DOI:
10.1039/D2DT01318E
(Paper)
Dalton Trans., 2022,
51, 11040-11047
Excellent yield of a variety of silicon-boron radicals and their reactivity†
Received
27th April 2022
, Accepted 1st June 2022
First published on 25th June 2022
Introduction
Compounds with low-valence silicon are heavily investigated for their high reactivity but challenging to obtain due to their very short lifetimes at ambient temperature. For many decades, silylenes have been considered to be short-lived species, either in the gas phase, in solution, or when trapped in frozen matrices.1,2 The silicon atom possesses both a nucleophilic and an electrophilic reactive site. These can be modelled as a high energy-lying lone pair orbital and as a low energy lying vacant pπ orbital, respectively.3 The isolation of the first stable silicon N-heterocyclic silylene (NHSi), analogous to Arduengo's N-heterocyclic carbene, was reported in 1994.4,5 Such compounds display a two-coordinate Si(II) center with one empty p-orbital and one electron pair, indicative of their electron acceptor and donor characteristics. Another important compound with a low-valence silicon is the amidinate-supported6 silylene LSiR (L = PhC(NtBu)2; R = halide or organic group).7 In comparison with NHSi, the silicon atom in LSiR is N,N′ chelated by both nitrogen atoms of the anionic ligand. This type of silylene is comparable to the acyclic silylene and represents a promising candidate for small molecule activation with unique reactivity and reaction patterns. Indeed, such reactions with LSiR were well studied with alkynes,8,9 chalcogens10 ketones,11 CO,12 and P4.13 The formation of adducts with boron compounds is usually obtained when LSiR reacts with a boron species, for example BH3, 9-BBN, or B(C6F5)3.14 Surprisingly, the reactivity of the amidinate silylene (PhC(NtBu)2SiCl) with boron compounds to synthesize silicon-boron radicals remained nearly unexplored.15 It is noteworthy that N-heterocyclic carbenes (NHC), silylenes, and cyclic alkyl amino carbenes (cAAC) have been employed as effective bases to stabilize neutral boryl radicals.16,17 In a previous publication, we have demonstrated and exploited the strong σ-donor and π-acceptor properties of cyclic alkyl(amino) carbenes (cAACs) for the stabilization of radical cations and radical anions of p-block elements.18 Encouraged by this study, we focused on the synthesis of silicon-boron radicals with a variety of interesting substituents. Herein, we report the synthesis of high yield radicals and facile reduction of X2BTip in the presence of LSiR, yielding three novel radical species LSi(NMe2)–B(Br)Tip (1), LSi(NMe2)–B(I)Tip (2) LSi(tBu)–B(I)Tip (3) [L = PhC(NtBu)2]. The subsequent reactions of the obtained radicals with Br2 give the disubstituted bromine compound LSi(tBu)–B(Br)Tip (4), while the addition of pure selenium results in decomposition, yielding a selenium–silicon compound LSi(tBu)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Se (5).
Se (5).
Results and discussion
The synthetic strategy of 1–3 involves the one-pot reaction of dibromo/iodo(2,4,6-triisopropylphenyl)borane with amidinate-silylene LSiNMe2 and LSitBu (L = PhC(NtBu)2) and 1.0 equivalent of potassium graphite. Diethyl ether was added at −78 °C and the reaction mixture was stirred overnight. Upon filtration of the solution under an inert atmosphere, the colorful clear solution was concentrated and stored at −30 °C. After one week, dark red or orange crystals of LSi(NMe2)–B(Br)Tip (1), LSi(NMe2)–B(I)Tip (2) and LSi(tBu)–B(I)Tip (3) were obtained, respectively, in 90%, 94% and 95% yields (Scheme 1).
 |
| | Scheme 1 Synthetic route for the synthesis of compounds 1–3. | |
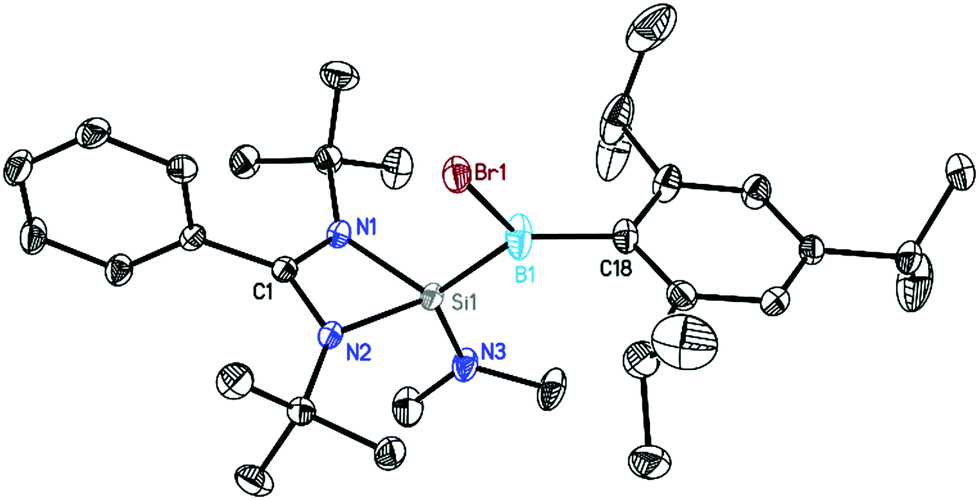
The crystals were subsequently analyzed by single-crystal X-ray diffraction (Fig. 1–3). Compounds 1–3 crystallized in the monoclinic space group P21/n with one molecule in the asymmetric unit. All three are similar in their overall geometry and feature a silicon–boron bond, as previously reported in analogous radical species.15a
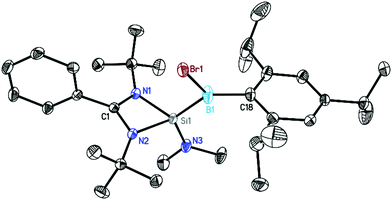 |
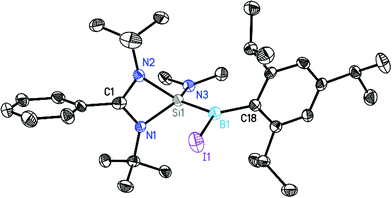
| | Fig. 1 Crystal structure of 1. The anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: N1–Si1 1.8475(12), N2–Si1 1.8407(13), N3–Si1 1.6946(13), Si1–B1 1.9292(19), B1–Br1 2.0247(19), B1–C18 1.590(2), N1–Si1–N2 70.86(6), N1–Si1–B1 118.41(7), B1–Si1–N3 119.56(7), Br1–B1–Si1 109.47(9). | |
 |
| | Fig. 2 Crystal structure of 2. The anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: N1–Si1 1.818(4), N2–Si1 1.877(5), N3–Si1 1.700(3), Si1–B1 1.930(5), B1–I1 2.221(5), B1–C18 1.573(6), N1–Si1–N2 70.66(16), N1–Si1–B1 119.9(2), B1–Si1–N3 119.2(2), I1–B1–Si1 114.6(2). | |
 |
| | Fig. 3 Crystal structure of 3. The anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: N1–Si1 1.852(3), N2–Si1 1.854(2), C1–Si1 1.906(2), Si1–B1 1.916(3), B1–I1 2.227(2), B1–C20 1.590(3), N1–Si1–N2 70.92(10), N1–Si1–B1 119.52(12), B1–Si1–C1 118.78(11), I1–B1–Si1 112.83(11). | |
The central silicon atom exhibits a distorted tetrahedral coordination environment due to the acute bite angle of the amidinate ligand. Additionally, the silicon atom is substituted by a NMe2 (1, 2) or tBu (3) group and by a B(X)Tip substituent (1: X = Br, 2, 3: X = I). It is noteworthy that, in agreement with the increase of ionic radii, the B1–Br1 distance (2.0247(19) Å) in 1 is shorter than the B1–I1 distance (2.221(5) Å) in the analogous complex 2. The boron atom shows an almost perfect trigonal planar environment with the sum of angles ranging between 359.69° and 359.98°. This should exclude the localization of the unpaired electron at the boron atom. The Si–B bond distances in 1–3 lie within the range of 1.916 Å to 1.930 Å which is on the shorter end of the reported Si–B single bonds (Fig. S22†) but very similar to that of the closely related radical compounds [LSi(tBu)–B(Br)Tip] (1.9136(14) Å)15a and [LSi(HMDS)–B(Br)Tip] (1.9641(19) Å).15a
The formation of radicals 1, 2 and 3 was further confirmed by electron paramagnetic resonance spectroscopy and LIFDI mass spectrometry. Compound 1 exhibits an EPR spectrum at g = 2.0083 with a clearly resolved boron coupling of 14.9 Gauss (11B, I = 3/2, 80.1% nat. abundance), signifying considerable spin density at B (Fig. 4). The iodine analogue 2 shows an unresolved EPR signal at 2.0028; the lack of resolution is attributed to slowed down mobility and increased spin orbit coupling involving the heavier element iodine. Compound 3 shows only partial EPR resolution (Fig. S1 and 2†). The LIFDI mass spectra in toluene solutions of 1, 2 and 3 show ions at m/z, 598.5, 516.9, and 657.3, respectively, corresponding to the formation of [LSi(NMe2)–B(Br)Tip], [LSi(NMe2)–B(I)Tip], and [LSi(tBu)–B(I)Tip] (Fig. S10–12†). Compounds 1 and 2 have a melting point in the range of 205–215 °C whereas 3 melts at 200–205 °C, as determined by differential scanning calorimetry. Moreover, NMR spectroscopy shows the absence of the resultant resonance, due to the radical formation. Quantum chemical calculations were carried out to explore the nature of the radical in compounds 1–3 at the M062X/def2-TZVPP//PBE/def2-TZVPP level of theory.19 The structural parameters in the optimized geometries showed good agreement with the experimental geometries (Fig. S23†). All three compounds feature a trigonal planar B centre coordinated to an N-amidinate silylene, a Tip group and a halogen atom X (X = Br in 1, I in 2 and 3). The molecular orbital (MO) analysis reveals that the singly occupied molecular orbital (SOMO) in compounds 1–3 is a π-type p-orbital largely localized on the B centre (Fig. 5a). The frontier MOs of compounds 2 and 3 are similar to those in 1 (Fig. S24†). Bond orbital analysis within the NBO framework shows 0.71 − 0.76 electron occupancy on the p-type orbital (99.7%-p character) on the B center.
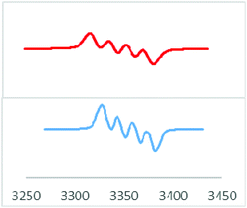 |
| | Fig. 4 EPR spectrum of 1 (bottom) with simulation (top) using a 11B coupling constant of 14.9 G. | |
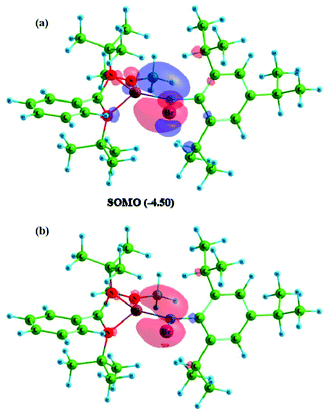 |
| | Fig. 5 Plot of (a) SOMO and (b) spin density of compound 1. Isosurface value for MO and spin density is 0.05 and 0.0045 respectively. The eigenvalue of the SOMO is given in parenthesis in eV. | |
The spin density plot (Fig. 5b) reaffirms the above observation. The natural spin density on B is 0.73 in 1, 0.75 in 2 and 0.70 in 3. The NHC and cAAC stabilized B-centred radical compounds showed spin density in the range of 0.41 − 0.50 at the B atom.20 Similar radical compounds LSi(L′)–B(Br)Tip (L′ = tBu (1); N(TMS)2 (2)) reported previously have 64 − 70% spin density on the B centre.15 This indicates that although the spin is largely localized on the B centre, a significant part of the spin density is delocalized onto the adjacent ligand framework through hyperconjugative interactions. The N-amidinate silylene and the Tip group act as radical sinks, stabilizing these B-centered radical compounds. This is supported by natural spin density values on the Si (0.14 − 0.21), N1 (0.02), and N2 (0.02) centres and Tip (0.03 − 0.04) group.
Second-order perturbation analysis indicates that the B radical centre is stabilized by hyperconjugative interactions with the Si–N and C–C skeleton of the N-amidinate ligand and Tip ring, respectively. The strength of the donation from the B radical centre to the Si–N σ*-orbitals is 28.13 kcal mol−1 for 1, 27.52 kcal mol−1 for 2 and 30.71 kcal mol−1 for 3. The extent of hyperconjugative donation from the B p-orbital to the C–C σ*-orbitals of the Tip ring is 13.42 kcal mol−1, 14.19 kcal mol−1 and 12.84 kcal mol−1 for 1–3 respectively. In all compounds, the strength of hyperconjugative donation towards the N-amidinate ligand is greater than that towards the Tip ring.
It is evident from the spin density analysis that the effect of halogen atom X (X = Br in 1, I in 2 and 3) on the radical density distribution is marginal. In contrast, the effect of the substituent R on the Si centre (R = NMe2 for 1 and 2, tBu for 3) is pronounced. This is because in compounds 1 and 2, the lone pair on the nitrogen atom of the NMe2 group is involved in hyperconjugative donation to the Si–N σ*-orbitals. Second-order perturbation analysis shows the strength of this donation to be 20.6 kcal mol−1 for 1 and 20.52 kcal mol−1 for 2. This hyperconjugative interaction competes with the hyperconjugative interaction from the B p-type radical orbital, whereas in compound 3 with tBu substitution, such an effect is absent. This results in more effective spin delocalization in compound 3 and in turn, a lower spin density value on B (0.70) in comparison with those of 1 and 2 (0.73 − 0.75). Topological analysis using the quantum theory of atoms in molecules indicates the polar covalent nature of the Si–B bond in compounds 1–3 (Fig. 6 and Fig. S27†).19 The low value of electron density (ρ(r) = 0.1213 − 0.1229 a.u) with negative values of Laplacian of electron density (∇2ρ(r) = −0.0495 to −0.0758 a.u) observed at the bond critical point (bcp) between the Si and B centres is characteristic of polar covalent interactions (Table 1 and Table S9†). This is also reflected in the respective positive and negative natural charges on the Si (1.79 − 1.86) and B (−0.29 to −0.33) centres (Table S7†). The ellipticity values (ε) in the range of 0.30 − 0.35 further support the back-bonding from the B centre to the Si centre in compounds 1–3. Furthermore, the Si–B radical was characterized by cyclic voltammetry (all potentials are given vs. Fc+/0). In dry and degassed acetonitrile, the Si–B radical undergoes an irreversible oxidation at Epa = 0.32 V, as well as a quasi-reversible reduction at Epc,1 = −1.34 V with the respective oxidation of the reverse scan at Epa,1 = −1.24 V, and an irreversible reduction at Epc,2 = −1.57 V (Fig. 7; for full CV, see the ESI, Fig. S28†).
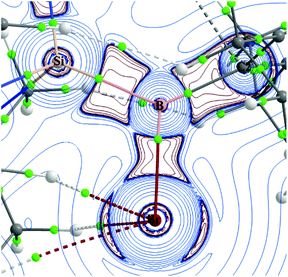 |
| | Fig. 6 Laplacian of electron density plotted in the Si–B–C2 plane of compound 1. Bond critical points (green dots), areas of charge concentration (red; ∇2ρ(r) < 0), charge depletion (blue) and bond paths are represented. | |
 |
| | Fig. 7 Cyclic voltammetry of the Si–B radical. | |
Table 1 Topological parameters of electron density at the bond critical points of selected bonds in compound 1. All values are in atomic unitsa
| |
Si–B |
B–Br |
B–C2 |
|
Electron density ρ(r), Laplacian of electron density ∇2ρ(r), energy density H(r), Lagrangian kinetic energy G(r), potential energy density V(r), and ellipticity ε.
|
|
ρ(r) |
0.1229 |
0.1000 |
0.1778 |
|
∇
2
ρ(r) |
−0.0758 |
−0.0757 |
−0.1841 |
|
H(r) |
0.1120 |
0.1040 |
0.2370 |
|
G(r) |
0.0738 |
0.0660 |
0.1445 |
|
V(r) |
−0.1666 |
−0.1508 |
−0.3350 |
|
ε
|
0.35 |
0.11 |
0.03 |
Our efforts began by exploring the reactivity of the Si–B radical with TEMPO and NO gas, but in both the reactions they showed a slow color change from red to light yellow at room temperature. However, we could not get clear NMR spectra and single-crystals for structural investigations. Furthermore the reactions of the Si–B radical with organic halides were studied but we could not observe any reactivity. Later on the Si–B radical resulted in oxidative addition products when we tried reactions with bromine and chalcogen (Se). Treatment of the silicon–boron radical with one equivalent of bromine in toluene at room temperature affords a yellow solution. The solution was further concentrated and stored at −30 °C in a freezer. After two weeks, yellow colored block-shaped crystals of dibrominated compound LSi(tBu)–B(Br2)Tip (4), (68% isolated yield) were obtained from a concentrated solution (Scheme 2). In the reaction of compound 4, we expected that the boron radical first generates the borenium cation (>B+) by oxidation of the Si–B radical followed by the addition of a bromine anion (Br−) to the borenium centre. The crystals of 4 were subsequently analyzed by single crystal X-ray diffraction (Fig. 8). Compound 4 crystallizes in the monoclinic space group P21/n with one complex molecule in the asymmetric unit. The tetrahedral environment of the boron atom is distorted by the steric hindrances induced by the tBu groups and the Tip ligand, resulting in angle variations between 96° (Br1–B1–Si1 angle) and 137° (C20–B1–Si1 angle). Compared to 1–3 and [LSi(tBu)–B(Br)Tip],15a the B–Si, B–C(Tip) and B–Br bond lengths are longer because of the fourfold silicon coordination in 4 compared to the threefold of the former.
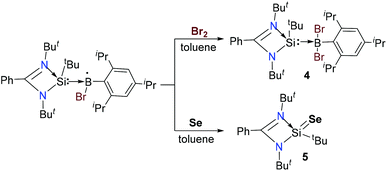 |
| | Scheme 2 Synthetic routes to 4 and 5. | |
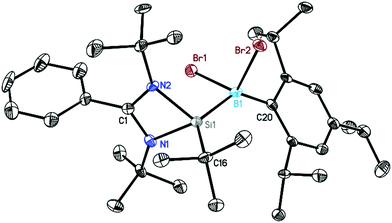 |
| | Fig. 8 Crystal structure of 4. The anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: N1–Si1 1.826(4), N2–Si1 1.847(3), Si1–B1 2.096(4), B1–Br1 2.106(4), B1–Br2 2.090(3), B1–C20 1.627(5), N1–Si1–N2 71.85(12), N1–Si1–B1 113.87(13), B1–Si1–C16 133.22(14), Br1–B1–Si1 96.11(15), Br2–B1–Si1 98.95(15), C20–B1–Si1 137.0(2). | |
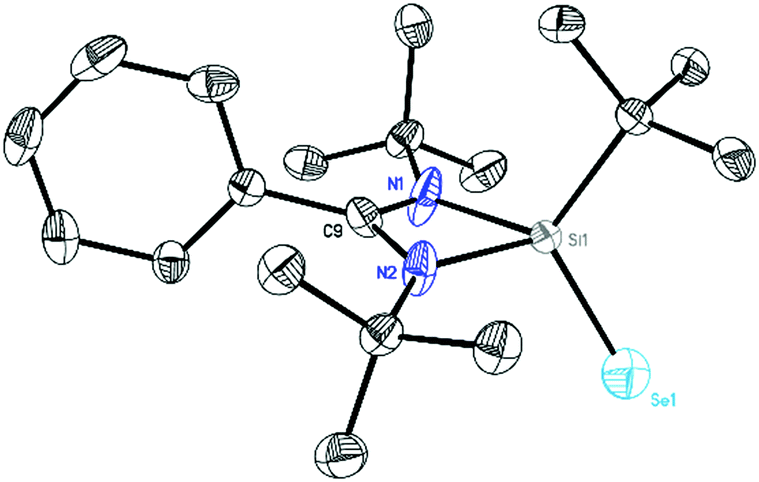
LSi(tBu)Se (5) was synthesized in 85% yield by reacting the Si–B radicals with one equivalent of selenium powder (Scheme 2). The color of the reaction mixture turned from red to light yellow. Product 5 was isolated as a colourless crystalline solid with good solubility in toluene. Subsequent analysis by single crystal X-ray diffraction reveals that the crystal contains two independent disordered molecules.
Compound 5 (Fig. 9) co-crystallizes in the monoclinic space group C2/c with the doubly protonated amidinate bromine salt and [L(H2)Si(tBu)Br] and a toluene molecule (see ESI Fig. S19–21†). The central silicon atom in 5 is chelated by the amidinate ligand and coordinated by a tBu group and a selenium atom. The Se1–Si1 bond is in the range of previously reported double bonds (2.1394(17) Å).21 The formation of compounds 4 and 5 was further confirmed by 1H, 13C, 11B and 29Si NMR spectroscopy. The 29Si NMR spectrum of 4 in C6D6 exhibits a resonance at δ −12.32 ppm for the Si(II) atoms, while compound 5 shows a signal in the 29Si NMR spectrum at 36.21 ppm, both in good agreement with silicon II and IV complexes (Fig. S6 and S9†).22
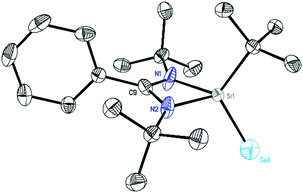 |
| | Fig. 9 Crystal structure of 5. The anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: N1–Si1 1.8990(15), N2–Si1 1.8367(15), Si1–Se1 2.1394(17), N1–Si1–N2 72.24(6), N1–Si1–Se1 114.65(8). | |
The 1H NMR spectra of 4 and 5 display sharp resonances for tBu (δ 1.21 and 1.13 ppm) and for phenyl protons (δ 6.87–7.27 ppm) each, which is fully consistent with their solid-state molecular structures established by single-crystal X-ray diffraction (Fig. S3 and S7†). The 11B NMR spectrum of 4 shows one resonance at δ −11.40 ppm which agrees with boron silicon adducts (Fig. S5†).23 The formation of 4 and 5 was further confirmed by LIFDI mass spectrometry. They display their molecular ion in the mass spectrum at m/z 671.4 and 397.7, corresponding to the formation of LSi(tBu)–B(Br2)Tip and LSi(tBu)Se, (Fig. S13 and 14†). 4 and 5 have melting points at 225–230 °C and 215–222 °C, respectively, as determined by differential scanning calorimetry.
Conclusions
In conclusion, we report amidinate-supported divalent silicon atoms that are capable of stabilizing silicon-boron radicals in high to excellent yields with different substituents. Furthermore we also showed the reactivity of these radicals with bromine and selenium which led to the dihalogenation of boron (4) and the oxidative addition of selenium to silicon (5). All radicals 1, 2 and 3 are highly stable at high temperatures in solution and towards air in the solid state. Quantum chemical calculations show that compounds 1–3 are primarily B-centred radical compounds that are stabilized via hyperconjugative interactions with the adjacent ligand framework. Compounds 4 and 5 are stable in solution and in the solid state under an inert atmosphere.
Experimental section
Materials and methods
Syntheses were carried out under an inert atmosphere of dinitrogen in oven-dried glassware using standard Schlenk techniques. All other manipulations were accomplished in a dinitrogen filled glove box. Solvents were purified by using an MBRAUN solvent purification system MB SPS-800. All chemicals were purchased from Aldrich and used without further purification. PhC(NtBu)2Si(tBu)–B(Br)Tip was prepared as reported in the literature.141H, 13C, and 29Si NMR spectra were recorded with a Bruker Avance DRX 500 spectrometer, using C6D6 as a solvent. Chemical shifts δ are given relative to SiMe4. EI–MS spectra were obtained using a Finnigan MAT 8230 spectrometer. Elemental analyses were performed at the Institut für Anorganische Chemie, Universität Göttingen. For elemental analysis, the compounds were kept under vacuum for six hours to remove the solvent molecules. Melting points were measured in a sealed glass tube on a Büchi B-540 melting point apparatus.
Synthesis
PhC(NtBu)2Si(NMe2)–B(Br)Tip (1).
To the mixture of LSiNMe2 (303 mg, 1 mmol), Br2BTip (376 mg, 1 mmol) and KC8 (135 mg, 1 mmol), 40 mL of diethyl ether was added at −78 °C, in a 100 mL round bottom flask. The reaction mixture was allowed to warm to room temperature slowly to give a bluish-purple solution of compound 1 after 12 hours of stirring. The solution was filtered and concentrated to 10 mL under a low vacuum. The filtered solution was stored at −30 °C in a freezer. After one week dark red-orange block-shaped X-ray quality crystals of 1 were obtained from the concentrated solution of diethyl ether (yield: 536 mg, 90%). MP: 205–210 °C to an orange liquid. Elemental analysis (%) calcd for C32H52BBrN3Si (596.32): C, 64.32; H, 8.77; N, 7.03; found: C, 63.90; H, 9.02; N, 7.54. MS (LIFDI, toluene): m/z = 596.5 ([M]+).
PhC(NtBu)2Si(NMe2)–B(I)Tip (2).
To the mixture of LSiNMe2 (303 mg, 1 mmol), I2BTip (482 mg, 1 mmol) and KC8 (135 mg, 1 mmol) 40 mL of diethyl ether was added at −78 °C, in a 100 mL round bottom flask. The reaction mixture was allowed to warm to room temperature slowly to give a bluish-purple solution of compound 2 after 12 hours of stirring. The solution was filtered and concentrated to 10 mL under low vacuum. The filtered solution was stored at −30 °C in a freezer. After one week dark red-orange block-shaped X-ray quality crystals of 2 were obtained from the concentrated solution of diethyl ether (yield: 630 mg, 98%). MP: 205–210 °C to an orange liquid. Elemental analysis (%) calcd for C32H52BBrN3Si (644.31): C, 69.63; H, 8.13; N, 6.51; Found: C, 69.92; H, 8.72; N, 6.14. MS (LIFDI, toluene): m/z = 516.9 ([M − I]+).
PhC(NtBu)2Si(tBu)–B(I)Tip (3).
A mixture of LSitBu (318 mg, 1 mmol), I2BTip (482 mg, 1 mmol) and KC8 (135 mg, 1 mmol) was placed in a 100 mL round bottom flask, and 40 mL of diethyl ether was added at −78 °C. The reaction mixture was allowed to warm to room temperature slowly to give a reddish-purple solution of compound 3 after 12 hours of stirring. The solution was filtered and concentrated to 10 mL under a high vacuum. The filtered solution was stored at −30 °C in a freezer. After one week dark red-orange block-shaped X-ray quality crystals of 3 were obtained from the concentrated solution of diethyl ether (yield: 624 mg, 95%). MP: 210–215 °C to an orange liquid. Elemental analysis (%) calcd for C34H55BIN2Si (657.33): C, 62.10; H, 8.43; N, 4.26; found: C, 62.45; H, 8.92; N, 4.62. MS (LIFDI, toluene): m/z = 657.4 ([M]+).
PhC(NtBu)2Si(tBu)–B(Br2)Tip (4).
To the toluene solution of PhC(NtBu)2Si(tBu)–B(Br)Tip (300 mg, 0.5 mmol), in a 50 mL round bottom flask, the Br2 (80 mg, 0.5 mmol) solution in toluene was added dropwise at a low temperature. The reaction mixture was allowed to stir at room temperature to give a yellow-colored solution of compound 4 after 12 hours. The solution was evaporated under a high vacuum and extracted in diethyl ether and concentrated to 5 mL under a low vacuum. The filtered solution was stored at −30 °C in a freezer. After two weeks dark yellow block-shaped X-ray quality crystals of 4 were obtained from the concentrated solution of diethyl ether (yield: 234 mg, 68%). MP: 225–230 °C to a colorless liquid. 1H NMR (500 MHz, C6D6, 298 K, ppm): δ = 1.21 (s, 18 H, CMe3), 1.33 (d, 6 H, CHMe2, J = 10 Hz), 1.48 (d, 6 H, CHMe2, J = 15 Hz), 1.53 (s, 9 H, CMe3), 1.70 (d, 6 H, CHMe2, J = 10 Hz), 2.97 (sep, 1 H, CHMe2), 3.47 (s, br, 2 H, CHMe2) 6.87–6.90 (m, 4 H, ArH), 6.94–6.96 (m, 1 H, ArH), 7.25–7.27 (m, 2 H, ArH). 13C NMR (126 MHz, C6D6, 298 K, ppm): δ = 23.96, 24.61, 26.08, 28.36, 31.97, 34.30, 34.73, 57.15, 66.24, 124.22, 128.71, 129.04, 129.85, 130.09, 130.11, 133.52, 151.35, 152.66, 183.52. 11B NMR (160 MHz, C6D6, 298 K, ppm): δ = 11.40. 29Si NMR (99 MHz, C6D6, 298 K, ppm): δ = −12.32. Elemental analysis (%) calcd for C34H55BBr2N2Si (688.26): C, 59.14; H, 8.03; N, 4.06; found: C, 59.62; H, 8.60; N, 4.42. MS (LIFDI, toluene): m/z = 672.4 ([M − CH4]+).
PhC(NtBu)2Si(tBu)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) Se (5).
Toluene solution was added to the mixture of PhC(NtBu)2Si(tBu)–B(Br)Tip (300 mg, 0.5 mmol) and Se powder (40 mg, 0.5 mmol) at room temperature in a 50 mL round bottom flask. The reaction mixture was allowed to stir overnight to give a yellow-colored solution of compound 5 after 20 h. The solution was filtered under a N2 atmosphere and then the filtered solution was stored at room temperature. After one-month colorless block-shaped X-ray quality crystals of 5 were obtained from the concentrated solution of toluene (yield: 170 mg, 86%). MP: 225–230 °C to a colorless liquid. 1H NMR (500 MHz, C6D6, 298 K, ppm): δ = 1.13 (s, 18 H, CMe3), 1.39 (s, 9 H, CMe3), 6.87–6.92 (m, 3 H, ArH), 6.97–6.99 (m, 1 H, ArH), 7.03–7.04 (m, 1 H, ArH). 13C NMR (126 MHz, C6D6, 298 K, ppm): δ = 24.02, 28.30, 31.84, 55.17, 128.06, 128.11, 128.16, 128.75, 130.48, 131.28, 175.28. 29Si NMR (99 MHz, C6D6, 298 K, ppm): δ = 36.21, no signals could be observed in 77Se NMR; elemental analysis (%) calcd for C19H32N2SeSi (396.15): C, 57.70; H, 8.16; N, 7.08; found: C, 58.07; H, 8.67; N, 7.53. MS (LIFDI, toluene): m/z = 397.7 ([M]+).
Se (5).
Toluene solution was added to the mixture of PhC(NtBu)2Si(tBu)–B(Br)Tip (300 mg, 0.5 mmol) and Se powder (40 mg, 0.5 mmol) at room temperature in a 50 mL round bottom flask. The reaction mixture was allowed to stir overnight to give a yellow-colored solution of compound 5 after 20 h. The solution was filtered under a N2 atmosphere and then the filtered solution was stored at room temperature. After one-month colorless block-shaped X-ray quality crystals of 5 were obtained from the concentrated solution of toluene (yield: 170 mg, 86%). MP: 225–230 °C to a colorless liquid. 1H NMR (500 MHz, C6D6, 298 K, ppm): δ = 1.13 (s, 18 H, CMe3), 1.39 (s, 9 H, CMe3), 6.87–6.92 (m, 3 H, ArH), 6.97–6.99 (m, 1 H, ArH), 7.03–7.04 (m, 1 H, ArH). 13C NMR (126 MHz, C6D6, 298 K, ppm): δ = 24.02, 28.30, 31.84, 55.17, 128.06, 128.11, 128.16, 128.75, 130.48, 131.28, 175.28. 29Si NMR (99 MHz, C6D6, 298 K, ppm): δ = 36.21, no signals could be observed in 77Se NMR; elemental analysis (%) calcd for C19H32N2SeSi (396.15): C, 57.70; H, 8.16; N, 7.08; found: C, 58.07; H, 8.67; N, 7.53. MS (LIFDI, toluene): m/z = 397.7 ([M]+).
Crystal structure determination
Crystals suitable for single crystal X-ray diffraction were mounted at a low temperature in inert oil under a nitrogen atmosphere by applying an X-Temp 2 device.24 Diffraction data were collected at 100 K with a Bruker D8 three circle diffractometer equipped with an incoatec Mo Microsource (1, 3–5) or a Bruker Mo rotating anode (2) with mirror optics (MoKα radiation, λ = 0.71073 Å). The data were integrated with SAINT25 and an empirical absorption correction was applied with SADABS26 (1–2, 4–5) or TWINABS (3).27 The structures were solved by SHELXT28 and refined on F2 using SHELXL29 in the graphical user interface ShelXle.30
Crystal data for 1 at 100(2) K
C32H52BBrN3Si, Mr = 597.57 g mol−1, 0.359 × 0.337 × 0.201 mm, monoclinic, P21/n, a = 11.102(2) Å, b = 14.901(2) Å, c = 21.157(3) Å, β = 102.55(2), V = 3416.4(9) Å3, Z = 4, μ (Mo Kα) = 1.262 mm−1, θmax = 28.413°, 133![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 144 reflections measured, 8563 independent (Rint = 0.0498), R1 = 0.0325 [I > 2σ(I)], wR2 = 0.0830 (all data), res. density peaks: 0.402 to −0.335 eÅ3, CCDC 2113990.†
144 reflections measured, 8563 independent (Rint = 0.0498), R1 = 0.0325 [I > 2σ(I)], wR2 = 0.0830 (all data), res. density peaks: 0.402 to −0.335 eÅ3, CCDC 2113990.†
Crystal data for 2 at 100(2) K
C32H52BIN3Si, Mr = 644.56 g mol−1, 0.426 × 0.224 × 0.122 mm, monoclinic, P21/n, a = 12.626(2) Å, b = 18.594(3) Å, c = 14.454(2) Å, β = 90.75(2), V = 3393.0(9) Å3, Z = 4, μ (Mo Kα) = 1.003 mm−1, θmax = 25.171°, 126965 reflections measured, 6038 independent (Rint = 0.0597), R1 = 0.0447 [I > 2σ(I)], wR2 = 0.0938 (all data), res. density peaks: 0.656 to −1.317 eÅ3, CCDC 2113991.†
Crystal data for 3 at 100(2) K
C34H55BIN2Si, Mr = 657.60 g mol−1, 0.255 × 0.146 × 0.125 mm, monoclinic, P21/n, a = 12.623(2) Å, b = 19.291(3) Å, c = 14.451(2) Å, β = 92.06(2), V = 3516.7(9) Å3, Z = 4, μ (Mo Kα) = 0.968 mm−1, θmax = 29.284°, 70008 reflections measured, 9573 independent (Rint = 0.0727), R1 = 0.0392 [I > 2σ(I)], wR2 = 0.0733 (all data), res. density peaks: 0.490 to −0.832 eÅ3, CCDC 2113992.†
Crystal data for 4 at 100(2) K
C34H55BBr2N2Si, Mr = 690.52 g mol−1, 0.276 × 0.231 × 0.127 mm, monoclinic, P21/n, a = 16.079(2) Å, b = 12.497(2) Å, c = 17.886(3) Å, β = 106.12(2), V = 3452.7(11) Å3, Z = 4, μ (Mo Kα) = 2.408 mm−1, θmax = 27.614°, 147210 reflections measured, 7978 independent (Rint = 0.0832), R1 = 0.0448 [I > 2σ(I)], wR2 = 0.1226 (all data), res. density peaks: 2.088 to −1.235 eÅ3, CCDC 2113993.†
Crystal data for 5 at 100(2) K
C41H65BrSeSi, Mr = 800.93 g mol−1, 0.350 × 0.236 × 0.122 mm, monoclinic, C2/c, a = 30.532(3) Å, b = 8.760(2) Å, c = 18.768(2) Å, β = 122.67(2), V = 4225.2(14) Å3, Z = 4, μ (Mo Kα) = 1.894 mm−1, θmax = 28.421°, 64030 reflections measured, 5315 independent (Rint = 0.0409), R1 = 0.0234 [I > 2σ(I)], wR2 = 0.0544 (all data), res. density peaks: 0.354 to −0.331 eÅ3, CCDC 2113994.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
D. S. acknowledges partial funding from the Danish National Research Foundation (DNRF93) Center of Materials Crystallography. C. M. L. thanks the Fonds der Chemischen Industrie for financial support for her Ph.D. studies. We thank Dr A. C. Stückl for measuring the EPR spectrum. Parvathy thanks the NITC for the research fellowship and CCMS for computational facility. Dr Parameswaran thanks the DST-SERB for research funding.
Notes and references
-
P. P. Gaspar and R. West, ed. Z. Rappoport and Y. Apeloig, John Wiley and Sons, The Chemistry of Organic Silicon Compounds, Chichester, 1998, vol. 2, Part 3, pp. 2463–2567.
-
(a) M. Haaf, T. A. Schmedake and R. West, Acc. Chem. Res., 2000, 33, 704–714 CrossRef CAS PubMed;
(b) B. Gehrhus and M. F. Lappert, J. Organomet. Chem., 2001, 618, 209–223 CrossRef;
(c) S. Yao, Y. Xiong and M. Driess, Organometallics, 2011, 30, 1748–1767 CrossRef CAS;
(d) M. Kira, Chem. Commun., 2010, 46, 2893–2903 RSC;
(e) R. Azhakar, R. S. Ghadwal, H. W. Roesky, H. Wolf and D. Stalke, Chem. Commun., 2012, 48, 4561–4563 RSC;
(f) R. S. Ghadwal, R. Azhakar and H. W. Roesky, Acc. Chem. Res., 2013, 46, 444–456 CrossRef CAS PubMed.
-
(a) R.-E. Li, J.-H. Sheu and M.-D. Su, Inorg. Chem., 2007, 46, 9245–9253 CrossRef CAS PubMed;
(b) P. W. Percival, J.-C. Brodovitch, M. Mozafari, A. Mitra, R. West, R. S. Ghadwal, R. Azhakar and H. W. Roesky, Chem. – Eur. J., 2011, 17, 11970–11973 CrossRef CAS PubMed;
(c) S. Inoue, J. D. Epping, E. Irran and M. Driess, J. Am. Chem. Soc., 2011, 133, 8514–8517 CrossRef CAS PubMed;
(d) S. Inoue, W. Wang, C. Präsang, M. Asay, E. Irran and M. Driess, J. Am. Chem. Soc., 2011, 133, 2868–2871 CrossRef CAS PubMed;
(e) R. S. Ghadwal, R. Azhakar, H. W. Roesky, K. Pröpper, B. Dittrich, C. Goedecke and G. Frenking, Chem. Commun., 2012, 48, 8186–8188 RSC.
- P. Jutzi, D. Kanne and C. Krüger, Angew. Chem., Int. Ed. Engl., 1986, 25, 164 CrossRef.
- M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belyakov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 1994, 116, 2691–2692 CrossRef CAS.
- D. Stalke, M. Wedler and F. T. Edelmann, J. Organomet. Chem., 1992, 431, C1–C5 CrossRef CAS.
-
(a) S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2010, 132, 1123–1126 CrossRef CAS PubMed.
-
(a) R. Azhakar, R. S. Ghadwal, H. W. Roesky, H. Wolf and D. Stalke, Organometallics, 2012, 31, 4588–4592 CrossRef CAS;
(b) R. Azhakar, R. S. Ghadwal, H. W. Roesky, H. Wolf and D. Stalke, Chem. Commun., 2012, 48, 4561–4563 RSC.
- S. S. Sen, H. W. Roesky, K. Meindl, D. Stern, J. Henn, A. C. Stückl and D. Stalke, Chem. Commun., 2010, 46, 5873–5875 RSC.
- N. Parvin, S. Pal, S. Khan, S. Das, S. K. Pati and H. W. Roesky, Inorg. Chem., 2017, 56, 1706–1712 CrossRef CAS PubMed.
- R. S. Ghadwal, S. S. Sen, H. W. Roesky, M. Granitzka, D. Kratzert, S. Merkel and D. Stalke, Angew. Chem.,
Int. Ed., 2010, 49, 3952–3955 CrossRef CAS PubMed.
- Y. Wang, A. Kostenko, T. J. Hadlington, M.-T. Luecke, S. Yao and M. Driess, J. Am. Chem. Soc., 2019, 141, 626–634 CrossRef CAS PubMed.
- S. S. Sen, S. Khan, H. W. Roesky, D. Kratzert, K. Meindl, J. Henn, D. Stalke, J.-P. Demers and A. Lange, Angew. Chem., Int. Ed., 2011, 50, 2322–2325 CrossRef CAS PubMed.
-
(a) A. Jana, D. Leusser, I. Objartel, H. W. Roesky and D. Stalke, Dalton Trans., 2011, 40, 5458–5463 RSC;
(b) A. Jana, R. Azhakar, S. P. Sarish, P. P. Samuel, H. W. Roesky, C. Schulzke and D. Koley, Eur. J. Inorg. Chem., 2011, 2011, 5006–5013 CrossRef CAS.
-
(a) M. Nazish, C. M. Legendre, S. K. Sarkar, J. Lücken, D. J. Goffitzer, M. Diefenbach, B. Schwederski, R. Herbst-Irmer, D. Stalke, M. C. Holthausen, W. Kaim and H. W. Roesky, Inorg. Chem., 2021, 60, 10100–10104 CrossRef CAS PubMed.
-
(a) S. Ueng, A. Solovyev, X. Yuan, S. J. Geib, L. Fensterbank, E. Lacôte, M. Malacria, M. Newcomb, J. C. Walton and D. P. Curran, J. Am. Chem. Soc., 2009, 131, 11256–11262 CrossRef CAS PubMed;
(b) S. Ueng, M. M. Brahmi, E. Derat, L. Fensterbank, E. Lacôte, M. Malacria and D. P. Curran, J. Am. Chem. Soc., 2008, 130, 10082–10083 CrossRef CAS PubMed;
(c) P. Bissinger, H. Braunschweig, A. Damme, I. Krummenacher, A. K. Phukan, K. Radacki and S. Sugawara, Angew. Chem., Int. Ed., 2014, 53, 7360–7363 CrossRef CAS PubMed;
(d) F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., 2014, 126, 13375–13379 CrossRef.
- For selected reviews, see:
(a) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC;
(b) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed;
(c) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed;
(d) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed;
(e) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed;
(f) M. H. Wang and K. A. Scheidt, Angew. Chem., Int. Ed., 2016, 55, 14912–14922 CrossRef CAS PubMed;
(g) J. P. Moerdyk, D. Schilter and C. W. Bielawski, Acc. Chem. Res., 2016, 49, 1458–1468 CrossRef CAS PubMed;
(h) C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020–3030 RSC.
-
(a) C. Mohapatra, S. Kundu, A. N. Paesch, R. Herbst-Irmer, D. Stalke, D. M. Andrada, G. Frenking and H. W. Roesky, J. Am. Chem. Soc., 2016, 138, 10429–10432 CrossRef CAS PubMed;
(b) S. Roy, K. C. Mondal and H. W. Roesky, Acc. Chem. Res., 2016, 49, 357–359 CrossRef CAS PubMed;
(c) B. Li, S. Kundu, A. C. Stückl, H. Zhu, H. Keil, R. Herbst-Irmer, D. Stalke, B. Schwederski, W. Kaim, D. M. Andrada, G. Frenking and H. W. Roesky, Angew. Chem., Int. Ed., 2017, 56, 397–400 CrossRef CAS PubMed;
(d) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed;
(e) S. Sinhababu, S. Kundu, A. N. Paesch, R. Herbst-Irmer, D. Stalke, I. Fernández, G. Frenking, A. C. Stückl, B. Schwederski, W. Kaim and H. W. Roesky, Chem. – Eur. J., 2018, 24, 1264–1268 CrossRef CAS PubMed;
(f) K. C. Mondal, H. W. Roesky, B. Dittrich, N. Holzmann, M. Hermann, G. Frenking and A. Meents, J. Am. Chem. Soc., 2013, 135, 15990–15993 CrossRef CAS PubMed;
(g) M. M. Siddiqui, S. K. Sarkar, M. Nazish, M. Morganti, C. Köhler, J. Cai, L. Zhao, R. Herbst-Irmer, D. Stalke, G. Frenking and H. W. Roesky, J. Am. Chem. Soc., 2021, 143, 1301–1306 CrossRef CAS PubMed.
- See ESI for computational details.†.
-
(a) R. Kinjo, B. Donnadieu, M. C. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610–613 CrossRef CAS PubMed;
(b) M. F. S. Valverde, P. Schweyen, D. Gisinger, T. Bannenberg, M. Freytag, C. Kleeberg and M. Tamm, Angew. Chem., Int. Ed., 2017, 56, 1135–1140 CrossRef PubMed;
(c) R. Bertermann, H. Braunschweig, R. D. Dewhurst, D. C. C. Hörl, D. C. T. Kramer and I. Krummenacher, Angew. Chem., Int. Ed., 2014, 53, 5453–5457 CrossRef CAS PubMed.
-
(a) M. Ghosh, P. Panwaria, S. Tothadi, A. Das and S. Khan, Inorg. Chem., 2020, 59, 17811–17821 CrossRef CAS PubMed;
(b) M. Nazish, M. M. Siddiqui, S. K. Sarkar, A. Münch, C. M. Legendre, R. Herbst-Irmer, D. Stalke and H. W. Roesky, Chem. – Eur. J., 2021, 27, 1744–1752 CrossRef CAS PubMed;
(c) S.-H. Zhang, H.-X. Yeong and C.-W. So, Chem. – Eur. J., 2011, 17, 3490–3499 CrossRef CAS PubMed.
- S. Sinhababu, M. M. Siddiqui, S. K. Sarkar, A. Münch, R. Herbst-Irmer, A. George, P. Parameswaran, D. Stalke and H. W. Roesky, Chem. – Eur. J., 2019, 25, 11422–11426 CrossRef CAS PubMed.
- J. Li, Y. Liu, S. Kundu, H. Keil, H. Zhu, R. Herbst-Irmer, D. Stalke and H. W. Roesky, Inorg. Chem., 2020, 59, 7910–7914 CrossRef CAS PubMed.
-
(a) T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615–619 CrossRef;
(b) D. Stalke, Chem. Soc. Rev., 1998, 27, 171–178 RSC.
- Bruker AXS Inc., Bruker Apex CCD, SAINT v8.37A, Bruker AXS Inst. Inc., WI, USA, Madison, 2017 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- M. Sevvana, M. Ruf, I. Usón, G. M. Sheldrick and R. Herbst-Irmer, Acta Crystallogr., Sect. D: Struct. Biol., 2019, 75, 1040–1050 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Parameswaran
Parvathy
a,
Parameswaran
Parvathy