
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New Cu(I) square grid-type and Ni(II) triangle-type complexes: synthesis and characterization of effective binders of DNA and serum albumins†‡
Martyna
Szymańska
a,
Maciej
Kubicki
 a,
Giovanni N.
Roviello
b,
Giuseppe
Consiglio
c,
Marta A.
Fik-Jaskółka
*a and
Violetta
Patroniak
a
a,
Giovanni N.
Roviello
b,
Giuseppe
Consiglio
c,
Marta A.
Fik-Jaskółka
*a and
Violetta
Patroniak
a
aFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland. E-mail: martafik@amu.edu.pl
bInstitute of Biostructures and Bioimaging – CNR, Area di Ricerca site and Headquartes, Via Tommaso De Amicis, 95, 80145 Napoli, Italy
cDipartimento di Scienze Chimiche, Università degli studi di Catania, viale A. Doria 6, I-95125 Catania, Italy
First published on 13th October 2022
Abstract
Multivalent molecules are a potential group of bioactive compounds endowed with high affinity and specificity in innovative biomolecule-targeting therapeutic approaches. Herein, we report on a new and versatile N,N,N,N-donor ligand L (1R,4R)-N1,N4-bis(quinolin-2-ylmethylene)cyclohexane-1,4-diamine with two coordinating quinoline moieties connected with trans-1,4-diaminocyclohexane. It coordinates Cu+ forming a [2 × 2] square grid-type complex C1 [Cu4L4]4+ and Ni2+ giving a triangle-type complex C2 [Ni3L3]6+. We screened their potential as versatile metal-based Serum Albumin (SA), double helical and G-quadruplex DNA binders taking advantage of their shape, size and stability effects using different spectroscopic experiments (UV-Vis, fluorescence, circular dichroism). The findings of our work suggest the potential utility of the metal complexes herein described in the context of the new drug discovery.
Introduction
The molecular self-assembly driven by the coordination of metal ions is a powerful tool providing access to a wide variety of metal complexes with extensive applications in host–guest chemistry,1 biomedicine,2 catalysis,3 electrooptical devices4,5 and magnetism.6 Owing to the wide range of ligands and metals compatible with the principles of supramolecular chemistry a wide range of discrete coordination complexes, including two- (2D) and three-dimensional (3D) architectures, with well-defined shapes and sizes has been emerged over the last years.7–9 Metal-based supramolecular scaffolds offer unique advantages over conventional organic molecules in the targeting of biorelevant molecules (proteins, nucleic acids) due to the stereochemical and geometrical diversity conferred through the metallic centre.10 In fact, the vital life processes heavily rely on the recognition phenomena between the relatively simple precursors folding into complex matter and became the paradigm of the supramolecular chemistry.9 The self-assembly can be achieved through the utilization of the directional H-bonding or other non-covalent interactions such as van der Waals, ion-ion, ion-dipole, π–π stacking and hydrophobic interactions. It can be also realized by the dynamic, predictable and directional coordination bonding. So far, a wide array of 2D grids, triangles, rectangles, helicates, 3D cubes, trigonal pyramids, adamantoids and other cages synthesized by this approach have been reported.11,12As mentioned above, the rational design empowers formation of aesthetically appealing architectures with functional applicability. The well-defined and thermodynamically stable scaffolds are of particular interest for their potential anticancer properties attained through various mechanisms. The metallo-supramolecular complexes-based drugs can be used in two main strategies – directly as drugs or as carriers for the drugs.13
Metallo-supramolecular complexes, both old and new, are currently being evaluated in conjunction with spectroscopic and biological techniques to probe their interactions with DNA and proteins, the relevant biomolecules. For this purpose, the metal ions are mainly used as core structural units, while the surrounding ligands are used for the binding with target molecules. Using the metal ions in a structural sheath allows for their use in cells, since the metals are unable to non-specifically coordinate to biomolecules and exert the toxic effect.10 A variety of biological interactions can be expected given the diversity of ligands. The interactions with negatively-charged DNA can arise due to the subtle non-coordinate interactions, such as electrostatic attraction on the surface of the helix, intercalation, groove binding as well as combination of all these modes.10,14,15 The double helix of DNA is a carrier of genetic information, therefore it is relevant from the therapeutic viewpoint, since the malfunction of DNA-related processes may lead to cells death and suppression of the tumor growth.16 The formation and stabilization of the GQ in the human telomeric sequences has also been shown to inhibit the activity of telomerase, a cancer-specific reverse transcriptase that is activated in 80–90% of tumors.17 Thus, the telomeric DNA GQ has been considered to be an attractive target for cancer therapeutic intervention.18–20
A therapeutic effect consists not only from the actual mechanism of the impairment of the processes in cancer cells but also from the transportation strategy. An important group of biomolecules are SA which act as the most significant modulators of plasma oncotic pressure and functions to transport a variety of substances. The ligands transported by SA include the endogenous ions, fatty acids, bilirubin, and the exogenous drugs as methotrexate, paclitaxel, warfarin, methadone, furosemide, and many others.21,22 The non-immunotoxicity, long circulation times and high accumulation in tumors are the attributes making SA good anticancer drug carriers.23 The SA are predominately present in the extravascular space rather than in intravascular. They return from the extravascular space into circulation through the lymphatic system.24 Since the solid tumors commonly possess an immature, highly permeable vasculature and lack the sufficient lymphatic drainage25 they tend to accumulate the macromolecules (as SA) within the tumor interstitium (it is known as the enhanced permeation and retention effect).26 This mechanism is used by the cancer cells to use albumin as the source of nutrients to sustain cancer cell proliferation.27 The same pathway can also be used for the drug delivery protocol bound to SA. Therefore, investigating the mechanism of binding of bioactive compounds with SAs is of a clear interest in the design of new anticancer molecules, to be used in targeted therapy and of their efficient delivery systems.
So far Drożdż et al. have showed the potency of multivalent cationic supramolecular Zn(II) grids as DNA binders.28,29 Domarco et al.30 demonstrated the usefulness of metal-directed self-assembly (MDSA) strategy for the rapid access of a family of square-shaped metallacycles of different sizes binding the unique pocket of the c-kit oncogene, an important target in the treatment of gastrointestinal tumors.31 Moreover, just in 2021 Zhang et al. presented a Pt(II) based metallatriangle inducing the PDT effect through ROS generation.32
Here we present a study on the design and synthesis of the metalosupramolecular multivalent square grid-type complex C1[Cu4L4]4+ and a triangle-type complex C2[Ni3L3]6+ where L is a chameleonic ditopic N,N,N,N-donor ligand N,N-bis(quinolin-2-ylmethylene)cyclohexane-1,4-diamine. The compounds were characterized by NMR, ESI-MS, EA and X-ray diffraction of monocrystals. Next, keeping in mind the multivalency of the complexes, we decided to study their potential for the binding with biorelevant molecules as DNA and Serum Albumin (SA) using some spectroscopic techniques (UV-Vis absorption, fluorimetry, circular dichroism).
Results and discussion
Concept, synthesis, characterization and stability of compounds
One of the advantages provided by supramolecular chemistry is obtaining a precisely chosen output from the input data. Another important parameter is the stability of the output figures, since it is directly translated onto their possible in vivo applications. At the same time the decomposition of compounds in certain conditions is desirable for the drug release in cells. As revealed before33–36 the properly designed Schiff base ligands may be suitable for the formation of various supramolecular scaffolds. Therefore, herein, we report on a specific assembly of a supramolecular square grid-type and triangle-type complexes from the tetradentate N,N,N,N-donor Schiff base ligand L N,N-bis(quinolin-2-ylmethylene)cyclohexane-1,4-diamine and metal ions Cu(I) and Ni(II), respectively. Ligand L was obtained by an efficient condensation reaction of 2 equiv. of 2-quinolinecarboxaldehyde with 1 equiv. of trans-1,4-diaminocyclohexane (Fig. S1‡). It was designed so as to possess two N,N-donor moieties separated by the cyclohexane ring. Such separator is rigid enough to play a role of the side wall of the molecular geometrical figures. As depicted in Fig. 3 (cf. 3.2.) a free ligand L exists as transoid conformer in solid state, while in complexes it can adopt, as expected, both cisoid and transoid conformation influencing the final complex arrangement.37–40 As revealed by X-ray diffraction method (cf. 3.2.) the complexation of 1 equiv. of Cu(I) ions results in the formation of a square grid-type complex C1 ([Cu4L4]4+) with two parallel ligands in a cisoid conformation and the other two as transoid isomers, while complexation of Ni(II) ions gives a triangle-type complex C2 ([Ni3L3]6+) with transoid conformers of L (Scheme 1). | ||
| Scheme 1 (a) Conformational isomerism of ligand L; (b) schematic demonstration of the formation of complexes C1 and C2 (the counterions in C2 where omitted for clarity). | ||
The reaction of L and Cu(I) is very selective as it leads to a single product – a [2 × 2] square grid-type complex C1. In ESI-MS spectrum of the C1 a characteristic peak appears at m/z = 2259 for {[Cu4L4](PF6)3}+ (Fig. S2‡), while for the triangle-type complex C2 only a part of the complex is visible at m/z = 459 for [NiL(ClO4)]+ (Fig. S3‡). Also the peaks at m/z = 393 and 415 for [HL]+ and [NaL]+ are present probably because of decomposition of the complex during ionization. The NMR titration experiment revealed that the [2 × 2] square grid-type complex C1 forms selectively among other possible products and that even excess of Cu(I) does not lead to complexes of different ratios (Fig. 1). During titration, 1H NMR spectra show a progressive shifting of the peaks due to the formation of the complex, especially prominent for the signal of imine proton –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– which is very close to the binding site of the ligand.
N– which is very close to the binding site of the ligand.
 | ||
| Fig. 1 Titration of ligand L (5 × 10−4 M in CD3CN) with Cu(CH3CN)4BF4 (5 × 10−3 M in CD3CN) at 27 °C. The titration was performed with Cu(MeCN)4BF4 instead of Cu(MeCN)4PF6 due to the better solubility of this salt in MeCN. | ||
The UV titration of ligand L with Cu(MeCN)4PF6 and Ni(ClO4)2·6H2O confirmed the formation and stabilization of the square grid-type structure of C1 and the triangle-type structure of C2 in acetonitrile. In both cases three isosbestic points were observed: at 298, 260 and 250 nm for C1 and at 305, 265 and 250 nm in case of C2. Importantly, no more changes in the UV-Vis spectrum is visible after addition of 1 equiv. and 1.33 equiv. of Cu(I) and Ni(II), respectively. (Fig. 2).
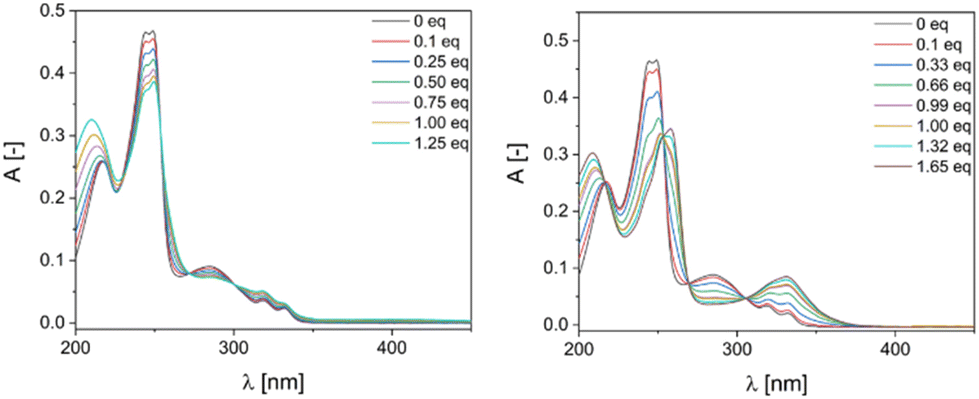 | ||
| Fig. 2 UV-Vis titration spectra of ligand in MeCN (5.1 μM) with (left) Cu(MeCN)4PF6 (0–1.25 equivalents of Cu(I)) and (right) Ni(ClO4)2·6H2O (0–1.65 equivalents of Ni(II)). | ||
The aim of this work was understanding the abilities of multivalent square grid-type complex C1 ([Cu4L4]4+) and triangle-type complex C2 ([Ni3L3]6+) to interact with some biomedically-relevant macromolecules – such as DNA and proteins, using Bovine Serum Albumin (BSA) as a model. Before performing the spectroscopic experiments, we evaluated the stability of the compounds within 2 hours (the time period was correlated with the duration of experiments) in the appropriate buffers. While the spectrum of ligand L seemed unchanged over time, the spectrum of C1 underwent subtle hypochromic changes related, according to our hypothesis, to the exchange of the outer coordination sphere of metal ions for the ligands abundant in the buffer (chlorides, hydroxyl, aqua etc.) leaving the grid scaffold undisrupted. Importantly, the stock solutions of C1 remain red for several weeks suggesting that the coordinated metal ions do not oxidize easily. Similarly, the triangle-type complex C2 seemed to exchange the outer coordination sphere, but also the inner labile ligands (acetonitrile molecules) for the other being abundant in the buffer solutions (Fig. S4 and S5‡). The lower stability of the more constrained structure of C2, in comparison to the structure of C1, found also reflection in the ESI-MS spectra reported above.
Description of the crystal structures
Perspective views of the molecule L, and cations from the structures C1 and C2 are shown in Fig. 3. The ligand molecule in the crystal structure is Ci-symmetrical as it lies across the inversion center in the space group P21/c; as a consequence, the two symmetry-related quinoline ring systems are exactly parallel. It is potentially tetradentate (N4) and in such denticity it exists in the Cu and Ni complexes C1 and C2.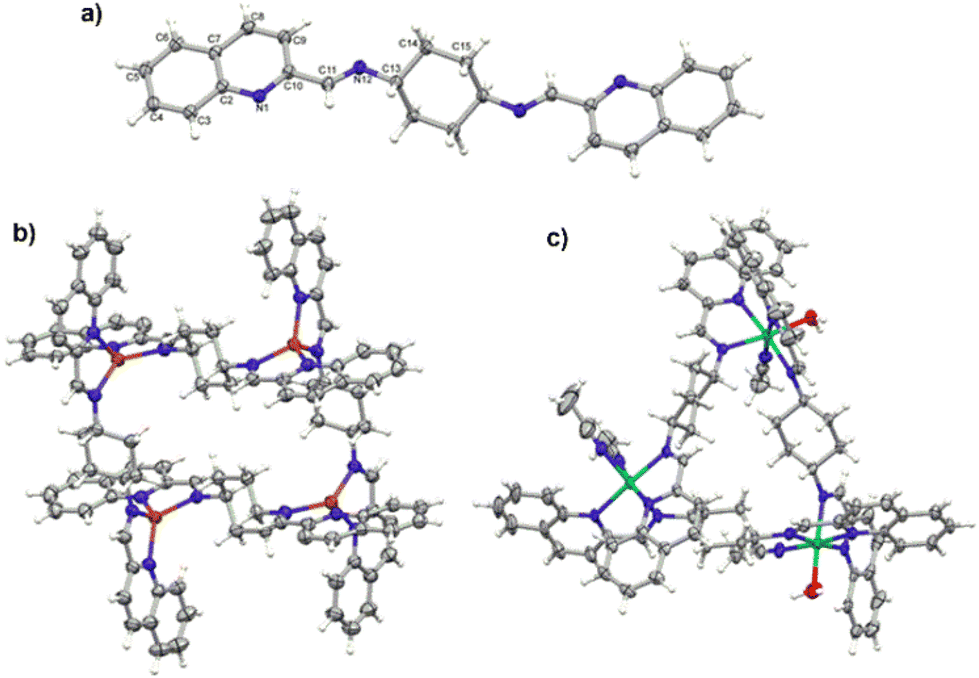 | ||
| Fig. 3 (a) Perspective view of the ligand L. The unlabeled part of the molecule is related to the labelled one by symmetry operation 1−x, 2−y, 1−z; (b) perspective view of the square grid-type complex C1; (c) perspective view of the triangle-type complex C2. All ellipsoids are drawn at the 50% probability level; hydrogen atoms are shown as spheres of arbitrary radii. | ||
The crystal structures of two cationic complexes show the flexibility of the ligand and its potential to create different architectures. The Cu(I) complex, C1, exists as the Ci-symmetrical, four-centered Cu4L44+ tetra-cation, with 4-coordinated Cu ions in the severely distorted tetrahedral environment (Table S1‡ lists the relevant geometrical data). In agreement with the requirements of this certain architecture the quinoline planes are almost parallel (cf. dihedral angles in Table S1‡).37–40 In turn, Ni(II) complex C2 makes triangular three-centered Ni3L3 structures, in which the Ni centers are six-coordinated in the quite regular octahedral geometry. As such a geometry requires additional ligands, the complex uses the solvent molecules. One of the Ni cations is additionally coordinated by two acetonitrile molecules (effectively N6), while the other two by one acetonitrile and one water molecule (N5O). In this case the particular geometry requires more twisted ligand conformation, and appropriately the dihedral angles between terminal quinoline rings are much larger.
The crystal structures also contain counterions and solvent molecules. In C1 there are four quite well-determined PF6 anions, four acetonitrile molecules and two toluene molecules, situation of C2 seems to be more complicated. The crystal structure suggests that besides the perchlorate anions (effectively 5.5), the half-occupied (at the inversion center), well-defined Ci-symmetrical octahedral anion is present; it has been interpreted as the PF6−, a result of the impurities. Additionally, one CH3CN and a number of at least partially occupied, disordered water molecules were found in this crystal structures.
Interestingly, even though a number of anions and water molecules were found, the structures are still quite porous, with the voids (up to 10% of the unit-cell volume) filled with the diffused solvent molecules.
DNA models as targets
The utilization of non-classical interactions of small molecules with nucleic acids can modulate the cellular interactions related to DNA and are of value as they manipulate the function of cells to produce a desired result, thereby allowing the diagnosis and/or treatment of disease.41,42 The spectroscopic properties of metal ions make them ideal not only from the design viewpoint, as scaffolds, but also as active sites.43 In our preliminary studies we employed several spectroscopic techniques to understand the interactions between the supramolecular Cu+ square grid-type complex C1 and Ni2+ triangle-type complex C2 with a duplex DNA and the GQ forming sequence Tel22 as a lower cost alternative for Pt2+ and Ru2+ compounds.First, in our study we investigated the affinity of C1, C2 and L toward the double helical DNA with use of absorption titration experiments (Fig. 4, Fig. S6‡). While C2 and L exhibited only weak interactions with the CT-DNA, the square grid-type complex C1 has shown to be a good DNA binder of a high affinity expressed in the binding constant Kb = 1.38 × 106; the standard Gibb's free energy is equal to −34.46 kJ mol−1.
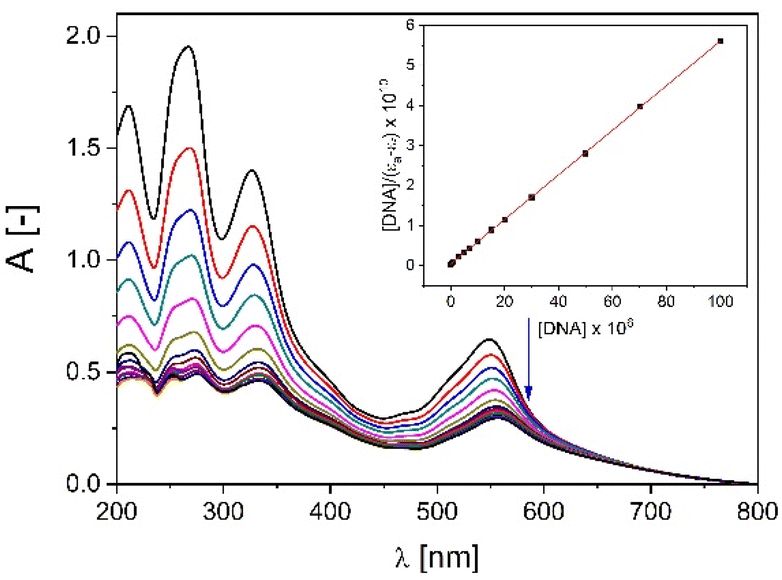 | ||
| Fig. 4 Absorption titration of C1 with increasing concentrations of CT-DNA (0–100 μM) in buffer Tris-HCl (5 mM Tris, 50 mM NaCl, pH = 7.22). Arrow shows hypochromic changes upon increasing CT-DNA concentration. Inset: plot of [DNA]/(εa − εf) versus [DNA]; ■, experimental data points; solid line, linear fitting of the data. | ||
Since such a large Kb suggests intercalation14 the competitive fluorescence quenching experiments with ethidium bromide (a classical intercalator) were conducted. As could be inferred, only the C1 pushed out the ethidium bromide from its fluorescent complex with DNA (Fig. 5a and Fig. S7‡). The interaction is expressed by the Stern–Volmer quenching constant equal to KSV = 7.12 × 104. The intercalation mode of binding has been further confirmed by the UV-thermal melting experiments which showed the stabilization of double helix by ca. 4 °C (Fig. 5b, Fig. S8 and S9‡).
 | ||
| Fig. 5 (a) Emission spectra of ethidium bromide bound to CT-DNA in the presence of increasing amounts of C1 (0–200 μM) in buffer Tris-HCl (5 mM Tris, 50 mM NaCl, pH = 7.22). Arrow shows the hypochromic changes upon increasing concentration of C1; inset: Stern–Volmer plot of I0/I − 1 vs. [C1] for the titration of EtBr-CT-DNA complex; ■, experimental data points; solid line, linear fitting of the data. (b) UV thermal melting curves of d(GTTAATCGCTGG) alone (green line) and with 4 eq. of C1 (black line) in cacodylate buffer (10 mM sodium cacodylate, 100 mM sodium chloride, pH = 7.23); DNA concentration is equal to 2.5 μM. (c) Structural conditions of C1 to act as bis-intercalator. Two parallel pairs of quinolines in transoid conformers of L are 7 Å apart. | ||
The intercalation describes the reversible insertion of a flat guest molecule into a lamellar host structure. DNA-drug intercalation is typically stabilized by significant π–π stackings, hydrophobic and polar interactions, as well as the electrostatic forces of cationic intercalators with polyanionic nucleic acid.44 By taking a closer look at the structure of cationic C1 one can type it is a potential example of a bis-intercalator due to its structural analogy to the bisantracyclines containing two daunorubicin molecules separated by 7 Å by a p- and m-xylylenyl linkers. Additionally, the linkers of the two daunorubicines line the minor groove of DNA. Such an arrangement on the two opposite sides of the metallogrid potentially could also allow it to cross-intercalate the DNA helixes (Fig. 5c). In our case the stiffened cyclohexane could also, potentially, interact on the surface of the helix and, moreover, the metal ions incorporated in the scaffold can induce the electrostatic interactions.45
Stabilization of GQ forming sequences to prevent its disassembly is a target of many studies30,46,47 therefore, taking advantage of the multivalency of metallosupramolecular complexes, we performed preliminary GQ binding (Fig. S10‡) and induction (induced circular dichroism, ICD) experiments (Fig. S10 and S11‡). Interestingly, the ICD spectra (the spectra given by the randomly folded Tel22 incubated with the given compound minus randomly folded Tel22, all in the absence of K+) suggest that the triangle-type complex C2 could induce a structure formation in the randomly folded telomeric Tel22 sequence (that is able to form hybrid type GQ in K+ solutions, Fig. S11‡), while C1 does not have any effect on the random DNA structure (Fig. S12‡). The final assembly is characterized by a spectrum with positive peaks at ca. 240 nm and 280 nm and negative peaks at ca. 260 nm and 300 nm (Fig. S10‡).48
Protein model as a target
Since the SA is a negatively charged protein it seemed to be reasonable to evaluate the cationic metallo-supramolecular square grid-type complex C1 (charge 4+) and triangle-type complex C2 (charge 6+) as binders. In our study we used BSA, since it is the most commonly used albumin model in in vitro experiments due to its similarity (76%) to Human Serum Albumin (HSA).49 The binding propensity of C1, C2 and L toward BSA was studied by fluorescence and CD spectroscopy. Due to the presence of neutral amino acids such as Trp, Tyr and Phe, the BSA protein shows a high fluorescence. The gradual diminution of the fluorescence band arising from the Trp-214 residue upon addition of potential drugs was monitored (Fig. 6) which suggest that they interact with the hydrophobic pocket of domain II. Analysis of the Stern–Volmer plots has shown that the square grid-type complex C1 (KSV = 8.0 × 104), triangle-type complex C2 (KSV = 1.1 × 105) and ligand L (KSV = 2.3 × 104) bind the BSA. The quenching rate kq was the highest for the C2 (1.1 × 1013 dm3 (mol s)−1, while for C1 and L it was and kq = 8.0 × 1012 dm3 (mol s)−1 and kq = 2.3 × 1012 dm3 (mol s)−1, respectively. One may assume it is due to the highest positive charge of triangle-type complex C2 equal to 6+. The affinity of our compounds toward BSA was characterized with the Scatchard method (Table 1, Fig. 7). While all compounds have similar number of binding sites (n) on BSA (ca. 1.0), C1 has a high Kb of ca. 106 and L a low Kb of ca. 103. Since the high Kb of drug–protein interaction can cause problems with release of the drug in the target region, the most moderate Kb of C2 equal to 105 seems to be the most promising in terms of the effective transportation and release to the tumor site by carrier proteins as SA.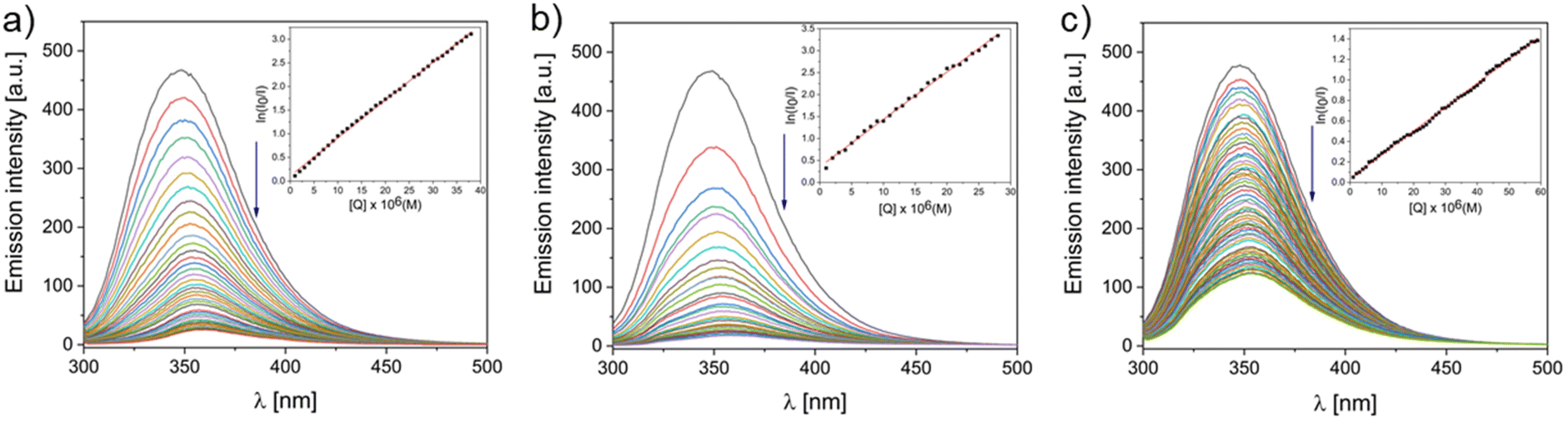 | ||
| Fig. 6 Fluorescent BSA (5 μM) titration with increasing concentrations of C1 [0–38 μM, (a)], C2 [0–28 μM, (b)] and L [0–59 μM, (c)] in PBS buffer (0.01 M phosphate buffer, 0.0027 M potassium chloride, 0.137 M sodium chloride pH = 7.4). Inset: plot of ln(I0/I) vs. [Q]; ■, experimental data points. | ||
 | ||
| Fig. 7 Scatchard plot of titration of BSA (5 μM) with C1 [0–15 μM, (a)], C2 [0–15 μM, (b)] and L [0–15 μM, (c)] in PBS buffer (0.01 M phosphate buffer, 0.0027 M potassium chloride, 0.137 M sodium chloride pH = 7.4). | ||
| Compound | n | K b |
|---|---|---|
| C1 [Cu4L4]4+ | 1.2 | 1.9 × 106 (R2 = 0.99) |
| C2 [Ni3L3]6+ | 1.0 | 4.1 × 105 (R2 = 0.97) |
| L | 0.8 | 5.2 × 103 (R2 = 0.99) |
Also, the conformational changes of BSA occurring upon binding were observed in the titration monitored by CD which is a precise and sensitive technique for the studies on secondary structures of proteins (Fig. 8). The bands at 209 nm (arising from n–π* transitions) and at 222 nm (from π–π* transitions) are characteristic for the α-helical structure of the BSA protein (ca. 55%).50,51 The high Kb of square grid-type complex C1 correlates well with the CD spectrum of BSA titrated with C1 in which an α-helix content dramatically drops to finally reach only 6%. Such distortion is undesirable, since they may lead to the impairment of the protein's functions. On the contrary, L was almost neutral for the BSA structure, while the triangle-type complex C2 moderately influenced the structure of BSA (decrease of α-helix content to 25%). Thus, together with its moderate Kb, one may assume it is able to be efficiently transported by SA.
 | ||
| Fig. 8 CD spectra of BSA (75 nM) in the presence of increasing concentration of C1 [0–37.50 μM, (a)], C2 [0–15.00 μM, (b)] and L [0–15.00 μM, (c)] in PBS buffer (0.01 M phosphate buffer, 0.0027 M potassium chloride, 0.137 M sodium chloride pH = 7.4). | ||
Conclusion
In conclusion, we were able to synthesize the [2 × 2] square grid-type complex C1[Cu4L4]4+ and triangle-type complex C2[Ni3L3]6+ with interwoven chameleonic N,N,N,N-donor ligand L as confirmed by X-ray. The versatility of such metallosupramolecular constructs prompted us to screen their specific interactions with relevant biomolecules – double stranded DNA, GQ and SA protein by means of several spectroscopic methods (UV-Vis absorption, fluorescence, circular dichroism). While the ligand L shows rather low affinity toward the tested molecules, we revealed that the square grid-type complex C1, possessing two parallel transoid ligands, attaches to the SA causing serious reduction of the α-helix content in the peptide and, according to our spectroscopic studies, intercalates double stranded DNA. On the other hand, the triangle-type complex C2 moderately binds SA and causes smaller structural perturbations in the peptide structure and also seems to induce a secondary structure in the unfolded GQ-forming sequence of Tel22. As a result, this preliminary study underlines the potential of multivalent metallo-supramolecular complexes in comparison to the free ligands in future drug design.Experimental
Materials and physical measurements
CT-DNA, BSA, ethidium bromide, Tris, PBS, metal salts and NaCl were supplied from Merck and used without further purification. Tel22 (d[AGGGTTAGGGTTAGGGTTAGGG]) was supplied from Genomed S. A. (Warsaw). ESI mass spectra for H2O solutions ∼10−4 M were measured using a Waters Micromass ZQ spectrometer. NMR spectra were run on a Varian Gemini 300 MHz (for ligand) or Mercury-plus 400 MHz (for C1) spectrometer and were calibrated against the residual protonated solvent signals with chemical shifts represented in ppm. Microanalyses were performed using Vario EL III CHN element analyzer. The FT-IR spectra were recorded in the range between 400 and 4000 cm−1 using Bruker FT-IR IFS 66 s−1 spectrometer. CT-DNA was dissolved in Tris Buffer (5 mM Tris HCl, 50 mM NaCl, pH 7.3) prior to use. The CT-DNA solution gave a ratio of UV absorbance of 1.82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 260 and 280 nm, indicating that the CT-DNA sample was sufficiently free from protein.52,53 CT-DNA concentration per nucleotide was determined from the UV absorbance at 260 nm using the extinction coefficient ε260 = 6600 dm3 mol−1 cm−1.54 BSA solution was prepared at a 75 nM concentration based on its molecular weight (MW 66 kDa) using PBS ([NaCl]: 137 mM; [KCl]: 2.7 mM; [Na2HPO4]: 10 mM; [KH2PO4]: 1.8 mM; pH 7.3) as a solvent for the spectral experiments. Additionally, BSA solution was checked spectroscopically and stored in the dark environment at 4 °C before usage. Electronic absorption titrations and melting experiments were performed on JASCO V-770 spectrophotometer equipped with a Peltier-thermo Cell Holder (water) PAC743R, in 10 × 10 mm quartz cells. Emission spectra in the competitive fluorescence titration experiments were measured at room temperature on an Agilent Technologies G9800A spectrofluorimeter in the range 540–720 nm with excitation and emission slits of 10 nm and excitation wavelength λexc = 467 nm in 10 × 10 mm quartz cells. CD spectra of BSA were obtained on a J-810 spectropolarimeter (Jasco Europe S.R.L., Cremella, Italy) equipped with a Peltier PTC-423S/15, using a Hellma (Milan, Italy) quartz cell (10 mm). In the investigation of BSA the spectra were measured in the 204–255 nm wavelength range at 15 °C. Freshly prepared stock solutions of complexes in MeCN [at concentration 5 × 10−3 M (for CD measurements) or 2 × 10−3 M (for other measurements)] were taken for all spectroscopic investigations.
:1 at 260 and 280 nm, indicating that the CT-DNA sample was sufficiently free from protein.52,53 CT-DNA concentration per nucleotide was determined from the UV absorbance at 260 nm using the extinction coefficient ε260 = 6600 dm3 mol−1 cm−1.54 BSA solution was prepared at a 75 nM concentration based on its molecular weight (MW 66 kDa) using PBS ([NaCl]: 137 mM; [KCl]: 2.7 mM; [Na2HPO4]: 10 mM; [KH2PO4]: 1.8 mM; pH 7.3) as a solvent for the spectral experiments. Additionally, BSA solution was checked spectroscopically and stored in the dark environment at 4 °C before usage. Electronic absorption titrations and melting experiments were performed on JASCO V-770 spectrophotometer equipped with a Peltier-thermo Cell Holder (water) PAC743R, in 10 × 10 mm quartz cells. Emission spectra in the competitive fluorescence titration experiments were measured at room temperature on an Agilent Technologies G9800A spectrofluorimeter in the range 540–720 nm with excitation and emission slits of 10 nm and excitation wavelength λexc = 467 nm in 10 × 10 mm quartz cells. CD spectra of BSA were obtained on a J-810 spectropolarimeter (Jasco Europe S.R.L., Cremella, Italy) equipped with a Peltier PTC-423S/15, using a Hellma (Milan, Italy) quartz cell (10 mm). In the investigation of BSA the spectra were measured in the 204–255 nm wavelength range at 15 °C. Freshly prepared stock solutions of complexes in MeCN [at concentration 5 × 10−3 M (for CD measurements) or 2 × 10−3 M (for other measurements)] were taken for all spectroscopic investigations.
Synthesis of ligand L N,N-bis(quinolin-2-ylmethylene)cyclohexane-1,4-diamine
trans-1,4-Diaminocyclohexane (51.80 mg, 0.45 mmol) was dissolved in 6 ml of EtOHabs. Then 2-quinolinecarboxaldehyde (2 equiv., 142.59 mg, 0.90 mmol) was added. The reaction was carried out in inert conditions with heating under reflux for 3 hours and then left for 21 hours at room temperature. The solution became brown and after a while a cream-colored precipitate formed. The solution was concentrated and the precipitation was filtered under reduced pressure. The precipitate was washed with cold EtOHabs. Yield 81.58% (Scheme 2).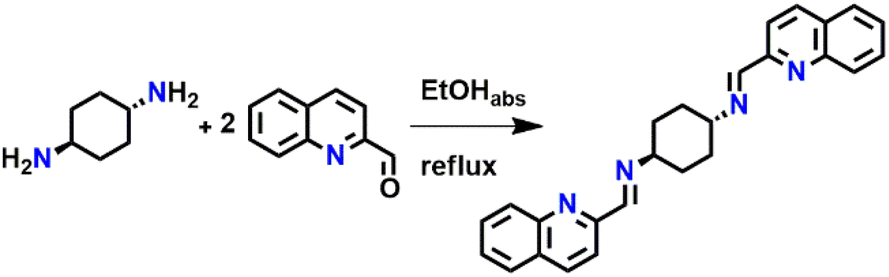 | ||
| Scheme 2 Synthesis of ligand (L). | ||
1H NMR (300 MHz, DMSO-d6): δ (ppm) = 8.60 (s, 1H); 8.44 (d, 1H); 8.07 (m, 3H), 7.82 (t, 1H); 7.67 (t, 1H); 1.82 (dt, 4H).
13C NMR (75 MHz, DMSO-d6): δ (ppm) = 160.47; 154.67; 147.24; 136.9; 130.16; 129.16; 128.36; 128.13; 127.65; 117.95; 67.84; 32.08 (2C).
ESI-MS(+) m/z (%): 393 (78) [HL]+; 254 (43) [HA]+; 415 (90) [L + Na]+; 807 (100) [2L + Na]+.
FT-IR (ATR): 3037 ν(C−H)arom; 2928, 2858 νs(C−H)aliphatic; 1642, 1596 ν(CC); 1463, 1449 ν(CN); 1085, 986, 957, 943, 9905, 791, 778, 757, 618 γ(C−H)arom cm−1.
Anal. calcd for (C26H24N4): C: 79.56, H: 6.17, N: 14.27%; found: C: 78.92, H: 6.11, N: 14.37%.
Synthesis of complex C1
The ligand L (21.00 mg, 0.053 mmol) was dissolved in 10 ml of MeCN giving a yellow solution. Then the Cu(MeCN)4PF6 salt (1 equiv., 19.94 mg, 0.053 mmol) was added and the solution turned into dark purple. The reaction was carried out for 24 hours. Then the solution was concentrated to ca. 2 ml on a rotary evaporator and 2 ml of Et2O were added. The dark purple precipitate formed was filtered under reduced pressure. Yield: 13.78%.A dark purple single crystal suitable for X-ray studies was obtained by a slow vapour diffusion of MeCN from the solution of complex [Cu4L4]4+ to toluene.
1H NMR (400 MHz, CD3CN-d6): δ (ppm) = 8.83 (s, 1H); 8.70 (d, 1H); 8.04 (d, 1H), 7.98 (d, 1H); 7.61 (t, 1H); 7.53 (d, 1H); 7.47 (t, 1H); 3.58 (s, 1H); 1.57 (m, 4H).
ESI-MS(+) m/z (%): 2259.72 (90) {[Cu4L4](PF6)3}+; 1057.29 (85) {[Cu4L4](PF6)2}2+; 656.84 (100) {[Cu4L4](PF6)}3+; 456.38 (30) [Cu4L4]4+.
FT-IR (ATR): 3060 ν(C−H)arom; 2932, 2860 νs(C−H)aliphatic; 1622, 1590 ν(CC); 1455, 1434 ν(CN); 1092, 1019, 996, 935, 783 γ(C−H)arom; 833, 750 (PF6−)cm−1.
Anal. calcd for [(C104H92Cu4N16)4+·4(PF6)−·6(C2H3N)]: C: 52.65, H: 4.19, N: 11.64%; found: C: 52.13, H: 4.22, N: 11.56%.
Synthesis of complex C2
The ligand L (21.23 mg, 0.054 mmol) was dissolved in 10 ml of MeCN giving a yellow solution. Then the Ni(ClO4)2·6H2O salt (1 equiv., 19.78 mg, 0.054 mmol) was added, the solution turned cloudy yellow-orange. The reaction was carried out for 24 hours. Then the solution was concentrated to ca. 2 ml on a rotary evaporator and 1 ml of Et2O was added. The brown precipitate formed was filtered under reduced pressure. Yield: 22.76%.A brown single crystal suitable for X-ray studies was obtained by a vapour slow diffusion of i-Pr2O into a solution of complex [Ni3L3]6+ in MeOH and MeCN (1:1 in volume).
ESI-MS(+) m/z (%): 393 (100%) [L + H]+; 449 (35) [L − H + Ni]+; 512 (30) [L + NiCO3]+; 549 (25) [L + NiClO4]+; 904 (5) [2L + NiCO3]+; 941 (5) [2L + NiClO4]+.
FT-IR (ATR): 3082 ν(C−H)arom; 2944, 2868 νs(C−H)aliphatic; 1618, 1593 ν(CC); 1466, 1436 ν(CN); 1000, 958, 950, 931, 904, 801, 772, 753 γ(C−H)arom; 1060; 620 (ClO4−) cm−1.
Anal. calcd for [2(C86H88N16Ni3O2)6+·11(ClO4)−·Cl−·2(C2H3N)·CH4O·7H2O]: C: 47.48, H: 4.50, N: 10.64%; found: C: 46.34, H: 4.54, N: 10.73%.
X-ray crystallography
Diffraction data were collected by the ω-scan technique: for L and C1 at 130(1) K on Rigaku SuperNova four-circle diffractometer with Atlas CCD detector, equipped with Nova microfocus CuKα radiation source (λ = 1.54178 Å), and for C2 at 100(1) K, on Rigaku XCalibur four-circle diffractometer with Eos CCD detector, with graphite-monochromatized MoKα radiation source (λ = 0.71073 Å). The data were corrected for Lorentz-polarization as well as for absorption effects.55 The structures were solved with SHELXT56 and refined with the full-matrix least-squares procedure on F2 by SHELXL-2013.57 All non-hydrogen atoms were refined anisotropically, hydrogen atoms were placed in idealized positions and refined as ‘riding model’ with isotropic displacement parameters set at 1.2 (1.5 for methyl and hydroxyl groups) times Ueq of appropriate carrier atoms. In C2 the restraints were used for both the geometry and displacement parameters, but no restraints were applied for the cationic parts of the structures. The well-defined, octahedrally symmetrical residual density located across the center of inversion was interpreted as a result of impurity – PF6 anion. Moreover, the structures of both complexes contained diffused electron density which fills the voids – these effects were taken into account by means of SQUEEZE procedure.58 In C1 this unmodeled electron density is probably a result of heavily disordered toluene molecule, in C2 the best description is a layer of a number of disordered water molecules. The relevant crystallographic data together with the details of structure refinement are listed in Table 2.| Compound | L | C1 | C2 |
|---|---|---|---|
| Formula | C26H24N | (C104H96Cu4N16)4+·4(PF6)−·6(C2H3N)·2(C7H8) + disordered solvent | 2(C86H88N16Ni3O2)6+·11(ClO4)−·PF6−·2(C2H3N)·CH4O·4H2O + disordered solvents |
| Formula weight | 392.49 | 2834.59 | 4532.73 |
| Crystal system | Monoclinic | Triclinic | Triclinic |
| Space group | P21/n | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a (Å) | 6.02703(15) | 12.9065(4) | 18.3597(7) |
| b (Å) | 13.2413(4) | 16.5279(5) | 18.4990(7) |
| c (Å) | 12.7941(4) | 16.7055(4) | 21.3107(8) |
| α (°) | 90 | 89.181(2) | 112.743(4) |
| β (°) | 91.236(2) | 76.218(2) | 91.517(3) |
| γ (°) | 90 | 82.422(2) | 117.701(4) |
| V (Å3) | 1020.80(5) | 3430.21(17) | 5718.5(4) |
| Z | 2 | 1 | 1 |
| D x (g cm−3) | 1.277 | 1.372 | 1.316 |
| F(000) | 416 | 1456 | 2342 |
| μ (mm−1) | 0.597 | 1.893 | 0.702 |
| Reflections | |||
| Collected | 4238 | 26696 |
45102 |
| Unique (Rint) | 2076 (0.0242) | 12344 (0.0297) |
20107 (0.0428) |
| With I > 2σ(I) | 1713 | 10191 |
13671 |
| R(F) [I > 2σ(I)] | 0.0393 | 0.0554 | 0.0818 |
| wR(F2) [I > 2σ(I)] | 0.1002 | 0.1541 | 0.2296 |
| R(F) [all data] | 0.0504 | 0.0667 | 0.1168 |
| wR(F2) [all data] | 0.1121 | 0.1630 | 0.2482 |
| Goodness of fit | 1.05 | 1.02 | 1.10 |
| Max/min Δρ (e Å−3) | 0.26/−0.17 | 0.99/−0.39 | 1.69/−0.81 |
| CCDC number | 2097988 | 2097989 | 2097990 |
NMR titration studies
A solution of ligand of concentration 5 × 10−4 M in CD3CN was prepared. Then 0.10; 0.25; 0.50; 0.75; 1.00; 1.25 equiv. of Cu(CH3CN)4BF4 (stock solution: 5 × 10−3 M in CD3CN) were added to the solution of ligand and spectra were measured.UV-Vis titration studies
A solution of L at concentration 5.1 × 10−6 M and solutions of Cu(CH3CN)4PF6 and Ni(ClO4)2·6H2O at concentrations 2 × 10−3 M were prepared in MeCN. 2.5 ml of the ligand solution was poured into a cuvette then the absorption was measured. Next the ligand solution was titrated with Cu(I) [0–1.25 equivalents of Cu+] or Ni(II) [0–1.65 equivalents of Ni2+] salt.DNA binding studies
| [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb (εb − εf), |
Gibb's standard free energy was calculated from the equation:60

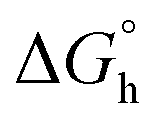 is the standard Gibb's free energy, and Kb is the intrinsic binding constant.
is the standard Gibb's free energy, and Kb is the intrinsic binding constant.
| I0/I = 1 + KSV[Q], |
Protein binding studies
| MRE = [observed CD/(Cpnl × 10)], [(deg × cm2)/dmol], |
α-Helical content of free and complexed BSA was calculated with the equation:51
| α-Helical (%) = [(−MRE209 − 4000)/(33000–4000)] × 100 |
000 is the value of the MRE for pure α-helix at 209 nm, while 4000 is the MRE value at 209 nm for random coil conformation and β-form cross.
| I0/I = e(Ksv[Q]) |
| log[(I0 − I)/I] = logKb + nlog[Q] |
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Conceptualization, data curation, formal analysis: MSz, MFJ; Funding aquisiton: MSz; Investigation, methodology: MSz, MK, GNR, GC, MFJ; Project administration: MFJ; Resources: MSz; Supervision: MFJ, VP; Validation: MSz, MK, GNR, GC, MFJ; Writing – original draft: MSz, MK, MFJ; Writing – review & editing: MSz, MK, GNR, GC, MFJ, VP.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MSz: the work was supported by the National Science Centre, Poland (grant no. 2020/37/N/ST4/00751) and grant no. POWR.03.02.00-00-I026/16 co-financed by the European Union through the European Social Fund under the Operational Program Knowledge Education Development. VP: the work was supported by IDUB-UAM (project no. 006/07/POB3/0006) “International support for AMU staff – international internships as part of the program – International Junior and Senior Exchange”.References
- M. D. Pluth and K. N. Raymond, Chem. Soc. Rev., 2007, 36, 161–171 RSC.
- H. Sepehrpour, W. Fu, Y. Sun and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14005–14020 CrossRef CAS PubMed.
- Q. Wang, G. Yang, Y. Fu, N. Li, D. Hao and S. Ma, ChemNanoMat, 2022, 8, e202100396 CAS.
- M. Ruben, J. Rojo, F. J. Romero-Salguero, L. H. Uppadine and J.-M. Lehn, Angew. Chem., Int. Ed., 2004, 43, 3644–3662 CrossRef CAS PubMed.
- H. Zhu, Q. Li, B. Shi, H. Xing, Y. Sun, S. Lu, L. Shangguan, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 17340–17345 CrossRef CAS PubMed.
- R. W. Hogue, S. Dhers, R. M. Hellyer, J. Luo, G. S. Hanan, D. S. Larsen, A. L. Garden and S. Brooker, Chem. – Eur. J., 2017, 23, 14193–14199 CrossRef CAS PubMed.
- Y. Sun and P. J. Stang, Aggregate, 2021, 2, e94 Search PubMed.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS PubMed.
- S. H. Hewitt and A. J. Wilson, Chem. Commun., 2016, 52, 9745–9756 RSC.
- J. W. Steed, D. R. Turner and K. Wallace, Core Concepts in Supramolecular Chemistry and Nanochemistry, John Wiley & Sons, West Sussex, U.K., 2007 Search PubMed.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, West Sussex, U.K., 2nd edn, 2009 Search PubMed.
- G. Yu, M. Zhang, M. L. Saha, Z. Mao, J. Chen, Y. Yao, Z. Zhou, Y. Liu, C. Gao, F. Huang, X. Chen and P. J. Stang, J. Am. Chem. Soc., 2017, 139, 15940–15949 CrossRef CAS PubMed.
- M. A. Fik, A. Gorczyński, M. Kubicki, Z. Hnatejko, A. Fedoruk-Wyszomirska, E. Wyszko, M. Giel-Pietraszuk and V. Patroniak, Eur. J. Med. Chem., 2014, 86, 456–468 CrossRef CAS PubMed.
- A. Adamski, M. A. Fik, M. Kubicki, Z. Hnatejko, D. Gurda, A. Fedoruk-Wyszomirska, E. Wyszko, D. Kruszka, Z. Dutkiewicz and V. Patroniak, New J. Chem., 2016, 40, 7943–7957 RSC.
- R. Huang and P.-K. Zhou, Signal Transduction Targeted Ther., 2021, 6, 254 CrossRef CAS PubMed.
- M. A. Muñoz-Lorente, A. C. Cano-Martin and M. A. Blasco, Nat. Commun., 2019, 10, 4723 CrossRef PubMed.
- M. Marzano, A. P. Falanga, D. Marasco, N. Borbone, S. D'Errico, G. Piccialli, G. N. Roviello and G. Oliviero, Mar. Drugs, 2020, 18, 49 CrossRef CAS PubMed.
- P. L. Scognamiglio, C. Platella, E. Napolitano, D. Musumeci and G. N. Roviello, Molecules, 2021, 26, 3558 CrossRef CAS PubMed.
- C. Vicidomini, F. Cioffi, K. Broersen, V. Roviello, C. Riccardi, D. Montesarchio, D. Capasso, S. Di Gaetano, D. Musumeci and G. N. Roviello, Future Med. Chem., 2019, 11, 285–302 CrossRef CAS PubMed.
- M. J. Hawkins, P. Soon-Shiong and N. Desai, Adv. Drug Delivery Rev., 2008, 60, 876–885 CrossRef CAS PubMed.
- A. M. Merlot, D. S. Kalinowski and D. R. Richardson, Front. Physiol., 2014, 5, 299 Search PubMed.
- B. Deka, T. Sarkar, S. Banerjee, A. Kumar, S. Mukherjee, S. Deka, K. K. Saikia and A. Hussain, Dalton Trans., 2017, 46, 396–409 RSC.
- T. W. Evans, Aliment. Pharmacol. Ther., 2002, 16, 6–11 CrossRef CAS PubMed.
- P. Carmeliet and R. K. Jain, Nature, 2000, 407, 249–257 CrossRef CAS PubMed.
- K. Greish, J. Drug Targeting, 2007, 15, 457–464 CrossRef CAS PubMed.
- C. Commisso, S. M. Davidson, R. G. Soydaner-Azeloglu, S. J. Parker, J. J. Kamphorst, S. Hackett, E. Grabocka, M. Nofal, J. A. Drebin, C. B. Thompson, J. D. Rabinowitz, C. M. Metallo, M. G. Vander Heiden and D. Bar-Sagi, Nature, 2013, 497, 633–637 CrossRef CAS PubMed.
- W. Drożdż, A. Walczak, Y. Bessin, V. Gervais, X.-Y. Cao, J.-M. Lehn, S. Ulrich and A. R. Stefankiewicz, Chem. – Eur. J., 2018, 24, 10802–10811 CrossRef PubMed.
- W. Drożdż, Y. Bessin, V. Gervais, X.-Y. Cao, J.-M. Lehn, A. R. Stefankiewicz and S. Ulrich, Chem. – Eur. J., 2018, 24, 1518–1521 CrossRef PubMed.
- O. Domarco, D. Lötsch, J. Schreiber, C. Dinhof, S. Van Schoonhoven, M. D. García, C. Peinador, B. K. Keppler, W. Berger and A. Terenzi, Dalton Trans., 2017, 46, 329–332 RSC.
- A. T. Phan, V. Kuryavyi, S. Burge, S. Neidle and D. J. Patel, J. Am. Chem. Soc., 2007, 129, 4386–4392 CrossRef CAS PubMed.
- Y. Zhang, X. Yan, L. Shi, M. Cen, J. Wang, Y. Ding and Y. Yao, Inorg. Chem., 2021, 60, 7627–7631 CrossRef CAS PubMed.
- W. Drożdż, C. Bouillon, C. Kotras, S. Richeter, M. Barboiu, S. Clément, A. R. Stefankiewicz and S. Ulrich, Chem. – Eur. J., 2017, 23, 18010–18018 CrossRef PubMed.
- A. R. Stefankiewicz, J. Harrowfield, A. M. Madalan and J.-M. Lehn, CrystEngComm, 2013, 15, 9128–9134 RSC.
- A. Gorczyński, M. Kubicki, K. Szymkowiak, T. Łuczak and V. Patroniak, RSC Adv., 2016, 6, 101888–101899 RSC.
- S. Napierała, M. Kubicki, V. Patroniak and M. Wałęsa-Chorab, Electrochim. Acta, 2021, 369, 137656 CrossRef.
- B. Schäfer, J.-F. Greisch, I. Faus, T. Bodenstein, I. Šalitroš, O. Fuhr, K. Fink, V. Schünemann, M. M. Kappes and M. Ruben, Angew. Chem., Int. Ed., 2016, 55, 10881–10885 CrossRef PubMed.
- V. Patroniak, J.-M. Lehn, M. Kubicki, A. Ciesielski and M. Wałęsa, Polyhedron, 2006, 25, 2643–2649 CrossRef CAS.
- V. Patroniak, P. Baxter, J.-M. Lehn, M. Kubicki, M. Nissinen and K. Rissanen, Eur. J. Inorg. Chem., 2003, 2003, 4001–4009 CrossRef.
- N. Suryadevara, A. Pausch, E. Moreno-Pineda, A. Mizuno, J. Bürck, A. Baksi, T. Hochdörffer, I. Šalitroš, A. S. Ulrich, M. M. Kappes, V. Schünemann, W. Klopper and M. Ruben, Chem. – Eur. J., 2021, 27, 15172–15180 CrossRef PubMed.
- H. Han and L. H. Hurley, Trends Pharmacol. Sci., 2000, 21, 136–142 CrossRef CAS PubMed.
- S. Mulliri, A. Laaksonen, P. Spanu, R. Farris, M. Farci, F. Mingoia, G. N. Roviello and F. Mocci, Int. J. Mol. Sci., 2021, 22, 6028 CrossRef CAS PubMed.
- B. J. Pages, D. L. Ang, E. P. Wright and J. R. Aldrich-Wright, Dalton Trans., 2015, 44, 3505–3526 RSC.
- N. J. Wheate, C. R. Brodie, J. G. Collins, S. Kemp and J. R. Aldrich-Wright, Mini-Rev. Med. Chem., 2007, 7, 627–648 CrossRef CAS PubMed.
- J. Portugal, D. J. Cashman, J. O. Trent, N. Ferrer-Miralles, T. Przewloka, I. Fokt, W. Priebe and J. B. Chaires, J. Med. Chem., 2005, 48, 8209–8219 CrossRef CAS PubMed.
- G. Zhou, X. Liu, Y. Li, S. Xu, C. Ma, X. Wu, Y. Cheng, Z. Yu, G. Zhao and Y. Chen, Oncotarget, 2016, 7, 14925–14939 CrossRef PubMed.
- F. Greco, D. Musumeci, N. Borbone, A. P. Falanga, S. D'Errico, M. Terracciano, I. Piccialli, G. N. Roviello and G. Oliviero, Molecules, 2022, 27, 2997 CrossRef CAS PubMed.
- M. A. Fik-Jaskółka, I. Pospieszna-Markiewicz, G. N. Roviello, M. Kubicki, W. Radecka-Paryzek and V. Patroniak, Inorg. Chem., 2021, 60, 2122–2126 CrossRef PubMed.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed.
- M. A. Fik-Jaskółka, A. F. Mkrtchyan, A. S. Saghyan, R. Palumbo, A. Belter, L. A. Hayriyan, H. Simonyan, V. Roviello and G. N. Roviello, Spectrochim. Acta, Part A, 2020, 229, 117884 CrossRef PubMed.
- M. A. Fik-Jaskółka, A. F. Mkrtchyan, A. S. Saghyan, R. Palumbo, A. Belter, L. A. Hayriyan, H. Simonyan, V. Roviello and G. N. Roviello, Amino Acids, 2020, 52, 755–769 CrossRef PubMed.
- N. Raman, K. Pothiraj and T. Baskaran, J. Mol. Struct., 2011, 1000, 135–144 CrossRef CAS.
- J. Marmur, J. Mol. Biol., 1961, 3, 208–218 CrossRef CAS.
- M. E. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- A. Technologies, CrysAlis PRO (Version 1.171.39.46), Agilent Technologies Ltd, 2018 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- L. Shivakumar, K. Shivaprasad and H. D. Revanasiddappa, Spectrochim. Acta, Part A, 2012, 97, 659–666 CrossRef CAS PubMed.
- D. Sabolová, M. Kožurková, T. Plichta, Z. Ondrušová, D. Hudecová, M. Šimkovič, H. Paulíková and A. Valent, Int. J. Biol. Macromol., 2011, 48, 319–325 CrossRef PubMed.
- B. C. Baguley and M. Le Bret, Biochemistry, 1984, 23, 937–943 CrossRef CAS PubMed.
- F. Shiri, M. Rahimi-Nasrabadi, F. Ahmadi and H. Ehrlich, Spectrochim. Acta, Part A, 2018, 203, 510–521 CrossRef CAS PubMed.
- V. M. Manikandamathavan, M. Thangaraj, T. Weyhermuller, R. P. Parameswari, V. Punitha, N. N. Murthy and B. U. Nair, Eur. J. Med. Chem., 2017, 135, 434–446 CrossRef CAS PubMed.
- X.-Z. Feng, Z. Lin, L.-J. Yang, C. Wang and C. I. Bai, Talanta, 1998, 47, 1223–1229 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Paolo Finocchiaro on the occasion of 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2097988–2097990. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02271k |
| This journal is © The Royal Society of Chemistry 2022 |