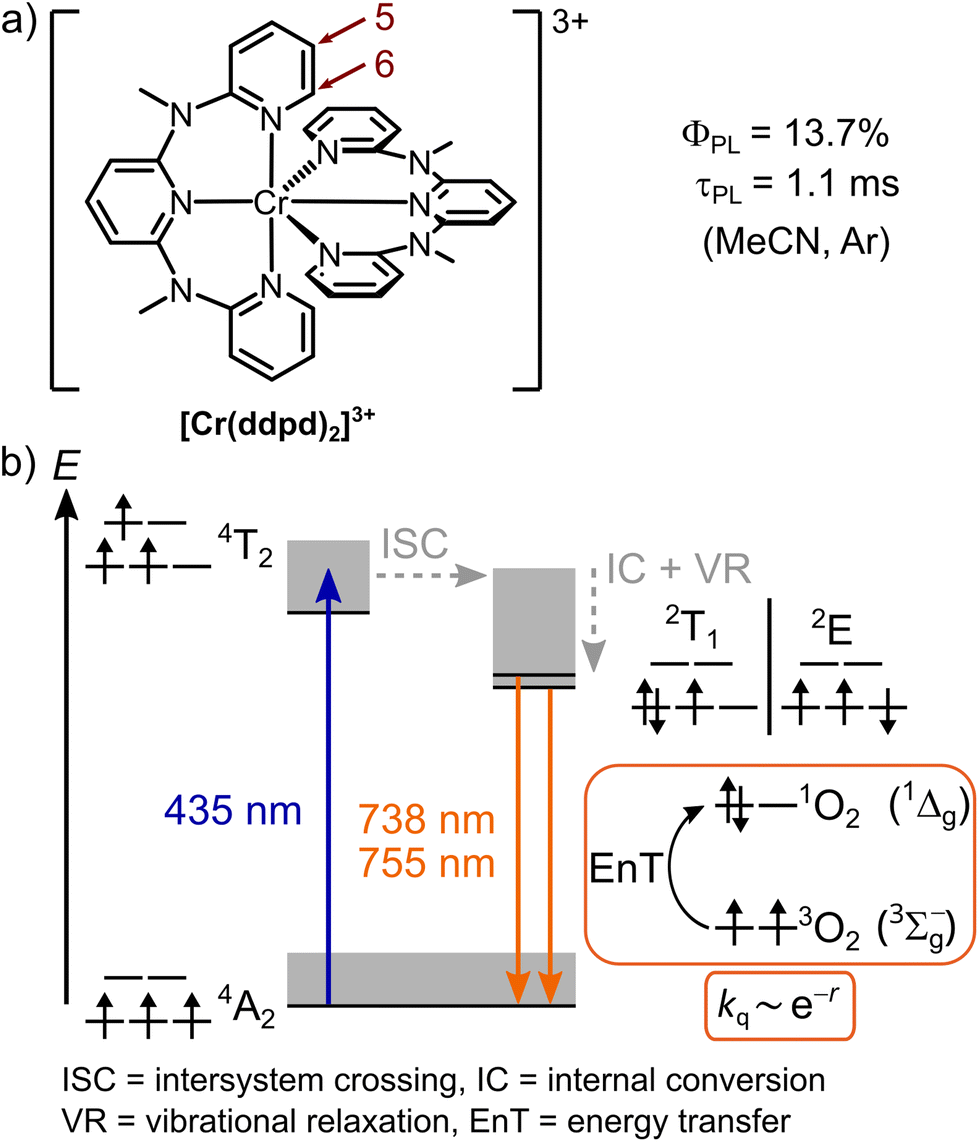 | ||
| Scheme 1 a) Molecular structure of [Cr5H]3+ including the atom numbering of the ligand and (b) schematic Jablonski diagram of a transition metal complex with d3 electron configuration in an octahedral environment with a large ligand field splitting. Relevant microstates and term symbols are indicated. The optical-spectroscopic data are given for [Cr5H]3+ in acetonitrile solution at room temperature. Dexter-type energy transfer from doublet excited states to triplet oxygen (3∑g−) is indicated by respective microstates and term symbols.5,11 The grey boxes indicate vibrational levels. | ||
Several unimolecular non-radiative decay pathways compete with the spin- and Laporte-forbidden radiative relaxation in octahedral chromium(III) complexes. This includes back-intersystem crossing to dissociative quartet states (4T2; Scheme 1b), excited state distortion,9 and multiphonon relaxation10 promoted by CH, NH or OH overtones from ligands or solvent molecules.3 Large ligand field splitting and rigid ligand frameworks promoting high octahedricity and ligand deuteration helped to increase the quantum yield and lifetime of the excited state of [Cr5H]3+ up to ϕArPL = 30% and τArPL = 2300 µs in O2-free deuterated acetonitrile solution at room temperature.12
The dual NIR emission of [Cr5H]3+ was already exploited for O2 and ratiometric temperature sensing as well as to optically probe hydrostatic pressure.13 The effective quenching of the chromium(III) emission by 3O2 leads to a high quantum yield of the formed singlet oxygen (Φ(1O2) ca. 61% in acetonitrile),14 enabling the usage of [Cr5H]3+ as 1O2-generating photosensitizer for organic photo-oxidation reactions14 and photodynamic therapy.15 However, the high sensitivity to 3O2 limits other potentially interesting applications of chromium(III) complexes as optical reporters, in bioimaging, and energy conversion schemes.
Mechanistically, quenching of long-lived electronically excited states with multiplicities differing from the ground state multiplicity by 3O2 follows the Dexter energy transfer mechanism.11 The doublet excited states of chromium(III) complexes are well suited to sensitize 1O2 generation due to their much higher energy than the 1O2 energy (ΔE = 0.6 eV), the favorable spin-statistics (67%), and the up to millisecond lifetimes of the excited doublet states.5 Based on the overlap of the donor and acceptor wavefunctions, the Dexter energy transfer rate constant kq decreases exponentially with the donor–acceptor distance,16 which is typically below 10 Å.17 To prevent this deactivation pathway, photoreactions, spectroscopic measurements and applications employing photoactive transition metal complexes are usually carried out under O2-free conditions.4,18 One strategy to protect the O2-sensitive luminophores from oxygen is to embed them in O2-impermeable matrices.18 For example, encapsulation of the molecular ruby in polystyrene nanoparticles coated with a silica shell and embedded in an oxygen-barrier poly(vinyl alcohol) film increased the quantum yield under air from ϕairPL < 1% to ϕairPL = 15.2%.6
In the present study, we follow a different approach exploiting a rational ligand design concept: the coordinating ddpd ligand is derivatized with substituents of increasing bulkiness to mitigate close contacts of the chromium(III) center and O2 and hence to reduce the overlap of their wavefunctions. The increased average Cr⋯O2 distance is expected to decrease the Dexter energy transfer rate constant kq, thereby reducing the O2-sensitivity of the chromium(III) complexes and increasing the photoluminescence quantum yields ϕairPL and lifetimes τairPL in air-saturated systems.
Results and discussion
DFT calculations
The position at the ddpd ligand closest to the chromium(III) center with arguably the strongest shielding effect is the 6-position (Scheme 1a). According to density functional theory (DFT) modeling, the 6-methyl substituted derivative ddpd6Me coordinates to chromium(III) giving [Cr(ddpd6Me)2]3+ ([Cr6Me]3+, Fig. 1b), however with significantly increased Cr–N distances to the terminal pyridines by 0.1 Å compared to the parent complex [Cr5H]3+ (Table S3†). Possibly, interligand steric congestion, as observed in iron(II) complexes with 6,6″-halogenated 2,2′:6′,2″-terpyridyl (tpy) ligands,20 hinders the successful coordination of ddpd6Me to the small chromium(III) ion (ionic radius 0.62 Å).21 Therefore, all attempts to coordinate ddpd6Me to chromium(III) to form [Cr6Me]3+ failed. The next suitable position for introducing a shielding substituent R is position 5 at the terminal pyridine rings to give the ddpd5R ligands (Scheme 1a). As these substituents R point away from the metal center, the substituent size clearly plays a key role to efficiently block the access of O2 to the chromium center (Fig. 1). Therefore, we prepared ddpd ligands with methyl, mesityl and 2,4,6-triisopropylphenyl substituents at position 5 (ddpd5Me, ddpd5Mes and ddpd5Tripp) and the corresponding chromium(III) complexes [Cr(ddpd5Me)2][BF4]3 ([Cr5Me][BF4]3), [Cr(ddpd5Mes)2][BF4]3 ([Cr5Mes][BF4]3) and [Cr(ddpd5Tripp)2][BF4]3 ([Cr5Tripp][BF4]3). Space-filling models of the DFT optimized complex geometries demonstrate the increasing steric demand of the ligand around the metal center (Fig. 1c–e). | ||
| Fig. 1 Space-filling models of (a) [Cr(ddpd)2]3+, (b) [Cr(ddpd6Me)2]3+, (c) [Cr(ddpd5Me)2]3+, (d) [Cr(ddpd5Mes)2]3+ and (e) [Cr(ddpd5Tripp)2]3+ from DFT calulcations demonstrating the increasing shielding of the chromium center (orange: Cr, blue: N, grey: C, white: H). Left: front view, right: top view. | ||
Synthesis and characterization
The ddpd5R ligands were synthesized according to the procedures outlined in Scheme 2 and analytically characterized (see ESI†).22 The methyl derivative ddpd5Me was prepared from 2,6-diaminopyridine and 2-bromo-5-methyl pyridine followed by N-methylation with methyl iodide (Scheme 2a). The required 2-(N-methyl)-amino-5-aryl pyridines py5Mes-NHMe and py5Tripp-NHMe were obtained from 2,5-dibromopyridine by amination of the 2-position followed by Suzuki coupling of the 5-position with the respective aryl boronic acid (Scheme 2b). N-Substitution of py5Mes-NHMe and py5Tripp-NHMe with 2,6-dibromopyridine yielded the tridentate ligands ddpd5Mes and ddpd5Tripp, respectively (Scheme 2b). Coordination of ddpd5Me to chromium(III) was successful using a procedure analogous to the preparation of [Cr5H][BF4]3 in MeCN/H2O giving [Cr5Me][BF4]3 (Scheme 2a).5 The aryl substituted ligands ddpd5Mes and ddpd5Tripp, however, are only poorly soluble in MeCN/H2O mixtures. Consequently, different solvents and a different chromium precursor were required. With [Cr(MeCN)4][BF4]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 in MeCN/CH2Cl2, these bulky ligands were successfully coordinated to chromium(II), yielding [Cr5Mes][BF4]3 and [Cr5Tripp][BF4]3, respectively, after oxidation (Scheme 2b). Slow diffusion of diethyl ether into concentrated acetonitrile solutions of [Cr5Me][BF4]3, [Cr5Mes][BF4]3 and [Cr5Tripp][BF4]3 yielded orange crystals suitable for X-ray diffraction analyses (Table 2). As derived from DFT optimized geometries and crystal structures (Tables 1, 2 and S1–S3†), in these complexes, a slight deviation from octahedral symmetry results with the largest substituent Tripp with the shape parameter19S(OC-6) > 0.5. In the solid state, the average Cr−N bond from the central pyridines to the metal center is shortened (2.042 Å to 2.023 Å), while the average Cr−N bond to the terminal pyridines is elongated from 2.045 Å to 2.053 Å. Simultaneously, the intramolecular N−Cr−N angles increasingly deviate from 90° and 180° within this series. This is expected as the size of the aromatic substituents forces the ligand scaffold to twist more strongly out of plane than with small substituents like R = H or Me. Due to the torsion induced by the bulky Tripp substituent, the dihedral angle between the central pyridine planes becomes smaller (∡10°, Table S1†) compared to the other ligands (∡14°–19°). The more efficient overlap of the pyridine and metal centered t2g orbitals presumably causes a stronger nephelauxetic effect and a bathochromic shift of the emission band for this complex (see below). Moreover, intermolecular π–π stacking occurs between the aromatic substituents (centroid-to-centroid distance in [Cr5Mes][BF4]3 = 4.961 Å, Fig. S21† and [Cr5Tripp][BF4]3 5.700 Å, Fig. S22†) and even intramolecular interligand π–π stacking was observed in [Cr5Tripp][BF4]3 (5.968 Å). This intramolecular π–π stacking might shield the chromium ion even more efficiently from the environment (Fig. 1e).
23 in MeCN/CH2Cl2, these bulky ligands were successfully coordinated to chromium(II), yielding [Cr5Mes][BF4]3 and [Cr5Tripp][BF4]3, respectively, after oxidation (Scheme 2b). Slow diffusion of diethyl ether into concentrated acetonitrile solutions of [Cr5Me][BF4]3, [Cr5Mes][BF4]3 and [Cr5Tripp][BF4]3 yielded orange crystals suitable for X-ray diffraction analyses (Table 2). As derived from DFT optimized geometries and crystal structures (Tables 1, 2 and S1–S3†), in these complexes, a slight deviation from octahedral symmetry results with the largest substituent Tripp with the shape parameter19S(OC-6) > 0.5. In the solid state, the average Cr−N bond from the central pyridines to the metal center is shortened (2.042 Å to 2.023 Å), while the average Cr−N bond to the terminal pyridines is elongated from 2.045 Å to 2.053 Å. Simultaneously, the intramolecular N−Cr−N angles increasingly deviate from 90° and 180° within this series. This is expected as the size of the aromatic substituents forces the ligand scaffold to twist more strongly out of plane than with small substituents like R = H or Me. Due to the torsion induced by the bulky Tripp substituent, the dihedral angle between the central pyridine planes becomes smaller (∡10°, Table S1†) compared to the other ligands (∡14°–19°). The more efficient overlap of the pyridine and metal centered t2g orbitals presumably causes a stronger nephelauxetic effect and a bathochromic shift of the emission band for this complex (see below). Moreover, intermolecular π–π stacking occurs between the aromatic substituents (centroid-to-centroid distance in [Cr5Mes][BF4]3 = 4.961 Å, Fig. S21† and [Cr5Tripp][BF4]3 5.700 Å, Fig. S22†) and even intramolecular interligand π–π stacking was observed in [Cr5Tripp][BF4]3 (5.968 Å). This intramolecular π–π stacking might shield the chromium ion even more efficiently from the environment (Fig. 1e).
 | ||
| Scheme 2 Syntheses of substituted ligands ddpd5R and corresponding chromium(III) complexes [Cr(ddpd5R)2][BF4]3. | ||
| [Cr5H]3+ | [Cr5Me]3+ | [Cr5Mes]3+ | [Cr5Tripp]3+ | |
|---|---|---|---|---|
| Cr–N1 | 2.059 | 2.056 | 2.062 | 2.064 |
| Cr–N2 | 2.046 | 2.048 | 2.038 | 2.041 |
| Cr–N3 | 2.059 | 2.056 | 2.057 | 2.060 |
| Cr–N4 | 2.059 | 2.055 | 2.068 | 2.062 |
| Cr–N5 | 2.046 | 2.047 | 2.045 | 2.044 |
| Cr–N6 | 2.059 | 2.055 | 2.059 | 2.060 |
| N1–Cr–N2 | 86.46 | 86.49 | 85.65 | 84.95 |
| N2–Cr–N3 | 86.46 | 86.42 | 86.97 | 85.11 |
| N1–Cr–N3 | 172.92 | 172.91 | 172.49 | 170.06 |
| N4–Cr–N5 | 86.46 | 86.46 | 86.06 | 85.07 |
| N5–Cr–N6 | 86.46 | 86.49 | 87.58 | 85.13 |
| N4–Cr–N6 | 172.92 | 172.95 | 173.13 | 170.20 |
| S(OC-6)19 | 0.26 | 0.26 | 0.32 | 0.52 |
|
[Cr5H][BF4]35 |
[Cr5Me][BF4]3 | [Cr5Mes][BF4]3 | [Cr5Tripp][BF4]3 | |
|---|---|---|---|---|
| Cr–N1 | 2.0485(18) | 2.0448(14) | 2.0577(17) | 2.0523(14) |
| Cr–N2 | 2.0393(18) | 2.0294(14) | 2.0268(17) | 2.0222(14) |
| Cr–N3 | 2.0394(19) | 2.0471(14) | 2.0623(18) | 2.0561(14) |
| Cr–N4 | 2.0446(17) | 2.0492(13) | 2.0579(18) | 2.0576(14) |
| Cr–N5 | 2.0444(18) | 2.0300(14) | 2.0215(17) | 2.0238(14) |
| Cr–N6 | 2.0485(18) | 2.0403(13) | 2.0549(18) | 2.0449(14) |
| N1–Cr–N2 | 85.13(8) | 85.83(6) | 87.63(7) | 84.91(6) |
| N2–Cr–N3 | 85.74(7) | 86.18(6) | 86.23(7) | 85.26(6) |
| N1–Cr–N3 | 170.86(8) | 172.00(5) | 173.44(7) | 170.02(6) |
| N4–Cr–N5 | 85.89(7) | 86.00(5) | 85.91(7) | 84.48(5) |
| N5–Cr–N6 | 84.99(7) | 86.08(5) | 86.74(7) | 84.98(6) |
| N4–Cr–N6 | 170.88(7) | 171.81(5) | 172.62(7) | 169.35(6) |
| Cr⋯F (BF4−) | 5.438(4) | 4.682(2) | 6.768(2) | 5.556(2) |
| Cr⋯N (MeCN) | 4.613(4) | 4.694(2) | 4.654(2) | 4.513(2) |
| S(OC-6)19 | 0.43 | 0.35 | 0.31 | 0.56 |
As a measure of the accessibility of the metal center within the ligand scaffold for subsequent luminescence studies without and in the presence of oxygen, the shortest Cr⋯F (BF4−) and Cr⋯N (MeCN) distances in the crystal structures were taken into account (Table S1 and Fig. S20–22†). Compared to the complex with R = Me, the distances to the counter ions increase, while they remain in the same range for MeCN contacts. This suggests better shielding from tetrahedral molecules than from linear molecules as well as a weaker electrostatic interaction between the counter ions and the cationic metal center in the solid state.
Optical properties of [Cr(ddpd5R)2][BF4]3
The absorption and photoluminescence spectra of [Cr5R][BF4]3 complexes with R = H, Me, Mes, Tripp were recorded in acetonitrile at room temperature under anaerobic conditions (Fig. 2a). The shapes of the respective absorption and emission spectra are relatively similar. Bathochromic shifts are noted for the 4A2 → 4T2/LMCT absorption band from 435 to 455 nm (Fig. S23a†) and for the spin-flip emission bands from 738 nm → 742 nm/775 nm → 794 nm (Table 3 and Fig. 2). The bathochromic shift of the 4A2 → 4T2 absorption band, combined with the more intense ligand-to-metal charge transfer absorption band, could be reproduced by time-dependent DFT calculations (Fig. S24†). The π–π* transitions of the ligands in the UV region display larger changes, in particular for complexes with aryl substituted ligands (Fig. 2a), which is already evident from the ligand absorption bands (Fig. S13†). The quantum yields ϕArPL and luminescence lifetimes τArPL of the complexes in Ar-saturated acetonitrile solution are very similar (Table 4). | ||
| Fig. 2 (a) Absorption and normalized emission spectra of [Cr5R][BF4]3 complexes with R = H (λexc = 435 nm, black), Me (λexc = 441 nm, blue), Mes (λexc = 450 nm, green) and Tripp (λexc = 454 nm, red) in Ar-saturated acetonitrile. Luminescence decays of [Cr5R][BF4]3 complexes with (b) R = Me, (c) R = Mes, (d) R = Tripp in air- and Ar-saturated acetonitrile solution at 293 K. | ||
| Complex | λ max/nm (ε/103 M−1 cm−1) | λ em/nm (λexc/nm) |
|---|---|---|
| [Cr5Me][BF4]3 | 444 (4), 323 (23, sh), 305 (27), 225 (53, sh) | 741, 779 (441) |
| [Cr5Mes][BF4]3 | 453 (3), 312 (22), 257 (31), 236 (44, sh) | 742, 782 (453) |
| [Cr5Tripp][BF4]3 | 455 (3), 319 (30), 240 (55, sh) | 740 (sh), 794 (455) |
|
[Cr5H][BF4]35,6 |
[Cr5Me][BF4]3 | [Cr5Mes][BF4]3 | [Cr5Tripp][BF4]3 | |
|---|---|---|---|---|
| λ obs/nm | 778 | 778 | 782 | 795 |
| ϕ ArPL/% | 13.7 | 13.9 | 12.1 | 11.3 |
| τ ArPL/µs | 1122 | 1258 | 1173 | 1131 |
| ϕ airPL/% | 0.8 | 0.8 | 2.2 | 5.1 |
| τ airPL/µs | 52 | 52 | 244 | 518 |
| ϕ ArPL/ϕPLair | 17 | 17 | 5 | 2 |
| τ ArPL/τairPL | 20 | 25 | 5 | 2 |
| K IntSV/hPa−1 | 0.116 | 0.135 | 0.023 | 0.012 |
| K LTSV/hPa−1 | 0.123 | 0.147 | 0.024 | 0.012 |
| k Intq/hPa s−1 | 103.3 | 107.4 | 19.4 | 10.7 |
| k LTq/hPa s−1 | 109.6 | 116.5 | 20.0 | 10.5 |
Oxygen quenching experiments
The photoluminescence of the complexes [Cr5R][BF4]3 is quenched in the presence of O2, yet to very different extents (Table 4, Fig. 2 and S25†). The luminescence properties of [Cr5H][BF4]3 and [Cr5Me][BF4]3 in air-saturated acetonitrile solution are very similar, demonstrating that the shielding by methyl groups in position 5 is not effective. The DFT modeling and XRD data of the complexes with R = H and Me confirm this assumption as the accessibility of the chromium(III) ion seems to be very similar for both complexes (Fig. 1). On the other hand, mesityl and 2,4,6-triisopropyl substituents in position 5 clearly affect the photoluminescence quantum yields and luminescence lifetimes of the chromium(III) complexes in air-saturated acetonitrile as reflected by ϕairPL = 2.2% and 5.1% and τairPL = 244 µs and 518 µs, respectively. Compared to R = H and Me, where the ratios of ϕArPL/ϕairPL and τArPL/τairPL amount to 17−25, for the Tripp substituted complex, these ratios drop remarkably to two (Table 4). This demonstrates the efficient shielding of the chromium(III) center from O2 by the bulky Tripp substituents.Compared to very recently reported chromium(III) complexes with 1,3-bis(2′-pyridylimino)-isoindoline ligands reported as showing long-lived NIR emission under inert and ambient conditions in acetonitrile (τArPL = 8.0 µs and 25.0 µs; τairPL = 4.5 µs and 8.1 µs),24 the here reported lifetimes of [Cr5Mes][BF4]3 and [Cr5Tripp][BF4]3 are larger by factors of 30 to 115.
Emission intensity and lifetime-based Stern–Volmer analyses with varying concentrations or partial pressures of O2 in acetonitrile solution gave linear plots with Stern–Volmer constants KIntSV and KLTSV matching within the measurement uncertainty. This confirms the diffusional quenching by O2 (Table 4, Fig. 3 and S25†). The KIntSV and KLTSV values drop significantly from KSV ≈ 0.13 hPa−1 for [Cr5H][BF4]3 and [Cr5Me][BF4]3 over 0.02 hPa−1 for [Cr5Mes][BF4]3 to 0.01 hPa−1 for [Cr5Tripp][BF4]3. Similarly, the quenching rate constants kq = KSV/τArPL decrease from ≈ 110 hPa s−1 for [Cr5H][BF4]3 and [Cr5Me][BF4]3 over 20 hPa s−1 for [Cr5Mes][BF4]3 to 11 hPa s−1 for [Cr5Tripp][BF4]3, equaling an overall shielding-related reduction by a factor of about 10. These results clearly demonstrate the superior shielding effect of the bulky ddpd5Tripp ligand.
 | ||
| Fig. 3 (a) Intensity- and (b) lifetime-based Stern–Volmer plots of the [Cr5R][BF4]3 complexes with O2 in acetonitrile at 293 K. | ||
Cooling the complexes under aerobic and anaerobic conditions increases the luminescence lifetimes (Fig. S26 and Table S4†) of the complexes [Cr5R][BF4]3 in acetonitrile solution, yet displaying a different degree of the temperature dependence for deaerated and atmospheric conditions (Fig. S27†). Under Ar-saturated conditions, for R = H, Me, Mes, the lifetimes are prolonged by a factor of f = 5–7 upon cooling from 338 to 278 K. For the bulky substituent R = Tripp, the temperature dependence is significantly smaller (factor of f = 2.4) under these conditions. This indicates a higher activation barrier for thermally induced non-radiative processes, probably because the substituent Tripp enforces a rigid structure of the respective chromium(III) complex. Under atmospheric conditions, the increase in the luminescence lifetimes upon lowering the temperature is less significant for all complexes (R = H: f ≈ 1.1, Me: f ≈ 1.3; Mes: f ≈ 2.5, Tripp: f ≈ 1.8). Especially for the complexes with the ddpd ligands bearing the small substituents R = H and Me, O2 quenching is now the most significant non-radiative decay process, as it is faster than other unimolecular, non-radiative (temperature-dependent) pathways.
Conclusion
This study reports the syntheses and structures of novel chromium(III) spin-flip emitters (molecular rubies) modified at position 5 of the tridentate chelate ligands with bulky substituents to protect the chromium(III) center from O2 quenching. Conceptually, the shielding of a spin-flip emitter from quenching by 3O2 is more straightforward than the shielding of a phosphorescent charge-transfer emitter due to the metal-localized and ligand-delocalized nature of the respective participating wavefunctions. While small methyl substituents barely show a protective effect, the aryl substituents mesityl and 2,4,6-triisopropylphenyl effectively shield the chromium(III) center from a close contact with O2. This reduces the Dexter-type energy transfer rate to O2 from kq ≈ 110 hPa s−1 to 11 hPa s−1 and consequently increases the photoluminescence quantum yields and lifetimes of the respective chromium(III) complexes from 0.8 to 5.1% and from 52 to 518 µs, respectively, in air-saturated acetonitrile solution at 293 K. In the future, these favorable luminescence features of the chromium(III) complexes with sterically demanding ddpd ligands can enable applications of shielded molecular rubies, for example as reporters in time-gated luminescence detection schemes25 or for circularly polarized NIR emission26 under aerobic conditions.Author contributions
L. S. performed the synthesis, characterization, DFT, UV-VIS, the quantum yield and lifetime measurements under Ar and air and temperature-dependent emission measurements under Ar and air, visualized the data and wrote the original draft. C. W. performed the oxygen-dependent Stern–Volmer studies and preliminary optical characterization. C. F. solved and refined the crystal structures. U. R.-G. supervised the Stern–Volmer studies and revised the manuscript. K. H. designed and supervised the project and finalized the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Dr Luca M. Carrella and Dr Dieter Schollmeyer for collection of XRD data and Dr Robert Naumann for support with the experimental setup for QY measurements. This work was supported by the Deutsche Forschungsgemeinschaft (RE 1203/23-1 and RE1203/23-2, HE 2778/10-2) and through grant INST 247/1018-1 FUGG to KH. Parts of this research were conducted using the supercomputer Elwetritsch and advisory services offered by the TU Kaiserslautern (https://elwe.rhrk.uni-kl.de) which is a member of the AHRP.References
- (a) C. Wegeberg and O. S. Wenger, JACS Au, 2021, 1, 1860–1876 CrossRef CAS PubMed; (b) C. Förster and K. Heinze, Chem. Soc. Rev., 2020, 49, 1057–1070 RSC.
- (a) P. A. Scattergood, in Organometallic Chemistry, ed. N. J. Patmore and P. I. P. Elliott, Royal Society of Chemistry, Cambridge, 2020, vol. 43, pp. 1–34 Search PubMed; (b) J.-R. Jiménez, B. Doistau, M. Poncet and C. Piguet, Coord. Chem. Rev., 2021, 434, 213750 CrossRef.
- S. Otto, M. Dorn, C. Förster, M. Bauer, M. Seitz and K. Heinze, Coord. Chem. Rev., 2018, 359, 102–111 CrossRef CAS.
- W. R. Kitzmann, J. Moll and K. Heinze, Photochem. Photobiol. Sci., 2022, 21, 1309–1331 CrossRef CAS PubMed.
- S. Otto, M. Grabolle, C. Förster, C. Kreitner, U. Resch-Genger and K. Heinze, Angew. Chem., Int. Ed., 2015, 54, 11572–11576 CrossRef CAS PubMed.
- C. Wang, W. R. Kitzmann, F. Weigert, C. Förster, X. Wang, K. Heinze and U. Resch-Genger, ChemPhotoChem, 2022, e202100296 CAS.
- F. Reichenauer, C. Wang, C. Förster, P. Boden, N. Ugur, R. Báez-Cruz, J. Kalmbach, L. M. Carrella, E. Rentschler, C. Ramanan, G. Niedner-Schatteburg, M. Gerhards, M. Seitz, U. Resch-Genger and K. Heinze, J. Am. Chem. Soc., 2021, 143, 11843–11855 CrossRef CAS PubMed.
- N. Sinha, J.-R. Jiménez, B. Pfund, A. Prescimone, C. Piguet and O. S. Wenger, Angew. Chem., Int. Ed., 2021, 60, 23722–23728 CrossRef CAS PubMed.
- (a) M. W. Perkovic, M. J. Heeg and J. F. Endicott, Inorg. Chem., 1991, 30, 3140–3147 CrossRef CAS; (b) A. M. McDaniel, H.-W. Tseng, N. H. Damrauer and M. P. Shores, Inorg. Chem., 2010, 49, 7981–7991 CrossRef CAS PubMed.
- E. Kreidt, C. Kruck and M. Seitz, in Handbook on the physics and chemistry of rare earths. Including actinides, ed. J.-C. G. Bünzli, V. K. Pecharsky, G.-Y. Adachi, K. A. Gschneider and E. LeRoy, North-Holland, Amsterdam, Oxford, 2018, vol. 53, pp. 35–79 Search PubMed.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- C. Wang, S. Otto, M. Dorn, E. Kreidt, J. Lebon, L. Sršan, P. Di Martino-Fumo, M. Gerhards, U. Resch-Genger, M. Seitz and K. Heinze, Angew. Chem., Int. Ed., 2018, 57, 1112–1116 CrossRef CAS PubMed.
- (a) S. Otto, N. Scholz, T. Behnke, U. Resch-Genger and K. Heinze, Chem. – Eur. J., 2017, 23, 12131–12135 CrossRef CAS PubMed; (b) S. Otto, J. P. Harris, K. Heinze and C. Reber, Angew. Chem., Int. Ed., 2018, 57, 11069–11073 CrossRef CAS PubMed; (c) C. Wang, S. Otto, M. Dorn, K. Heinze and U. Resch-Genger, Anal. Chem., 2019, 91, 2337–2344 CrossRef CAS PubMed.
- S. Otto, A. M. Nauth, E. Ermilov, N. Scholz, A. Friedrich, U. Resch-Genger, S. Lochbrunner, T. Opatz and K. Heinze, ChemPhotoChem, 2017, 1, 344–349 CrossRef CAS.
- U. Basu, S. Otto, K. Heinze and G. Gasser, Eur. J. Inorg. Chem., 2019, 37–41 CrossRef CAS.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- V. Balzani, G. Bergamini, S. Campagna and F. Puntoriero, in Photochemistry and Photophysics of Coordination Compounds I, ed. V. Balzani and S. Campagna, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, vol. 280, pp. 1–36 Search PubMed.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- P. Alemany, D. Casanova, S. Alvarez, C. Dryzun and D. Avnir, in Reviews in Computational Chemistry, ed. A. L. Parrill and K. B. Lipkowitz, Wiley, Hoboken, NJ, 2017, pp. 289–352 Search PubMed.
- S. M. Fatur, S. G. Shepard, R. F. Higgins, M. P. Shores and N. H. Damrauer, J. Am. Chem. Soc., 2017, 139, 4493–4505 CrossRef CAS PubMed.
- U. Müller, Anorganische Strukturchemie, Vieweg+Teubner, Wiesbaden, 6th edn, 2008 Search PubMed.
- (a) C. Förster, M. Dorn, T. Reuter, S. Otto, G. Davarci, T. Reich, L. Carrella, E. Rentschler and K. Heinze, Inorganics, 2018, 6, 86 CrossRef; (b) J. Tasseroul, M. M. Lorenzo-Garcia, J. Dosso, F. Simon, S. Velari, A. de Vita, P. Tecilla and D. Bonifazi, J. Org. Chem., 2020, 85, 3454–3464 CrossRef CAS; (c) A. Breivogel, C. Förster and K. Heinze, Inorg. Chem., 2010, 49, 7052–7056 CrossRef CAS PubMed.
- R. A. Heintz, J. A. Smith, P. S. Szalay, A. Weisgerber and K. R. Dunbar, in Inorganic syntheses, ed. D. Coucouvanis, Wiley-Interscience, New York, 2002, vol. 33, pp. 75–121 Search PubMed.
- N. Sawicka, C. J. Craze, P. N. Horton, S. J. Coles, E. Richards and S. J. A. Pope, Chem. Commun., 2022, 58, 5733–5736 RSC.
- W. Yang and S.-L. Chen, J. Innovative Opt. Health Sci., 2020, 13, 2030006 CrossRef CAS.
- (a) J.-R. Jiménez, B. Doistau, C. M. Cruz, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, J. Am. Chem. Soc., 2019, 141, 13244–13252 CrossRef PubMed; (b) C. Dee, F. Zinna, W. R. Kitzmann, G. Pescitelli, K. Heinze, L. Di Bari and M. Seitz, Chem. Commun., 2019, 55, 13078–13081 RSC; (c) J.-R. Jiménez, M. Poncet, S. Míguez-Lago, S. Grass, J. Lacour, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, Angew. Chem., Int. Ed., 2021, 60, 10095–10102 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2125019–2125021. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02950b |
| This journal is © The Royal Society of Chemistry 2022 |