
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst supports based on ZnO–ZnAl2O4 nanocomposites with enhanced selectivity and coking resistance in isobutane dehydrogenation†
Anna N.
Matveyeva
 *a,
Shamil O.
Omarov
a,
Alexey V.
Nashchekin
b,
Vadim I.
Popkov
a and
Dmitry Yu.
Murzin
*c
*a,
Shamil O.
Omarov
a,
Alexey V.
Nashchekin
b,
Vadim I.
Popkov
a and
Dmitry Yu.
Murzin
*c
aLaboratory of Materials and Processes for Hydrogen Energy, Ioffe Institute, Politekhnicheskaya ul. 28, St Petersburg 194021, Russia. E-mail: anna.matveyeva@mail.ioffe.ru
bFederal Joint Research Center “Material science and characterization in advanced technology”, Ioffe Institute, Politekhnicheskaya ul. 26, St Petersburg 194021, Russia
cLaboratory of Industrial Chemistry and Reaction Engineering, Åbo Akademi University, Henriksgatan 2, Turku/Åbo 20500, Finland. E-mail: dmurzin@abo.fi
First published on 22nd July 2022
Abstract
Development of coking resistant supports and catalysts for hydrocarbons conversion is challenging, especially when using such acidic materials as alumina. Apparently, this problem can be mitigated by using spinels that are less acidic, being, however, thermally stable. In this study, a series of ZnO–ZnAl2O4 nanocomposites with different ZnO loading were prepared by urotropine-nitrate combustion synthesis to be used as supports for isobutane dehydrogenation catalysts. The nanocomposites were characterized by XRD, SEM, N2-physisorption analysis, EDS, H2-TPR, TPD of NH3 and tested in isobutane dehydrogenation. Spinels with small amounts of ZnO displayed higher acidity and specific surface areas than samples with a higher ZnO content (30–40 mol%). At the same time, the maximum activity and the lowest selectivity to by-products (CH4 and C3H6) after 10 min of the reaction were observed for the nanocomposite containing 20 mol% of ZnO. The obtained nanocomposites have demonstrated better resistance to coking compared to commercial alumina.
1. Introduction
Olefins are one of the key building blocks of modern society with a multitude of daily-use products coming from their transformations.1 Among them, isobutene is used for the production of isooctane, methacrolein, synthetic resins, etc. In the recent years, there has been a drift away from the traditional cracking based on naphtha and heavy oil to processes relying on light alkanes due to emergence of a large number of sources of light alkanes, including the shale gas.2 Subsequently abundant sources of light alkanes, coupled with the growing global demand for light alkenes,2d have pushed forward the use of alternative technologies for the production of light alkenes, namely direct conversion of alkanes to alkenes through environmentally friendly, economical, and efficient dehydrogenation.3In industrial settings the catalysts, commonly used for dehydrogenation of alkanes, are mainly Pt and CrOx supported on alumina.2d However, because of the high prices and low availability of platinum, and toxicity of CrVI species,4 development of alternative, low cost, and environmentally friendly catalysts is of great importance. In this respect, other catalysts based on gallia,5 nickel,6 cobalt,7 zinc oxide,8etc. are currently being actively investigated for dehydrogenation of isobutane to isobutene.
Dehydrogenation requires a relatively high temperature (above 500 °C) to obtain a high yield of alkenes. Under these harsh reaction conditions, catalyst deactivation due to particle sintering and coke formation is inevitable.2a
Catalyst supports, their specific surface area, acid–base properties and thermal stability play an important role in catalytic performance and coke formation. In the recent years, many researchers have undertaken numerous attempts to use such materials as SiO2,6 ZrO2,9 and zeolites10 for isobutane dehydrogenation, instead of the traditionally applied alumina, which is even considered to be a dehydrogenation catalyst per se.11
In the case of zinc oxide, catalytic behaviour depends on the state of this oxide: bulk ZnO, dispersed (supported) Zn2+, or complex oxides containing Zn2+, for example, spinel-type oxides or Zn-modified zeolites. Bulk ZnO is considered to be almost inactive in dehydrogenation.12 At the same time, catalysts with active Zn sites (Zn2+) are promising candidates considering their low costs and generally low toxicity of Zn.8a,13
A particular attention is paid to nanocrystalline materials, such as various spinels, which are of great interest for catalysis due to their low surface acidity, high thermal stability and mechanical resistance, and sufficient ability to disperse metals.14 The first characteristic is very important in alkane dehydrogenation because low acidity of the spinel support allows minimizing the undesirable side reactions (such as cracking and coke formation).14a The latter characteristic explains a rather widespread utilization of spinels for Pt and Pd catalysts. Dispersion of platinum, as well as the mean particle size and particle size distribution of metal nanoparticles, are often critical for catalytic activity and selectivity.14d
Subsequently, it is clearly of relevance to explore the properties of zinc-aluminum spinels having an excess of zinc oxide. In such a composite, active Zn2+ particles can be present both in the spinel and bulk ZnO, and some synergy effect between Zn2+ and Al3+ can be anticipated.14e
A low surface area, which is a disadvantage of spinel materials for catalytic applications, can be improved by choosing an appropriate synthesis method. For example, ZnAl2O4 can be synthesized by the solid-state reaction or the ceramic method;14l,15 wet chemical routes such as the sol–gel technique,16 precipitation or coprecipitation;17 and other techniques, such as hydrothermal methods,18 mechano-chemical methods,14g solution combustion synthesis (SCS),19 including microwave-initialized SCS;20 molten salts synthesis,21 vibrational milling method,22etc.
Combustion synthesis is one of the most efficient, simple and fast methods for the synthesis of simple and complex metal oxides, which makes it possible to obtain highly pure and chemically homogeneous powders with nanosized particles.23 There are two main initiation modes in SCS: the volume combustion and the self-propagating combustion modes.23a The first one is much more commonly used, because in this case the entire combustion mass is uniformly heated until ignition. Most often, a heating plate or a muffle furnace is used to initiate the combustion process. Another emerging and promising initiation type is to use a microwave oven. However, the microwave initiation has some limitations, such as a high cost of the equipment, the low size limit of the combustion reactor and impossibility to monitor the particle growth during combustion.
After initialization the combustion reaction is self-sustaining allowing a very fast attainment of temperatures required for crystallization and the phase formation. The metal precursors (often metal nitrates) and the so-called fuels (mainly organic compounds such as urea, citric acid, glycine) are mixed in an appropriate stoichiometric ratio. This controls combustion following the principles of propellant chemistry, causing a very rapid and exothermic chemical reaction to form the oxide powder. In addition, nanocomposites of different compositions can be easily prepared using the combustion method by varying the reagents in the initial reaction solution.
In this study, ZnAl2O4 spinels with an excess of ZnO, prepared by solution combustion synthesis, were used for the first time in isobutane dehydrogenation, allowing to obtain high selectivity and resistance to coking.
2. Experimental
2.1. Synthetic procedures
Al(NO3)3·9H2O (97%), Zn(NO3)2·6H2O (98%), and urotropine ((CH2)6N4, 99.5%) were purchased from Lenreactiv (Russia).An appropriate amount of urotropine and the corresponding nitrates were dissolved in ca. 200 ml of bidistilled water for the preparation of Al2O3, ZnO and xZnO–yZnAl2O4 (where x is the molar fraction of ZnO, y is the molar fraction of ZnAl2O4). The weight of the reagents was taken to ensure synthesis of 4 g of a sample. One batch of each material was made. Balancing the reaction was done through an online balancer (WebQC.org). For example, for the synthesis of a sample containing 30 mol% ZnO, the reaction will be presented as: 1800Al(NO3)3·9H2O + 1170Zn(NO3)2·6H2O + 1075C6H12N4 = 9(ZnAl2O4)100·(ZnO)30 + 6020N2 + 6450CO2 + 29670H2O.
The obtained solution was heated to boiling on an electric plate (1 kW) and, after complete evaporation of water, was burned in a glow combustion mode with the formation of a solid. This substance was crushed and heated in a muffle to 700 °C with a heating rate of 5 °C min−1, and kept at this temperature for 2 h.
2.2. Characterization
XRD analysis was done using XRD-6100 diffractometer (Shimadzu) with CuKα radiation (λ1 = 1.54056 Å) and Ni-filter at 40 kV, 30 mA, scan speed of 1° min−1, step width of 0.02°, exposition of 1.2 s and D (divergence)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S (scatter):R (receiving) = 1:1:0.3. The diffraction data were analyzed by the Rietveld's powder diffraction profile-fitting technique to determine the crystal structural parameters and phase composition.24 FullProf software was used for the refinement.25 The structural parameters for ZnAl2O4 with the space group Fd
:S (scatter):R (receiving) = 1:1:0.3. The diffraction data were analyzed by the Rietveld's powder diffraction profile-fitting technique to determine the crystal structural parameters and phase composition.24 FullProf software was used for the refinement.25 The structural parameters for ZnAl2O4 with the space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m and ZnO with the space group P63mc were taken from the Crystallography Open Database (CIF No. 9013642 and No. 2300450, respectively). The Pseudo-Voigt function was used for the simulation of the peak shape. The mean crystallite size was calculated using the Scherrer equation.
m and ZnO with the space group P63mc were taken from the Crystallography Open Database (CIF No. 9013642 and No. 2300450, respectively). The Pseudo-Voigt function was used for the simulation of the peak shape. The mean crystallite size was calculated using the Scherrer equation.
N2-Physisorption analysis was performed on ASAP 2020 (Micromeritics). The specific surface area, the total pore volume and the pore size distribution were determined from N2 adsorption isotherms applying the BET equation and a standard NLDFT analysis. The samples were degassed at 200 °C under vacuum up to the residual pressure 30 Pa for 4 h.
SEM micrographs were obtained in SE and BSE modes using JSM-7001F microscope (JEOL) equipped with Oxford instruments INCA PentaFETx for energy dispersed X-ray spectroscopy (EDS). EDS was also performed on VEGA 3 SBH microscope (Tescan) using Oxford instruments INCAx-act.
NH3-TPD and H2-TPR were performed on Chemosorb (SOLO, Russia) with a thermal conductivity detector. For NH3-TPD a sample calcined at 700 °C (ca. 160 mg) was pretreated in a flow of He (99.9999 vol% purity) at 700 °C for 40 min, then cooled to 110 °C. The sample saturated with ammonia from a flow of 5 vol% NH3/He mixture (20 ml min−1) at 110 °C for 30 min was thereafter purged with a He flow (20 ml min−1) for 30 min. Ammonia desorption was carried out by heating the sample to 700 °C at a rate of 10° min−1 in He flow (20 ml min−1).
For H2-TPR a sample (ca. 100 mg) was pretreated in Ar (99.998 vol% purity) at 300 °C for 30 min and heated thereafter to 900 °C at a ramp rate of 10° min−1 under 10 vol% H2/Ar mixture (18 ml min−1 Ar and 2 ml min−1 H2). Isopropanol, cooled in liquid nitrogen to the temperature at which it began to crystallize, was used as a water trap. CuO was used as a reference for calibration of hydrogen consumption.
Thermogravimetry and DTA analysis were done using DTG-60A (Shimadzu) by heating 30–60 mg of a sample in air to 700 °C at a rate of 10 °C min−1. The error of weight measurements was 1% for TGA and 1 μV for DTA.
2.3. Reactor set-up and catalytic tests
The experiments were performed in a fixed-bed reactor at 1 atm. The reactor was a cylindrical quartz tube, 370 mm in length, 15 mm in inner diameter, in which the sample was placed on a diffuser grid made of SiO2. A thermocouple pocket with a diameter of 5 mm was located in the center of the tube. The gases were fed into the reactor from top to bottom.Approximately 200 mg of the powder sample (the fraction below 50 μm) was initially heated in the air flow (100 ml min−1) to 550 °C. At this temperature, it was kept for 1 hour and then flushed with He (30 ml min−1) for 15 min. Hereafter, 40 vol% iso-C4H10 (99.5 mol% purity, KINEF refinery (Russia)) in He (99.999 vol% purity) was fed with a total flow of 50 ml min−1. After dehydrogenation, the reactor was purged by helium, followed by regeneration at 650 °C in the air for 30 min.
Separate experiments were performed in neat isobutane (20 ml min−1) with a sample weight of 1 g.
2.4. Analysis of reaction products and calculations
The reaction products were analyzed using gas chromatography GC-2010 Plus (Shimadzu) equipped with a FID and a capillary column (Supelco Petrocol DH (ID: 24160, 100 m × 0.25 mm, film thickness 0.5 μm, stationary phase: nonpolar bonded poly(dimethylsiloxane)). Helium was used as the carrier gas (100 ml min−1). Analysis was performed at the column temperature of 35 °C for 15 min followed by heating to 55 °C at a rate of 1 °C min−1. Processing of chromatograms was performed by Dragon DHA software, which calculates the composition of hydrocarbons in mol, vol and wt%. The molar fraction of hydrogen was considered to be equal to the sum of C4-alkenes according to the stoichiometry of dehydrogenation. Then the gas mixture was normalized to 100%.Isobutane conversion X (%), the yield of isobutene Y (mol%) and selectivity to isobutene S (mol%) were calculated as:
 | (1) |
 | (2) |
 | (3) |
The yields of methane and propene were calculated similarly, only dividing them by two, since one molecule of isobutane gives two molecules: methane and propene.
To calculate the amount of coke deposited on the sample, the volume of COx gases released during regeneration was recorded with a TEST gas analyzer (BonAir Ltd, Russia) equipped with an infrared sensor. The carbon mass balance closure was 99.5–100%.
2.5. Accelerated coking
A schematic representation of the experimental technique is shown in Fig. 1. An industrial chromia–alumina catalyst (2 g) was loaded in the main part of the reactor to obtain a large amount of isobutene for accelerated coking. A mixture of quartz with the test sample (0.5 g) was placed downstream the diffuser grid.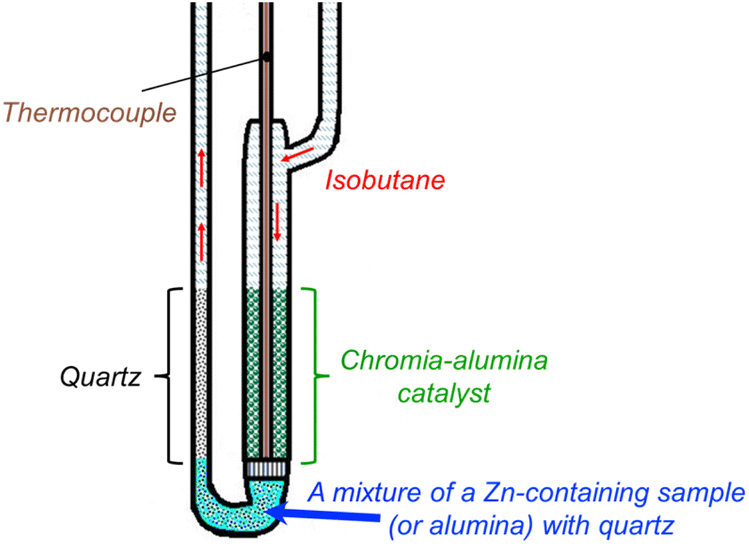 | ||
| Fig. 1 Schematic representation of the reactor with two catalytic layers for accelerated coking. | ||
The reactor was heated to 550 °C in a flow of air and kept at this temperature for 1 hour. After purging with an inert gas, neat isobutane was fed to the inlet at a volumetric flow rate of 20 ml min−1. The duration of the process was 50 min at a temperature in the layer of a chromia–alumina catalyst of 550 °C. Presence of this made it possible to subject the test sample to a mixture of isobutane with isobutene because selectivity to isobutene is high and the fraction of by-products is insignificant.
3. Results and discussion
3.1. Characterization
 | ||
| Fig. 2 XRD patterns of synthesized materials calcined at 700 °C (where ○ – ZnAl2O4 JCPDS card no. 290967; * – ZnO JCPDS card no. 193696). | ||
| N | Sample | Phase composition (Rietveld refinement), mol% | D (ZnAl2O4), nm | D (ZnO), nm | Reliability factorsc (Rwp/Re) | |
|---|---|---|---|---|---|---|
| ZnO | ZnAl2O4 | |||||
| a Hereinafter referred as ZnAl2O4. b Hereinafter referred as 3.5ZnO–96.5ZnAl2O4. c The ratio of the weighted (Rwp) and expected (Re) R-factors characterizes goodness of fit, if the value is equal to one or constant the refinement procedure is complete. | ||||||
| 1 | ZnAl2O4 no. 2a | 0.0 | 100.0 | 11.3 | — | 1.5 |
| 2 | ZnAl2O4 no. 1b | 3.5 | 96.5 | 11.2 | 29.0 | 1.8 |
| 3 | 10ZnO–90ZnAl2O4 | 9.2 | 90.8 | 11.5 | 22.3 | 1.7 |
| 4 | 20ZnO–80ZnAl2O4 | 18.2 | 81.8 | 12.5 | 18.5 | 1.6 |
| 5 | 30ZnO–70ZnAl2O4 | 23.5 | 76.5 | 11.1 | 15.9 | 1.6 |
| 6 | 40ZnO–60ZnAl2O4 | 35.5 | 64.5 | 11.0 | 18.3 | 1.6 |
Zinc oxide reflections are visible in XRD patterns for all materials except ZnAl2O4. Therefore, in the latter case, it was not possible to apply the Rietveld refinement. In general, considering the errors of the method, a good agreement between the expected and the measured phase composition was achieved. The data on the crystallinity of ZnAl2O4 are in agreement with the literature, reporting that ZnAl2O4 obtained by the solvothermal method and calcined at the same temperature, has the mean crystallite size of ca. 11 nm.14h Most likely, the mean size of the spinel crystallites does not change with increasing ZnO content, and the value obtained at 20 mol% of ZnO is an outlier.
Regarding the mean size of ZnO crystallites, the obtained data are in some contradiction with the data given in ref. 26, where with an increase of ZnO content from 20 to 40 mol% in the ZnO–ZnAl2O4 nanocomposites (prepared by SCS assisted with microwaves), the mean nanocrystallite size increases from ca. 22 to 28 nm. In this study, a decrease in the size of the ZnO crystallites can be a result of another fuel applied during synthesis by SCS, as the fuel type plays an important role.23a From the physical point of view, the difference between the used fuels is the standard enthalpy of combustion,27 resulting in variations of the heat supply during their combustion and subsequently in the particle size of the synthesized materials. Moreover, a very high calcination temperature (1000 °C) can be a reason for the difference in the mean crystallite size, as in the case of ref. 26.
EDS analysis was performed to determine the elemental composition of the obtained nanocomposites with the results presented in Table 2. According to these data, the Zn/Al atomic ratios are slightly higher than the expected ones, which can be explained by a low depth of microanalysis (0.5–1 μm) and a large difference in the atomic masses of the elements. EDS mapping analysis displayed in Fig. 3 indicates a uniform distribution of Zn and Al across the surface.
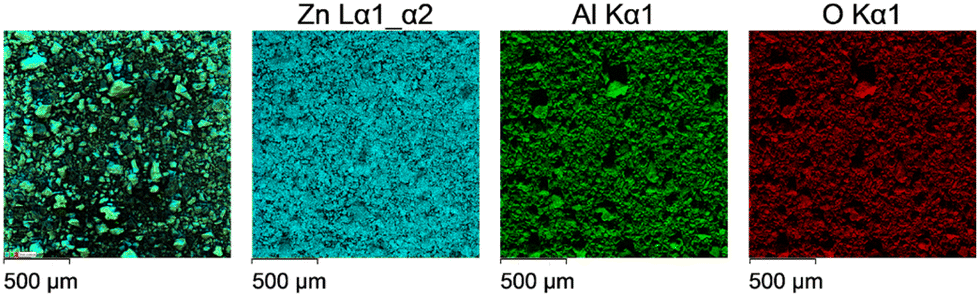 | ||
| Fig. 3 Energy-dispersive X-ray spectroscopy elemental maps (Zn, Al and O) for 30ZnO–70ZnAl2O4. | ||
| N | Sample | Element composition (according to EDS analysis)a, at% | Zn/Al atomic ratio | ||
|---|---|---|---|---|---|
| Al | O | Zn | |||
| a Data were obtained using Tescan microscope. | |||||
| 1 | ZnAl2O4 | 24.38 | 60.92 | 14.71 | 1:1.66 |
| 2 | 3.5ZnO–96.5ZnAl2O4 | 23.97 | 63.11 | 12.91 | 1:1.86 |
| 3 | 5ZnO–95ZnAl2O4 | 24.91 | 59.31 | 15.78 | 1:1.58 |
| 4 | 10ZnO–90ZnAl2O4 | 24.08 | 59.80 | 16.13 | 1:1.49 |
| 5 | 20ZnO–80ZnAl2O4 | 23.04 | 62.11 | 14.85 | 1:1.55 |
| 6 | 30ZnO–70ZnAl2O4 | 22.62 | 59.24 | 18.14 | 1:1.25 |
 | ||
| Fig. 4 SEM images of the synthesized materials. Top images (a–c): secondary electron analysis – topography; rest images (d–i): backscattered electron analysis – composition. | ||
According to SEM data, some of the randomly selected spherical ZnO particles of ZnAl2O4 are ca. 10 nm, which tended to agglomerate to diminish the total surface free energy and form a stable system.28 Similar data on the morphology of ZnO–ZnAl2O4 nanocomposites prepared by the same method were obtained in ref. 28, namely ZnO nanopowders exhibited clusters of ultrafine particles, while flakes and plates of fine particles were present for ZnAl2O4 nanopowders. In addition, as in the current study, with increasing ZnO loading up to 30 mol%, zinc oxide particles are distributed across the nanocomposite more uniformly compared to a case with a lower ZnO content (20 mol%).
Based on the temperature profiles obtained in ref. 29, the combustion temperature during synthesis of ZnO–ZnAl2O4 nanocomposites should not exceed 1000 °C. The currently accepted phase diagram for the ZnO–Al2O3 binary mixture indicates that the phase separation at the ZnO excess and 1000 °C leads to solid phases of zinc aluminate and zincite. The reason for formation of ZnO on the spinel surface may be that it is lighter than ZnAl2O4. After the spinel formation, residual gases from fuel decomposition and water vapor could push the oxide to the surface. Compared to other material preparation methods, it is unlikely that impregnation of the spinel with such large amounts of ZnO is feasible or that zinc oxide particles can be formed on the surface by coprecipitation.
Textural characteristics for the obtained samples summarized in Table 3 illustrate that with an increase in the ZnO content, a decrease in the specific surface area and the total pore volume was observed. The maxima of specific surface area (Ssp.) and the total pore volume are ca. 60 m2 g−1 and 0.2 cm3 g−1, respectively. In general, these values exceed the data obtained for samples prepared by traditional methods. For example, ZnAl2O4 obtained by coprecipitation had 40 m2 g−1 and 0.12 cm3 g−1,14g while Ssp. is 50 m2 g−1 for ZnAl2O4 obtained by the sol–gel method.14l On the other hand, it is possible to achieve a very high specific surface area for ZnAl2O4 reaching 150 m2 g−1 by solvothermal synthesis at 700 °C.30 However, this method is characterized by extremely difficult synthesis conditions (pressure of 25 bar) and high costs of the reagents.
| N | Sample | BET surface area, m2 g−1 | Pore volume, cm3/g | Pore size, nm |
|---|---|---|---|---|
| 1 | ZnAl2O4 | 59 | 0.18 | 12.4 |
| 2 | 5ZnO–95ZnAl2O4 | 55 | 0.20 | 14.2 |
| 3 | 10ZnO–90ZnAl2O4 | 46 | 0.17 | 14.6 |
| 4 | 20ZnO–80ZnAl2O4 | 45 | 0.19 | 17.3 |
| 5 | 30ZnO–70ZnAl2O4 | 40 | 0.12 | 11.9 |
| 6 | 40ZnO–60ZnAl2O4 | 37 | 0.12 | 12.6 |
The adsorption isotherms obtained at the temperature of liquid nitrogen (77 K) and the pore size distribution for these samples are displayed in Fig. 5. According to the IUPAC classification, nitrogen adsorption–desorption isotherms for the nanocomposites are unambiguously classified as type IVa, which is characteristic of materials with a mesoporous pore size.
 | ||
| Fig. 5 N2 adsorption–desorption isotherms and particle size distribution for the samples calcined at 700 °C. | ||
The isotherm hysteresis loops can be attributed to the H1 type for porous materials consisting of well-defined cylindrical-like pore channels or agglomerates of approximately uniform spheres. All obtained composites exhibit a broad monomodal pore distribution predominantly in the mesoporous range (inset in Fig. 5), while the pore size distribution for ZnAl2O4 is somewhat closer to the bimodal one with maxima at ca. 10 and 15 nm. It is possible that the sample particles per se are non-porous, while small volumes of mesopores are formed due to secondary porosity, namely, aggregation of small crystalline particles. According to the t-plot analysis, the samples do not contain micropores.
 | ||
| Fig. 6 NH3-TPD profiles for the samples calcined at 700 °C (a) and acidity dependence on the ZnO content (b and c). The acid site density was calculated as the acidity in μmol g−1 divided by the specific surface area from Table 3. The specific surface area of spinel was calculated as 59 × (mol% of ZnAl2O4 in ZnO–ZnAl2O4)/100. | ||
Total acidity of ZnAl2O4 in μmol g−1 obtained in this study is somewhat lower than that of zinc aluminate prepared by the hydrothermal method.32 Such acidity difference could be caused by the presence of small amounts of ZnO (according to SEM data) and a higher calcination temperature compared to the prior work.32
With increasing ZnO loading, an increase in the acid site density is also observed (Fig. 6c), which is associated with a faster decrease in the specific surface area compared to the total acidity (in percentage from the initial value). A correlation analysis of the obtained dependences showed that zinc oxide, being itself acidic, gradually fills the surface of the spinel (Fig. 6c, blue dots). On the other hand, when considering the entire surface of the sample, it was found that acidity reaches a constant level for 30–40 mol% of ZnO (Fig. 6c, red dots), which means that there is a layering of zinc oxide particles on top of each other.
In order to quantify acid sites of varying strength, the TPD profiles were fitted with four Gaussian peaks (Fig. S2†). A more detailed analysis of the changes in the acid sites shows that the amount of weak sites decreases until reaching 20 mol% ZnO, not changing thereafter (Table 4 and Fig. S3a†). Medium acid sites, on the contrary, do not change at a low content of ZnO starting to decrease at 30 mol% of zinc oxide. The dependence of strong acid sites on the zinc oxide content is not very clear; and it is even possible that their amounts does not change. Regarding the acid sites density, it is challenging to single out any features, because there is a trend of its increase for all types of sites (Fig. S3b†). Although it can be noted that a high density of weak Lewis acid sites on the surface of the catalyst is beneficial for dehydrogenation reaction and could suppress cracking avoiding thereby carbon deposition.
| Sample | Weak acid sites (μmol g−1) | Medium acid sites (μmol g−1) | Strong acid sites (μmol g−1) |
|---|---|---|---|
| According to NH3-TPD. | |||
| ZnAl2O4 | 54.3 | 57.8 | 37.8 |
| 10ZnO–90ZnAl2O4 | 52.1 | 60.9 | 30.2 |
| 20ZnO–80ZnAl2O4 | 43.6 | 57.9 | 33.5 |
| 30ZnO–70ZnAl2O4 | 42.9 | 46.0 | 39.9 |
| 40ZnO–60ZnAl2O4 | 43.8 | 47.6 | 28.6 |
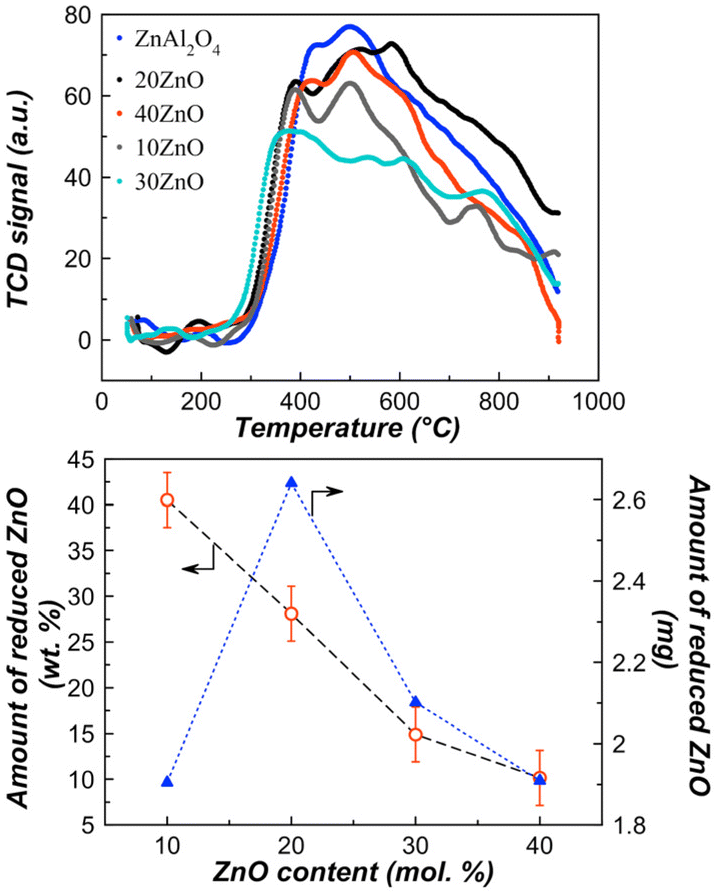 | ||
| Fig. 7 H2-TPR profiles for the samples calcined at 700 °C (top) and dependence of the amount of reduced ZnO on the ZnO content (bottom). | ||
The assumption about reduction of zinc oxide to zero-valent zinc is confirmed by a decrease in the zinc content in the samples after TPR, in particular, for 20ZnO–ZnAl2O4 it decreased by ca. 21 at% compared to a freshly prepared sample. This means that a significant part of Zn0 left the sample at high temperatures due to zinc volatility.
3.2. Activity in isobutane dehydrogenation
Dependence of the isobutane conversion on the ZnO content is shown in Fig. 8 and S4.† Conversion values can be reproduced with minor deviations after 50 min of the reaction and regeneration of samples under air (Fig. S4a†). These deviations are related to precision of gas sampling and fluctuations in the gas flow. Fig. S4b† shows a slight difference in the volume fraction of hydrogen during isobutane dehydrogenation in the first and second cycles. | ||
| Fig. 8 Isobutane conversion depending on the ZnO content, obtained after 10, 30 and 50 min of dehydrogenation in the 1st cycle. Reaction conditions: 550 °C, 20 ml min−1 of isobutane:He = 40:60. | ||
The initial activities of all studied samples are higher than after 30 and 50 minutes of dehydrogenation (Fig. 8), clearly reflecting deactivation. The timing of isobutane test data namely 10, 30, 50 min was selected to harmonize sampling with the GC analysis, i.e. the retention times of the reaction products. As can be seen in the activity vs. time on stream profiles (Fig. 8), the activity decline is slowing down still not reaching the steady state after 50 min.
A sample containing 20 mol% ZnO has a more significant drop in conversion after 30 min. Obviously, for this case the deactivation constant (kd), determined by fitting isobutane conversion dependence on TOS in accordance with the equation:
| a = a + a(t)e−kdt), | (4) |
At the same time, differences in conversion between the samples are significantly leveled out and no longer depend on the ZnO content after 50 min. Deactivation, in addition to coking, can also be associated with a partial reduction of Zn2+ sites.
Contrary to all other samples synthesized by the same method, composites with 30 and 40% of ZnO exhibited low isobutane conversion, while neat zinc oxide and alumina (not shown in Fig. 8 and S4,† initial conversion was ca. 2%) are practically inactive.
It can be noted that dependence of conversion on the ZnO loading after 10 min of dehydrogenation passes through a maximum, being linear below 20 mol% ZnO. A similar change in the isobutane conversion depending on the ZnO content was observed for a mixture of ZnO and Ga2O3.33 With an increase in the ZnO content, conversion was first elevated and then decreased when the ZnO/Ga2O3 molar ratio exceeded 50.
To explain maximum activity depending on ZnO loading, it is necessary to understand the nature of active sites. The following types of sites are supported to be present on the catalyst surface: Zn2+ and Al3+ of ZnAl2O4, Zn2+ from ZnO, according to ref. 14e. There are also oxygen vacancies, which can affect the process. According to the SEM results, with an increase in the ZnO content, the spinel surface is gradually filled up, which means that with a certain excess of ZnO, the Al3+ sites overlap resulting in less oxygen vacancies. It has been suggested that proximity of Zn2+ and Al3+ sites plays an important role, since the basic Al-bound oxygen is essential for the heterolytic cleavage of the C–H bond, according to theoretical calculations.13c,34 In a recent work,8a it was shown that ZnAl2O4 with tetrahedral Zn2+ sites located in close proximity to Al sites gives an increased activity and selectivity in propane dehydrogenation compared to 30ZnO/SiO2 reference.
Thus, a following explanation can be suggested for the observed activity dependence on the ZnO loading. Addition of ZnO to ZnAl2O4 promotes an increase in isobutane dehydrogenation activity until spinel covering by agglomerated particles reaches a “critical” value. Moreover, an increase in ZnO loading increases the weak acid site density positively affecting dehydrogenation.
On the other hand, a degree of zinc oxide reduction is decreasing with more ZnO, which can have a negative effect, contributing less to removal of hydrogen from the reaction mixture. Although isobutane conversion does not reach equilibrium values (ca. 52% at 550 °C),35 it can be concluded that removal of hydrogen from the reaction mixture should promote the forward reaction and an increase in isobutane conversion.
A high initial activity may be related to a high initial concentration of Zn2+ active sites, which are gradually reduced under the reaction conditions. According to the TPR data (Fig. 7, bottom), the largest reduction of ZnO (in absolute value) occurred in a sample containing 20 mol% zinc oxide, therefore this catalyst exhibited a faster decrease in conversion. Because ZnO is only partially reduced, the residual zinc contributes directly to conversion after 50 minutes of the reaction. A similar effect was also found in ref. 13d, where the authors noted that catalysts can reach the same isobutane conversion because the concentration of Zn2+ reaches the same value in all samples. In this case, the stationary concentration of Zn2+ sites was determined both by disappearance of the existing Zn2+ sites due to their reduction and formation of new ones due to interactions of ZnO particles with the support surface.
Because zinc tends to volatilize from the sample after reduction, the zinc content after dehydrogenation was also measured. According to the data presented in Table 5, it can be concluded that after 50 min of dehydrogenation, the Zn content is close to the initial one. However, high-temperature regeneration with air reduces the zinc content by ca. 10 at%. It can be hypothesized that lowering the regeneration temperature can minimize this effect.
| N | Conditions | Areas | O k | Al k | Zn l |
|---|---|---|---|---|---|
| Data were obtained using JEOL microscope. | |||||
| Initial | — | 1 | 54.0 | 27.5 | 18.5 |
| 2 | 54.7 | 26.5 | 18.8 | ||
| 1 | After 50 min of isobutane dehydrogenation at 550 °C | 1 | 53.3 | 28.7 | 18.0 |
| 2 | 53.8 | 27.7 | 18.5 | ||
| 2 | After 50 min of isobutane dehydrogenation at 550 °C and 30 min air regeneration at 650 °C | 1 | 63.8 | 21.2 | 15.0 |
| 2 | 61.2 | 21.8 | 17.0 | ||
| 3 | After H2-TPR | 1 | 52.3 | 32.4 | 15.4 |
| 2 | 55.8 | 30.3 | 13.9 | ||
3.3. Selectivity and the reaction scheme
Experimentally obtained selectivity to isobutene is quite high, being ca. 90% for both ZnO-containing composites and ZnAl2O4, indicating that spinel itself is active for dehydrogenation (Fig. 9a). With an increase of the ZnO content, selectivity to isobutene, similar to isobutane conversion, first was elevated and then decreased once the ZnO content ratio reached 20 mol%.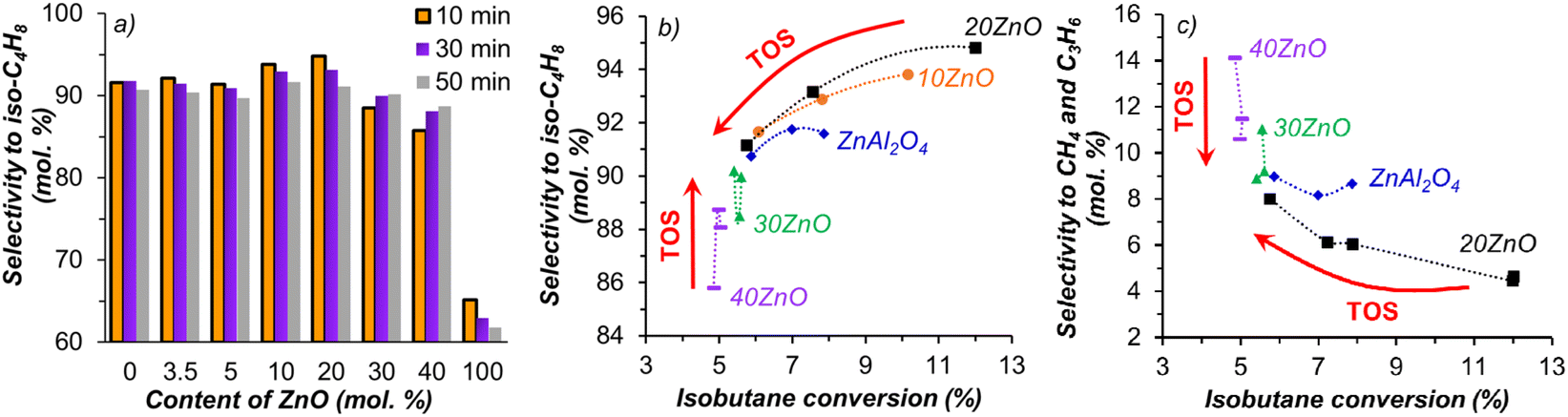 | ||
| Fig. 9 Selectivity to isobutene depending on the ZnO content, obtained after 10, 30 and 50 min of dehydrogenation in the 1st cycle (a); selectivity to iso-C4H8 (b) and selectivity to CH4 and C3H6 (c) versus isobutane conversion at different reaction time. Reaction conditions: 550 °C, 20 ml min−1 of isobutane:He = 40:60. TOS – time-on-stream. | ||
Less significantly than conversion, selectivity, nevertheless, decreases with increasing time-on-stream, which can be related to the loss of Zn mentioned above. Only for two samples containing 30 and 40% ZnO, selectivity was marginally dependent on conversion (Fig. 9b). It should be noted that the use of fine particles (<50 μm) ensured absence mass and heat transfer processes limitations. Curves presented in Fig. 9b are typical for parallel-consecutive reactions, and were reported earlier for other systems.35,36 However, in contrast to this work, deactivation was not observed for gallia/alumina catalysts and in the presence of chromia/alumina catalysts selectivity to isobutene only slightly decreased with isobutane conversion from 10 to 40%.36 Selectivity to methane and propene was changing with isobutane conversion (Fig. 9c) in an opposite way to isobutene selectivity, which indicates that dehydrogenation and cracking are parallel reactions.
For explaining the obtained regularities over the ZnO–ZnAl2O4 nanocomposites, a mechanistic scheme of isobutane dehydrogenation, shown in Fig. 10, was proposed. As noted above, the combination and proximity of Zn2+ (especially dispersed, since it is well reduced by hydrogen) and aluminum cations improve the course of dehydrogenation (path a in Fig. 10).
 | ||
| Fig. 10 A scheme of isobutane dehydrogenation on ZnO–ZnAl2O4 nanocomposites. Unstable compounds are presented in brackets. | ||
ZnAl2O4 and the samples containing 10 and 20 mol% ZnO with a relatively high initial isobutane conversion and selectivity to isobutane are also characterized by more enhanced coke formation (Fig. 11a). Compared to samples with a higher ZnO content, these samples also have stronger surface acidity, which favor alkene adsorption, increasing its residence time and surface concentration.36 As a consequence, adsorbed isobutene molecules can interact with each other to form carbonaceous deposits. All strong acid sites (created apparently by aluminum cations) are gradually covered by coke causing a decrease in conversion (paths c and d in Fig. 10). In addition, Al3+ sites apparently promote cracking of isobutane to methane and propene (path b in Fig. 10), since selectivity to the latter decreases with an increasing ZnO content (Fig. 11b). In the absence of uncovered Al3+ sites, Zn2+ also promotes isobutane cracking.
 | ||
| Fig. 11 Carbon content on the samples after 50 min of dehydrogenation (a). Selectivity to methane and propene and their ratios (b), depending on the ZnO content, obtained after 10 min of dehydrogenation in the 1st cycle. Reaction conditions: 550 °C, 20 ml min−1 of isobutane:He = 40:60. | ||
ZnAl2O4 and the samples containing 10 and 20 mol% ZnO with a relatively high initial isobutane conversion and selectivity to isobutane are also characterized by more enhanced coke formation (Fig. 11a). Compared to samples with a higher ZnO content, these samples also have stronger surface acidity, which favor alkene adsorption, increasing its residence time and surface concentration.36 As a consequence, adsorbed isobutene molecules can interact with each other to form carbonaceous deposits. All strong acid sites (created apparently by aluminum cations) are gradually covered by coke causing a decrease in conversion (paths c and d in Fig. 10). In addition, Al3+ sites apparently promote cracking of isobutane to methane and propene (path b in Fig. 10), since selectivity to the latter decreases with an increasing ZnO content (Fig. 11b). In the absence of uncovered Al3+ sites, Zn2+ also promotes isobutane cracking.
At a low conversion, when the concentration of the intermediate product (isobutene) is low, side reactions give the main contribution to the dependence of selectivity on conversion. This is confirmed by a selectivity increase to methane and propene, when the ZnO content increases from 20 to 100% ZnO (Fig. 11b). Moreover, the propene to methane ratio was almost constant below 20 mol% ZnO (Fig. 11b), increasing with a further increase in the ZnO loading. A high yield of propene compared to other by-products of isobutane dehydrogenation was also found for ZnO–ZrO2 systems.12 It can be assumed that isobutene dimerizes and immediately cracks with the formation of propene and isoamylene. The latter also leads to propene during hydrogenolysis in lieu of no strong acid sites on the surface required for its strong adsorption (paths e–f in Fig. 10). This assumption is in line with weak acidity of zinc oxide exhibiting mainly Brønsted acid sites, which promote isobutene dimerization.37
3.4. Coking tests
In experiments with the dilution presented above, the amount of coke formed on the sample surface was not large enough; therefore, additional experiments to explore the coking behaviour with a longer contact time (in neat isobutane) were carried out. Alumina was chosen as a reference for coking tests, since it is a traditional support for dehydrogenation catalysts, and the obtained composites containing spinel can also be considered as supports. Moreover, recently it has been argued that unmodified bulk alumina can be used as a potential efficient catalyst for dehydrogenation of isobutane11a and propane.11b Alumina was compared with the sample showing the best conversion and selectivity – 20ZnO–ZnAl2O4.Alumina itself did not display high activity (change in conversion from 10 to 50 min from 5.4 to 3.7%) compared to 20ZnO–ZnAl2O4 with an initial conversion value of 33.6% (at 10 min), which decreased by 36.6% at 50 min of time-on-stream. After dehydrogenation of neat isobutane (in the absence of a chromia-alumina catalyst), it was not possible to detect an exothermic peak of coke combustion for alumina (Fig. 12c), although the color of the sample (light brown) and a weight loss (Fig. 12a and b) indicated its presence. Such low coke content can be explained by low conversion and low yields of alkenes, which are the main precursors of coke.
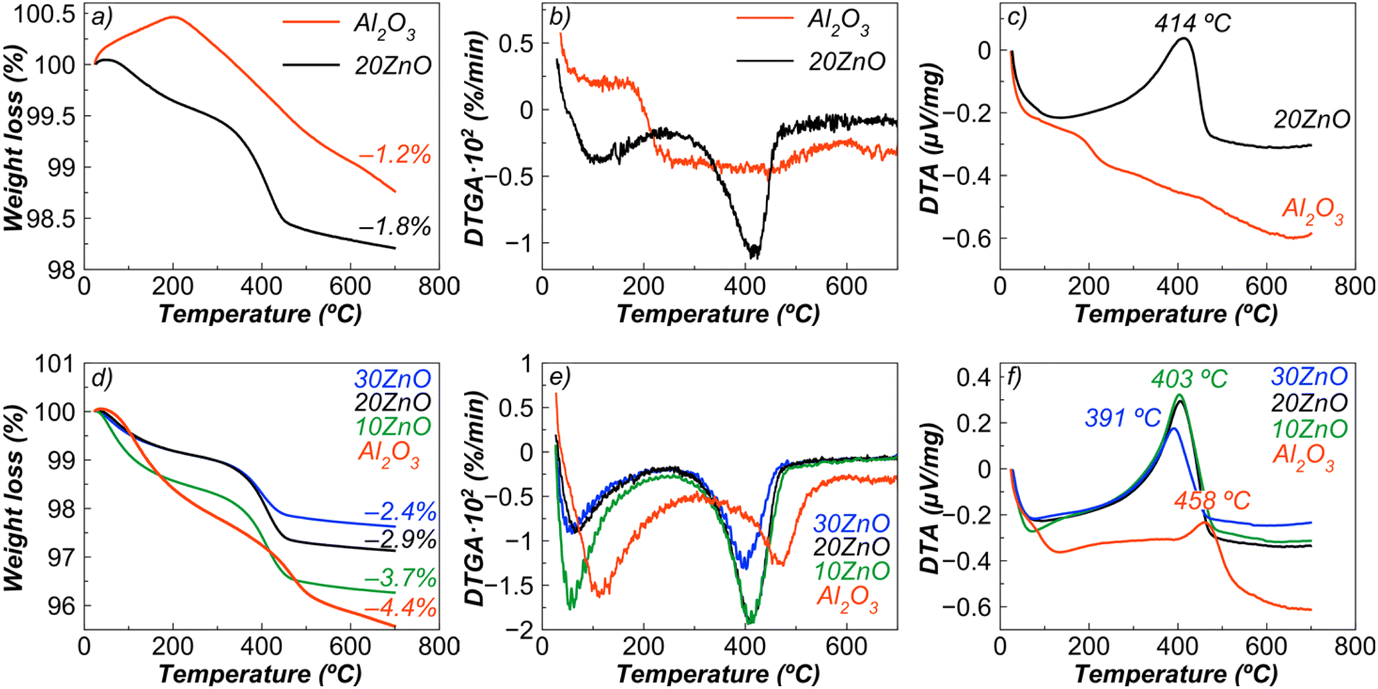 | ||
| Fig. 12 Differential thermal and thermogravimetric analysis data of γ-Al2O3 (obtained by calcining pseudo-boehmite (from Catapal) at 700 °C, specific surface area is 212 m2 g−1) and ZnO–ZnAl2O4 composites (designated as 10ZnO, 20ZnO, 30ZnO), obtained in the absence (a, b and c) and presence (d, e and f) of industrial chromia–alumina catalyst. Reaction conditions: 550 °C, 20 ml min−1 of isobutane. | ||
A different situation was seen for the ZnO-containing sample. This sample has a slightly higher coke content than alumina (Fig. 12a) implying better resistance to coke formation taking also into account that a large content of alkenes was passing through the catalyst bed because of a relatively high conversion (33.6–12.3%) and high selectivity (86.3–92.2 mol%) compared to alumina.
To create equal conditions for coking of Zn-containing composites and alumina, a highly active and selective industrial catalyst based on chromia was introduced in the reactor. Selectivity to isobutene, as a rule, for the chromia–alumina system reaches almost 100% (at 550 °C); therefore, it can be assumed that at conversion of ca. 50%, coking will be carried out with a 50% isobutane/50% isobutene mixture.
Fig. 12d illustrates that alumina tends to carbonize more than ZnO–ZnAl2O4 nanocomposites, producing more condensed and graphite-like coke. In addition, when the coked alumina or spinel with a low ZnO content (10 mol%) is heated to 250 °C, it released a larger amount of easily desorbed reaction products. As can be seen from Fig. 12d, addition of ZnO reduced the coke deposition on the tested sample.
All results from the catalytic and coking tests show that ZnO–ZnAl2O4 composites can successfully replace alumina supports for dehydrogenation reactions, because these systems tend to form isobutene with high selectivity and resist coke formation.
Nevertheless, conversion values obtained for ZnO–ZnAl2O4 systems do not exceed those given in the literature for some other systems under the same experimental conditions, in particular Cr10Zr90Ox.38 In terms of the space time yield (STY) of isobutene formation, performance of 20ZnO–80ZnAl2O4 is superior to an industrially relevant reference material – Cr2O3/Al2O339 (Table S1†).
4. Conclusions
In the present study, dehydrogenation of isobutane to isobutene in the presence of nanocomposites based on ZnAl2O4 spinel with an excess of zinc oxide was investigated. Such materials were synthesized by the urotropin-nitrate solution combustion synthesis, which has allowed an easy control of the phase composition.ZnAl2O4 spinel containing traces of ZnO exhibits moderate activity in isobutane dehydrogenation. Among the nanocomposites, a sample containing 20 mol% ZnO exhibited maximum isobutane conversion and selectivity to isobutene. At this ZnO concentration, Al3+ sites on the surface are still accessible, as shown by SEM, being necessary for dehydrogenation to proceed.
An increase in ZnO loading decreases acidity positively affecting dehydrogenation, diminishing at the same time reducibility of surface zinc oxide, which can have a negative effect, contributing to a lesser extent to the removal of hydrogen from the reaction mixture. It is shown that, depending on the ZnO content, the type of reactions occurring in isobutane dehydrogenation changes. The predominance of parallel reactions is replaced by the predominance of consecutive ones with an increase in the content of zinc oxide up to 40 mol%.
For the sake of a fair comparison of various catalysts in the kinetic regime differential conditions were used thus the isobutane conversion for all tested samples was only ca. 10% at 550 °C. Not surprisingly the conversion per se does not exceed the dehydrogenation performance of other catalytic systems described in the literature. Nevertheless, compared with other materials, such as alumina, which is widely used as a support for dehydrogenation catalysts, the obtained ZnO–ZnAl2O4 nanocomposites are characterized by high selectivity to isobutene and resistance to coke formation even under severe dehydrogenation conditions (at a high contact time).
Author contributions
AM – conceptualization, investigation, visualization, writing – original draft. ShO – formal analysis, investigation, visualization. AN – investigation. VP – conceptualization. DM – conceptualization, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to research assistant Kirill Martinson for EDS analysis performed at the facilities of the Engineering Centre of St Petersburg State Institute of Technology (Technical University); research assistant Maria Chebanenko for N2 physisorption measurements at the Laboratory of New Inorganic Materials of Ioffe Institute.Notes and references
- D. F. Rodríguez-Vallejo, G. Guillén-Gosálbez and B. Chachuat, ACS Sustainable Chem. Eng., 2020, 8, 3072–3081 CrossRef.
- (a) S. R. Docherty, L. Rochlitz, P.-A. Payard and C. Copéret, Chem. Soc. Rev., 2021, 50, 5806–5822 RSC; (b) Z. Nawaz, Rev. Chem. Eng., 2015, 31, 413–436 CAS; (c) T. Ridha, Y. Li, E. Gençer, J. J. Siirola, J. T. Miller, F. H. Ribeiro and R. Agrawal, Processes, 2016, 6, 139 CrossRef; (d) J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- Z. Song, J. Wang, X. Zhang, F. Liu and L. Zhang, Mol. Catal., 2021, 515, 111944 CrossRef CAS.
- Y. Wang, H. Su, Y. Gu, X. Song and J. Zhao, OncoTargets Ther., 2017, 10, 4065–4079 CrossRef PubMed.
- A. N. Matveyeva, N. A. Zaitseva, N. A. Pakhomov and D. Yu. Murzin, Catal. Sci. Technol., 2020, 10, 7719–7723 RSC.
- Q. Zhu, H. Zhang, S. Zhang, G. Wang, X. Zhu and C. Li, Ind. Eng. Chem. Res., 2019, 58, 7834–7843 CrossRef CAS.
- G. Wang, C. Gao, X. Zhu, Y. Sun, C. Li and H. Shan, ChemCatChem, 2014, 6, 2305–2314 CrossRef CAS.
- (a) M. Nadjafi, A. M. Kierzkowska, A. Armutlulu, R. Verel, A. Fedorov, P. M. Abdala and C. R. Müller, J. Phys. Chem. C, 2021, 125, 14065–14074 CrossRef CAS; (b) S. Arndt, B. Uysal, A. Berthold, T. Otrebma, Y. Aksu, M. Driess and R. Schomäcker, J. Nat. Gas Chem., 2012, 21, 581–594 CrossRef CAS.
- T. P. Otroshchenko, V. A. Kondratenko, U. Rodemerck, D. Linke and E. V. Kondratenko, Catal. Sci. Technol., 2017, 7, 4499–4510 RSC.
- S. Vaezifar, H. Faghihian and M. Kamali, Korean J. Chem. Eng., 2011, 28, 370–377 CrossRef CAS.
- (a) U. Rodemerck, E. V. Kondratenko, T. Otroshchenko and D. Linke, Chem. Commun., 2016, 52, 12222 RSC; (b) P. Wang, Z. Xu, T. Wang, Y. Yue, X. Bao and H. Zhu, Catal. Sci. Technol., 2020, 10, 3537 RSC.
- Y. Liu, C. Xia, Q. Wang, L. Zhang, A. Huang, M. Ke and Z. Song, Catal. Sci. Technol., 2018, 8, 4916–4924 RSC.
- (a) N. M. Schweitzer, B. Hu, U. Das, H. Kim, J. Greeley, L. A. Curtiss, P. C. Stair, J. T. Miller and A. S. Hock, ACS Catal., 2014, 4, 1091–1098 CrossRef CAS; (b) S. Han, D. Zhao, T. Otroshchenko, H. Lund, U. Bentrup, V. A. Kondratenko, N. Rockstroh, S. Bartling, D. E. Doronkin, J.-D. Grunwaldt, U. Rodemerck, D. Linke, M. Gao, G. Jiang and E. V. Kondratenko, ACS Catal., 2020, 10, 8933–8949 CrossRef CAS; (c) G. Liu, L. Zeng, Z.-J. Zhao, H. Tian, T. Wu and J. Gong, ACS Catal., 2016, 6, 2158–2162 CrossRef CAS; (d) A. I. Serykh and Y. A. Agafonov, Mol. Catal., 2020, 493, 111055 CrossRef CAS.
- (a) S. A. Bocanegra, A. Guerrero-Ruiz, S. R. de Miguel and O. A. Scelza, Appl. Catal., A, 2004, 277, 11–22 CrossRef CAS; (b) S. de Miguel, A. Ballarini and S. Bocanegra, Appl. Catal., A, 2020, 590, 117315 CrossRef; (c) B. K. Vu, M. B. Song, I. Y. Ahn, Y.-W. Suh, D. J. Suh, W.-I. Kim, H.-L. Koh, Y. G. Choi and E. W. Shin, Appl. Catal., A, 2011, 400, 25–33 CrossRef CAS; (d) W. Walerczyk, M. Zawadzki and J. Okal, Appl. Surf. Sci., 2011, 257, 2394–2400 CrossRef CAS; (e) A. Ye, Z. Li, J. Ding, W. Xiong and W. Huang, ACS Catal., 2021, 11, 10014–10019 CrossRef CAS; (f) M. Nilsson, K. Jansson, P. Jozsa and L. J. Pettersson, Appl. Catal., B, 2009, 86, 18–26 CrossRef CAS; (g) A. D. Ballarini, S. A. Bocanegra, A. A. Castro, Se. R. de Miguel and O. A. Scelza, Catal. Lett., 2009, 129, 293–302 CrossRef CAS; (h) T. Sirikajorn, O. Mekasuwandumrong, P. Praserthdam, J. G. Goodwin Jr. and J. Panpranot, Catal. Lett., 2008, 126, 313–318 CrossRef CAS; (i) H. Grabowska, M. Zawadzki and L. Syper, Appl. Catal., A, 2004, 265, 221–327 CrossRef CAS; (j) L. Chen, X. Sun, Y. Liu, K. Zhou and Y. Li, J. Alloys Compd., 2004, 376, 257–261 CrossRef CAS; (k) J. Wrzyszcz, M. Zawadzki, A. Trzeciak and J. Ziółkowski, J. Mol. Catal. A: Chem., 2002, 189, 203–210 CrossRef CAS; (l) M. A. Valenzuela, J.-P. Jacobs, P. Bosch, S. Reijne, B. Zapata and H. H. Brongersma, Appl. Catal., A, 1997, 148, 315–324 CrossRef CAS; (m) G. Aguilar-Ríos, M. A. Valenzuela, H. Armendariz, P. Salas, J. M. Domínguez, D. R. Acosta and I. Schifter, Appl. Catal., A, 1992, 90, 25–34 CrossRef.
- N. J. van der Laag, M. D. Snel, P. C. M. M. Magusin and G. de With, J. Eur. Ceram. Soc., 2004, 24, 2417–2424 CrossRef CAS.
- (a) A. Chaudhary, A. Mohammad and S. M. Mobin, Mater. Sci. Eng., B, 2018, 227, 136–144 CrossRef CAS; (b) Y. Wu, J. Du, K.-L. Choy, L. L. Hench and J. Guo, Thin Solid Films, 2005, 472, 150–156 CrossRef CAS.
- (a) S. Suwanboon, P. Amornpitoksuk, T. Rattana and C. Randorn, Ceram. Int., 2020, 46, 21958–21977 CrossRef CAS; (b) T. K. Parya, R. K. Bhattacharyya, S. Banerjee and U. B. Adhikari, Ceram. Int., 2010, 36, 1211–1215 CrossRef CAS; (c) S. Farhadi and S. Panahandehjoo, Appl. Catal., A, 2010, 382, 293–302 CrossRef CAS.
- S. Ishii, T. Nakane, S. Uchida, M. Yoshida and T. Naka, J. Asian Ceram. Soc., 2018, 6, 7–12 CrossRef.
- (a) R. Ianos, S. Borcanescu and R. Lazau, Chem. Eng. J., 2014, 240, 260–263 CrossRef CAS; (b) C. T. Alves, A. Oliveira, S. A. V. Carneiro, A. G. Silva, H. M. C. Andrade, S. A. B. Vieira de Melo and E. A. Torres, Fuel Process. Technol., 2013, 106, 102–107 CrossRef CAS.
- (a) M. Shahmirzaee, M. S. Afarani, A. I. Nejhad and A. M. Arabi, Part. Sci. Technol., 2019, 37, 110–117 CrossRef CAS; (b) R. Yuvasravana, P. P. George and N. Devanna, Mater. Today: Proc., 2017, 4, 10664–10671 Search PubMed; (c) H. P. Macedo, R. L. B. A. Medeiros, A. L. Medeiros, A. A. S. Oliveira, G. P. Figueredo, M. A. F. Melo and D. M. A. Melo, Mater. Res., 2017, 20, 29–33 CrossRef; (d) M. Shahmirzaee, M. S. Afarani, A. M. Arabi and A. I. Nejhad, Res. Chem. Intermed., 2017, 43, 321–340 CrossRef CAS.
- Z. Li, S. Zhang and W. Lee, J. Eur. Ceram. Soc., 2007, 27, 3407–3412 CrossRef CAS.
- W. Mekprasart, K. Boonyarattanakalin, W. Pecharapa and K. N. Ishihara, Mater. Today: Proc., 2018, 5, 14126–14130 CAS.
- (a) F. Deganello and A. K. Tyagi, Prog. Cryst. Growth Charact. Mater., 2018, 64, 23–61 CrossRef CAS; (b) A. Varma, A. S. Mukasyan, A. S. Rogachev and K. V. Manukyan, Chem. Rev., 2016, 116, 14493–14586 CrossRef CAS PubMed.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
- M. Shahmirzaee, M. S. Afarani, A. M. Arabi and A. I. Nejhad, Res. Chem. Intermed., 2017, 43, 321–340 CrossRef CAS.
- T. S. Cam, T. A. Vishnievskaia and V. I. Popkov, Rev. Adv. Mater. Sci., 2020, 59, 131–143 CAS.
- S. Suwanboon, P. Amornpitoksuk and A. Sukolrat, Ceram. Int., 2011, 37, 1359–1365 CrossRef CAS.
- E. Leal, B. B. Dantas, L. S. Neiva, R. H. G. A. Kiminami and A. C. F. M. Costa, Mater. Sci. Forum, 2012, 727–728, 1260–1265 CAS.
- W. Staszak, M. Zawadzki and J. Okal, J. Alloys Compd., 2010, 492, 500–507 CrossRef CAS.
- J. P. H. Li, A. A. Adesina, E. M. Kennedy and M. Stockenhuber, Phys. Chem. Chem. Phys., 2017, 19, 26630–26644 RSC.
- J. Wrzyszcz, M. Zawadzki, J. Trawczyński, H. Grabowska and W. Miśta, Appl. Catal., A, 2001, 210, 263–269 CrossRef CAS.
- G. Wang, C. Li and H. Shan, Catal. Sci. Technol., 2016, 6, 3128–3136 RSC.
- C. Coperet, Chem. Rev., 2010, 110, 656–680 CrossRef CAS PubMed.
- A. N. Matveyeva, Sh. O. Omarov, D. A. Sladkovskiy and D. Y. Murzin, Chem. Eng. J., 2019, 372, 1194–1204 CrossRef CAS.
- T. Otroshchenko, V. Kondratenko, U. Rodemerck, D. Linke and E. V. Kondratenko, Catal. Sci. Technol., 2017, 7, 4499–4510 RSC.
- J. M. Fernández-Morales, E. Castillejos, E. Asedegbega-Nieto, A. B. Dongil, I. Rodríguez-Ramos and A. Guerrero-Ruiz, Nanomaterials, 2020, 10, 1235 CrossRef PubMed.
- T. Otroshchenko, J. Radnik, M. Schneider, U. Rodemerck, D. Linke and E. V. Kondratenko, Chem. Commun., 2016, 52, 8164–8167 RSC.
- A. N. Matveyeva, N. A. Zaitseva, P. Mäki-Arvela, A. Aho, A. K. Bachina, S. P. Fedorov, N. A. Pakhomov and D. Yu. Murzin, Ind. Eng. Chem. Res., 2018, 57, 927–938 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt02088b |
| This journal is © The Royal Society of Chemistry 2022 |