
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isoelectronic Pt(II) complexes of cyclometalating C^N^N ligands with phenyl/(benzo)thiophenyl and pyridyl/(benzo)thiazolyl moieties†
Maren
Krause
 a,
Joshua
Friedel
a,
Stefan
Buss
b,
Dana
Brünink
c,
Annemarie
Berger
a,
Cristian A.
Strassert
*b,
Nikos L.
Doltsinis
*c and
Axel
Klein
*a
a,
Joshua
Friedel
a,
Stefan
Buss
b,
Dana
Brünink
c,
Annemarie
Berger
a,
Cristian A.
Strassert
*b,
Nikos L.
Doltsinis
*c and
Axel
Klein
*a
aUniversität zu Köln, Department für Chemie, Institut für Anorganische Chemie, Greinstraße 6, D-50939 Köln, Germany. E-mail: krause.maren@gmx.net; jfried20@uni-koeln.de; annemarie.berger@uni-koeln.de; simonmartin.schmitz@gmail.com; axel.klein@uni-koeln.de
bWestfälische Wilhelms-Universität Münster, Institut für Anorganische und Analytische Chemie, CiMIC, CeNTech, Heisenbergstraße 11, D-48149 Münster, Germany. E-mail: s_buss14@uni-muenster.de; cstra_01@uni-muenster.de
cWestfälische Wilhelms-Universität Münster, Institut für Festkörpertheorie and Center for Multiscale Theory and Computation, Wilhelm-Klemm-Straße 10, 48149 Münster, Germany. E-mail: dana.bruenink@googlemail.com; nikos.doltsinis@wwu.de
First published on 30th September 2022
Abstract
A series of cyclometalated Pt(II) complexes [Pt(C^N^N)X] (X = Cl, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh, CCC6F5) was synthesised from the protoligands HC^N^N containing either phenyl (ph), naphthyl (na) or (benzo)thiophenyl (b(th)) C aryl functions and either pyridyl (py) or (benzo)thiazolyl ((b)tz) peripheral N units, alongside the central 4-phenyl-pyridyl (ppy) or tBu2-phenyl-pyridyl (tbppy) N group. Depending on the combination of the peripheral N or C aryl building blocks, these square planar complexes reveal very different electrochemical, UV-vis absorption and emission behaviour. The reversible reductions shift anodically along the series th/py < ph/tz ≈ th/tz < ph/btz while the irreversible oxidations shift cathodically along the series Cl ≈ CCC6F5 < CCPh. Similar trends were observed for the long-wavelength UV-vis absorption and photoluminescence properties. The emission maxima range from 605 to 675 nm at 298 K in CH2Cl2 solution and from 555 to 655 nm at 77 K in glassy frozen CH2Cl2/MeOH matrices. Large differences in amplitude-weighted average lifetimes τav (up to 0.9 μs at 298 K, up to 12 μs at 77 K) and photoluminescence quantum yields ΦL (up to 0.15 at 298 K and up to 0.82 at 77 K) were found. TD-DFT calculations showed that the decomposition of the triplet excited states into LC (π–π*, centred in the individual parts of the C^N^N ligand) and LLCT (π–π*, between the individual parts of the C^N^N ligand + X–π* from coligand to C^N^N) contributions for the ligand-centred states as well as MLCT (dPt-to-π*C^N^N) and LMCT (pCl or πCCR-to-dPt) character for the charge-transfer states involving the metal is beneficial to assess the participation of the individual heteroaryl groups of the C^N^N ligands. In view of the modular synthesis of these ligands, this will allow the realisation of tailor-made Pt(II) triplet emitters in future work.
CPh, CCC6F5) was synthesised from the protoligands HC^N^N containing either phenyl (ph), naphthyl (na) or (benzo)thiophenyl (b(th)) C aryl functions and either pyridyl (py) or (benzo)thiazolyl ((b)tz) peripheral N units, alongside the central 4-phenyl-pyridyl (ppy) or tBu2-phenyl-pyridyl (tbppy) N group. Depending on the combination of the peripheral N or C aryl building blocks, these square planar complexes reveal very different electrochemical, UV-vis absorption and emission behaviour. The reversible reductions shift anodically along the series th/py < ph/tz ≈ th/tz < ph/btz while the irreversible oxidations shift cathodically along the series Cl ≈ CCC6F5 < CCPh. Similar trends were observed for the long-wavelength UV-vis absorption and photoluminescence properties. The emission maxima range from 605 to 675 nm at 298 K in CH2Cl2 solution and from 555 to 655 nm at 77 K in glassy frozen CH2Cl2/MeOH matrices. Large differences in amplitude-weighted average lifetimes τav (up to 0.9 μs at 298 K, up to 12 μs at 77 K) and photoluminescence quantum yields ΦL (up to 0.15 at 298 K and up to 0.82 at 77 K) were found. TD-DFT calculations showed that the decomposition of the triplet excited states into LC (π–π*, centred in the individual parts of the C^N^N ligand) and LLCT (π–π*, between the individual parts of the C^N^N ligand + X–π* from coligand to C^N^N) contributions for the ligand-centred states as well as MLCT (dPt-to-π*C^N^N) and LMCT (pCl or πCCR-to-dPt) character for the charge-transfer states involving the metal is beneficial to assess the participation of the individual heteroaryl groups of the C^N^N ligands. In view of the modular synthesis of these ligands, this will allow the realisation of tailor-made Pt(II) triplet emitters in future work.
Introduction
The interesting cyclometalated Pt(II) complexes as triplet emitters have been recognised and developed by rational design in recent years for applications in luminescent materials and for biological applications.1–20 The d8-configured metal ion in combination with π-accepting ligands (low lying π* orbitals) provides long-lived metal-to-ligand charge transfer (MLCT) excited states whereas heteroaromatic ligands allow the admixture of π–π* (IL) configurations to yield mixed MLCT/IL excited states.1–22 The so-called ‘dark’ d–d* states, as their population leads to rapid radiationless decay of excited molecules, can be suppressed through rigidity of both the organic ligand and the square planar coordination along with a strong ligand field.5,10,12,15–17,22–27 Thus, bidentate,1,3–8,13,14,16–18,20–22 tridentate,1,6–8,10,13,15–20,23–38 or tetradentate1,2,6–9,11–13,16–18,31,39–41 cyclometalated heteroaromatic ligands have been frequently used.1–28Amongst them, the tridentate variants C^N^N, N^C^N, and C^N^C require a fourth coligand (or ancillary ligand) to complete the square planar coordination environment. The fourth coordination site is frequently considered to be prone to distortion and thus enhancing radiationless decay. Recent work has shown that careful choice of these coligands allows for fine-tuning of the triplet emission properties, as high quantum yields and long lifetime have been reported.1,15,17,25,31,34,36 For the optimisation of the photoluminescence properties of such complexes, combined experimental and theoretical studies are helpful.5,6,8,9,12,15,18,21,23–28 Variation of these ligands, including substitution of the heteroaromatic units has previously allowed the generation of complexes with photoluminescence (PL) quantum yields ΦL of up to 100% and a broad range of emission energies.1,2,6,8–17,20,24,27,31,34 Unfortunately, in many cases, structurally very similar derivatives of these complexes failed to perform in the same way and the reasons remained unclear.31 DFT modelling has allowed emission energies to be rationalised; however, quantum yields are less predictable.4,9,10,12,15,24,26,29,41,42 Very recently, DFT calculations have started to seek quantitative insight into the character and dynamic behaviour of the excited states of transition metal complexes,12,21,41–46 also allowing the rationalisation of differences in quantum yields.
A prominent example where structurally very similar complexes show very different luminescence efficiencies was recently published by Che et al.31 reporting on [Pt(C^N^N)X] (X = Cl or CCC6F5) complexes (Scheme 1, A) with cyclometalating C^N^N ligands in which the C aryl unit included various substituted phenyl (R-ph), naphthyl (na), thiophenyl (th), or benzothiophenyl (bth) groups, whereas the central N unit involved always the 4-(3,5-di-tert-butyl-phenyl)pyridyl (tbppy) group and the peripheral N group pyridyl (py), 1-quinolinyl (1-quin), 2-quinolinyl (2-quin) or 3-isoquinolinyl (3-isoquin). In this series of complexes, the efficiency expressed by ΦL varied from <0.001 to 0.99 (thus almost unity) in CH2Cl2 at 298 K.31 This extended an earlier study by Che et al. on complexes replacing the C aryl group by R-ph, na and th, a central 4-(4-carboxy)phenyl-pyridyl group and a peripheral pyridyl unit (Scheme 1, B). These complexes achieved ΦL of only 0.002 to 0.01, while the N^C^N derivative (N = py, C = 4(4-carboxy)phenyl-phenyl) showed a ΦL of 0.44 in CHCl3.32 Very recently Huo et al. reported series of [Pt(C^N^N)X] (X = Cl, CCPh, or Ph) containing a pyrazolyl (pz) peripheral N group, a pyridyl central N group and various C aryl groups including R-ph, na, th, and bth (Scheme 1, C).30 As in the case of the recent work by Che et al.,31 the ΦL at 298 K in this extensive work ranged from <0.0001 to 0.62 with [Pt(ph(py)pz)CCPh] being the most potent derivative within this extensive study.
 | ||
| Scheme 1 Structures of previously reported complexes [Pt(C^N^N)X] (A to D),23,30–32 and those under study (E to G) (th = thiophenyl, bth = benzothiophenyl, py = pyridyl, 1-quin = 1- quinolinyl, 2-quin = 2-quinolinyl, 3-isoquin = 3-isoquinolinyl, R-ph = substituted phenyl, na = naphthyl, pyz = pyridazolyl, tz = thiazolyl, btz = benzothiazolyl. | ||
Although there is now a rich body of experimental data available, no obvious correlation between the individual C and N units and high ΦL or other desirable photoluminescence properties has emerged. The recent work by Che et al. provided limited insights into the excited states through (TD)-DFT calculations on two selected complexes.31 The authors attribute the vast differences in the luminescence properties to different contributions to the emission from 3MLCT (dPt-to-π*C^N^N), 3π–π*, and XLCT (pCl-to-π*) excited states for the chlorido complexes and 3MLCT, 3π–π* and 3L′LCT (πCC–π*) for the alkynyl derivatives,31 following established ideas.39,47 In a subsequent DFT study, the benchmarking complex [Pt(ph(tbppy)3-isoquin)(CCC6F5)] (ΦL = 0.99 at 298 K in solution) was compared to the three derivatives containing na (instead of ph), py (instead of 3-isoquin), and Cl (instead of CCC6F5).43 When breaking down the contributions to the excited triplet T1 state, the calculations showed a remarkably high participation of the Pt dxz orbital to the highest occupied molecular orbital (HOMO) for the parent complex alongside with a high contribution of the CCC6F5 moiety; thus a high 3MLCT and 3L′LCT character of the triplet excited state can be assessed. Further, the S1 and S2 states are composed of two effective excitations, namely HOMO−1 → LUMO and HOMO → LUMO resulting in the largest radiative rate constant kr in this series of complexes. Further, the energy barrier between the 3MC and the T1 is high for the parent complex and the non-radiative rate constant knr very small. The same is basically true for the Cl derivative, but not for the na and py derivatives,43 which matches qualitatively well the observed rate-constants and ΦL.31
We recently studied the electronic states of the Pt(C^N^N) complexes [Pt(ph(tbppy)tz)Cl] and [Pt(ph(py)tz)Cl] alongside with their corresponding Pd(II) derivatives using a variety of DFT-based tools (Scheme 1, D).23 This motivated us to apply these methods also to the excited states properties and dynamics on a broader series of [Pt(C^N^N)X] complexes, and we herein report a series of Pt(II) complexes [Pt(C^N^N)X] with the coligands X = Cl, CCPh, CCC6F5 as well as cyclometalating tridentate C^N^N ligands based on 2-(6-arylpyridin-2-yl)thiazoles and 2-(6-arylpyridin-2-yl)thiophenes (Scheme 1E to G and Table 1). We have synthesised 16 new complexes and studied experimentally their electrochemical and photophysical properties in detail. The C aryl group was varied using phenyl (ph), naphthyl (na), thiophenyl (th), or benzothiophenyl (bth). Phenylpyridine (ppy) or its 3,5-di-tert-butyl derivative (tbppy) represent the central N core and the peripheral N group was varied using pyridyl (py), thiazolyl (tz), or benzothiazolyl (btz). Although they contain different groups, they form pairs of formally isoelectronic complexes, e.g. [Pt(ph(ppy)tz)X] and [Pt(th(ppy)py)X], as the thiazolyl and thiophenyl as well as the phenyl and pyridyl groups are isoelectronic. The central phenylpyridyl unit had turned out to be beneficial for the ability of such cyclometalated complex to act as efficient triplet emitters.10,31–34,36,41 For the variation of the coligand X, we expected to increase the ligand-field splitting (LFS) when going from the Cl coligand to phenylacetylido (CCPh) and pentaflourophenylacetylido (CCC6F5) ligands. The stronger σ-donor character of these organic ligands destabilises the otherwise thermally accessible MC-states towards improved emission efficiencies.1,17,30–32,34,36
| No. | Classb | [Pt(C^N^N)X] | C aryl | N central | N peripheral | Coligand |
|---|---|---|---|---|---|---|
| a See Scheme 1; phenyl (ph)/naphthyl (na)/thiophenyl (th)/benzothiophenyl (bth), 4-phenyl-pyridyl (ppy)/3,5-tBu2-phenyl-pyridyl (tbppy), pyridyl (py)/thiazolyl (tz)/benzothiazolyl (btz). b Classes of complexes defined for the discussion. | ||||||
| 1 | A | [Pt(ph(ppy)tz)Cl]23 | ph | ppy (R1 = H) | tz | Cl |
| 2 | [Pt(ph(tbppy)tz)Cl]23 | ph | tbppy (R1 = 3,5-tBu2) | tz | Cl | |
| 3 | [Pt(ph(tbppy)tz)(CCPh)] | ph | tbppy (R1 = 3,5-tBu2) | tz | CCPh |
|
| 4 | [Pt(ph(tbppy)tz)(CCC6F5)] | ph | tbppy (R1 = 3,5-tBu2) | tz | CCC6F5 |
|
| 5 | [Pt(na(tbppy)tz)Cl] | na | tbppy (R1 = 3,5-tBu2) | tz | Cl | |
| 6 | [Pt(na(tbppy)tz)(CCPh)] | na | tbppy (R1 = 3,5-tBu2) | tz | CCPh |
|
| 7 | [Pt(ph(ppy)btz)Cl] | ph | ppy (R1 = H) | btz | Cl | |
| 8 | [Pt(ph(tbppy)btz)Cl] | ph | tbppy (R1 = 3,5-tBu2) | btz | Cl | |
| 9 | [Pt(ph(tbppy)btz)(CCPh)] | ph | tbppy (R1 = 3,5-tBu2) | btz | CCPh |
|
| 10 | B | [Pt(th(ppy)py)Cl] | th | ppy (R1 = H) | py | Cl |
| 11 | [Pt(th(tbppy)py)Cl] | th | tbppy (R1 = 3,5-tBu2) | py | Cl | |
| 12 | [Pt(th(tbppy)py)(CCPh)] | th | tbppy (R1 = 3,5-tBu2) | py | CCPh |
|
| 13 | [Pt(th(tbppy)py)(CCC6F5)] | th | tbppy (R1 = 3,5-tBu2) | py | CCC6F5 |
|
| 14 | [Pt(bth(ppy)py)Cl] | bth | ppy (R1 = H) | py | Cl | |
| 15 | [Pt(bth(tbppy)py)Cl] | bth | tbppy (R1 = 3,5-tBu2) | py | Cl | |
| 16 | [Pt(bth(tbppy)py)(CCPh)] | bth | tbppy (R1 = 3,5-tBu2) | py | CCPh |
|
| 17 | C | [Pt(th(tbppy)tz)Cl] | th | tbppy (R1 = 3,5-tBu2) | tz | Cl |
| 18 | [Pt(th(tbppy)tz)(CCPh)] | th | tbppy (R1 = 3,5-tBu2) | tz | CCPh |
|
As in our previous study,23 we calculated UV-vis absorption and emission spectra using (TD)-DFT methods and decomposed the triplet emitting states into LC (π–π*, centred in the individual parts of the C^N^N ligand), LLCT (π–π*, between the individual parts of the C^N^N ligand and X–π*, between the coligand and the C^N^N), MLCT (dPt-to-π*C^N^N), and LMCT (pCl or πCCR-to-dPt) contributions. The decomposition of ligand-centred states into LC and LLCT character goes beyond previous DFT analyses of such complexes31,42–44 and turned out very helpful to assess adequately the contributions of the individual heteroaryl groups of the C^N^N ligands.
Results and discussion
Synthesis and characterisation
The HC^N^N protoligands (ligand precursors prior to cyclometalation) were assembled from the corresponding chalcones using the versatile Kröhnke synthesis (further details are found in the ESI†). The Pt(II) chlorido complexes were synthesised from the HC^N^N protoligands and K2[PtCl4] with up to 98% yield as orange to red solids. For excellent yields, a sufficient stirring and long reaction times with up to 3 days are required. The chlorido coligand was exchanged for alkynides under Sonogashira-like conditions in yields ranging from 69 to 96%. The compounds were analysed and characterised through elemental analysis, 1H NMR spectroscopy, and EI-MS(+) or HR-ESI-MS(+) (see Experimental section in the ESI†). The solubility of most of the complexes in common organic solvents is generally low but is significantly increased by decoration of the phenylpyridine with tert-butyl groups.Crystal and molecular structures
The two compounds [Pt(ph(tbppy)btz)(CCPh)] (9) and [Pt(th(ppy)py)Cl]·CD2Cl2 (10·CH2Cl2) were obtained as single crystals. Their crystal and molecular structures were studied by X-ray diffractometry experiments. Details of the structure solution and refinement (Table S1, ESI†) as well as figures showing the crystal structures (Fig. S1 to S4, ESI†) can be found in the ESI.† Both complexes show intermolecular π-stacking interactions with short centroid⋯centroid distances around 3.5 Å (Fig. S4†), which is characteristic for such complexes.10,13,15,24–28,30,31,33
Both complexes show planar geometries around the Pt atoms and almost planar C^N^N ligand cores (Fig. 1). The pending phenyl or di-tert-butyl-phenyl groups appear twisted towards this plane. Also, the phenyl group of the CCPh ligand in 9 shows a deviation of 65.8(3)° from the coordination plane (data in Table S2†).
 | ||
| Fig. 1 ORTEP plot of the Pt(II) complex in [Pt(ph(tbppy)btz)(CCPh)] (9) (left) and [Pt(th(ppy)py)Cl]·CH2Cl2 (10·CH2Cl2) (right) with ellipsoids at 50% probability. Co-crystallised CD2Cl2 and H atoms were omitted for clarity. | ||
Electrochemistry and DFT-calculated frontier orbitals of the complexes
The complexes showed a first reversible one-electron reduction wave in the range from −1.5 to −1.8 V (Fig. 2 and 3, Fig. S5 to S22, Table S4†). The first wave is usually followed by a second fully or partly reversible reduction wave at very low potentials (−2.1 to −2.5 V). A similar behaviour is observed for the protoligands HC^N^N at potentials lying about 0.8 V lower than those corresponding to the complexes (Fig. S23 to S26 and Table S3†). | ||
| Fig. 2 Cyclic voltammograms of [Pt(bth(tbppy)py)Cl] (15) in 0.1 M nBu4NPF6/THF (A) and /CH2Cl2 (B). | ||
 | ||
| Fig. 3 Redox potentials of [Pt(C^N^N)Cl] (A) and [Pt(C^N^N)CCPh] (B) complexes. The bars represent the experimental potentials (see Table S4, ESI†). | ||
The reduction potentials are very similar for the (benzo)thiophenyl complexes having an opposite thiazolyl moiety (Fig. 3A) and they are approximately invariant for the progression phenyl – naphthyl – thiophenyl (Fig. 3B). For the variation of the peripheral N group, the potentials increase along the series pyridine < thiazolyl < benzothiazolyl (Fig. 3B), while variation of the central N unit from ppy to tbppy leads only to subtle changes. Exchange of the coligand has almost no influence on the reduction potentials. We thus assign the first reductions as essentially taking place at the bipyridine and pyridyl(thiazolyl) moieties, in line with results for comparable complexes,10,13,23,24,33,35,48–57 with DFT calculated lowest unoccupied molecular orbitals (LUMO) for the phenyl complexes [Pt(ph(ppy)tz)Cl] (1) and [Pt(ph(tbppy)tz)Cl] (2),23 and calculations on the thiophenyl derivatives [Pt(th(tbppy)py)Cl] (11) and [Pt(th(tbppy)py)(CCPh)] (12) (Fig. S29†). The second reduction waves are separated by about 0.6 to 0.7 V, indicating that the second reduction events are also ligand-centred and a PtII → PtI reduction being unlikely.
On the anodic side, irreversible and in part poorly defined first oxidation waves are recorded in the range 0.1 to 0.9 V. Potentials for comparable Cl and CCC6F5 complexes are very similar; complexes with CCPh show a cathodic shift in agreement with a stronger σ donor ability of CCPh compared with Cl. In the case of fluoro substituents in CCC6F5, the σ donor ability is decreased making its overall impact on the electron density comparable to Cl. Very different oxidation potentials were found for variations in the C aryl group. Within the series of Cl complexes possesing a central tbppy unit, the potentials increase along the series bth < ph < th (Fig. 3A); however, for the oxidation waves, also the central py, ppy, or tbppy units seem to be of importance and a simple correlation is not obvious. The same is true for the complexes containing CCPh coligands, where all three groups seem to contribute, as indicated by the very different potentials for the two complexes containing the ph(tbppy)tz and ph(tbppy)btz ligand (Fig. 3B). Reports for similar complexes suggest essentially metal (PtII/PtIII) based oxidation processes.2,10,13,23,24,33,48–57 Our results agree with this hypothesis but also show strong contributions from the coligand and the C aryl group.
Our series of complexes covers a vast range of electrochemical gaps from 1.7 to 2.6 eV (Fig. 3, Table S4, ESI†) between the first reductions ranging from about −1.5 to −1.8 V and the first oxidations ranging from about 0.1 to 0.9 V. Thus, the gaps are essentially governed by the oxidation potentials. The smallest gaps were found for [Pt(ph(tbppy)btz)CCPh] (9) (1.71 eV) and [Pt(bth(tbppy)py)CCPh] (16) (1.90 eV) which are complexes bearing the same coligand but structurally very different, although isoelectronic, C^N^N ligands. The largest gaps were those of the th(tbppy)py (11) (2.62 eV) and ph(tbppy)tz (2) (2.31 V) complexes with the Cl coligand, which are also very different but isoelectronic. The similarity of the gaps for both pairs of complexes is probably the result of several electron-donating and -accepting effects compensating each other.
Experimental and TD-DFT calculated UV-vis absorption spectra
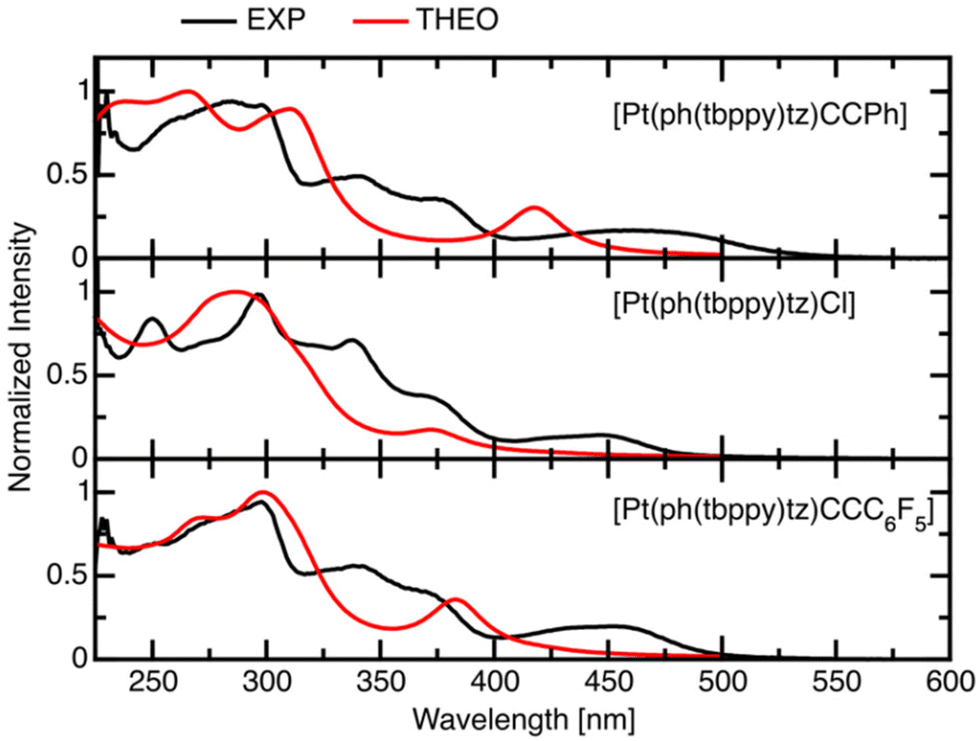
At a first glance, all complexes show similar absorption spectra with structured and intense bands in the range 250 to 400 nm, along with a partially structured band system from 400 to 500 nm with medium intensity and weak absorption bands in the 500 to 600 nm range (Fig. 4 and 5 and S31 to S44;† selected data in Table 2, full data in Table S6†). The intense UV bands appear also for the protoligands HC^N^N (Fig. S30, Table S5†) and can thus be assigned to transitions into states with predominant π–π* character. | ||
| Fig. 4 TD-DFT-calculated UV-vis absorption spectra (THEO, red) compared to experimental spectra (EXP, black) for all complexes with the CCPh coligand. | ||
 | ||
| Fig. 5 TD-DFT calculated UV-vis absorption spectra (THEO, red) of compared to experimental spectra (EXP, black) of [Pt(ph(tbppy)tz)CCPh] (3), [Pt(ph(tbppy)tz)Cl] (2), and [Pt(ph(tbppy)tz)CCC6F5] (4). | ||
| Complex | T /K |
λ
abs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) /nm
/nm |
λ
em
/nm |
τ
av
/μs |
Φ L ± 0.02/± 0.05e | k R/105 s−1f | k NR/105 s−1g | |
|---|---|---|---|---|---|---|---|---|
a Ar-Purged CH2Cl2 solution at 298 K or frozen glassy matrix of CH2Cl2/MeOH (1:1) at 77 K.
b Absorption data including molar absorption coefficients in Table S6, ESI.†
c
λ
exc = 350 nm, sh = shoulder.
d
λ
exc = 376 nm, amplitude-weighted average lifetime (the single exponential components and relative amplitudes are listed in Table S7†), the corresponding uncertainties are found in the ESI.†59
e The uncertainties for the glassy matrices are higher due to the measurement set up.
f Average radiative deactivation rate constants from the amplitude-weighted average lifetimes and their uncertainties are calculated as follows:  ; ;  .59
g Average radiationless deactivation rate constants from the amplitude-weighted average lifetimes and their uncertainties are calculated as follows: .59
g Average radiationless deactivation rate constants from the amplitude-weighted average lifetimes and their uncertainties are calculated as follows: 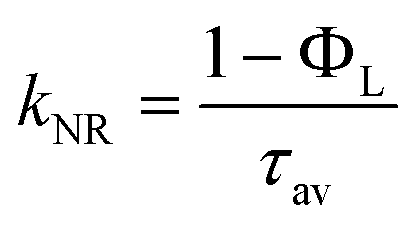 ; ;  .59
h From ref 23. .59
h From ref 23.
|
||||||||
| A | [Pt(ph(tbppy)tz)Cl] (2) h | 298 | 301, 340, 370, 446 | 608 | 0.71 | 0.07 | 1.0 ± 0.3 | 13.0 ± 0.4 |
| 77 | 300, 333, 360, 429 | 557, 603, 662sh | 4.01 | 0.22 | 0.55 ± 0.14 | 2.0 ± 0.2 | ||
| [Pt(ph(tbppy)tz)CCPh] (3) |
298 | 302, 346, 380, 443sh, 473, 495sh | 626, 645sh | 0.39 | 0.06 | 1.5 ± 0.7 | 24 ± 3 | |
| 77 | 336, 370, 425, 438, 468 | 558, 605, 660sh | 3.50 | 0.62 | 1.8 ± 0.2 | 1.1 ± 0.3 | ||
| [Pt(ph(tbppy)tz)CCC6F5] (4) |
298 | 345, 370, 445sh, 465 | 605, 635sh | 0.91 | 0.12 | 1.3 ± 0.3 | 9.7 ± 0.6 | |
| 77 | 340, 365, 420, 445 | 555, 600, 655sh, 725sh | 3.53 | 0.49 | 1.39 ± 0.19 | 1.4 ± 0.3 | ||
| [Pt(na(tbppy)tz)CCPh] (6) |
298 | 311, 335sh, 370, 398, 420sh, 468 | 635 | 0.41 | 0.10 | 2.5 ± 0.6 | 22.1 ± 1.4 | |
| 77 | 330, 395, 437, 463 | 555, 595, (650sh) | 3.97 | 0.82 | 2.1 ± 0.2 | 0.5 ± 0.3 | ||
| [Pt(ph(tbppy)btz)Cl] (8) | 298 | 353, 464 | 635, 675sh | 0.21 | < 0.02 | ≤ 2 | 47.6 ± 1.4 | |
| 77 | 346, 425, 452 | 600, 656, 710sh | 1.59 | 0.07 | 0.4 ± 0.3 | 5.8 ± 0.6 | ||
| [Pt(ph(tbppy)btz)CCPh] (9) |
298 | 360, 470, 500sh | 655 | 0.11 | < 0.02 | 1.8 ± 1.8 | 87 ± 5 | |
| 77 | 360, 470, 500sh | 600, 650, 700sh | 1.70 | 0.18 | 1.1 ± 0.3 | 4.8 ± 0.5 | ||
| B | [Pt(th(tbppy)py)Cl] (11) | 298 | 315, 370, 440 | 617, 660sh | 1.81 | 0.07 | ≤ 0.4 | 9.6 ± 0.5 |
| 77 | 375, 425, 470sh, 490sh | 595, 645, 700 | 11.6 | 0.58 | 0.50 ± 0.08 | 0.36 ± 0.15 | ||
| [Pt(th(tbppy)py)CCPh] (12) |
298 | 300, 373, 440, 475sh | 620, 660sh | 1.63 | 0.14 | 0.86 ± 0.13 | 5.28 ± 0.17 | |
| 77 | 360, 425sh, 460 | 590, 640, 700sh | 8.25 | 0.79 | 0.96 ± 0.10 | 0.25 ± 0.14 | ||
| [Pt(th(tbppy)py)CCC6F5] (13) |
298 | 312, 370, 425sh, 445 | 620, 660sh | 2.18 | 0.15 | 0.7 ± 0.1 | 3.90 ± 0.13 | |
| 77 | 350–375, 430 | 595, 650, 705 | 8.7 | 0.79 | 0.91 ± 0.09 | 0.24 ± 0.13 | ||
| [Pt(bth(tbppy)py)Cl] (15) | 298 | 350, 395, 440sh | 675, 730 | 1.07 | 0.03 | 0.28 ± 0.19 | 9.1 ± 0.3 | |
| 77 | 375, 480 | 655, 720, 745 | 4.16 | 0.13 | 0.31 ± 0.13 | 2.1 ± 0.2 | ||
| [Pt(bth(tbppy)py)CCPh] (16) |
298 | 360, 380sh, 405sh, 475 | 675, 740 | 1.11 | 0.04 | 0.36 ± 0.18 | 8.7 ± 0.3 | |
| 77 | 365, 400sh, 460 | 650, 720, 785 | 3.57 | 0.24 | 0.67 ± 0.16 | 2.1 ± 0.3 | ||
| C | [Pt(th(tbppy)tz)Cl] (17) | 298 | 368, 450 | 655 | 0.22 | <0.02 | ≤1.9 | 45 ± 3 |
| 77 | 364, 430, 503sh | 610, 660, 725sh | 4.17 | 0.13 | 0.31 ± 0.13 | 2.1 ± 0.2 | ||
| [Pt(th(tbppy)tz)CCPh] (18) |
298 | 375, 470 | 658 | 0.25 | <0.02 | ≤1.6 | 39.6 ± 1.1 | |
| 77 | 365, 425, 454 | 604, 657, 725sh | 4.19 | 0.35 | 0.84 ± 0.15 | 1.6 ± 0.2 | ||
With the exception of complexes 5, 7, 8, 14, and 15, all structures were optimised in the electronic ground state using DFT (for images, see Fig. S28 and S29†). The theoretical structures match very well with the experimentally obtained geometries for the complexes 9 and 10 and other derivatives. Based on these geometries, UV-vis absorption spectra were calculated using TD-DFT and compared with the experimental spectra. Fig. 4 compares all complexes carrying the CCPh coligand and shows a good match between calculated and experimental data in the UV range spanning from 200 to 350 nm, while for the visible range (350 to 500 nm) the calculated long-wavelength bands were slightly blue-shifted compared with the experimental bands. The weak transitions in the 500 to 600 nm range might thus be assigned to spin-forbidden absorption processes into the triplet manifold.
The long-wavelength absorption bands are markedly altered with the C^N^N variations, with maxima ranging from 435 to 505 nm. The change of coligands generally leads to red-shifts in the maxima along the series Cl < CCC6F5 < CCPh (Fig. 5, Fig. S36 and S40†) in agreement with the influence of the electron-withdrawing F substituents, whereas the Cl complexes appear even further blue-shifted. This is fully in line with previous reports.31,43,57,58 The highest energies for these absorption bands were found for the th(tbppy)py (11) and th(tbppy)tz (17) (436 nm) complexes bearing the Cl ligand. By far, the lowest energy was recorded at 505 nm for [Pt(ph(tbppy)btz)CCPh] (9), which obviously combines preferential C aryl and peripheral N groups for a low-energy absorption. However, the four “second-best” candidates [Pt(bth(tbppy)py)CCPh] (16) (469 nm), [Pt(th(tbppy)tz)CCPh] (18) (468 nm), [Pt(ph(tbppy)tz)CCPh] (3) (466 nm), and [Pt(ph(tbppy)btz)Cl] (8) (466 nm) are structurally very dissimilar, but their comparable energies might be the result of several effects and a best combination of C aryl and peripheral N groups cannot be deduced.
Experimental photoluminescence spectra
The photoluminescence (PL) spectra of all complexes (Fig. 6, and Fig. S37 to S106†) are characterised by broad emission bands at 298 K which partially gain vibrational structure upon cooling to 77 K. The PL properties are summarised in Table 2 (for the complete set of data with the uncertainties as well as the multiexponential decay components, see Fig. S54 to S106 and Table S7†). The excitation spectra (data in Table 2) mirror their absorption counterparts (Table S6†). | ||
Fig. 6 Normalised photoluminescence spectra (λexc = 350 nm) of all classes of complexes with focus on the influence of the co-ligand; top = classA: [Pt(ph(tbppy)tz)Cl] (2) (black), [Pt(ph(tbppy)tz)C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CPh] (3) (red) and [Pt(ph(tbppy)tz)CCC6F5] (4) (blue); centre = classB: [Pt(th(tbppy)py)Cl] (11) (black), [Pt(th(tbppy)py)CCPh] (12) (red) and [Pt(th(tbppy)py)CCC6F5] (13) (blue); bottom = class C: [Pt(th(tbppy)tz)Cl] (17) (black) and [Pt(th(tbppy)tz)CCPh] (18) (red). Measured in Ar-purged CH2Cl2 at 298 K (left) or frozen glassy CH2Cl2/MeOH matrices at 77 K (right). CPh] (3) (red) and [Pt(ph(tbppy)tz)CCC6F5] (4) (blue); centre = classB: [Pt(th(tbppy)py)Cl] (11) (black), [Pt(th(tbppy)py)CCPh] (12) (red) and [Pt(th(tbppy)py)CCC6F5] (13) (blue); bottom = class C: [Pt(th(tbppy)tz)Cl] (17) (black) and [Pt(th(tbppy)tz)CCPh] (18) (red). Measured in Ar-purged CH2Cl2 at 298 K (left) or frozen glassy CH2Cl2/MeOH matrices at 77 K (right). | ||
The change from the central ppy to tbppy has a marginal influence on the emissive properties of the complexes, in line with our recent report on [Pt(ph(ppy)py)Cl] (1) and [Pt(ph(tbppy)py)Cl] (2).23 However, the superior solubility of the tbppy was remarkable and we focused on these derivatives for the photophysical measurements, thus omitting complexes 1, 10 and 14 in Table 2. Also very generally, in CH2Cl2 solutions at 298 K, the triplet states are quenched by dissolved O2, as evidenced by the τav and ΦL values, which are increased upon purging with argon (see also Table S7†).
The complex [Pt(ph(tbppy)tz)Cl] (2) benchmarks the classA (Table 2) of complexes containing a peripheral phenyl (ph) or naphthyl (na) C aryl group as well as a peripheral N (benzo)thiazolyl, and shows a broad emission peaking at λmax = 608 nm (Fig. 6) with an incipient vibronic progression and amplitude-averaged lifetime τav of 0.714 μs with a photoluminescence quantum yield of ΦL = 0.07 at 298 K. The quantum yield is markedly increased for the CCC6F5 derivative 4 (600 nm, 3.53 μs, ΦL = 0.12) and also slightly for the naphthyl complex [Pt(na(tbppy)tz)CCPh] (6) (595 nm, 3.97 μs, ΦL = 0.10). Counterintuitively and probably due to additional rotovibrational modes, the non-radiative rate constant kNR at 298 K is increased from ∼13 × 105 s−1 to ∼24 × 105 s−1 when going from Cl to CCPh (2 → 3, 5 → 6, 8 → 9) and dropped when going to CCC6F5 (∼10 × 105 s−1, 4); however, the radiative rate constants kR are increased for both types of alkynyl complexes. The marked red-shifts of the emission maxima when introducing benzothiazolyl (1 → 7) instead of thiazolyl (2 → 8) (Fig. S44†) goes along with a reduction of ΦL, which seems to be mainly caused by the markedly increased kNR values, while the kR values drop only slightly. This can be attributed to the energy-gap law (EGL). In contrast, the change from phenyl to naphthyl (3 → 6) has almost no impact on the spectra, but markedly increases the ΦL. In frozen glassy matrices (CH2Cl2:MeOH; 1:1) at 77 K, the quantum yields are generally higher. Here, starting from 2 (Φ = 0.22), variation of ph to na or from Cl to CCPh allowed us to increase the quantum yield up to a ΦL of 0.82 (6), thus matching the highest values from the previous studies.30–32
For [Pt(th(tbppy)py)Cl] (11), as a starting point for classB (Table 2), for the complexes that contain a (benzo)thiophenyl C unit and a pyridine peripheral N group, a vibronic progression is visible but not completely pronounced with λmax = 617 nm, τav = 1.809 μs and ΦL = 0.07 at 298 K (Fig. 6), which is similar to the benchmark from classA, the complex [Pt(ph(tbppy)tz)Cl] (2). Both complexes constitute structural isomers in view of their atoms and their ring systems, but the variation of ph vs. th and tz vs. py might strongly affect the HOMO and the LUMO energies as well as the photoluminescence properties (the reasons will be discussed in the DFT-section, vide infra).
Thus, this similarity represents probably a cancellation of several effects. The markedly different behaviour at 77 K with a much larger ΦL for the thiophenyl (th) complex 17 (classC) points into this direction. As in classA complexes, going from thiophenyl (10 → 13) to the corresponding benzothiophenyl (14 → 16) derivatives leads to a red shift (Fig. S39†). For classB complexes, this has a stronger impact than the change tz → btz and we assume a higher 3π–π* character of the excited state for the (b)th complexes in line with the more pronounced vibronic progression. The τav, as well as the ΦL dropped (τav = 1.809 μs/ΦL = 0.07 to τav = 1.069 μs/ΦL = 0.03 in the case of [Pt(R(tbppy)py)Cl] (R = bth (15) vs. th (11)), most likely due to the energy-gap law leading to faster radiationless decays with increased kNR-values. An interesting exception is the naphthyl complex [Pt(na(tbppy)tz)CCPh] (6). When compared to the ph analogue 3, a smaller red shift at 298 K along with a higher kR is found, whereas at 77 K the emission appears very slightly blue shifted with a higher ΦL (0.82 vs. 0.62).
When going from fluid CH2Cl2 at 298 K to frozen glassy CH2Cl2/MeOH matrices at 77 K, the emission spectra show a pronounced vibronic progression and extended lifetimes. This can be attributed to the loss of solvent stabilisation. Nonetheless, the kR values remain essentially unaffected, whereas for the complexes displaying a bigger impact, the values remain in the same order of magnitude. The kNR-values, on the other hand, are all dramatically decreased due to the suppression of otherwise thermally accessible MC-mediated deactivation pathways.
Class
C complexes combine the thiophenyl ring as a C aryl donor and a thiazolyl as the peripheral N donor in the tridentate ligand. In general, the broad yet unstructured emission maxima appear red-shifted to λmax = 655 nm at 298 K, if compared with their extendend benzo-fused analogues while resembling classB complexes in this regard (Fig. S51†). However, upon cooling to 77 K, the properties are similar to classA derivatives. Besides a red-shift, the photophysical properties of [Pt(th(tbppy)tz)Cl] (17) at 298 K resemble those of [Pt(ph(tbppy)btz)Cl] (8); in frozen glassy matrices, the values are the same as for [Pt(bth(tbppy)py)Cl] (15) with a pronounced blue-shifted emission. The change of the coligand to yield [Pt(th(tbppy)tz)CCPh] (18) does not influence the emission wavelength, which resembles the behaviour of classB complexes. The overall values for [Pt(th(tbppy)tz)Cl] (17) can be best compared to [Pt(ph(tbppy)btz)CCPh] (9) at 298 K. So again, several effects compensate each other. Due to the loss of solvent stabilisation upon cooling (leading to more 3π–π*-character), the values at 77 K resemble again classB (e.g. [Pt(bth(tbppy)py)CCPh] (16)). All in all, classC complexes display photophysical properties lying between classes A and B, where at 298 K the MLCT-character is dominant, even though the coligand does not influence the emission maxima. In frozen glassy matrices, the loss of MLCT-character seems strong enough to resemble classB.
It is very interesting to compare our results with those reported for the very similar C^N^N-based Pt(II) complexes shown in Scheme 1 A (data in Table S8†).31 As for our series, some of these complexes show relatively poor photoluminescene efficiency at 298 K with ΦL reaching up to about 0.1 (such as in the case of [Pt(na(tbppy)py)Cl]), which fits perfectly into our series of structures. However, Table S8† shows derivatives with greatly enhanced ΦL, which makes them worth describing (and comparing) more in detail. The introduction of 2-quinolinyl or 1-isoquinolinyl replacing the peripheral N pyridine unit (Scheme 1A) resulted in two complexes [Pt(ph(tbppy)2-quin)Cl] and [Pt(ph(tbppy)1-isoquin)Cl] with highly red-shifted emission profiles and low ΦL (classD in Table S8†). In contrast to this, the introduction of the 3-isoquinolinyl group gave emitters with ΦL up to 0.99 for (classD). As in our study, the introduction of the CCC6F5 coligand turned out to be beneficial in the benchmarking complex [Pt(ph(tbppy)3-isoquin)(CCC6F5)] (ΦL = 0.99). At the same time, the emission of these complexes is markedly blue-shifted if compared with our derivatives containing peripheral N pyridyl or thiazolyl groups, which accounts at least for a part of the enhanced ΦL (energy gap law). Interestingly, the variation of the C aryl phenyl group through substitution does not vary essentially the emission energy but markedly the ΦL (from 0.14 to 0.79, classE in Table S8†). Introduction of thiophenyl instead of phenyl shifts the emission maxima closer to 600 nm (with high ΦL); however, in the presence of benzothiophenyl (bth), the emission spectra are red-shifted to over 640 nm and the ΦL values drop dramatically below 0.1 (classF). The authors explain these differences for two selected complexes invoking different contributions to the excited states, namely 3MLCT (d-to-π*(C^N^N)), 3π–π*(C^N^N), and XLCT (pCl-to-π*(C^N^N) for the chlorido complexes and 3MLCT, 3π–π* and 3L′LCT (π(CC-π*(C^N^N) for the alkynyl derivatives.31 The following TD-DFT-calculated emission spectra and their decomposition into different components for the excited triplet state (T1) shed more light on these assumptions for our series of complexes.
When comparing the best candidates of previous studies with our series, the complexes 11, 12, and 13 evolve as strong candidates with ΦL of 0.60 and more at 77 K along with very long average lifetimes τav (∼8–12 μs) at emission energies far lower than the benchmarking complex [Pt(ph(tbppy)3-isoquin)(CCC6F5)].31 Taken the relatively low emission energy, the high ΦL, and a long τav (876 μs), complex 13 is the most interesting candidate out of our series for applications, such as sensor arrays or optoelectronics, followed by complex 12 and 11.
(TD)-DFT calculations on the excited states and emission spectra
Vibrationally resolved emission spectra from the T1 state were calculated for selected complexes. In some cases, only the 0–0 transition energy (corrected for zero-point and thermal free energy contributions) could be computed due to numerical problems caused by insufficient overlap between the S0 and T1 wavefunctions. Fig. 7 shows a comparison of the calculated results at 300 K with the experimental emission spectra at 77 K and at RT for all complexes with the CCPh coligand. While the calculated curves match the experimental RT data quite well, they are considerably red-shifted compared to some of the experimental spectra at 77 K, in particular [Pt(ph(tbppy)tz)(CCPh)] (3) and [Pt(na(tbppy)tz)(CCPh)] (6).
 | ||
| Fig. 7 DFT-calculated emission spectra (THEO, red) compared to experimental spectra (EXP) at 77 K (black) and RT (blue) for all complexes with the CCPh coligand. Calculated 0–0 transitions are shown as vertical dashed green lines. | ||
Fig. S107† shows the influence of the central ligand (ppy versus tbppy) on the emission spectrum by comparing complexes [Pt(ph(ppy)tz)Cl] (1) and [Pt(ph(tbppy)tz)Cl] (2). As the calculated data confirm, the emission is affected very little by changing the central ligand from ppy to tbppy. When comparing the effect of different coligands (X = Cl, CCPh, and CCC6F5) on the emission spectra for [Pt(ph(tbppy)tz)X] complexes (Fig. S108†), the experimental spectra exhibit a very small blue-shift (less than 3 nm at 77 K) going from CCPh via Cl to CCC6F5, while the calculations produce a much more pronounced blue-shift, especially for CCC6F5. It is also worth noting that the calculated spectra of complexes 2 and 3 are in reasonable agreement with the experimental RT spectra, the experimental spectra at 77 K being considerably blue-shifted. For complex 4, however, the calculated spectrum matches almost exactly the experimental data at 77 K. A very different picture emerges from a similar analysis for [Pt(th(tbppy)py)X] complexes (Fig. S109†). Here, the calculated spectra are practically unaffected by changing the coligand, which is in accord with the experimental data. Furthermore, there is a much smaller difference between the experimental spectra at 77 K and at RT, the latter being in very good agreement with the calculations.
To understand the reasons for the different behaviours of classA and classB compounds in the calculations, we analysed in detail the character of the respective T1 → S0 transitions by performing a correlated electron–hole pair analysis.60 Fig. S110 to S113† show the decomposition of the emissive T1 states into the charge transfer contributions, namely MLCT (dPt-to-π*C^N^N) and LMCT (pCl or πCCR-to-dPt), as well as the ligand-centred excitations, namely LC (π–π* in one of the components of the C^N^N ligand) and LLCT (π–π* between different components of the C^N^N ligand), along with MC excitations possessing metal-centred character (d–d*). This goes beyond the previously presented decomposition into MLCT, π–π*, and XLCT (for Cl as coligand or L′LCT (for the alkynyl derivatives),29,31,36,42–44,47,57,58 and appears necessary in view of the individual contributions of the different parts of the C^N^N ligand, as discussed above.
The charge-transfer character (both MLCT and LLCT) increases significantly when changing the C aryl group from ph to na (3 → 6). Going from ph to th (3 → 18), on the other hand, slightly decreases the MLCT contribution whilst significantly increasing the LLCT character (Fig. S110†). The comparison of 12 and 16 reveals that changing th for bth considerably reduces the MLCT participation. Introducing py as a peripheral N group instead of tz also decreases the overall CT contributions, therefore increasing the LC character (Fig. S110†).
The effect of the main ligand on the character of the emissive T1 state is illustrated by comparing [Pt(ph(ppy)tz)Cl] (1) to [Pt(ph(tbppy)tz)Cl] (2) (Fig. S111†). Rather surprisingly, there are some sizable differences between the two compounds. Most notably, the MLCT character is higher for tbppy, while at the same time the LLCT contribution is lower. The analysis of the character of the emissive T1 states and the influence of the coligand for classA complexes (Fig. S110 and S112†) shows that variations between the different coligands are small. The MLCT contribution is largest for Cl and smallest for CCC6F5, whereas the opposite is true for the LLCT character. An analogous analysis for classB derivatives (Fig. S113†) reveals that generally the LC contribution is considerably larger and the MLCT character much smaller than for classA, which could partially explain why the accuracy of the calculated emission spectra is seemingly higher for classB than for classA (Fig. S112 and S113†). Moreover, the influence of the coligand is smaller than in the case of classA.
In addition, we have determined for all calculated complexes (from the TD-DFT eigenvectors) which molecular orbitals make the largest contributions to the emissive T1 states. All single-electron molecular orbital configurations that contribute at least 10% to the main configurations describing the emissive T1 states are listed in Table S9.† Visualisations of all involved orbitals can be found in Fig. 8 and Fig. S114 to S121.† In none of the compounds, the emissive T1 state corresponds to a pure HOMO–LUMO configuration; in some cases, other MO excitations are even more dominant. All contributing MOs have a pronounced metal component in addition to an extensive ligand part.
 | ||
| Fig. 8 Molecular orbitals of complexes 2 (top), 6 (middle) and 13 (bottom) with the largest contributions to the main monoelectronic excitations describing the emissive T1 state. | ||
A closer look reveals, that extending the C aryl group from ph to na (Fig. 8) or from th to bth (Fig. S119 and S120†) fully extents the π system and thus HOMO components. The same is true for the extension of tz to btz (Fig. 8 and Fig. S117†). Both the Cl and the CCPh coligands contribute exclusively to HOMO−n orbitals, including the HOMO for CCPh, while the CCC6F5 also contributes to the LUMO (Fig. 8) underpinning the special role of this coligand.31,36,43,53,54,57,58
Conclusions and outlook
Through a broad variation of the versatile cyclometalating anionic −C^N^N ligands and the ancillary ligands (or coligands) X in the Pt(II) complexes [Pt(C^N^N)X] (X = Cl, CCPh, or CCC6F5), we aimed to probe for the best combination of the potentially electron-donating peripheral C aryl group, the π accepting peripheral N group, and the coligand, while keeping the central N core (4-(3,5-di-tert-butyl)phenyl)pyridyl constant. Variations at the C aryl included phenyl (ph), naphthyl (na), thiophenyl (th), and benzothiophenyl (bth), while the peripheral N group was thiazolyl (tz), benzothiazolyl (btz), or pyridyl (py). Similar [Pt(C^N^N)X] complexes had been previously reported, some of them showing high photoluminescence (PL) quantum yields at 289 K in solution, but no straightforward conclusion on structure–properties relations was drawn so far. We thus embarked to produce the above described series of [Pt(C^N^N)X] complexes and studied them in detail through an experimental and in-depth (TD)-DFT study.
Through variation of the donating character of the C aryl group and the π accepting abilities of the N^N moiety varied through the peripheral N group our complexes covered a broad range of electrochemical gaps from 1.7 to 2.6 eV between the first reductions and the first oxidations. At the same time similar gaps were found for pairs of complexes which contain structurally very different C^N^N ligands which is probably the result of electron-donating and -accepting effects compensating each other.
The experimental long-wavelength UV-vis absorption bands are markedly altered with the C^N^N variations with maxima ranging from 435 to 505 nm. The change in coligands generally leads to red-shifts in the maxima along the series Cl < CCC6F5 < CCPh. TD-DFT calculated spectra agree qualitatively well with the observed trends for the long-wavelength absorption bands, while the bands in the UV range were very well matched by the calculations.
Photoluminescence (PL) spectra at 298 K in CH2Cl2 show broad emissions peaking between 605 and 675 nm with quantum yields ΦL ranging from <0.02 to 0.15. At 77 K, in frozen glassy matrices (CH2Cl2/MeOH 1:1), the PL spectra gain markedly in structure and show a general blue-shift of the maximum peak compared with the maximum of the broad bands at 298 K. The first maxima are found between 555 and 655 nm. As expected, the amplitude-weighted lifetimes τav increase from 0.39 to 2.18 μs at 298 K to 3.50 to 11.6 μs at 77 K.
The TD-DFT calculated emission spectra matched very well with the experimental spectra and the deconvolution showed excited states with pronounced LC (π–π*, centred in the individual parts of the C^N^N ligand) and LLCT (π–π*, between the individual parts of the C^N^N ligand) character, while MLCT (dPt-to-π*C^N^N) (15%) and LMCT (pCl or πCCR-to-dPt) contributions are markedly lower.
In conclusion we can qualitatively confirm the often made quote that efficient triplet emitting Pt(II) complexes show a high contribution (>50%) of ligand-centred states and a good contribution (>25%) of MLCT and LMCT states to the emitting triplet state. This combination makes the intersystem crossing fast and gives high quantum yields by boosting the phosphorescence rate from the lowest triplet state. At the same time, high ligand-centred contributions lead to high emission energies and can be easily traced by a high degree of vibronic progression of the emission bands. In turn, MLCT and LMCT contributions generally shorten the lifetimes. In our study we have deconvoluted the ligand-centred contributions to LC and LLCT, which has allowed to trace the contributions to individual parts of the C^N^N ligand. Thus, we have identified several very interesting candidates for future applications. But more importantly, since the C^N^N ligands can be readily composed of different C aryl, central N, and peripheral N groups in a modular fashion, we can provide a rational to synthesise efficient triplet emitting Pt(II) complexes. For the central N group, the (4-(3,5-di-tert-butyl)phenyl)-pyridyl was confirmed to be very useful. For the C aryl and peripheral N groups, the choice depends on the desired emission energy. The simple rule that the replacement of the Cl coligand by phenylacetylido (CCPh) or pentaflourophenylacetylido (CCC6F5) generally improves the emission efficiencies is not true in this case. The increased σ-donor character of the acetylido groups has to be well-matched with the σ-donor and π-acceptor properties of the C^N^N ligand to achieve efficient triplet emission. All in all, the approach of using these modular tridentate ligands + coligand, instead of sophisticated (laborous, low-yields) tetradentate ligands, has turned out to be very successful in providing efficiently phosphorescent Pt(II) complexes. To evaluate their potential application, further PL measurements on the promising candidates (including those of previous studies) in solid matrices (e.g. PMMA) are necessary.
Experimental section
Materials
Reagents were purchased from Merck, Alfa Aesar, Acros Organics or abcr and used without further purification. Solvents (CH2Cl2, THF, toluene, diethyl ether and CH3CN) were dried using an MBRAUN MB SPS-800 solvent purification system. Reactions which required the absence of air or moisture were performed under standard Schlenk technique. The protoligands and the Pt complexes are air-stable solids.Syntheses
The synthesis and detailed characterisation of the ligand precursors, the HC^N^N protoligands and the Pt(II) complexes is provided in the ESI† alongside with NMR spectra of ligands and Pt complexes (Fig. S001 to S074†).Instrumentation
1H, 13C and 19F NMR spectra were recorded on a Bruker Avance II 300 MHz (1H: 300.13 MHz, 13C: 75.47 MHz) – double resonance (BBFO) 5 mm observe probehead with z-gradient coil, Bruker Avance 400 MHz (1H: 400.13 MHz, 13C: 100.61 MHz, 19F: 376.50 MHz) using a triple resonance 1H, 19F, BB inverse probe head or Bruker Avance II 600 MHz spectrometer (1H: 600.13 MHz, 13C: 150.93 MHz) with a triple resonance (TBI) 5 mm inverse probehead with z-gradient coil using a triple resonance. The unambiguous assignment of the 1H resonances was obtained from 1H NOESY and 1H COSY experiments. 2D NMR experiments were performed using standard pulse sequences from the Bruker pulse program library. Chemical shifts are relative to tetramethylsilane (TMS) respectively. UV-vis absorption spectra were recorded with Varian Cary 05E or Cary 50 scan spectrophotometers. Photoluminescence spectra at room temperature were recorded with a Spex FluoroMax-3 spectrometer. A PicoQuant FluoTime 300 spectrometer was used for lifetime measurements. Lifetime analysis was performed using the commercial FluoFit software. The quality of the fit was assessed by minimising the reduced chi-squared function. Photoluminescence quantum yields (ΦL) were determined with a Hamamatsu Photonics absolute PL quantum yield measurement system (C9920-02), equipped with a L9799-01 CW xenon light source, monochromator, photonic multichannel analyser and integrating sphere (an uncertainty of ±5% is estimated for ΦL). All solvents were of spectroscopic grade. Both aerated samples and Ar-purged CH2Cl2 solutions were measured at 298 K in. Elemental analyses were obtained using a HEKAtech CHNS EuroEA 3000 analyzer. EI-MS spectra were measured with a Finnigan MAT 95, and HR-ESI-MS using a THERMO Scientific LTQ Orbitrap XL. Electrochemical measurements were carried out in 0.1 M nBu4NPF6/THF solution using a three-electrode configuration (glassy carbon working electrode, Pt counter electrode, Ag/AgCl reference electrode) and a Metrohm Autolab PGSTAT30 or μStat 400 potentiostats. The ferrocene/ferrocenium couple served as internal reference. For electrochemical measurements extra dry solvents were used: extra dry DMF and CH2Cl2 was purchased from Acros Organics and stored over molecular sieve. THF was dried and destilled over Na/K (70/30) prior to use.Single crystal X-ray diffractometric analysis
The data collection for [Pd(th(ppy)py)Cl]·CH2Cl2 (10·CH2Cl2) was performed at T = 170(2) K on a STOE IPDS II diffractometer (STOE and Cie., Darmstadt, Germany) with Mo-Kα radiation (λ = 0.71073 Å) employing ω–φ–2θ scan technique. The structures were solved by dual space methods (SHELXT-2015)61 and refined by full-matrix least-squares techniques against F2 (SHELXL-2017/1)62,63 with F02 ≥ 2σ(F02). The numerical absorption correction (X-RED V1.31; STOE and Cie, 2005)64 were performed after optimising the crystal shapes using X-SHAPE V1.06 (Stoe & Cie, 1999).65 The structure of [Pt(ph(tbppy)btz)(CCPh)] (9) was measured using synchrotron radiation (λ = 0.56076 Å) at 100(2) K, solved and refined without absorption correction as mentioned above. All non-hydrogen atoms were treated anisotropically; hydrogen atoms were included by using appropriate riding models. The dataset for 9 is generally poor and A alerts appeared in the checkcif. The problem is due to poor quality of the crystal. Nevertheless, we are convinced that the structure solution represents the geometric details of complex 9. CCDC 2081667 [Pd(th(ppy)py)Cl]·CD2Cl2 and 2084095 [Pt(ph(tbppy)btz)(CCPh)]† contains the full crystallographic data.
DFT calculations
The optimised geometries and the vibrational Franck–Condon spectra were determined with the quantum chemistry package Gaussian 09 Rev. D.0166 using the CAM-B3LYP functional67 together with Grimme's D3 dispersion correction with Becke–Johnson damping.68 The SDD basis set applies an effective core potential for the Pt atoms,69 while the D95 basis set is used for H, C, N, and O atoms.70To compute vibrational Franck–Condon spectra, the method of Barone et al.71,72 with Kohn–Sham DFT-based geometry optimisations in the S0 and T1 states followed by frequency analysis calculations was used.
All energy corrections were done by adding zero-point vibrational energies and thermal free energy contributions. To calculate the overlap integrals for the vibronic spectra, the transitions are divided into classes Cn, where n is the number of the excited normal modes in the final electronic state. The number of quanta per mode was limited to 100 while the number of quanta for combinations of two modes was restricted to 65. The maximum number of computed integrals was set to 1.5 × 108. A maximum of 20 classes was calculated. The line spectrum was broadened with the aid of Gaussian functions with a half-width at half-maximum of 400 cm−1.
The solvent CH2Cl2 was taken into account by the polarisable continuum model (PCM) in an integral equation formalism framework73 with atomic radii from the universal force field model (UFF).74 To characterise the emissive T1 state, the T1 geometry was first optimised with Kohn–Sham DFT with multiplicity 3 followed by TD-DFT calculations of the T1 state. TD-DFT calculations of the 40 lowest excited singlet states were performed at the optimised S0 geometry. Finally, a Lorentzian broadening with a half-width at half-maximum (HWHM) of 10 nm was used for each transition.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The Deutsche Forschungsgemeinschaft [DFG Priority Programme 2102 “Light-controlled Reactivity of Metal Complexes”, DO 768/5-1 and 5-2, STR 1186/6-1 and 6-2, and KL1194/16-1 and 16-2] is acknowledged for funding of this project. We also like to thank Prof. Dr Mathias Schäfer, Department of Chemistry, University of Cologne for HR-ESI-MS measurements and the Regional Computing Center of the University of Cologne (RRZK) for providing computing time on the DFG-funded High Performance Computing (HPC) system CHEOPS as well as for the support. We also thank Ms Rose Jordan, Department of Chemistry, University of Cologne for assistance with the crystal structure solutions. Furthermore, computer time at the HPC cluster PALMA II at WWU is gratefully acknowledged. CAS would like to acknowledge the Cluster of Excellence Cells in Motion (DFG EXC 1003) for financial support.References
- I. O. Koshevoy, M. Krause and A. Klein, Non-Covalent Intramolecular Interactions through Ligand-Design Promoting Efficient Luminescence from Transition Metal Complexes, Coord. Chem. Rev., 2020, 405, 213094 CrossRef.
- G. Cheng, Y. Kwak, W.-P. To, T.-L. Lam, G. S. M. Tong, M.-K. Sit, S. Gong, B. Choi, W. Choi, C. Yang and C.-M. Che, High-Efficiency Solution-Processed Organic Light-Emitting Diodes with Tetradentate Platinum(II) Emitters, ACS Appl. Mater. Interfaces, 2019, 11, 45161–45170 CrossRef PubMed.
- G. Millán, N. Giménez, R. Lara, J. R. Berenguer, M. T. Moreno, E. Lalinde, E. Alfaro-Arnedo, I. P. López, S. Piñeiro-Hermida and J. G. Pichel, Luminescent Cycloplatinated Complexes with Biologically Relevant Phosphine Ligands: Optical and Cytotoxic Properties, Inorg. Chem., 2019, 58, 1657–1673 CrossRef PubMed.
- A. I. Solomatina, P. S. Chelushkin, T. O. Abakumova, V. A. Zhemkov, M. Kim, I. Bezprozvanny, V. V. Gurzhiy, A. S. Melnikov, Y. A. Anufrikov, I. O. Koshevoy, S.-H. Su, P.-T. Chou and S. P. Tunik, Reactions of Cyclometalated Platinum(II) [Pt(N^C)(PR3)Cl] Complexes with Imidazole and Imidazole-Containing Biomolecules: Fine-Tuning of Reactivity and Photophysical Properties via Ligand Design, Inorg. Chem., 2019, 58, 204–217 CrossRef CAS.
- L. Liu, X. Wang, F. Hussain, C. Zeng, B. Wang, Z. Li, I. Kozin and S. Wang, Multiresponsive Tetradentate Phosphorescent Metal Complexes as Highly Sensitive and Robust Luminescent Oxygen Sensors: Pd(II) Versus Pt(II) and 1,2,3-Triazolyl Versus 1,2,4-Triazolyl, ACS Appl. Mater. Interfaces, 2019, 11, 12666–12674 CrossRef CAS.
- X. Wang and S. Wang, Phosphorescent Pt(II) Emitters for OLEDs: From Triarylboron-Functionalized Bidentate Complexes to Compounds with Macrocyclic Chelating Ligands, Chem. Rec., 2019, 19, 1–18 CrossRef.
- Y. Zhang, Y. Wang, J. Song, J. Qu, B. Li, W. Zhu and W.-Y. Wong, Near-Infrared Emitting Materials via Harvesting Triplet Excitons: Molecular Design, Properties, and Application in Organic Light Emitting Diodes, Adv. Opt. Mater., 2018, 6, 1800466 CrossRef.
- C. Cebrián and M. Mauro, Recent advances in phosphorescent platinum complexes for organic light-emitting diodes, Beilstein J. Org. Chem., 2018, 14, 1459–1481 CrossRef PubMed.
- S. Wilde, D. Ma, T. Koch, A. Bakker, D. Gonzalez-Abradelo, L. Stegemann, C. D. Daniliuc, H. Fuchs, H. Gao, N. L. Doltsinis, L. Duan and C. A. Strassert, Toward Tunable Electroluminescent Devices by Correlating Function and Submolecular Structure in 3D Crystals, 2D-Confined Monolayers, and Dimers, ACS Appl. Mater. Interfaces, 2018, 10, 22460–22473 CrossRef PubMed.
- M. Krause, D. Kourkoulos, D. González-Abradelo, K. Meerholz, C. A. Strassert and A. Klein, Luminescent PtII Complexes of Tridentate Cyclometalating 2,5-Bis(aryl)-pyridine Ligands, Eur. J. Inorg. Chem., 2017, 2017, 5215–5223 CrossRef.
- T. Fleetham, G. Li and J. Li, Phosphorescent Pt(II) and Pd(II) Complexes for Efficient, High-Color-Quality, and Stable OLEDs, Adv. Mater., 2017, 29, 1601861 CrossRef PubMed.
- T.-T. Feng, F.-Q. Bai, L.-M. Xie, Y. Tang and H.-X. Zhang, Theoretical study and design of highly efficient platinum(II) complexes bearing tetradentate ligands for OLED, RSC Adv., 2016, 6, 11648–11656 RSC.
- K. Li, G. S. M. Tong, Q. Wan, G. Cheng, W.-Y. Tong, W.-H. Ang, W.-L. Kwong and C.-M. Che, Highly phosphorescent platinum(II) emitters: photophysics, materials and biological application, Chem. Sci., 2016, 7, 1653–1673 RSC.
- T. Strassner, Phosphorescent Platinum(II) Complexes with C^C* Cyclometalated NHC Ligands, Acc. Chem. Res., 2016, 49, 2680–2689 CrossRef PubMed.
- J. Sanning, L. Stegemann, P. R. Ewen, C. Schwermann, C. G. Daniliuc, D. Zhang, N. Lin, L. Duan, D. Wegner, N. L. Doltsinis and C. A. Strassert, Colour-tunable asymmetric cyclometalated Pt(II) complexes and STM-assisted stability assessment of ancillary ligands for OLEDs, J. Mater. Chem. C, 2016, 4, 2560–2565 RSC.
- J. Zhao, W. Wu, J. Sun and S. Guo, Triplet photosensitizers: from molecular design to applications, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- V. W.-W. Yam and K. M.-C. Wong, Luminescent metal complexes of d6, d8 and d10 transition metal centres, Chem. Commun., 2011, 47, 11579–11592 RSC.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, The triplet state of organo-transition metal compounds. Triplet harvesting and singlet harvesting for efficient OLEDs, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- J. A. G. Williams, The coordination chemistry of dipyridylbenzene: N-deficient terpyridine or panacea for brightly luminescent metal complexes?, Chem. Soc. Rev., 2009, 38, 1783–1801 RSC.
- J. A. G. Williams, S. Develay, D. L. Rochester and L. Murphy, Optimising the luminescence of platinum(II) complexes and their application in organic light emitting devices (OLEDs), Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef.
- J. Föller, D. H. Friese, S. Riese, J. M. Kaminski, S. Metz, D. Schmidt, F. Würthner, C. Lambert and C. M. Marian, On the photophysical properties of IrIII, PtII, and PdII (phenylpyrazole) (phenyldipyrrin) complexes, Phys. Chem. Chem. Phys., 2020, 22, 3217–3233 RSC.
- S. Poirier, H. Lynn, C. Reber, E. Tailleur, M. Marchivie, P. Guinneau and M. R. Probert, Variation of M⋯H−C Interactions in Square-Planar Complexes of Nickel(II), Palladium(II), and Platinum(II) Probed by Luminescence Spectroscopy and X-ray Diffraction at Variable Pressure, Inorg. Chem., 2018, 57, 7713–7723 CrossRef CAS.
- M. Krause, R. von der Stück, D. Brünink, S. Buss, N. L. Doltsinis, C. A. Strassert and A. Klein, Platinum and palladium complexes of tridentate −C^N^N (phen-ide)-pyridine-thiazol ligands – A case study involving spectroelectrochemistry, photoluminescence spectroscopy and TD-DFT calculations, Inorg. Chim. Acta, 2021, 518, 120093 CrossRef.
- T. Eskelinen, S. Buss, S. K. Petrovskii, E. V. Grachova, M. Krause, L. Kletsch, A. Klein, C. A. Strassert, I. O. Koshevoy and P. Hirva, Photophysics and Excited State Dynamics of Cyclometalated [M(C^N^N)(CN)] (M = Ni, Pd, Pt) Complexes: A Theoretical and Experimental Study, Inorg. Chem., 2021, 60, 8777–8789 CrossRef.
- M. Hebenbrock, D. González-Abradelo, A. Hepp, J. Meadowcroft, N. Lefringhausen, C. A. Strassert and J. Müller, Influence of the ancillary ligands on the luminescence of platinum(II) complexes with a triazole-based tridentate C^N^N luminophore, Inorg. Chim. Acta, 2021, 516, 119988 CrossRef.
- S. C. Gangadharappa, I. Maisuls, D. A. Schwab, J. Kösters, N. L. Doltsinis and C. A. Strassert, Compensation of Hybridization Defects in Phosphorescent Complexes with Pnictogen-Based Ligands-A Structural, Photophysical, and Theoretical Case-Study with Predictive Character, J. Am. Chem. Soc., 2020, 142, 21353–21367 CrossRef PubMed.
- S. Garbe, M. Krause, A. Klimpel, I. Neundorf, P. Lippmann, I. Ott, D. Brünkink, C. A. Strassert, N. L. Doltsinis and A. Klein, Cyclometalated Pt Complexes of CNC Pincer Ligands: Luminescence and Cytotoxic Evaluation, Organometallics, 2020, 39, 746–756 CrossRef.
- Y. Luo, Z. Chen, J. Hu, Z. Xu, Q. Meng and D. Tang, Small substituent groups as geometric controllers for tridentate platinum(II) complexes to effectively suppress non-radiative decay processes, Phys. Chem. Chem. Phys., 2019, 21, 2764–2770 RSC.
- Y.-T. Liu, Y.-R. Li, X. Wang and F.-Q. Bai, Theoretical investigation of N^C^N-coordinated Pt(II) and Pd(II) complexes for long-lived two-photon photodynamic therapy, Dyes Pigm., 2017, 142, 55–61 CrossRef.
- R. Mroz, D. A. K. Vezzu, B. Wallace, D. Ravindranathan, J. Carroll, R. D. Pike and S. Huo, A Comparative Study on Phosphorescent Cycloplatinated Complexes Based on Tridentate C^N^N-Coordinating Ligands and Phenylethynyl or Phenyl Ligand, Chin. J. Org. Chem., 2018, 38, 171–182 CrossRef CAS.
- P.-K. Chow, G. Cheng, G. S. M. Tong, W.-P. To, W.-L. Kwong, K.-H. Low, C.-C. Kwok, C. Ma and C.-M. Che, Luminescent Pincer Platinum(II) Complexes with Emission Quantum Yields up to Almost Unity: Photophysics, Photoreductive C-C Bond Formation, and Materials Applications, Angew. Chem., Int. Ed., 2015, 54, 2084–2089 CrossRef CAS PubMed.
- P. Wu, E. L.-M. Wong, D.-L. Ma, G. S.-M. Tong, K.-M. Ng and C.-M. Che, Cyclometalated Platinum(II) Complexes as Highly Sensitive Luminescent Switch-On Probes for Practical Application in Protein Staining and Cell Imaging, Chem. – Eur. J., 2009, 15, 3652–3656 CrossRef CAS PubMed.
- S. C. F. Kui, F.-F. Hung, S.-L. Lai, M.-Y. Yuen, C.-C. Kwok, K.-H. Low, S. S.-Y. Chui and C.-M. Che, Luminescent Organoplatinum(II) Complexes with Functionalized Cyclometalated C^N^C Ligands: Structures, Photophysical Properties, and Material Applications, Chem. – Eur. J., 2012, 18, 96–109 CrossRef CAS.
- L. Wang, Y. Zhang, J. Li, H. He and J. Zhang, Influence of primary and auxiliary ligand on spectroscopic properties and luminescent efficiency of organoplatinum(II) complexes bearing functionalized cyclometalated C^N^C ligands, Dalton Trans., 2014, 43, 14029–14038 RSC.
- I. Maisuls, C. Wang, M. E. Gutierrez Suburu, S. Wilde, C.-G. Daniliuc, D. Brünink, N. L. Doltsinis, S. Ostendorp, G. Wilde, J. Kösters, U. Resch-Genger and C. A. Strassert, Ligand-controlled and nanoconfinement-boosted luminescence employing Pt(II) and Pd(II) complexes: from color-tunable aggregation-enhanced dual emitters towards self-referenced oxygen reporters, Chem. Sci., 2021, 12, 3270–3281 RSC.
- D. Qiu, J. Wu, Z. Xie, Y. Cheng and L. Wang, Synthesis, photophysical and electrophosphorescent properties of mononuclearPt(II) complexes with arylamine functionalized cyclometalating ligands, J. Organomet. Chem., 2009, 694, 737–746 CrossRef.
- M. Hruzd, N. le Poul, M. Cordier, S. Kahlal, J.-Y. Saillard, S. Achelle, S. Gauthier and F. Robin-le Guen, Luminescent cyclometalated alkynylplatinum(II) complexes with 1,3-di(pyrimidin-2-yl)benzene ligands: synthesis, electrochemistry, photophysics and computational studies, Dalton Trans., 2022, 51, 5546–5560 RSC.
- G. De Soricellis, F. Fagnani, A. Colombo, C. Dragonetti and D. Roberto, Exploring the potential of N^C^N cyclometalated Pt(II) complexes bearing 1,3-di(2-pyridyl)benzene derivatives for imaging and photodynamic therapy, Inorg. Chim. Acta, 2022, 541, 121082 CrossRef.
- W. Cai, H. Zhang, X. Yan, A. Zhao, R. He, M. Li, Q. Meng and W. Shen, What accounts for the color purity of tetradentate Pt complexes? A computational analysis, Phys. Chem. Chem. Phys., 2019, 21, 8073–8080 RSC.
- M. Cnudde, D. Brünink, N. L. Doltsinis and C. A. Strassert, Tetradentate N^N°N^N-type luminophores for Pt(II) complexes: Synthesis, photophysical and quantum-chemical investigation, Inorg. Chim. Acta, 2021, 518, 120090 CrossRef.
- L. Wang, J. Wen, H. He and J. Zhang, The reasons for ligand-dependent quantum yields and spectroscopic properties of platinum(II) complexes based on tetradentate O^N^C^N ligands: a DFT and TD-DFT study, Dalton Trans., 2014, 43, 2849–2858 RSC.
- G. S.-M. Tong and C.-M. Che, Emissive or Nonemissive? A Theoretical Analysis of the Phosphorescence Efficiencies of Cyclometalated Platinum(II) Complexes, Chem. – Eur. J., 2009, 15, 7225–7237 CrossRef PubMed.
- B. Yang, S. Huang and J. Wang, How do ligands influence the quantum yields of cyclometalated platinum(II) complexes, theoretical research study, Phys. Chem. Chem. Phys., 2017, 19, 23454–23460 RSC.
- W. Li, J. Wang, X. Yan, H. Zhang and W. Shen, Electronic structures and photophysical properties of phosphorescent platinum(II) complexes with tridentate C^N*N cyclometalated ligands, Appl. Organomet. Chem., 2018, 32, e3929 CrossRef.
- A. Heil and C. M. Marian, Structure−Emission Property Relationships in Cyclometalated Pt(II) β-Diketonate Complexes, Inorg. Chem., 2019, 58, 6123–6136 CrossRef CAS.
- H. Kurz, K. Schötz, I. Papadopoulos, F. W. Heinemann, H. Maid, D. M. Guldi, G. Hörner and B. Weber, A Fluorescence-Detected Coordination-Induced Spin State Switch, J. Am. Chem. Soc., 2021, 143, 3466–3480 CrossRef CAS.
- M. L. Clark, S. Diring, P. Retailleau, D. R. McMillin and R. Ziessel, Spectroscopic Properties of Orthometalated Platinum(II) Bipyridine Complexes Containing Various Ethynylaryl Groups, Chem. – Eur. J., 2008, 14, 7168–7179 CrossRef.
- V. Lingen, A. Lüning, C. Strauss, I. Pantenburg, G. B. Deacon, G. Meyer and A. Klein, Platinum Complexes with the SC6F4H-4 Ligand – Synthesis, Spectroscopy and Structures, Inorg. Chim. Acta, 2014, 423, 152–162 CrossRef.
- G.-J. Zhang, X. Gan, Q.-Q. Xu, Y. Chen, X.-J. Zhao, B. Qin, X.-J. Lv, S.-W. Lai, W.-F. Fu and C.-M. Che, Photophysical and electrochemical properties of platinum(II) complexes bearing a chromophore–acceptor dyad and their photocatalytic hydrogen evolution, Dalton Trans., 2012, 41, 8421–8429 RSC.
- M.-Y. Yuen, S. C. F. Kui, K.-H. Low, C.-C. Kwok, S. S.-Y. Chui, C.-W. Ma, N. Zhu and C.-M. Che, Synthesis, Photophysical and Electrophosphorescent Properties of Fluorene-Based Platinum(II) Complexes, Chem. – Eur. J., 2010, 16, 14131–14141 CrossRef PubMed.
- K. Li, T. Zou, Y. Chen, X. Guan and C.-M. Che, Pincer-Type Platinum(II) Complexes Containing N-Heterocyclic Carbene (NHC) Ligand: Structures, Photophysical and Anion-Binding Properties, and Anticancer Activities, Chem. – Eur. J., 2015, 21, 7441–7453 CrossRef PubMed.
- R. P.-L. Tang, K. M.-C. Wong, N. Zhu and V. W.-W. Yam, Luminescence, electrochemistry and host–guest properties of dinuclear platinum(II) terpyridyl complexes of sulfur-containing bridging ligands, Dalton Trans., 2009, 2009, 3911–3922 RSC.
- A. Klein, J. van Slageren and S. Záliš, Co-ligand involvement in ground and excited states of electron rich polypyridyl platinum(II) complexes, Eur. J. Inorg. Chem., 2003, 2003, 1917–1938 CrossRef.
- S.-C. Chan, M. C. W. Chan, Y. Wang, C.-M. Che, K.-K. Cheung and N. Zhu, Organic Light-Emitting Materials Based on Bis(arylacetylide)platinum(II) Complexes Bearing Substituted Bipyridine and Phenanthroline Ligands: Photo- and Electroluminescence from 3MLCT Excited States, Chem. – Eur. J., 2001, 7, 4180–4190 CrossRef CAS PubMed.
- J. van Slageren, A. Klein, S. Záliš and D. J. Stufkens, Resonance Raman spectra of d6 metal-diimine complexes reflect changes in metal-ligand interaction and character of electronic transitions, Coord. Chem. Rev., 2001, 219–221, 937–955 CrossRef CAS.
- T.-C. Cheung, K.-K. Cheung, S.-M. Peng and C.-M. Che, Photoluminescent cyclometallated diplatinum(II,II) complexes: photophysical properties and crystal structures of [PtL(PPh3)]ClO4 and [Pt2L2(μ-dppm)][ClO4] (HL = 6-phenyl-2,2′-bipyridine, dppm = Ph2PCH2PPh2), J. Chem. Soc., Dalton Trans., 1996, 1996, 1645–1651 RSC.
- W. Lu, B.-X. Mi, M. C. W. Chan, Z. Hui, C.-M. Che, N. Zhu and S.-T. Lee, Light-Emitting Tridentate Cyclometalated Platinum(II) Complexes Containing σ-Alkynyl Auxiliaries: Tuning of Photo- and Electrophosphorescence, J. Am. Chem. Soc., 2004, 126, 4958–4971 CrossRef PubMed.
- W. Lu, M. C. W. Chan, N. Zhu, C.-M. Che, Z. He and K.-Y. Wong, Structural Basis for Vapoluminescent Organoplatinum Materials Derived from Noncovalent Interactions as Recognition Components, Chem. – Eur. J., 2003, 9, 6155–6166 CrossRef PubMed.
- A. Sillen and Y. Engelborghs, The Correct Use of “Average” Fluorescence Parameters, Photochem. Photobiol., 1998, 67, 475–486 Search PubMed.
- F. Plasser, TheoDORE: A toolbox for a detailed and automated analysis of electronic excited state computations, J. Chem. Phys., 2020, 152(8), 84108 CrossRef PubMed.
- G. M. Sheldrick, ShelXT – Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, SHELXL-2017/1, Program for the Solution of Crystal Structures, University of Göttingen, Göttingen, Germany, 2017 Search PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- STOE X-RED, Data Reduction Program, Version 1.31/Windows, STOE & Cie, Darmstadt, Germany, 2005 Search PubMed.
- STOE X-SHAPE, Crystal Optimisation for Numerical Absorption Correction, Version 1.06/Windows, STOE & Cie, Darmstadt, Germany, 1999 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian 09, Revision D.01. 2009, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange−correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 293, 51–57 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Energy-adjusted ab initio pseudopotentials for the second and third row transition elements, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- T. H. Dunning and P. J. Hay, in Modern Theoretical Chemistry, ed. H. F. Schaefer III, Plenum, New York, 1976, Vol. 3, pp. 1–28 Search PubMed.
- V. Barone, J. Bloino and M. Biczysko, Vibrationally-resolved electronic spectra in GAUSSIAN 09, GAUSSIAN 09 Revision A.02, 2009, 1–20 Search PubMed.
- V. Barone, J. Bloino, M. Biczysko and F. Santoro, Fully Integrated Approach to Compute Vibrationally Resolved Optical Spectra: From Small Molecules to Macrosystems, J. Chem. Theory Comput., 2009, 5, 540–554 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef PubMed.
- W. M. Skid, UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis procedures and NMR spectra. Figures with structural information, cyclic voltammograms, UV-vis absorption and photoluminescence spectra (incl. time-resolved decays) alongside with tables containing complete crystallographic and structural data as well as electrochemical potentials, UV-vis absorption and photoluminescence details. CCDC 2208874 and 2084095 for [Pt(ph(tbppy)btz)(CCPh)] and [Pt(th(ppy)py)Cl]·CD2Cl2, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02688k |
| This journal is © The Royal Society of Chemistry 2022 |