
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTitanium complexes with unsymmetrically substituted imidazolin-2-iminato ligands†
Marvin
Koneczny
a,
Arife Büsra
Erol
a,
Marc
Mauduit
 b,
Moris S.
Eisen
c and
Matthias
Tamm
*a
b,
Moris S.
Eisen
c and
Matthias
Tamm
*a
aInstitut für Anorganische und Analytische Chemie, Technische Universität Braunschweig, Hagenring 30, 38106 Brauschweig, Germany. E-mail: m.tamm@tu-bs.de
bUniv Rennes, Ecole Nationale Supérieure de Chimie de Rennes, CNRS, ISCR-UMR 6226, F-35000 Rennes, France
cSchulich Faculty of Chemistry, Technion – Israel Institute of Technology, Technion City, 32000 Haifa, Israel
First published on 8th July 2022
Abstract
The reaction of the unsymmetrical N-heterocyclic carbenes 1-(2,4,6-trimethylphenyl)-3-(adamantyl)imidazolin-2-ylidene (IAdMes, 1a) and 1-(2,6-diisopropylphenyl)-3-(adamantyl)imidazolin-2-ylidene (IAdDipp, 1b) with trimethylsilyl azide furnished the 2-(trimethylsilylimino)imidazolines 2a (ImAdMesNSiMe3) and 2b (ImAdDippNSiMe3). Desilylation by stirring in methanol gave the corresponding imidazolin-2-imines 3a (ImAdMesNH) and 3b (ImAdDippNH). 2a and 2b were treated with [TiCl4(THF)2] (THF = tetrahydrofuran) and [CpTiCl3] (Cp = η5-C5H5) to form the mono- and bis(imidazolin-2-iminato) titanium(IV) complexes [(ImAdRN)TiCl3] (4, R = Mes, Dipp), [Cp(ImAdRN)TiCl2] (5, R = Mes, Dipp), and [(ImAdRN)2TiCl2] (6, R = Mes, Dipp). The crystal structures of all compounds except 2b were determined by X-ray diffraction analysis.
Introduction
Imidazolin-2-imines (ImRNH) and their deprotonated, anionic counterparts, imidazolin-2-imides (ImRN−), have become important additions to the class of nitrogen donor ligands after their introduction by Kuhn and co-workers.1,2 The widespread use of these systems was largely triggered by Tamm et al. through the development of a versatile synthetic protocol based on the reaction of N-heterocyclic carbenes (NHCs) of the imidazolin-2-ylidene-type with trimethylsilyl azide followed by desilylation.3–5 The intermediate silylated imines ImRNSiMe3 may serve as suitable precursors for the preparation of imidazolin-2-iminato transition metal complexes, which feature short metal–nitrogen bonds owing to the ability of these ligands to act as strong 2σ-/4π-electron donors, preferably to early transition metals and/or metals in higher oxidation states.6 Accordingly, numerous homogeneous d0-metal (pre-)catalysts with ancillary imidazolin-2-iminato ligands have been developed, with titanium(IV) complexes of type I and their use in olefin polymerisation representing the most prominent application (Fig. 1).4,7–9 Related titanium complexes with additional aryloxo,10,11 amido,11,12 and guanidinate ligands13 were also reported, while imidazolidin-2-iminato-titanium complexes represent a closely related class of olefin polymerisation and oligomerisation catalysts.14 In contrast, zirconium and hafnium imidazolin-2-iminato complexes have so far found significantly less application in homogeneous catalysis.15 Furthermore, imidazolin-2-iminato-supported vanadium(V) imido complexes of type II were employed in ethylene (co-)polymerisation and ring-opening metathesis polymerisation,16 whereas molybdenum(VI) and tungsten(VI) alkylidyne complexes of type III served as highly active alkyne metathesis catalysts.17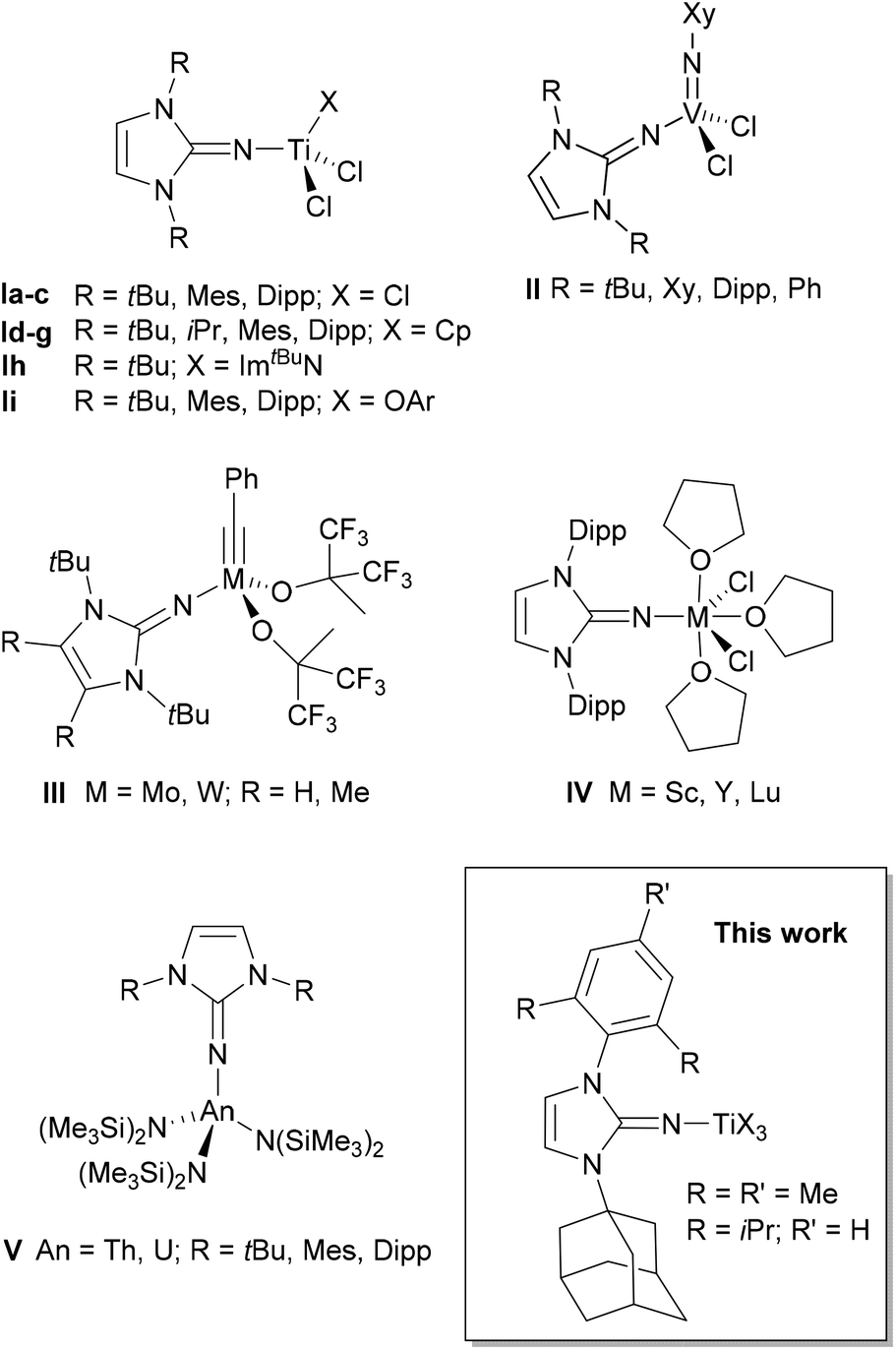 | ||
| Fig. 1 Selected examples for imidazolin-2-iminato complexes. Mes = 2,4,6-trimethylphenyl, Dipp = 2,6-diisopropylphenyl, Xy = 2,6-dimethylphenyl. | ||
Very short metal–nitrogen bonds were also found in rare-earth metal imidazolin-2-iminato complexes such as IV,18–20 which were employed as hydroamination and hydrosilylation catalysts.21 Recently, the dysprosium(III) complex [(ImDippN)DyCl2(THF)3] was presented as a suitable single-molecule magnet.22 Moreover, numerous actinide complexes such as the thorium(IV) and uranium(IV) complexes V were prepared,23 with these and related complexes serving as highly active catalysts for ring-opening polymerisation, hydroelementation, and aldehyde disproportionation (Tishchenko) reactions.24 Uranium(III) complexes were also reported and used for the generation of a photochemically generated terminal uranium nitride.25
All of the compounds shown in Fig. 1, like most of the known imidazolin-2-iminato complexes, have a symmetrical N,N-substitution pattern, whereas unsymmetrical systems are rare.26 Recently, however, Eisen and co-workers presented unsymmetrical benzimidazolin-2-iminato ligands and their application primarily in the development of organoactinide catalysts.27 These systems contain a combination of bulky N-aryl and small N-alkyl substituents to create more accessible, sterically less encumbered catalysts. In search for unsymmetrical imidazolin-2-iminato ligands with two different, yet bulky N-substituents, we identified the carbenes 1-(2,4,6-trimethylphenyl)-3-(adamantyl)imidazolin-2-ylidene (IAdMes, 1a) and 1-(2,6-diisopropylphenyl)-3-(adamantyl)imidazolin-2-ylidene (IAdDipp, 1b), introduced by Mauduit, Baslé and co-workers,28,29 as suitable starting materials for the synthesis of the corresponding imidazolin-2-imines. These NHC ligands have been used, for example, for the preparation of highly Z-selective olefin metathesis ruthenium catalysts,30 and in principle, the corresponding imidazolin-2-iminato ligands might also pave the way to their application in asymmetric homogeneous catalysis. Accordingly, we wish to present herein the isolation and structural characterisation of the free carbenes IAdMes (1a) and IAdDipp (1b) as well as their use as starting materials for the preparation of unsymmetrical imidazolin-2-imines ImAdMesNR (2a, R = SiMe3; 3a, R = H) and ImAdDippNR (2b, R = SiMe3; 3b, R = H). In addition, the silylated imines were used for the synthesis of a series of mono- and bis(imidazolin-2-iminato) titanium(IV) complexes to allow comparison with their well-established symmetrical congeners of type I (Fig. 1).
Results and discussion
Preparation and characterisation of imidazolin-2-imine ligands
Based on the unsymmetrical N-substituted imidazolium salts established by Mauduit and Baslé,28,29 the corresponding free carbenes 1a and 1b can be generated by deprotonation with KOtBu in THF (Scheme 1). Since the free NHCs were previously always generated in situ, full analytical data will be provided herein. The carbenes 1a and 1b show typical sets of signals for the protons in the 4,5-position of the heterocycle in the 1H NMR spectra as doublets with chemical shifts of 6.88/6.50 ppm (1a) and 6.84/6.63 ppm (1b). Significantly deshielded 13C NMR resonances at 216.1 ppm (1a) and 217.0 ppm (1b) clearly indicate the formation of the free carbenes. The 2-(trimethylsilylimino)imidazolines 2a and 2b were prepared by treatment of the corresponding imidazolin-2-ylidenes 1a and 1b with trimethylsilyl azide in boiling toluene following the established procedure.3–52a and 2b were isolated as pale yellow solids after bulb-to-bulb distillation of the crude brownish viscous oils and subsequent washing with or crystallisation from n-hexane. The product formation can be followed by 1H NMR spectroscopy, as there is a clear upfield shift of the resonances of the hydrogen atoms in the 4,5-position (backbone) to 6.16/5.66 ppm (2a) and 6.19/5.89 ppm (2b), consistent with the observations for the symmetrically substituted analogues.5 In addition, the relatively narrow multiplets observed for the distal CH2 groups in the carbene starting materials broaden and evolve into doublets of doublets in agreement with the presence of diastereotopic hydrogen atoms. The resonances of the trimethylsilyl protons appear at 0.09 ppm (2a) and 0.07 ppm (2b). In the 13C NMR spectra, the resonances for the NCN atoms are shifted to higher field by ca. −75 ppm compared to the free carbenes and are found at 141.2 ppm (2a) and 141.0 ppm (2b). Desilylation of 2a and 2b was accomplished by stirring their methanol solutions for short periods of time (30–120 min). The 1H NMR spectra exhibit broad signals for the NH hydrogen atoms at 4.46 ppm (3a) and 4.38 ppm (3b), while the resonances of the carbene moieties remain rather unaffected in comparison with 2a and 2b. In contrast, a significant downfield shift of ca. 10 ppm can be found for the NCN resonances in the 13C NMR spectra, resulting in chemical shifts of 153.1 ppm (3a) and 154.4 ppm (3b).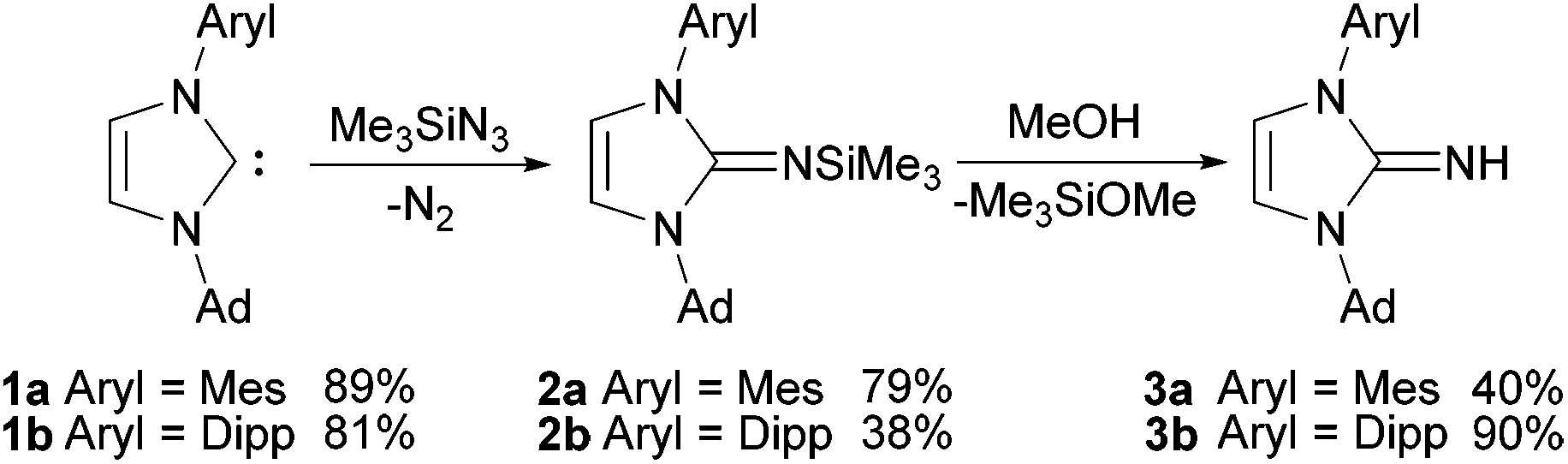 | ||
| Scheme 1 Preparation of the unsymmetrical N-substituted imidazolin-2-imines. Mes = 2,4,6-trimethylphenyl, Dipp = 2,6-diisopropylphenyl. | ||
To compare the new ligands with the previously established ones,3,5 the molecular structures of 1a, 1b, 2a, 3a, and 3b were established by X-ray diffraction analysis; single crystals of 2b were not obtained due to its high solubility in n-hexane. Pertinent structural parameters are assembled in Table 1, and presentations of the structures are depicted in Fig. 2. 1a and 1b exhibit N–C–N angles of 102.05(3)° and 101.48(11)°, which are as expected significantly smaller than those reported for the imidazolium salt ([IAdMesH]Cl = 108.56(12)°)28 and clearly indicate the formation of free carbenes. The C1–N1 bond length of 1.2782(10) Å in 2a agrees well with the values reported for the corresponding imines ImMesNSiMe3 (1.267(2) Å)5 and ImtBuNSiMe3 (1.275(3) Å)3 and falls in the range expected for C–N double bonds.31 The trimethylsilyl (TMS) group in 2a points towards the mesityl substituent, and accordingly, the C1–N1–Si1 angle of 142.68(7)° is similar to 147.2(1)° in ImMesNSiMe3, whereas significantly larger angles of 155.4(1)° and 169.3(2)° were observed for ImDippNSiMe3 and ImtBuNSiMe3, respectively.3,5 The position of hydrogen atoms in 3a and 3b could be freely refined, both facing the aryl group. The C1–N1 bond lengths are 1.2966(14) Å (3a) and 1.2891(13) Å (3b), which falls in the range reported for other imidazolin-2-imines,5e.g., 1.284(2) Å in ImMesNH32 and 1.289(2) Å in ImDippNH.20
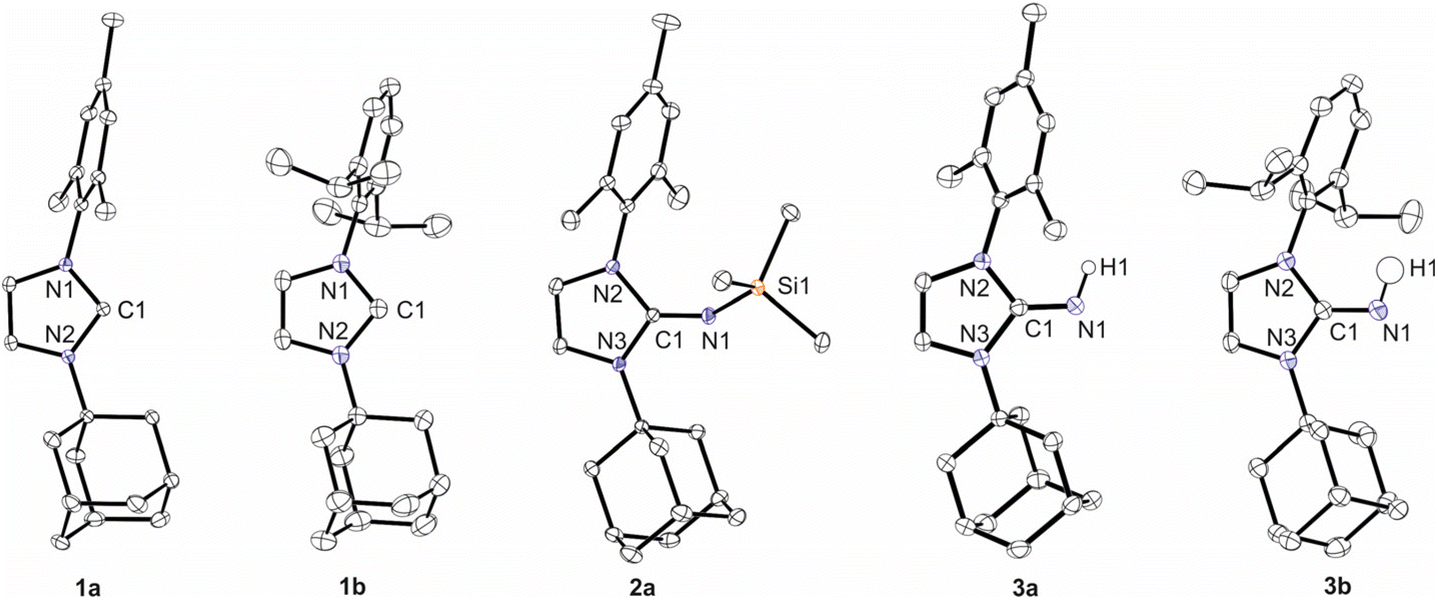 | ||
| Fig. 2 Molecular structures of 1a, 1b, 2a, 3a and 3b with thermal displacement parameters drawn at 50% probability. Hydrogen atoms, except H1, are omitted for clarity. For further information on the X-ray diffraction analysis please refer to the ESI.† | ||
| C1–N1 | N1–Si1/H1 | N2–C1–N3 | C1–N1–Si1 | |
|---|---|---|---|---|
| 1a | 1.3739(5) | 102.05(3) | ||
| 1.3651(5) | ||||
| 1b | 1.3709(18) | 101.48(11) | ||
| 1.3627(18) | ||||
| 2a | 1.2782(10) | 1.6823(7) | 103.99(6) | 142.68(7) |
| 3a | 1.2966(14) | 0.902(16) | 104.55(9) | |
| 3b | 1.2891(13) | 0.926(14) | 104.78(8) |
Preparation and characterisation of titanium imidazolin-2-iminato complexes
To introduce the new unsymmetrically N-substituted imidazolin-2-imines to transition metal chemistry, a series of titanium complexes was prepared, following the approach established since the earliest reports in the field.2–4 Thus, the silylated imines 2a and 2b were treated with titanium chloride precursors in toluene solution to afford the corresponding titanium complexes 4–6, with Me3SiCl formation as the driving force of the reaction (Scheme 2). Treatment with [TiCl4(THF)2] or [CpTiCl3] (Cp = η5-cyclopentadienyl) at room temperature gave the mono(imidazolin-2-iminato) complexes 4a/4b or 5a/5b as orange-red crystalline solids after stirring for 16 h, followed by evaporation and washing with n-hexane. The preparation of the bis(imidazolin-2-iminato) complexes 6a/6b required more forcing conditions, and the reaction of [TiCl4(THF)2] with two equivalents of 2a/2b in toluene at 100 °C afforded 6a/6b as orange crystalline solids after similar work-up. The significantly harsher reaction conditions required for the introduction of the second ligand point to the possibility of sequential introduction of two different imidazolin-2-iminato ligands in the future. The 1H and 13C NMR spectra of the complexes 4–6 are very similar, with little influence of metal coordination on the NHC signals. For the Cp complexes 5a/5b, the signals at 6.05/5.90 ppm and 114.8/115.3 ppm can be assigned to the C5H5 hydrogen and carbon atoms, respectively. It should be noted that the room-temperature NMR spectra of the bis(imidazolin-2-iminato) complexes 6a and 6b reveal dynamic behaviour in solution, since only one set of signals for the NHC fragment is observed in each case. Therefore, a variable-temperature 1H NMR study was conducted for 6b, which led to a splitting of the isopropyl signals below a coalescence temperature of about −40 °C (see ESI†). This evolves the original two doublets and one septet (or quartet–quartet) into four doublets and two septets, which can be attributed to the presence of diastereotopic isopropyl groups in either C2- or Cs-symmetric conformers (vide infra).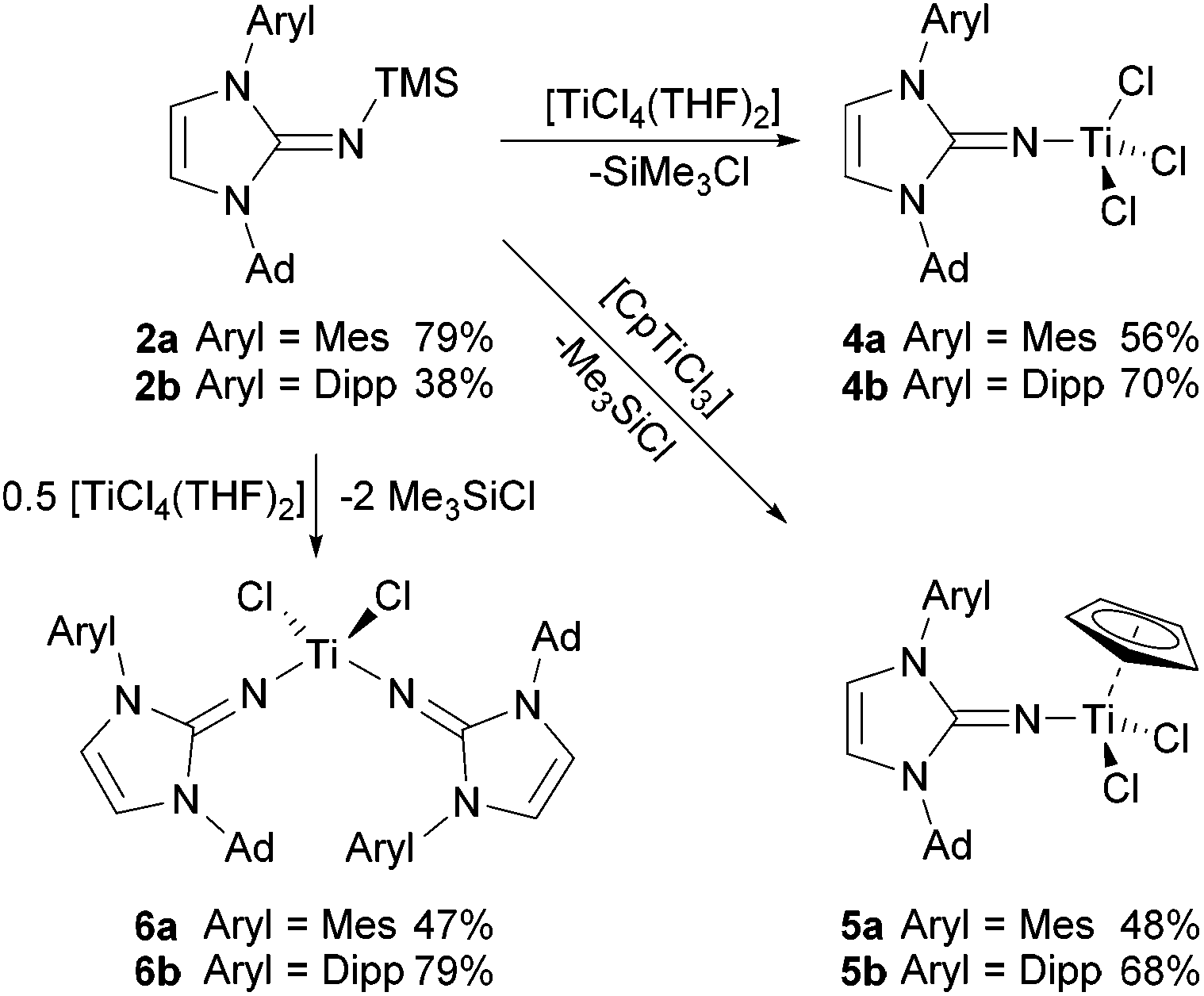 | ||
| Scheme 2 Preparation of titanium complexes bearing ImAdArylN ligands. Ad = adamantly, Mes = 2,4,6-trimethylphenyl, Dipp = 2,6-diisopropylphenyl. | ||
The new complexes 4–6 were additionally characterised by X-ray diffraction analysis, and the resulting molecular structures are presented in Fig. 3. Pertinent structural data are assembled in Table 2. The titanium atoms in the TiCl3 complexes 4a/4b exhibit only slightly distorted tetrahedral geometries and short Ti–N distances of 1.7347(5) Å (4a) and 1.7316(17) Å (4b), which are in good agreement with the values reported for [(ImMesN)TiCl3] and [(ImDippN)TiCl3] and indicate strong π-donation towards the metal atom.9 The Ti1–N1–C1 angles are 162.58(5)° in 4a and 173.04(16)° in 4b, with the former deviating significantly from linearity, presumably due to different steric demands of the mesityl and adamantyl substituents. For the cyclopentadienyl complexes 5, slightly longer Ti1–N1 bond lengths of 1.7758(16) Å (5a) and 1.7760(5) Å (5b) together with large Ti1–N1–C1 angles of 170.26(14)° (5a) and 170.86(5)° (5b) are found, which agrees well with the structural characteristics established for related systems, e.g., 1.765(3) Å and 170.7(2)° in [Cp(ImtBuN)TiCl2]3 as well as 1.778(2) Å and 175.8(1)° in [Cp(ImDippN)TiCl2].4 The bis(imidazolin-2-iminato) complexes 6a and 6b exhibit the longest Ti–N distances of complexes 4–6, namely 1.7951(15)/1.7867(16) Å (6a) and 1.7994(18)/1.8067(18) Å (6b), which is slightly longer compared to 1.788(2)/1.790(2) Å in [(ImtBuN)2TiCl2].8 The Ti–N–C angles of 166.51(13)°/166.94(13)° (6a) and 167.14(17)°/167.64(17)° (6b) indicate slightly stronger bending, which can be ascribed to the asymmetric N,N-substitution pattern of the imidazolin-2-iminato ligands. Above all, the latter adopt C2-symmetric orientations in the solid state with the aryl and adamantyl substituents of two the ligands facing in opposite directions. Accordingly, it is reasonable to assume that these chiral conformations also exist in solution, albeit only at low temperatures (vide supra).
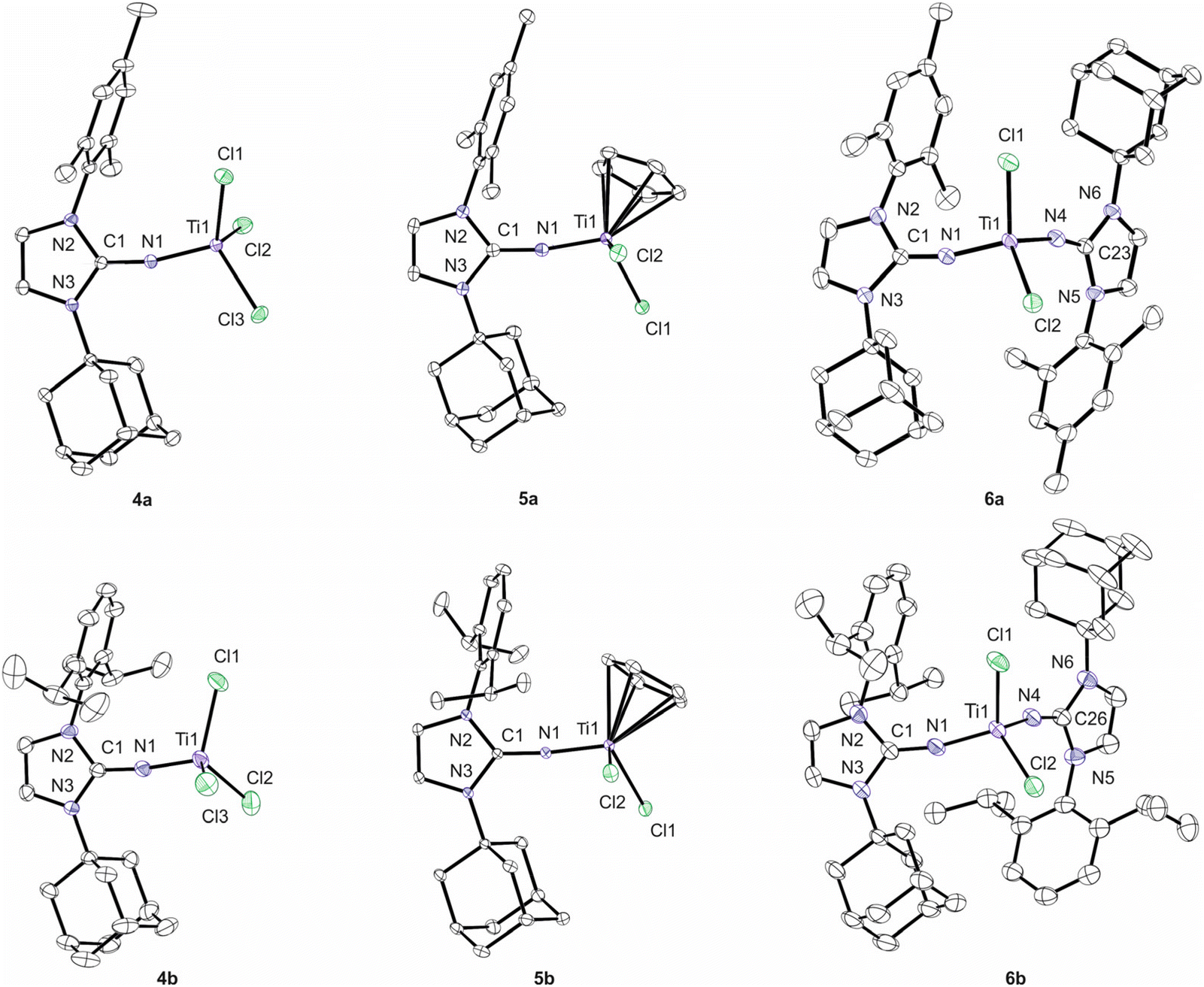 | ||
| Fig. 3 Molecular structures of 4a, 4b·CH2Cl2·Solv., 5a, 5b, 6a·0.5CH2Cl2 and 6b·CH2Cl2 with thermal displacement parameters drawn at 50% probability. Hydrogen atoms and co-crystallised solvent molecules are omitted for clarity. The molecular structure of 4b contains disordered solvent molecules along a tube-shaped solvent accessible void. For further information on the X-ray diffraction analysis please refer to the ESI.† | ||
| C1–N1 | Ti1–N1 | Ti1–N1–C1 | N2–C1–N3 | |
|---|---|---|---|---|
| 4a | 1.3265(7) | 1.7347(5) | 162.58(5) | 107.41(4) |
| 4b | 1.330(3) | 1.7316(17) | 173.04(16) | 106.89(17) |
| 5a | 1.319(2) | 1.7758(16) | 170.26(14) | 105.99(15) |
| 5b | 1.3189(8) | 1.7760(5) | 170.86(5) | 106.13(5) |
| 6a | 1.304(2) | 1.7951(15) | 166.51(13) | 105.89(14) |
| 1.302(2) | 1.7867(16) | 166.94(13) | 105.69(15) | |
| 6b | 1.304(3) | 1.7994(18) | 167.14(17) | 105.69(18) |
| 1.302(3) | 1.8067(18) | 167.64(17) | 105.24(18) |
Conclusions
Imidazolin-2-imines with an unsymmetrical aryl-adamantyl substitution pattern were prepared from the free N-heterocyclic carbenes IAdMes (1a) and IAdDipp (1b). The reactions of the silylated imines ImAdMesNSiMe3 (2a) and ImAdDippNSiMe3 (2b) with [TiCl4(THF)2] and [CpTiCl3] provided a series of mono- and bis(imidazolin-2-iminato) titanium complexes 4–6 with release of trimethylsilyl chloride. All titanium complexes feature short metal–nitrogen bonds and large Ti–N–C angles (162°–173°), confirming the ability of these systems to act as electron-rich imido-type N-donor ligands. These complexes could serve as efficient olefin polymerisation pre-catalysts, with the asymmetry introduced by the unsymmetrical substitution pattern possibly enabling their application in stereospecific olefin polymerisation.33 This aspect will be under investigation in due course. In general, these ligands enrich the portfolio of available imidazolin-2-imine systems, which are not only widely used ligands in transition metal,6 rare earth metal,19 and actinide chemistry,23 but have also found considerable attention in main group element chemistry.34Experimental details
Materials and methods
All operations with air- and moisture-sensitive compounds were performed in a glove box under a dry argon atmosphere (MBraun GmbH, MB20G) or on a vacuum line using Schlenk techniques. Solvents were either purified via standard methods35 or obtained by purification with an Mbraun SPS and subsequently stored over Molecular Sieves (3–4 Å). Deuterated solvents were distilled from Na/K or CaH2, and degassed prior to use. The 1H and 13C{1H} NMR spectra were recorded on Bruker AVII300, Bruker AVIIHD400, Bruker AVIIIHD500 and Bruker AVII600 spectrometers at room temperature. 1H and 13C{1H} NMR spectra were referenced against the (residual) solvent signals.36 Chemical shifts are reported in ppm (parts per million). Coupling constants (J) are reported in Hertz (Hz), and splitting patterns are indicated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), sept (septet) and br (broad). NMR assignments were made using additional 2D NMR experiments. Elemental analysis was carried out with a Vario Micro Cube System. Unless otherwise indicated, all starting materials were obtained from Sigma-Aldrich, ABRC, TCI, Acros or Fluka and were purified if necessary. [IAdMesH]Cl28,29 and [IAdDippH]Cl28,29 were prepared according to literature procedures.Synthetic procedures
1 H NMR (500 MHz, C6D6): δ = 6.88 (d, 3JHH = 1.6 Hz, 1H, CH-backbone), 6.83–6.79 (m, 2H, m-Mes), 6.50 (d, 3JHH = 1.8 Hz, 1H, CH-backbone), 2.28 (d, 3JHH = 2.8 Hz, 6H, CH2-Ad), 2.15 (s, 3H, p-CH3), 2.13 (s, 6H, o-CH3), 2.02 (br. s, 3H, CH-Ad), 1.66–1.52 (br. m, 6H, CH2-Ad).
13 C NMR (126 MHz, C6D6): δ = 216.1 (s, N–C–N), 140.0 (s, i-Mes), 136.9 (s, p-Mes), 135.5 (s, o-Mes), 129.0 (s, m-Mes), 119.4 (s, CH-backbone), 114.8 (s, CH-backbone), 56.2 (s, Cq-Ad), 44.9 (s, CH2-Ad), 36.6 (s, CH2-Ad), 30.3 (s, CH-Ad), 21.0 (s, p-CH3), 18.2 (s, o-CH3).
EA (%) calc. for C22H28N2 (320 g mol−1): C 82.45, H 8.81, N 8.74; found: C 82.57, H 8.893, N 8.74.
1 H NMR (300 MHz, C6D6): δ = 7.29–7.22 (m, 1H, p-Dipp), 7.16–7.11 (m, 2H, m-Dipp), 6.84 (d, 3JHH = 1.6 Hz, 1H, CH-backbone), 6.63 (d, 3JHH = 1.6 Hz, 1H, CH-backbone), 2.84 (sept., 3JHH = 7.0 Hz, 2H, CH(CH3)2), 2.22 (m, 6H, CH2-Ad), 1.97 (br. s, 3H, CH-Ad), 1.54 (br. s, 6H, CH2-Ad), 1.22 (d, 3JHH = 6.8 Hz, 6H, CH(CH3)2), 1.11 (d, 3JHH = 6.8 Hz, 6H, CH(CH3)2).
13 C NMR (75 MHz, C6D6): δ = 217.0 (s, NCN), 146.4 (s, o-Dipp), 139.9 (s, i-Dipp), 128.7 (s, p-Dipp), 123.6 (s, m-Dipp), 120.6 (s, CH-backbone), 114.6 (s, CH-backbone), 56.2 (s, Cq-Ad), 44.8 (s, CH2-Ad), 36.6 (s, CH2-Ad), 30.3 (s, CH-Ad), 28.6 (s, CH(CH3)2), 24.6 (s, CH(CH3)2), 24.0 (s, CH(CH3)2).
EA (%) calc. for C25H34N2 (363 g mol−1): C 82.82, H 9.45, N 7.73; found: C 82.67, H 9.481, N 7.21.
1 H NMR (500 MHz, C6D6): δ = 6.79–6.77 (m, 2H, m-Mes), 6.16 (d, 3JHH = 3.0 Hz, 1H, CH-backbone), 5.66 (d, 3JHH = 3.1 Hz, 1H, CH-backbone), 2.36 (d, 3JHH = 2.7 Hz, 6H, CH2-Ad), 2.13 (s, 6H, o-CH3), 2.10 (s, 3H, p-CH3), 2.03 (br. s., 3H, CH-Ad), 1.71–1.55 (br. m, 6H, CH2-Ad), 0.09 (s, 9H, Si(CH3)3).
13 C NMR (126 MHz, C6D6): δ = 141.2 (s, NCN), 138.1 (s, p-Mes), 137.7 (s, o-Mes), 134.8 (s, i-Mes), 129.3 (s, m-Mes), 110.2 (s, CH-backbone), 109.3 (s, CH-backbone), 55.7 (s, Cq-Ad), 39.7 (s, CH2-Ad), 36.7 (s, CH2-Ad), 30.2 (s, CH-Ad), 21.1 (s, p-CH3), 18.3 (s, o-CH3), 3.5 (s, Si(CH3)3).
EA (%) calc. for C25H37N3Si (408 g mol−1): C 73.66, H 9.15, N 10.31; found: C 73.95, H 9.032, N 10.00.
1 H NMR (400 MHz, C6D6): δ = 7.24–7.19 (m, 1H, p-Dipp), 7.13–7.10 (m, 2H, m-Dipp), 6.19 (d, 3JHH = 3 Hz, 1H, CH-backbone), 5.89 (d, 3JHH = 3 Hz, 1H, CH-backbone), 3.01 (sept, 3JHH = 6.89 Hz, 2H, CH(CH3)2), 2.33 (d, 3JHH = 2.7 Hz, 6H, CH2-Ad), 2.01 (br. s, 3H, CH-Ad), 1.69–1.53 (br. m., 6H, CH2-Ad), 1.33 (d, 3JHH = 6.9 Hz, 6H, CH(CH3)2), 1.11 (d, 3JHH = 6.9 Hz, 6H, CH(CH3)2), 0.07 (s, 9H, Si(CH3)3).
13 C NMR (100 MHz, C6D6): δ = 148.3 (s, o-Dipp), 141.0 (NCN), 135.5 (s, i-Dipp), 129.5 (s, p-Dipp), 124.2 (s, m-Dipp), 112.3 (s, CH-backbone), 108.7 (s, CH-backbone), 55.7 (s, Cq-Ad), 39.8 (s, CH2-Ad), 36.7 (s, CH2-Ad), 30.2 (s, CH-Ad), 28.7 (s, CH(CH3)2), 25.0 (s, CH(CH3)2), 23.1 (s, CH(CH3)2), 3.5 (s, Si(CH3)3).
EA (%) calc. for C28H43N3Si (450 g mol−1): C 74.78, H 9.64, N 9.34, found: C 75.27, H 9.481, N 9.29.
1 H NMR (400 MHz, C6D6): δ = 6.71 (m, 2H, m-Mes), 6.16 (d, 3JHH = 2.9 Hz, 1H, CH-backbone), 5.66 (d, 3JHH = 2.8 Hz, 1H, CH-backbone), 4.46 (s, 1H, NH), 2.53 (br.s, 6H, CH2-Ad), 2.09 (s, 6H, o-CH3), 2.07 (s, 3H, p-CH3), 2.03 (br. s., 3H, CH-Ad),1.72–1.52 (br. m, 6H, CH2-Ad).
13 C NMR (100 MHz, C6D6): δ = 153.1 (s, NCN), 138.2 (s, p-Mes), 138.0 (s, o-Mes), 132.9 (s, i-Mes), 129.7 (s, m-Mes), 109.9 (s, CH-backbone), 109.4 (s, CH-backbone), 55.9 (s, Cq-Ad), 39.7 (s, CH2-Ad), 36.7 (s, CH2-Ad), 30.2 (s, CH-Ad), 21.0 (s, p-CH3), 18.0 (s, o-CH3).
EA (%) calc. for C22H29N3 (336 g mol−1): C 78.76, H 8.71, N 12.53; found: C 78.94, H 8.774, N 12.46.
1 H NMR (400 MHz, C6D6): δ = 7.23–7.18 (m, 1H, p-Dipp), 7.12–7.08 (m, 2H, m-Dipp), 6.19 (d, 3JHH = 2.8 Hz, 1H, CH-backbone), 5.82 (d, 3JHH = 2.8 Hz, 1H, CH-backbone), 4.38 (br. s, 1H, NH), 3.04 (sept, 3JHH = 6.9 Hz, 2H, CH(CH3)2), 2.51 (br. s, 6H, CH2-Ad), 2.01 (br. s, 3H, CH-Ad), 1.70–1.49 (br. m, 6H, CH2-Ad), 1.23 (d, 3JHH = 6.9 Hz, 6H, CH(CH3)2), 1.12 (d, 3JHH = 6.9 Hz, 6H, CH(CH3)2).
13 C NMR (100 MHz, C6D6): δ = 154.4 (s, NCN), 149.3 (s, o-Dipp), 133.0 (s, i-Dipp), 129.9 (s, p-Dipp), 124.6 (s, m-Dipp), 111.4 (s, CH-backbone), 109.3 (s, CH-backbone), 55.9 (s, Cq-Ad), 39.7 (s, CH2-Ad), 36.7 (s, CH2-Ad), 30.2 (s, CH-Ad), 28.8 (s, CH(CH3)2), 24.3 (s, CH(CH3)2), 23.9 (s, CH(CH3)2).
EA (%) calc. for C25H35N3 (378 g mol−1): C 79.53, H 9.34, N 11.13, found: C 79.66, H 9.353, N 10.48.
1 H NMR (500 MHz, CDCl3): δ = 7.02–7.00 (m, 2H, m-Mes), 6.75 (d, 3JHH = 2.5 Hz, 1H, CH-backbone), 6.45 (d, 3JHH = 2.4 Hz, 1H, CH-backbone), 2.48 (br. d, 3JHH = 2.7 Hz, 6H, CH2-Ad), 2.34 (s, 3H, p-CH3), 2.32 (br. s, 3H, CH-Ad), 2.13 (s, 6H, o-CH3), 1.94–1.75 (br. m, 6H, CH2-Ad).
13 C NMR (126 MHz, CDCl3): δ = 142.9 (s, NCN), 140.6 (s, p-Mes), 136.3 (s, o-Mes), 130.9 (s, i-Mes), 129.6 (s, m-Mes), 115.0 (s, CH-backbone), 110.4 (s, CH-backbone), 60.7 (s, Cq-Ad), 41.0 (s, CH2-Ad), 35.7 (s, CH2-Ad), 29.9 (s, CH-Ad), 21.3 (s, p-CH3), 17.8 (s, o-CH3).
EA (%) calc. for C22H28Cl3N3Ti (489 g mol−1): C 54.07, H 5.78, N 8.60; found: C 54.04, H 6.180, N 8.20.
1 H NMR (600 MHz, CDCl3): δ = 7.52–7.48 (m, 1H, p-Dipp), 7.33–7.31 (m, 2H, m-Dipp), 6.77 (d, 3JHH = 2.5 Hz, 1H, CH-backbone), 6.48 (d, 3JHH = 2.5 Hz, 1H, CH-backbone), 2.53 (d, 3JHH = 2.6 Hz, 6H, CH2-Ad), 2.47 (sept, 3JHH = 6.9 Hz, 2H, CH(CH3)2), 2.34 (br. s, 3H, CH-Ad), 1.91–1.75 (br. m, 6H, CH2-Ad), 1.37 (d, 3JHH = 6.8 Hz, 6H, CH(CH3)2), 1.12 (d, 3JHH = 6.9 Hz, 6H, CH(CH3)2).
13 C NMR (151 MHz, CDCl3): δ = 146.4 (s, o-Dipp), 143.5 (s, NCN), 131.3 (s, p-Dipp), 130.8 (s, i-Dipp), 124.8 (s, m-Dipp), 115.8 (s, CH-backbone), 110.2 (s, CH-backbone), 60.9 (s, Cq-Ad), 41.2 (s, CH2-Ad), 35.8 (s, CH2-Ad), 29.9 (s, CH-Ad), 29.1 (s, CH(CH3)2), 25.0 (s, CH(CH3)2), 23.3 (s, CH(CH3)2).
EA (%) calc. for C25H34Cl3N3Ti (531 g mol−1): C 56.57, H 6.46, N 7.92, found: C 56.24, H 6.585, N 7.42.
1 H NMR (600 MHz, C6D6): δ = 6.80–6.78 (m, 2H, m-Mes), 6.05 (s, 5H, Cp), 5.87 (d, 3JHH = 2.7 Hz, 1H, CH-backbone), 5.38 (d, 3JHH = 2.5 Hz, 1H, CH-backbone), 2.21 (br. d, 3JHH = 2.7 Hz, 6H, CH2-Ad), 2.10 (s, 6H, o-CH3), 2.08 (s, 3H, p-CH3), 2.06 (br. s, 3H, CH-Ad), 1.91–1.50 (br. m, 6H, CH2-Ad).
13 C NMR (151 MHz, C6D6): δ = 144.4 (s, NCN), 139.8 (s, p-Mes), 136.8 (s, o-Mes), 132.7 (s, i-Mes), 129.6 (s, m-Mes), 114.8 (s, Cp), 113.7 (s, CH-backbone), 109.8 (s, CH-backbone), 59.4 (s, Cq-Ad), 40.5 (s, CH2-Ad), 35.8 (s, CH2-Ad), 30.2 (s, CH-Ad), 21.0 (s, p-CH3), 18.2 (s, o-CH3).
EA (%) calc. for C27H33Cl2N3Ti (518 g mol−1): C 62.56, H 6.42, N 8.11; found: C 62.19, H 6.019, N 7.87.
1 H NMR (500 MHz, CDCl3): δ = 7.57–7.53 (m, 1H, p-Dipp), 7.41–7.39 (m, 2H, m-Dipp), 6.69 (d, 3JHH = 2.7 Hz, 1H, CH-backbone), 6.35 (d, 3JHH = 2.7 Hz, 1H, CH-backbone), 5.90 (s, 5H, Cp), 2.67 (sept, 3JHH = 6.9 Hz, 2H, CH(CH3)2), 2.49 (d, 3JHH = 2.7 Hz, 6H, CH2-Ad), 2.27 (br. s, 3H, CH-Ad), 1.98–1.68 (br. m, 6H, CH2-Ad), 1.40 (d, 3JHH = 6.8 Hz, 6H, CH(CH3)2), 1.10 (d, 3JHH = 6.9 Hz, 6H, CH(CH3)2).
13 C NMR (126 MHz, CDCl3): δ = 147.3 (s, o-Dipp), 144.0 (s, NCN), 132.8 (s, i-Dipp), 131.1 (s, p-Dipp), 124.7 (s, m-Dipp), 115.7 (s, CH-backbone), 115.3 (s, Cp), 109.7 (s, CH-backbone), 60.0 (s, Cq-Ad), 40.9 (s, CH2-Ad), 35.8 (s, CH2-Ad), 30.0 (s, CH-Ad), 28.9 (s, CH(CH3)2), 25.3 (s, CH(CH3)2), 23.0 (s, CH(CH3)2).
EA (%) calc. for C30H39Cl2N3Ti (560 g mol−1): C 64.30, H 7.01, N 7.50; found: C 63.85, H 7.032, N 7.14.
1 H NMR (500 MHz, CDCl3): δ = 6.87–6.84 (m, 2H, m-Mes), 6.46 (d, 3JHH = 2.8 Hz, 1H, CH-backbone), 6.04 (d, 3JHH = 2.7 Hz, 1H, CH-backbone), 2.25 (s, 3H, p-CH3), 2.18 (br. d, 3JHH = 2.5 Hz, 6H, CH2-Ad), 2.09 (s, 9H, o-CH3 + CH-Ad), 1.80–1.58 (br. m, 6H, CH2-Ad).
13 C NMR (126 MHz, CDCl3): δ = 141.9 (s, NCN), 137.8 (s, p-Mes), 136.6 (s, o-Mes), 132.7 (s, i-Mes), 128.7 (s, m-Mes), 112.4 (s, CH-backbone), 108.6 (s, CH-backbone), 57.8 (s, Cq-Ad), 39.9 (s, CH2-Ad), 35.8 (s, CH2-Ad), 29.7 (s, CH-Ad), 21.1 (s, p-CH3), 17.9 (s, o-CH3).
EA (%) calc. for C44H56Cl2N6Ti (788 g mol−1): C 67.09, H 7.17, N 10.67; found: C 67.06, H 7.535, N 10.30.
1 H NMR (500 MHz, CDCl3): δ = 7.31–7.25 (m, 1H, p-Dipp), 7.18–7.14 (m, 2H, m-Dipp), 6.45 (d, 3JHH = 2.7 Hz, 1H, CH-backbone), 6.14 (d, 3JHH = 2.7 Hz, 1H, CH-backbone), 2.78 (sept, 3JHH = 6.9 Hz, 2H, CH(CH3)2), 2.16 (br. s, 6H, CH2-Ad), 2.09 (br. s, 3H, CH-Ad), 1.82–1.57 (br. m, 6H, CH2-Ad), 1.33 (d, 3JHH = 7.0 Hz, 6H, CH(CH3)2), 1.02 (d, 3JHH = 6.9 Hz, 6H, CH(CH3)2).
13 C NMR (126 MHz, CDCl3): δ = 147.0 (s, o-Dipp), 142.5 (s, NCN), 133.0 (s, i-Dipp), 129.1 (s, p-Dipp), 123.7 (s, m-Dipp), 114.3 (s, CH-backbone), 108.2 (s, CH-backbone), 58.4 (s, Cq-Ad), 40.3 (s, CH2-Ad), 35.9 (s, CH2-Ad), 29.9 (s, CH-Ad), 28.8 (s, CH(CH3)2), 25.4 (s, CH(CH3)2), 23.2 (s, CH(CH3)2).
EA (%) calc. for C50H68Cl2N6Ti·0.5 (CH2Cl2) (872 g mol−1): C 66.34, H 7.61, N 9.19, found: C 66.84, H 7.703, N 8.91.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. T. and M. E. thank the German Israel Foundation (GIF) for continuous support in the years 2010–2012 and 2015–2017.Notes and references
- (a) N. Kuhn, R. Fawzi, M. Steimann, J. Wiethoff, D. Bläser and R. Boese, Z. Naturforsch., B: J. Chem. Sci., 1995, 50, 1779–1784 CrossRef CAS; (b) N. Kuhn, M. Göhner, M. Grathwohl, J. Wiethoff, G. Frenking and Y. Chen, Z. Anorg. Allg. Chem., 2003, 629, 793–802 CrossRef CAS.
- N. Kuhn, R. Fawzi, M. Steimann and J. Wiethoff, Z. Anorg. Allg. Chem., 1997, 623, 769–774 CrossRef CAS.
- M. Tamm, S. Randoll, T. Bannenberg and E. Herdtweck, Chem. Commun., 2004, 876–877 RSC.
- M. Tamm, S. Randoll, E. Herdtweck, N. Kleigrewe, G. Kehr, G. Erker and B. Rieger, Dalton Trans., 2006, 459–467 RSC.
- M. Tamm, D. Petrovic, S. Randoll, S. Beer, T. Bannenberg, P. G. Jones and J. Grunenberg, Org. Biomol. Chem., 2007, 5, 523–530 RSC.
- (a) X. Wu and M. Tamm, Coord. Chem. Rev., 2014, 260, 116–138 CrossRef CAS; (b) A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed; (c) P. Raja, S. Revathi and T. Ghatak, Eng. Sci., 2020, 12, 23–37 CAS.
- (a) S. H. Stelzig, M. Tamm and R. M. Waymouth, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6064–6070 CrossRef CAS; (b) K. Nomura, H. Fukuda, W. Apisuk, A. G. Trambitas, B. Kitiyanan and M. Tamm, J. Mol. Catal. A: Chem., 2012, 363–364, 501–511 CrossRef CAS; (c) W. Apisuk, A. G. Trambitas, B. Kitiyanan, M. Tamm and K. Nomura, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2575–2580 CrossRef CAS.
- M. Sharma, H. S. Yameen, B. Tumanskii, S.-A. Filimon, M. Tamm and M. S. Eisen, J. Am. Chem. Soc., 2012, 134, 17234–17244 CrossRef CAS PubMed.
- D. Shoken, M. Sharma, M. Botoshansky, M. Tamm and M. S. Eisen, J. Am. Chem. Soc., 2013, 135, 12592–12595 CrossRef CAS PubMed.
- (a) D. Shoken, L. J. W. Shimon, M. Tamm and M. S. Eisen, Organometallics, 2016, 35, 1125–1131 CrossRef CAS; (b) K. Naktode, S. Das, H. P. Nayek and T. K. Panda, Inorg. Chim. Acta, 2017, 456, 24–33 CrossRef CAS.
- S. Das, J. Bhattacharjee and T. K. Panda, Dalton Trans., 2019, 48, 7227–7235 RSC.
- (a) K. Naktode, S. Das, H. P. Nayek and T. K. Panda, J. Coord. Chem., 2018, 71, 4148–4163 CrossRef CAS; (b) K. Naktode, S. Das, J. Bhattacharjee, H. P. Nayek and T. K. Panda, Inorg. Chem., 2016, 55, 1142–1153 CrossRef CAS PubMed.
- (a) J. R. Aguilar-Calderón, A. J. Metta-Magaña, B. Noll and S. Fortier, Angew. Chem., Int. Ed., 2016, 55, 14101–14105 CrossRef PubMed; (b) J. R. Aguilar-Calderón, J. Murillo, A. Gomez-Torres, C. Saucedo, A. Jordan, A. J. Metta-Magaña, M. Pink and S. Fortier, Organometallics, 2020, 39, 295–311 CrossRef.
- (a) W. P. Kretschmer, C. Dijkhuis, A. Meetsma, B. Hessen and J. H. Teuben, Chem. Commun., 2002, 608–609 RSC; (b) K. Nomura, H. Fukuda, S. Katao, M. Fujiki, H. J. Kim, D.-H. Kim and S. Zhang, Dalton Trans., 2011, 40, 7842–7849 RSC; (c) I. Haas, C. Hübner, W. P. Kretschmer and R. Kempe, Chem. – Eur. J., 2013, 19, 9132–9136 CrossRef CAS PubMed; (d) K. Nomura, S. Patamma, H. Matsuda, S. Katao, K. Tsutsumi and H. Fukuda, RSC Adv., 2015, 5, 64503–64513 RSC; (e) K. Nomura, H. Fukuda, H. Matsuda, S. Katao and S. Patamma, J. Organomet. Chem., 2015, 798, 375–383 CrossRef CAS; (f) Z.-Q. Zhang, J.-T. Qu, S. Zhang, Q.-P. Miao and Y.-X. Wu, Polym. Chem., 2018, 9, 48–59 RSC; (g) T. Dietel, F. Lukas, W. P. Kretschmer and R. Kempe, Science, 2022, 375, 1021–1024 CrossRef CAS PubMed.
- (a) A. Glöckner, T. Bannenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chem., 2012, 51, 4368–4378 CrossRef PubMed; (b) J. Klosin, P. P. Fontaine, R. Figueroa, S. D. McCann and D. Mort, Organometallics, 2013, 32, 6488–6499 CrossRef CAS; (c) M. Khononov, N. Fridman, M. Tamm and M. S. Eisen, Eur. J. Org. Chem., 2020, 3153–3160 CrossRef CAS.
- (a) S. Zhang, M. Tamm and K. Nomura, Organometallics, 2011, 30, 2712–2720 CrossRef CAS; (b) K. Nomura, B. K. Bahuleyan, K. Tsutsumi and A. Igarashi, Organometallics, 2014, 33, 6682–6691 CrossRef CAS; (c) K. Nomura, B. K. Bahuleyan, S. Zhang, P. M. V. Sharma, S. Katao, A. Igarashi, A. Inagaki and M. Tamm, Inorg. Chem., 2014, 53, 607–623 CrossRef CAS PubMed.
- (a) S. Beer, C. G. Hrib, P. G. Jones, K. Brandhorst, J. Grunenberg and M. Tamm, Angew. Chem., Int. Ed., 2007, 46, 8890–8894 CrossRef CAS PubMed; (b) S. Beer, K. Brandhorst, J. Grunenberg, C. G. Hrib, P. G. Jones and M. Tamm, Org. Lett., 2008, 10, 981–984 CrossRef CAS PubMed; (c) B. Haberlag, X. Wu, K. Brandhorst, J. Grunenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem. – Eur. J., 2010, 16, 8868–8877 CrossRef CAS PubMed; (d) B. Haberlag, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2012, 51, 13019–13022 CrossRef CAS PubMed; (e) S. Lysenko, C. G. Daniliuc, P. G. Jones and M. Tamm, J. Organomet. Chem., 2013, 744, 7–14 CrossRef CAS; (f) B. Haberlag, M. Freytag, P. G. Jones and M. Tamm, Adv. Synth. Catal., 2014, 356, 1255–1265 CrossRef CAS; (g) Z. S. Qureshi, A. Hamieh, S. Barman, N. Maity, M. K. Samantaray, S. Ould-Chikh, E. Abou-Hamad, L. Falivene, V. D'Elia, A. Rothenberger, I. Llorens, J.-L. Hazemann and J.-M. Basset, Inorg. Chem., 2017, 56, 861–871 CrossRef CAS PubMed.
- (a) T. K. Panda, S. Randoll, C. G. Hrib, P. G. Jones, T. Bannenberg and M. Tamm, Chem. Commun., 2007, 5007–5009 RSC; (b) A. G. Trambitas, J. Yang, D. Melcher, C. G. Daniliuc, P. G. Jones, Z. Xie and M. Tamm, Organometallics, 2011, 30, 1122–1129 CrossRef CAS.
- A. G. Trambitas, T. K. Panda and M. Tamm, Z. Anorg. Allg. Chem., 2010, 636, 2156–2171 CrossRef CAS.
- T. K. Panda, A. G. Trambitas, T. Bannenberg, C. G. Hrib, S. Randoll, P. G. Jones and M. Tamm, Inorg. Chem., 2009, 48, 5462–5472 CrossRef CAS PubMed.
- (a) A. G. Trambitas, T. K. Panda, J. Jenter, P. W. Roesky, C. Daniliuc, C. G. Hrib, P. G. Jones and M. Tamm, Inorg. Chem., 2010, 49, 2435–2446 CrossRef CAS PubMed; (b) A. G. Trambitas, D. Melcher, L. Hartenstein, P. W. Roesky, C. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chem., 2012, 51, 6753–6761 CrossRef CAS PubMed.
- B.-C. Liu, N. Ge, Y.-Q. Zhai, T. Zhang, Y.-S. Ding and Y.-Z. Zheng, Chem. Commun., 2019, 55, 9355–9358 RSC.
- S. Revathi, P. Raja, S. Saha, M. S. Eisen and T. Ghatak, Chem. Commun., 2021, 57, 5483–5502 RSC.
- (a) I. S. R. Karmel, N. Fridman, M. Tamm and M. S. Eisen, J. Am. Chem. Soc., 2014, 136, 17180–17192 CrossRef CAS PubMed; (b) I. S. R. Karmel, M. Botoshansky, M. Tamm and M. S. Eisen, Inorg. Chem., 2014, 53, 694–696 CrossRef CAS PubMed; (c) I. S. R. Karmel, N. Fridman, M. Tamm and M. S. Eisen, Organometallics, 2015, 34, 2933–2942 CrossRef CAS; (d) I. S. R. Karmel, M. Khononov, M. Tamm and M. S. Eisen, Catal. Sci. Technol., 2015, 5, 5110–5119 RSC; (e) I. S. R. Karmel, M. Tamm and M. S. Eisen, Angew. Chem., Int. Ed., 2015, 54, 12422–12425 CrossRef CAS PubMed.
- M. Yadav, A. Metta-Magaña and S. Fortier, Chem. Sci., 2020, 11, 2381–2387 RSC.
- (a) J. T. Goettel, H. Gao, S. Dotzauer and H. Braunschweig, Chem. – Eur. J., 2020, 26, 1136–1143 CrossRef CAS PubMed; (b) T. Glöge, F. Aal, S.-A. Filimon, P. G. Jones, J. Michaelis de Vasconcellos, S. Herres-Pawlis and M. Tamm, Z. Anorg. Allg. Chem., 2015, 641, 2204–2214 CrossRef.
- (a) W. Zhao, B. Wang, B. Dong, H. Liu, Y. Hu, M. S. Eisen and X. Zhang, Organometallics, 2020, 39, 3983–3991 CrossRef CAS; (b) H. Liu, M. Khononov, N. Fridman, M. Tamm and M. S. Eisen, Inorg. Chem., 2017, 56, 3153–3157 CrossRef CAS PubMed; (c) H. Liu, N. Fridman, M. Tamm and M. S. Eisen, Organometallics, 2017, 36, 4600–4610 CrossRef CAS; (d) H. Liu, N. Fridman, M. Tamm and M. S. Eisen, Organometallics, 2017, 36, 3896–3903 CrossRef CAS; (e) H. Liu, M. Khononov, N. Fridman, M. Tamm and M. S. Eisen, J. Organomet. Chem., 2018, 857, 123–137 CrossRef CAS; (f) H. Liu, K. Kulbitski, M. Tamm and M. S. Eisen, Chem. – Eur. J., 2018, 24, 5738–5742 CrossRef CAS PubMed; (g) H. Liu, M. Khononov, N. Fridman, M. Tamm and M. S. Eisen, Inorg. Chem., 2019, 58, 13426–13439 CrossRef CAS PubMed; (h) M. Khononov, H. Liu, N. Fridman, M. Tamm and M. S. Eisen, Organometallics, 2020, 39, 3021–3033 CrossRef CAS.
- P. Queval, C. Jahier, M. Rouen, I. Artur, J.-C. Legeay, L. Falivene, L. Toupet, C. Crévisy, L. Cavallo, O. Baslé and M. Mauduit, Angew. Chem., Int. Ed., 2013, 52, 14103–14107 CrossRef CAS PubMed.
- R. Tarrieu, A. Dumas, J. Thongpaen, T. Vives, T. Roisnel, V. Dorcet, C. Crévisy, O. Baslé and M. Mauduit, J. Org. Chem., 2017, 82, 1880–1887 CrossRef CAS PubMed.
- (a) A. Dumas, R. Tarrieu, T. Vives, T. Roisnel, V. Dorcet, O. Baslé and M. Mauduit, ACS Catal., 2018, 8, 3257–3262 CrossRef CAS; (b) J. Morvan, T. McBride, I. Curbet, S. Colombel-Rouen, T. Roisnel, C. Crévisy, D. L. Browne and M. Mauduit, Angew. Chem., Int. Ed., 2021, 60, 19685–19690 CrossRef CAS PubMed.
- (a) F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC; (b) J. March, Advanced Organic Chemistry, John Wiley & Sons, Inc, New York, 4th edn, 1992 Search PubMed.
- C. Camp, L. C. E. Naested, K. Severin and J. Arnold, Polyhedron, 2016, 103, 157–163 CrossRef CAS.
- H. H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1143–1170 CrossRef CAS.
- T. Ochiai, D. Franz and S. Inoue, Chem. Soc. Rev., 2016, 45, 6327–6344 RSC.
- W. L. F. Armarego and C. L. L. Chai, Purification of laboratory chemicals, Elsevier/Butterworth-Heinemann, Amsterdam, London, 7th edn, 2013 Search PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, crystallographic details. CCDC 2174864–2174874. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt01740g |
| This journal is © The Royal Society of Chemistry 2022 |