
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a polyaminocarboxylate-based aluminum complex and its structural studies using 1H{13C}-HMBC NMR and a Karplus-type function†
Karel D.
Klika
a,
Rana
Alsalim
b,
Mohammad
Eftekhari
c and
Ata
Makarem‡
 *b
*b
aGerman Cancer Research Center (DKFZ), Molecular Structure Analysis, INF 280, 69120 Heidelberg, Germany
bUniversity of Hamburg, Institute of Pharmacy, Bundesstraße 45, 20146 Hamburg, Germany. E-mail: ata.makarem@chemie.uni-hamburg.de
cUniversity of Neyshabur, Department of Chemistry, Neyshabur, Iran
First published on 5th August 2022
Abstract
The HBED chelator is used to stabilize small and hard metal ions such as Fe3+, Ti4+, Ga3+ and Al3+ in both medicine and industry. While the coordination of hexadentate HBED4− is known in the case of Fe3+, Ti4+ and Ga3+, it is unknown in the case of the small Al3+ ion since its corresponding complex has never been fully characterized. Thus, in this work the coordination pattern in a newly synthesized aluminum HBED-based complex ([Al–HBED–NN]−Na+) was determined using 2D NMR in conjunction with DFT calculations.
While X-ray crystallography remains the preferred method for determining the coordination of metal complexes, the requirement to obtain X-ray quality crystals can sometimes not be met.1 In such instances NMR can be an alternative means to determine the coordination pattern and geometry of complexes.2–5 HMBC in particular is an especially useful NMR technique for structural analyses as it reveals multiple-bond connectivity between protons and X nuclei.2N,N′-Bis(2-hydroxybenzyl)ethylenediamine-N,N′-diacetic acid (HBED) is a popular polyaminocarboxylate-type chelator which can tightly bind to metal ions via its 6 coordinating atoms (two phenolate groups, two amine groups and two carboxylate groups) resulting in three possible octahedral isomers,6–12 each as a racemate.13 From an application point of view, it is important to distinguish these isomers from each other since isomeric structures can exhibit distinct (bio)chemical profiles.8,14 However, the content of the predominant complex species in the HBED chelation depends on a number of factors such as the reaction conditions, particularly the temperature, as well as the nature of the metal in terms of its size, valence, hardness/softness, etc.3,7
Although the synthesis and application of the Al–HBED hexacoordinate complex ([Al–HBED]−) have already been described in basic and applied research, it has not yet been investigated from a structural point of view; and even characterization data to support its existence have not been reported.15–18 Only in one case was the occurrence of [Al–HBED]− reported in the research literature where it was detected by mass spectroscopy as a byproduct, but the compound was not isolated and characterized as a single substance.19 However, in contrast to [Al–HBED]−, other HBED hexacoordinate scaffolds such as [Fe–HBED]−, [Ti–HBED] and [Ga–HBED]− have had their structures determined.3,11,12 Herein, we employed NMR in conjunction with DFT calculations to determine the coordination of the Al–HBED cage using the newly synthesized [Al–HBED–NN]−Na+ complex (Scheme 1).
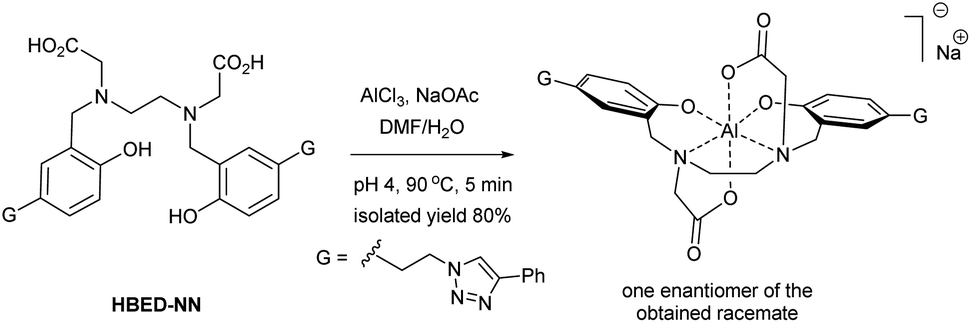 | ||
| Scheme 1 Synthesis of the [Al–HBED–NN]− complex at elevated temperature as a single, racemic isomer. | ||
For the preparation of the [Al–HBED–NN]−Na+ complex, the HBED–NN chelator (Scheme 1) bearing 1,2,3-triazolyl terminal groups was first prepared according to literature.3 As mentioned, metal chelation in general is highly dependant on the reaction conditions, such as the temperature, pH, etc., often resulting in a mixture of complex species, isomers, side products and conformers with similar physical properties and spectra. Consequently, the ensuing analysis of the chromatographic and spectroscopic data can be challenging in order to elucidate the reaction progress and optimize the process to a single product.7,8,20–22 For example, the upfield aliphatic region of 1H NMR spectra in chelate systems especially is usually crowded with various multiplets and broad peaks.3,22–24 This challenge in the case of underivatized HBED has been detailed in the literature.7 In contrast, the HBED–NN ligand facilitates convenient reaction monitoring by 1H NMR since the sharp triazolyl singlet at 8.4 ppm (in DMSO) occurs in an uncrowded downfield region.3 Generally, triazolyl residue protons are very sensitive to structural modification, even to quite distant changes elsewhere in the molecule to which their δH's respond accordingly.3–5 Thus, by utilizing the proton triazolyl singlet for reaction monitoring, the conversion and chemoselectivity of chelation is able to be followed and duly optimized.
Fortunately, due to the multiple permutations of the reaction conditions that we had conducted on the analogous Ga3+ complex,3 the optimal conditions found in that study were utilized for the [Al–HBED–NN]− complex realizing its preparation in a straightforward manner (Scheme 1). Indeed, the chelation of the HBED–NN ligand to Al3+ was found to be high yielding (80% yield) when the reaction was conducted at an elevated temperature (90 °C) to conveniently produce a single geometric isomer after only 5 min. The isolated and purified [Al–HBED–NN]−Na+ product was fully characterized by NMR while HR-ESI-MS provided an ion of m/z 753.2730 Da (calcd for C40H38AlN8O6, 753.2730 Da) to confirm its fabrication (see ESI†).
The three possible isomers for the isolated [Al–HBED–NN]− complex – isomers I (carboxylate groups cis, phenolate groups trans), II (carboxylate groups trans, phenolate groups cis) and III (carboxylate groups cis, phenolate groups cis) – are depicted in Scheme 2. For both isomers I and II, the two halves of the ligand are equivalent due to the presence of a two-fold rotation axis while isomer III lacks any symmetry element. Thus, while isomer III should exhibit 36 unique carbon signals and 34 unique proton signals in the respective spectra, isomers I and II should only exhibit half these numbers due to the equivalent halves of the ligand. On the basis of the 1H and 13C NMR spectra, isomer III can be immediately ruled out since in the 13C NMR spectrum only 18 signals are present and in the 1H NMR spectrum only 17 signals are present (see ESI†).
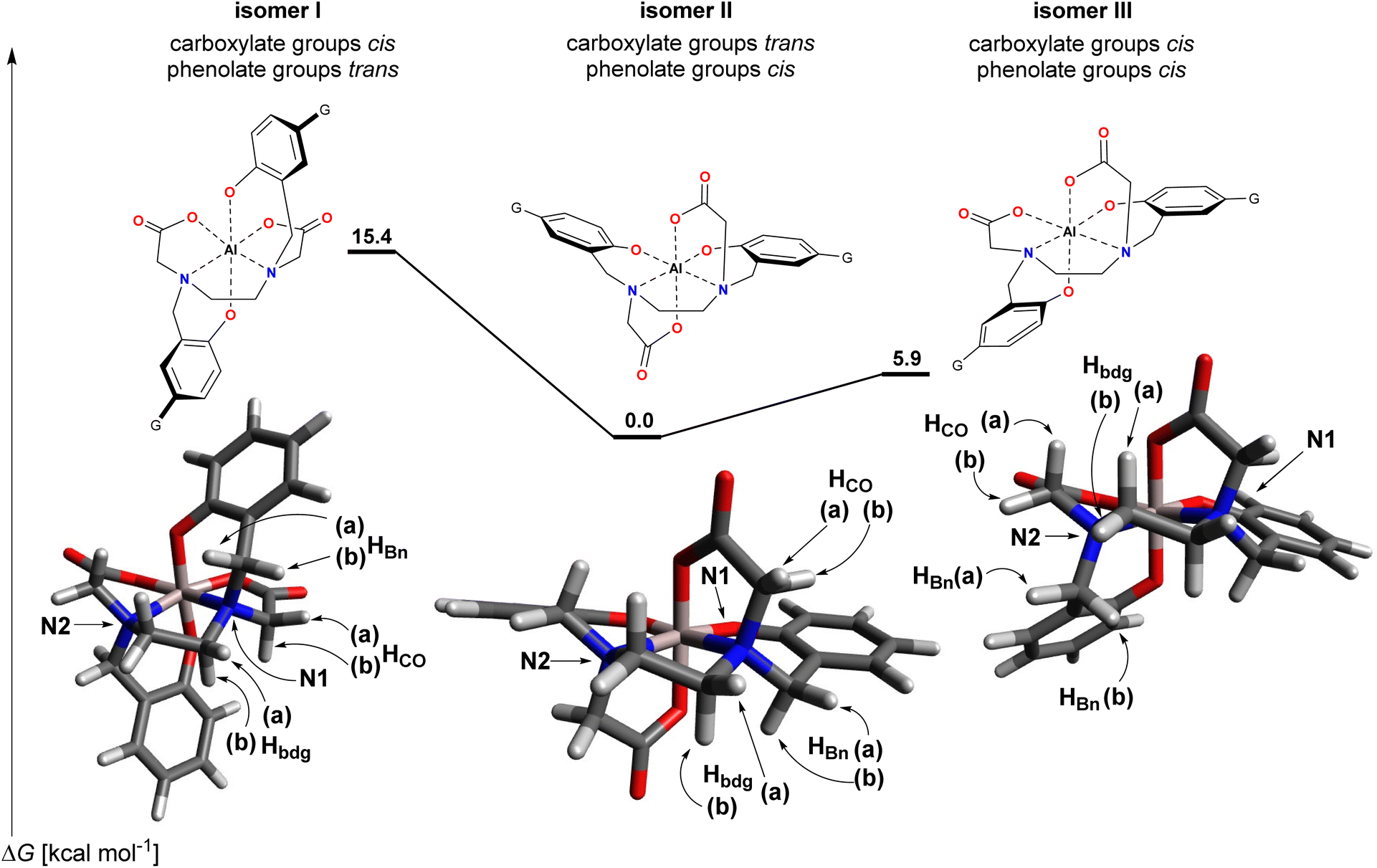 | ||
| Scheme 2 Structures of the three possible isomers and their DFT-calculated models for the [Al–HBED–NN]− complex. N.b. Each isomer depicted here is one of a pair of enantiomers and for the sake of convenience, the G groups were not included in the calculations and hence are missing from the depicted DFT-calculated structures. Methylene protons that are oriented upwards in the depictions are designated as a, while methylene protons oriented downwards are designated as b. | ||
The strategy applied here to distinguish between isomers I and II basically follows that applied previously3 where the distinction between the isomers relies on an examination of a comprehensive set of possible vicinal proton–carbon correlations in a HMBC spectrum, not only in terms of correlations observed, but also correlations not observed. Pertinent 3JH,C's, viz. those within the coordination sphere, are evaluated to be either large or small depending on the dihedral angle between the nuclei concerned. If 3JH,C was anticipated to be large, e.g. >5 Hz, then a correlation should be observed in a HMBC spectrum optimized for a long-range coupling of 8 Hz; while values of 3JH,C < 5 Hz would otherwise not be expected to give rise to an observable correlation.
The size evaluation of 3JH,C is dependent on vicinal proton–carbon nuclei following a Karplus-type function.25 The well-known Karplus-type relationships for vicinal couplings between nuclei are functions relating 3J's with the dihedral angles (α) between the nuclei.26,27 They since have been developed for various systems (e.g., H–C–C–C, H–C–O–C, F–C–C–H, P–C–C–H, H–C–N+–H and H–C–C–N)25,28 however, they all exhibit quite similar trajectories, viz., 3J is a maximum when α = 0° or 180° due to maximum overlap of orbitals and a minimum when α = 90° due to the orbitals being perpendicular to one another. The dihedral angle is not the only factor determining the size of 3J, other factors such as bond length, order and angle as well as the presence of electronegative/-positive substituents etc. also have an influence; but for the comparison of systems containing the same atoms, Karplus-type relationships are very useful for evaluating vicinal coupling–dihedral angle relationships. The Karplus-type function for the 3JH,C's in propane is depicted in Fig. 1, used here as the function for the vicinal proton–carbon relationship.29 For this analysis, a value of 5 Hz is defined as the cutoff between small and large couplings and from the plot in Fig. 1 it can be discerned that large 3JH,C's occur when α < 31.5° or α > 135°.
 | ||
| Fig. 1 Theoretical plot of 3JH,Cvs. the dihedral angle between the proton and carbon nuclei for propane, used as the function here. | ||
For the structural evaluation, all 16 vicinal proton–carbon relationships involving the methylene protons within the coordination sphere were taken into consideration. To evaluate the expected size of the 3JH,C's from the dihedral angles, the structures of isomers I–III were optimized by DFT calculations at the b3lyp/6-31 g(d,p)//b3lyp/6-31 g(d,p) level of theory with DMSO simulated as the solvent by a polarized continuum medium (Scheme 2). This level of theory was considered appropriate to attain geometries sufficiently reliable for the ensuing analysis. The starting geometry for isomer II was developed from the X-ray crystallographic structure of [Ti–HBED],12 while isomers I and III were modified accordingly from the optimized geometry of isomer II. For the sake of convenience and to reduce computational cost, the terminal G groups were not included in the calculations as they were not considered to have any undue influence on the geometry of the coordination sphere.
The ΔG values for isomers I–III obtained from the DFT calculations overwhelmingly support isomer II as the thermodynamically favored structure (isomers I and III were computed to be higher in energy by 15.4 and 5.9 kcal mol−1, respectively). In both isomers I and III, there appears to be a possible steric interaction between an acetyl segment and a phenyl ring, and as well, for isomer I there appears to be an additional possible steric interaction between the ethylenediamine bridge and the other phenyl ring. These possible steric interactions could account for some of the energy difference with respect to isomer II. Moreover, taking the preferred bond angles as those present in isomer II where the angles are effectively identical for the two halves of the ligand, for bond angles starting from carbon and ending in the Al3+ ion, isomer I had more distorted bond angles (taken as a deviation >4°, relative to the value in isomer II) than isomer III (6 vs. 4, respectively). Furthermore, for both isomers I and III, a majority of the distorted bond angles were in that half of the ligand experiencing possible steric interaction with the acetyl segment. By comparison for the X-ray crystallographic structure of [Ti–HBED],12 only two degenerate angles were significantly different to those in isomer II. For bond angles with the Al3+ ion as the central atom and only comparing angles within the one ligand half, there was nothing of note between the four structures, and similarly for the bond lengths. A listing of the bond angles and bond lengths considered is presented in Table S5.†
Of note, calculated energies are highly sensitive to the environment and method of calculation and can change markedly. Moreover, it may be the kinetic product that is formed initially and if interconversion between the isomers is inexorably slow, the kinetic product only may be isolated. While it is noted that multiple isomers have been claimed to be observed in solution for the larger Ga3+ ion,7,8 perhaps only one isomer is overwhelmingly favored thermodynamically for Al3+. A possible reason for this is the smaller size of Al3+ which might be exacerbating the bond angle strain and steric interactions in isomers I and III. This would account for their large DFT-calculated energies. However, the geometries for the immediate atoms of the coordination sphere of these quite rigid complexes will not fundamentally alter due to the environment or higher levels of theory. The purpose of the DFT calculations was to use the optimized geometries for structural evaluation by NMR which now follows.
For isomer II, the corresponding dihedral angles calculated for the two halves are identical to one decimal place. Despite the differences calculated for isomer I between the two halves of the ligand, it is expected that rapid fluxional motion will interconvert the two halves of the ligand rendering them equivalent on the NMR timescale; thus the 3JH,C values were averaged for the structural evaluation. For isomer III – included for the sake of completeness though already eliminated from the candidate structures – interconversion of the two halves of the ligand is not possible and was treated in two ways: firstly by averaging of the coupling values and; secondly, by consideration that if one dihedral angle of a methylene pair were to provide a large 3JH,C value, it is taken that a correlation would be expected for both that proton and its corresponding partner in the other half of the ligand. Of note, in the 1H NMR spectrum, the four protons of the ethylenediamine bridge segment are effectively isochronous; so, if one equivalent pair of protons in the ethylenediamine bridge were to provide a large 3JH,C value, it is taken that a correlation would be expected for the ethylenediamine bridge-methylene signal. The result of this is that one relationship for each isomer is rendered indeterminate (or in other words, a possible correlation expected to be present is masked by a possible correlation expected to not be present), leaving a total of 15 relationships that can be utilized for structural evaluation.
From a compilation of the evaluated dihedral angles, selected relationships and their expected vs. experimental results are presented in Table 1 for isomers I–III (a full listing of the evaluated dihedral angles are presented in Tables S1–S4†). For isomer I, only in five cases was there a match between expected and experimental results and thus isomer I is eliminated as a candidate structure for the Al–HBED hexacoordinate cage (Table S2†). Interestingly, in the case of isomer III, both approaches described above only provide two contradictory predictions to experimental observations (Tables S3 and S4†). This is provided of course that corresponding protons from each half of the ligand were in fact isochronous; thus, isomer III can also be eliminated as a candidate structure on the basis of this analysis. On the other hand, for isomer II, all 15 expected results were in concert with experimental results consisting of seven observed correlations and eight possible correlations not observed (Table S1†). The complete match between observed and unobserved correlations undoubtedly confirms that the structure of [Al–HBED–NN]− isolated from the reaction has the carboxylate groups trans to each other and thus the coordination of HBED to Al3+ follows that of Fe3+ in [Fe–HBED]−, Ti4+ in [Ti–HBED] and Ga3+ in [Ga–HBED]−.
|
|
||||
|---|---|---|---|---|
| Entry | Nuclei | Dihedral angle, αa | Large 3JH,C expected? | Expectation vs. observationb |
a N1 and N2 refer to the ligand half.
b ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)  = consistent; = consistent;  = inconsistent.
c Despite the medium size of α, a correlation is expected for entry 3 due to the overlap of Hbdg (a) and (b). = inconsistent.
c Despite the medium size of α, a correlation is expected for entry 3 due to the overlap of Hbdg (a) and (b).
|
||||
| 1 | HCO (b), Cbdg | Isomer I: 30° (N1), 40° (N2) | No |

|
| Isomer II: 146° (N1, N2) | Yes |

|
||
| Isomer III: 153° (N1), 92° (N2) | No |

|
||
| 2 | HCO (a), Cbdg | Isomer I: 89° (N1), 81° (N2) | No |

|
| Isomer II: 28° (N1, N2) | Yes |

|
||
| Isomer III: 34° (N1), 28° (N2) | Yes |

|
||
| 3 | Hbdg (a), CCO | Isomer I: 85° (N1), 94° (N2) | Noc |

|
| Isomer II: 46° (N1, N2) | Noc |

|
||
| Isomer III: 158° (N1), 34° (N2) | Yes |

|
||
| Hbdg (b), CCO | Isomer I: 34° (N1), 23° (N2) | Yes |

|
|
| Isomer II: 163° (N1, N2) | Yes |

|
||
| Isomer III: 41° (N1), 84° (N2) | Noc |

|
||
| 4 | Hbdg (a), CBn | Isomer I: 40° (N1), 29° (N2) | No |

|
| Isomer II: 79° (N1, N2) | No |

|
||
| Isomer III: 32° (N1), 159° (N2) | Yes |

|
||
| Hbdg (b), CBn | Isomer I: 158° (N1), 146° (N2) | Yes |

|
|
| Isomer II: 38° (N1, N2) | No |

|
||
| Isomer III: 84° (N1), 41° (N2) | No |

|
||

It is worth noting that the structural determination analysis presented here can still be successfully performed without stereochemical assignment of the methylene protons, i.e. assigning the two protons of each methylene group. However, not stereochemically assigning the protons reduces the number of relationships that can be used for the analysis by eight (six relationships for the second evaluation approach in the case of isomer III). For isomer I this makes a sizeable difference as six contrarian predictions to isomer II are lost, though four contrarian predictions remain to enable a distinction between isomers I and II. Beguilingly, not stereochemically assigning the protons makes no difference in either of the two evaluation approaches for isomer III as no contrarian predictions to isomer II are lost.
Conclusions
A new aluminum HBED-type hexacoordinate complex has been prepared and fully characterized, and the coordination geometry of HBED4− to Al3+ proven for the first time. Only for isomer II was there a complete match between observed correlations consistent with prediction and possible correlations that were not observed but also in line with prediction. The coordination of the hexadentate HBED chelator to Al3+ follows that of Fe3+, Ti4+ and Ga3+ in which the carboxylate ligands are trans to each other, and perhaps because of the small size of Al3+ only one isomer may be thermodynamically possible. The strategy of examining a comprehensive set of vicinal proton–carbon relationships in conjunction with DFT calculations – both for those correlations expected to be present as well as those expected to be absent in a 8 Hz-optimized HMBC spectrum – has once again proven successful.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge the Institute of Cancer Research in London, the Chemistry Department of the University of Hamburg (Germany), the German Cancer Research Center, Prof. Klaus Kopka, Prof. Wolfgang Maison and Prof. Achim Oberg for support of this work.This work was partly funded by a grant from the German Academic Exchange Service (DAAD – project number: 57407594).
Notes and references
- M. Ladd and R. Palmer, in Structure Determination by X-ray Crystallography, Springer US, Boston, MA, 2013 Search PubMed.
- R. M. Silverstein, F. X. Webster, D. J. Kiemle and D. L. Bryce, in Spectrometric Identification of Organic Compounds, John Wiley & Sons, New York, NY, 2014 Search PubMed.
- A. Makarem, K. D. Klika, G. Litau, Y. Remde and K. Kopka, J. Org. Chem., 2019, 84, 7501–7508 CrossRef CAS PubMed.
- A. Makarem, PhD Thesis, Ruprecht Karls Universität Heidelberg, 2015.
- A. Makarem, R. Berg, F. Rominger and B. F. Straub, Angew. Chem., Int. Ed., 2015, 54, 7431–7435 CrossRef CAS PubMed.
- (a) J. Schuhmacher, G. Klivényi, R. Matys, M. Stadler, T. Regiert, H. Hauser, J. Doll, W. Maier-Borst and M. Zöller, Cancer Res., 1995, 55, 115–123 CAS; (b) A. Makarem, M. K. Sarvestani, K. D. Klika and K. Kopka, Synlett, 2019, 1795–1798 CAS; (c) A. Makarem, M. Konrad, C. Liolios and K. Kopka, Synlett, 2018, 1239–1243 CAS; (d) K. D. Klika, C. Da Pieve, K. Kopka, G. Smith and A. Makarem, Org. Biomol. Chem., 2021, 19, 1722–1726 RSC; (e) C. Liolios, A. Shegani, I. Roupa, C. Kiritsis, A. Makarem, M. Paravatou-Petsotas, M. Pelecanou, P. Bouziotis, M. Papadopoulos, K. Kopka and I. Pirmettis, Bioorg. Chem., 2020, 100, 103855 CrossRef CAS PubMed.
- M. I. Tsionou, C. E. Knapp, C. A. Foley, C. R. Munteanu, A. Cakebread, C. Imberti, T. R. Eykyn, J. D. Young, B. M. Paterson, P. J. Blower and M. T. Ma, RSC Adv., 2017, 7, 49586–49599 RSC.
- M. Eder, O. Neels, M. Müller, U. Bauder-Wüst, Y. Remde, M. Schäfer, U. Hennrich, M. Eisenhut, A. Afshar-Oromieh, U. Haberkorn and K. Kopka, Pharmaceuticals, 2014, 7, 779–796 CrossRef CAS PubMed.
- J. Schuhmacher, G. Klivényi, W. E. Hull, R. Matys, H. Hauser, H. Kalthoff, W. H. Schmiegel, W. Maier-Borst and S. Matzku, Nucl. Med. Biol., 1992, 19, 809–824 CAS.
- H. Chen, X. Tang, X. Gong, D. Chen, A. Li, C. Sun, H. Lin and J. Gao, Chem. Commun., 2020, 56, 4106–4109 RSC.
- S. K. Larsen, B. G. Jenkins, N. G. Memon and R. B. Lauffer, Inorg. Chem., 1990, 29, 1147–1152 CrossRef CAS.
- A. D. Tinoco, C. D. Incarvito and A. M. Valentine, J. Am. Chem. Soc., 2007, 129, 3444–3454 CrossRef CAS PubMed.
- M. Zöller, J. Schuhmacher, J. Reed, W. Maier-Borst and S. Matzku, J. Nucl. Med., 1992, 33, 1366–1372 Search PubMed.
- Y. Ning, Y.-W. Liu, Z.-S. Yang, Y. Yao, L. Kang, J. L. Sessler and J.-L. Zhang, J. Am. Chem. Soc., 2020, 142, 6761–6768 CrossRef CAS PubMed.
- C. Kropf, S. Schaefer, C. Umbreit and K. Hegetschweiler, Eur Pat, EP3795665A1, 2021 Search PubMed.
- R. A. Yokel and H. B. Kostenbauder, Toxicol. Appl. Pharmacol., 1987, 91, 281–294 CrossRef CAS PubMed.
- O. Krokhin, H. Hoshino, O. Shpigun and T. Yotsuyanagi, J. Chromatogr. A, 2000, 895, 255–261 CrossRef CAS PubMed.
- K. S. Rajan, S. Mainer, N. L. Rajan and J. M. Davis, J. Inorg. Biochem., 1981, 14, 339–350 CrossRef CAS PubMed.
- F. Cleeren, J. Lecina, E. M. F. Billaud, M. Ahamed, A. Verbruggen and G. M. Bormans, Bioconjugate Chem., 2016, 27, 790–798 CrossRef CAS PubMed.
- X. Wang, M. de Guadalupe Jaraquemada-Peláez, C. Rodríguez-Rodríguez, Y. Cao, C. Buchwalder, N. Choudhary, U. Jermilova, C. F. Ramogida, K. Saatchi, U. O. Häfeli, B. O. Patrick and C. Orvig, J. Am. Chem. Soc., 2018, 140, 15487–15500 CrossRef CAS PubMed.
- S. Martin, R. Tönnesmann, I. Hierlmeier, S. Maus, F. Rosar, J. Ruf, J. P. Holland, S. Ezziddin and M. D. Bartholomä, J. Med. Chem., 2021, 64, 4960–4971 CrossRef CAS PubMed.
- N. Choudhary, H. Scheiber, J. Zhang, B. O. Patrick, M. de Guadalupe Jaraquemada-Peláez and C. Orvig, Inorg. Chem., 2021, 60, 12855–12869 CrossRef CAS PubMed.
- E. W. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260–290 RSC.
- A. D. Zubenko, A. A. Shchukina and O. A. Fedorova, Synthesis, 2020, 1087–1095 CAS.
- D. L. Pavia, G. M. Lampman, G. S. Kriz and J. R. Vyvyan, in Introduction to Spectroscopy, Cengage Learning, Belmont, CA, 2008 Search PubMed.
- M. Karplus, J. Chem. Phys., 1959, 30, 11–15 CrossRef CAS.
- M. Karplus, J. Am. Chem. Soc., 1963, 85, 2870–2871 CrossRef CAS.
- B. Coxon, ChemInform, 2010, 41, 15–18 Search PubMed.
- R. Wasylishen and T. Schaefer, Can. J. Chem., 1972, 50, 2710–2712 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Methodologies and experimental procedures, characterization data for the synthesized compounds, and copies of spectra. See DOI: https://doi.org/10.1039/d2dt01702d |
| ‡ Project leader. |
| This journal is © The Royal Society of Chemistry 2022 |