
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterisation of group 8 tris(1-benzyl-1,2,3-triazol-4-yl)-p-anisolylmethane complexes†
Samuel
Francis
 ,
Craig R.
Rice
,
Paul A.
Scattergood
and
Paul I. P.
Elliott
*
,
Craig R.
Rice
,
Paul A.
Scattergood
and
Paul I. P.
Elliott
*
Department of Chemical Sciences & Centre for Functional Materials, University of Huddersfield, Queensgate, Huddersfield, HD1 3DH, UK. E-mail: p.i.elliott@hud.ac.uk
First published on 22nd August 2022
Abstract
The tris(1,2,3-triazol-4-yl)methane framework offers a highly versatile architecture for ligand design, yet the coordination chemistry of this class of ligand remains largely unexplored. We report here the synthesis and characterisation of the homoleptic complexes [M(ttzm)2](PF6)2 (ttzm = tris(1-benzyl-1,2,3-triazol-4-yl)-p-anisolylmethane; M = Fe (Fe), Ru (Ru), Os (Os)). Initial attempts to prepare Ru by reaction of [Ru(p-cymene)Cl2]2 and ttzm also led to the isolation of the heteroleptic complex [Ru(p-cymene)(ttzm)](PF6)2. The structures of [Ru(p-cymene)(ttzm)](PF6)2, [Fe(ttzm)2]2+ (as its BPh4− salt) and Os were solved by X-ray diffraction. The homoleptic Fe(II) and Os(II) containing cations adopt distorted octahedral geometries due to the steric interactions between the ansiole and triazole rings of the ttzm ligands. The homoleptic complexes all adopt a low-spin d6 configuration and exhibit reversible M(II)/M(III) processes (+0.35 to +0.72 V vs. Fc/Fc+). These oxidation processes are cathodically shifted relative to those of related hexatriazole donor based complexes with density functional theory (DFT) calculations showing the metal d-orbitals are destabilised through a π-donor contribution from the triazole rings. The complexes all show prominent UV-visible absorption bands between 350 and 450 nm assigned to transitions of 1MLCT character. Whilst none of the homoleptic complexes are emissive in room temperature fluid solutions, Os is emissive at 77 K in an EtOH/MeOH glass (λmax 472 nm).
Introduction
Tris(pyrazoly)borate (Tp) ligands, originally pioneered by Trofimenko,1 have seen widespread use in coordination chemistry and have led to a wealth of coordination and organometallic chemistry involving the d-block elements.2–5 The basic ligand structure has been modified to modulate the steric and electronic properties of the tripodal ligand, for example, with replacement of the pyrazolyl N-donor with N-heterocyclic carbene donor moieties6,7 that have enabled access to first row transition metal complexes with exciting photophysical properties.8,9Tris(pyrazolyl)alkane ligands as neutral analogues of the Tp framework have also been explored.10,11 For example, Meyer12 and Thomas have explored the coordination chemistry of tris(pyrazolyl)methane ligands with Ru(II) with the Thomas group reporting achiral DNA-light switch complexes when combined in heteroleptic complexes with DNA-intercalating dipyridophenazine-type ligands.13 Analogous neutral tris(pyridyl)ethane (tpe) ligands have also recently attracted interest with Heinze and co-workers reporting the homoleptic Cr(III) complex [Cr(tpe)2]3+ which exhibits efficient near-IR phosphorescence stemming from the highly octahedral coordination geometry.14 This leads to sufficiently large ligand field splitting such that the 4T2g dd excited state lies above the 2Eg state enabling efficient intersystem crossing and microsecond-to-millisecond timescale phosphorescence. The design of new rigid tripodal ligands which provide highly octahedral coordination geometries is therefore a current area of topical interest.
1,2,3-Triazoles, formed through copper-catalysed alkyne/azide cycloaddition (CuAAC),15,16 have become a useful ligand design motif for coordination chemistry.17–19 For example, tris(triazol-4-ylmethyl)amine based ligands may coordinate in a substituent-dependent tripodal facial tridentate fashion through all three triazole donors (e.g. [Fe(TPTA)3](BF4)2 where TPTA = tris(1-phenyl-1,2,3-triazol-4-ylmethyl)amine20) or through two of the triazoles and the central amine donors (e.g. [M(TBTA)3](BF4)2 where M = Fe, Co and TBTA = tris(1-benzyl-1,2,3-triazol-4-ylmethyl)amine21,22). The ligand may also bind as a tetradentate ligand featuring a meridional arrangement of the triazole donors.23–26
Pericàs and co-workers have reported the more rigid tripodal ligand system, tris(1,2,3-triazolyl)methanol.27–30 This ligand has been used to form Cu(I) complexes which are themselves potent CuAAC catalysts.29 Beyond copper31 there has only been reported a Au(I) complex,32 though the ligand has been used to stabilise nanoparticles of a range of d-block metals.32–34
The tris(triazolyl)methane framework is a highly versatile architecture which can be readily modified to tune the solubility and physicochemical properties of resultant complexes. Firstly, there is broad substrate scope for the preparation of the azide and thus the substituent R1 (Scheme 1). Secondly, the hydroxyl group can be readily derivatised through simple base-assisted nucleophilic displacement enabling alkylation29 through etherification (R2) or immobilisation.27 Further, Chauvin and co-workers demonstrated that the tris(triazolyl)methanol can be dehydrated with trifuoroacetic anhydride to yield a carbocation intermediate which may then be trapped by a suitable nucleophile (Nuc).35
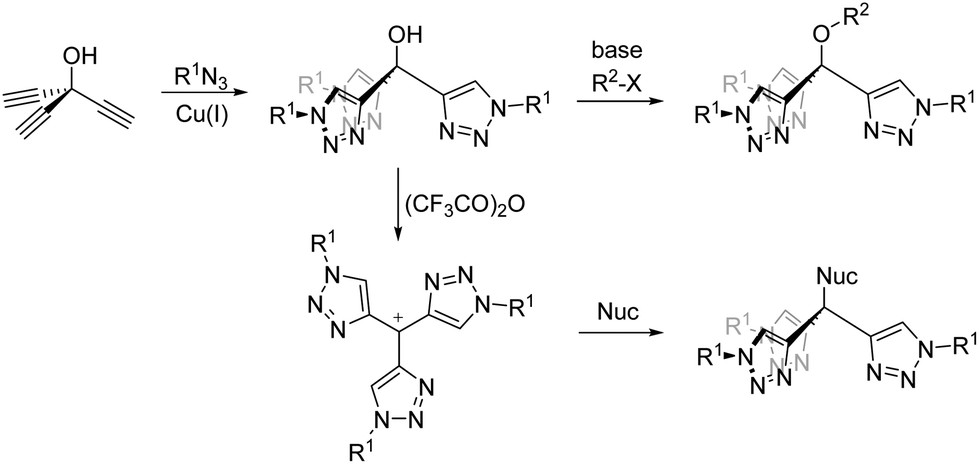 | ||
| Scheme 1 Synthetically versatile routes to tris(triazolyl)methane ligands. | ||
Given the interests of our group in triazole coordination chemistry and the photophysics and photochemistry of resultant complexes18,19 we decided to elaborate on the previously reported synthetic routes to the ligand and its coordination chemistry. In the current contribution we therefore report the synthesis and characterisation of the homoleptic complexes of the group 8 metal ions Fe(II), Ru(II) and Os(II) of the ligand tris(1-benzyl-1,2,3-triazol-4-yl)-p-anisolylmethane and discuss the electrochemical and photophysical properties in relation to previously reported tris-bidentate hexatriazole complexes [M(btz)3]2+ where btz is a bitriazolyl ligand.36,37
Results & discussion
Synthesis and characterisation
The ligand tris(1-benzyl-1,2,3-triazol-4-yl)-p-anisolylmethane (ttzm) was prepared according to literature procedures.28 Briefly, Cu(I)-catalysed coupling of trisethynylmethanol with benzylazide furnished the precursor tris(1-benzyl-1,2,3-triazol-4-yl)methanol. Subsequently, treatment with trifuoroacetic anhydride enabled dehydration and in situ formation of a tristriazolylmethyl carbocation, which then underwent electrophilic addition to anisole to yield the final ligand (Fig. S1 and S2†).35The complex [Fe(ttzm)2](PF6)2 (Fe) was prepared by reaction of FeCl2 with two equivalents of the ligand in MeOH/water and was isolated as its hexafluorophosphate salt (Scheme 2). The complex is diamagnetic demonstrating the low-spin character of the d6 Fe(II) centre. The 1H NMR spectrum (Fig. S3†) exhibits a pair of roofed doublet resonances for the anisole rings at δ 7.20 and 7.92 of the two ttzm ligands and a singlet resonance for three equivalent triazole ring protons at δ 7.94 suggesting free rotation of the anisole ring on the NMR timescale. Electrospray mass spectrometry enables observation of the dication [Fe(ttzm)2]2+ (m/z 621.2308) along with the monocationic ion-pair {[Fe(ttzm)2]PF6}+ (m/z 1387.4146), confirming formation of the homoleptic bis-ttzm complex.
 | ||
| Scheme 2 Synthesis of homoleptic complexes Fe, Ru and Os and the heteroleptic complex [Ru(p-cymene)(ttzm)]2+ (conditions: (a) (i) trifuoroacetic anhydride, dichloromethane, (ii) anisole, (b) where M = Fe, (i) FeCl2, MeOH/water, 90 °C, (ii) NH4PF6; M = Ru, (i) [Ru(p-cymene)Cl2]2, DMF, 160 °C, (ii) NH4PF6, MeOH; M = Os, (i) (NH4)2OsCl6, ethylene glycol, 200 °C, (ii) NH4PF6, water, and (c) (i) [Ru(p-cymene)Cl2]2, EtOH, 80 °C, (ii) NH4PF6. | ||
In our initial attempt to prepare the Ru(II) analogue, the precursor complex [Ru(p-cymene)Cl2]2 was heated to reflux in EtOH with two equivalents of ttzm per Ru(II) centre. Treatment with ammonium hexafluorophosphate then led to the isolation of a pale-yellow solid. 1H NMR spectroscopic analysis of this material revealed methyl, isopropyl and aromatic resonances for a p-cymene ligand as well as signals for a coordinated ttzm ligand (Fig. S5†). This suggests formation of the heteroleptic complex [Ru(p-cymene)(ttzm)](PF6)2, which was corroborated by electrospray mass spectrometry. One singlet resonance is observed for the three triazole ring protons at δ 7.81 suggesting fast rotation of the cymene ligand as well as the anisole ring in solution on the NMR timescale. Crystals of X-ray diffraction quality were grown and the structure of the cation is depicted in Fig. 1. The structure confirms the tripodal coordination character of the ttzm ligand with Ru–N bond lengths of 2.0544(13) to 2.0897(13) Å. The N–Ru–N bond angles range between 78.77(5) to 82.22(5)°, somewhat smaller than the ideal 90° bond angle for a tripodal ligand and may reflect the steric requirements of the p-cymene ligand. Ru–C bond lengths to the p-cymene ligand are typical and lie between 2.1859(16) and 2.2231(17) Å. The complex shows two irreversible electrochemical reduction processes at −1.48 and −1.93 V vs. Fc+/Fc and is non-emissive at room temperature, but exhibits a structured emission band at 77 K with λmax = 537 nm (Fig. S7†).
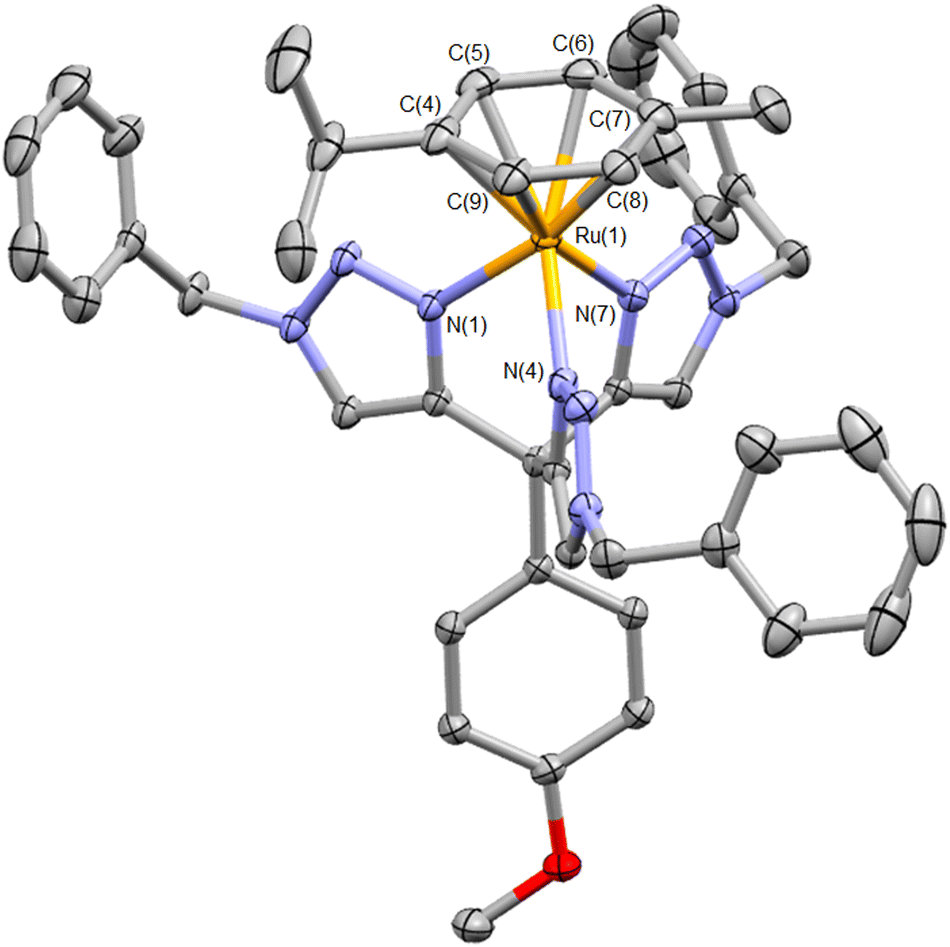 | ||
| Fig. 1 Molecular structure of the cation [Ru(ttzm)(p-cymene)]2+ (hydrogen atoms, counterions and solvent of crystallisation omitted for clarity. Ellipsoids at 50% probability). Selected bond lengths (Å) and angles (°): Ru(1)–N(1) 2.0874(12); Ru(1)–N(4) 2.0544(13); Ru(1)–N(7) 2.0897(13); Ru(1)–C(4) 2.2053(16); Ru(1)–C(5) 2.1859(16); Ru(1)–C(6) 2.2026(17); Ru(1)–C(7) 2.2231(17); Ru(1)–C(8) 2.1954(16); Ru(1)–C(9) 2.1865(17); N(1)–Ru(1)–N(4) 82.22(5); N(1)–Ru(1)–N(7) 78.77(5); N(4)–Ru(1)–N(7) 81.92(5) (CCDC 2156982†). | ||
The homoleptic complex [Ru(ttzm)2](PF6)2 (Ru) was successfully prepared by repeating the reaction but by heating to reflux using dimethylformamide as a solvent to enable higher reaction temperatures and facilitate displacement and replacement of the p-cymene ligand. The appearance of the 1H NMR spectrum (Fig. S8†) closely resembles that of the Fe(II) analogue and electrospray mass spectrometry allowed observation of a dication with m/z 644.2172. The Os(II) analogue, [Os(ttzm)2](PF6)2 (Os) was prepared by reaction of two equivalents of ttzm with (NH4)2[OsCl6] in refluxing ethylene glycol. 1H NMR data (Fig. S10†) closely match those of its Ru(II) and Fe(II) analogues with a dication of m/z 689.2453 being observed by mass spectrometry.
Crystals of X-ray diffraction quality were grown for Os, however crystals for Fe could not be obtained whilst crystals for Ru were disordered. Anion metathesis enabled successful growth of crystals of the tetraphenylborate salt of the Fe(II) complex. The structures of the cations [Fe(ttzm)2]2+ and [Os(ttzm)2]2+ are depicted in Fig. 2a and b, respectively. The cations adopt distorted octahedral geometries with cis N–Fe–N angles varying between 85.93(7) to 94.07(7)° and cis N–Os–N angles between 82.16(9) and 97.84(9)°. The Fe–N(triazole) bond lengths are similar to those reported by Sarkar for the homoleptic low-spin complex [Fe(TBTA)2](BF4)2.21,22
 | ||
| Fig. 2 Molecular structures of the cations [Fe(ttzm)2]2+ (a) and [Os(ttzm)2]2+ (b). Hydrogen atoms, counterions and solvent of crystallisation omitted for clarity. Ellipsoids at 50% probability. In each case only half of the cation is present within the asymmetric unit with the remainder generated by symmetry. (Selected bond lengths (Å) and angles (°) for [Fe(ttzm)2]2+: Fe(1)–N(1) 1.9351(15); Fe(1)–N(4) 1.9413(15); Fe(1)–N(7) 1.9646(16); N(1)–Fe(1)–N(1′) 180.0; N(4)–Fe(1)–N(4′) 180.0; N(7)–Fe(1)–N(7′) 180.0; N(1)–Fe(1)–N(4) 86.25(6); N(1)–Fe–(7) 86.82(6); N(4)–Fe(1)–N(7) 85.93(7); N(1)–Fe(1)–N(4) 93.75(6); N(1)–Fe–(7′) 93.18(6); N(4)–Fe(1)–N(7′) 94.07(7) (CCDC 2178375†). For [Os(ttzm)2]2+: Os(1)–N(3) 2.066(2); Os(1)–N(4) 2.048(3); Os(1)–N(7) 2.049(2); N(3)–Os(1)–N(3′) 180.00(9); N(4)–Os(1)–N(4′) 180.0; N(7)–Os(1)–N(7′) 180.00(15); N(3)–Os(1)–N(4) 84.93(9); N(3′)–Os(1)–N(4) 95.07(9); N(3)–Os(1)–N(7) 82.16(9); N(3′)–Os(1)–N(7) 97.84(9); N(4)–Os(1)–N(7) 85.81(9); N(4′)–Os(1)–N(7) 94.19(9) (CCDC 2156981†)). | ||
The Fe–N bond to the triazole ring whose plane is approximately perpendicular to that of the anisole ring is shorter at 1.9351(15) Å than the other two Fe–N bonds (1.9413(15) & 1.9646(16) Å). The Canisole–C–Ctriazole bond angle for the triazole ring arranged perpendicularly to the anisole ring is 108.32(15)° whilst the corresponding angles for the other two triazole rings are larger at 112.10(16) and 117.02(15)°. These differences stem from steric interactions between the anisole and triazole rings which breaks the three-fold symmetry of the Ctz3 moiety that might be expected and results in a distortion of the cation such that the Canisole–Ctz3 bonds are not colinear with each other or the Fe(II) centre.
Despite this distortion, the [Fe(ttzm)2]2+ cation exhibits a much lower distortion parameter Σ (defined as the sum of the deviations of the twelve cis N–Fe–N bond angles from the ideal of 90°) of only 44.04° compared to that of the high-spin cation [Fe(TPTA)2]2+ (Σ = 130.8°)20 or the low-spin cation [Fe(TBTA)2]2+ as its BF4− salt (Σ = 71.7°).22 These differences are likely contributed to by the larger chelate ring size in the TPTA complex and the methylene spacers in both TPTA and TBTA which will afford a greater degree of flexibility compared to the more structurally rigid ttzm framework. The Os(II) cation exhibits a comparable distortion to that observed for [Fe(ttzm)2]2+. Os–N bond lengths are longer than those for the Fe(II) complex due to the larger ion and consistent with those in other Os(II) 1,2,3-triazole-based complexes,23,38 lying between 2.048(3) and 2.066(2) Å.
The geometries of the cations [M(ttzm)2]2+ (M = Fe, Ru, Os, Fig. S12,† coordinates in ESI†) where optimised using density functional theory (DFT). In each case the benzyl substituents were replaced by methyl, firstly as previous studies have indicated that the nature of the N-substituent has minimal contribution to the photophysical properties,18,19 and secondly, to reduce computational expense. The Fe–N bonds for the Fe(II) complex are between 1.949 & 1.972 Å, slightly longer but in good agreement with experimentally determined Fe–N bond lengths, whilst the Ru–N bond lengths in the Ru(II) analogue are longer, lying between 2.042 & 2.067 Å. For the osmium-containing cation, calculated Os–N bond lengths are between 2.050 and 2.073 Å and again similar to those observed crystallographically. These calculations reproduce in each case the distortion induced by the steric effects of the anisole ring that is observed experimentally for Fe and Os.
Electrochemistry
The three homoleptic complexes were subject to cyclic voltammetry analysis in acetonitrile. All three complexes display a reversible M(II)/M(III) oxidation process (Table 1, Fig. S13†). This appears at +0.72 V (versus Fc/Fc+ = 0.0 V) for Fe and is shifted to more positive potential for Ru consistent with the lower energy of the 4d orbitals of Ru(II) compared to the 3d orbitals of Fe(II). For Os this process is significantly cathodically shifted and appears at +0.35 V consistent with the screening of the increased nuclear charge by the 4f core orbitals. Interestingly, the Ru(II)/Ru(III) oxidation process for Ru is shifted by 0.15 V to more negative potential compared to that of the related hexatriazole donor complex [Ru(btz)3]2+ (+1.01 V vs. Fc/Fc+)37 but is 0.1 V more positive than that of [Ru(TBTA)(btz)]2+.25 The Os(II)/Os(III) couple for Os is cathodically shifted by 0.11 V compared to that for [Os(btz)3]2+ (+0.46 V vs. Fc/Fc+).36 This suggests that the d-orbitals in the ttzm complexes are destabilised relative to those in the homoleptic btz complexes for the same ions.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 EtOH/MeOH glass)
:1 EtOH/MeOH glass)
| Complex | E ox/V (Epa − Epc/mV) | E red/V | λ abs /nm (ε/dm3 mol−1 cm−1) | λ em /nm (77 K)b |
|---|---|---|---|---|
| a Irreversible. b λ ex = 357 nm. | ||||
| Fe | +0.72 (110) | −2.53a | 487 (140), 343 (14470), 305 sh (7560), 282 (5390), 276 (5560) |
— |
| Ru | +0.86 (76) | −2.68a | 339 (24560), 283 (12480) |
— |
| Os | +0.35 (70) | −2.61a | 413 sh (11720), 393 (17480), 359 (25800), 345 (24000), 317 (15060), 283 (8610), 276 (8690) |
453, 472, 498 (sh) |
Irreversible processes assigned to ligand-based reduction are observed at the very edge of the available electrochemical window at −2.53 to −2.68 V (vs. Fc/Fc+) for Fe, Ru and Os. This suggests a high energy ligand-centred LUMO in these complexes which is consistent with previously reported hexatriazole-based coordination complexes such as [Ru(btz)3]2+ and [Os(btz)3]2+ where the LUMO lies approximately 1 eV higher in energy than those of their bpy analogues36,37 and data for [Ru(TBTA)(btz)]2+.25
DFT calculations confirm the nature of the frontier orbitals in the complexes (Fig. 3 and S14–S16†). As anticipated the LUMO is localised on the triazole rings. The HOMO is indeed dominated by the metal centre but in each case is represented by a degenerate pair of orbitals of dπ–Lπ* character. This suggests that the ttzm ligand may be acting as a π-donor, leading to the destabilisation of the metallic d-orbitals inferred electrochemically (vide supra). For Fe and Os this pair sits above HOMO−2 which in each case has dz2 character. In the case of Ru, HOMO−2, HOMO−3, and HOMO−4 all have contributions from the dz2 orbital but mixed with additional contributions from the anisole moiety π-system (Fig. S15†). Below the set of metal-based orbitals for Fe and Os lie orbitals with significant contributions from the anisole moiety (HOMO−3 to HOMO−7). HOMO−8 and orbitals lower in energy have significant triazole π character.
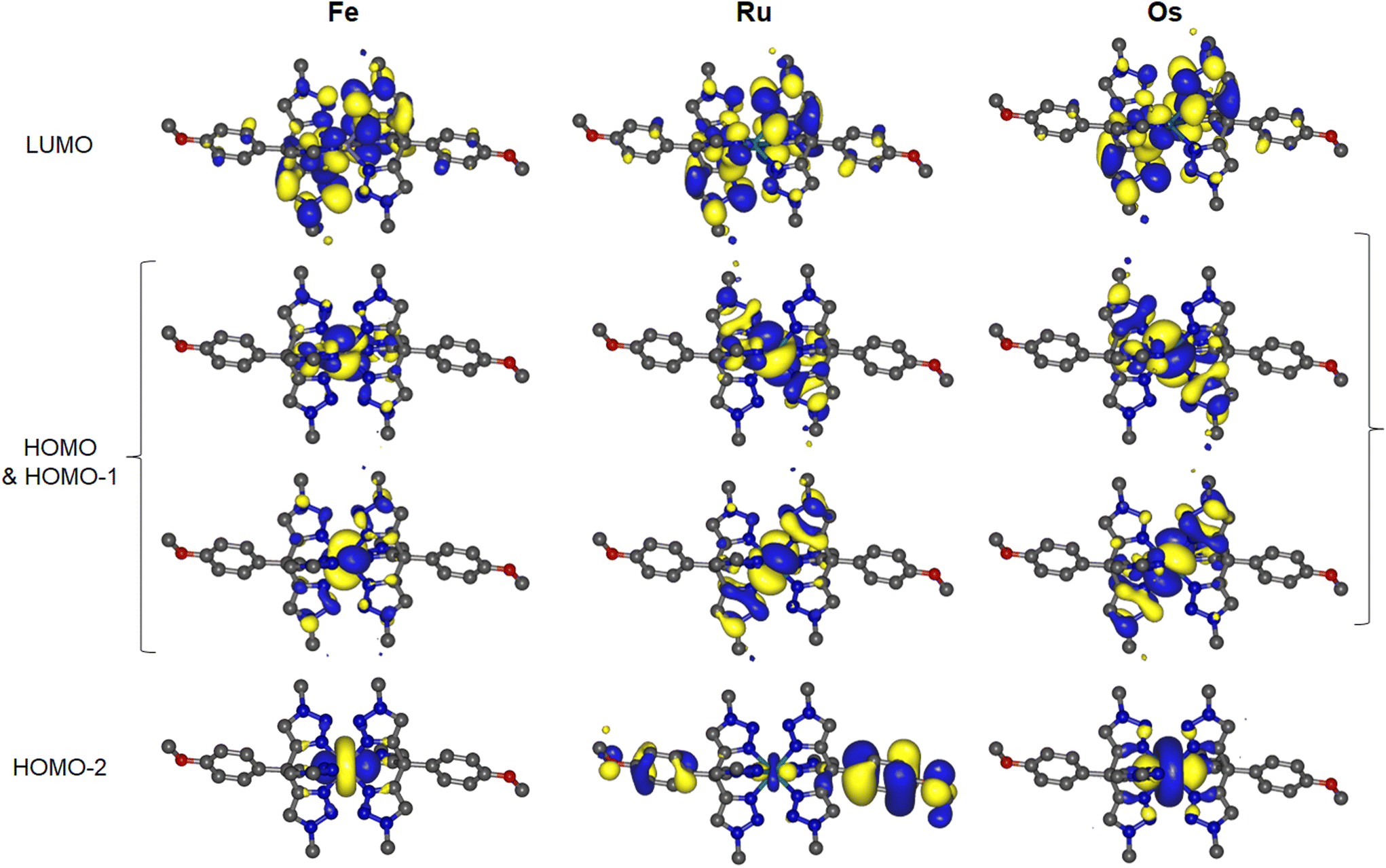 | ||
| Fig. 3 Plots of HOMO−2 to LUMO orbitals of Fe, Ru and Os from DFT calculations (isosurfaces at 0.02 a.u.). | ||
Above the LUMO, LUMO+1 to LUMO+5 for the complexes have significant triazole π* character whilst LUMO+6 to LUMO+9 have anisole π* character. The first antibonding M–N dσ* orbital appears for Fe, Ru and Os as LUMO+10, LUMO+14 and LUMO+24 respectively. The LUMO-dσ* energy gap increases from Fe to Os from 1.00 to 2.93 eV. This is consistent with the increase in M–N bond strength and larger ligand field splitting expected on descending the group.
Photophysical properties
The UV-visible absorption spectra of all three complexes (Fig. 4) contain intense absorptions below 250 nm assigned to singlet ligand-centred (1LC) transitions with bands at lower energy between 300 and 450 nm assigned to metal-to-ligand charge transfer transitions (1MLCT).37,39 In addition to these bands, the spectra of the three complexes share a common vibrationally resolved feature which appears superimposed on other absorption bands between 250 and 280 nm. The identically positioned vibrational progressions also appear in the spectrum of the free ligand (Fig. S17†) and are therefore independent of the metal ion. | ||
| Fig. 4 UV-visible absorption spectra of the complexes Fe, Ru and Os in acetonitrile solution (solid lines) and the normalised emission spectrum of Os (λex = 357 nm, 1MLCT absorption maximum) in an EtOH/MeOH glass at 77 K (dashed line). Inset: expansion of absorption spectrum for Fe showing dd transition band. | ||
The limited optical absorption in the visible region for Fe and Ru is consistent with the absorption spectra of other triazole-containing complexes due to the high energy of ligand-centred unoccupied orbitals.25,37 The 1MLCT absorption band for Fe appears at lower energy compared to that of Ru which is in line with the observed anodic shift in the M(II)/M(III) process on moving from Fe to Ru due to the lower energy of the Ru d-orbital-centred HOMO. The absorption spectrum for Os features a more complex set of absorption bands that appear red-shifted compared to those of Fe and Ru. This is consistent with the cathodic shift in the Os(II)/Os(III) redox potential compared to the other complexes, destabilisation of the 5d orbital-localised HOMO and diminished HOMO–LUMO gap. A very weak absorption feature is also evident for Fe at 487 nm (ε = 140 dm3 mol−1 cm−1) which is assigned to the metal-centred 1A1 → 1T1 dd transition.
On the low energy side of the most prominent MLCT absorption feature for Os (359 nm) is a sharp absorption feature at 393 nm. Based on the photophysical behaviour of other Os(II) complexes,36,40 and the similarity to the absorption spectrum of [Os(btz)3]2+ the main feature is assigned to transitions of predominantly 1MLCT character whilst the lower energy feature is assigned as having significant contributions from spin-forbidden ground state to 3MLCT state excitation. The observation of these spin-forbidden transitions in absorption spectra of Os(II) complexes is well documented and arises from the high spin–orbit coupling constant associated with the Os centre.41–43
Absorption spectra were simulated using time dependent DFT calculations at the same level of theory and using the same basis set as used for geometry optimisation (ESI†). Calculated spectra are in good agreement with those determined experimentally (Fig. S18†), reproduce the relative positioning and confirm the character of transitions for the 1MLCT bands between 300 and 400 nm. These lower energy transitions predominantly involve transfer of an electron from the metal ion to the triazole moieties in each case. However, whilst the S1 states of Ru (343 nm) and Os (384 nm) have 1MLCT character, the S1 state of Fe appears at significantly lower energy (471 nm) and is of 1MC character in agreement with the experimental observation of the 1A1 → 1T1 dd transition. The first state with 1MLCT character for Fe is S6 (349 nm).
TDDFT data also support the assignment of the high energy transitions between 200 and 250 nm as arising from 1LC excitations. Between the two features (250–300 nm) calculations reveal a number of transitions of 1MLCT character in which the excited electron is transferred to the anisole rings of the ligands. Additional transitions in this region are also evident arising from intraligand charge transfer from the anisole moieties to the triazole rings which may account for the ligand associated vibrationally resolved feature observed experimentally.
None of the complexes are emissive in room temperature fluid solutions in agreement with behaviour of their btz analogues.36,37 The high energy of the triazole-based ligand localised LUMO resulting in high energy 3MLCT states will enable facile population of 3MC dd states and rapid non-radiative deactivation to the ground state. In fact, for [Ru(btz)3]2+ DFT studies indicated that both the S1 and T1 states of the complex were of MC state character in contrast to the bpy analogue which has MLCT character for the S1 and T1 states.37,44 Neither Fe nor Ru are emissive at 77 K in frozen solution glass matrices. The energies of spin-forbidden vertical transitions to triplet states were also calculated by TDDFT at the ground state geometries (ESI†). The T1 state for Fe has 3MC character, again appearing at low energy (642 nm). Whilst the T1 state of Ru is calculated to have 3MLCT character, non-radiative deactivation via highly accessible 3MC states is clearly efficient, even at 77 K.
Conversely, Os is emissive at 77 K in an EtOH/MeOH frozen glass matrix, giving rise to a vibronically structured emission band with λemmax 472 nm. The excitation profile recorded under these conditions closely matches the electronic absorption spectrum (Fig. S19†). Further, with the free ligand not showing any absorbance at the wavelength of excitation (Fig. S17†), we can rule out emission arising from any free ligand. In our previous work, TDDFT data indicated lowest energy transitions of MLCT character for [Os(btz)3]2+ which is also emissive at 77 K but is non-emissive at room temperature. However, the heavily destabilised 1/3MLCT states compared to those of [Os(bpy)3]2+ will similarly result in rapid population of 3MC states and non-radiative deactivation in solution.36 We therefore assign the T1 state for Os as having significant 3MLCT state character (the T1 state in the Frank–Condon region indeed has 3MLCT character), resulting in the observed cryogenic emission, but which likely lies in close proximity to deactivating 3MC states.
Given our previous work reporting the ligand-release photochemistry of ruthenium(II) and osmium(II) complexes containing 1,2,3-triazole based ligands we briefly investigated the photostability of the three complexes reported here. For the heteroleptic complexes [Ru(bpy)2(btz)]2+ and [Ru(bpy)(btz)2]2+,39,45–48 as well as the homoleptic complex [Os(btz)3]2+,38 significant changes are observed in 1H NMR spectra of samples within a matter in minutes with irradiation using the Hg emission lines from a domestic fluorescent lamp. Using this method, and on a similar timescale and concentration, 1H NMR spectra of Fe, Ru and Os in d3-acetonitrile are largely unchanged. This comparative photostability might be attributed to the tripodal terdentate character of the ligands and resultant rigidity that will limit conformational freedom for photodissociation, instead favouring ground state recovery from MC states. This is in contrast to the seemingly more facile photo-dechelation and solvent coordination observed for [Ru(TBTA)(btz)]2+ and [Os(TBTA)(DMSO)Cl]+.23,25 Significant changes are observed in 1H NMR spectra after exposure to light for 24 hours or more, showing that the complexes do in fact undergo photodecomposition. However, this is evidently far less efficient than for the photoreactive btz complexes alluded to above.
Conclusions
The chemistry for forming 1,2,3-triazoles provides highly versatile avenues for ligand design that have been widely exploited.17–19 Given the rich coordination chemistry associated with facially coordinating tripodal tris(pyrazolyl)borate and tris(pyrazolyl)methane type ligands, it is somewhat surprising that very little coordination chemistry has been reported for the tridentate tris(1,2,3-triazolyl)methane framework. The ligand structure is highly amenable to modification through the triazole substituents and the substituent on the central carbon atom of the ligand. Whilst copper(I) and gold(I) complexes have been reported, and ligands based on this core architecture have been used to stabilise the surface of metallic nanoparticles, the wider coordination chemistry of this attractive ligand has yet to be fully explored.We have reported here the coordination chemistry of this ligand architecture with the group 8 metals for the homoleptic complexes [M(ttzm)2]2+ (M = Fe, Ru & Os). The iron and ruthenium complexes are non-emissive, most likely stemming from low-lying ligand field excited states. Indeed, the S1 and T1 states of [Fe(ttzm)2]2+ are calculated to be of metal-centred character. The osmium complex, on the other hand, exhibits luminescence at 77 K, assigned to a 3MLCT state, similarly to the previously reported hexatriazole complex [Os(btz)3]2+ (btz = 1,1′-dibenzyl-bi-1,2,3-triazol-4-yl). Despite the presence of low-lying MC states, the complexes are relatively photostable in comparison to previously reported btz complexes.
We are currently continuing our investigations of the basic coordination chemistry of the ttzm ligand framework, as well as the resultant photophysical and electrochemical properties of its complexes across the transition metal elements. We will report results from these on-going studies elsewhere in due course.
Experimental section
General methods
Where specified, reactions were carried out under an atmosphere of dry nitrogen gas, using standard Schlenk techniques. The ligand precursor tris(1-benzyl-1H-1,2,3-triazol-4-yl)methanol was prepared according to literature procedures.28 Reagents were obtained from Sigma-Aldrich, Alfa Aesar, Acros Organics and Fluorochem and were used without any further purification. Bruker Ascend spectrometers (400 and 600 MHz) were used to collect NMR spectra, where all chemical shifts are reported relative to the residual solvent signal. In the assignments of resonances for the anisole ring, ortho and meta positions are with respect to the position of the methoxy substituent. Mass spectra were collected using Agilent 6530 QTOF or Bruker micro-Q-TOF spectrometers. UV-visible absorption spectra were recorded on an Agilent Cary-60 spectrophotometer using quartz cuvettes with a path length of 1.0 cm. 77 K emission spectra of complexes were recorded on a Horiba Fluoromax-4 spectrophotometer using (4:1) EtOH/MeOH frozen glass solutions. Cyclic voltammetry studies were undertaken using a PalmSens EmStat3 potentiostat with PSTrace electrochemical software using a glassy carbon disc working electrode, Pt wire counter electrode, Ag/AgCl reference electrode and (nBu4N)(PF6) electrolyte in acetonitrile. All voltammograms were calibrated relative to the Fc+/Fc couple as an internal reference.
Synthesis of 4,4′,4′′-((4-methoxyphenyl)methanetriyl)tris(1-benzyl-1H-1,2,3-triazole)
Tris(1-benzyl-1H-1,2,3-triazol-4-yl)methanol (2.07 g, 4.12 mmol) was suspended in anhydrous DCM under an atmosphere of nitrogen. The suspension was cooled to 0 °C and trifluoroacetic anhydride (0.69 mL, 4.96 mmol) was added dropwise. An intensely yellow-coloured solution was immediately formed, which was left to stir for one hour at 0 °C. Anisole (0.45 mL, 4.12 mmol) was added and the solution turned a brown colour, slowly becoming pale yellow as the reaction progressed. The solution was stirred at low temperature for approximately 3 hours, before being allowed to return to room temperature, with stirring, overnight. The reaction was quenched with (1 M) aqueous NaOH (100 mL) and the product was extracted with DCM (2 × 100 mL). Organic phases were combined, washed with brine (∼50 mL), dried over MgSO4 and volatiles were removed under vacuum. The crude product was attained as a yellow oil. Purification by column chromatography over silica gel (4% MeOH/CH2Cl2) yielded the product as a pale yellow, microcrystalline solid (1.81 g, 74%).1H NMR (400 MHz, CDCl3) δ 3.73 (s, 3H, –OCH3), 5.46 (s, 6H, –CH2Ph), 6.72 (d, J = 8.94 Hz, 2H, Ho-anisole), 6.95 (d, J = 8.89 Hz, 2H, Hm-anisole), 7.23 (m, 6H, Ho-phenyl), δ 7.32 (m, 9H, Hm,p-phenyl), 7.50 (s, 3H, Htriazole); 13C NMR (101 MHz, CDCl3) δ 46.43, 54.25, 55.36, 113.58, 124.29, 128.13, 128.72, 129.17, 129.50, 134.75, 137.03, 152.53, 158.47; HRMS calcd for C35H32N9O 594.2724, found m/z 594.2726 [MH]+ and for C35H31N9NaO 616.2544 found m/z 616.2546 [MNa]+.
Synthesis of [Fe(ttzm)2](PF6)2 (Fe)
The ligand ttzm (0.12 g, 0.205 mmol) was dissolved in MeOH (5 mL) and stirred at room temperature. FeCl2·4H2O (0.021 g, 0.107 mmol) was dissolved in water (5 mL), before being added to the ligand solution which became red in colour instantly and which quickly turned to a suspension. The mixture was heated to 90 °C (reforming a solution in the process) and was allowed to stir at this temperature for one hour. NH4PF6 (0.19 g, 1.18 mmol) was added to the solution, causing instant precipitation of a pink solid. The mixture was stirred at high temperature for a further twenty minutes, before being allowed to cool to room temperature. This mixture was filtered and the solids were washed with water, MeOH and diethyl ether. The product was dried in vacuo for 20 minutes (0.14 g, 88%).1H NMR (400 MHz, CD3CN) δ 3.92 (s, 3H, –OCH3), 5.37 (s, 6H, –CH2Ph), 7.06 (d, J = 3.73 Hz, 6H, Ho-phenyl), 7.20 (d, J = 8.72 Hz, 2H, Ho-anisole), 7.29 (m, 9H, Hm,p-phenyl), 7.92 (d, J = 8.85 Hz, 2H, Hm-anisole), 7.94 (s, 3H, Htriazole); 13C NMR (150 MHz, CD3CN) δ 47.08, 56.21, 56.37, 116.13, 124.83, 125.52, 128.59, 129.79, 129.96, 130.22, 135.48, 151.97, 161.49; HRMS (ESI) calcd for [FeC70H62N18O2]2+ 621.2321 found m/z 621.2308 [M]2+.
Synthesis of [Fe(ttzm)2](BPh4)2
The iron complex was also prepared by an analogous route as its BPh4− salt from FeBr2 (0.026 g, 0.122 mmol), ttzm (0.133 g, 0.224 mmol) and NaBPh4 (0.254 g, 0.741 mmol) to yield a pink solid (0.173 g, 82%). The BPh4− salt showed limited solubility precluding full NMR characterisation but did enable crystals of diffraction quality to be grown from the vapour diffusion of diethylether into a concentrated dimethylformamide solution.Synthesis of [Ru(p-cymene)(ttzm)](PF6)2
The ligand ttzm (0.21 g, 0.349 mmol) and [Ru(p-cymene)Cl2]2 (0.051 g, 0.084 mmol) were combined in a two-necked flask under an atmosphere of nitrogen. Deaerated EtOH (40 mL) was added and the mixture was stirred at 80 °C overnight, in the dark. The resultant yellow solution was allowed to cool to room temperature before being concentrated under reduced pressure. NH4PF6 (0.35 g, 2.14 mmol) was added and the mixture was allowed to stir for 20 minutes at r.t. during which time precipitation of a yellow solid was observed. Diethyl ether was added to ensure precipitation of all product. The mixture was filtered and solids were washed with cold water, EtOH and diethyl ether. The solids were dried in vacuo for 10 minutes and then oven dried at 65 °C for a further 10 minutes (0.15 g, 94%). X-ray quality crystals were attained by slow evaporation of an acetone solution.1H NMR (400 MHz, CD3CN) δ 0.97 (d, J = 6.94 Hz, 6H, iPr CH3), δ 2.34 (s, 3H, cymene CH3), 3.02 (sept, J = 6.90 Hz, H, iPr –CHMe2), 3.88 (s, 3H, –CH3), 5.53 (s, 6H, –CH2Ph), 5.93 (d, J = 6.37 Hz, 2H, Hcymene), 6.15 (d, J = 6.42 Hz, 2H, Hcymene), 7.13 (d, J = 9.02 Hz, 2H, Ho-anisole), 7.38 (s, 15H, Ph), 7.61 (d, J = 9.00 Hz, 2H, Hm-anisole), 7.81 (s, 3H, Htriazole); 13C NMR (150 MHz, CD3CN) δ 19.34, 22.08, 31.14, 45.17, 56.38, 56.77, 87.83, 89.86, 107.16, 110.34, 116.14, 123.94, 125.93, 129.88, 130.00, 130.05, 130.14, 134.45, 144.46, 161.66; HRMS calcd for [RuC45H45N9O](PF)6+ 974.2427, found m/z 974.2420 [M − PF6]+ and calcd for [RuC45H45N9O]2+ 414.6390, found m/z 414.6397 [M]2+.
Synthesis of [Ru(ttzm)2](PF6)2 (Ru)
The ligand ttzm (0.2019 g, 0.340 mmol) and [Ru(p-cymene)Cl2]2 (0.0514 g, 0.0839 mmol) were dissolved in deaerated dimethylformamide (12 mL) under a nitrogen atmosphere. The red/orange solution was then heated to 160 °C and stirred overnight in the dark. A beige suspension was observed which was filtered after cooling. The collected solid was dissolved in MeOH and an excess of NH4PF6 was added resulting in precipitation. Diethyl ether was added to precipitate further material from solution and the mixture was filtered. The collected solids were washed with water, MeOH and diethyl ether and dried in vacuo for 5 minutes before being oven dried at 65 °C for 15 minutes (0.1034 g, 39%).1H NMR (400 MHz, CD3CN) δ 3.93 (s, 3H, –OCH3), 5.38 (s, 6H, –CH2Ph), 7.09 (m, 6H, Ho-phenyl), 7.20 (d, J = 8.94 Hz, 2H, Ho-anisole), 7.29 (m, 9H, Hm,p-phenyl), 7.91 (s, 3H, Htriazole), 7.93 (d, J = ∼8.9 Hz, 2H, Hm-anisole, partially overlapped with triazole proton resonance); 13C NMR (101 MHz, CD3CN) δ 47.49, 56.14, 56.38, 116.06, 125.32, 125.82, 128.69, 129.82, 129.97, 130.40, 135.37, 147.65, 161.49; HRMS calcd for [RuC-70H62N18O2]2+ 644.2168, found m/z 644.2172 [M]2+.
Synthesis of [Os(ttzm)2](PF6)2 (Os)
The ligand ttzm (0.2030 g, 0.342 mmol) was combined with (NH4)2OsCl6 (0.0748 g, 0.170 mmol) and ethylene glycol (8 mL) under a nitrogen atmosphere. The mixture was heated to 200 °C and stirred overnight in the dark. The dark solution was allowed to cool to room temperature and a solution of NH4PF6 (0.2188 g, 1.34 mmol) in water (25 mL) was added and the mixture stirred for 30 minutes resulting in precipitation. The mixture was filtered and the collected solids were washed with water and diethyl ether. The crude material was dried in vacuo for 10 minutes and oven-dried at 65 °C for 30 minutes. The product was then purified by column chromatography (silica gel, DCM/MeCN 6:1) yielding pure material as a yellow powder (0.058 g, 20%). X-ray quality crystals were obtained by vapour diffusion of diisopropyl ether into an acetonitrile/acetone solution.
1H NMR (400 MHz, CD3CN) δ 3.94 (s, 3H, –OCH3), 5.37 (s, 6H, –CH2Ph), 7.10 (m, 6H, Ho-phenyl), 7.22 (d, J = 8.94 Hz, 2H, Ho-anisole), 7.29 (m, 9H, Hm,p-phenyl), 7.92 (s, 3H, Htriazole), 7.96 (d, J = 8.95 Hz, 2H, Hm-anisole); 13C NMR (101 MHz, CD3CN) δ 48.36, 56.15, 56.38, 116.06, 124.96, 125.60, 128.69, 129.82, 129.96, 130.37, 135.36, 145.52, 161.50; HRMS calcd for [OsC70H62N18O2]2+ 689.2453, found m/z 689.2453 [M]2+.
Computational details
The geometries of the ground states of the complexes were optimised using density functional theory using the B3LYP hybrid functional49,50 (20% Hartree–Fock) for Ru and Os in the Orca 4.2.1 software package.51,52 The B3LYP* functional (15% Hartree–Fock) was used for Fe.53,54 Def2-ECP effective core potential and def2/j auxiliary basis set was used for ruthenium and osmium with def2-tzvp(-f) basis sets used for all other atoms.55 All calculations were conducted using Grimme's D3-BJ dispersion correction56,57 along with the SMD implicit solvation model (acetonitrile).58 In these DFT calculations the resolution-of-identity (RI) approximation for hybrid functionals (as implemented in ORCA) was employed to calculate the Coulomb energy term using the Ahlrichs/Weigend Def2-TZV basis as the auxiliary basis set and the exchange term by the so-called ‘chain-of-spheres exchange’ (COSX) algorithm. Benzyl substituents were replaced by methyl groups as these will have little impact on the photophysical properties and also saves on computational expense. Calculated optical absorption spectra were determined by time dependent DFT for the first 100 vertical excitations. Molecular orbitals were visualised using the Gabedit software package with isosurfaces set to 0.02.Single crystal X-ray diffraction
Crystals of X-ray diffraction quality of [Ru(p-cymene)(ttzm)](PF6)2 and [Os(ttzm)2](PF6)2 were obtained by slow evaporation of an acetone and slow diffusion of diisopropylether into an acetonitrile/acetone solution respectively. Crystals of [Fe(ttzm)2](BPh4)2 were grown by vapour diffusion of diethylether into a dimethylformamide solution. Data were collected at 150 K on a Bruker D8 Venture diffractometer equipped with a graphite monochromated Mo(Kα) radiation source and a cold stream of N2 gas. Solutions were generated by conventional Patterson heavy atom or direct methods and refined by full-matrix least-squared on F2 data, using SHELXS-97 and SHELXL software respectively.59 Absorption corrections were applied based upon multiple and symmetry-equivalent measurements using SADABS.60 One of the hexafluorophosphate counter ions displayed some rotational disorder and this was refined over two positions using the PART instruction in the l.s. refinement. Crystallographic data is available as ESI† or can be downloaded from the Cambridge Crystallographic Datacentre (CCDC 2156982, 2156981 & 2178375†).Crystal data for CCDC 2156982,† C51H45F12N9O3P2Ru, M = 1222.97, triclinic, a = 11.4134(4) Å, b = 11.7793(4) Å, c = 21.3297(7) Å, α = 105.174(1), β = 93.806(1), γ = 98.627(1), V = 2719.49(16) Å3, T = 150 K, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, 20754 reflections measured, 17315 independent reflections (Rint = 0.0396). The final R1 values were 0.0402 (I > 2σ(I)). The final wR(F2) values were 0.0976 (I > 2σ(I)). The final R1 values were 0.0539 (all data). The final wR(F2) = 0.1046 (all data). The goodness of fit on F2 was 1.046. Largest peak and hole (e Å−3) 0.759/−0.782.
, Z = 2, 20754 reflections measured, 17315 independent reflections (Rint = 0.0396). The final R1 values were 0.0402 (I > 2σ(I)). The final wR(F2) values were 0.0976 (I > 2σ(I)). The final R1 values were 0.0539 (all data). The final wR(F2) = 0.1046 (all data). The goodness of fit on F2 was 1.046. Largest peak and hole (e Å−3) 0.759/−0.782.
Crystal data for CCDC 2156981,† C78H68F12N22O2OsP2, M = 1825.68, triclinic, a = 11.866(3) Å, b = 12.902(3) Å, c = 12.921(3) Å, α = 95.897(10), β = 94.850(9), γ = 92.366(14), V = 1958.2(9) Å3, T = 150 K, space group P, Z = 1, 11883 reflections measured, 11329 independent reflections (Rint = 0.0567). The final R1 values were 0.0395 (I > 2σ(I)). The final wR(F2) values were 0.0993 (I > 2σ(I)). The final R1 values were 0.0445 (all data). The final wR(F2) = 0.1032 (all data). The goodness of fit on F2 was 1.046. Largest peak and hole (e Å−3) 1.484/−1.405.
Crystal data for CCDC 2178375,† C118H102B2FeN18O2, M = 1881.64, orthorhombic, a = 21.3382(7) Å, b = 17.6313(5) Å, c = 25.4672(7) (3) Å, α = 90, β = 90, γ = 90, V = 9581.3(5) Å3, T = 150 K, space group Pbca, Z = 4, 13426 reflections measured, 9608 independent reflections (Rint = 0.0626). The final R1 values were 0.0628 (I > 2σ(I)). The final wR(F2) values were 0.1267 (I > 2σ(I)). The final R1 values were 0.0951 (all data). The final wR(F2) = 0.1391 (all data). The goodness of fit on F2 was 1.090. Largest peak and hole (e Å−3) 0.448/−0.494.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank the University of Huddersfield and EPSRC Doctoral Training Partnership for Materials Science (EP/T51813X/1) for supporting this work.References
- S. Trofimenko, J. Am. Chem. Soc., 1967, 89, 3170–3177 CrossRef CAS.
- E. Becker, S. Pavlik and K. Kirchner, in Advances in Organometallic Chemistry, ed. R. West, A. F. Hill and M. J. Fink, Academic Press, 2008, vol. 56, pp. 155–197 Search PubMed.
- C. Kammerer and G. Rapenne, Eur. J. Inorg. Chem., 2016, 2016, 2214–2226 CrossRef CAS.
- B. A. McKeown, J. P. Lee, J. Mei, T. R. Cundari and T. B. Gunnoe, Eur. J. Inorg. Chem., 2016, 2016, 2296–2311 CrossRef CAS.
- C. G. Young, Eur. J. Inorg. Chem., 2016, 2016, 2357–2376 CrossRef CAS.
- A. P. Forshaw, R. P. Bontchev and J. M. Smith, Inorg. Chem., 2007, 46, 3792–3794 CrossRef CAS PubMed.
- R. Frankel, U. Kernbach, M. Bakola-Christianopoulou, U. Plaia, M. Suter, W. Ponikwar, H. Noth, C. Moinet and W. P. Fehlhammer, J. Organomet. Chem., 2001, 617–618, 530–545 CrossRef CAS.
- K. S. Kjær, N. Kaul, O. Prakash, P. Chábera, N. W. Rosemann, A. Honarfar, O. Gordivska, L. A. Fredin, K.-E. Bergquist, L. Häggström, T. Ericsson, L. Lindh, A. Yartsev, S. Styring, P. Huang, J. Uhlig, J. Bendix, D. Strand, V. Sundström, P. Persson, R. Lomoth and K. Wärnmark, Science, 2019, 363, 249–253 CrossRef PubMed.
- S. Kaufhold, N. W. Rosemann, P. Chábera, L. Lindh, I. Bolaño Losada, J. Uhlig, T. Pascher, D. Strand, K. Wärnmark, A. Yartsev and P. Persson, J. Am. Chem. Soc., 2021, 143, 1307–1312 CrossRef CAS PubMed.
- C. Pettinari and R. Pettinari, Coord. Chem. Rev., 2005, 249, 525–543 CAS.
- H. R. Bigmore, S. C. Lawrence, P. Mountford and C. S. Tredget, Dalton Trans., 2005, 635–651, 10.1039/B413121E.
- A. Llobet, P. Doppelt and T. J. Meyer, Inorg. Chem., 1988, 27, 514–520 CrossRef CAS.
- C. Metcalfe, H. Adams, I. Haq and J. A. Thomas, Chem. Commun., 2003, 1152–1153, 10.1039/B300436H.
- S. Treiling, C. Wang, C. Förster, F. Reichenauer, J. Kalmbach, P. Boden, J. P. Harris, L. M. Carrella, E. Rentschler, U. Resch-Genger, C. Reber, M. Seitz, M. Gerhards and K. Heinze, Angew. Chem., Int. Ed., 2019, 58, 18075–18085 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- J. D. Crowley and D. A. McMorran, in Click Triazoles, ed. J. Košmrlj, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 31–83, DOI:10.1007/7081_2011_67.
- P. A. Scattergood and P. I. P. Elliott, Dalton Trans., 2017, 46, 16343–16356 RSC.
- P. A. Scattergood, A. Sinopoli and P. I. P. Elliott, Coord. Chem. Rev., 2017, 350, 136–154 CrossRef CAS.
- D. Schweinfurth, S. Demeshko, M. G. Sommer, S. Dechert, F. Meyer and B. Sarkar, Eur. J. Inorg. Chem., 2016, 2016, 2581–2585 CrossRef CAS.
- M. Noessler, D. Hunger, F. Reichert, M. Winkler, M. Reimann, J. Klein, S. Suhr, L. Suntrup, J. Beerhues, M. Kaupp, J. van Slageren and B. Sarkar, Dalton Trans., 2021, 50, 18097–18106 RSC.
- D. Schweinfurth, S. Demeshko, S. Hohloch, M. Steinmetz, J. G. Brandenburg, S. Dechert, F. Meyer, S. Grimme and B. Sarkar, Inorg. Chem., 2014, 53, 8203–8212 CrossRef CAS PubMed.
- J. Klein, U. Albold, L. Suntrup and B. Sarkar, ChemPhotoChem, 2018, 2, 357–361 CrossRef CAS.
- D. Schweinfurth, S. Demeshko, M. M. Khusniyarov, S. Dechert, V. Gurram, M. R. Buchmeiser, F. Meyer and B. Sarkar, Inorg. Chem., 2012, 51, 7592–7597 CrossRef CAS PubMed.
- F. Weisser, S. Plebst, S. Hohloch, M. van der Meer, S. Manck, F. Fuehrer, V. Radtke, D. Leichnitz and B. Sarkar, Inorg. Chem., 2015, 54, 4621–4635 CrossRef CAS PubMed.
- F. Weisser, H. Stevens, J. Klein, M. van der Meer, S. Hohloch and B. Sarkar, Chem. – Eur. J., 2015, 21, 8926–8938 CrossRef CAS PubMed.
- L. Maestre, E. Ozkal, C. Ayats, A. Beltran, M. M. Diaz-Requejo, P. J. Perez and M. A. Pericas, Chem. Sci., 2015, 6, 1510–1515 RSC.
- S. Özçubukçu, E. Ozkal, C. Jimeno and M. A. Pericàs, Org. Lett., 2009, 11, 4680–4683 CrossRef PubMed.
- E. Ozkal, P. Llanes, F. Bravo, A. Ferrali and M. A. Pericas, Adv. Synth. Catal., 2014, 356, 857–869 CrossRef CAS.
- E. Ozkal, S. Özçubukçu, C. Jimeno and M. A. Pericàs, Catal. Sci. Technol., 2012, 2, 195–200 RSC.
- N. Khunoad, N. Krittametaporn, S. Pornsuwan, P. Kongsaeree, S. Demeshko and P. Sangtrirutnugul, Inorg. Chim. Acta, 2019, 488, 141–144 CrossRef CAS.
- C. Wang, L. Salmon, Q. Li, M. E. Igartua, S. Moya, R. Ciganda, J. Ruiz and D. Astruc, Inorg. Chem., 2016, 55, 6776–6780 CrossRef CAS PubMed.
- S. Ampawa, N. Krittametaporn, T. Ungpittagul, K. Phomphrai and P. Sangtrirutnugul, Appl. Organomet. Chem., 2019, 33, e5238 CrossRef CAS.
- C. Wang, R. Ciganda, L. Salmon, D. Gregurec, J. Irigoyen, S. Moya, J. Ruiz and D. Astruc, Angew. Chem., Int. Ed., 2016, 55, 3091–3095 CrossRef CAS PubMed.
- M. Oukessou, Y. Génisson, D. El Arfaoui, A. Ben-Tama, E. M. El Hadrami and R. Chauvin, Tetrahedron Lett., 2013, 54, 4362–4364 CrossRef CAS.
- D. A. W. Ross, P. A. Scattergood, A. Babaei, A. Pertegás, H. J. Bolink and P. I. P. Elliott, Dalton Trans., 2016, 45, 7748–7757 RSC.
- C. E. Welby, S. Grkinic, A. Zahid, B. S. Uppal, E. A. Gibson, C. R. Rice and P. I. P. Elliott, Dalton Trans., 2012, 41, 7637–7646 RSC.
- P. A. Scattergood, D. A. W. Ross, C. R. Rice and P. I. P. Elliott, Angew. Chem., Int. Ed., 2016, 55, 10697–10701 CrossRef CAS PubMed.
- P. A. Scattergood, U. Khushnood, A. Tariq, D. J. Cooke, C. R. Rice and P. I. P. Elliott, Inorg. Chem., 2016, 55, 7787–7796 CrossRef CAS PubMed.
- B. J. Pankuch, D. E. Lacky and G. A. Crosby, J. Phys. Chem., 1980, 84, 2061–2067 CrossRef CAS.
- A. Ito, T. E. Knight, D. J. Stewart, M. K. Brennaman and T. J. Meyer, J. Phys. Chem. A, 2014, 118, 10326–10332 CrossRef CAS PubMed.
- E. M. Kober and T. J. Meyer, Inorg. Chem., 1982, 21, 3967–3977 CrossRef CAS.
- F. Felix, J. Ferguson, H. U. Güdel and A. Ludi, Chem. Phys. Lett., 1979, 62, 153–157 CrossRef CAS.
- I. M. Dixon, J. L. Heully, F. Alary and P. I. P. Elliott, Phys. Chem. Chem. Phys., 2017, 19, 27765–27778 RSC.
- R. Z. Boota, S. J. O. Hardman, G. P. Ashton, C. R. Rice, P. A. Scattergood and P. I. P. Elliott, Inorg. Chem., 2021, 60, 15768–15781 CrossRef CAS PubMed.
- C. E. Welby, G. K. Armitage, H. Bartley, A. Sinopoli, B. S. Uppal and P. I. P. Elliott, Photochem. Photobiol. Sci., 2014, 13, 735–738 CrossRef CAS PubMed.
- C. E. Welby, G. K. Armitage, H. Bartley, A. Wilkinson, A. Sinopoli, B. S. Uppal, C. R. Rice and P. I. P. Elliott, Chem. – Eur. J., 2014, 20, 8467–8476 CrossRef CAS PubMed.
- C. E. Welby, C. R. Rice and P. I. P. Elliott, Angew. Chem., Int. Ed., 2013, 52, 10826–10829 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- M. Reiher, O. Salomon and B. A. Hess, Theor. Chem. Acc., 2001, 107, 48–55 Search PubMed.
- M. Reiher, Inorg. Chem., 2002, 41, 6928–6935 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- SHELXTL Program System, Vers 5.1, Bruker Analytical X-ray Instruments Inc., Madison, WI, 1998 Search PubMed.
- G. M. Sheldrick, SADABS: A Program for Absorption Correction with the Siemens SMART System, University of Göttingen (Germany), 1996 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of the ttzm ligand and its complexes, cyclic voltammetry traces, ttzm ligand absorption spectrum, optimised ground state geometries and calculated molecular orbitals, and summarised time-dependent DFT data. CCDC 2156981 ([Os(ttzm)2](PF6)2), 2156982 [(Ru(p-cymene)(ttzm)](PF6)2) and 2178375 ([Fe(ttzm)2](BPh4)2). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02503e |
| This journal is © The Royal Society of Chemistry 2022 |