
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterisation and reactivity of group 2 complexes with a thiopyridyl scorpionate ligand†
Matthew P.
Stevens
 a,
Emily
Spray
a,
Iñigo J.
Vitorica-Yrezabal
b,
Kuldip
Singh
a,
Vanessa M.
Timmermann
a,
Lia
Sotorrios
c,
Stuart A.
Macgregor
*c and
Fabrizio
Ortu
*a
a,
Emily
Spray
a,
Iñigo J.
Vitorica-Yrezabal
b,
Kuldip
Singh
a,
Vanessa M.
Timmermann
a,
Lia
Sotorrios
c,
Stuart A.
Macgregor
*c and
Fabrizio
Ortu
*a
aSchool of Chemistry, University of Leicester, University Road, LE1 7RH Leicester, UK. E-mail: fabrizio.ortu@leicester.ac.uk
bDepartment of Chemistry, The University of Manchester, Oxford Road, M13 9PL Manchester, UK
cInstitute of Chemical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. E-mail: s.a.macgregor@hw.ac.uk
First published on 15th July 2022
Abstract
Herein we report the reactivity of the proligand tris(2-pyridylthio)methane (HTptm) with various Alkaline Earth (AE) reagents: (1) dialkylmagnesium reagents and (2) AE bis-amides (AE = Mg–Ba). Heteroleptic complexes of general formulae [Mg(Tptm)(R)] (R = Me, nBu; Tptm = {C(S–C5H4N)3}−) and [AE(Tptm)(N′′)] (AE = Mg–Ba; N′′ = {N(SiMe3)2}−) were targeted from the reaction of HTptm with R2Mg or [AE(N′′)2]2. Reaction of the proligand with dialkylmagnesium reagents led to formation of [{Mg(κ3C,N,N–C{Bu}{S–C5H4N}2)(μ-S–C5H4N)}2] (1) and [{Mg(κ3C,N,N–C{Me}{S–C5H4N}2)(μ-OSiMe3)}2] (2) respectively, as a result of a novel transfer of an alkyl group onto the methanide carbon with concomitant C–S bond cleavage. However, reactivity of bis-amide precursors for Mg and Ca did afford the target species [AE(Tptm)(N′′)] (3-AE; AE = Mg–Ca), although these proved susceptible to ligand degradation processes. DFT calculations show that alkyl transfer in the putative [Mg(Tptm)(nBu)] (1m′) system and amide transfer in 3-Ca is a facile process that induces C–S bond cleavage in the Tptm ligand. 3-Mg and 3-Ca were also tested as catalysts for the hydrophosphination of selected alkenes and alkynes, including the first example of mono-hydrophosphination of 4-ethynylpyridine which was achieved with high conversions and excellent regio- and stereochemical control.
1. Introduction
The organometallic chemistry of the Alkaline Earth (AE) metals has undergone a true renaissance over the last three decades.1–4 A lot of the interest around these metals arises from their high abundance in the Earth's crust and low toxicity, with the notable exceptions of beryllium and radium.5 The abundances of the group 2 metals are far greater than those of precious metals commonly employed in industrial processes, e.g. the Fischer–Tropsch process (Ru),6 the Monsanto process (Rh),7 the Wacker process (Pd)8 and oil reforming (Pt).9 A lot of effort is being put into breaking the reliance of industrial chemistry on precious metals and to move instead towards more Earth-abundant materials.10 Over the last four decades AE metal complexes have been studied as catalysts for a variety of reactions including hydroelementation, polymerisation and hydrogenation.11–14 Besides catalytic applications, the organometallic chemistry of magnesium is extremely well established,3 whilst analogous chemistry with the heavy AEs has historically lagged behind. Nonetheless, a lot of progress has been achieved in the isolation of heavy Grignard reagents in recent years.2,15 Heavy AE organometallics are difficult to handle16,17 and suffer with multiple degradation pathways, including solvent degradation and Wurtz coupling.18–21 The main reasons for this high reactivity are the strong electropositive character, large ionic radii and the very polarised AE–C bonds.22 Strategies for stabilising heavy AE organometallics usually involve a combination of steric and electronic stabilisation, a typical example being the use of bulky silyl substituents around the carbon donor.23–25 In general, the use of monodentate ligands is extremely challenging for the heavy AEs due to their high steric demands26 and a lot of effort has been put into using multidentate ligands, which include β-diketiminates,27 macrocycles,28 and tripodal scorpionates.29,30 C-Donor functionalities can also be incorporated into multidentate ligands for the stabilisation of AE organometallics, such as bis(phosphoranyl)-methanide and -methanediide ligands,31–39 and various tripodal systems.40,41 In 2008 Kitano et al. reported the use of the scorpionate ligand tris(2-pyridylthio)methanide ({C(S–C5H4N)3}−; Tptm) for the stabilisation of the heteroleptic zinc complexes [Zn(Tptm)(X)] (X = Cl, Br) via salt elimination.42 Parkin and co-workers further developed this chemistry by including various functionalities (e.g. alkyl, alkoxide, azide)43–45 and also extended this to heavier group 12 analogues.46 Group 12 and AE metals have very similar coordination chemistry owing to their dominant and stable +2 oxidation state and similar ionic radii (6-coordinate ionic radii: Zn 0.74 Å, Mg 0.72 Å, Cd 0.95 Å, Ca 1.00 Å).47 Therefore, we decided to investigate the use of Tptm as a supporting ligand for obtaining heteroleptic AE complexes which could function as catalysts in hydroelementation reactions. Herein, we report the synthesis of AE derivatives supported by Tptm obtained via protonolysis routes, together with some unexpected reactivity pathways affecting the ligand scaffold and applications in catalytic hydrophosphination reactions.2. Synthesis and NMR spectroscopic characterisation
Heteroleptic complexes of general formulae [Mg(Tptm)(R)] (R = Me, nBu) and [AE(Tptm)(N′′)] (AE = Mg–Ba; N′′ = {N(SiMe3)2}−) were targeted from the reaction of HTptm with R2Mg (R = Me, nBu) or [AE(N′′)2]2. Reactions of the proligand with dialkylmagnesium reagents were carried out in diethyl ether at room temperature; unexpectedly we could not obtain the target [Mg(Tptm)(R)] complexes, and the dimeric species [{Mg(κ3C,N,N–C{Bu}{S–C5H4N}2)(μ-S–C5H4N)}2] (1) and [{Mg(κ3C,N,N–C{Me}{S–C5H4N}2)(μ-OSiMe3)}2] (2) were isolated instead (Scheme 1). This indicated a novel alkyl migration onto the methanide carbon accompanied by C–S bond cleavage of the Tptm scaffold. Interestingly, these alkyl migrations are not observed for the analogous complex [Zn(κ3-Tptm)(Me)] reported by Parkin and co-workers, thus highlighting the unique role of the metal in promoting this pathway.43 Complex 1 was obtained consistently from this reaction (27% crystalline yield) upon repeated attempts, and separated from a mixture of products which we could not unequivocally identify (vide infra; also see ESI, Fig. S5 and S6†). In the case of the reaction with Me2Mg, 2 was likely formed from the degradation of a species analogous to 1 in the presence of silicone grease and was isolated in very small yields. Also in this case it was not possible to identify other species obtained from the reaction (see ESI, Fig. S7 and S8†). Nonetheless, reactivity involving bis-amide precursors of Mg and Ca afforded target species [AE(Tptm)(N′′)] (3-AE; AE = Mg–Ca). In the case of 3-Ca, the reaction and follow-up purification steps had to be carried out at low temperature (<−10 °C) due to its temperature instability. Such sensitivity is exacerbated in the case of heavier analogues 3-Sr and 3-Ba. The former of these could not be isolated in pure form, despite strict temperature control and quick work-up times (see ESI, Fig. S30 and S31†), whilst reaction between HTptm and [Ba(N′′)2]2 led to an intractable mixture (see ESI, Fig. S32†). Despite the well-recorded problems of using ethereal solvents with heavy organo-AE metal complexes,48,49 no issues of this type were observed in these systems and we ascribe the thermal instability to the availability of facile decomposition pathways for the supporting ligand (vide infra). | ||
| Scheme 1 Synthesis of 1–3via protonolysis between HTptm and nBu2Mg or [Ae(N′′)2]2 (AE = Mg–Ba). | ||
1H NMR analysis of 1 revealed a set of signals in the aromatic region (6.0–8.8 ppm) which could be assigned to the newly formed methanide {κ3C,N,N–C(Bu)(S–C5H4N)2}− ligand and the bridging thiopyridyl ligand, whilst signals belonging to the butyl fragment were also identified in the alkyl region (0.8–3.0 ppm). 1H NMR analysis of the crude mixture prior to recrystallization reveals that several species are present, which include unreacted nBu2Mg, 1 and 4-Mg (vide infra), together with other species that we could not unequivocally identify. Interestingly, 1 is only a minor component of the crude mixture, therefore it could not be excluded that its formation takes place during the crystallisation procedure. In the case of 2, analysis of crude products prior to recrystallisation afforded a spectrum similar to 1, with the additional presence of signals which can be assigned to various decomposition products, including [Mg{κ3N,N-CH(S–C5H4N)}2] (5) (vide infra and ESI, Fig. S6 and S7†).
3-Mg and 3-Ca were fully characterised via multinuclear NMR spectroscopy (Fig. S8–S10 and S11–S13†). The 1H NMR spectrum of 3-Mg shows a set of three multiplets in the aromatic region (δH = 8.98–9.04, 6.45–6.55 and 6.28–6.33 ppm) and with relative integration values of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:3 corresponding to the protons of the pyridyl fragments of the Tptm ligand. Additionally, one strong signal is present in the aliphatic region (δH = 0.41 ppm, 9H) which can be attributed to the protons of the N′′ ligand. 13C{1H} data are also in good agreement with the proposed formulation, revealing five distinct environments for the pyridyl carbon atoms (δC = 118.6, 120.4, 138.0, 151.8 and 164.8 ppm). We were also able to identify the signal of the methanide carbon donor (δC = 34.2 ppm), which is very difficult to detect and was not reported previously for the analogous complex [Zn(Tptm)(N′′)].50 Finally, the 29Si{1H} spectrum of 3-Mg shows one single resonance at −10.5 ppm. The multinuclear NMR spectra of 3-Ca are analogous to those of 3-Mg, though the resonance of the methanide carbon in the 13C{1H} is significantly shifted upfield (δC = 5.6 ppm); additionally, the SiMe3 signal in the 29Si{1H} NMR spectrum is shifted with respect to 3-Mg, resonating at −14.7 ppm. In both 3-Mg and 3-Ca, the 29Si{1H} signal is noticeably downfield shifted from the corresponding [AE(N′′)2]2 signals at 11.6 and −1.3 ppm respectively.51
:6:3 corresponding to the protons of the pyridyl fragments of the Tptm ligand. Additionally, one strong signal is present in the aliphatic region (δH = 0.41 ppm, 9H) which can be attributed to the protons of the N′′ ligand. 13C{1H} data are also in good agreement with the proposed formulation, revealing five distinct environments for the pyridyl carbon atoms (δC = 118.6, 120.4, 138.0, 151.8 and 164.8 ppm). We were also able to identify the signal of the methanide carbon donor (δC = 34.2 ppm), which is very difficult to detect and was not reported previously for the analogous complex [Zn(Tptm)(N′′)].50 Finally, the 29Si{1H} spectrum of 3-Mg shows one single resonance at −10.5 ppm. The multinuclear NMR spectra of 3-Ca are analogous to those of 3-Mg, though the resonance of the methanide carbon in the 13C{1H} is significantly shifted upfield (δC = 5.6 ppm); additionally, the SiMe3 signal in the 29Si{1H} NMR spectrum is shifted with respect to 3-Mg, resonating at −14.7 ppm. In both 3-Mg and 3-Ca, the 29Si{1H} signal is noticeably downfield shifted from the corresponding [AE(N′′)2]2 signals at 11.6 and −1.3 ppm respectively.51
3. Structural analysis
Compound 1 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and features two 6-coordinate Mg centres in a slightly distorted octahedral arrangement (Fig. 1). Interestingly, two independent molecules are present in the asymmetric unit, which feature significantly different metrics. In both cases, the dimeric arrangement of the molecule is supported by the presence of two bridging thiopyridyl ligands: each of these fragments binds one metal cation in a bidentate fashion [Mg(1)–N(3) 2.215(3) Å; Mg(1)–S(3) 2.6431(15) Å; Mg(2)–N(6) 2.195(3) Å; Mg(2)–S(6) 2.6182(15) Å] and bridges to the adjacent Mg centre via the S donor [Mg(1)i–S(3) 2.6484(15) Å; Mg(2)i–S(6) 2.6772(15) Å]. The tridentate ligand {κ3C,N,N–C(Bu)(S–C5H4N)2}− displays a similar binding mode to Tptm in the zinc complex [Zn(Tptm)(N′′)] reported by Parkin and co-workers (vide infra).50 The structural motif of 1 is analogous to that of 2, in which the bridging bidentate thiopyridyl ligand is replaced by a monodentate siloxide, {OSiMe3}− (Fig. S27†).
and features two 6-coordinate Mg centres in a slightly distorted octahedral arrangement (Fig. 1). Interestingly, two independent molecules are present in the asymmetric unit, which feature significantly different metrics. In both cases, the dimeric arrangement of the molecule is supported by the presence of two bridging thiopyridyl ligands: each of these fragments binds one metal cation in a bidentate fashion [Mg(1)–N(3) 2.215(3) Å; Mg(1)–S(3) 2.6431(15) Å; Mg(2)–N(6) 2.195(3) Å; Mg(2)–S(6) 2.6182(15) Å] and bridges to the adjacent Mg centre via the S donor [Mg(1)i–S(3) 2.6484(15) Å; Mg(2)i–S(6) 2.6772(15) Å]. The tridentate ligand {κ3C,N,N–C(Bu)(S–C5H4N)2}− displays a similar binding mode to Tptm in the zinc complex [Zn(Tptm)(N′′)] reported by Parkin and co-workers (vide infra).50 The structural motif of 1 is analogous to that of 2, in which the bridging bidentate thiopyridyl ligand is replaced by a monodentate siloxide, {OSiMe3}− (Fig. S27†).
 | ||
| Fig. 1 Crystal structure of 1. Ellipsoids are set at 50% probability level and hydrogens omitted for clarity. The asymmetric unit contains two independent molecules, only one of them has been reproduced here. Symmetry operation used to generate equivalent atoms: i = 1 − x, 1 − y, −z. C: black; N: blue; Mg: aquamarine; S: yellow. Selected bond lengths (Å) and angles (°): Mg(1)–C(1) 2.212(4), Mg(1)–N(1) 2.149(3), Mg(1)–N(2) 2.204(3), Mg(1)–N(3) 2.177(3), Mg(1)–S(3) 2.6431(15), Mg(1)–S(3)i 2.6773(18), N(1)–Mg(1)–N(2) 86.45(12), N(2)–Mg(1)–N(3) 86.84(12), C(1)–Mg(1)–S(3) 172.82(12), N(2)–Mg(1)–S(3)i 177.58(10). Selected bond lengths (Å) and angles (°) of second molecule of 1: Mg(2)–C(21) 2.209(4), Mg(2)–N(4) 2.184(3), Mg(2)–N(5) 2.202(3), Mg(2)–N(6) 2.195(3), Mg(2)–S(6) 2.6182(15), Mg(2)–S(6)i 2.6772(15), N(4)–Mg(2)–N(5) 92.46(11), N(2)–Mg(1)–N(3) 84.95(11), C(21)–Mg(2)–S(6) 176.29(11), N(5)–Mg(2)–S(6)i 175.06(9). | ||
Recrystallisation of 3-Mg from boiling toluene gave X-ray quality crystals which led to the determination of the expected molecular structure (Fig. 2). 3-Mg crystallises in the monoclinic P21/c space group, with the metal centre positioned inside the steric pocket of the scorpionate Tptm ligand and coordinated axially by {N(SiMe3)2}−. As can be seen, the Mg–C bond is similar in length [2.303(4) Å] to the Mg–C bond length in [Mg(TismiPrBenz)(NPh2)] scorpionate complex [2.373(3) Å].52
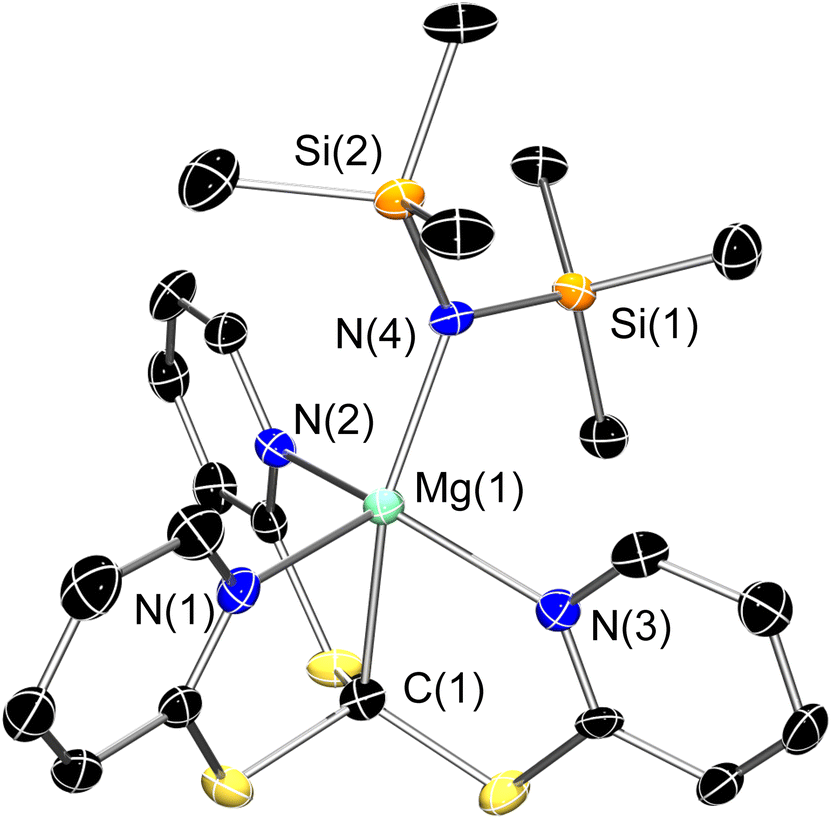 | ||
| Fig. 2 X-ray crystal structure of 3-Mg. Ellipsoids are set at 50% probability level, hydrogen atoms have been excluded for clarity. The asymmetric unit contains two molecules with identical connectivity and statistically identical metrics; one of these has been excluded for clarity. C: black; N: blue; S: yellow; Mg: aquamarine; Si: orange. Selected bond lengths (Å) and angles (°): Mg(1)–C(1) 2.303(4), Mg(1)–N(1) 2.220(4), Mg(1)–N(2) 2.181(3), Mg(1)–N(3) 2.215(4), Mg(1)–N(4) 2.061(3), N(1)–Mg(1)–N(2) 106.50(13), N(2)–Mg–N(3) 94.90(13), C(1)–Mg–N(4) 158.70(15). | ||
The Mg–Npy bond distances [2.181(3)–2.220(4) Å] are similar to typical Mg–N distances within closely related tris(pyrazolyl)-methane and -methanide systems [2.092(6)–2.315(4) Å],53 whilst the Mg–N bond to the {N(SiMe3)2}− ligand [2.061(3) Å] is a little longer than other Mg–N distances in analogous complexes [1.961(2)–1.987(2) Å].54,55 The geometry around magnesium is best described as distorted square pyramidal. Interestingly, the analogous complex [Zn(Tptm)(N′′)] reported by Parkin and co-workers displays Tptm acting as a k3 donor, with one of the pyridyl pendant arms flipped below the steric pocket.50
4. Decomposition studies
3-Mg is stable at room temperature, however, signs of decomposition at higher temperatures were observed (discolouration of the solution) during recrystallizations from toluene, and also during catalytic studies (vide infra). A 1H NMR time course was acquired at 100 °C in d8-toluene, from which a half-life of 104.1 ± 0.3 minutes was calculated (Fig. S133†). 3-Ca was observed to possess distinct temperature sensitivity during synthesis, and an analogous 1H NMR time course experiment (Fig. 3) was conducted in d6-benzene at 25 °C and monitored following signal a (δH 8.09 ppm, doublet). We analysed the kinetics of this decomposition, but found it possessed a non-integer reaction order thereby rendering extraction of meaningful kinetic information about this decomposition very difficult. This doublet slowly decreases in intensity and a new signal at 8.09 (A) gradually rises in intensity, but later plateaus out. This was attributed to the precipitation of a decomposition product, which is corroborated by the observation of a precipitate at the bottom of the NMR tube. Similar decreases in intensity are seen for the signals at 6.62 (d), 6.49 (c), and 6.25 (b) ppm, together with increases and subsequent plateauing for the signals at 7.06 (D), 6.74 (C), and 6.36 (B) ppm. Much as for 3-Ca, 3-Mg also shows the co-dependent increase and decrease in the most downfield signals, i.e. 9.01 (a) and 8.91 (A) for 3-Mg and a major decomposition product analogous to that observed for 3-Ca respectively (see Fig. S133†). | ||
| Fig. 3 1H NMR time course of the decomposition of 3-Ca at 298 K in d6-benzene, zoomed in the region between 6.0–9.0 ppm. Spectra were recorded every 10 minutes for 140 minutes. | ||
During attempts to crystallise 3-Ca from a solution in DME, we isolated crystals of decomposition product [Ca(S–C5H4N)2(DME)2] (4-Ca·DME2) (Fig. S28†). This compound was then obtained from the direct reaction of 2-mercaptopyridine with [Ca(N′′)2]2. NMR characterisation of 4-Ca revealed that this species is present in the thermal decomposition of 3-Ca, and presumably a similar decomposition pathway takes place for the other analogues 3-AE (for completeness we also deliberately obtained 4-Mg from the reaction of 2-mercaptopyridine with nBu2Mg – see Experimental Section).
5. Computational studies
We have used DFT calculations to study the unexpected formation of bimetallic Mg dimer 1 and the decomposition of 3-Ca to 4-Ca. Both processes involve cleavage of CMethanide–S bonds and, in the case of the Mg-butyl, a well-defined transfer of the alkyl group onto the methanide carbon. We therefore considered the Mg-butyl system first and started with the targeted heteroleptic complex [Mg(Tptm)(nBu)] (1m′). The computed structure of 1m′ shows a distorted trigonal bipyramidal (TBP) geometry with the methanide and alkyl carbons in axial positions (see Fig. 4). A transition state for butyl group migration, TS(1m′-1m), was located at +12.1 kcal mol−1 and displays another distorted TBP structure in which two pyridyl moieties (bound through N2 and N3) now occupy the axial positions. This places the butyl ligand, C1 and the remaining pyridyl moiety in equatorial positions and as a result the widened C1–Mg–N1 angle (138.8°) can no longer accommodate the C1–S1 bond which therefore breaks (C1⋯S1 = 3.26 Å). TS(1m’-1m) also features a C(S–NC5H4)2 carbene-like ligand with an sp2-hybridised carbon (sum of angles at C1 = 360.0°). A shortening of the Mg–C1 distance is also seen (2.21 Å in TS(1m′-1m)cf. 2.38 Å in 1m′). TS(1m′-1m) leads to the formation of 1m, the monomeric precursor to 1, in which the new C1–C2 bond is fully formed (1.55 Å) and N1 on the free 2-thiopyridide ligand has moved to the position vacated by the butyl such that the sulfur of that group occupies the axial position trans to C1 (Mg–S1 = 2.65 Å). This combined butyl migration/C–S cleavage and rearrangement is both readily accessible kinetically with a barrier of only 12.1 kcal mol−1 and significantly exergonic (ΔG = −40.3 kcal mol−1). The subsequent dimerisation of 1m to form 1 is also computed to be very favourable (ΔG = −60.7 kcal mol−1). | ||
| Fig. 4 Computed structures of 1m′, TS(1m′-1m) and 1m with key distances in Å and free energies in kcal mol−1. Aromatic H hidden for clarity. | ||
We postulate that an analogous ligand transfer process, this time of an amide, could also be involved in the decomposition of 3-Ca to 4-Ca. In this case amide transfer is computed to involve a two-step process viaI(3-4-Ca)1 at +12.8 kcal mol−1 (see Fig. 5). This species resembles TS(1m′-1m) in the Mg-butyl system in that one C–S bond has broken and a carbene-like C(S–C5H4N)2 ligand is in place. In the second step viaTS(3-4-Ca)2 (+19.7 kcal mol−1) the C1–N4 bond is formed; the resultant intermediate, I(3-4-Ca)2, is analogous to 1m in the Mg system, with the exception that the movement of the newly formed thiopyridide ligand is restricted by the bulky {N(SiMe3)2}− group. From I(3-4-Ca)2 formation of 4-Ca must involve cleavage of a second C–S bond and this would leave behind a C(S–C5H4N){N(SiMe3)2} carbene ligand. Intriguingly, Westerhausen and co-workers have observed a similar degradation process of a tris(pyrazoyl)methanide ligand,40,56 in which C–N bond cleavage is accompanied by formation of tetra-substituted alkene. The analogous process in the present case would form 4-Ca with the C(S–C5H4N){N(SiMe3)2} carbene ligand dimerising to an alkene. Indeed, cleavage of the C1–S2 bond in I(3-4-Ca)2 proceeds with a minimal barrier of 2.9 kcal mol−1 to give I(3-4-Ca)3 at −7.7 kcal mol−1 which is an adduct of 4-Ca with the C(S–C5H4N){N(SiMe3)2} ligand. Dissociation and dimerization of the carbene liberates 4-Ca and forms the tetra-substituted alkene 6 at −21.0 kcal mol−1. The decomposition of 3-Ca to 4-Ca and alkene therefore has a modest overall barrier of 19.7 kcal mol−1 and is exergonic by 21.0 kcal mol−1, consistent with a facile process that proceeds at room temperature.57 The equivalent barrier for the initial amide transfer and C–S bond cleavage process in 3-Mg was computed to be 29.7 kcal mol−1, consistent with the much slower degradation of this system seen experimentally.
 | ||
| Fig. 5 (Top) Free energy profile of the decomposition 3-Ca to provide 4-Ca and tetrasubstituted ethylene, as computationally found. (Bottom) Geometries of intermediates I(3-4-Ca)1, I(3-4-Ca)2 and I(3-4-Ca)3. Aromatic H hidden for clarity. | ||
6. Catalytic studies
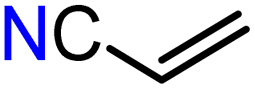
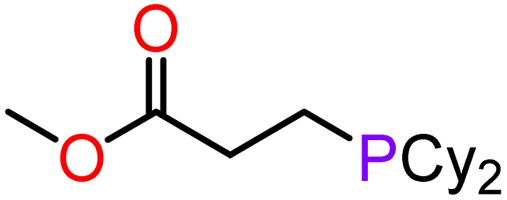
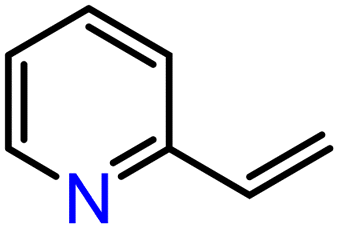
With compounds 3-Mg and 3-Ca in hand we decided to explore their reactivity in hydroelementation reactions, which have been extensively studied with group 2 catalysts.11,12,58 In particular, we decided to focus on hydrophosphination reactions, as these are suitable for amide complexes 3-AE which are capable of deprotonating protic E–H bonds. Additionally, group 2-catalysed hydrophosphinations of alkenes and alkynes have been extremely well studied,59–64 thus providing a benchmark for the performances of our new systems. 3-Mg and 3-Ca were initially tested as pre-catalysts for the hydrophosphination of selected terminal alkenes with diphenylphosphine (HPPh2) and dicyclohexylphosphine (HPCy2), to give α-addition (branched, Markovnikov, A) and β-addition (linear, anti-Markovnikov, B) products (Scheme 2). Various terminal alkenes were screened (styrene, methyl acrylate, acrylonitrile, 2-vinylpyridine, and 4-vinylpyridine) to test functional group tolerance and catalytic efficiency (Table 1 and S3†). The reactions were carried out on NMR scales in deuterated (d6-benzene) or non-deuterated (THF) solvents under various conditions, though reactions involving 3-Ca could only be carried out at room temperature due to its thermal instability (vide supra). Different solvents were chosen to ascertain if the presence of coordinating solvents would interfere with the catalytic cycle, or if changes in polarity would affect the reaction rate. Additionally we carried out selected controlled experiments: (1) in the absence of catalyst; (2) in the presence of [Ca(N′′)2]2; (3) in the presence of known decomposition products, 4-Mg and 4-Ca (see Table S6†). Integration of the 31P{1H} NMR signals enabled the quantification of yields and selectivity between linear anti-Markovnikov and branched Markovnikov products (see ESI†). | ||
| Scheme 2 Catalytic hydrophosphination of alkenes with 3-AE (AE = Mg, Ca). | ||
| Substrate | Entry | Catalyst | Conditions | Conv. (%) | Product ratio (α:β) |
Product |
|---|---|---|---|---|---|---|

|
1 | 3-Mg | C6D6 80 °C, 29 h | 23 | 0:100 |

|
| 2 | 3-Ca | — | — | — | — | |
|
3 | 3-Mg | C6D6 80 °C, 29 h | 47 | 0:100 |

|
| 4 | 3-Ca | C6D6 r.t., 1 h | 5 | 0:100 |

|
|

|
5 | 3-Mg | C6D6 80 °C, 29 h | 23 | 17:83 |

|
| 6 | 3-Mg | THF 60 °C, 70 h | 40 | 27:73 |
|
|
| 7 | 3-Ca | — | — | — | — | |
|
8 | 3-Mg | THF 60 °C, 29 h | 36 | 9:91 |
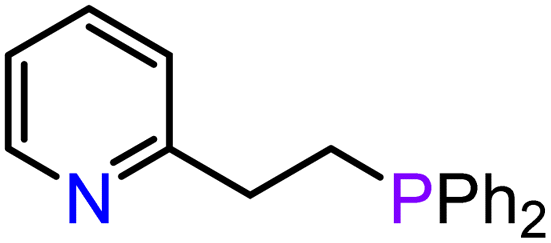
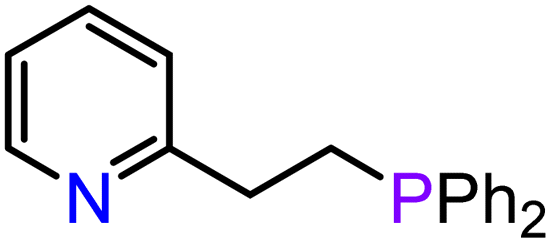
|
| 9 | 3-Ca | THF r.t., 1 h | 20 | 9:91 |
|
|

|
10 | 3-Mg | THF r.t., 100 h | 62 | 17:83 |

|
| 11 | 4-Mg | THF 60 °C, 135 h | 71 | 93:7 |

|
|
| 12 | 3-Ca | C6D6 r.t., 1 h | 37 | 0:100 |

|
For the reactions carried out with HPPh2, poor conversions were obtained with most of the alkene substrates employed in this study, though excellent selectivities were obtained in most cases. However, moderate conversions were observed in the hydrophosphination of 4-vinylpyridine with HPPh2 when 3-Mg was employed as pre-catalyst (62% yield, entry 10, Table 1). Interestingly, 4-Mg gave even better conversions (71%, entry 11, Table 1) than 3-Mg, though leading to the formation of the α-addition product. Additionally, comparable conversions were obtained with 3-Ca or 4-Ca (3-Ca, 37%: entry 12, Table 1; 4-Ca, 36%: entry 4, Table S6†). When the more electron-rich HPCy2 was used, significant conversion to the desired hydrophosphination products was only observed in the case of methyl acrylate (entry 6, Table 1) with 3-Mg as pre-catalyst, albeit with poor selectivity. Accuracy of the 31P{1H} integrations was confirmed through repeated screenings of hydrophosphination of 4-vinylpyridine with and without hexamethylbenzene as an internal standard.65 Notably, the anti-Markovnikov product was by far the dominant species across the board, with the sole exception of the hydrophosphination of 2-vinylpyridine catalysed by 3-Ca in benzene (entry 20, Table S3†), yielding over five times as much of the Markovnikov isomer as anti-Markovnikov. In the 31P{1H} NMR spectra, we also observed several smaller signals clustered around the Markovnikov product resonance (δP −21 to −22 ppm), which we have tentatively assigned as oligomerisation or polymerisation products.66
Since 3-Ca seemed to be quite effective at catalysing the polymerisation of the substrate, we tried reducing the pre-catalyst loading from 10 mol% to 2.5 mol%, with a corresponding reduction in yield of desired hydrophosphination product without a reduction in the yield of oligomeric products. Another possibility is that these products could be formed via anionic polymerisation initiated by {PPh2}−, though this could not be proved experimentally.67 We also determined approximate yields of these soluble phosphorus-containing oligomers via31P{1H} NMR spectroscopy (Tables S3 and S4†). Simple polymerisation of the alkene could not be detected in the 1H NMR spectra. A test with 4-vinylpyridine and 3-Mg in the absence of phosphine revealed neither any change in the 1H NMR spectra nor precipitate formation even after 10 days. Additionally, upon combination of a solution of 3-Ca with methyl acrylate, the immediate formation of yellow precipitate was observed, which upon closer inspection revealed a gel-like consistency, thus suggesting a polymerisation reaction had occurred; this is a likely occurrence, as calcium-initiated polymerisation of methyl acrylate has been previously reported.68 No alkene signals were visible in the 1H NMR spectra, and the 31P{1H} spectra did not change over time and contained only one resonance, which corresponded to HPPh2 (see ESI†). Analogous behaviour was not observed with 3-Mg, indicating its greater tolerance towards ester functionality. It can be noted that both catalysts tolerated nitrile groups (Fig. 4; entries 12–16, Table S3†). However, attempts to use fumaronitrile led to instant decomposition of the pre-catalyst and formation of a brown precipitate which could not be identified.
On the basis of the results obtained with alkene substrates, we decided to trial 3-AE (AE = Mg, Ca) as catalysts for the hydrophosphination of selected alkynes (phenylacetylene, 1,3-diethynylbenzene, 4-ethynyltoluene, methyl propiolate, 2-ethynylpyridine, 4-ethynylpyridine, and 4-ethynylanisole) with HPPh2 (Scheme 3, Fig. 6, Table 2 and Table S5†). 3-Mg and 4-Ca provided poor to moderate catalytic activity with most of the alkynes screened in this work. However, excellent results were obtained for the hydrophosphination of 4-ethynylpyridine when 3-Mg is employed as pre-catalyst, leading to high conversions (96%) with excellent regio- and stereoselectivity (entry 13, Table 2); this is in contrast with the catalytic performance of 3-Ca which gave only 40% conversion to the hydrophosphination product. Nonetheless, 3-Ca showed excellent catalytic activity in the hydrophosphination of 2-ethynylpyridine, which gave 95% conversion (by 31P{1H} NMR) to β-addition products (C and D, Scheme 3; entry 12, Table 2; Fig. 6) with excellent regioselectivity. In these reactions we also noticed the presence of trace amounts of double addition product (E) and dehydrocoupling product Ph2P–PPh2 (F) with all substrates.69 It is noteworthy that Webster and co-workers reported the double hydrophosphination of 4-ethynylpyridine catalysed by K(N′′).69 Nonetheless, to the best of our knowledge, our study is the first report of catalytic mono-hydrophosphination of 4-ethynylpyridine; the resulting novel vinyl phosphine, 4-(2-(diphenylphosphaneyl)vinyl)pyridine (Fig. 6), was isolated as the parent phosphine oxide in excellent yield (88% – see ESI, page S32†). Overall 3-Mg and 3-Ca do not perform as well as previously reported AE catalysts, such as [Ca(BDIDipp)N(SiMe3)2(THF)] (BDIDipp = {HC(C(Me)2N-2,6-iPr2C6H3)2}−), [Ca{N(SiMe3)2}(THF)2]59 and [Ca{2-NC(Ph)NArC6H4CHNAr}{N(SiMe3)2}(THF)],63 which can all catalyse the hydrophosphination of styrene with extremely high conversions (95%, 92% and >99% respectively). The latter complex was also used by Cui and Hu to catalyse the hydrophosphination of 2-ethynylpyridine, giving slightly better conversion (>99%) and selectivity (E:Z = 69:31) compared to 3-Ca.63
 | ||
| Scheme 3 Catalytic hydrophosphination of alkynes with 3-AE (AE = Mg, Ca). | ||
 | ||
| Fig. 6 Selected phosphines obtained from hydrophosphination reactions with alkenes and alkynes, including yields, selectivities and reaction conditions. Both α- and β-addition products are illustrated for relevant entries. All catalyst loadings were 10 mol%. | ||
| Substrate | Entry | Catalyst | Conditions | Conv. (%) | Product ratio (α:β/E:Z) |
Product |
|---|---|---|---|---|---|---|
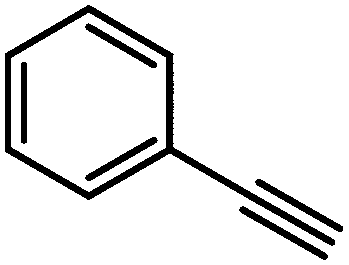
|
1 | 3-Mg | THF 60 °C, 135 h | 9 | 7:93/7:93 |

|
| 2 | 3-Ca | THF r.t., 1 h | 6 | 18:86/21:79 |

|
|
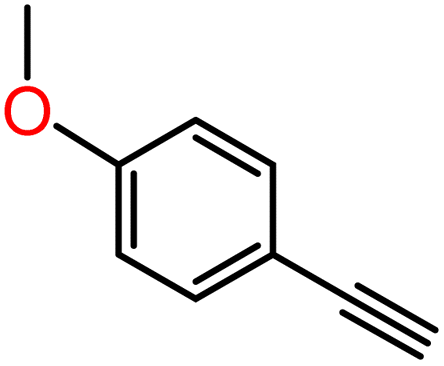
|
3 | 3-Mg | THF 60 °C, 135 h | 5 | 0:100/0:100 |

|
| 4 | 3-Ca | THF r.t., 1 h | 3 | 0:100/0:100 |

|
|

|
5 | 3-Mg | THF 60 °C, 135 h | 8 | 0:100/76:24 |

|
| 6 | 3-Ca | THF r.t., 1 h | 4 | 0:100/74:26 |

|
|

|
7 | 3-Mg | THF r.t., 96 h | 4 | 0:100/49:51 |

|
| 8 | 3-Ca | THF r.t., 1 h | 8 | 30:70/39:61 |

|
|
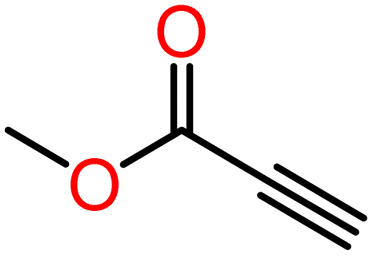
|
9 | 3-Mg | THF 60 °C, 135 h | 18 | 11:89/77:23 |

|
| 10 | 3-Ca | THF r.t., 1 h | 11 | 4:96/84:16 |

|
|
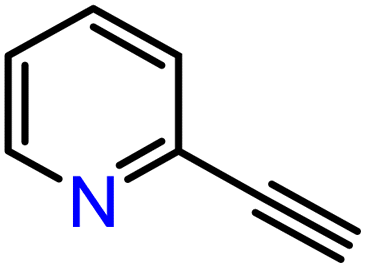
|
11 | 3-Mg | THF 60 °C, 135 h | 7 | 0:100/49:51 |

|
| 12 | 3-Ca | THF r.t., 1 h | 95 | 0:100/46:54 |
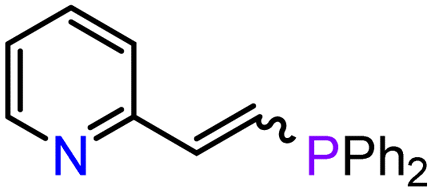
|
|
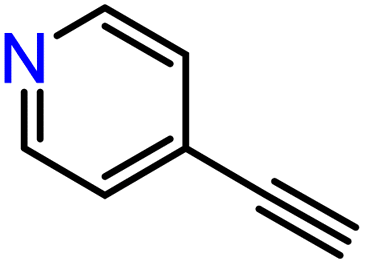
|
13 | 3-Mg | THF r.t., 72 h | 96 | 0:100/96:4 |
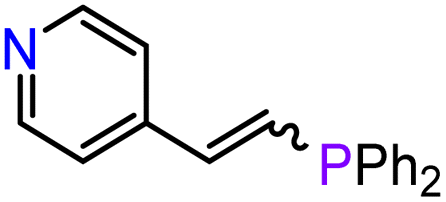
|
| 14 | 3-Ca | THF r.t., 1 h | 40 | 0:100/34:66 |

|
These hydrophosphination reactions are likely to occur via typical intermolecular hydroelementation mechanism in which the first step consists of protonolysis between the pre-catalyst and the phosphine.59 To shed some light on the generally low conversion rates, we attempted to prepare the proposed catalytic intermediate [Mg(Tptm)(PPh2)] via protonolysis between 3-Mg and HPPh2, however on an NMR scale, very little conversion was observed in the 31P{1H} NMR spectra even under forcing conditions (12 h at 50 °C, Fig. S19 and S20†). As a result, we chose not to scale this up to a preparative scale and instead opted to react Mg(PPh2)2 with HTptm in THF (Scheme 4). However, 1H and 31P{1H} NMR analysis of the crude mixture revealed that the target phosphido complex was not obtained, revealing instead the presence of 4-Mg, together with several other unknown components (Fig. S28 and S29†). Crystallisation of the reaction mixture led to the isolation of a small crystalline crop of material (<10 mg) which was identified via X-ray studies as [Mg(Bptm)2] (Bptm = {κ3C,N,N-CH(S–C5H4N)2}−) (5) and the diphosphine Ph2P–PPh2 co-crystallise together (Scheme 4 and Fig. 7).
 | ||
| Scheme 4 Attempted synthesis of [Mg(Tptm)(PPh2)] and resultant formation of 5, Ph2P–PPh2, and 4-Mg. | ||
 | ||
| Fig. 7 Crystal structure of 5·Ph2P–PPh2. Ellipsoids are set at 50% probability level and hydrogens omitted for clarity. The asymmetric unit contains half a molecule of each; the full molecules have been reproduced here. Symmetry operation used to generate equivalent atoms: i = −x, +1/2 y, −1/2 z. C: black; N: blue; Mg: aquamarine; P: purple; S: yellow. | ||
It is unclear how this diphosphine may have formed, however this is a known occurrence in hydrophosphination catalysis.70–7231P{1H} NMR spectra of the crude reaction mixture reveal two resonances, while the bulk recrystallised material contained but one signal corresponding to the diphosphine at −15.1 ppm. We tentatively attribute the resonance at 25.1 ppm in the 31P{1H} NMR spectrum of the crude to tetraphenyldiphosphane dioxide (Ph2P(O)–P(O)Ph2), which is likely formed from adventitious oxygen or moisture.73 The formation of 5·Ph2P–PPh2 from this reaction is in agreement with the observation of free Ph2P–PPh2 in some of our catalytic studies. Therefore, these findings shed light on another possible decomposition pathway for this family of catalysts which could be responsible for some of the poor catalytic performances.
7. Conclusions
In conclusion we have shown the varying reactivity of HTptm with AE reagents. When alkylmagnesium reagents are employed, this leads to degradation of the ligand through cleavage of a C–S bond which is replaced by the alkyl nucleophile (methyl or butyl) and forms bimetallic dimers 1 and 2. Despite this, heteroleptic amide complexes 3-AE can be obtained by reacting HTptm with bis-amide AE reagents, [AE(N′′)2]2. However, these complexes become increasingly unstable descending the group, which poses some limitations in the development of reactivity studies. DFT calculations characterise the facile transfer of anionic alkyl and amide ligands onto the methanide carbon and that this induces C–S bond cleavage. We were also able to directly identify several components of these decomposition pathways, including the formation of dehydrocoupling product Ph2P–PPh2 co-crystallised with decomposition product 5. Nonetheless, compound 3-Ca displayed excellent catalytic activity and regioselectivity in the hydrophosphination of 2-ethynylpyridine, whilst 3-Mg gave excellent conversions for 4-ethynylpyridine, together with excellent regio- and stereoselectivity. We are currently investigating the reactivity of 3-Mg and 3-Ca further and expanding the scope of their catalytic applications, together with modifications of Tptm in order to increase the steric protection around the metal centre and also impart more stability to the ligand backbone.8. Experimental section
General methods
THF, Et2O, toluene and hexane were passed through columns containing molecular sieves, then stored over molecular sieves (THF) or over a potassium mirror (Et2O, hexane, toluene) and thoroughly degassed prior to use. Anhydrous 1,2-dimethoxyethane (DME) was purchased from Sigma Aldrich, dried and stored over molecular sieves and thoroughly degassed prior to use. For NMR spectroscopy, C6D6, C4D8O, and C7D8 were dried by refluxing over K, and then vacuum transferred and degassed by three freeze–pump–thaw cycles before use. NMR spectra were recorded on either a Bruker AVIII HD 400 or Bruker AVIII 500 spectrometer operating at 400.07/500.13 (1H), 100.60/125.78 (13C{1H}), 79.48 (29Si{1H}), or 161.98 (31P{1H}) MHz. To achieve a greater signal to noise ratio, the 29Si{1H} NMR spectra were acquired with a DEPT24 where available or otherwise with a DEPT45 pulse sequence. In some cases, the recycle delay between scans was set to a value of up to 60 s to improve the spectra. Solvent suppression procedures were implemented in the acquisition of 1H NMR data run in C4H8O, suppressing signals at chemical shifts 1.79 and 3.72 ppm. The solvent suppression procedure used a 1D version of Bruker library pulse program noesyprph which made use of presaturation at the aforementioned chemical shifts during the relaxation delay and mixing time. NMR spectra were recorded at 298 K unless otherwise stated, and were referenced to residual solvent signals in the case of 1H and 13C{1H} experiments. FTIR spectra were recorded on a Bruker Alpha II spectrometer with Platinum-ATR module. Elemental microanalyses were carried out by Elemental Microanalysis Ltd. nBu2Mg, nBuLi, 2-mercaptopyridine, and bromoform were used as received. Hexamethyldisilazane was dried over activated molecular sieves, stored over the same and thoroughly degassed before use. MgI2, CaI2, SrI2 and BaI2 were baked at 300 °C under vacuum for 4 hours before use. HTptm,50 [AE(N′′)2]2,74,75 Mg(PPh2)2,76,77 and Me2Mg78 and were prepared according to literature procedures. Diphenylphosphine, dicyclohexylphosphine, phenylacetylene, 2-ethynylpyridine, 4-ethynylanisole, 4-ethynyltoluene, and 1,3-diethynylbenzene were degassed via three freeze–pump–thaw cycles before use. Styrene, methyl acrylate, acrylonitrile, 2-vinylpyridine, 4-vinylpyridine, and methyl propiolate were dried over calcium hydride then vacuum transferred and degassed by three freeze–pump–thaw cycles before use. Hexamethylbenzene and 4-ethynylpyridine were dried under vacuum for 4 hours before use. Most phosphines obtained in the catalytic studies have been previously reported;71,79–83 novel phosphines have been isolated as parent oxides and their characterisation data is reported in the ESI (page S33).†Synthesis
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 1583, 1552, 1411, 1260, 1002, 794, 748, 720, 638, 481, 409 cm−1.
= 1586, 1555, 1457, 1412, 1235, 1129, 1045, 992, 880, 808, 755, 657, 635, 606, 485, 414 cm−1.
= 1583, 1552, 1411, 1260, 1002, 794, 748, 720, 638, 481, 409 cm−1.
= 1586, 1555, 1457, 1412, 1235, 1129, 1045, 992, 880, 808, 755, 657, 635, 606, 485, 414 cm−1.
A very small crop of crystals of 4-Ca·2DME (ca. 10 mg) were obtained from a concentrated DME solution of 3-Ca (ca. 200 mg in 6 mL) at room temperature (vide infra).
3-Ca: 1H NMR (400 MHz, 298 K, C6D6): 0.47 (18H, s, SiCH3), 6.32 (3H, t, 3JHH = 6.3 Hz, C5H4N–CHb), 6.52 (3H, t, 3JHH = 7.3 Hz, C5H4N–CHd), 6.61 (3H, d, 3JHH = 8.2 Hz, C5H4N–CHc), 8.75 (3H, d, 3JHH = 5.0 Hz, C5H4N–CHa) ppm. 1H NMR (400 MHz, 263 K, d8-toluene): 0.47 (18H, s, SiCH3), 6.32 (3H, t, 3JHH = 6.3 Hz, C5H4N–CHb), 6.52 (3H, t, 3JHH = 7.3 Hz, C5H4N–CHd), 6.61 (3H, d, 3JHH = 8.2 Hz, C5H4N–CHc), 8.75 (3H, d, 3JHH = 5.0 Hz, C5H4N–CHa) ppm. 13C{1H} NMR (100 MHz, 263 K, C7D8): 5.62 (CaC), 6.21 (Si(CH3)3), 118.3 (C5H4N–CbH), 120.8 (C5H4N–CcH), 136.7 (C5H4N–CdH), 148.4 (C5H4N–CaH), 167.3 (C5H4N–CeS) ppm. 29Si{1H} NMR (79.5 MHz, 263 K, C7D8): −14.7 (SiCH3) ppm. Anal. calcd for C22H30CaN4S3Si2: C, 48.67%; H, 5.57%; N, 10.32%. Found: C, 48.84%; H, 5.16%; N, 10.22%. FTIR: = 1581, 1550, 1457, 1408, 1236, 1124, 1043, 878, 810, 751, 659, 633, 563, 481 cm−1.
4-Ca: 1H NMR (400 MHz, 298 K, C4H8O): 6.46 (2H, t, 3JHH = 6.1 Hz, C5H4N-CHb), 7.04 (2H, td, 3JHH = 7.5 Hz, 4JHH = 1.8 Hz, C5H4N–CHc), 7.14 (2H, d, 3JHH = 8.2 Hz, C5H4N–CHd) 7.81 (2H, d, 3JHH = 4.8 Hz, C5H4N–CHa) ppm. 13C{1H} NMR (100 MHz, C4H8O): 113.3 (C5H4N–CbH), 129.4 (C5H4N–CcH), 134.6 (C5H4N–CdH), 146.2 (C5H4N–CaH), 180.5 (C5H4N–CHe) ppm. Anal. calcd for C14H18N2S2Ca: C, 46.13%; H, 3.10%; N, 10.76%. Found: C, 45.59%; H, 3.24%; N, 10.18%. FTIR = 1583, 1545, 1447, 1410, 1267, 1131, 1084, 1044, 998, 749, 725, 633, 488, 444, 407 cm−1.
4-Ca·2DME: 1H NMR (400 MHz, 298 K, C6D6/C4D8O, 4:1): 3.12 (12H, s, DME-CH3), 3.32 (8H, s, DME-CH2), 6.33 (2H, t, 3JHH = 6.0 Hz, C5H4N–CHb), 6.85 (2H, t, 3JHH = 7.5 Hz, C5H4N–CHd) 7.98 (2H, d, 3JHH = 5.3 Hz, C5H4N–CHa), 7.68 (2H, d, 3JHH = 8.3 Hz, C5H4N–CHc) ppm. 13C{1H} NMR (100 MHz, 298 K, C6D6): 58.8 (DME-CH3), 72.2 (DME-CH2), 114.0 (C5H4N–CbH), 130.2 (C5H4N–CcH), 135.3 (C5H4N–CdH), 146.4 (C5H4N–CaH) ppm; Note: signal for C5H4N–CeS not observed. FTIR: = 2963, 2365, 2335, 1578, 1556, 1537, 1458, 1407, 1260, 1128, 1098, 1068, 1019, 864, 796, 760, 726 cm−1.
4-Mg: 1H NMR (400 MHz, 298 K, C4H8O): 6.44 (2H, td, 3JHH = 5.5 Hz, 4JHH = 2.5 Hz, C5H4N–CHb), 7.06–7.14 (4H, m, C5H4N–CHc,d), 7.85 (2H, d, 3JHH = 5.2 Hz, C5H4N–CHa) ppm. 13C{1H} NMR (100 MHz, C4H8O): 114.0 (C5H4N–CbH), 127.2 (C5H4N–CcH), 135.9 (C5H4N–CdH), 145.2 (C5H4N–CaH), 177.5 (C5H4N–CHe) ppm.
4-Mg·DME: (400 MHz, 298 K, C6D6): 2.94 (6H, s, DME-CH3), 3.04 (4H, s, DME-CH2), 6.13 (2H, t, 3JHH = 6.0 Hz, C5H4N–CHb), 6.70 (2H, t, 3JHH = 7.4 Hz, C5H4N–CHd), 7.53 (2H, d, 3JHH = 8.2 Hz, C5H4N–CHc), 8.07 (2H, d, 3JHH = 3.5 Hz, C5H4N–CHa) ppm. 13C{1H} NMR (100 MHz, C6D6): 30.2 (DME-CH3), 58.9 (DME-CH2), 114.8 (C5H4N–CbH), 127.9 (C5H4N–CcH), 136.8 (C5H4N–CdH), 145.8 (C5H4N–CaH), 178.4 (C5H4N–CHe) ppm. Anal. calcd for C14H18N2S2O2Mg: C, 50.23%; H, 5.42%; N, 8.37%. Found: C, 49.76%; H, 5.41%; N, 8.17%. FTIR = 1586, 1537, 1438, 1404, 1263, 1132, 1058, 1002, 864, 776, 756, 726, 642 cm−1.
Computational details
DFT calculations were run with Gaussian 16 (rev. A.03).85 Geometry optimizations and thermodynamic corrections were performed with the BP86 functional86,87 with Mg, Ca, Si and S centres described by Stuttgart RECPs and associated basis sets88 and 6-31G** basis sets for all other atoms.89,90 A set of d-orbital polarization functions was added to Ca (ζd = 1.245), Si (ζd = 0.284) and S (ζd = 0.503).91 All stationary points were fully characterized via analytical frequency calculations as either minima (all positive frequencies) or transition states (one negative frequency), and the latter were characterized via IRC calculations and subsequent geometry optimizations to confirm the adjacent minima. Electronic energies were recomputed with the def2-TZVP basis sets92,93 and included corrections for Et2O solvent (PCM approach)94 and dispersion (Grimme's D3 parameter set with Becke–Johnson damping, BJD3).95 Details of all computed structures are provided in the ESI† and as a separate XYZ file.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Leicester and the College of Life Science and Engineering for a PhD scholarship (M. P. S.), Royal Society (RGS\R2\202097) and Engineering and Physical Sciences Research Council (EP/W00691X/1 and EP/T019867/1) for funding. X-ray diffraction at Leicester was also supported by the Engineering and Physical Sciences Research Council (EP/V034766/1). Additional research data supporting this publication are available from Mendeley at https://dx/doi.org/10.17632/rjjjsv8krf.2.References
- J. S. Alexander, M. Guino-o, M. F. Zuniga, R. C. Hahn and K. Ruhlandt-Senge, Alkaline Earth Metals: Organometallic Chemistry, John Wiley & Sons, Ltd., Hoboken, New Jersey, 2006 Search PubMed.
- M. Westerhausen, A. Koch, H. Görls and S. Krieck, Chem. – Eur. J., 2017, 23, 1456–1483 CrossRef CAS.
- M. Westerhausen, Angew. Chem., Int. Ed., 2001, 40, 2975–2977 CrossRef CAS PubMed.
- S. Krieck and M. Westerhausen, Inorganics, 2017, 5, 8–11 CrossRef.
- W. M. Haynes, D. R. Lide and T. J. Bruno, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, Florida, 97th edn, 2017 Search PubMed.
- H. Schulz, Appl. Catal., A, 1999, 186, 3–12 CrossRef CAS.
- J. H. Jones, Platinum Met. Rev., 2000, 44, 94–105 CAS.
- R. Jira, Angew. Chem., Int. Ed., 2009, 48, 9034–9037 CrossRef CAS PubMed.
- M. R. Rahimpour, M. Jafari and D. Iranshahi, Appl. Energy, 2013, 109, 79–93 CrossRef CAS.
- W. D. Buchanan, D. G. Allis and K. Ruhlandt-Senge, Chem. Commun., 2010, 46, 4449–4465 RSC.
- S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- S. Harder, Early Main Group Metal Catalysis: Concepts and Reactions, Wiley-VCH Verlag, Weinheim, 2020 Search PubMed.
- M. M. D. Roy, A. A. Omaña, A. S. S. Wilson, M. S. Hill, S. Aldridge and E. Rivard, Chem. Rev., 2021, 121, 12784–12965 CrossRef PubMed.
- M. Westerhausen, Coord. Chem. Rev., 2008, 252, 1516–1531 CrossRef CAS.
- A. Koch, Q. Dufrois, M. Wirgenings, H. Görls, S. Krieck, M. Etienne, G. Pohnert and M. Westerhausen, Chem. – Eur. J., 2018, 24, 16840–16850 CrossRef.
- M. Westerhausen, M. Gärtner, R. Fischer, J. Langer, L. Yu and M. Reiher, Chem. – Eur. J., 2007, 13, 6292–6306 CrossRef CAS PubMed.
- N. Kawabata, A. Matsumura and S. Yamashita, Tetrahedron, 1973, 29, 1069–1071 CrossRef CAS.
- J. Langer, M. Köhler, R. Fischer, F. Dündar, H. Görls and M. Westerhausen, Organometallics, 2012, 31, 6172–6182 CrossRef CAS.
- J. Langer, M. Köhler, H. Görls and M. Westerhausen, J. Organomet. Chem., 2014, 751, 563–567 CrossRef CAS.
- M. Köhler, J. Langer, R. Fischer, H. Görls and M. Westerhausen, Chem. – Eur. J., 2013, 19, 10497–10500 CrossRef PubMed.
- M. Westerhausen, J. Langer, S. Krieck, R. Fischer, H. Görls and M. Köhler, Top. Organomet. Chem., 2013, 45, 29–72 CrossRef CAS.
- C. Eaborn, S. A. Hawkes, P. B. Hitchcock and J. D. Smith, Chem. Commun., 1997, 1961–1962 RSC.
- M. Köhler, A. Koch, H. Görls and M. Westerhausen, Organometallics, 2016, 35, 242–248 CrossRef.
- F. G. N. Cloke, P. B. Hitchcock, M. F. Lappert, G. A. Lawless and B. Royo, J. Chem. Soc., Chem. Comm., 1991, 18, 724–726 RSC.
- D. L. Kays, Chem. Soc. Rev., 2016, 1004–1018 RSC.
- A. S. S. Wilson, M. S. Hill and M. F. Mahon, Organometallics, 2019, 38, 351–360 CrossRef CAS.
- J. Okuda, D. Schuhknecht, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2020, 59, 310–314 CrossRef PubMed.
- X. Shi, C. Hou, L. Zhao, P. Deng and J. Cheng, Chem. Commun., 2020, 56, 5162–5165 RSC.
- B. M. Wolf, C. Stuhl, C. Maichle-Mössmer and R. Anwander, J. Am. Chem. Soc., 2018, 140, 2373–2383 CrossRef CAS PubMed.
- L. Orzechowski and S. Harder, Organometallics, 2007, 26, 5501–5506 CrossRef CAS.
- S. Marks, M. Kuzdrowska, P. W. Roesky, L. Annunziata, S. M. Guillaume and L. Maron, ChemPlusChem, 2012, 77, 350–353 CrossRef CAS.
- M. S. Hill and P. B. Hitchcock, Chem. Commun., 2003, 3, 1758–1759 RSC.
- L. Orzechowski, G. Jansen and S. Harder, J. Am. Chem. Soc., 2006, 128, 14676–14684 CrossRef CAS PubMed.
- L. Orzechowski and S. Harder, Organometallics, 2007, 26, 2144–2148 CrossRef CAS.
- T. K. Panda, A. Zulys, M. T. Gamer and P. W. Roesky, J. Organomet. Chem., 2005, 690, 5078–5089 CrossRef CAS.
- C. P. Sindlinger, S. R. Lawrence, D. B. Cordes, A. M. Z. Slawin and A. Stasch, Inorganics, 2017, 5, 1–13 CrossRef.
- J. Guo, J. S. Lee, M. C. Foo, K. C. Lau, H. W. Xi, K. H. Lim and C. W. So, Organometallics, 2010, 29, 939–944 CrossRef CAS.
- P. Wei and D. W. Stephan, Organometallics, 2003, 601–604 CrossRef CAS.
- C. Müller, A. Koch, H. Görls, S. Krieck and M. Westerhausen, Inorg. Chem., 2015, 54, 635–645 CrossRef.
- M. Rauch and G. Parkin, J. Am. Chem. Soc., 2017, 139, 18162–18165 CrossRef CAS PubMed.
- K. Kitano, N. Kuwamura, R. Tanaka, R. Santo, T. Nishioka, A. Ichimura and I. Kinoshita, Chem. Commun., 2008, 1314–1316 RSC.
- W. Sattler and G. Parkin, J. Am. Chem. Soc., 2011, 133, 9708–9711 CrossRef CAS PubMed.
- W. Sattler, S. Ruccolo and G. Parkin, J. Am. Chem. Soc., 2013, 135, 18714–18717 CrossRef CAS PubMed.
- W. Sattler and G. Parkin, Chem. Sci., 2012, 3, 2015–2019 RSC.
- N. Chakrabarti, S. Ruccolo and G. Parkin, Inorg. Chem., 2016, 55, 12105–12109 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Found. Crystallogr., 1976, 32, 751–767 CrossRef.
- J. Langer, M. Köhler, R. Fischer, F. Dündar, H. Görls and M. Westerhausen, Organometallics, 2012, 31, 6172–6182 CrossRef CAS.
- M. Köhler, J. Langer, R. Fischer, H. Görls and M. Westerhausen, Chem. – Eur. J., 2013, 19, 10497–10500 CrossRef PubMed.
- W. Sattler and G. Parkin, J. Am. Chem. Soc., 2011, 133, 9708–9711 CrossRef CAS PubMed.
- M. Westerhausen, Inorg. Chem., 1991, 30, 96–101 CrossRef CAS.
- M. Rauch, R. C. Roberts and G. Parkin, Inorg. Chim. Acta, 2019, 494, 271–279 CrossRef CAS.
- T. Kloubert, C. Müller, S. Krieck, T. Schlotthauer, H. Görls and M. Westerhausen, Eur. J. Inorg. Chem., 2012, 5991–6001 CrossRef CAS.
- M. G. Cushion, J. Meyer, A. Heath, A. D. Schwarz, I. Fernández, F. Breher and P. Mountford, Organometallics, 2010, 29, 1174–1190 CrossRef CAS.
- A. P. Dove, V. C. Gibson, P. Hormnirun, E. L. Marshall, J. A. Segal, A. J. P. White and D. J. Williams, Dalton Trans., 2003, 3088–3097 RSC.
- S. Krieck, A. Koch, K. Hinze, C. Müller, J. Lange, H. Görls and M. Westerhausen, Eur. J. Inorg. Chem., 2016, 2016, 2332–2348 CrossRef CAS.
- Carbene loss from I(3-4-Ca)3 is readily accessible with a computed bond dissociation of only 21.4 kcal mol−1.
- C. Brinkmann, A. G. M. Barrett, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2012, 134, 2193–2207 CrossRef CAS PubMed.
- M. R. Crimmin, A. G. M. Barrett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2007, 26, 2953–2956 CrossRef CAS.
- M. R. Crimmin, A. G. M. Barrett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2008, 27, 497–499 CrossRef CAS.
- B. Liu, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2012, 19, 2784–2802 CrossRef PubMed.
- B. Liu, J. F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2012, 18, 13259–13264 CrossRef CAS PubMed.
- H. Hu and C. Cui, Organometallics, 2012, 31, 1208–1211 CrossRef CAS.
- J. Pahl, T. E. Stennett, M. Volland, D. M. Guldi and S. Harder, Chem. – Eur. J., 2019, 25, 2025–2034 CrossRef CAS PubMed.
- In one case (entry 23, Table S3†), we obtained a significantly worse yield of hydrophosphination product in the presence of the internal standard. In the absence of hexamethylbenzene, the yield was 62% by 31P{1H} NMR spectroscopy, which drops to approximately 6% in the presence of hexamethylbenzene.
- C. Scriban, I. Kovacik and D. S. Glueck, Organometallics, 2015, 24, 4871–4874 CrossRef.
- E. M. W. Tsang and S. Holdcroft, Alternative Proton Exchange Membranes by Chain-Growth Polymerization, Elsevier B.V., Amsterdam, 2012 Search PubMed.
- B. B. Noble and M. L. Coote, J. Polym. Sci., 2020, 58, 52–61 CrossRef CAS.
- N. T. Coles, M. F. Mahon and R. L. Webster, Chem. Commun., 2018, 54, 10443–10446 RSC.
- R. Nolla-Saltiel, A. M. Geer, L. J. Taylor, O. Churchill, E. S. Davies, W. Lewis, A. J. Blake and D. L. Kays, Adv. Synth. Catal., 2020, 362, 3148–3157 CrossRef CAS.
- M. O. Shulyupin, M. A. Kazankova and I. P. Beletskaya, Org. Lett., 2002, 4, 761–763 CrossRef CAS PubMed.
- R. L. Melen, Chem. Soc. Rev., 2016, 45, 775–788 RSC.
- J. E. Nycz and R. Musiol, Heteroat. Chem., 2006, 17, 310–316 CrossRef CAS.
- A. R. Kennedy, R. E. Mulvey and R. B. Rowlings, J. Am. Chem. Soc., 1998, 120, 7816–7824 CrossRef CAS.
- J. D. Leng, C. A. P. Goodwin, I. J. Vitorica-Yrezabal and D. P. Mills, Dalton Trans., 2018, 47, 12526–12533 RSC.
- K. Issleib and H.-J. Deylig, Chem. Ber., 1964, 67, 946–951 CrossRef.
- M. Gärtner, H. Görls and M. Westerhausen, Inorg. Chem., 2008, 47, 1397–1405 CrossRef PubMed.
- R. I. Yousef, B. Walfort, T. Rüffer, C. Wagner, H. Schmidt, R. Herzog and D. Steinborn, J. Organomet. Chem., 2005, 690, 1178–1191 CrossRef CAS.
- M. M. I. Basiouny, D. A. Dollard and J. A. R. Schmidt, ACS Catal., 2019, 9, 7143–7153 CrossRef CAS.
- L. Liu, Y. Wang, Z. Zeng, P. Xu, Y. Gao, Y. Yin and Y. Zhao, Adv. Synth. Catal., 2013, 355, 659–666 CrossRef CAS.
- A. Di Giuseppe, R. De Luca, R. Castarlenas, J. J. Pérez-Torrente, M. Crucianelli and L. A. Oro, Chem. Commun., 2016, 52, 5554–5557 RSC.
- R. Nolla-Saltiel, A. M. Geer, L. J. Taylor, O. Churchill, E. S. Davies, W. Lewis, A. J. Blake and D. L. Kays, Adv. Synth. Catal., 2020, 362, 3148–3157 CrossRef CAS.
- P. Cheruku, A. Paptchikhine, T. L. Church and P. G. Andersson, J. Am. Chem. Soc., 2009, 131, 8285–8289 CrossRef CAS PubMed.
- C. A. P. Goodwin, A. Smith, F. Ortu, I. J. Vitorica-Yrezabal and D. P. Mills, Dalton Trans., 2016, 45, 6004–6014 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision A.03), Gaussian Inc., Pittsburgh, PA, 2016 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- W. J. Hehre, K. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- A. Höllwarth, M. Böhme, S. Dapprich, A. W. Ehlers, A. Gobbi, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 237–240 CrossRef.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Weigend, A. Köhn and C. Hättig, J. Chem. Phys., 2002, 116, 3175–3183 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2012, 32, 1456–1465 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2097915–2097920 and 2170882. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02012b |
| This journal is © The Royal Society of Chemistry 2022 |