
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclometalated iridium complexes based on monodentate aminophosphanes†
Marco
Palmese
 ,
Jesús J.
Pérez-Torrente
and
Vincenzo
Passarelli
*
,
Jesús J.
Pérez-Torrente
and
Vincenzo
Passarelli
*
Departamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), Universidad de Zaragoza-CSIC, C/Pedro Cerbuna 12, ES-50009 Zaragoza, Spain. E-mail: passarel@unizar.es
First published on 25th July 2022
Abstract
Monodentate aminophosphanes HNP [NH(4-tolyl)PPh2] and SiMe3NP [SiMe3N(4-tolyl)PPh2] react with [Ir(μ-Cl)(cod)]2 affording tetra- or pentacoordinate complexes of formula [IrCl(L)n(cod)] (L = HNP, n = 1, 2; L = SiMe3NP, n = 1). The reaction of [IrCl(SiMe3NP)(cod)] with carbon monoxide smoothly renders [Ir(CO)3(SiMe3NP)2][IrCl2(CO)2]. The reaction of HNP or SiMe3NP with [Ir(CH3CN)2(cod)][PF6] yields the cyclometalated iridium(III)-hydride derivatives [IrH{κ2C,P-NR(4-C6H3CH3)PPh2}(cod)(CH3CN)][PF6] (R = H, SiMe3) as a result of the intramolecular oxidative addition of the tolyl C2–H bond to iridium. The straighforward formation of [IrH{κ2C,P-SiMe3N(4-C6H3CH3)PPh2}(cod)(CH3CN)]+ was observed when the reaction was monitored by NMR spectroscopy at 233 K, whereas a more complex reaction sequence was observed in the formation of [IrH{κ2C,P-NH(4-C6H3CH3)PPh2}(cod)(CH3CN)]+, including the formation of [IrH{κ2C,P-NH(4-C6H3CH3)PPh2}(HNP)(cod)]+ and [Ir(cod)(HNP)2]+. The “mixed” complex [IrH{κ2C,P-SiMe3N(4-C6H3CH3)PPh2}(HNP)(cod)]+ was obtained upon reaction of [IrH{κ2C,P-NH(4-C6H3CH3)PPh2}(cod)(CH3CN)][PF6] with SiMe3NP at 233 K. Finally, the reaction of [Ir(CH3CN)2(coe)2][PF6] with SiMe3NP or HNP resulted in the formation of [Ir(CH3CN)2(SiMe3NP)2][PF6] and [IrH{κ2C,P-NH(4-C6H3CH3)PPh2}(HNP)2(CH3CN)][PF6], respectively. Both the OC-6-35 and the OC-6-52 isomers of [IrH{κ2C,P-NH(4-C6H3CH3)PPh2}(HNP)2(CH3CN)]+ – featuring facial and meridional dispositions of the phosphorus atoms, respectively – were isolated depending on the reaction solvent. Several compounds described herein catalyse the dehydrogenation of formic acid in DMF, [IrCl(HNP)2(cod)] being the most active, with TOF1 min of about 2300 h−1 (5 mol% catalyst, 50 mol% sodium formate, DMF, 80 °C).
Introduction
Ligand design is a longstanding, generally accepted strategy which allows for the preparation of new compounds with tailored properties. In this regard, aminophosphanes have been attracting attention due to their modular synthesis from amines and chlorophosphanes,1 allowing facile structural modifications. As far as aminophosphanes of general formula NHRPR2 or NR2PR2 are concerned, different coordination modes have been reported, so far (Scheme 1). As a matter of fact, in mononuclear complexes, the expected monodentate κP coordination2 has been observed along with the η2 coordination,3 whereas the 1κN,2κP coordination4 has been reported in dinuclear complexes (Scheme 1-top). Additionally, a very limited number of examples of κ2C,P cyclometalated aminophosphano ligands have been reported, when NR2 is N-indolyl5a,b or N-carbazolyl,5b or in the case that a phenyl group2f,5c is attached to nitrogen (Scheme 1-bottom). In these cases, a five member metalacycle M–P–N–C–C forms as a result of the CH activation. Interestingly, aminophosphano complexes have found application as drugs,2d catalysts,2b,g,3a,b and redox-active multimetallic systems.4e,h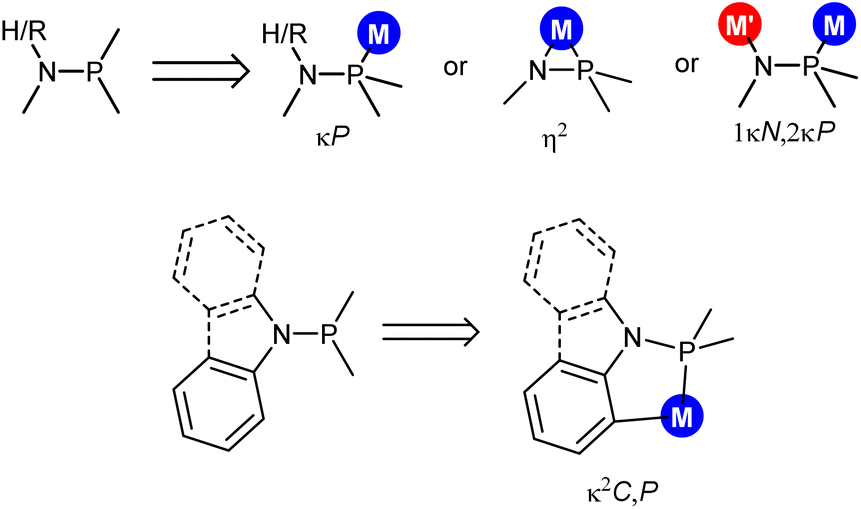 | ||
| Scheme 1 Coordination modes of aminophosphanes NHRPR2 or NR2PR2. | ||
Relevant to this paper, we have recently described2a the synthesis of the iridium(I) complex [IrCl(HNP)2(tfbb)] [HNP = NH(4-tolyl)PPh2; tfbb = tetrafluorobenzobarrelene] and its reaction with trimethyl phosphite, reporting that one of the peripheral NH groups is able to establish an intramolecular NH⋯O hydrogen bond that triggers the elimination of methylchloride and the formation of the phosphonato complex [Ir{PO(OMe)2}(HNP)2(tfbb)], thus showing that the HNP-Ir scaffold is able to activate small molecules via hydrogen bonding (Scheme 2).2a
 | ||
| Scheme 2 Reaction of [IrCl(HNP)2(tfbb)] with trimethyl phosphite.2a | ||
With this in mind, we envisioned that Ir-HNP derivatives could act as catalysts for the dehydrogenation of formic acid. Actually, a number of homogeneous catalysts based on iridium6 have proved to be extremely active, and mechanistic studies have revealed that hydrogen bonds between peripheral NH groups and formic acid play a crucial role in stabilizing both intermediates and transition states involved in the formation of dihydrogen and/or carbon monoxide.7 As an example, Fig. 1 shows the transition states TSA and TSB proposed for the dehydrogenation of formic acid catalysed by [IrHCl(LPCP)]6d and [IrCp*Cl(LCN)],6g,i respectively, highlighting the role of NH ancillary groups in the formation of both carbon dioxide (TSA) or dihydrogen (TSB).
 | ||
| Fig. 1 Selected iridium catalysts (left) for the formic acid dehydrogenation and related transition states (right).6d,g,i | ||
On this background, we decided to delve into the preparation of new iridium complexes with the aminophosphano ligand NH(4-tolyl)PPh2 (HNP) and its silylated analogue SiMe3N(4-tolyl)PPh2 (SiMe3NP). Specifically, herein we describe the synthesis of iridium(I) complexes of formula [IrCl(RNP)n(cod)] (R = H, SiMe3; n = 1, 2) as well as cyclometalated iridium(III) derivatives resulting from the intramolecular CH oxidative addition of the C2–H bond of the tolyl group to the iridium centre. In addition, a selection of the prepared complexes has been tested as catalysts for the dehydrogenation of formic acid, guessing that the peripheral NH group(s) might foster the catalytic process.
Results and discussion
Synthesis of SiMe3N(4-tolyl)PPh2 (SiMe3NP)
The synthesis of SiMe3N(4-tolyl)PPh2 (SiMe3NP) was carried out following a route similar to that reported for SiMe2{N(4-tolyl)PPh2}2.8 Indeed, the parent aminophosphane NH(4-tolyl)PPh2 was sequentially treated with n-BuLi and SiMe3Cl affording SiMe3NP (Scheme 3). As a consequence of the H/SiMe3 substitution, a well-shaped 1H doublet is observed at 0.47 ppm for the SiMe3 moiety (4JHP = 1.0 Hz) instead of the 1H signal of the NH hydrogen atom of HNP, and a significant downfield shift of the 31P{1H} NMR signal of SiMe3NP (δP 50.5 ppm) with respect to HNP (δP 28.7 ppm) was observed. | ||
| Scheme 3 Synthesis of SiMe3NP. | ||
The crystal structure of SiMe3NP (Fig. 2) shows an almost planar geometry of the nitrogen atom 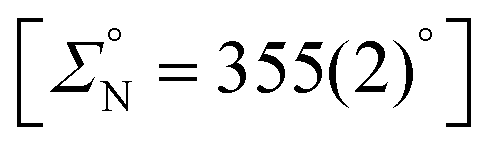 and an almost perpendicular arrangement of the C16–C17–C18–C19–C20–C21 aromatic ring with respect to the nitrogen plane [Si–N–C16–C17 −116.94(10)°]. Also, bond lengths Si–N [1.7574(10) Å], P–N [1.7167(10) Å] and C16–N [1.4383(14) Å] are similar to those reported for the related aminophosphane SiMe2{N(4-tolyl)PPh2}2 (Si–N 1.751 Å, av.; P–N 1.723 Å, av.; C–N 1.446 Å, av.).9 On these grounds, similar to what proposed for SiMe2{N(4-tolyl)PPh2}2, back-donation should exist mainly between nitrogen and phosphorus and should primarily be responsible for the planar geometry of the nitrogen atom in SiMe3NP (vide infra).
and an almost perpendicular arrangement of the C16–C17–C18–C19–C20–C21 aromatic ring with respect to the nitrogen plane [Si–N–C16–C17 −116.94(10)°]. Also, bond lengths Si–N [1.7574(10) Å], P–N [1.7167(10) Å] and C16–N [1.4383(14) Å] are similar to those reported for the related aminophosphane SiMe2{N(4-tolyl)PPh2}2 (Si–N 1.751 Å, av.; P–N 1.723 Å, av.; C–N 1.446 Å, av.).9 On these grounds, similar to what proposed for SiMe2{N(4-tolyl)PPh2}2, back-donation should exist mainly between nitrogen and phosphorus and should primarily be responsible for the planar geometry of the nitrogen atom in SiMe3NP (vide infra).
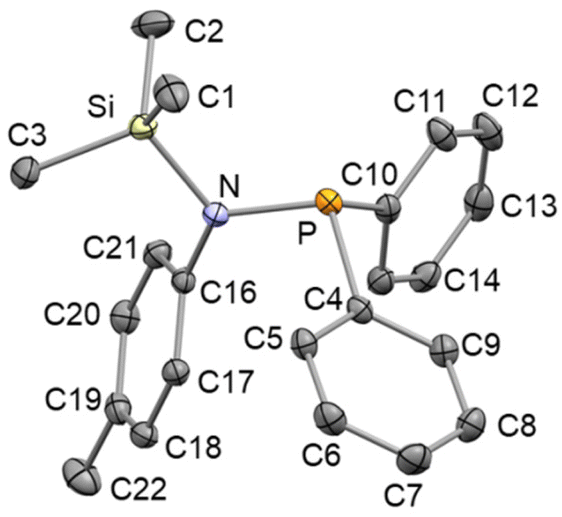 | ||
| Fig. 2 ORTEP plot of SiMe3N(4-tolyl)PPh2 (SiMe3NP). Hydrogen atoms are omitted for clarity and thermal ellipsoids are at 50% probability. Selected bond lengths (Å) and angles (°) are: P–N 1.7167(10), Si–N 1.7574(10), N–C16 1.4383(14), C16–N–P 122.30(7), C16–N–Si 118.88(7), P–N–Si 117.22(5), Si–N–C16–C17 −116.94(10). | ||
Preparation of iridium(I) compounds
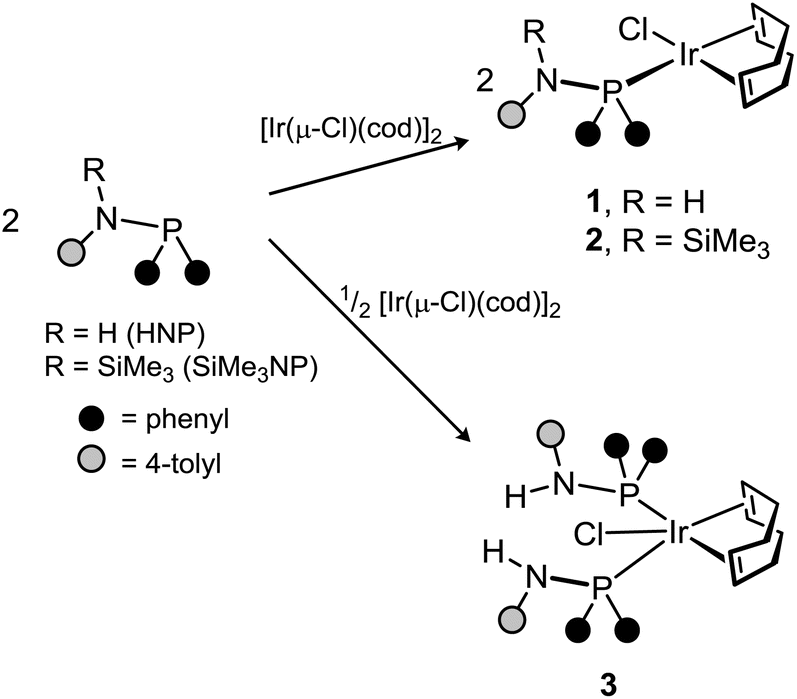
Both HNP and SiMe3NP react with [Ir(μ-Cl)(cod)]2 (P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ir = 1) affording the tetracoordinate iridium complexes [IrCl(L)(cod)] (L = HNP, 1; SiMe3NP, 2) (Scheme 4).
:Ir = 1) affording the tetracoordinate iridium complexes [IrCl(L)(cod)] (L = HNP, 1; SiMe3NP, 2) (Scheme 4).
 | ||
| Scheme 4 Reaction of HNP or SiMe3NP with [Ir(μ-Cl)(cod)]2. | ||
The crystal structure of 1 and 2 are given in Fig. 3 showing a distorted square planar geometry at iridium with a cis disposition of the chlorido and aminophosphano ligands. The coordination environments in 1 and 2 are virtually superimposable, nevertheless 1 and 2 differ in terms of intramolecular short contacts. Indeed, a NH⋯Cl hydrogen bond was observed in 1, whereas an anagostic CH⋯Ir interaction10 was observed in 2 (Fig. 3). As a consequence, a significantly different dihedral angle Cl–Ir–N–P is observed [1, 36.05(13)°; 2, −51.40(6)°].
 | ||
| Fig. 3 ORTEP plot of IrCl(cod)(HNP) (1, top) and IrCl(cod)(SiMe3NP) (2, bottom). Most hydrogen atoms are omitted for clarity and thermal ellipsoids are at 50% probability. Selected bond lengths (Å) and angles (°) are: 1, Ir–P 2.2850(9), Ir–Cl 2.3488(9), P–N 1.675(3), C20–C21 1.384(5), C25–C24 1.414(5), Ir–Ct2 1.98682(17), Ir–Ct1 2.08157(17), N–H 0.96(3), N–C13 1.417(4), P–Ir–Cl 88.31(3), C13–N–P 132.4(3), C13–N–H 113(3), P–N–H 115(3), Cl–Ir–P–N 36.05(13), N–H⋯Cl hydrogen bond: H⋯Cl 2.48(4), N⋯Cl 3.156(3), N–H 0.960(19), Cl–H–N 127(4); Ct1, centroid of C20 and C21; Ct2, centroid of C24 and C25. 2, N–P 1.6926(12), N–Si 1.7814(12), N–C16 1.4419(18), P–Ir 2.3028(4), Ir–Cl 2.3634(4), C28–C27 1.4260(19), C24–C23 1.394(2), Ct1–Ir 2.08575(16), Ct2–Ir 1.98914(14), P–N–Si 129.23(7), C16–N–P 118.78(9), C16–N–Si 111.98(9), P–Ir–Cl 89.136(12), Ct2–Ir–Ct1 86.812(6), C17–C16–N–Si 83.66(17), N–P–Ir–Cl −51.46(6). CH⋯Ir contact: H1c⋯Ir 2.56(3), C1⋯Ir 3.371(2), C1–H1c 1.076(19), Ir–H1c–C30 131.1(9). Ct1, centroid of C23 and C24; Ct2, centroid of C27 and C28. | ||
The NMR spectra of 1 and 2 suggest that the square planar structure observed in the solid state is preserved in solution. Indeed, for both compounds one 31P{1H} NMR signal is observed (δP 43.9, 1; 62.2 ppm, 2) along with two olefinic resonances for the cod ligand (δH 2.81 and 5.63, 1; 2.39 and 5.48 ppm, 2; see ESI, Fig. SI5† for selected NMR data with the proposed assignment).
The pentacoordinate derivative [IrCl(HNP)2(cod)] (3) was obtained when [Ir(μ-Cl)(cod)]2 was reacted with HNP (Ir:HNP molar ratio 1:2, Scheme 4). In contrast, the higher steric hindrance of SiMe3NP prevents the formation of the pentacoordinate complex [IrCl(SiMe3NP)2(cod)], even if SiMe3NP is added in excess. The 1H and 13C{1H} NMR spectra of 3 indicate that the compound is fluxional in solution even at 213 K. Indeed, at that temperature one broad 1H resonance was observed for the four olefinic hydrogen atoms of the cod ligand (δH 3.37 ppm) and two 1H resonances at 2.36 and 1.91 ppm for the endo and exo methylene hydrogen atoms of the cod ligand. By the same token, two 13C{1H} resonances at 69.0 ppm (t, 2JCP = 7.5 Hz) and at 32.5 ppm (br) were observed for the olefinic and methylene carbon atoms of the cod ligand. In this regard, DFT calculations indicate that 3 may adopt two stable configurations, namely TBPY-5-1311 (I,‡Fig. 4) and SPY-5-1311 (II, Fig. 4), with similar energies (TBPY-5-13 → SPY-5-13; ΔG = −1.6 kcal mol−1). Thus, reasonably the fast equilibrium TBPY-5-13 ⇄ SPY-5-13 should exchange the olefinic ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH moieties as well as the CH2 groups, consequently accounting for the averaged spectra observed for 3.
CH moieties as well as the CH2 groups, consequently accounting for the averaged spectra observed for 3.
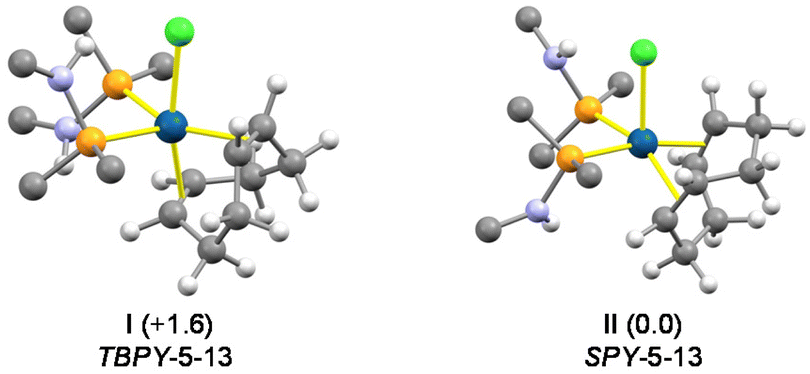 | ||
| Fig. 4 View of the calculated structures of the TBPY-5-13 and SPY-5-13 configurations of 3. The relative Gibbs free energies are given in parentheses (kcal mol−1). For clarity, only ipso carbon atoms of tolyl and phenyl groups are shown. Blue, iridium; gray, carbon; orange, phosphorus; green, chlorine; violet, nitrogen; white, hydrogen. | ||
The CH⋯Ir interaction observed in 2 (vide supra) is reminiscent of the CH⋯Ir short contact in the related square planar intermediate [IrCl(SiNP)(CO)]12 resulting from the carbonylation of IrCl(SiNP)(cod) (Scheme 5-top). Taking into account that [IrCl(SiNP)(CO)] smoothly undergoes an intramolecular oxidative addition of the SiCH2–H bond to iridium, the reaction of 2 with CO was carried out, and, in contrast to our expectations, the formation of 4[IrCl2(CO)2] was observed (Scheme 5-bottom). The IR spectrum of a CH2Cl2 solution of 4[IrCl2(CO)2] shows five CO stretching bands (see ESI, Fig. SI3†) confirming the presence of the anion [IrCl2(CO)2]− [2055 (s), 1971 (s) cm−1] and the pentacoordinated cation 4+ [2080 (w), 2008 (s); 1998 (s) cm−1].§ Accordingly, the 13C{1H} triplet at 173.5 ppm (2JCP = 11.1 Hz) and the singlet at 169.1 ppm were assigned to the carbonyl ligands of 4+ and [IrCl2(CO)2]−, respectively, and the 31P{1H} NMR spectrum contains one singlet at 42.9 ppm. Despite the fact that 4[IrCl2(CO)2] is thermally stable in the solid state and was obtained as an analytically pure sample, it is thermally unstable in CD2Cl2 and slowly evolves to unidentified species, rendering mixtures still containing about 85 mol% (31P{1H} NMR) of 4[IrCl2(CO)2] after 15 h at room temperature.
 | ||
| Scheme 5 Carbonylation reaction of [IrCl(SiNP)(cod)] (A, previous work12) and [IrCl(SiMe3NP)(cod)] (2, B, this work). | ||
Cyclometalation reactions
The reactivity of HNP and SiMe3NP towards cationic iridium(I) complexes bearing labile ligands has been explored. In the following subsections, the reactions of HNP and SiMe3NP with [Ir(CH3CN)2(cod)][PF6] or [Ir(CH3CN)2(coe)2][PF6] will be discussed.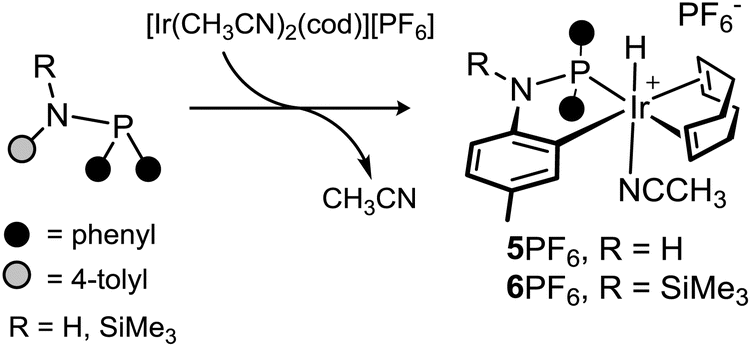 | ||
| Scheme 6 Formation of 5PF6 and 6PF6. | ||
The crystal structure of 6PF6 was determined by single crystal X-ray diffraction measurements. Its asymmetric unit contains four crystallographically independent pairs of the ions OC-6-24-[IrH{κ2C,P-SiMe3N(4-C6H3CH3)PPh2}(cod)(CH3CN)]+ and PF6− (see ESI, Fig. SI1†). As for the cations, despite the fact that they are chemically equivalent, they differ in the configuration of the metal centre. Actually, the metal centre of 6+ is stereogenic and two of the cations of the asymmetric unit exhibit C configuration11 at the metal, whereas the remaining two exhibit A configuration11 (ESI, Fig. SI1†). When comparing the cations of the asymmetric unit, related bond lengths and angles are similar, regardless of the configuration at the metal centre. Therefore, for the sake of brevity, only the structure of one of the cation exhibiting C configuration (Fig. 5) will be discussed. A distorted octahedral geometry at the metal centre is observed with a trans disposition of the hydrido and acetonitrile ligands. The five member Ir1–P1–N1–C113–C114 metallacycle is planar and exhibits a bite angle P1–Ir1–C114 of 81.14(14)° nicely fitting two cis coordination sites at the metal centre.
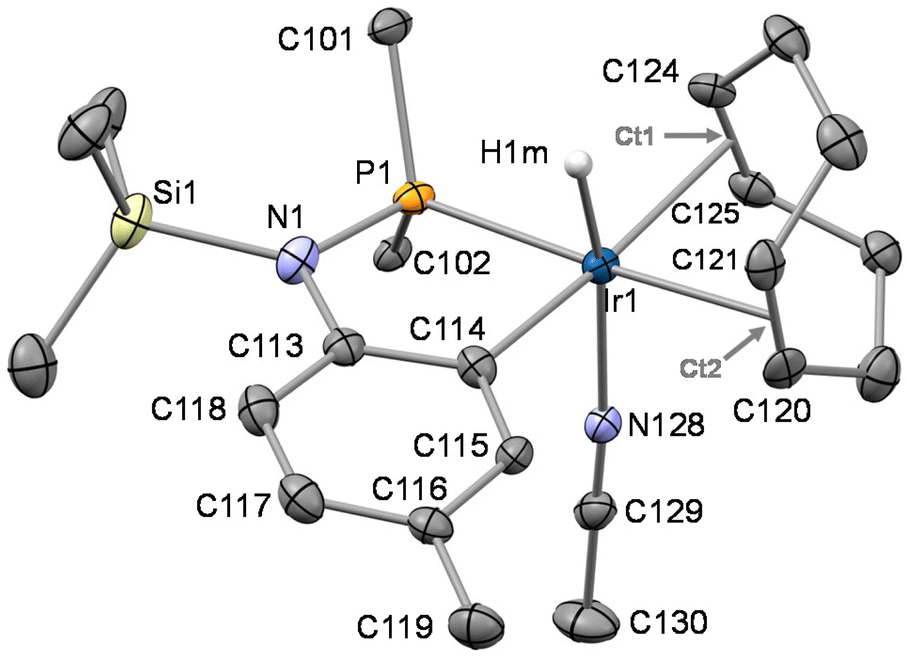 | ||
| Fig. 5 ORTEP plot of CIr-OC-6-24-[IrH{κ2C,P-SiMe3N(4-C6H3CH3)PPh2}(cod)(CH3CN)]+ in 6PF6. For clarity, most hydrogen atoms are omitted and only the ipso carbon atoms of the phenyl groups are shown. Thermal ellipsoids are at 50% probability. Selected bond lengths (Å) and angles (°) are: C114–Ir1 2.075(4), C113–C114 1.410(6), C113–N1 1.455(6), N1–P1 1.681(4), N1–Si1 1.796(4), N128–Ir1 2.127(4), P1–Ir1 2.2640(14), Ir1–H1m 1.596(10), C120–C121 1.390(7), C124–C125 1.380(6), Ir1–Ct1 2.1909(5), Ir1–Ct2 2.1971(6), C114–Ir1–H1m 80(2), P1–Ir1–H1m 87(2), C114–Ir1–P1 81.14(14), N128–Ir1–H1m 168(2), Ct1–Ir1–Ct2 82.52(2), C113–N1–P1 113.2(3), C113–N1–Si1 121.2(3), P1–N1–Si1 125.5(2). | ||
In view of the bond lengths C113–N1 [1.455(6) Å], N1–P1 [1.681(4) Å], and N1–Si1 [1.796(4) Å] and taking into account the planarity of the nitrogen atom N1  , the metalation of the tolyl substituent in 6+ and the consequent coplanarity of the tolyl moiety and the N1–P1–Si–C113 fragment appear not to bring about any substantial difference in the electronic distribution at the nitrogen atom with respect to SiMe3NP and 2. In this regard, the delocalization indexes13 (DI, aka Fuzzy Bond Orders, FBO) were calculated for SiMe3NP, 2 and 6+ (Fig. 6) showing that the nitrogen–phosphorus back-donation is still operative in 6+ and the nitrogen–carbon bond basically holds its single bond character, similar to SiMe3NP and 2, where the aromatic ring lies almost perpendicular to the N–Si–C–P plane.
, the metalation of the tolyl substituent in 6+ and the consequent coplanarity of the tolyl moiety and the N1–P1–Si–C113 fragment appear not to bring about any substantial difference in the electronic distribution at the nitrogen atom with respect to SiMe3NP and 2. In this regard, the delocalization indexes13 (DI, aka Fuzzy Bond Orders, FBO) were calculated for SiMe3NP, 2 and 6+ (Fig. 6) showing that the nitrogen–phosphorus back-donation is still operative in 6+ and the nitrogen–carbon bond basically holds its single bond character, similar to SiMe3NP and 2, where the aromatic ring lies almost perpendicular to the N–Si–C–P plane.
 | ||
| Fig. 6 Delocalization index (DI) for the Si–N, C–N and P–N bonds in SiMe3NP, 2 and 6+. | ||
The solution structure of 6+ is similar to that observed in the solid state. In particular, the 1H signal of the hydrido ligand is a doublet at −16.43 ppm with a 2JHP of 11.2 Hz indicating a mutual cis disposition of the hydrido ligand and phosphorus. Also, four non-equivalent olefinic hydrogen atoms are observed for the cod ligand (δH 5.54, 5.37, 4.69, 4.09 ppm) and a 31P{1H} singlet is observed at 73.9 ppm downfield-shifted with respect to SiMe3NP (50.5 ppm) and 2 (62.2 ppm), reasonably as a consequence of the oxidation state +3 of the metal centre. The metalation of the tolyl group brings about a 1H pattern consisting of two mutually coupled doublets at 7.14 and 6.91 ppm (3JHH = 8.2 Hz) and one broad singlet at 6.72 ppm assigned to the aromatic hydrogen atoms of the [Ir{κ2C,P-SiMe3N(4-C6H3CH3)P}] moiety. Also, the 13C{1H} signal of the metalated carbon atom (δC 126.8 ppm) is a doublet as a consequence of the carbon–phosphorus coupling (2JCP = 3.5 Hz). Finally the 1H singlet at 1.80 ppm indicates the presence of coordinated acetonitrile. The NMR data for 5PF6 are analogous to those discussed for 6PF6 suggesting that the solution structure of 5PF6 should be similar. It is worth a mention that, at variance with 2, the NH 1H signal of 5+ is a broad singlet (Δν1/2 = 5.1 Hz) at 5.22 ppm, which suggests that the cyclometalation causes a reduced hydrogen-phosphorus coupling constant. Fig. SI6 (ESI†) shows the detailed assignment of 1H, 13C{1H} and 31P{1H} signals relevant for the solution structure elucidation of 5+ and 6+.
The formation of 5+ and 6+ was monitored by NMR spectroscopy. In the case of 6+, the straightforward and clean formation of 6+ was observed even at 233 K, no intermediate being detected by 1H and 31P{1H} NMR spectroscopy. On the other hand, when monitoring the formation of 5+, a more complex reaction sequence was brought to light (Scheme 7). Actually, upon adding HNP to [Ir(CH3CN)2(cod)][PF6] (1:1 molar ratio) at 233 K,¶ part of [Ir(CH3CN)2(cod)]+ remains unreacted (as seen in the 1H NMR spectrum), whereas HNP reacts completely. In addition, beside 5+, the square planar complex [Ir(cod)(HNP)2]+ (7+) and the hydrido derivative [IrH{κ2C,P-NH(4-C6H3CH3)PPh2}(HNP)(cod)]+ (8+) were observed (Scheme 7). Fig. 7 shows the 31P{1H} NMR spectrum of the reaction mixture at 233 K along with selected NMR data relevant for the identification of 7+ and 8+. Upon raising the temperature up to 298 K (in 10 min approximately), 5+ is observed in solution as the only product. On these grounds, the reaction of [Ir(CH3CN)2(cod)]+ with HNP is likely to yield 5+ reasonably via the putative intermediate [Ir(cod)(HNP)(CH3CN)]+ (III+) which undergoes an intramolecular CH oxidative addition to iridium (vide infra). On the other hand, the formation of [Ir(cod)(HNP)2]+ (7+) and [IrH{κ2C,P-HN(4-C6H3CH3)PPh2}(HNP)(cod)]+ (8+) indicates that the putative intermediate [Ir(cod)(HNP)(CH3CN)]+ (III+) undergoes a substitution reaction, in which HNP replaces CH3CN rendering [Ir(cod)(HNP)2]+ (7+), which in turn undergoes the intramolecular CH oxidative addition to iridium, yielding 8+.
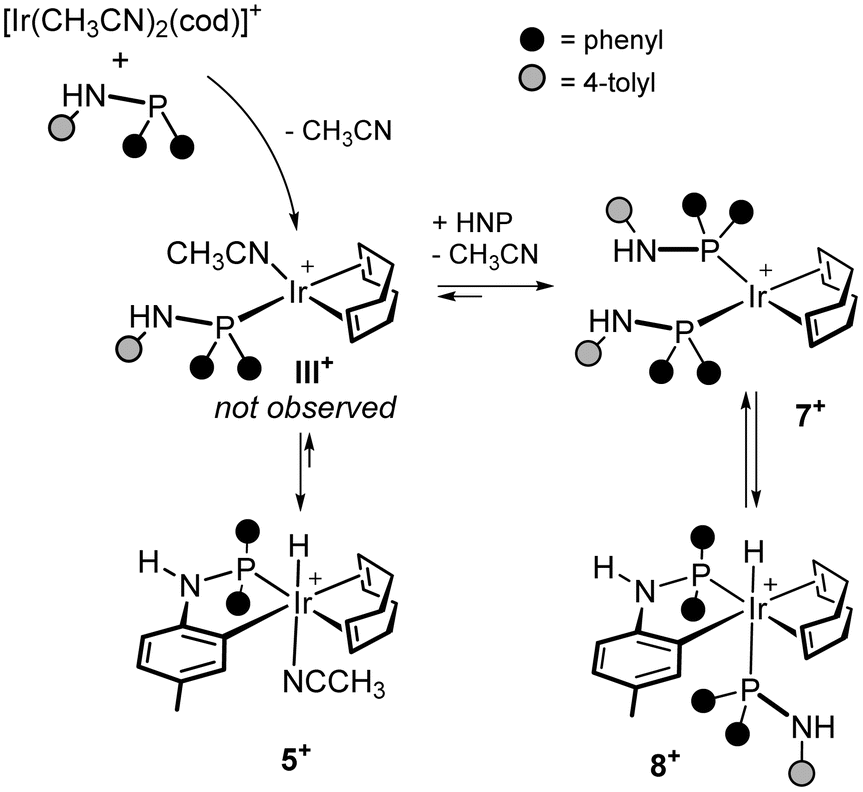 | ||
| Scheme 7 Reaction sequence for the reaction of [Ir(CH3CN)2(cod)]+ with HNP. | ||
 | ||
| Fig. 7 (Top) 31P{1H} NMR of the CD2Cl2 solution resulting from the reaction of [Ir(CH3CN)2(cod)][PF6] and HNP (1:1) at 233 K, with the proposed assignment; (bottom) selected 1H, 13C{1H} and 31P{1H} NMR data for 7+ and 8+ (δ are given in ppm, J in Hz). | ||
Admittedly, once formed, 5+ might react with HNP rendering 8+ and releasing CH3CN (Scheme 7). Nonetheless, the 1H–1H EXSY spectrum of the reaction mixture at 233 K shows exchange peaks between [Ir(cod)(HNP)2]+ (7+) and [IrH{κ2C,P-HN(4-C6H3CH3)PPh2}(HNP)(cod)]+ (8+) but no exchange peaks have been observed between 8+ and 5+, suggesting that the reaction 5+ + HNP → 8+ + CH3CN may not be viable at 233 K whereas the equilibrium 7+ ⇄ 8+ should be operative at that temperature.||
Finally, it is worth a mention that the CH oxidative addition to iridium in 1+ is totally regiospecific. In fact, the isomers IV+ (OC-6-25) and V+ (OC-6-35, Fig. 8) were not detected in the reaction mixture. As a confirmation, IV+ was calculated to be less stable than 5+ (ΔGrel = +6.6 kcal mol−1) whereas the structure of V+ could not even be located on the potential energy surface (PES), reasonably due to the steric congestion between the PPh2 moiety and the cod ligand, leading to the dissociation of the CH3CN ligand and a severe distortion of the resulting square pyramidal structure (ΔGrel = +20.6 kcal mol−1).
 | ||
| Fig. 8 Isomers of [IrH{κ2C,P-NH(4-C6H3CH3)PPh2}(cod)(CH3CN)]+. | ||
Interestingly, the cyclometalation of the tolyl group can be reverted by reacting 5+ or 6+ with chloride (Scheme 8). Actually, the reaction of 5PF6 or 6PF6 with tetrabutylammonium chloride (1:1) cleanly and immediately affords 1 and 2, respectively.** On the other hand, 5PF6 and 6PF6 were also obtained via chloride abstraction upon treating 1 and 2, respectively, with TlPF6 in CH3CN (Scheme 8). On these grounds, the C–H oxidative addition to iridium in [IrX(RNP)(cod)]n+ (X = Cl, n = 0; X = CH3CN, n = 1, R = H, SiMe3) is subtly controlled by the ancillary ligand X. Actually, it should be hampered by the chlorido ligand and facilitated when the acetonitrile ligand is present, instead. With this in mind, the Gibbs free energy profiles of the cyclometalation reaction for 1 and [Ir(cod)(HNP)(CH3CN)]+ (III+) were explored by means of DFT calculations (Fig. 9). Despite the fact that the activation barriers are easily accessible at room temperature for both 1 (cf.TS_1-VI, ΔGact = +17.5 kcal mol−1) and III+ (cf.TS_III+-5+, ΔGact = +11.5 kcal mol−1), the relative stability of the hydrido derivative vs. the related square complex are different. As a matter of fact, in agreement with the observed outcome of the reactions given in Scheme 8, the putative hydrido complex [IrHCl{κ2C,P-NH(4-C6H3CH3)PPh2}(cod)] (VI) is less stable than the parent square planar complex [IrCl(HNP)(cod)] (1), whereas [IrH{κ2C,P-NH(4-C6H3CH3)PPh2}(cod)(CH3CN)]+ (5+) is more stable than [Ir(cod)(HNP)(CH3CN)]+ (III+, Fig. 9), which indicates that switching from X = CH3CN to X = Cl− in [IrX(RNP)(cod)]n+ makes the oxidative addition unfavourable.
 | ||
| Scheme 8 Reversible Formation of 5PF6 and 6PF6. | ||
 | ||
| Fig. 9 (Top) Gibbs free energy profile for the formation of [IrHX{κ2C,P-HN(4-C6H3CH3)PPh2}(cod)]n+ (X = CH3CN, 5+; Cl−, II) along with the calculated Gibbs free energies (kcal mol−1, M06/def2tzvp//B3PW91-GD3BJ/def2svp, CH2Cl2, 298 K, 1 atm). (bottom) View of TS_I+-5+ and TS_1-II along with selected interatomic distances (Å). For clarity, most hydrogen atoms have been omitted and phenyl groups are shown in a wireframe style. Blue, iridium; gray, carbon; orange, phosphorus; green, chlorine; violet, nitrogen; white, hydrogen. | ||
For the sake of comparison, the energy profile of the reaction 7+ → 8+ was also calculated (Fig. SI46 ESI†) confirming that this transformation is a fast equilibrium under the explored reaction conditions.
In view of the reaction sequence discussed in Scheme 7, the reaction of 5+ with HNP (1:1) was carried out aiming at preparing 8+. Surprisingly, on a preparative scale an inseparable mixture of products was obtained, and monitoring the reaction at 233 K evidenced the formation of a mixture of products mainly containing 8+ and a new thermally unstable product, which was independently prepared at 233 K and identified in situ as [Ir(HNP)3(cod)]+ (9+).†† In addition, when the reaction mixture was heated up to room temperature, 8+ disappeared and a mixture of products was obtained including, among others, hydrido derivatives containing three HNP ligands. This evidence suggests that the outcome of the reaction of 5+ with HNP is spoiled by the reversibility of the CH oxidative addition as well as by the formation of substitution products containing up to three HNP ligands per iridium.
Also, the reaction of 5+ with SiMe3NP was carried out, envisioning that the higher steric hindrance of SiMe3NP could prevent the formation of unwanted products (Scheme 9). As a matter of fact, this reaction smoothly takes place at 233 K in 6 h rendering the unexpected compound 10+ (along with residual amounts of unreacted 5+, Fig. 10). Notably, the formation of 10+ (instead of VII+, Scheme 9) indicates that 5+ does not simply exchange the CH3CN ligand with the incoming SiMe3NP, but also that, at some point, the CH reductive elimination rendering HNP and the following CH oxidative addition at SiMe3NP take place. The 1H and 13C{1H} signals assigned to the tolyl groups (see ESI, Fig. SI7†) confirm the presence of the [Ir{κ2C,P-SiMe3N(4-C6H3CH3)P}] moiety along with the κP HNP ligand. Also the 2JHP coupling constants (105.5, 12.4 Hz) of the 1H signal at −10.18 ppm of the Ir–H moiety and the 2JPP constant (22.2 Hz) support the structure proposed for 10+.
 | ||
| Scheme 9 Reaction of 5+ with SiMe3NP. | ||
 | ||
| Fig. 10 31P{1H} NMR spectrum of 10+ (prepared in situ) with traces of residual 5+ (6 mol% approx.). | ||
Finally, for the sake of comparison, the relative stability of 10+ and of the expected isomer VII+ was calculated by DFT methods showing that 10+ is 31.5 kcal mol−1 more stable than VII+, reasonably because of the high steric congestion that exists between the trimethylsilyl group and the metalated tolyl group (Fig. 11). In our hands, any attempt to isolate 10+ was unsuccessful due to its limited thermal stability.
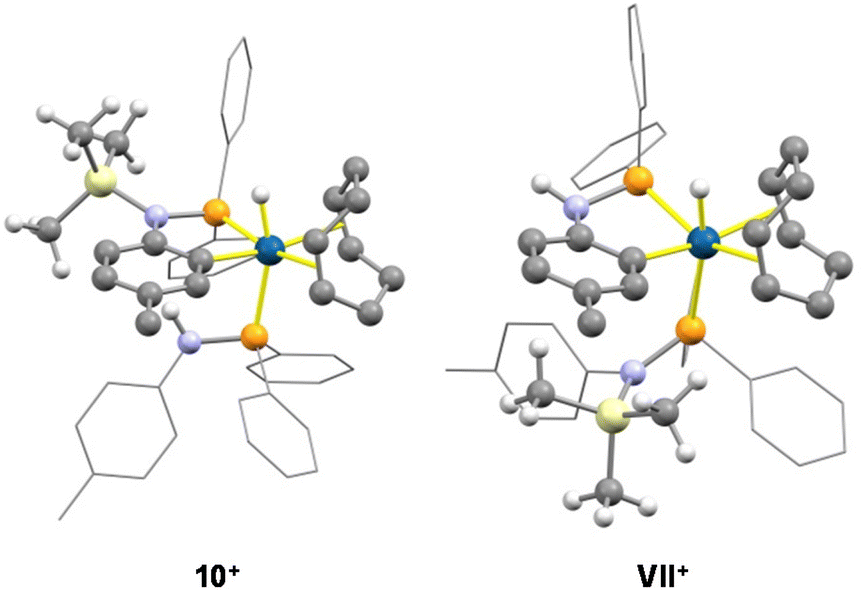 | ||
| Fig. 11 Calculated structures of 10+ and VII+ (B3PW91-GD3BJ/def2svp). For clarity, most hydrogen atoms are omitted and most tolyl and phenyl groups are shown in a wireframe style. Blue, iridium; gray, carbon; orange, phosphorus; yellow, silicon; violet, nitrogen; white, hydrogen. Blue, iridium; gray, carbon; orange, phosphorus; yellow, silicon; violet, nitrogen; white, hydrogen. | ||
:2 molar ratio) affording the square planar complex [Ir(CH3CN)2(SiMe3NP)2]+ (11+), isolated as the hexafluorophosphate salt, as a result of the substitution of the coe ligands. In agreement with the square planar geometry at the metal centre, one 31P{1H} singlet at 61.9 ppm is observed for the two equivalent phosphorus atoms, along with two 1H singlets at 1.42 and 0.03 ppm for the equivalent acetonitrile ligands and the trimethyl silyl groups, respectively. On the other hand, the reaction of [Ir(CH3CN)2(coe)2][PF6] with HNP entails the intramolecular C–H oxidative addition to iridium, yielding the iridium(III) hydrido isomers 12+ (OC-6-35) and 13+ (OC-6-52) of formula [IrH{κ2C,P-NH(4-C6H3CH3)PPh2}(HNP)2(CH3CN)]+, isolated as the hexafluorophosphate salts when the reaction was carried out in CH3CN or CH2Cl2, respectively (Scheme 10). More specifically, in CD2Cl2, 12+ is the kinetic product observed in the first place (31P{1H}NMR), whereas 13+ is finally obtained as the thermodynamic product. On the contrary, in CD3CN, only 12+ forms, 13+ being not observed (31P{1H} NMR). Accordingly, when a CD2Cl2 solution of 12+ was allowed to stand at room temperature for 12 h, the clean conversion of 12+ into 13+ takes place (ESI, Fig. SI45†).
 | ||
| Scheme 10 Reaction of [Ir(CH3CN)2(coe)2]+ with HNP or SiMe3NP. | ||
The solid state structure of 12PF6 was determined by means of single crystal X-ray diffraction measurements (Fig. 12). The asymmetric unit of 12PF6 contains two crystallographically independent pairs of the ions OC-6-35-[IrH{κ2C,P-NH(4-C6H3CH3)PPh2}(HNP)2(CH3CN)]+ and PF6− (ESI, Fig. S12†). Similar to 6+, 12+ contains a stereogenic metal centre, and the cations of the asymmetric unit are chemically equivalent, but exhibit different configurations at the metal centre. When comparing bond lengths and angles, similar values are observed for the two enantiomers. Therefore, for the sake of brevity, only the crystal structure of the enantiomer C is discussed in detail (Fig. 12). An octahedral environment is observed at the metal centre with a facial disposition of the three phosphorus atoms P1, P2 and P3. The remaining coordination sites are occupied by the hydrido ligand H1m (trans to P3), the acetonitrile ligand (trans to P1) and the carbon atom C14 of the metalated tolyl group (trans to P2). Similar to 6+, the [Ir{κ2C,P-NH(4-C6H3CH3)P}] moiety of 12+ is planar and the bond lengths N1–P1 [1.680(4) Å] and C13–N1 [1.412(5) Å] indicate a nitrogen–phosphorus back-donation with a nitrogen–carbon single bond.
 | ||
| Fig. 12 ORTEP plot of CRh-OC-6-35-[IrH{κ2C,P-HN(4-C6H3CH3)PPh2}(HNP)2(CH3CN)][PF6] (CRh-12PF6). For clarity most hydrogen are omitted and only ipso carbon atoms of the phenyl and tolyl groups are shown. Thermal ellipsoids are at 50% probability. Selected bond lengths (Å) and angles (°) are: N58–Ir1 2.062(3), P1–Ir1 2.2484(10), P2–Ir1 2.3387(10), P3–Ir1 2.4027(9), Ir1–H1m 1.54(4), C14–Ir1 2.103(4), C13–C14 1.407(5), C13–N1 1.412(5), N1–P1 1.680(4), C14–Ir1–P1 81.14(11), P1–Ir1–H1m 83.1(19), C14–Ir1–H1m 84.1(18), P3–Ir1–H1m 172.6(19), P1–Ir1–P2 98.90(4), P1–Ir1–P3 97.96(3), P2–Ir1–P3 105.03(3), N58–Ir1–P1 166.85(9). | ||
Based on the NMR spectra of 12+, its solid state structure should be maintained in solution. Specifically, three 31P{1H} signals are observed at 49.8 (P1), 19.8 (P2) and 11.2 ppm (P3) with 2JPP coupling constants (16.9, 11.5 Hz) indicating a mutually cis disposition. Accordingly, three 1H signals are observed for the non-equivalent NH groups (δH 6.28, HNP1; 5.17, HNP2; 4.64 ppm, HNP3). The 1H signal of the hydrido ligand at −11.83 ppm is observed as a doublet of triplets with 2JHP constants (150.5, 16.2 Hz) underpinning the presence of two phosphorus atoms at cis positions (P1 and P2) and one (P3) at the trans position. The 1H and 13C{1H} signals assigned to the tolyl group are similar to those observed for 6+, thus confirming its metalation (see ESI Fig. SI8† for selected 1H, 13C{1H} and 31P{1H} data and the proposed assignment).
Also, the solution structure of 13+ was established by means of multinuclear NMR spectroscopy. Relevant for the solution structure elucidation, the 31P{1H} spectrum contains two signals at 43.4 and 19.7 ppm assigned to the metalated Ir{κ2C,P-NH(4-C6H3CH3)P} moiety and the equivalent HNP ligands, respectively. Accordingly, two 1H signals are observed for the NH moieties at 5.57 (1H) and 5.18 ppm (2H). The hydrido ligand lies trans to the phosphorus atom of the metalated HNP ligand originating a 1H doublet of triplets at −9.86 ppm (2JHP = 133.0, 18.6 Hz). Like for 12+, the 1H and 13C{1H} signals assigned the tolyl groups confirm the presence of one metalated tolyl group (see ESI Fig. SI8†).
As for the formation of 12+ and 13+, DFT calculations were performed in order to shed light on the underlying reaction sequence. Provided that [Ir(CH3CN)2(SiMe3NP)2]+ (11+) forms upon reaction of [Ir(CH3CN)2(coe)2]+ with SiMe3NP, the related complex [Ir(CH3CN)2(HNP)2]+ (VIII+) is assumed as the starting point for the formation of 12+ (Fig. 13A). Two routes leading from VIII+ to 12+ can be envisioned. On one hand (Fig. 13A, red arrows), the oxidative addition of the CH bond to iridium in III+ leads to the iridium(III) hydrido derivatives IX+ or X+, depending on the regiochemistry of the CH oxidative addition. The barriers for the formation of IX+ (TS_VIII+-IX+, +16.6 kcal mol−1) and X+ (TS_VIII+-X+,‡‡ ΔEact ≈ +16.7 kcal mol−1) are accessible at room temperature. On their turn, IX+ or X+ might undergo a substitution reaction exchanging CH3CN with HNP. Indeed, the CH3CN ligand trans to carbon atom (IX+) or hydrido ligand (X+) are expected to be labile and prone to being replaced by the incoming HNP ligand, thus rendering 12+. On the other hand (Fig. 13A, blue arrows), VIII+ might convert into the square planar intermediate XII+via a presumably barrierless, exergonic associative mechanism§§ going through the pentacoordinate derivative XI+. Subsequently, XII+ should undergo the intramolecular CH oxidative addition and, depending on the implied tolyl group and on the regiochemistry of the CH oxidative addition itself, 12+, XIII+ or XIV+ might form. Notably, both XIII+ and XIV+ are calculated to be less stable than 12+, in agreement with the fact that neither of them was obtained. Also the transition state TS_XII+-12+ (Fig. 13B) leading from XII+ to 12+ was calculated to be accessible at room temperature (Fig. 13A, ΔGact = +12.5 kcal mol−1). With this in mind, both routes are accessible under experimental conditions. Nonetheless, from an energetic point of view, starting from VIII+, the route viaIX+ and X+ (red arrows) goes up to approx. +17 kcal mol−1 (corresponding to the transition states TS_VIII+-IX+ and TS_VIII+-X+) and then downhill to 12+, whereas the route viaXI+ (blue arrows) goes through two exergonic initial steps (VIII+ → IX+, ΔG = −22.5 kcal mol−1; IX+ → X+, ΔG = −1.5 kcal mol−1) followed by the CH oxidative addition rendering 12+, which is feasible under experimental conditions (ΔGact = +12.5 kcal mol−1, TS_XII+-12+vs. XII+). As a result, even if the route viaIX+ and X+ (red arrows) is viable, the reaction sequence going through XII+ (blue arrows) is more favourable and should be operative under experimental conditions.
 | ||
| Fig. 13 (A) Reaction sequences for the formation of 12+ and 13+ with the relative Gibbs free energy (kcal mol–1, B3PW91-GD3BJ/def2svp) of the proposed intermediates and transition states. (B) Calculated structure for the transition state TS_XII+-11+ along with selected interatomic distances (Å, B3PW91-GD3BJ/def2svp). For clarity, most hydrogen atoms are omitted and phenyl and selected tolyl groups are shown in a wireframe style. (C) Proposed reaction sequence for the isomerization of 12+ to 13+ (M06/def2tzvp//B3PW91-GD3BJ/def2svp). | ||
As far as the formation of 13+ is concerned, the meridional disposition of the phosphorus and carbon atoms and of the hydrido ligand within the IrH{κ2C,P-HN(4-C6H3CH3)PPh2} moiety rules out that 13+ might result from the straightforward CH oxidative addition to iridium.¶¶
Thus, the transformation of 12+ into 13+ should be the result of a genuine isomerization process which alters the disposition of the ligands at the metal centre, and more than likely is triggered by the dissociation of CH3CN when 12+ is dissolved in CH2Cl2. On this basis, Fig. 13C depicts a plausible reaction sequence (green arrows) for the conversion of 12+ into 13+ along with the calculated relative Gibbs free energy of the proposed intermediates. In the first place, the dissociation of CH3CN from 12+ renders the pentacoordinated iridium complex XV+ which stepwise isomerise to XVII+viaXVI+. Finally, 13+ forms upon reaction of XVII+ with CH3CN.
Catalytic dehydrogenation of formic acid
The iridium(I) derivatives 1–3 and 11PF6, and the iridium(III) compounds 5PF6, 6PF6, and 13PF6 were tested as catalysts for formic acid dehydrogenation. Preliminary catalytic tests (1 mol% catalyst, 10 mol% NaHCOO, in DMF at 80 °C) revealed that IrCl(HNP)2(cod) (3) is by far the most active catalyst (Table 1, entries 1–7) reaching TOF1 min of about 1700 h−1 and complete conversion to H2 and CO2 after 18 min, CO not being observed in the resulting gas mixture (IR). In this regard, these preliminary data point out that the presence of two aminophosphano ligands, each one with an available NH group, may be responsible for the good catalytic performance of 3. When using 1 mol% of 3 as a catalyst, increasing the amount of sodium formate up to 50 mol% does not increase the activity of the catalyst (Table 1, entry 8). Notably, when 5 mol% of 3 along with 50 mol% of sodium formate was used a short induction time was clearly observed (Fig. 14) and a TOF1 min of about 2300 h−1 (Table 1, entry 9) was reached, which indicates that its activity is similar to that reported for the iridium complexes [IrCp*(H2O)(2,2′-bipyridine-4,4′-diol)]2+ (ref. 6k) and [IrCp*Cl{1,2-(NH)2C6H10}]+.6g Also, at a 5 mol% catalyst loading and 50 mol% of sodium formate, 3 showed a decreasing activity upon consecutively adding formic acid, without any induction period (C–F, Fig. 14). When triethylamine was used as a base instead of sodium formate (Table 1, entries 10 and 11), a lower activity was observed for 3 (TOF1 min 1000 h−1, Fig. 14). | ||
| Fig. 14 Conversion vs. time profiles for the formic acid dehydrogenation catalysed by 3 (5% catalyst, 50% base, DMF, 80 °C). | ||
a
| Entry | Catalyst | Cat. mol% | Base | Base mol% | Conv.%b, time | TOF1 minc (h−1) |
|---|---|---|---|---|---|---|
| a Reaction conditions: 20 mL of DMF, 20 μL of HCOOH (0.529 mmol), NaHCOO or NEt3 (10 or 50 mol%), iridium catalyst (1 or 5 mol%), 353 K. b Based on the pressure of the resulting gas mixture. c TOF1 min is calculated as TON at 1 min divided by 1 min. | ||||||
| 1 | 1 | 1% | NaHCOO | 10% | 38%, 40 min | 400 |
| 2 | 2 | 1% | NaHCOO | 10% | 68%, 40 min | 240 |
| 3 | 3 | 1% | NaHCOO | 10% | 92%, 18 min | 1700 |
| 4 | 5PF6 | 1% | NaHCOO | 10% | 37%, 40 min | 240 |
| 5 | 6PF6 | 1% | NaHCOO | 10% | 30%, 40 min | 150 |
| 6 | 11PF6 | 1% | NaHCOO | 10% | 94%, 35 min | 500 |
| 7 | 13PF6 | 1% | NaHCOO | 10% | 95%, 18 min | 680 |
| 8 | 3 | 1% | NaHCOO | 50% | 97%, 18 min | 1640 |
| 9 | 3 | 5% | NaHCOO | 50% | 99%, 0.8 min | 2330 |
| 10 | 3 | 1% | NEt3 | 10% | 96%, 25 min | 1000 |
| 11 | 3 | 5% | NEt3 | 50% | 98%, 2 min | 900 |
Conclusions
Monodentate aminophosphanes HNP [NH(4-tolyl)PPh2] and SiMe3NP [SiMe3N(4-tolyl)PPh2] react with iridium(I) precursors affording a varied family of iridium(I) and cyclometalated iridium(III) complexes, exhibiting κP or κ2C,P coordination modes. The tetra- or pentacoordinate iridium(I) complexes [IrCl(κP-RNP)n(cod)] (R = H, n = 1, 2; R = SiMe3, n = 1) result from bridge splitting at [Ir(μ-Cl)(cod)]2, the steric hindrance of the incoming aminophosphane being decisive in the outcome of the reaction. On the other hand, octahedral iridium(III) complexes can also be obtained as a result of the intramolecular CH oxidative addition of the C2–H bond of the tolyl substituent of both HNP and SiMe3NP to iridium(I) rendering the [IrH{κ2C,P-RN(4-C6H3CH3)P}] moiety. The calculated activation barrier of the CH oxidative addition is relatively small and the related CH reductive elimination can be smoothly triggered (Scheme 11). Unexpectedly, complex reaction sequences have been observed as a consequence of the reversibility of the CH oxidative addition and/or competing substitution reactions.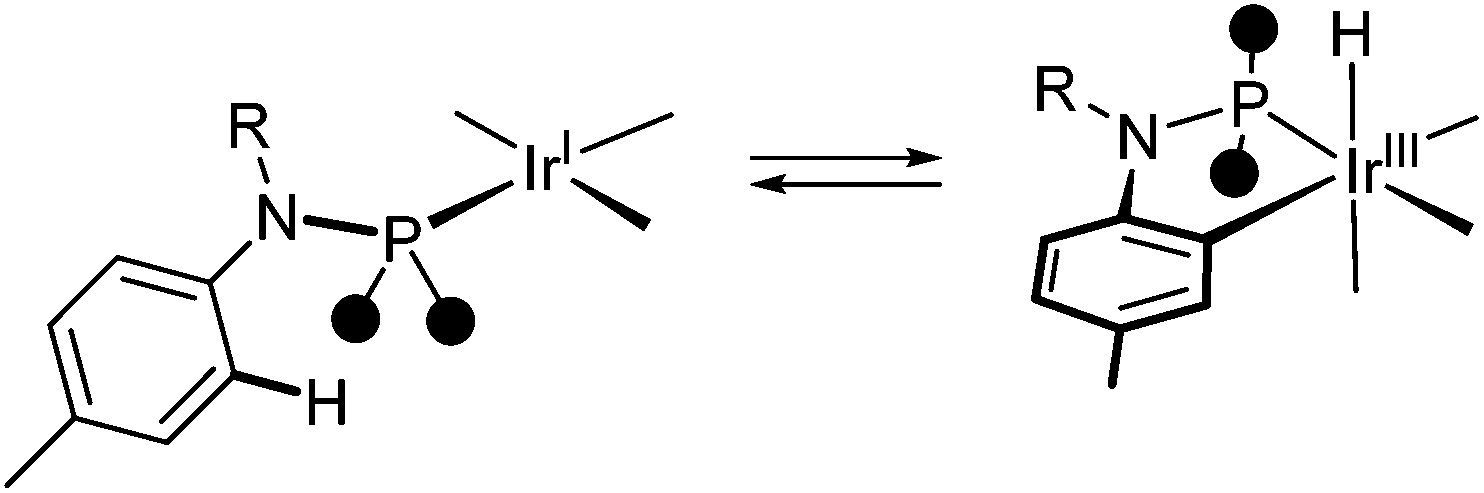 | ||
| Scheme 11 Reversible CH oxidative addition to iridium. | ||
Both iridium(I) and iridium(III) derivatives catalyze the dehydrogenation of formic acid in DMF, the iridium(I) compound [IrCl(HNP)2(cod)] being the most active species. A detailed study on the reaction mechanism operative in the dehydrogenation of formic acid catalysed by 3 is under run in order to elucidate the role of the NH groups as well as the influence of the structure of the iridium-aminophosphane platform on the observed catalytic activity.
Experimental
General section
All the operations were carried out using standard Schlenk tube techniques under an atmosphere of pre-purified argon or in a Braun glove-box under argon. Organic solvents were dried by standard procedures and distilled under argon or obtained oxygen- and water-free from a Solvent Purification System (Innovative Technologies). The compounds NH(4-tolyl)PPh2 (HNP),8 [Ir(μ-Cl)(cod)]2,14 and [Ir(CH3CN)2(cod)][PF6]15 were prepared according to the literature. [Ir(CH3CN)2(coe)2][PF6] was obtained as a spectroscopically pure solid following a procedure similar to that reported by Milstein for [Ir(acetone)2(coe)2][PF6].16 NMR spectra were recorded with Bruker spectrometers (AV300, AV400, and AV500) and are referred to SiMe4 (1H, 13C{1H}) and H3PO4 (31P{1H}). The proposed 1H, 13C{1H}, and 31P{1H} assignment relies on the combined analysis of 1D [1H, 1H{31P}, 13C{1H}-apt, 31P{1H}] and 2D NMR spectra (1H–1H COSY, 1H–1H NOESY, 1H–13C HSQC, 1H–13C HMBC, 1H–31P HMBC). For 5PF6 and 6PF6, olefinic carbon and hydrogen atoms are labelled as “up” or “down” if they point towards the hydride or if they do not, respectively, and as “trans” or “cis” if the HCCH moiety lies trans to phosphorus or cis to it, respectively. For 12PF6 and 13PF6, labels P1, P2, and P3 are used for non-equivalent phosphorus atoms, and, accordingly, superscript labels “tol-P1/2/3” and “PhP1/2/3” are used for hydrogen and carbon atoms belonging to the tolyl and phenyl groups attached/linked to the phosphorus atom P1/P2/P3. As for the reaction monitored by NMR spectroscopy at low temperature, the working temperature of 233 K was chosen in order to avoid the precipitation of reactants and/or products. C, H, and N analyses were carried out on a Perkin-Elmer 2400 CHNS/O analyzer. Infrared spectra were recorded on a Thermo Nicolet Avatar 360 FT-IT spectrometer on CH2Cl2 solutions using KBr windows (1 mm path).
Synthesis of SiMe3N(4-tolyl)PPh2 (SiMe3NP)
A solution of NH(4-tolyl)PPh2 (2.43 g, 8.34 mmol, 291.33 g mol−1) in THF (10 mL) and toluene (25 mL) at 198 K was treated with n-BuLi (1.6 M in hexane, 5.2 mL, 8.3 mmol). The mixture was allowed to warm up to room temperature (1 h) and SiMe3Cl (1.06 mL, 8.35 mmol, 0.856 g mL−1, 108.64 g mol−1) was added. After stirring overnight at room temperature, the suspension was heated at 333 K for 3 h. All the volatiles were removed in vacuo and the residue was extracted with CH2Cl2. The liquid phase was evaporated up to 3 mL and added with hexane (10 mL), affording the precipitation of a colourless solid, which was filtered off and washed with diethyl ether, dried in vacuo, and finally identified as SiMe3N(4-tolyl)PPh2 (1.61 g, 53% yield). Found: C 73.01, H 6.99, N 3.78. Calcd for C22H26NPSi (363.51 g mol−1): C 72.69, H 7.21, N 3.85. 1H NMR (C6D6, 298 K): δH 7.66–7.52 (4H, o-PPh), 7.23–7.14 (6H tot; 4H m-PPh, 2H p-PPh), 6.75 (d, 2H, 3JHH = 8.8 Hz, C3Htol), 6.73 (d, 2H, 3JHH = 8.8 Hz, C2Htol), 2.07 (s, 3H, CH3tol), 0.47 (d, 9H, 3JHP = 1.0 Hz, SiCH3). 13C{1H} NMR (C6D6, 298 K): δC 143.4 (d, 2JCP = 8.0 Hz, C1, tol), 140.7 (d, 1JCP = 17.7 Hz, C1, PhP), 134.7 (C4, tol), 134.2 (d, 2JCP = 21.1 Hz, C2, PhP), 130.9 (d, 3JCP = 1.3 Hz, C2, tol), 129.6 (C3, tol), 129.3 (C4, PhP C3, tol), 128.7 (d, 3JCP = 5.9 Hz, C3, PhP), 21.3 (CH3tol), 2.2 (d, 3JCP = 8.4 Hz, SiCH3), 31P{1H} NMR (C6D6, 298 K): δP 50.5 (s).![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) O), 169.1 (Cl2IrO), 142.8 (C1, tol), 138.3 (C4, tol), 134.9 (t, 1JCP = 34.0 Hz, C1, PhP), 133.7 (C4, PhP), 133.3 (t, 2JCP = 6.4 Hz, C2, PhP), 131.11 (C3, tol), 131.08 (d, 3JCP = 3.0 Hz C2, tol), 130.0 (t, 3JCP = 5.6 Hz, C3, PhP), 21.7 (CH3tol), 3.4 (d, 3JCP = 0.5 Hz, SiCH3). 31P{1H} NMR (CD2Cl2, 298 K): δP 42.9 (s).
O), 169.1 (Cl2IrO), 142.8 (C1, tol), 138.3 (C4, tol), 134.9 (t, 1JCP = 34.0 Hz, C1, PhP), 133.7 (C4, PhP), 133.3 (t, 2JCP = 6.4 Hz, C2, PhP), 131.11 (C3, tol), 131.08 (d, 3JCP = 3.0 Hz C2, tol), 130.0 (t, 3JCP = 5.6 Hz, C3, PhP), 21.7 (CH3tol), 3.4 (d, 3JCP = 0.5 Hz, SiCH3). 31P{1H} NMR (CD2Cl2, 298 K): δP 42.9 (s).
Method (2): An acetonitrile solution (8 mL) of [IrCl(HNP)(cod)] (227 mg, 0.361 mmol, 627.18 g mol−1) was added with TlPF6 (126 mg, 0.361 mmol, 349.94 g mol−1). The resulting pale yellow suspension was stirred for 1 h and evaporated in vacuo, affording a colorless solid that was extracted with dichloromethane (3 × 3 mL). The resulting pale yellow liquid phase was evaporated up to 1 mL and added with diethyl ether (5 mL), affording a colourless solid which was filtered off, washed with diethyl ether (3 × 5 mL), dried in vacuo and finally identified as [IrH{κ2C,P-HN(4-C6H3CH3)PPh2}(cod)(CH3CN)][PF6] (252 mg, 89% yield). Found: C 44.95, H 4.21, N 3.69. Calcd for C29H33F6IrN2P2 (777.74 g mol−1): C 44.78, H 4.28, N 3.60. 1H NMR (CD2Cl2, 298 K): δH 7.79 (dd, 2H, 3JHP = 12.9 Hz, 3JHH = 7.3 Hz, 2H o-PPh), 7.71–7.66 (3H tot; 2H, m-PPh; 1H, p-PPh), 7.65–7.55 (5H tot; 2H, o-PPh; 2H, m-PPh; 1H, p-PPh), 6.88 (d, 1H, 3JHH = 7.8 Hz, C6Htol), 6.83 (d, 1H, 3JHH = 7.8 Hz, C5Htol), 6.70 (br, 1H, C3Htol), 5.77 (m, 1H, Csp2Hcod, up-trans), 5.40 (m, 1H, Csp2Hcod down-trans), 5.22 (br, 1H, NH), 4.76 (dd, 3JHP = 9.5 Hz, 3JHH = 7.4 Hz 1H, Csp2Hcod up-cis), 4.61 (m, 1H, Csp2Hcod down-cis), 2.68 (m, 6H, Csp3Hcod), 2.49 (m, 2H, Csp3Hcod), 2.23 (s, 3H, CH3tol), 1.92 (s, 3H, CH3CN), −16.07 (d, 1H, 2JHP = 10.6 Hz, IrH). 13C{1H} NMR (CD2Cl2, 298 K): δC 151.6 (d, 2JCP = 16.2 Hz, C1, tol), 135.2 (C3, tol), 133.6 (d, 4JCP = 2.8 Hz, C4, PhP), 133.2 (d, 2JCP = 12.5 Hz, C2, PhP), 132.8 (d, 4JCP = 2.9 Hz, C4, PhP), 131.21 (d, 2JCP = 11.2 Hz, C2, PhP), 131.19 (d, 1JCP = 60.4 Hz, C1, PhP), 130.4 (d, 1JCP = 60.2 Hz, C1, PhP), 130.2 (d, 3JCP = 11.8 Hz, C3, PhP), 130.13 (d, 2JCP = 11.2 Hz, C3, PhP), 130.09 (C4, tol), 127.4 (br, C5, tol), 122.2 (CH3N), 122.0 (d, 2JCP = 2.9 Hz, C2, tol) 111.5 (d, 3JCP = 14.5 Hz, C6, tol), 100.6 (d, 2JCP = 10.6 Hz, Csp2, cod up-trans), 98.8 (d, 2JCP = 12.7 Hz, Csp2, cod down-trans), 92.93 (br, Csp2, cod up cis), 92.87 (Csp2, cod down-cis), 33.3 (d, 3JCP = 2.2 Hz, Csp3, cod), 32.4 (br, Csp3, cod), 30.3 (d, 3JCP = 2.8 Hz, Csp3, cod), 28.8 (d, 3JCP = 1.3 Hz, Csp3, cod), 21.2 (CH3tol), 2.31 (d, 3JCP = 1.2 Hz, SiCH3), 3.6 (H3CN). 31P{1H} NMR (CD2Cl2, 298 K): δP 62.5 (s, NP), −144.7 (hept, 1JPF = 710.1 Hz, PF6−).
Method (2): An acetonitrile solution (5 mL) of [IrCl{SiMe3N(4-C6H4CH3)PPh2}(cod)] (157 mg, 0.224 mmol, 699.18 g mol−1) was added with TlPF6 (78.5 mg, 0.224 mmol, 349.94 g mol−1). The resulting red solution was stirred for 1 h and evaporated in vacuo, affording a pale red solid which was eventually extracted with dichloromethane (2 × 5 mL). The resulting red liquid phase was evaporated up to 2 mL and added with hexane (5 mL), affording a pale red solid which was filtered off, washed with hexane (3 × 5 mL), dried in vacuo and finally identified as [IrH{κ2C,P-SiMe3N(4-C6H3CH3)PPh2}(cod)(CH3CN)][PF6] (154 mg, 81% yield). Found: C 44.99, H 4.95, N 3.22. Calcd for: C32H41F6IrN4P2Si (849.92 g mol−1): C 45.22, H 4.86, N 3.30. 1H NMR (CD2Cl2, 298 K): δH 7.89 (m, 2H, o-PPh), 7.73 (td, 2H, 3JHH = 7.3 Hz, 4JHP = 2.9 Hz m-PPh), 7.69–7.49 (6H tot; 2H, o-PPh; 2H, m-PPh; 2H, p-PPh), 7.14 (d, 1H, 3JHH = 8.2 Hz, C6Htol), 6.91 (d, 1H, 3JHH = 8.2 Hz, C5Htol), 6.72 (br, 1H, C3Htol), 5.54 (m, 1H, Csp2Hcod up-trans), 5.37 (m, 1H, Csp2Hcod down-trans), 4.69 (m, 1H, Csp2Hcod down-cis), 4.09 (t, 1H, 3JHH = 7.8 Hz, Csp2Hcod up-cis), 2.76 (m, 2H, Csp3Hcod), 2.50 (m, 4H, Csp3Hcod), 2.25 (s, 3H, CH3tol), 2.12 (m, 2H, Csp3Hcod), 1.80 (s, 3H, CH3CN), 0.14 (s, 9H, SiCH3), −16.43 (d, 1H, 2JHP = 11.2 Hz, IrH). 13C{1H} NMR (CD2Cl2, 298 K): δC 154.8 (d, 2JCP = 16.2 Hz, C1, tol), 135.7 (C3, tol), 132.8 (d, 1JCP = 73.7 Hz, C1, PhP), 132.6 (d, 4JCP = 2.8 Hz, C4, PhP), 132.1 (d, 4JCP = 2.9 Hz, C4, PhP), 131.8 (d, 2JCP = 11.8 Hz, C2, PhP), 131.2 (d, 1JCP = 74.7 Hz, C1, PhP),131.1 (d, 3JCP = 10.5 Hz, C2, PhP), 130.5 (C4, tol), 129.8 (d, 3JCP = 11.0 Hz, C3, PhP), 129.3 (d, 2JCP = 11.9 Hz, C3, PhP), 127.1 (d, 2JCP = 3.5 Hz, C2, tol), 126.2 (C5, tol), 121.4 (CH3N), 114.4 (d, 3JCP = 16.1 Hz, C6, tol), 101.1 (d, 2JCP = 10.6 Hz, Csp2, cod down-trans), 100.6 (d, 2JCP = 12.2 Hz, Csp2, cod up-trans), 94.9 (d, 2JCP = 12.2 Hz, Csp2, cod up-trans), 94.4 (Csp2, cod down-cis), 32.7 (d, 3JCP = 2.5 Hz, Csp3, cod), 31.5 (d, 3JCP = 1.2 Hz, Csp3, cod), 29.7 (d, 3JCP = 2.7 Hz, Csp3, cod), 28.1 (d, 3JCP = 2.0 Hz, Csp3, cod), 20.7 (CH3tol), 2.64 (d, 3JCP = 1.2 Hz, SiCH3), 2.63 (H3CN). 31P{1H} NMR (CD2Cl2, 298 K): δP 73.9 (s, NP), −144.7 (hept, 1JPF = 710.1 Hz, PF6−).
N), 21.5 (CH3tol), 4.2 (H3CN), 2.7 (SiCH3). 31P{1H} NMR (CD2Cl2, 298 K): δP 61.8 (s, 2P, NP), −144.7 (hept, 1JPF = 710.1 Hz, PF6−).
N), 119.6 (d, 3JCP = 5.3 Hz, C2, tol-P3), 119.0 (d, 3JCP = 5.4 Hz, C2, tol-P2), 112.2 (d, 3JCP = 14.0 Hz, C6, tol-P1), 21.4 (CH3tol-P1), 20.8 (CH3tol-P3, CH3tol-P2), 3.8 (H3CN). 31P{1H} NMR (CD3CN, 298 K): δP 49.8 (t, 1P, 2JCP = 11.5 Hz, NP1), 19.8 (dd, 1P, 2JCP = 16.0 Hz, 2JCP = 11.5 Hz, NP2), 11.2 (dd, 1P, 2JCP = 16.0 Hz, 2JCP = 11.5 Hz, NP3), −144.7 (hept, 1JPF = 710.1 Hz, PF6−).
N), 118.8 (C3, tol-P2/3), 113.5 (dd, 2JCP = 10.0 Hz, 2JCP = 1.6 Hz, C2, tol-P1), 111.7 (d, 3JCP = 13.4 Hz, C6, tol-P1), 21.3 (CH3tol-P1), 20.8 (CH3tol-P2/3), 2.5 (H3CN). 31P{1H} NMR (CD2Cl2, 298 K): δP 43.4 (t, 1P, 2JPP = 16.0 Hz, NP1), 19.7 (d, 2P, 2JPP = 16.0 Hz, NP2/3), −144.7 (hept, 1JPF = 710.1 Hz, PF6−).
Method (2): A CD2Cl2 solution (0.4 mL) of 12PF6 (14.8 mg, 0.0118 mmol, 1252.21 g mol−1) was allowed to stand at room temperature for 12 h in a standard 5 mm NMR tube. 1H and 31P{1H} NMR indicated the clean and complete conversion of OC-6-34-[IrH{κ2C,P-HN(4-C6H3CH3)PPh2}(HNP)2(CH3CN)]+ to OC-6-52-[IrH{κ2C,P-HN(4-C6H3CH3)PPh2}(HNP)2(CH3CN)]+.
General procedure for the catalytic dehydrogenation of formic acid
The catalytic tests were carried out on a Man on the Moon kit (Series X102, https://www.manonthemoontech.com/x102-gas-evolu-tion.html), employing a reactor with a total volume of 19 mL. A DMF solution (1.7 mL) of formic acid (20 μL, 0.529 mmol) and sodium formate or trimethylamine (10 mol%, 0.0529 mmol; 50 mol%, 0.264 mmol) was introduced into the reactor and placed in a thermostated oil bath at 353 K. After 10 min approx., a solution of the catalyst (1 mol%, 0.00529 mmol; 5 mol%, 0.0264 mmol) in 0.3 mL of DMF was added and the recording of the curve pressure vs. time started (cf.Table 1).DFT calculations
Molecular structure optimizations and frequencies calculations were carried out with the Gaussian09 program (revision D.01)17 using the method B3PW91,18 including the D3 dispersion correction scheme by Grimme with Becke–Johnson damping.19 The def2-SVP20 basis and pseudo potential were used for all atoms and the “ultrafine” grid was employed in all calculations. Stationary points were characterized by vibrational analysis. The structures were optimized in dichloromethane (298 K, 1 atm) using the PCM method.21 In order to improve the accuracy of the calculated energies, in selected cases, single point energy calculations were carried out on the optimized structures of intermediates and transition states using the method M06,22 the def2-TZVP20 basis and pseudo potentials, where appropriate, and the SMD model23 for the solvent (dichloromethane). Delocalization indexes (DI) were calculated using Multiwfn.24Crystal structure determination
Single crystals of SiMe3NP, 1, 2, and 6PF6 were obtained by slow evaporation of dichloromethane solutions of the compounds; single crystals of 12PF6 were grown by slow diffusion of diethyl ether into a CH3CN solution of the compound. X-ray diffraction data were collected at 100(2) K on a SMART APEX (1), APEX DUO (2, 6PF6) or D8 VENTURE (SiMe3NP, 12PF6) Bruker diffractometers with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) using ω-scans (and φ-scans for SiMe3NP and 12PF6). Intensities were integrated and corrected for absorption effects with SAINT-PLUS25 and SADABS26 programs, both included in APEX4 package. The structures of 1, 6PF6, and 12PF6 were solved by the Patterson method with SHELXS-9727 and refined by full matrix least-squares on F2 with SHELXL-201428 under WinGX.29
The structure of 2 was solved with the ShelXS-201330 solution program using direct methods and by using Olex2 1.5-dev31 as the graphical interface. The model was refined with olex2.refine 1.5-dev32 using full matrix least squares minimisation on F2. Eventually the crystal structure of 2 was refined using NoSpherA2, an implementation of NOn-SPHERical Atom-form-factors in Olex2.33 NoSpherA2 implementation of Hirshfeld atom refinement makes use of tailor-made aspherical atomic form factors calculated on-the-fly from a Hirshfeld-partitioned electron density (ED), not from spherical-atom form factors. The ED is calculated from a gaussian basis set single determinant SCF wavefunction – either Hartree–Fock or DFT using selected funtionals – for a fragment of the crystal. This fragment can be embedded in an electrostatic crystal field by employing cluster charges or modelled using implicit solvation models, depending on the software used. The following options were used: software, Orca 5.0; partitioning, NoSpherA2; int. accuracy, normal; method, r2scan; basis set: x2c-tzvp; charge: 0; multiplicity: 1; relativistic: dkh2.
Crystal data and structure refinement for SiMe3N(4-tolyl)PPh2 (SiMe3NP)
C22H26NPSi, 363.50 g mol−1, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 8.6970(3) Å, b = 11.2407(4) Å, c = 11.9717(4) Å, α = 117.9100(10)°, β = 97.2470(10)°, γ = 95.1980(10)°, V = 1010.90(6) Å, Z = 2, Dcalc = 1.194 g cm3, μ = 0.200 mm−1, F(000) = 388, colourless prism, 0.190 × 0.180 × 0.120 mm, θmin/θmax 2.053/28.276°, −11 ≤ h ≤ 11, −14 ≤ k ≤ 13, −15 ≤ l ≤ 15, reflections collected/independent 39891/4998 [R(int) = 0.0300], Tmax/Tmin 0.7457/0.7210, data/restraints/parameters 4998/0/230, GooF(F2) 1.051, R1 = 0.0327 [I > 2σ(I)], wR2 = 0.0876 (all data), largest diff. peak/hole 0.368/−0.261 e Å−3. CCDC deposit number 2178754.†
, a = 8.6970(3) Å, b = 11.2407(4) Å, c = 11.9717(4) Å, α = 117.9100(10)°, β = 97.2470(10)°, γ = 95.1980(10)°, V = 1010.90(6) Å, Z = 2, Dcalc = 1.194 g cm3, μ = 0.200 mm−1, F(000) = 388, colourless prism, 0.190 × 0.180 × 0.120 mm, θmin/θmax 2.053/28.276°, −11 ≤ h ≤ 11, −14 ≤ k ≤ 13, −15 ≤ l ≤ 15, reflections collected/independent 39891/4998 [R(int) = 0.0300], Tmax/Tmin 0.7457/0.7210, data/restraints/parameters 4998/0/230, GooF(F2) 1.051, R1 = 0.0327 [I > 2σ(I)], wR2 = 0.0876 (all data), largest diff. peak/hole 0.368/−0.261 e Å−3. CCDC deposit number 2178754.†
Crystal data and structure refinement for [IrCl(HNP)(cod)] (1)
C27H30ClIrNP, 627.14 g mol−1, monoclinic, P21/c, a = 18.6429(17) Å, b = 10.8746(10) Å, c = 12.3111(11) Å, β = 104.9190(10)°, V = 2411.7(4) Å3, Z = 4, Dcalc = 1.727 g cm−3, μ = 5.728 mm−1, F(000) = 1232, orange prism, 0.250 × 0.180 × 0.160 mm, θmin/θmax 2.188/28.755°, −24 ≤ h ≤ 25, −14≤k ≤ 14, −16≤l ≤ 16, reflections collected/independent 33669/5870 [R(int) = 0.0444], Tmax/Tmin 0.7458/0.6264, data/restraints/parameters 5870/1/285, GooF(F2) 1.028, R1 = 0.0270 [I > 2σ(I)], wR2 = 0.0604 (all data), largest diff. peak/hole 1.495/−0.949 e Å−3. CCDC deposit number 2178757.†
Crystal data and structure refinement for [IrCl(SiMe3NP)(cod)] (2)
C30H38ClIrNPSi, 699.37 g mol−1, triclinic, P, a = 9.4160(10) Å, b = 10.5553(11) Å, c = 14.9182(16) Å, α = 86.0960(10)°, β = 74.7700(10)°, γ = 76.9410(10)°, V = 1393.6(3) Å3, Z = 2, Dcalc = 1.667 g cm−3, μ = 5.007 mm−1, F(000) = 696, orange prism, 0.300 × 0.190 × 0.180 mm, θmin/θmax 1.41/28.37°, −11 ≤ h ≤ 12, −14 ≤ k ≤ 14, 0 ≤ l≤19, reflections collected/independent 18124/6925 [R(int) = 0.0158], Tmin/Tmax 0.3302/0.2449, data/restraints/parameters 6925/21/658, GooF(F2) 1.064, R1 = 0.0115 [I > 2σ(I)], wR2 = 0.0261 (all data), largest diff. peak/hole 1.092/−0.443 e Å−3. CCDC deposit number 2178753.†
Crystal data and structure refinement for [IrH{κ2C,P-SiMe3N(4-C6H3CH3)PPh2}(cod)(CH3CN)][PF6] (6PF6)
C32H41F6IrN2P2Si, 849.90 g mol−1, triclinic, P, a = 15.413(5) Å, b = 16.528(6) Å, c = 28.517(10) Å, α = 106.793(6)°, β = 94.609(7)°, γ = 102.683(8)°, V = 6704(4) Å3, Z = 8, Dcalc = 1.684 g cm−3, μ = 4.173 mm−1, F(000) = 3376, yellow prism, 0.210 × 0.150 × 0.050 mm, θmin/θmax 0.755/26.372°, −19 ≤ h ≤ 19, −20 ≤ k ≤ 20, −35 ≤ l ≤ 35, reflections collected/independent 121535/27407 [R(int) = 0.0464], Tmax/Tmin 0.5959/0.4686, data/restraints/parameters 27407/4/1621, GooF(F2) 1.018, R1 = 0.0309 [I > 2σ(I)], wR2 = 0.0695 (all data), largest diff. peak/hole 2.061/−0.848 e Å−3. CCDC deposit number 2178755.†
Crystal data and structure refinement for [IrH{κ2C,P-HN(4-C6H3CH3)PPh2}(HNP)2(CH3CN)][PF6] (12PF6)
2[C59H57IrN4P3][PF6]·3 CH3CN·C4H10O, 2701.61 g mol−1, monoclinic, P21/c, a = 24.8378(10) Å, b = 23.4102(8) Å, c = 20.6295(8) Å, β = 91.556(2)°, V = 11990.8(8) Å3, Z = 4, Dcalc = 1.497 g cm−3, μ = 2.399 mm−1, F(000) = 5472, colourless prism, 0.190 × 0.040 × 0.020 mm3, θmin/θmax 1.975/25.681°, index ranges −30 ≤ h ≤ 30, −28 ≤ k ≤ 28, −25 ≤ l ≤ 25, reflections collected/independent 337915/22758 [R(int) = 0.0705], Tmax/Tmin 0.8621/0.7473, data/restraints/parameters 22758/10/1504, GooF(F2) 1.056, R1 = 0.0339 [I > 2σ(I)], wR2 = 0.0860 (all data), largest diff. peak/hole 3.415/−0.872 e Å−3. CCDC deposit number 2178756.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministerio de Ciencia e Innovación MCIN/AEI/10.13039/501100011033, under the project PID2019-103965GB-I00, and the Departamento de Ciencia, Universidad y Sociedad del Conocimiento del Gobierno de Aragón (group E42_20R) is gratefully acknowledged. Authors are grateful for the reviewer's insightful comments on the refinement of the crystal structure of 2 using non-spherical form factors.References
- (a) G. Ewart, D. S. Payne, A. L. Porte and A. P. Lane, J. Chem. Soc., 1962, 3984 RSC; (b) H. H. Sisler and N. L. Smith, J. Org. Chem., 1961, 26, 611 CrossRef CAS; (c) W. A. Hart and H. H. Sisler, Inorg. Chem., 1964, 3, 617–622 CrossRef CAS.
- (a) V. Passarelli, J. J. Pérez-Torrente and L. A. Oro, Dalton Trans., 2016, 45, 951–962 RSC; (b) J. Jansa, T. Řezníček, L. Dostál, Z. Růžičková, F. Bureš and R. Jambor, Appl. Organomet. Chem., 2016, 30, 1036–1042 CrossRef CAS; (c) S. Pavlik, K. Mereiter, R. Schmid and K. Kirchner, Organometallics, 2003, 22, 1771–1774 CrossRef CAS; (d) A. D. Burrows, M. F. Mahon and M. T. Palmer, J. Chem. Soc., Dalton Trans., 2000, 1669–1677 RSC; (e) L. Melounková, M. Syková, R. Jirásko, R. Jambor, R. Havelek, E. Peterová, J. Honzíček and J. Vinklárek, New J. Chem., 2021, 45, 19506–19514 RSC; (f) P. Chen, Y. Li, Z. Chen, W. Du and Y. Chen, Angew. Chem., Int. Ed., 2020, 59, 7083–7088 CrossRef CAS PubMed; (g) K. G. Gaw, M. B. Smith, J. B. Wright, A. M. Z. Slawin, S. J. Coles, M. B. Hursthouse and G. J. Tizzard, J. Organomet. Chem., 2012, 699, 39–47 CrossRef CAS; (h) M. Aydemir, A. Baysal, N. Gürbüz, I. Özdemir, B. Gümgüm, S. Özkar, N. Çaylak and L. T. Yildirim, Appl. Organomet. Chem., 2010, 24, 17–24 CrossRef CAS; (i) S. Priya, M. S. Balakrishna and J. T. Mague, J. Organomet. Chem., 2003, 679, 116–124 CrossRef CAS.
- (a) Y. Chen, D. Song, J. Li, X. Hu, X. Bi, T. Jiang and Z. Hou, ChemCatChem, 2018, 10, 159–164 CrossRef CAS; (b) H. Lin, Y. Li, J. Wang, M. Zhang, T. Jiang, J. Li and Y. Chen, Appl. Organomet. Chem., 2021, 35, e6345, DOI:10.1002/aoc.6345.
- Selected references are: (a) B. S. Mitchell, W. Kaminsky and A. Velian, Inorg. Chem., 2021, 60, 6135–6139 CrossRef CAS PubMed; (b) A. Aloisi, É. Crochet, E. Nicolas, J.-C. Berthet, C. Lescot, P. Thuéry and T. Cantat, Organometallics, 2021, 40, 2064–2069 CrossRef CAS; (c) J. A. Kephart, A. C. Boggiano, W. Kaminsky and A. Velian, Dalton Trans., 2020, 49, 16464–16473 RSC; (d) H. Zhang, G. P. Hatzis, C. E. Moore, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2019, 141, 9516–9520 CrossRef CAS PubMed; (e) J. A. Kephart, B. S. Mitchell, A. Chirila, K. J. Anderton, D. Rogers, W. Kaminsky and A. Velian, J. Am. Chem. Soc., 2019, 141, 19605–19610 CAS; (f) K. M. Gramigna, D. A. Dickie, B. M. Foxman and C. M. Thomas, ACS Catal., 2019, 9, 3153–3164 CrossRef CAS; (g) M. L. Bin Ismail and C.-W. So, Chem. Commun., 2019, 55, 2074–2077 RSC; (h) B. A. Barden, G. Culcu, J. P. Krogman, M. W. Bezpalko, G. P. Hatzis, D. A. Dickie, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2019, 58, 821–833 CrossRef CAS PubMed; (i) A. J. Ayres, A. J. Wooles, M. Zegke, F. Tuna and S. T. Liddle, Inorg. Chem., 2019, 58, 13077–13089 CrossRef CAS PubMed.
- (a) K. A. Grice, W. Kaminsky and K. I. Goldberg, Inorg. Chim. Acta, 2011, 369, 76–81 CrossRef CAS; (b) D. Wang, M. Li, X. Chen, M. Wang, Y. Liang, Y. Zhao, K. N. Houk and Z. Shi, Angew. Chem., Int. Ed., 2021, 60, 7066–7071 CrossRef CAS PubMed; (c) K. G. Gaw, A. M. Z. Slawin and M. B. Smith, Organometallics, 1999, 18, 3255–3257 CrossRef CAS.
- Selected references are: (a) A. Luque-Gómez, S. García-Abellán, J. Munarriz, V. Polo, V. Passarelli and M. Iglesias, Inorg. Chem., 2021, 60, 15497–15508 CrossRef PubMed; (b) N. Onishi, R. Kanega, E. Fujita and Y. Himeda, Adv. Synth. Catal., 2019, 361, 289–296 CrossRef CAS; (c) S.-M. Lu, Z. Wang, J. Wang, J. Li and C. Li, Green Chem., 2018, 20, 1835–1840 RSC; (d) S. Cohen, V. Borin, I. Schapiro, S. Musa, S. De-Botton, N. V. Belkova and D. Gelman, ACS Catal., 2017, 7, 8139–8146 CrossRef CAS; (e) S. Siek, D. B. Burks, D. L. Gerlach, G. Liang, J. M. Tesh, C. R. Thompson, F. Qu, J. E. Shankwitz, R. M. Vasquez, N. Chambers, G. J. Szulczewski, D. B. Grotjahn, C. E. Webster and E. T. Papish, Organometallics, 2017, 36, 1091–1106 CrossRef CAS PubMed; (f) C. Fink and G. Laurenczy, Dalton Trans., 2017, 46, 1670–1676 RSC; (g) J. Li, J. Li, D. Zhang and C. Liu, ACS Catal., 2016, 6, 4746–4754 CrossRef CAS; (h) J. J. A. Celaje, Z. Lu, E. A. Kedzie, N. J. Terrile, J. N. Lo and T. J. Williams, Nat. Commun., 2016, 7, 11308 CrossRef CAS PubMed; (i) J. H. Barnard, C. Wang, N. G. Berry and J. Xiao, Chem. Sci., 2013, 4, 1234–1244 RSC; (j) J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed; (k) Y. Himeda, Green Chem., 2009, 11, 2018–2022 RSC.
- M. Iglesias and L. A. Oro, Eur. J. Inorg. Chem., 2018, 2125–2138 CrossRef CAS.
- V. Passarelli and F. Benetollo, Inorg. Chem., 2011, 50, 9958–9967 CrossRef CAS PubMed.
- M. Palmese, V. Passarelli and J. J. Pérez-Torrente, Dalton Trans., 2022, 51, 7142–7153 RSC.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS PubMed.
- Nomenclature of Inorganic Chemistry, IUPAC Recommendations 2005, the Red Book, compiled by N. G. Connelly, R. M. Hartshorn, T. Damhus and A. T. Hutton, Published for the International Union of Pure and Applied Chemistry by The Royal Society of Chemistry, Thomas Graham House, Science Park, Milton Road, Cambridge CB4 0WF, UK ISBN 0-85404-438-8.
- V. Passarelli, J. J. Pérez-Torrente and L. A. Oro, Inorg. Chem., 2014, 53, 972–980 CrossRef CAS PubMed.
- E. Matito, J. Poater, M. Solá, M. Duran and P. Salvador, J. Phys. Chem. A, 2005, 109, 9904–9910 CrossRef CAS PubMed.
- J. L. Herde, J. C. Lambert and C. V. Senoff, Inorg. Synth., 1974, 15, 18–20 CAS.
- V. W. Day, W. G. Klemperer and D. J. Main, Inorg. Chem., 1990, 29, 2345–2355 CrossRef CAS.
- R. Dorta, R. Goikhman and D. Milstein, Organometallics, 2003, 22, 2806–2809 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, C. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- J. P. Perdew, in Electronic Structure of Solids ‘91, ed. P. Ziesche and H. Eschrig, Akademie Verlag, Berlin, 1991 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- SAINT+: Area-Detector Integration Software, version 6.01, Bruker AXS, Madison, WI, 2001 Search PubMed.
- G. M. Sheldrick, SADABS program, University of Göttingen, Göttingen, Germany, 1999 Search PubMed.
- G. M. Sheldrick, SHELXS 97, Program for the Solution of Crystal Structure, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 339–341 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59–71 CrossRef CAS PubMed.
- F. Kleemiss, O. V. Dolomanov, M. Bodensteiner, N. Peyerimhoff, M. Midgley, L. J. Bourhis, A. Genoni, L. A. Malaspina, D. Jayatilaka, J. L. Spencer, F. White, B. Grundkoetter-Stock, S. Steinhauer, D. Lentz, H. Puschmann and S. Grabowsky, Chem. Sci., 2021, 12, 1675–1692 RSC.
- M. J. Church, M. J. Mays, R. N. F. Simpson and F. P. Stefanini, J. Chem. Soc. (A), 1970, 2909–2914 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystal structures, IR and NMR spectra, NMR data, coordinates of calculated structures. CCDC 2178753–2178757. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02081e |
| ‡ Note that all non-observed compounds (either expected or calculated) are labelled with sequential roman numerals, whereas sequential arabic numerals have been used exclusively for observed/isolated compounds, including isomers. |
| § For the sake of comparison, the IR spectrum of the related complex [Ir(CO)3(PPh3)2]+ shows one weak band at 2074 cm–1 (A1 mode) and two strong bands at 2010 and 2018 cm–1 (split E mode), cf. ref. 34 |
| ¶ A CD2Cl2 (0.5 mL) solution of [Ir(CH3CN)2(cod)][PF6] (0.031 mmol) at 233 K was added with a CD2Cl2 solution (0.5 mL) of HNP (0.031 mmol) and introduced into an NMR spectrometer at 233 K. |
| || As far as the equilibrium 7+ ⇄ 8+ is concerned, 1H–1H EXSY crosspeaks have been observed between the following groups of signals (see Fig. 7 for labelling): (a) 2.01 (MeII8+), 2.10 (Me 7+), 2.20 (MeI8+), (b) 5.96 (NHI8+), 5.57 (NHII8+), 4.43 (NH 7+); (b) –9.90 (IrH 8+), 5.87 (C2HII8+), 7.19 (C6HI8+); (c) 4.36 (CHcod7+), 5.29 (CdH 8+), 4.01 (CbH 8+); d) 6.14 (C3HII8+), 6.59 (C3HI8+), 6.72 (C3H 7+), 6.96 (C5HI8+)]. |
| ** A CD2Cl2 solution (0.5 mL) of 5PF6 or 6PF6 (0.021 mmol) was added with [NBu4]Cl (0.021 mmol). Soon after mixing, the clean formation of 1 and 2, respectively, was observed (31P{1H} NMR spectroscopy). |
| †† [Ir(HNP)3(cod)]+ (9+) was prepared by reaction of HNP and [Ir(cod)(CH3CN)2][PF6] (Ir:HNP = 1:3) at 223 K in CD2Cl2 as a thermally unstable compound and was spectroscopically identified in situ in the presence of about 20 mol% of unidentified by-products. At 233 K, its 31P{1H} NMR spectrum shows a broad signal at 18.5 ppm (Δν1/2 = 26 Hz). Accordingly, one 1H singlet is observed at 2.19 ppm for the methyl moiety of the tolyl groups and one broad 1H signal at 3.41 ppm is observed for the cod HCCH moieties. On this ground, [Ir(HNP)3(cod)]+ is likely to undergo a fluxional process exchanging the three HNP ligands as well as averaging the coordinated cod ligand. In this regard, similar to 3, an equilibrium between square pyramidal (SPY-3-12) and trigonal bipyramidal structures (TBPY-5-12) may account for the observed solution behaviour. |
| ‡‡ The transition state TS_VIII+-X+ could not be located on the PES. So, the reported activation barrier was estimated (ΔEact ≈ +16.7 kcal·mol–1) scanning the C–H coordinate of the carbon–hydrogen bond undergoing oxidative addition to the iridium centre. |
| §§ The dissociative route was also considered and finally discarded since the tricoordinate intermediate [Ir(HNP)2(CH3CN)]+ was calculated to lie at +14.5 kcal mol–1vs.VIII+. |
| ¶¶ Actually, the meridional disposition of the hydrido ligand, the phosphorus and carbon (tolyl) atoms within the IrH{κ2C,P-HN(4-C6H3CH3)PPh2} moiety may only result from a parallel arrangement of the reacting C–H bond and the iridium–phosphorus bond, which is incompatible with the topology of the Ir(HNP) fragment. In other words, the tether of the tolyl group to the phosphorus atom restricts the reacting C–H bond to lying perpendicular to the iridium–phosphorus bond (of the aminophosphano ligand undergoing the CH oxidative addition), which may uniquely affords a mutually cis disposition of the hydrido ligand, and the phosphorus and carbon (tolyl) atoms (cf.TS_XII+-12+, Fig. 13B). |
| This journal is © The Royal Society of Chemistry 2022 |