
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbonyl hypoiodites from pivalic and trimesic acid and their silver(I) intermediates†
Jas S.
Ward
 *a,
Jevgenija
Martõnova
b,
Laura M. E.
Wilson
a,
Eric
Kramer
a,
Riina
Aav
b and
Kari
Rissanen
a
*a,
Jevgenija
Martõnova
b,
Laura M. E.
Wilson
a,
Eric
Kramer
a,
Riina
Aav
b and
Kari
Rissanen
a
aUniversity of Jyvaskyla, Department of Chemistry, Jyväskylä 40014, Finland. E-mail: james.s.ward@jyu.fi
bDepartment of Chemistry and Biotechnology, School of Science, Tallinn University of Technology, Tallinn, Estonia
First published on 5th September 2022
Abstract
The first tris(O–I–N) carbonyl hypoiodites have been synthesised based on trimesic acid and pyridine or 4-methylpyridine, with their structures definitively confirmed by single crystal X-ray diffraction (SCXRD). The more soluble carbonyl hypoiodites based on pivalic acid have also been studied via NMR, SCXRD, and computational analyses, enabling the study of the direct silver(I) precursor and intermediates of the resulting carbonyl hypoiodites generated using a range of substituted pyridines.
Introduction
Halogen bonding, though not as popular as the closely related hydrogen bonding, offers advantages such as being far more directional than hydrogen bonding, in that it enforces linear symmetry due to its electronic origins as a σ-hole interaction.1 This reliable, high-degree of linear directionality has found much success in endeavours such as toward synthesising supramolecular architectures utilising self-assembling methodologies.2–4The recent revisitation of carbonyl hypoiodites has revitalised interest in these species, both in themselves and their potential use as iodination reagents.5,6 The use of halogen(I) reagents, such as the ubiquitous Barluenga's reagent, [I(pyridine)2]BF4, to effect iodination and oxidation reactions has been well established for over two decades.7–10 Halogen(I) (also known as halonium) ions are the incarnation of a fully ionised halogen atom to a formally cationic state, X+ (X = Cl, Br, I). Despite their reactivity, a growing number of these halogen(I) complexes have been characterised in the solid state by X-ray diffraction studies, with the largest subset of those incorporating the comparatively more stable iodine(I) species (as stability follows the trend: I+ > Br+ ≫ Cl+).11–13
Owing to their ease of preparation from suitable pairs of halogen bond (XB) donors (e.g., haloimides, halosulfonimides) and acceptors (e.g., pyridine derivatives, pyridine-N-oxides), neutral N–I⋯N and N–I⋯O–N halogen-bonded systems have enjoyed widespread study.14–19 The closely related anionic dioxoiodane complexes, [O–I–O]−, are also known and have been previously utilised as organic reagents.20
Similarly, hypoiodites (R-OI; R = alkyl) also have a well-documented history of being used as iodination reagents.21,22 However, these reagents are often generated in situ,23 with their exact structures only being speculated, such as with “tBuOI”,24,25 where molecular weight measurements suggest another species may exist in equilibrium with the carbonyl hypoiodite form, and makes discerning the structure–reactivity relationships of these reagents difficult. Similarly, the carbonyl hypoiodite iodine monoacetate, CH3C(O)OI, has only been characterised by 1H NMR spectroscopy and spectrophotometrically,26,27 despite a proven track record of being used as an iodination reagent.27 The handful of examples of trapped or stabilised carbonyl hypoiodites,5,6,28 where the carbonyl hypoiodite is stabilised by a Lewis base via halogen bonding, for which the solid-state structures are known are therefore a valuable resource to better understand such relationships. Comparisons between the ion-pair iodine(I) complexes and overall neutral stabilised carbonyl hypoiodites have revealed the two subsets of species possess structurally similar I–N bond lengths in the solid-state, characteristic of iodine(I) ions.5,6
As iodination reagents, halogen(I) ions are consumed stoichiometrically, making the potential of multi-iodination reagents attractive targets to increase atom economy and efficiency of their use. Halogen(I) complexes of the form [N–I–N]+ like Barluenga's reagent maintain good solubility in organic solvents, despite being an ion-pair complex, owing to their stabilising Lewis bases, mono-cationic charge, and the identity of the anion, which overall greatly contributes to their utility as reagents. Unfortunately, the inclusion of a two or more cationic [N–I–N]+ centres in one complex, for which only a single bis([N–I–N]+) example exists,4 would be expected to be concomitant with a decrease in solubility in organic solvents, that would become increasingly prominent as more [N–I–N]+ moieties were incorporated.
However, this is not an issue for carbonyl hypoiodites, which incorporate a neutral O–I–N moiety, with four examples of bis(O–I–N) compounds being reported in the solid-state to date (Fig. 1),6,28 all of which maintained good solubility in dichloromethane (DCM). This is not including a fifth example of a bis(O–I–N) compound (a structural isomer of two of the other examples), which was only able to be characterised by NMR studies.6
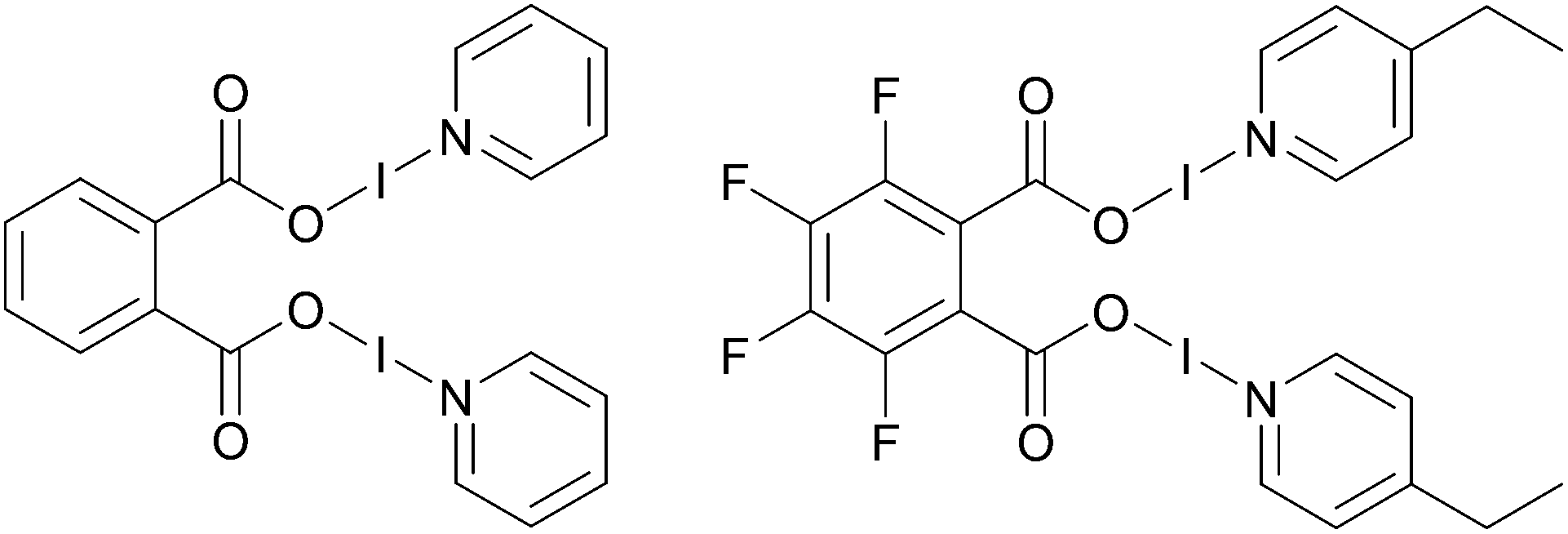 | ||
| Fig. 1 Two examples of structurally characterised bis(O–I–N) compounds.6,28 | ||
The potential of bis(O–I–N) as multi-iodination reagents are currently untested, undoubtedly due to their rarity in the literature. The silver(I) intermediates of the relatively straightforward one-pot process for the synthesis of carbonyl hypoiodites are not fully understood, as in contrast to the analogous formation of [N–I–N]+ halogen(I) complexes, the silver(I) precursors and intermediates are effectively insoluble in the solvents used. Therefore, a better understanding of the silver(I) intermediates involved in the formation of carbonyl hypoiodites would facilitate the study of these species, and enable other scaffolds to be incorporated into their design.
Results and discussion
Derivatives of pivalic acid
The carboxylic acid pivalic acid, tBuC(O)OH, was selected for these studies, as the organic-solvent solubility imparted to its derivatives by the tBu substituent was envisioned to be sufficient to overcome the ensuing insolubility of its respective silver(I) precursor (1) and intermediates (1a–1d; Scheme 1). Compound 1 was formed in excellent yields through deprotonation with NaOH, followed by subsequent Na+ to Ag+ cation exchange upon reaction with AgNO3, and could be easily prepared in bulk. However, 1 was effectively insoluble in DCM and MeCN, with good solubility only being observed in dimethyl sulfoxide (DMSO).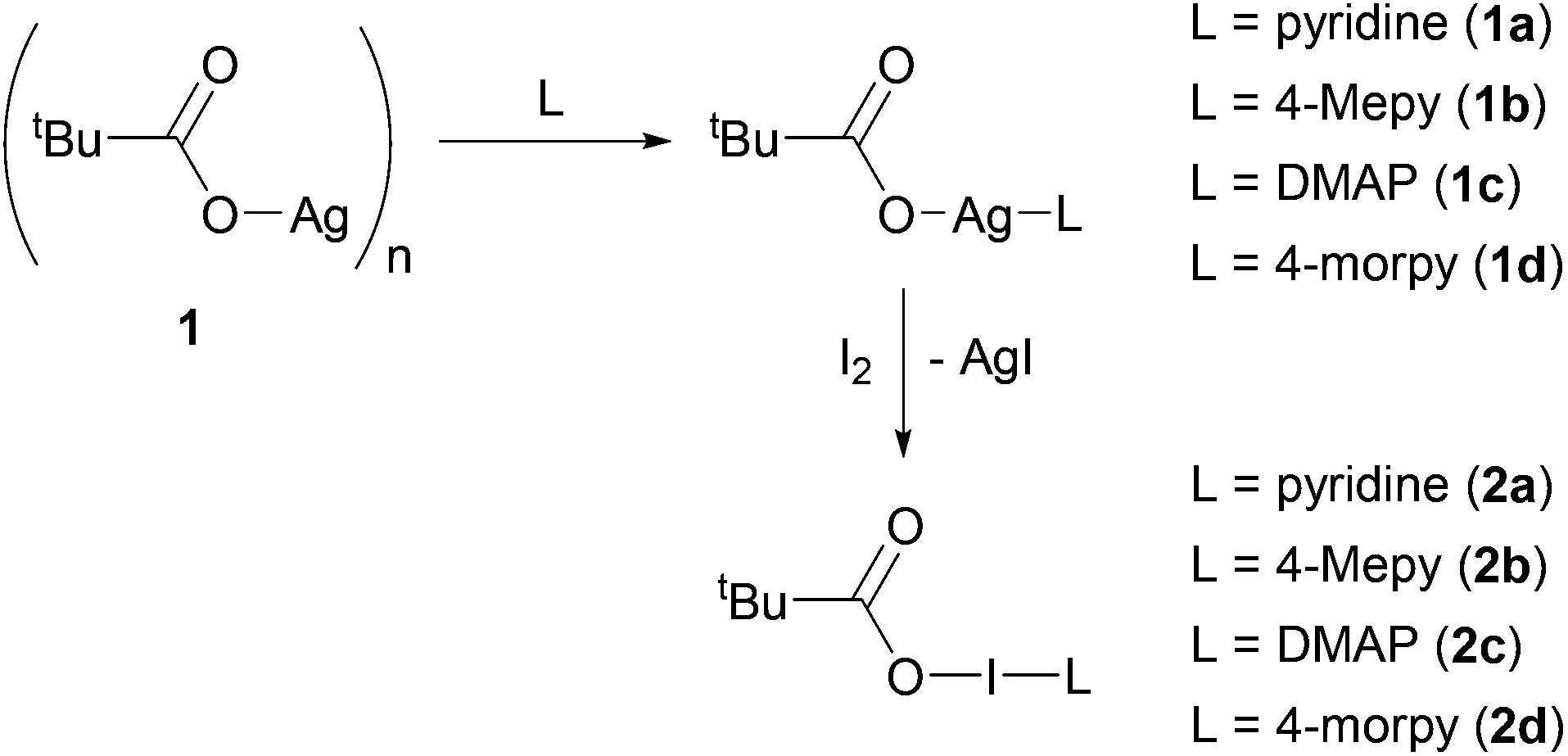 | ||
| Scheme 1 The procedure to synthesise carbonyl hypoiodite derivatives of pivalic acid. | ||
The solid-state structure of 1 revealed 1D polymeric chains comprised of dimeric subunits (Ag–Ag = 2.8613(8) and 2.8649(8) Å; Ag–O = 2.170(6)–2.255(6) Å) arrayed into bilayers possessing extensive Ag⋯Ag argentophilic interactions (2.9479(7)–3.0662(8) Å)29,30 and Ag⋯O (2.457(5)–2.530(5) Å) interactions (Fig. 2), as has been previously observed in related systems.31 These 1D polymeric chains explain the poor solubility of 1, given that a significant network of intra- and intermolecular bonds would need to be broken in order to liberate a single monomeric unit.
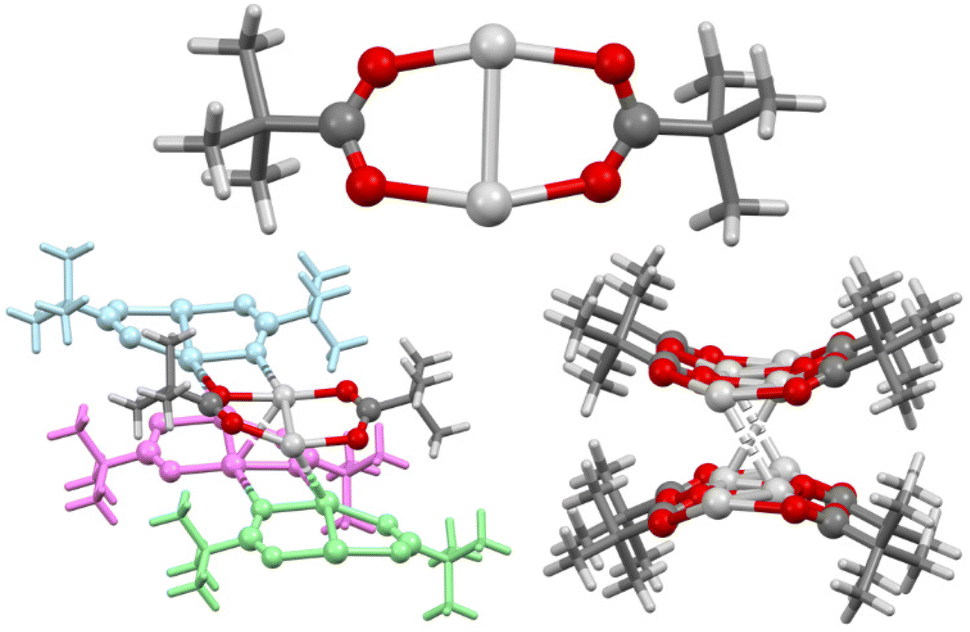 | ||
| Fig. 2 The X-ray crystal structure of a dimeric unit of 1 (top), the intermolecular bonding between four dimeric units of 1 (with three of the dimeric units block coloured for clarity; bottom left), and the view along the axis of the polymer chains showing the bilayer structure (bottom right; all alkyl groups simplified for clarity). Colour key: light grey = silver, red = oxygen, dark grey = carbon, white = hydrogen. | ||
The silver(I) intermediates (1a–1d) formed upon addition of a stoichiometric amount of Lewis base to 1 (1a = pyridine; 1b = 4-Mepy; 1c = DMAP; 1d = 4-morpy; where 4-Mepy = 4-methylpyridine, DMAP = dimethylaminopyridine, 4-morpy = 4-morpholinopyridine), displayed improved DCM and MeCN solubility in comparison to 1, though none were observed to have stoichiometric ratios of their tBu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Lewis base chemical shifts in their respective 1H NMR spectra, again necessitating the use of DMSO as the NMR solvent to ensure complete dissolution of the components. Unfortunately, 1a and 1b, the weaker Lewis bases, were observed to decompose in the DMSO media such that only their 1H NMR spectra could be reliably obtained. However, 1c and 1d displayed increased stability in DMSO, permitting more extensive NMR studies to be conducted.
:Lewis base chemical shifts in their respective 1H NMR spectra, again necessitating the use of DMSO as the NMR solvent to ensure complete dissolution of the components. Unfortunately, 1a and 1b, the weaker Lewis bases, were observed to decompose in the DMSO media such that only their 1H NMR spectra could be reliably obtained. However, 1c and 1d displayed increased stability in DMSO, permitting more extensive NMR studies to be conducted.
The 1H NMR spectra of 1a–1d displayed effectively negligible downfield changes in their chemical shifts compared to their respective neat Lewis bases and 1, all within the range of 0.01–0.09 ppm. This is in contrast to the formation of the [N–Ag–N]+ silver(I) precursors which display more noticeable perturbations in the chemical shifts,32 though unsurprising given the contrasting solvents (DMSO rather than DCM) and the potential coordinating nature of DMSO to the silver(I) centre being in competition with the Lewis bases in solution. The carboxylate carbon atoms were also probed by 13C NMR studies for 1c (182.2 ppm) and 1d (182.1 ppm), though again only negligible changes were observed from 1 (181.8 ppm). However, the directly-involved pyridinic nitrogen atoms were probed by 1H–15N HMBC studies and revealed much more indicative changes for 1c (−144.6 ppm) and 1d (−132.3 ppm) when compared to their corresponding Lewis base components of DMAP (−105.3 ppm) and 4-morpy (−94.7 ppm). The resulting coordination-induced shifts of −39.3 (1c) and −37.6 (1d) ppm are reminiscent of, but slightly smaller than, the large coordination shifts observed for [N–Ag–N]+ complexes, e.g., the 15N NMR coordination shift of the pyridinic nitrogen atoms (in DCM) for pyridine (−67.7 ppm) to [Ag(pyridine)2]PF6 (−122.7 ppm) is −55.0 ppm, and similarly for DMAP (−108.9 ppm) to [Ag(DMAP)2]PF6 (−169.8 ppm) is −60.9 ppm.32
The single crystal X-ray structures were determined for both 1b and 1c, albeit only at an isotropic level of accuracy for 1b which prevented detailed interrogation of the crystallographic parameters, but nonetheless was able to confirm the stoichiometry, identity, and linear nature of the O–Ag–N moiety (Fig. 3), fully analogous to the expected linear O–I–N bonding motif induced by the I+ centre. In contrast, 1c was observed as a discrete dimer (Fig. 4), possessing a similar, but slightly shorter (2.7854(8) Å), argentophilic Ag–Ag bond and pair of bridging pivalate molecules (Ag–O = 2.225(3) and 2.230(4) Å) as was found in 1, though in this instance with no additional intermolecular interactions of any significance. As a result of the neutral nature of 1 and 1a–1d, mass spectrometry analysis was also unable to provide any additional insights, with none of the structures being observed as either 2-coordinate silver(I) ‘monomers’ or 4-coordinate silver(I) dimers. The linear, 2-coordinate [N–Ag–N]+ precursors of [N–I–N]+ complexes are common in the literature,32–35 as are 4-coordinate AgN4 species,36,37 though 3-coordinate AgN3 examples have also been reported.38–40 Similarly, whilst the O–Ag–N motif is present in the literature,41,42 as are other examples of expanded coordination spheres incorporating similar ligands, suggesting that the adaptable nature of the Ag+ centre can accommodate a myriad of potential structures and coordination modes, which are somewhat independent of the identity of the ligands involved.
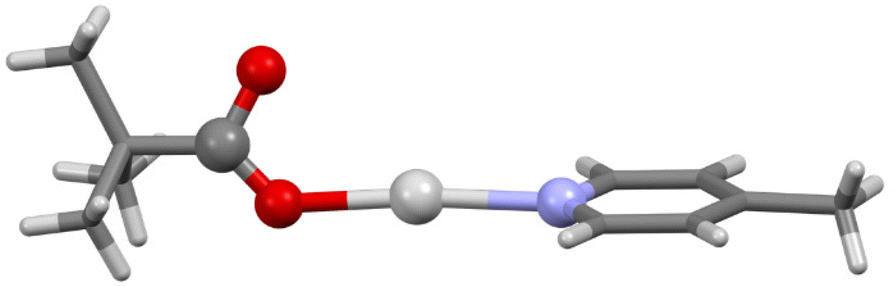 | ||
| Fig. 3 The X-ray determined connectivity model of 1b (aryl and alkyl groups simplified for clarity) showing the 2-coordinate silver(I) centre with a linear O–Ag–N geometry, analogous to the linear O–I–N geometry observed for stabilised hypoiodite compounds. Colour key: light grey = silver, red = oxygen, blue = nitrogen, dark grey = carbon, white = hydrogen. | ||
 | ||
| Fig. 4 The X-ray crystal structure of (1c)2 (aryl and alkyl groups simplified for clarity). Colour key: light grey = silver, red = oxygen, blue = nitrogen, dark grey = carbon, white = hydrogen. | ||
The silver(I) species are important precursors for the synthesis of carbonyl hypoiodites, as they provide a strong driving force for the reaction (via loss of AgI). However, the coordinative geometry of these silver(I) species is relevant to their subsequent use as precursors for the synthesis of carbonyl hypoiodites, as prior studies have found that more coordinatively saturated silver(I) species (≥4) have proven unreactive toward I2 when trying to synthesise more traditional [N–I–N]+ complexes.37,40
The subsequent conversions of 1a–1d to their stabilised carbonyl hypoiodite analogues 2a–2d upon reaction with one equivalent of elemental iodine were attempted in DMSO, however, mixtures were observed in all cases, necessitating that 2a–2d be synthesised from 1 in DCM and not DMSO, including their subsequent solution studies. This solvent change, though unfortunate with respect to drawing direct comparisons to the previously discussed 1 and 1a–1d without the need to consider the influence of the solvents, is useful given that nearly all modern NMR studies performed on carbonyl hypoiodites and halogen(I) complexes have been performed in deuterated DCM (or a closely-related chlorinated solvent).
The 1H–15N HMBC determined 15N NMR chemical shifts revealed large coordination shifts of the pyridinic nitrogen atoms going from the free ligands to the stabilised carbonyl hypoiodites 2b–2d.‡ The complexation-induced chemical shift change, ΔδN, defined here as δ(15NO–I–N)-δ(15NL), with O–I–N being the stabilised carbonyl hypoiodite compounds 2b–2d, and L being their corresponding free substituted pyridines (4-Mepy, DMAP, or 4-morpy, respectively), all determined in CD2Cl2 for the aforementioned reasons. The ΔδN values ranged from −79.3 ppm for the complexation of 4-Mepy (−75.1 ppm) in 2b (−154.4 ppm), to −94.2 ppm for the complexation of 4-morpy (−99.1 ppm) in 2d (−193.3 ppm), reflecting in all cases a dramatic change in the environment of the nitrogen atoms (Fig. S54†). These large coordination shifts correspond with those observed for other O–I–N compounds,5,6 as well as for [N–I–N]+ complexes which have also been observed to demonstrate analogously large values, e.g., [I(DMAP)2]PF6 had a ΔδN value of −108.9 ppm,32 which closely approximates the ΔδN value observed for 2c of −92.9 ppm. This indicates that despite the difference of a ligand between [I(DMAP)2]PF6 and 2c, the influence on the pyridinic nitrogen atom in the DMAP, upon coordination to the I+ centre, is predominantly the same between the two species.
The solid-state structures for the stabilised carbonyl hypoiodites 2b–2d were determined by single crystal X-ray crystallography (Fig. 5), with only 2a remaining elusive, agreeing with the solution-state observations. The I–O bond lengths for 2b (2.166(3) Å), 2c (2.167(3), 2.169(4), 2.175(3) Å; three crystallographically independent molecules present in the asymmetric unit cell), and 2d (2.155(3), 2.170(3) Å; two crystallographically independent molecules present in the asymmetric unit cell) were all effectively indistinguishable within the 3σ tolerance. The I–N bond lengths for 2b (2.282(4) Å), 2c (2.254(3), 2.268(4), 2.270(4) Å), and 2d (2.271(4), 2.292(3) Å) showed slightly greater variability, though all still very much within the known range of all reported [N–I–N]+ iodine(I) complexes§ of 2.23(1)–2.316(6) Å.42,43 The O–I–N bond angles for 2b (174.7(1)°), 2c (172.7(1), 173.3(1), 175.8(1)°), and 2d (174.13(9), 174.2(1)°), all stray beyond the 180° linear ideal and also, with the exception of 175.8(1)°, the most deviated example reported for [N–I–N]+ iodine(I) complexes of 175.2(2)° for [I(4-trifluoromethylpyridine)2]BF4.34 However, these angles are consistent with those reported for analogous O–I–N compounds bearing the same or similar 4-substituted pyridines and benzoyl moieties.5
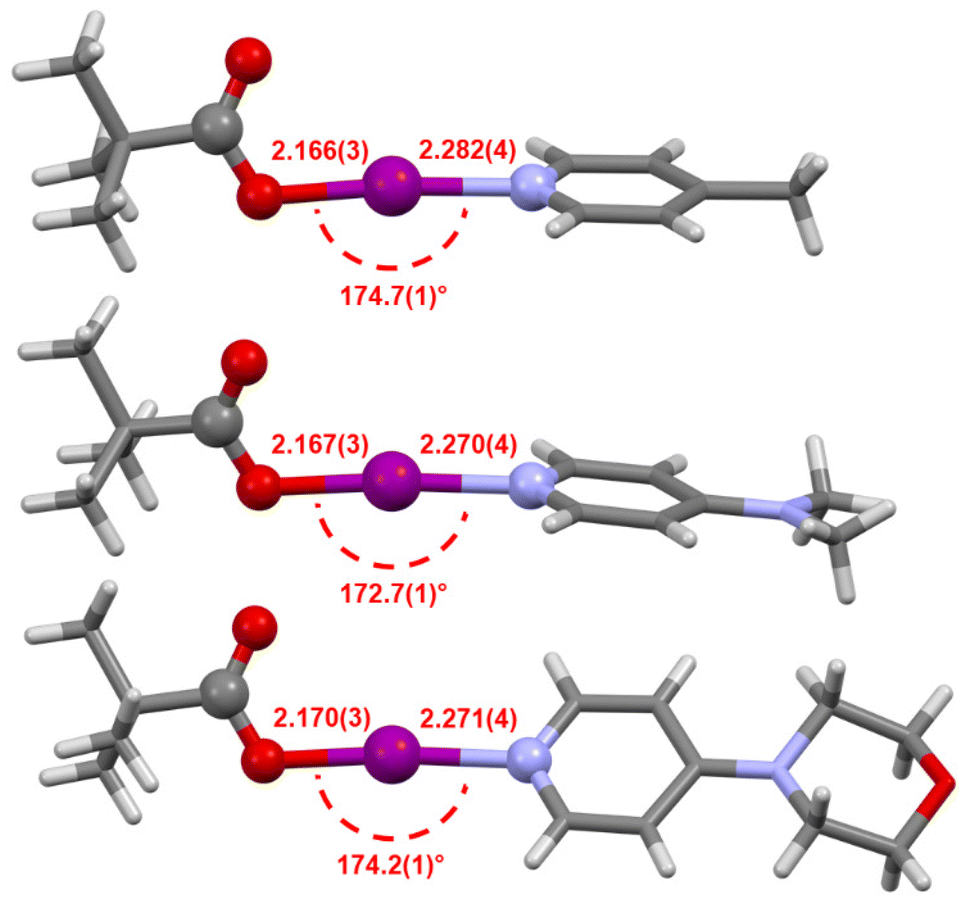 | ||
| Fig. 5 The X-ray crystal structures of 2b (top), 2c (middle), and 2d (bottom), annotated with their I–O and I–N bond lengths (in Å), and O–I–N angles (aryl and alkyl groups simplified for clarity; additional crystallographically independent molecules of 2c and 2d not shown). | ||
The computational structures of 2a–2d were calculated at the M06-2X/def2-TZVP level of theory44 using the SPARTAN18 program45 with dichloromethane (dielectric = 8.82) as the solvent using the conductor like polarizable continuum model (C-PCM)46,47 to confirm the higher reactivity observed experimentally for 2a (Fig. 6). The computational models of 2b–2d all showed good agreement with their crystallographic structures (Table S3†), validating the level of theory used. The model of 2a had an I–N bond length of 2.318 Å, which is at the extreme end of the range of those observed for solid-state [N–I–N]+ complexes,43 supporting that the pyridine ligand, a weaker Lewis base than those in the other complexes, would only be weakly bound and therefore more likely to be labile in solution as was observed experimentally.
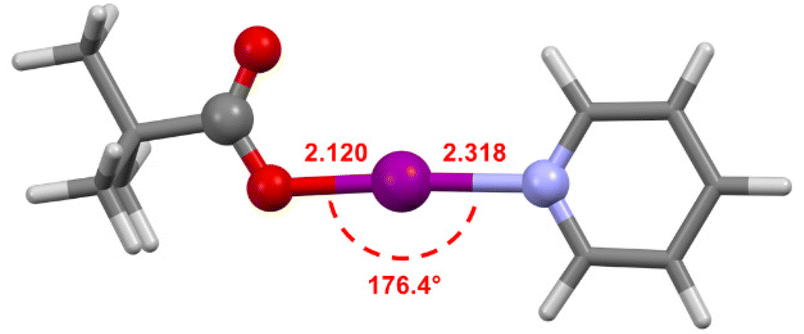 | ||
| Fig. 6 The computationally generated geometry of 2a calculated at the M06-2X/def2-TZVP level of theory, annotated with the I–O and I–N bond lengths (in Å), and the O–I–N bond angle. | ||
There are limited solid-state examples for a direct comparison to 2b and 2d, though differing substituent of the carbonyl group in 2c can be directly compared with (acetyl-OI)(DMAP) (I–O = 2.20(1) Å; I–N = 2.24(1) Å; O–I–N = 172.9(5)°) and (benzoyl-OI)(DMAP) (I–O = 2.210(3) Å; I–N = 2.241(3) Å; O–I–N = 178.1(1)°).5 As the comparison of these three compounds shows, 2c has the shortest I–O bond lengths and the longest I–N bond lengths. This is consistent with the electronic influence of the carbonyl substituent, with the tBu group of the pivalyl in 2c being the most electron donating, which could be equated to 2c having the least iodopyridinium character of the three compounds. Compound 2c lies between the two literature examples with respect to O–I–N bond angles, but these angles are undoubtedly heavily influenced by other factors than the electronic contributions of the substituents, such as packing effects, making this a less informative metric to consider in comparison to the bond lengths. Comparisons could also be drawn with the literature complex [NnBu4][AcO–I–succinimide],48 despite its anionic charge, given that it contains the same linear O–I–N motif. However, the I–O (2.293(2) and 2.299(2) Å) and I–N (2.166(2) and 2.168(2) Å) bond lengths of that complex clearly reflect that it contains significant covalent character in its I–N bond,5 and is therefore best described as an acetate anion coordinating to a neutral molecule of N-iodosuccinimide.
Derivatives of trimesic acid
The installation of multiple O–I–N moieties into a single compound, as previously discussed, is a worthwhile endeavour toward potentially more atom-economical iodinating reagents. Toward this goal, trimesic acid was seen as a natural progression from the benzene-dicarboxylic acids that have found prior success in synthesising bis(O–I–N) compounds. The silver(I) precursor Ag3(trimesate) (3) was synthesised in an analogous fashion to 1, adjusting for the 3:1 stoichiometry, and was obtained in excellent yields. Unlike for compound 1, no suitable solvent could be found to even partially dissolve 3, even at elevated temperatures, precluding solution studies. This apparent insolubility extended to the 3:1 stoichiometric pyridine (3a) and 4-Mepy (3b) derivatives of 3, though a huge excess of pyridine doped with a modest amount of DMSO was able to generate [Ag3(trimesate)(py)7]n (3a·(py)4; Fig. S4†). This polymeric structure demonstrated a 7:1 pyridine:trimesate ratio, wherein two of the carboxylate groups of each trimesate formed a 1D chain with bridging Ag+ centres coordinated to two carboxylates and two molecules of pyridine, whilst the third carboxylate of each trimesate would terminate as a Ag+ centre coordinated to one oxygen atom and three molecules of pyridine.
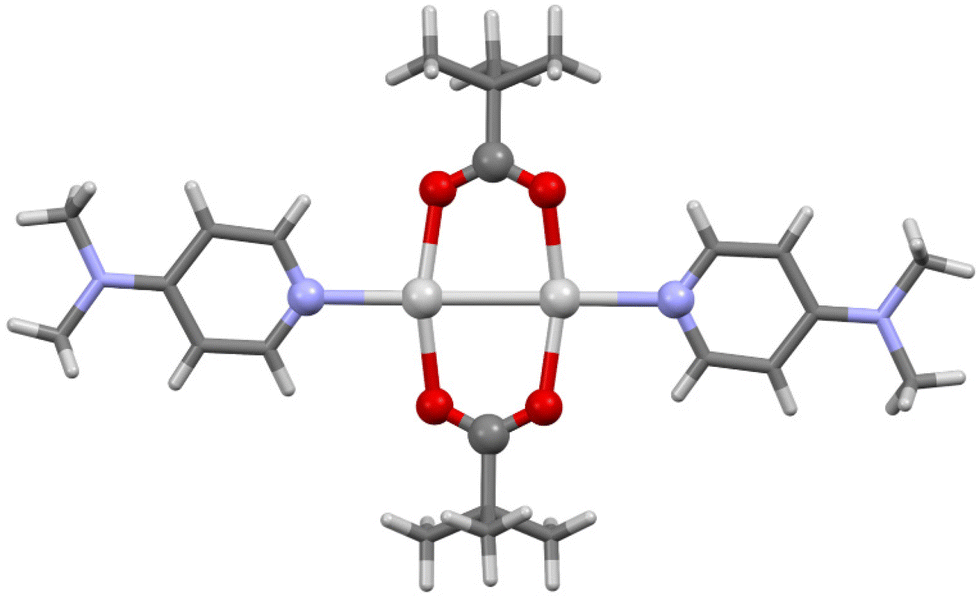
The stabilised tris(O–I–N) carbonyl hypoiodites of trimesic acid with pyridine (4a) or 4-Mepy (4b) were successfully prepared in a one-pot reaction in DCM, despite the near insolubility of the silver(I) precursor and intermediates, the first and only examples of tris(O–I–N) compounds to date. The 1H NMR studies of 4a and 4b confirmed the 3:1 pyridine/4-Mepy:trimesyl ratio, but 1H–15N HMBC studies to determine the 15N NMR chemical shifts could not be adequately obtained before decomposition was observed. Rapidly isolated samples of 4a and 4b, which were white solids, also clearly showed noticeable discolouration from decomposition within minutes, even under an inert atmosphere, preventing more detailed NMR studies. However, despite their highly reactive nature, the identity of 4a and 4b were definitively confirmed by single crystal X-ray diffraction studies (Fig. 7).
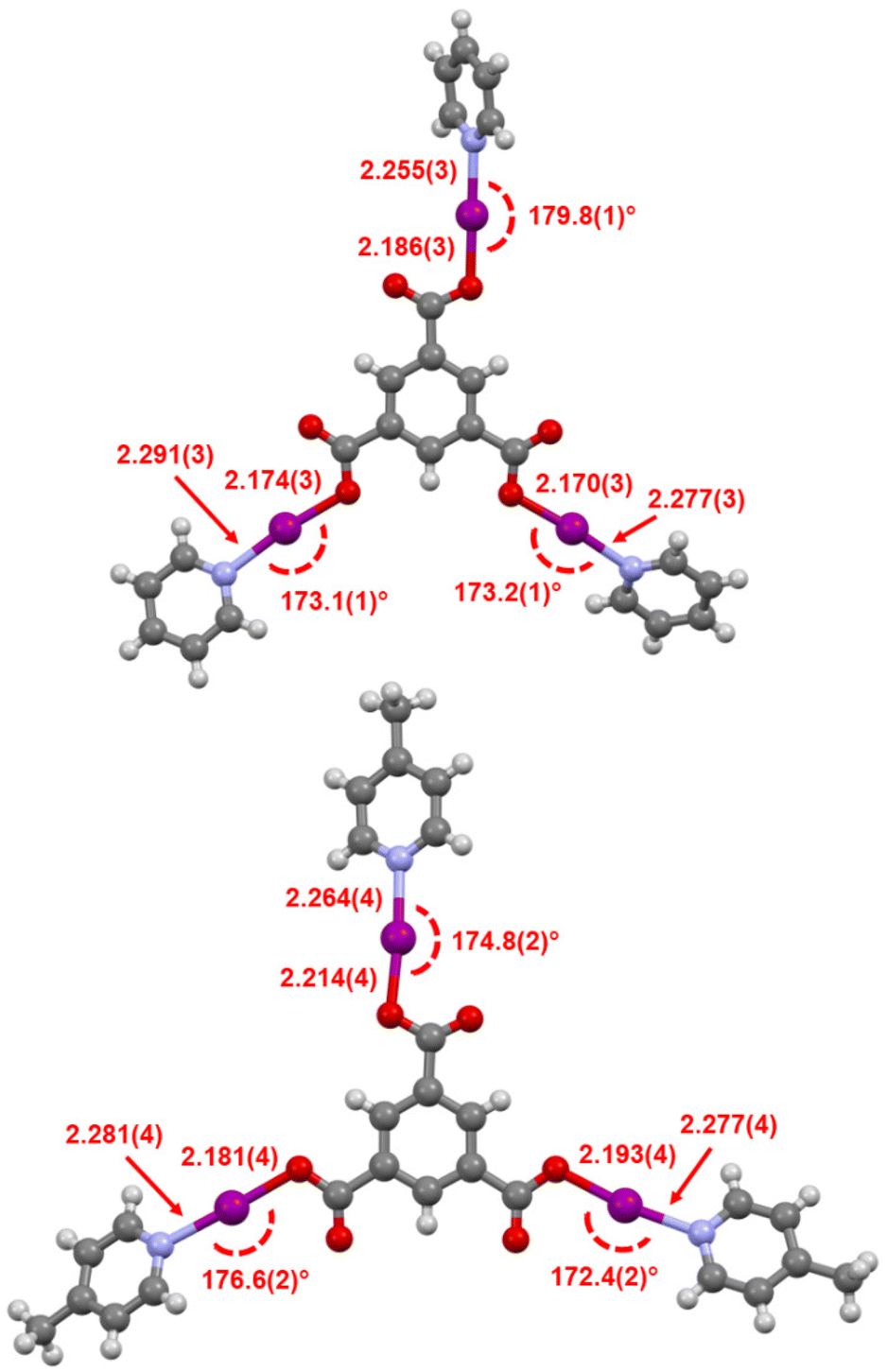 | ||
| Fig. 7 The X-ray crystal structures of 4a (top) and 4b (bottom), annotated with their I–O and I–N bond lengths (in Å) and O–I–N angles. | ||
Both 4a and 4b were found to possess monoclinic symmetry and were solved in the space group P21/n, and whilst 4b had no additional accoutrements, surprisingly 4a was found to also include a single molecule of H2O (per molecule of 4a) as a solvate, intermolecularly bridging the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of two neighbouring molecules of 4a. The I–O and I–N bond lengths of 4a (I–O = 2.170(3), 2.174(3), 2.186(3) Å; I–N = 2.255(3), 2.277(3), 2.291(3) Å) and 4b (I–O = 2.181(4), 2.193(4), 2.214(4) Å; I–N = 2.264(4), 2.277(4), 2.281(4) Å) reveal a slight elongation of the average I–O bond length in 4b (2.196 Å) versus4a (2.177 Å), concordant with the stronger Lewis base in 4b inducing more iodopyridinium (O⋯I–N) character in the O–I–N bonds. However, this is not clearly reflected in the I–N bond lengths of the two species, the ranges of which effectively overlap, as the two Lewis bases are still relatively quite similar in their basicities. This effect of influencing the electronic character of the O–I–N halogen bond between the two extremes of a pyridine-stabilised carbonyl hypoiodite (O–I⋯N) or a carboxylate-iodopyridinium ion pair (O⋯I–N) has been previously explored with other carbonyl hypoiodites,5 with only minor differences in the I–O and I–N bond lengths being noted when varying the strength of Lewis base used. Similar to the relatively large range of O–I–N angles observed for 2b–2d, 4a (173.1(1), 173.2(1), 179.8(1) Å) and 4b (172.4(2), 174.8(2), 176.6(2) Å) were also observed to demonstrate significant variability in their O–I–N angles, again likely a reflection of the packing, rather than electronic, effects.
O groups of two neighbouring molecules of 4a. The I–O and I–N bond lengths of 4a (I–O = 2.170(3), 2.174(3), 2.186(3) Å; I–N = 2.255(3), 2.277(3), 2.291(3) Å) and 4b (I–O = 2.181(4), 2.193(4), 2.214(4) Å; I–N = 2.264(4), 2.277(4), 2.281(4) Å) reveal a slight elongation of the average I–O bond length in 4b (2.196 Å) versus4a (2.177 Å), concordant with the stronger Lewis base in 4b inducing more iodopyridinium (O⋯I–N) character in the O–I–N bonds. However, this is not clearly reflected in the I–N bond lengths of the two species, the ranges of which effectively overlap, as the two Lewis bases are still relatively quite similar in their basicities. This effect of influencing the electronic character of the O–I–N halogen bond between the two extremes of a pyridine-stabilised carbonyl hypoiodite (O–I⋯N) or a carboxylate-iodopyridinium ion pair (O⋯I–N) has been previously explored with other carbonyl hypoiodites,5 with only minor differences in the I–O and I–N bond lengths being noted when varying the strength of Lewis base used. Similar to the relatively large range of O–I–N angles observed for 2b–2d, 4a (173.1(1), 173.2(1), 179.8(1) Å) and 4b (172.4(2), 174.8(2), 176.6(2) Å) were also observed to demonstrate significant variability in their O–I–N angles, again likely a reflection of the packing, rather than electronic, effects.
The comparison of 4a and (benzoyl-OI)(pyridine) (I–O = 2.14(1), 2.14(1) Å; I–N = 2.29(1), 2.31(1) Å),¶28 which can be considered the respective mono(O–I–N) analogue of 4a, does indicate that the presence of the two additional O–I–N moieties has a tangible impact on the I–O and I–N bond lengths in 4a, with an elongation of the I–O and shortening of the I–N bond lengths when compared to the average values in 4a (I–O(average) = 2.177 Å; I–N(average) = 2.274 Å). This would suggest that the two additional O–I–N moieties in 4a are causing the trimesyl group to be more electron withdrawing than the benzoyl group, inducing comparatively more iodopyridinium character in 4a. This is an interesting observation, as the inclusion, placement, and proximity of multiple O–I–N moieties had contributed to making compound 4a more reactive, and such factors will invariably have an impact on future scaffold design for other multi(O–I–N) compounds.
The silver(I) precursors of pyromellitic acid (benzene-1,2,4,5-tetracarboxylic acid), Ag4(pyromellitate), and mellitic acid (benzenehexacarboxylic acid), Ag6(mellitate), were also successfully prepared, and attempts were made to prepare the respective tetrakis(O–I–N) and hexakis(O–I–N) stabilised carbonyl hypoiodites. However, the reactivity of these species was found to be extreme to the point that the reactions displayed clear signs of decomposition even before complete addition of the elemental iodine.
Conclusions
The silver(I) derivatives of pivalic acid were synthesised to try and better understand the species present and ultimately facilitate the synthesis of a greater range of carbonyl hypoiodite compounds in the future. However, despite the high solubility of pivalic acid and its derivatives due to the inclusion of a tBu substituent, the ruinously poor solubility of the silver(I) precursor 1 or its pyridine-based derivatives 1a–1d, still proved a strong obstacle, with potentially even more soluble carboxylic acid starting materials needing to be considered in future studies. However, the use of the strongly polar solvent dimethyl sulfoxide (DMSO) permitted detailed solution-state studies to be conducted on the complexes incorporating the stronger Lewis bases DMAP (1c) and 4-morpy (1d), which displayed similar coordination shifts as their analogous [N–I–N]+ iodine(I) counterparts. The respective stabilised carbonyl hypoiodites were successfully synthesised (2a–2d), and all but the least stable 2a (which was supported by computational calculations), were characterised in the solid state.The first examples of tris(O–I–N) compounds, 4a and 4b, derivatives of trimesic acid, were also successfully prepared and, despite their observed reactivity, were definitely characterised in the solid-state by X-ray crystallography. The two structures revealed metrics comparable to their mono(O–I–N) analogues, which belied their observed increased reactivity, possibly indicating that additional factors contribute to the stability of stabilised hypoiodites. This was also supported by the attempts to synthesise tetrakis(O–I–N) and hexakis(O–I–N) stabilised hypoiodites being met with immediate failure due to greatly increased reactivities. However, the successful first report of tris(O–I–N) compounds presents a significant step forward in the field of halogen(I) chemistry, and heralds the emergence of more efficient iodination reagents potentially being developed in the future.
Experimental
General considerations
All reagents and solvents were obtained from commercial suppliers and used without further purification. For structural NMR assignments, 1H NMR and 1H–15N NMR correlation spectra were recorded on a Bruker Avance III 500 MHz spectrometer at 25 °C in CD2Cl2, or at 30 °C in (CD3)2SO (DMSO melting point = 19 °C). Chemical shifts are reported on the δ scale in ppm using the residual solvent signal as internal standard (CH2Cl2 in CD2Cl2: δH 5.32, δC 53.84; (CH3)2SO in (CD3)2SO: δH 2.50, δC 39.52), or for 1H–15N NMR spectroscopy, to an external CD3NO2 standard. For the 1H NMR spectroscopy, each resonance was assigned according to the following conventions: chemical shift (δ) measured in ppm, observed multiplicity, observed coupling constant (J Hz), and number of hydrogens. Multiplicities are denoted as: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and br (broad). For the 1H–15N HMBC spectroscopy, spectral windows of 4 ppm (1H) and 300 ppm (15N) were used, with 1024 points in the direct dimension and 512 increments used in the indirect dimension, with subsequent peak shape analysis being performed to give the reported 15N NMR resonances. The NMR data for neat pyridine and neat DMAP in CD2Cl2 have been previously reported.32The single crystal X-ray data for 1, 1b, 2c, 2e, 4a, and 4b were collected at 120 K using an Agilent SuperNova dual wavelength diffractometer with an Atlas detector using mirror-monochromated Cu-Kα (λ = 1.54184 Å) radiation. The single crystal X-ray data for (1c)2, 2b and 3a·(py)4 were collected at 120 K using an Agilent SuperNova diffractometer with an Eos detector using mirror-monochromated Mo-Kα (λ = 0.71073 Å) radiation. The program CrysAlisPro49 was used for the data collection and reduction on the SuperNova diffractometers. All structures were solved by intrinsic phasing (SHELXT)50 and refined by full-matrix least squares on F2 using Olex2,51 utilising the ShelXL-2015 module.52 Anisotropic displacement parameters were assigned to non-H atoms and isotropic displacement parameters for all H atoms were constrained to multiples of the equivalent displacement parameters of their parent atoms with Uiso(H) = 1.2Ueq (aromatic; cyclic alkyl) or Uiso(H) = 1.5Ueq (acyclic alkyl) of their respective parent atoms.
Synthetic and characterisation details, including annotated NMR assignments, of all new compounds are included in the accompanying ESI.†
Author contributions
J.S.W. was responsible for the conceptualisation of the research and provided supervision, as well as performing the investigation of the solution and X-ray studies. J.M., L.M.E.W., and E.K. were responsible for the preparation of crystallographic samples and solution studies. The visualisation and writing of the manuscript were undertaken by J.S.W., with R.A. and K.R. contributing to the reviewing and editing. J.S.W., R.A., and K.R. were responsible for the acquisition of funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the University of Jyväskylä mass spectrometry service for their time and expertise, and gratefully acknowledge the Magnus Ehrnrooth Foundation (J. S. W.), the ERDF DORA travel grant (J. M.), the Estonian Research Council (R.A. grant no. PRG399), the Academy of Finland (K.R. grant no. 317259), and the University of Jyväskylä, Finland for financial support.References
- J. Pancholi and P. D. Beer, Coord. Chem. Rev., 2020, 416, 213281 CrossRef CAS.
- L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem., Int. Ed., 2016, 55, 14033–14036 CrossRef CAS PubMed.
- L. Turunen, U. Warzok, C. A. Schalley and K. Rissanen, Chem, 2017, 3, 861–869 CAS.
- A. Vanderkooy, A. K. Gupta, T. Földes, S. Lindblad, A. Orthaber, I. Pápai and M. Erdélyi, Angew. Chem., Int. Ed., 2019, 58, 9012–9016 CrossRef CAS PubMed.
- S. Yu, J. S. Ward, K.-N. Truong and K. Rissanen, Angew. Chem., Int. Ed., 2021, 60, 20739–20743 CrossRef CAS.
- E. Kramer, S. Yu, J. S. Ward and K. Rissanen, Dalton Trans., 2021, 50, 14990–14993 RSC.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- J. Barluenga, J. M. González, M. A. Garcia-Martin, P. J. Campos and G. Asensio, J. Chem. Soc., Chem. Commun., 1992, 1016–1017 RSC.
- J. Ezquerra, C. Pedregal, C. Lamas, J. Barluenga, M. Pérez, M. A. García-Martín and J. M. González, J. Org. Chem., 1996, 61, 5804–5812 CrossRef CAS.
- G. Espuña, G. Arsequell, G. Valencia, J. Barluenga, M. Pérez and J. M. González, Chem. Commun., 2000, 1307–1308 RSC.
- E. S. Stoyanov, I. V. Stoyanova, F. S. Tham and C. A. Reed, J. Am. Chem. Soc., 2010, 132, 4062–4063 CrossRef CAS.
- A. Karim, M. Reitti, A.-C. C. Carlsson, J. Gräfenstein and M. Erdélyi, Chem. Sci., 2014, 5, 3226–3233 RSC.
- L. Turunen and M. Erdélyi, Chem. Soc. Rev., 2020, 49, 2688–2700 RSC.
- O. Makhotkina, J. Lieffrig, O. Jeannin, M. Fourmigué, E. Aubert and E. Espinosa, Cryst. Growth Des., 2015, 15, 3464–3473 CrossRef CAS.
- E. Aubert, E. Espinosa, I. Nicolas, O. Jeannin and M. Fourmigué, Faraday Discuss., 2017, 203, 389–406 RSC.
- K. Raatikainen and K. Rissanen, CrystEngComm, 2011, 13, 6972–6977 RSC.
- K. Raatikainen and K. Rissanen, Chem. Sci., 2012, 3, 1235–1239 RSC.
- R. Puttreddy, O. Jurček, S. Bhowmik, T. Mäkelä and K. Rissanen, Chem. Commun., 2016, 52, 2338–2341 RSC.
- R. Puttreddy, J. M. Rautiainen, T. Mäkelä and K. Rissanen, Angew. Chem., Int. Ed., 2019, 58, 18610–18618 CrossRef CAS PubMed.
- K. Muñiz, B. García, C. Martínez and A. Piccinelli, Chem. – Eur. J., 2017, 23, 1539–1545 CrossRef.
- D. D. Tanner and G. C. Gidley, J. Am. Chem. Soc., 1968, 90, 808–809 CrossRef CAS.
- J. Barluenga, F. González-Bobes and J. M. González, Angew. Chem., Int. Ed., 2002, 41, 2556–2558 CrossRef CAS PubMed.
- R. Kawasumi, S. Narita, K. Miyamoto, K. Tominaga, R. Takita and M. Uchiyama, Sci. Rep., 2017, 7, 17967 CrossRef PubMed.
- R. Montoro and T. Wirth, Org. Lett., 2003, 5, 4729–4731 CrossRef CAS PubMed.
- D. D. Tanner, G. C. Gidley, N. Das, J. E. Rowe and A. Potter, J. Am. Chem. Soc., 1984, 106, 5261–5267 CrossRef CAS.
- J. L. Courtneidge, J. Lusztyk and D. Pagé, Tetrahedron Lett., 1994, 35, 1003–1006 CrossRef CAS.
- T. Hokamp, A. T. Storm, M. Yusubov and T. Wirth, Synlett, 2018, 415–418 CAS.
- H. Hartl and M. Hedrich, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1981, 36, 922–928 CrossRef.
- S. Sculfort and P. Braunstein, Chem. Soc. Rev., 2011, 40, 2741–2760 RSC.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- Z.-H. Yan, X.-Y. Li, L.-W. Liu, S.-Q. Yu, X.-P. Wang and D. Sun, Inorg. Chem., 2016, 55, 1096–1101 CrossRef CAS PubMed.
- J. S. Ward, G. Fiorini, A. Frontera and K. Rissanen, Chem. Commun., 2020, 56, 8428–8431 RSC.
- M. Bedin, A. Karim, M. Reitti, A.-C. C. Carlsson, F. Topić, M. Cetina, F. Pan, V. Havel, F. Al-Ameri, V. Sindelar, K. Rissanen, J. Gräfenstein and M. Erdélyi, Chem. Sci., 2015, 6, 3746–3756 RSC.
- A.-C. C. Carlsson, K. Mehmeti, M. Uhrbom, A. Karim, M. Bedin, R. Puttreddy, R. Kleinmaier, A. A. Neverov, B. Nekoueishahraki, J. Gräfenstein, K. Rissanen and M. Erdélyi, J. Am. Chem. Soc., 2016, 138, 9853–9863 CrossRef CAS PubMed.
- S. Yu, E. Kalenius, A. Frontera and K. Rissanen, Chem. Commun., 2021, 57, 12464–12467 RSC.
- K. V. Goodwin, D. R. McMillin and W. R. Robinson, Inorg. Chem., 1986, 25, 2033–2036 CrossRef CAS.
- S. Yu, P. Kumar, J. S. Ward, A. Frontera and K. Rissanen, Chem, 2021, 7, 948–958 CAS.
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, J. Chem. Soc., Dalton Trans., 1998, 1943–1946 RSC.
- D. A. Beauchamp and S. J. Loeb, Supramol. Chem., 2005, 17, 617–622 CrossRef CAS.
- J. S. Ward, A. Frontera and K. Rissanen, Inorg. Chem., 2021, 60, 5383–5390 CrossRef CAS PubMed.
- X. Zhang, L. W. Liu, Z. C. Hao and G. H. Cui, Transit. Met. Chem., 2016, 41, 459–466 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr. Sect. B, 2016, 72, 171–179 CrossRef CAS PubMed.
- J. S. Ward, K.-N. Truong, M. Erdélyi and K. Rissanen, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2022, DOI:10.1016/B978-0-12-823144-9.00043-1.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Spartan'18, Wavefunction Inc., Irvine CA, USA, 2018 Search PubMed.
- X. Zhang and J. M. Herbert, J. Phys. Chem. B, 2014, 118, 7806–7817 CrossRef CAS PubMed.
- A. W. Lange and J. M. Herbert, Chem. Phys. Lett., 2011, 509, 77–87 CrossRef CAS.
- A. J. Guzmán Santiago, C. A. Brown, R. D. Sommer and E. A. Ison, Dalton Trans., 2020, 49, 16166–16174 RSC.
- CrysAlisPro, Agilent Technologies Ltd, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, NMR and X-ray crystallographic. CCDC 2173463–2173470. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt01988d |
| ‡ The 15N NMR chemical shift could not be accurately determined for 2a due to decomposition being observed during the 1H–15N HMBC experiment. |
| § Incorporating sp2-nitrogen Lewis bases, which form by far the largest subset of reported [N–I–N]+ iodine(I) complexes. |
| ¶ The bond lengths and esds given are those reported in the original 1981 publication, though are in slight contrast to the bond lengths reported in the CSD (refcode: BZPRIA) for which no esds are given. |
| This journal is © The Royal Society of Chemistry 2022 |