
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntroduction of a triphenylamine substituent on pyridyl rings as a springboard for a new appealing brightly luminescent 1,3-di-(2-pyridyl)benzene platinum(II) complex family†
Alessia
Colombo
 a,
Giulia
De Soricellis
a,
Francesco
Fagnani
a,
Claudia
Dragonetti
*a,
Massimo
Cocchi
b,
Bertrand
Carboni
c,
Véronique
Guerchais
c and
Daniele
Marinotto
d
a,
Giulia
De Soricellis
a,
Francesco
Fagnani
a,
Claudia
Dragonetti
*a,
Massimo
Cocchi
b,
Bertrand
Carboni
c,
Véronique
Guerchais
c and
Daniele
Marinotto
d
aDipartimento di Chimica, Università degli Studi di Milano, UdR INSTM di Milano, via C. Golgi 19, 20133 Milan, Italy. E-mail: claudia.dragonetti@unimi.it
bIstituto per la Sintesi Organica e la Fotoreattività (ISOF), Consiglio Nazionale delle Ricerche (CNR), via P. Gobetti 101, 40129 Bologna, Italy
cUniversité de Rennes 1, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France
dIstituto di Scienze e Tecnologie Chimiche (SCITEC) “Giulio Natta”, Consiglio Nazionale delle Ricerche (CNR), via C. Golgi 19, 20133 Milan, Italy
First published on 20th July 2022
Abstract
The preparation and characterization of three new complexes, namely [Pt(1,3-bis(4-triphenylamine-pyridin-2-yl)-4,6-difluoro-benzene)Cl] ([PtL1Cl]), [Pt(1,3-bis(4-triphenylamine-pyridin-2-yl)-5-triphenylamine-benzene)Cl] ([PtL2Cl]), and [Pt(1,3-bis(4-triphenylamine-pyridin-2-yl)-5-mesityl-benzene)Cl] ([PtL3Cl]), is reported. All of them are highly luminescent in dilute deaerated dichloromethane solution (Φlum = 0.88–0.90, in the yellow-green region; the λmax,em in nm for the monomers are: 562, 561 and 549 for [PtL1Cl], [PtL2Cl] and [PtL3Cl], respectively).[PtL1Cl] is the most appealing, being characterized by a very long lifetime (103.9 μs) and displaying intense NIR emission in concentrated deaerated solution (Φlum = 0.66) with essentially no “contamination” by visible light < 600 nm. This complex allows the fabrication of both yellow-green and deep red/NIR OLEDs; OLED emissions are in the yellow-green (CIE = 0.38, 0.56) and deep red/NIR (CIE = 0.65, 0,34) regions, for [PtL1Cl] 8 wt% (with 11% ph/e EQE) and pure [PtL1Cl] (with 4.3% ph/e EQE), respectively.
1. Introduction
There is a growing interest in the study of square planar platinum(II) complexes for various applications like bioimaging,1–8 electroluminescent devices,9–21 sensing,21–23 and nonlinear optics.24–28 The significant spin–orbit coupling related to the platinum atom helps intersystem crossing, and consequently emission of light from triplet excited states, a performance that is further improved by the introduction of Pt–C bonds.29–31 Because of this strong spin–orbit coupling, an internal electroluminescence quantum efficiency of one can be achieved in devices, since both the singlet and triplet excitons generated by electron–hole recombination (of which the ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) can be induced to emit (triplet harvesting).32–34 For this reason, square planar platinum(II) complexes have become a centre of interest for the preparation of organic light-emitting diodes (OLEDs) which earned a pivotal position for solid-state lighting applications and full-color flat-panel display.35,36 Due to their geometrical structure which favours the creation of aggregates or excimers by Pt–Pt or ligand–ligand intermolecular interactions,20,37–42 they are a useful tool for the fabrication of deep-red to near infrared (NIR) OLEDs which have unique applications in optical communication, night-vision readable displays, and photodynamic therapy.20,43–45 Moreover, platinum(II) aggregates, characterized by low-energy red to NIR emissions and by relatively long phosphorescence lifetimes, are especially attractive for application in biological imaging with the aim of improving the accuracy of medical diagnosis.1–8 It is worth pointing out that finding red-emissive materials is a big challenge since the “energy gap law” predicts that the non-radiative decay rate constant increases exponentially with decreasing optical energy gap (ΔEopt).46
:3) can be induced to emit (triplet harvesting).32–34 For this reason, square planar platinum(II) complexes have become a centre of interest for the preparation of organic light-emitting diodes (OLEDs) which earned a pivotal position for solid-state lighting applications and full-color flat-panel display.35,36 Due to their geometrical structure which favours the creation of aggregates or excimers by Pt–Pt or ligand–ligand intermolecular interactions,20,37–42 they are a useful tool for the fabrication of deep-red to near infrared (NIR) OLEDs which have unique applications in optical communication, night-vision readable displays, and photodynamic therapy.20,43–45 Moreover, platinum(II) aggregates, characterized by low-energy red to NIR emissions and by relatively long phosphorescence lifetimes, are especially attractive for application in biological imaging with the aim of improving the accuracy of medical diagnosis.1–8 It is worth pointing out that finding red-emissive materials is a big challenge since the “energy gap law” predicts that the non-radiative decay rate constant increases exponentially with decreasing optical energy gap (ΔEopt).46
In the last two decades it turned out that platinum(II) chlorido complexes with a cyclometalated terdentate 1,3-bis(pyridin-2-yl)benzene ligand (bpyb), which permits a rigid N^C^N coordination environment, are among the best emitters.47–52 For example, in deaerated CH2Cl2, [Pt(bpyb)Cl] is characterized by an emission quantum yield (Φlum) of 0.60,47 a value much larger than that observed for [Pt(N^C-2-phenylpyridine)(N-2-phenylpyridine)Cl] or the parent N^N^C-coordinated isomer.49 Such a superiority has been attributed to the presence of the Pt–C bond, which causes a high ligand-field strength, and to the rigidity of the N^C^N ligand, which hinders distortions that would cause non-radiative decay.49
An appealing aspect of these platinum(II) chlorido compounds is that the introduction of suitable substituents, in the phenyl or pyridyl rings of the cyclometalated 1,3-bis(pyridin-2-yl)benzene ligand, allows to control the colour of the emission, keeping large quantum yields.14 As a matter of fact, time-dependent density functional theory (TD-DFT) calculations put in evidence that the frontier orbitals are localised on diverse portions of the molecule, such that their energies can be altered practically independently.53–55 Thus, in this kind of complexes in which the triplet state at lowest-energy has mainly HOMO → LUMO character, the phenyl ring makes the principal contribution to the HOMO whereas the pyridyl rings dominate in the LUMO. Accordingly, the introduction of electron-donating substituents in the phenyl ring (which raises the energy of the HOMO) and electron-accepting substituents in the pyridyl rings (which lowers the LUMO energy, especially in para of the N atoms) will allow to red-shift the emission, whereas electron-accepting substituents in the phenyl ring and electron-donating substituents in the pyridyl rings will lead to a blue-shift. The experimentally observed tendencies follow this design well.3
Thus, many derivatives of [Pt(1,3-bis(pyridin-2-yl)-5-R-benzene)Cl] (R = H) have been prepared and well characterized. For example, incorporation of an ester substituent at the 5 position of the central benzene ring leads to a blue-shift of the monomer emission (from 491 to 481 nm) whereas there is a progressive red-shift on going from R = mesityl (501 nm) to CH3 (505 nm), tolyl (516 nm), thienyl (548) or dimethylaminophenyl (588 nm).48 The study of the effect of the nature of the substituents at the 4 position of pyridyl rings has been mainly carried out with [Pt(1,3-bis(4-R-pyridin-2-yl)-4,6-difluoro-benzene)Cl] (R = H) as parent complex; as expected, there is a blue-shift of the monomer emission energy on going from R = CF3 (496 nm)15 to H (472 nm)50 and NMe2 (453 nm).56 All these complexes are usually characterized by high quantum yields (Φlum = 0.50–0.87) and relative long lifetimes (τ = 5–10 μs).
In this context, we were curious to see the effect of the introduction of a polarizable π-delocalized bulky substituent on the phosphorescence properties of the [Pt(1,3-bis(pyridin-2-yl)-4,6-difluoro-benzene)Cl] complex. Therefore, we prepared the complex with a triphenylamine substituent at the 4 position of each pyridine [PtL1Cl] (Scheme 1). This new complex is even more emissive than its known parent derivatives, in the yellow-green or deep red/NIR region depending on its concentration in solution. It is characterized by an unexpected long lifetime and can be used to prepare efficient yellow-green and deep red/NIR OLEDs. The related new complexes with a cyclometalated 5-triphenylamine-benzene [PtL2Cl] or 5-mesityl-benzene [PtL3Cl], prepared for comparison, are also highly emissive leading to a new fascinating brightly luminescent family of cyclometalated 1,3-di-(2-pyridyl)benzene platinum(II) complexes.
 | ||
| Scheme 1 Chemical structures of the investigated complexes. | ||
2. Experimental
2.1 Synthesis of [PtLnCl]
The synthetic procedure is reported here. See details and full characterization in ESI.†:1 to 8:2) and a clear yellow lacquer was obtained (42% yield).
2.2 Photophysical characterization
Electronic absorption spectra were obtained at room temperature in dichloromethane, by means of a Shimadzu UV3600 spectrophotometer and quartz cuvettes with 1 cm optical path length. Absolute photoluminescence quantum yields, Φ, were measured using a C11347 Quantaurus Hamamatsu Photonics K.K spectrometer, equipped with a 150 W Xenon lamp, an integrating sphere and a multichannel detector. Steady state and time-resolved fluorescence data were recorded with a FLS980 spectrofluorimeter (Edinburg Instrument Ltd). See details in ESI.†2.3 Procedure for OLED fabrication with [PtL1Cl]
OLEDs were fabricated by growing a sequence of thin layers on clean glass substrates pre-coated with a 120 nm-thick layer of indium tin oxide (ITO) with a sheet resistance of 20 U per square. A 2 nm-thick hole-injecting layer of MoOx was deposited on top of the ITO by thermal evaporation under high vacuum of 10−6 hPa. The other layers were deposited in succession by thermal evaporation under high vacuum: first, 4,4′,4′′-tris(N-carbazolyl)triphenylamine (TCTA, 50 nm) as exciton blocking layers; second, the emitting layer (EML) evaporated by co-deposition of [PtL1Cl] and (bis-4-(N-carbazolyl)phenyl)phenylphosphine oxide (BCPO) to form a 30 nm-thick blend film (8 wt% Pt complex:92 wt% BCPO), or by single deposition of the [PtL1Cl] complex only, to form a 30 nm neat film; third, 2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole (TPBi, 30 nm) as an electron-transporting and hole-blocking layer. This was followed by thermal evaporation of the cathode layer consisting of 0.5 nm thick LiF and a 100 nm thick aluminium cap.
The current–voltage characteristics were measured with a Keithley Source-Measure unit, model 236, under continuous operation mode, while the light output power was measured with an EG&G power meter, and electroluminescence (EL) spectra recorded with a StellarNet spectroradiometer. All measurements were carried out at room temperature under argon atmosphere and were reproduced for many runs, excluding any irreversible chemical and morphological changes in the devices.
3. Results and discussion
3.1 Preparation of the new pro-ligands HLn and related [PtLnCl] complexes
The new pro-ligands 1,3-bis(4-(triphenylamino)pyridin-2-yl)-4,6-diR1-5-R2-benzene (HL1: R1 = F, R2 = H; HL2: R1 = H, R2 = triphenylamino; HL3: R1 = H, R2 = mesityl) were prepared by Suzuki–Miyaura cross-coupling of the corresponding boronic acid pinacol ester with 2-chloro-4-(triphenylamino)pyridine (see Scheme 2). Reaction of these pro-ligands with K2PtCl4 led to the desired [PtLnCl] complexes in 82–93% yields (see Experimental and ESI†). | ||
| Scheme 2 Synthesis of [PtLnCl]. | ||
3.2 Photophysical properties in solution and in solid state
The absorption spectra of [PtL1Cl], [PtL2Cl] and [PtL3Cl] complexes in dichloromethane solution at different concentrations are shown in Fig. 1 and relevant values are given in Table 1.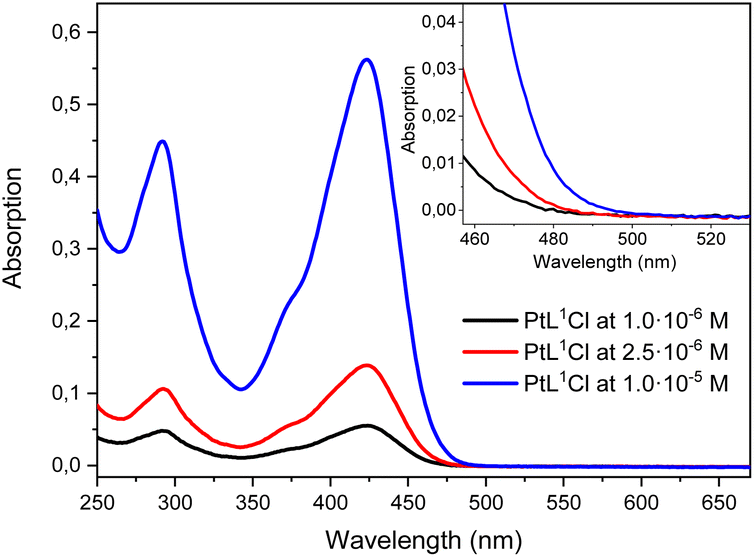 | ||
| Fig. 1 Absorption spectra at different concentrations of [PtL1Cl], [PtL2Cl] and [PtL3Cl] in CH2Cl2 at 298 K. The weak bands at longer wavelengths are shown on an expanded scale for clarity. | ||
| Complex | λ max,abs/nm [ε/M−1 cm−1] | λ max,em/nmb | ϕ lum | τ/μs |
|---|---|---|---|---|
| Monomer [excimer/aggregate] | Degassed [aerated] | |||
| a At 2 × 10−6 M. b Excitation at 422 nm. c Excimer and Aggregate at 2.4 × 10−4 M. | ||||
| [PtL1Cl] | 293 [4.5 × 104] | 562 [696]c | 0.90 [0.059] | 103.9 |
| 423 [5.6 × 104] | ||||
| [PtL2Cl] | 296 [7.5 × 104] | 561 [704]c | 0.88 [0.051] | 6.6 |
| 425 [7.1 × 104] | ||||
| 502 [0.7 × 103] | ||||
| [PtL3Cl] | 295 [5.9 × 104] | 549 [727]c | 0.89 [<0.01] | 50.0 |
| 424 [8.0 × 104] | ||||
| 495 [1.0 × 103] | ||||
All three complexes show intense absorption bands at 260–320 nm which can be attributed to intraligand 1π–π* transitions of the N^C^N ligand by analogy with related Pt(II) complexes;57 moreover, intense bands are observed at around 350–470 nm which are attributed, by comparison with related cyclometalated Ir(III) complexes,58 to an intraligand charge transfer (ILCT) from the triphenylamine moiety to the pyridyl rings; these strong latter bands cover the absorption bands (350–460 nm) of the charge-transfer transitions involving the cyclometalated ligand and the metal.57 In addition, very weak absorption bands at lower energy (485–520 nm) are distinguishable in the absorption spectra of [PtL2Cl] and [PtL3Cl] complexes which are connected to direct population of 3π–π* states facilitated by the high spin–orbit coupling associated with the Pt(II) center. Although a similar transition is also expected for the [PtL1Cl] complex, no absorption band is detected up to 5 × 10−5 M, probably due to its low molar extinction coefficient (see Fig. S4 in ESI†).
Complexes [PtL1Cl], [PtL2Cl] and [PtL3Cl] are highly luminescent in dilute deaerated dichloromethane solution (2.5 × 10−6 M), with a luminescence quantum yield of 0.90, 0.88 and 0.89, respectively. [PtL1Cl] displays a luminescence quantum yield which is similar or superior to that reported for the parent [Pt(1,3-bis(4-R-pyridin-2-yl)-4,6-difluoro-benzene)Cl] complexes with different R groups in para of the pyridine rings (Φlum = 0.80, 0.59, 0.87, 0.71, and 0.60, for R = H,50 CF3,15 Me,38 OMe54 and NMe2,56 respectively). Besides, [PtL3Cl] has a higher luminescence quantum yield than the related [Pt(1,3-bis(pyridin-2-yl)-mesityl-benzene)Cl] complex (Φlum = 0.62).48
The three complexes, at room temperature in dilute aerated dichloromethane solution, are very efficiently quenched by oxygen: the quantum yields of [PtL1Cl], [PtL2Cl] and [PtL3Cl] are 15, 17 and at least 89 times lower in air-equilibrated dichloromethane, respectively (Table 1); given the efficacy of oxygen quenching, efficient production of singlet oxygen – the 1Δg state of O2 – can be anticipated.
Upon excitation at around 422 nm, dilute dichloromethane solutions (1.0 × 10−6–2.5 × 10−6 M) of complexes [PtL1Cl], [PtL2Cl] and [PtL3Cl] show intense phosphorescent bands in the yellow-green region with a maximum wavelength at 562, 561 and 549 nm (Fig. 2), respectively.
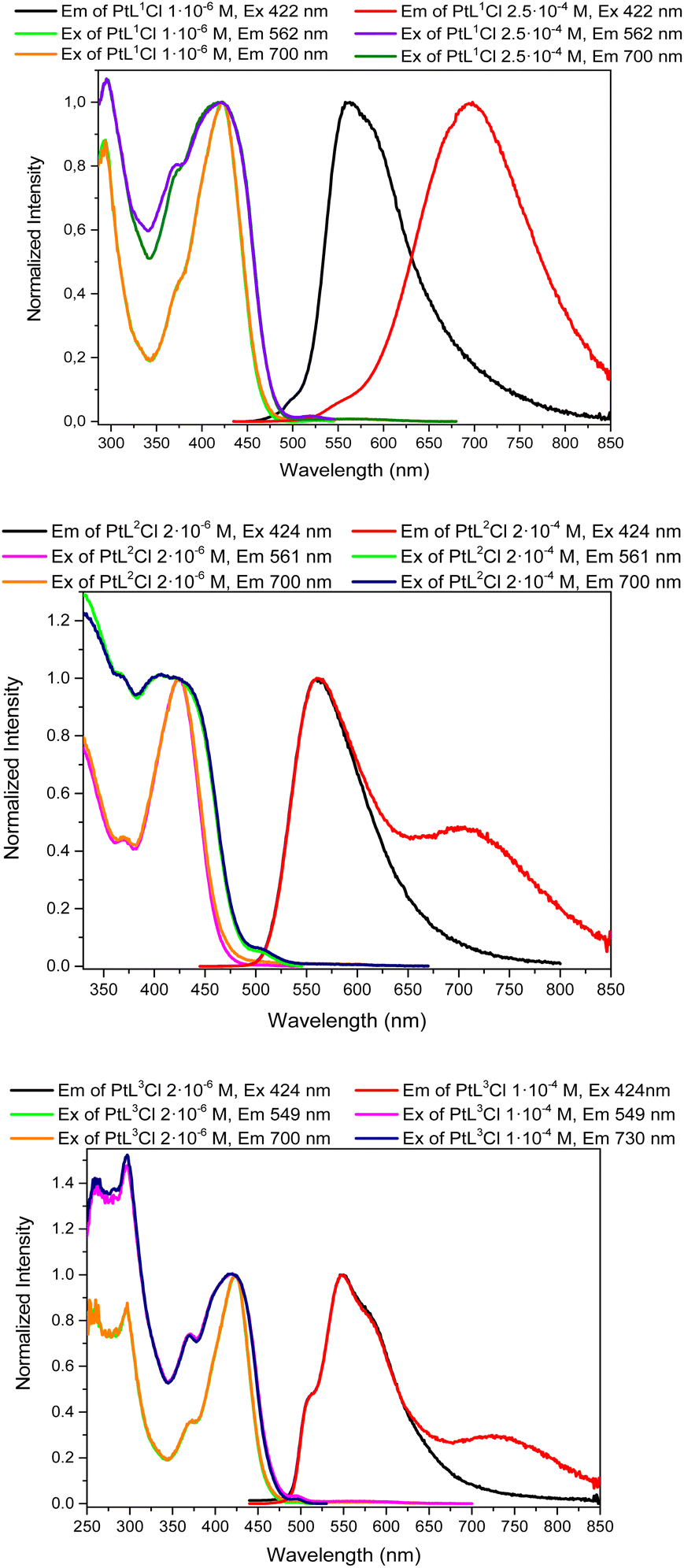 | ||
| Fig. 2 Normalized emission and excitation spectra of diluted and concentrated solutions of [PtL1Cl], [PtL2Cl] and [PtL3Cl] in CH2Cl2 at 298 K. | ||
Thus, the substitution with the triphenylamine at the 4 position of each pyridine causes a strong red shift in the emission of complexes [PtL1Cl] and [PtL3Cl] with respect to the related [Pt(1,3-bis(4-R-pyridin-2-yl)-4,6-difluoro-benzene)Cl] (R = H) (472 nm)50 and [Pt(1,3-bis(pyridin-2-yl)-5-R-benzene)Cl] (R = mesityl) (501 nm)48 complexes, the strongest shift (90 nm) being observed for [PtL1Cl].
When the concentration of the [PtL1Cl], [PtL2Cl] and [PtL3Cl] complexes is increased up to around 2.4 × 10−4 M, new broad structureless emission bands arise at 696, 704 and 727 nm, respectively. These bands at lower energy can be ascribed to the emission from bi-molecular emissive excited states (excimers and aggregates) of the platinum(II) complexes, as previously reported.52,57,59 Remarkably, the [PtL1Cl] complex displays intense deep red/NIR emission (Φlum = 0.66) in concentrated deaerated solution with essentially no “contamination” by visible light < 600 nm. To our knowledge this represents the highest quantum yield in the deep red/NIR region reported for a platinum(II) complex in solution.
It is interesting to point out that an increase of the concentration of the three complexes doesn't lead to a strong decrease of the luminescence quantum yield in deareated dichloromethane solution (see Tables S1, S2 and S3 in the ESI†), in contrast to the behavior of other N^C^N-platinum(II) complexes.52 Such a superior performance could be reasonably attributed to the presence of the sterically hindered triphenylamine substituents.
Lifetime measurements were performed on dilute deareated dichloromethane solutions at room temperature at the maximum emission wavelength of the [PtL1Cl], [PtL2Cl] and [PtL3Cl] complexes, exciting around 424 nm. Whereas [PtL2Cl] has a lifetime (6.6 μs) in the typical range reported for N^C^N-platinum(II) complexes, [PtL1Cl] and [PtL3Cl] are characterized by an unexpected very long lifetime. Thus [PtL1Cl] has a lifetime (103.9 μs) 16 times longer than that of the related [Pt(1,3-bis(pyridin-2-yl)-4,6-difluoro-benzene)Cl] complex (τ = 6.6 μs),50 while the lifetime of [PtL3Cl] (50.0 μs) is 7 times longer than that of [Pt(1,3-bis(pyridin-2-yl)-5-mesityl-benzene)Cl] (τ = 7.2 μs).48
The longer lifetimes of [PtL1Cl] and [PtL3Cl] with respect to [PtL2Cl] can be associated to the structural variation of the substituent at the central aryl ring; in particular, we note that increasing the electron deficiency of the substituent (Ph3N < mesityl < F2Ph) makes the lifetime of the complex longer. These large lifetimes are of particular relevance, being desirable for many applications such as bioimaging.1–8
Of the three new complexes analysed in solution at room temperature, [PtL1Cl] is particularly appealing for its interesting photophysical properties such as high luminescent quantum yield, long lifetime and an emission that strongly depends on its concentration. Therefore, we decided to characterize the photophysical properties of the [PtL1Cl] complex both in solution at low temperature (77 K, Fig. S15–S18†) and in the solid state as: blend in a polymethyl methacrylate (PMMA) matrix (0.5% w/w), neat thin film and powder; the preparation of the films can be found in the general information section of the ESI.†Table 2 reports the luminescence data for complex [PtL1Cl] at the solid state.
The normalized emission and excitation spectra at 77 K of the [PtL1Cl] complex at 2.4 × 10−4 M are shown in Fig. S15 (ESI†). The emission spectra of the [PtL1Cl] display a structured phosphorescent band with a maximum wavelength at 546 nm (τ = 79.1 μs) and a broad structureless emission band at 684 nm (τ = 1.4 μs) which increases up to 730 nm (τ = 1.1 μs) by varying the excitation wavelength from 442 to 560 nm. The structured band is attributed to the monomers of the complex and, by comparison to the room temperature measurements (see Fig. S13†), it increases in intensity with respect to the broad structureless emission band at 684 nm. This behavior of the monomer signal is generally related to presence of excimers (emission at 684 nm); in fact, at low temperature the monomers have a reduced mobility and therefore a lower probability of excimer formation. The emission band at 730 nm is attributed to aggregates of the complex, as confirmed from the different excitation spectra recorded at 546 nm and 730 nm. It is interesting to note that the excitation spectrum at 546 nm and 684 nm are not equal in shape because the emission band at 730 nm partially overlaps to the excimer emission band.
The normalized absorption spectra of the neat and blend in PMMA thin films are shown in Fig. S19 (ESI†). The absorption spectrum of the thin film in PMMA is quite similar to that observed in solution. This is probably due to the weak intermolecular interactions felt by the [PtL1Cl] molecules into the polymeric matrix. On the other hand, the neat film displays a broader absorption spectrum, due to the presence of excimers/aggregates.
The normalized emission and excitation spectra of the [PtL1Cl] complex in PMMA film are given in Fig. S20 (ESI†). Upon excitation at 421 nm, the blend of [PtL1Cl] in PMMA matrix shows intense phosphorescent bands with a maximum emission wavelength at 541 nm (τ = 35.0 μs), blue-shifted by ca. 21 nm with respect to the dilute dichloromethane solution. Moreover, a new band is observed at lower energy (ca. 670–740 nm) that increases by varying the excitation wavelength from 421 to 480 nm. This latter band can be ascribed mainly to the emission from aggregates of the [PtL1Cl] complex, because of the limited mobility of the molecules into PMMA matrix, which prevents a high excimer formation. Furthermore, in support of this assignment, we note as the excitation spectra recorded at 541 nm and 700 nm are different in shape.
The normalized emission and excitation spectra of [PtL1Cl] as neat film are given in Fig. S23 (ESI†). Regardless of the excitation wavelength, the neat film shows only one phosphorescent band with a maximum emission wavelength at 688 nm. The spectral position of this band is similar to that observed in solution at 2.5 × 10−4 M at room and low temperature (see Fig. 2 and S15), therefore, it can be ascribed to the emission from bi-molecular emissive excited states (excimers and aggregates) of the [PtL1Cl] complex. Moreover, lifetime measurements performed at 691 nm, exciting at 404 nm, show two values: τ1 = 94.71 ns and τ2 = 342.27 ns (see ESI†).
The normalized emission and excitation spectra of the powder of the [PtL1Cl] complex at room temperature and 77 K are shown in Fig. S25 and S29 (ESI†), respectively. The emission spectra at room temperature are characterized by a broad structureless band at 652 nm (τ = 0.74 μs) with shoulders at 535 (τ = 0.56 μs) and 580 nm (τ = 0.87 μs) that decrease in intensity by varying the excitation wavelength from 460 to 500 nm.
At 77 K, the emission spectra of the powder shown a more intense structured band at 552 nm (τ = 33.7 μs) and a structureless band at 667 nm (τ = 0.99 μs), which is red-shifted by ca. 15 nm with respect to the powder at room temperature, (see Fig. 3). The peaks at 552 and 580 nm can be attributed to the monomer excitonic states of the complex by comparison to the emission spectra of the [PtL1Cl] in PMMA film (see Fig. 3). Interestingly, the emission spectra have a very similar behavior to these observed for a concentrated solution (2.4 × 10−4 M) at 77 K (see Fig. S33, ESI†); therefore, it is reasonable to assume that peak at 667 nm is mainly composed by excimer emission. However, since the excitation spectra at 552 nm and 667 nm are different from each other, it is not possible to exclude the presence of aggregates.
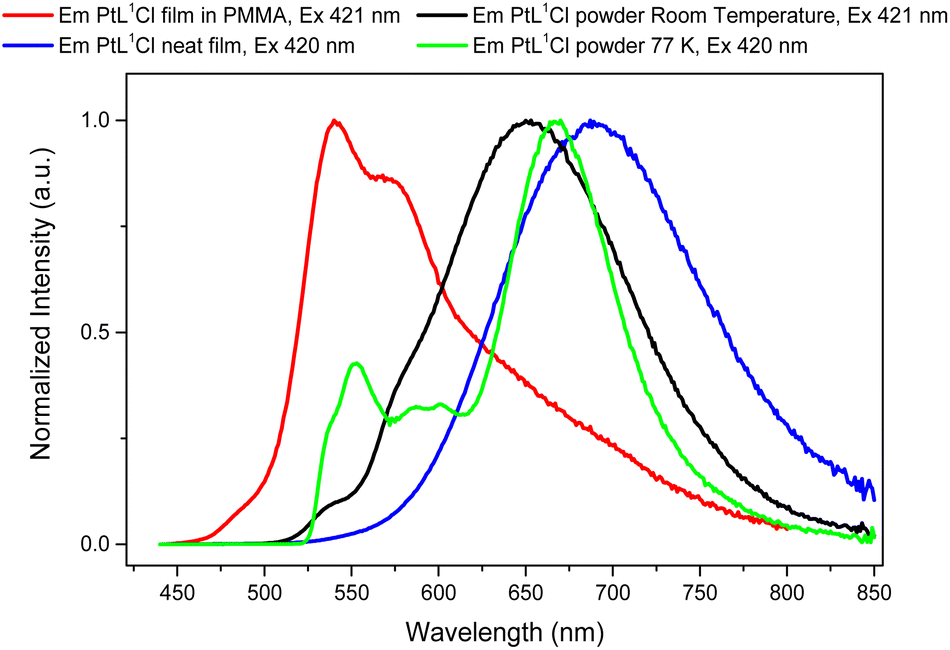 | ||
| Fig. 3 Normalized emission spectra of [PtL1Cl] complex in solid state. | ||
The luminescent quantum yield of the neat and PMMA thin film is 0.05 and 0.56, respectively, while is 0.11 for the powder. Clearly, although a significant quantum yield is maintained in the PMMA film, the higher presence of excimers and aggregates in the neat film and powder causes a strong quenching of the quantum yield.
3.3 Fabrication of OLEDs based on [PtL1Cl]
Highly luminescent platinum(II) chlorido complexes with a cyclometalated 1,3-di(2-pyridyl)benzene ligand have been used as emitters in multilayer OLEDs, affording high external quantum efficiencies.16 They showed their great potential for tailoring the emission colour of OLEDs. As a matter of fact, they are characterized by two phosphorescence bands, located in the bluish green (monomeric emission) and red (excimer/aggregate emission) regions of the visible spectrum, which contributions to the total emission can be controlled by the concentration of the platinum complex in the host matrix used for the fabrication of the blend emissive layer of the OLED. Thus, it was reported that [Pt(1,3-bis(pyridin-2-yl)-4,6-difluoro-benzene)Cl] allows the preparation of bluish green (5 wt% of complex in the emitting layer) and red (100 wt% of complex) OLEDs, the introduction of CH3 groups in para of the pyridine rings leading to better quantum efficiencies.38Substitution of the methyl groups with electron-donating dimethylamino substituents causes a blue-shift of the monomer emission, allowing the preparation of blue (3–5 wt% complex within the emitting layer), white (20 wt%) and amber (100 wt%) OLEDs.56 Similar results were obtained with less donor methoxy groups in para of the pyridines54 whereas the use of electron-withdrawing CF3 groups allows to prepare green-yellow OLEDs, at the concentration of 5 wt% in the emitting layer, and deep red OLEDS, when used as neat-film emitter.15
In the present work, OLEDs were fabricated using as emitting layer either a matrix of BCPO matrix hosting the [PtL1Cl] complex (8 wt%) or a film of pure [PtL1Cl]. Holes were injected from the indium tin oxide anode and passed through a 50 nm thick transporting layer made of 4,4′,4′′-tris(N-carbazolyl)triphenylamine TCTA.
Electrons were injected from an Al/LiF cathode and transported to the emitting layer (EML) by means of a layer of 2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole) (TPBi, 30 nm thick). Charges recombined in the 30 nm thick EML made of pure [PtL1Cl] or of a BCPO matrix hosting the [PtL1Cl] (8 wt%). EL spectra of the OLEDs are shown in Fig. 4. OLED emissions are in the yellow-green (CIE = 0.38, 0.56) and deep red (CIE = 0.65, 0.34) regions, for [PtL1Cl] 8 wt% and pure [PtL1Cl], respectively.
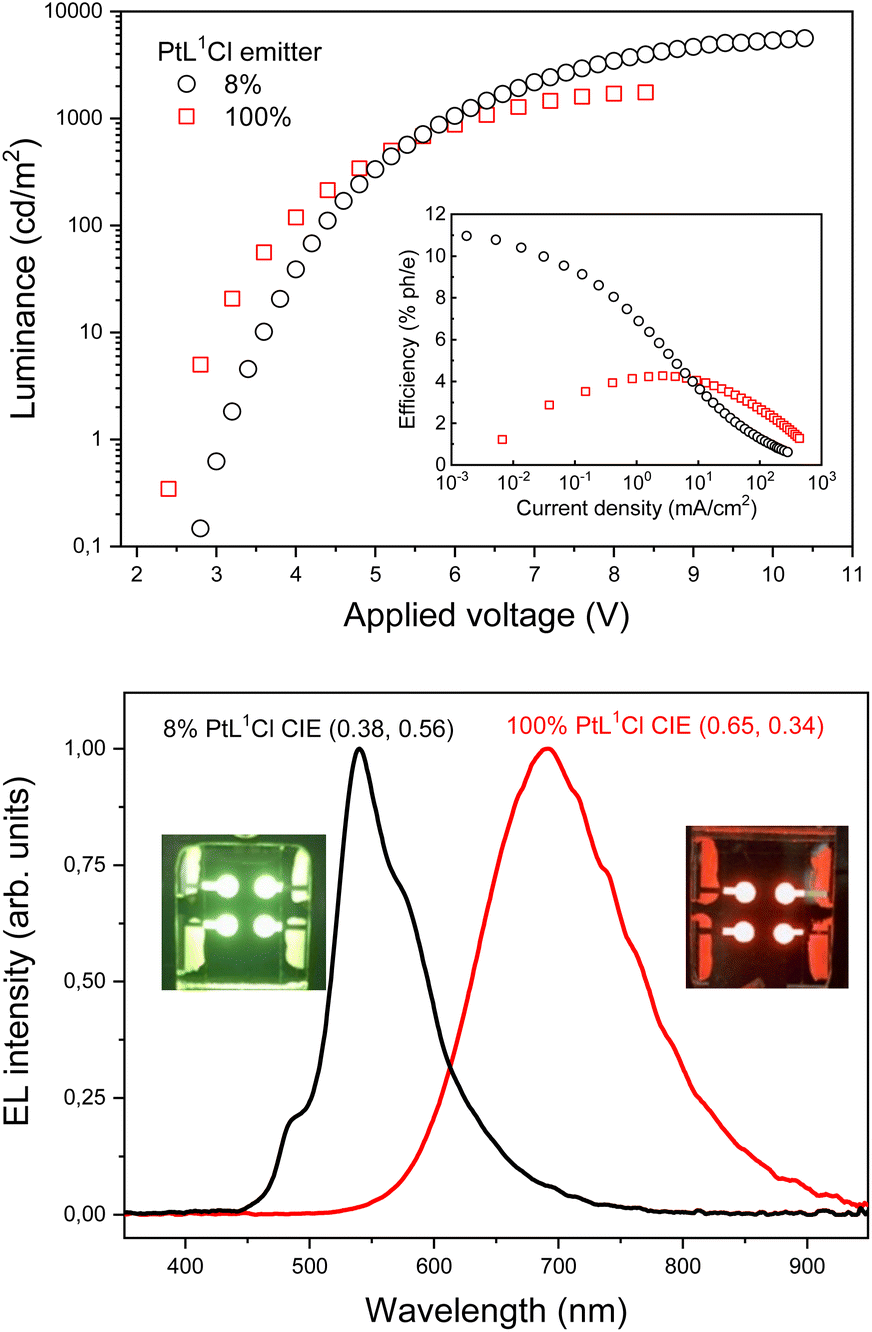 | ||
| Fig. 4 (a) Luminance vs. applied voltage and external EL efficiency vs. current density (inset) of the OLEDs. (b) Electroluminescence spectra at 7 V of OLEDs. In the inset there are photos of yellow-green and deep red OLEDs based on [PtL1Cl]. | ||
There is no substantial contribution to the EL emission bands from the electron-transporting (hole-blocking) or TCTA binder layers, in accordance with a good charge carrier confinement within the EML and complete energy transfer from the excited states of BCPO (formed by charge carrier recombination) to the Pt complex.
The luminance and external EL efficiency as function of current density and applied voltage of both OLEDs are shown in Fig. 4a. The EL efficiency shows the typical roll-off at high currents due to exciton–exciton and/or exciton/charge interaction and to high field induced exciton dissociation.60 Nevertheless, for [PtL1Cl] pure film as EML the efficiency remains in excess of 1.5% for all range of current density while EL efficiency drops off an order of magnitude when [PtL1Cl] has been employed as the emitting dopants (inset of Fig. 4a). Clearly, the shorter luminescence lifetime of aggregate [PtL1Cl] (high concentration) compared to [PtL1Cl] monomer (low concentration) ensures that there is a lower probability of the excited state being quenched. Although the maximum brightness of 1750 cd m−2 achieved for [PtL1Cl] pure film as EML is apparently less than the value of 5610 cd m−2 for [PtL1Cl] as dopant in BCPO (see Table 3), it should be noted that this is a photometric (as opposed to radiometric) measurement of brightness factors in the eye's sensitivity, which falls to zero beyond 700 nm. In fact, EL intensity of OLED based on [PtL1Cl] pure film reaches 10 mW cm−2, a good result compared to the best deep red/NIR OLEDs (see Table 3).61–65
| PtL1 Cl | Turn-on voltage (V) | Maximum elettroluminescence efficiencies @max @100 cd m−2 @1000 cd m−2 | Maximum elettroluminescence intensity | Color paramiters | |||||
|---|---|---|---|---|---|---|---|---|---|
| EQE (%ph/e) | Current efficiency (cd/A) | Power efficiency (lm/W) | B (cd m−2) | P (mW cm−2) | CIE (x;y) | Dominat wavelength (nm) purity (%) | CCT (°K) | ||
| 8% | 3.0 | 11.0 | 35.5 | 37.3 | 5610 | 3.8 | (0.38;0.56) | 563 | 5860 |
| 8.1 | 26.0 | 18.6 | 84 | ||||||
| 4.0 | 12.9 | 6.8 | |||||||
| 100% | 2.6 | 4.3 | 1.3 | 1.4 | 1750 | 10.0 | (0.65;0.34) | 607 | 1000 |
| 4.1 | 1.3 | 1.0 | 99 | ||||||
| 2.5 | 0.8 | 0.4 | |||||||
4. Conclusion
In conclusion, a new brightly luminescent family of 1,3-di-(2-pyridyl)benzene platinum(II) complexes bearing a triphenylamine substituent on the pyridyl rings has been discovered. It was shown how the color of the phosphorecence can be tuned by the nature of the substituents on the benzene ring, keeping quantum yields near unity thanks to the presence of the triphenylamine substituents on the pyridines. The novel [Pt(1,3-bis(4-triphenylamine-pyridin-2-yl)-4,6-difluoro-benzene)Cl] complex is particularly fascinating: in diluted deaerated solution it displays intense yellow-green emission (Φlum = 0.90) with an unexpected very long lifetime (103.9 μs) whereas in concentrated deaerated solution it displays intense deep red/NIR emission (Φlum = 0.66) with essentially no “contamination” by visible light < 600 nm. To our knowledge this represents the highest quantum yield in the deep red/NIR region reported for a platinum(II) complex in solution. Thus, our new complex, which allowed the preparation of efficient yellow-green and deep red/NIR OLEDs, represents surely a useful tool for bio-imaging.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We deeply thank Prof. Dominique Roberto for fruitful discussions. The National Interuniversity Consortium of Materials Science and Technology (PROGETTO INSTM21MIROBERTO), the Università degli Studi di Milano (PSR2020_DIP_005_PI_ACOLO), CNR are acknowledged for financial support in Italy.Notes and references
- V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186–202 RSC.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef CAS.
- E. Baggaley, S. W. Botchway, J. W. Haycock, H. Morris, I. V. Sazanovich, J. A. G. Williams and J. A. Weinstein, Chem. Sci., 2014, 5, 879–886 RSC.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Struct. Bonding, 2015, 165, 205–256 CrossRef CAS.
- A. Colombo, F. Fiorini, D. Septiadi, C. Dragonetti, F. Nisic, A. Valore, D. Roberto, M. Mauro and L. De Cola, Dalton Trans., 2015, 44, 8478–8487 RSC.
- V. W.-W. Yam and A. S.-Y. Law, Coord. Chem. Rev., 2020, 414, 213298 CrossRef CAS.
- A. S.-Y. Law, L. C.-C. Lee, K. K.-W. Lo and V. W.-W. Yam, J. Am. Chem. Soc., 2021, 143, 5396–5405 CrossRef CAS PubMed.
- W. Sotoyama, T. Satoh, N. Sawatari and H. Inoue, Appl. Phys. Lett., 2005, 86, 153505–153507 CrossRef.
- M. Cocchi, D. Virgili, V. Fattori, D. L. Rochester and J. A. G. Williams, Adv. Funct. Mater., 2007, 17, 285–289 CrossRef CAS.
- X. Yang, Z. Wang, S. Madakuni, J. Li and G. E. Jabbour, Adv. Mater., 2008, 20, 2405–2409 CrossRef CAS.
- W. Y. Wong and C. L. Ho, Chem. Soc. Rev., 2009, 253, 1709–1758 CAS.
- C. M. Che, C. C. Kwok, S. W. Lai, A. F. Rausch, W. J. Finkenzeller, N. Y. Zhu and H. Yersin, Chem. – Eur. J., 2010, 16, 233–247 CrossRef CAS PubMed.
- J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401–2425 CrossRef CAS.
- E. Rossi, L. Murphy, P. L. Brothwood, A. Colombo, C. Dragonetti, D. Roberto, R. Ugo, M. Cocchi and J. A. G. Williams, J. Mater. Chem., 2011, 21, 15501–15510 RSC.
- L. F. Gildea and J. A. G. Williams, Iridium and platinum complexes for OLEDs, in Organic Light-Emitting Diodes: Materials, Devices and Applications, ed. A. Buckley, Woodhead, Cambridge, 2013 Search PubMed.
- F. Nisic, A. Colombo, C. Dragonetti, D. Roberto, A. Valore, J. M. Malicka, M. Cocchi, G. R. Freeman and J. A. G. Williams, J. Mater. Chem. C, 2014, 2, 1791–1800 RSC.
- C. Cebrian and M. Mauro, Beilstein J. Org. Chem., 2018, 14, 1459–1481 CrossRef CAS PubMed.
- X. Yang, H. Guo, X. Xu, Y. Sun, G. Zhou, W. Ma and Z. Wu, Adv. Sci., 2019, 6, 1801930 CrossRef PubMed.
- W.-C. Chen, C. Sukpattanacharoen, W.-H. Chan, C.-C. Huang, H.-F. Hsu, D. Shen, W.-Y. Hung, N. Kungwan, D. Escudero, C.-S. Lee and Y. Chi, Adv. Funct. Mater., 2020, 30, 2002494 CrossRef CAS.
- A. Haque, H. El Moll, K. M. Alenezi, M. S. Khan and W. Y. Wong, Materials, 2021, 14, 4236–4261 CrossRef CAS PubMed.
- A. S.-Y. Law, M. C.-L. Yeung and V. W.-W. Yam, ACS Appl. Mater. Interfaces, 2017, 9, 41143–41150 CrossRef CAS PubMed.
- Y. Y. Ning, G. Q. Jin, M. X. Wang, S. Gao and J. L. Zhang, Curr. Opin. Chem. Biol., 2022, 66, 102097–102107 CrossRef CAS PubMed.
- E. Rossi, A. Colombo, C. Dragonetti, S. Righetto, D. Roberto, R. Ugo, A. Valore, J. A. G. Williams, M. G. Lobello, F. De Angelis, S. Fantacci, I. Ledoux-Rak, A. Singh and J. Zyss, Chem. – Eur. J., 2013, 19, 9875–9883 CrossRef CAS PubMed.
- A. Colombo, C. Dragonetti, D. Marinotto, S. Righetto, D. Roberto, S. Tavazzi, M. Escadeillas, V. Guerchais, H. Le Bozec, A. Boucekkine and C. Latouche, Organometallics, 2013, 32, 3890–3894 CrossRef CAS.
- H. Zhao, E. Garoni, T. Roisnel, A. Colombo, C. Dragonetti, D. Marinotto, S. Righetto, D. Roberto, D. Jacquemin, J. Boixel and V. Guerchais, Inorg. Chem., 2018, 57(12), 7051–7063 CrossRef CAS PubMed.
- S. Attar, F. Artizzu, L. Marchik, D. Espa, L. Pilia, M. F. Casula, A. Serpe, M. Pizzotti, A. Orbelli-Biroli and P. Deplano, Chem. – Eur. J., 2018, 24, 10503–10512 CrossRef CAS PubMed.
- A. Colombo, C. Dragonetti, V. Guerchais and D. Roberto, Coord. Chem. Rev., 2021, 446, 214113–213133 CrossRef CAS.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- P. T. Chou, Y. Chi, M. W. Chung and C. C. Lin, Coord. Chem. Rev., 2011, 255, 2653–2665 CrossRef CAS.
- E. A. Wood, L. F. Gildea, D. S. Yufit and J. A. G. Williams, Polyhedron, 2021, 207, 115401–115409 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forest, Nature, 1998, 395, 151 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, M. E. Thompson and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 14422 CrossRef CAS.
- H. Yersin, Top. Curr. Chem., 2004, 241, 1–26 CrossRef CAS.
- M. Koden, OLED Displays and Lighting, John Wiley & Sons, NJ, 2016 Search PubMed.
- D. Ma, in Handbook of Advanced Lighting Technology, ed: R. Karlicek, C.-C. Sun, G. Zissis and R. Ma, Springer International Publishing, Cham, 2016, pp. 1–34 Search PubMed.
- V. Adamovich, J. Brooks, A. Tamayo, A. M. Alexander, P. I. Djurovich, B. W. D'Andrade, C. Adachi, S. R. Forrest and M. E. Thompson, New J. Chem., 2002, 26, 1171–1178 RSC.
- M. Cocchi, J. Kalinowski, V. Fattori, J. A. G. Williams and L. Murphy, Appl. Phys. Lett., 2009, 94, 073309–073311 CrossRef.
- W. Mroz, C. Botta, U. Giovanella, E. Rossi, A. Colombo, C. Dragonetti, D. Roberto, R. Ugo, A. Valore and J. A. G. Williams, J. Mater. Chem., 2011, 21, 8653–8661 RSC.
- E. Rossi, A. Colombo, C. Dragonetti, D. Roberto, F. Demartin, M. Cocchi, P. Brulatti, V. Fattori and J. A. G. Williams, Chem. Commun., 2012, 48, 3182–3184 RSC.
- P. Brulatti, V. Fattori, S. Muzzioli, S. Stagni, P. P. Mazzeo, D. Braga, L. Maini, S. Milita and M. Cocchi, J. Mater. Chem. C, 2013, 1, 1823–1831 RSC.
- Y. C. Wei, S. F. Wang, Y. Hu, L. S. Liao, D. G. Chen, K. H. Chang, C. W. Wang, S. H. Liu, W. H. Chan, J. L. Liao, W. Y. Hung, T. H. Wang, P. T. Chen, H. Hsu, F. Y. Chi and P. T. Chou, Nat. Photonics, 2020, 14, 570–577 CrossRef CAS.
- P. P. Ghoroghchian, P. R. Frail, K. Susumu, D. Blessington, A. K. Brannan, F. S. Bates, B. Chance, D. A. Hammer and M. J. Therien, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2922–2927 CrossRef CAS PubMed.
- L. Yuan, W. Lin, S. Zhao, W. Gao, B. Chen, L. He and S. Zhu, J. Am. Chem. Soc., 2012, 134, 13510–13523 CrossRef CAS PubMed.
- G. Qian and Z. Y. Wang, Chem. – Asian J., 2010, 5, 1006–1029 CrossRef CAS PubMed.
- J. V. Caspar and T. J. Meyer, J. Phys. Chem., 1983, 87, 952–957 CrossRef CAS.
- J. A. G. Williams, A. Beeby, S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609–8611 CrossRef CAS PubMed.
- S. J. Farley, D. L. Rochester, A. L. Thompson, J. A. K. Howard and J. A. G. Williams, Inorg. Chem., 2005, 44, 9690–9703 CrossRef CAS PubMed.
- J. A. G. Williams, Chem. Soc. Rev., 2009, 38, 1783–1801 RSC.
- A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2012, 51, 312–319 CrossRef CAS PubMed.
- A. Rodrigue-Witchel, D. L. Rochester, S. B. Zhao, K. B. Lavelle, J. A. G. Williams, S. Wang, W. B. Connick and C. Reber, Polyhedron, 2016, 108, 151–155 CrossRef CAS.
- C. Dragonetti, F. Fagnani, D. Marinotto, A. Di Biase, D. Roberto, M. Cocchi, S. Fantacci and A. Colombo, J. Mater. Chem. C, 2020, 8, 7873–7881 RSC.
- D. L. Rochester, S. Develay, S. Zális and J. A. G. Williams, Dalton Trans., 2009, 1728–1741 RSC.
- M. Cocchi, J. Kalinowski, L. Murphy, J. A. G. Williams and V. Fattori, Org. Electron., 2010, 11, 388–396 CrossRef CAS.
- W. Sotoyama, T. Satoh, H. Sato, A. Matsuura and N. Sawatari, J. Phys. Chem. A, 2005, 109, 9760–9766 CrossRef CAS PubMed.
- L. Murphy, P. Brulatti, V. Fattori, M. Cocchi and J. A. G. Williams, Chem. Commun., 2012, 48, 5817–5819 RSC.
- F. Fagnani, A. Colombo, C. Dragonetti, D. Roberto and D. Marinotto, Inorg. Chim. Acta, 2022, 532, 120744 CrossRef CAS.
- C. Hierlinger, D. B. Cordes, A. M. Z. Slawin, A. Colombo, C. Dragonetti, S. Righetto, D. Roberto, D. Jacquemin, E. Zysman-Colman and V. Guerchais, Dalton Trans., 2018, 47, 8292 RSC.
- J. Kalinowski, M. Cocchi, L. Murphy, J. A. G. Williams and V. Fattori, Chem. Phys., 2010, 378, 47–57 CrossRef CAS.
- J. Kalinowski, W. Stampor, J. Szmytkowski, D. Virgili, M. Cocchi, V. Fattori and C. Sabatini, Phys. Rev. B, 2006, 74, 85316 CrossRef.
- M. Cocchi, D. Virgili, V. Fattori, J. A. G. Williams and J. Kalinowski, Appl. Phys. Lett., 2007, 90, 023506 CrossRef.
- U. Balijapalli, R. Nagata, N. Yamada, H. Nakanotani, M. Tanaka, A. D'Aléo, V. Placide, M. Mamada, Y. Tsuchiya and C. Adachi, Angew. Chem., Int. Ed., 2021, 60, 8477–8482 CrossRef CAS PubMed.
- Y. Zhang, J. Miao, J. Xiong, K. Li and C. Yang, Angew. Chem., 2022, 61, e202113718 CAS.
- L. Cao, J. Li, Z.-Q. Zhu, L. Huang and J. Li, ACS Appl. Mater. Interfaces, 2021, 13, 60261–60268 CrossRef CAS PubMed.
- M. Z. Shafikov, P. Pander, A. V. Zaytsev, R. Daniels, R. Martinscroft, F. B. Dias, J. A. G. Williams and V. N. Kozhevnikov, J. Mater. Chem. C, 2021, 9, 127–135 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of the complexes, films preparation, and photoluminescence investigations. See DOI: https://doi.org/10.1039/d2dt01792j |
| This journal is © The Royal Society of Chemistry 2022 |