
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fundamental workings of chemical substitution at the A-site of perovskite oxides— a 207Pb NMR study of Ba-substituted PbZrO3†
Sonja
Egert
 a,
Jurij
Koruza
b,
Hergen
Breitzke
a,
Changhao
Zhao
c,
Barbara
Malič
d,
Gerd
Buntkowsky
*a and
Pedro B.
Groszewicz
*e
a,
Jurij
Koruza
b,
Hergen
Breitzke
a,
Changhao
Zhao
c,
Barbara
Malič
d,
Gerd
Buntkowsky
*a and
Pedro B.
Groszewicz
*e
aEduard Zintl Institute for Inorganic and Physical Chemistry, Technical University of Darmstadt, Darmstadt 64287, Germany. E-mail: gerd.buntkowsky@chemie.tu-darmstadt.de
bInstitute for Chemistry and Technology of Materials, Graz University of Technology, Graz 8010, Austria
cDepartment of Materials and Earth Sciences, Nonmetallic Inorganic Materials, Technical University of Darmstadt, Darmstadt 64287, Germany
dElectronic Ceramics Department, Jožef Stefan Institute, Ljubljana, Slovenia
eDepartment of Radiation Science and Technology, Delft University of Technology, Delft 2629JB, Netherlands. E-mail: p.groszewicz@tudelft.nl
First published on 9th November 2022
Abstract
Lead zirconate (PbZrO3, PZ) is a prototype antiferroelectric (AFE) oxide from which state-of-the-art energy storage materials are derived by chemical substitutions. A thorough understanding of the structure–property relationships of PZ-based materials is essential for both performance improvement and the design of more environmentally friendly replacements. (Pb1−xBax)ZrO3 (PBZ) can serve as a model system for studying the effect of A-site substitution in the perovskite lattice, with barium destabilizing the AFE state. Here, the two-dimensional 207Pb solid-state NMR spectra of PZ and PBZ were recorded to analyze the local structural role of barium substitution. At low substitution levels, 207Pb NMR spectroscopy reveals the presence of Pb–O bond length disorder. Upon crossing the threshold value of x for the macroscopic phase transition into a ferroelectric (FE) state, the barium cations cause local-scale lattice expansions in their vicinity, resulting in the collapse of two lead lattice sites into one. The stabilization of the larger volume site coincides with the favoring of larger lead displacements. We also observed more covalent bonding environments which may originate from the lower polarizability of the barium cations, facilitating the formation of stronger Pb–O bonds in their vicinity. From the local structural point of view, we propose that the substitution-induced AFE → FE phase transition is therefore related to an increasing correlation of larger lead displacements in larger oxygen cavities as the barium content increases. Our results also highlight 207Pb NMR spectroscopy as a valuable method for the characterization of the structure–property relationships of PbZrO3-based AFE and FE oxides.
Introduction
Antiferroelectric (AFE) oxides constitute a promising class of functional materials suitable for energy-storage applications1 or ferroic cooling.2,3 Among this class, compositions based on lead zirconate (PbZrO3, PZ) are considered to be state-of-the-art compositions. PZ was the first material for which antiferroelectricity was described4–6 and is arguably the best-studied AFE perovskite.7,8 It exhibits an orthorhombic symmetry with the space group Pbam below the Curie temperature TC = 230 °C.9,10 Antiparallel displacements of lead cations,11,12 as well as long-range ordered a−a−c0 rotations of oxygen octahedra,8,13 are characteristic of its room temperature phase. Despite the long history of PZ, its structure and properties are far from being fully understood, with previous publications highlighting the complexity of the phase transition to the ferroelectric (FE) state.14–17 An intermediate phase4 is observable only under specific conditions.18 In addition, the question of disordered lead displacements even in the ground state has been raised.19For practical applications of bulk ceramic AFEs, the AFE → FE phase transition must be both inducible by an electric field and reversible. While PZ is oftentimes regarded as a prototype AFE material, its field-induced phase transition is indeed only observable for single crystals20 or at elevated temperatures.5 This is because under ambient conditions the forward-switching field EAF necessary to induce the transition typically exceeds the breakdown field of the ceramic.1,10 Since the 1950s, efforts have been made to lower EAF by chemical substitution at both the A- and B-sites of the perovskite lattice.10,21
The effects of substituting Pb2+ with larger isovalent Ba2+ ions in (Pb1−xBax)ZrO3 (PBZ) solid solutions have previously been reviewed in detail.8,21 While EAF is indeed lowered,6,22 the AFE phase is destabilized at the same time, causing the appearance of an intermediate, rhombohedral FE phase with a polar 〈111〉 axis at elevated temperatures.6,22,23 The field-induced transition between the two phases is then accompanied by an increase in lattice volume.24 It has been proposed that at x > 0.175, the FE phase is stabilized at room temperature,25,26 and relaxor behavior has been reported for x > 0.3.25,27,28 The destabilizing effect of barium has been ascribed to the larger ions inducing parallel displacements in the structure, thus increasing the symmetry and driving the transition from AFE to FE.22 It was also suggested that Ba2+ ions elongate the Zr–O distances in their local surroundings, resulting in an instability of the Zr4+ ions and the formation of permanent electric dipoles.25 In addition, evidence of partial disorder of the Pb2+ displacements was found in PBZ above 130 K.25
Environmental concerns about PZ-based functional ceramics have motivated the search for lead-free replacements.29 A thorough understanding of the structure–property relationships could contribute to smart material design, but established compositions such as (Pb,La)(Zr,Sn,Ti)O3 are complex. The study of PBZ as a simpler model system in comparison with the parent structure of PZ can give valuable insights into the isolated effect of A-site modification under the influence of a destabilizing ion. In a similar fashion, the structures of pure30 and modified31 NaNbO3 are of interest for the development of functional, lead-free ceramics.
With solid-state nuclear magnetic resonance (NMR) spectroscopy, structural information on a local scale can be obtained, including, for example, bond lengths or the distortion of local environments.31,32 This is valuable especially for disordered materials such as solid solutions and chemically substituted oxides. Due to the sensitivity of NMR spectroscopy to the local structure, in particular, this technique is able to provide insights into the immediate surroundings of cations in the lattice under the influence of chemical substitution, which can be supplemented with XRD and electrical measurements in order to include the average lattice structure and the macroscopic properties in a more complete picture of the material. While diffraction methods can only give averaged information in this case, NMR spectroscopy often provides insights into the presence of different local environments or parameter distributions.
Due to its high chemical shift (CS) dispersion, 207Pb NMR is specifically sensitive to changes in the chemical environment of lead cations. An overview of 207Pb NMR studies can be found in the review by Dybowski and Neue.38 As the CS interaction is typically the dominant influence in 207Pb spectra, empirical correlations between the structural and CS parameters are established for a series of lead-containing compounds.34–37,40 This makes it a suitable probe for the local environment of Pb and the effects of neighboring cation substituents on it, which play an essential role in the functional properties of PZ-based materials. Accessibility of this information has previously been demonstrated for PbZrO3-based perovskites.33–36 One of the challenges of the method lies in the high electron number of lead, which accounts for large polarizabilities and causes even small deviations of the electron cloud from spherical symmetry to result in large chemical shift anisotropies (CSA).37 Hence, broad line shapes or complicated spinning sideband (SSB) patterns under magic angle spinning (MAS) are characteristic aspects.38 Furthermore, the sensitivity of the chemical shift to the probe temperature is to be expected,39 and the spin–lattice relaxation times for known lead perovskite oxides have been found to vary in a broad range between 1 s and 160 s.35
While the effects of barium substitution on the global structure and phase transition temperatures of PbZrO3 are reported in previous literature, rationalization of the chemical and structural reasons for this effect remained elusive. This fact has motivated our current search for a local structural origin of the changes brought about by barium, particularly in relation to the chemistry of this substituent in perovskite oxides. In this work, we analyzed the 207Pb NMR spectra of pure PZ, (Pb0.94Ba0.06)ZrO3 (PBZ06), and (Pb0.88Ba0.12)ZrO3 (PBZ12) in order to determine how the local structure of Pb2+ responds to the modification with Ba2+. These experiments are unique in that they reveal considerable local structural disorder upon chemical substitution, which is interpreted in terms of the distribution of the shortest Pb–O bond lengths. Furthermore, our results indicate an increased Pb–O bond covalency in the FE structure, attributed to the lower polarizability of barium cations, thus providing experimental evidence for the mechanism which leads to the AFE destabilization caused by Ba2+. The large CSA prevented the determination of NMR parameters at moderate MAS rates, which are necessary for spin sintered ceramic sample pieces. This experimental challenge has been overcome by employing the 2D-PASS (Two-Dimensional Phase-Adjusted Spinning Sidebands) method.41 Here, we used it to separate overlapping SSB patterns from different lead sites and to obtain purely isotropic spectra at 8 kHz MAS. In addition, the CSA patterns of two different Pb2+ sites were extracted.
Experimental
PbZrO3 (PZ), Pb0.94Ba0.06ZrO3 (PBZ06), and Pb0.88Ba0.12ZrO3 (PBZ12) were prepared by solid state synthesis. The starting powders PbO (Sigma, 99.9% purity), ZrO2 (TZ-0, Tosoh), and BaCO3 (Alfa, 99.8% purity) were mixed in a stoichiometric ratio using a planetary mill and calcined twice at 850 °C for 2 h with intermediate milling. The synthesized powders were pressed into pellets using uniaxial and isostatic pressure and subsequently sintered at 1250 °C for 2 h. The sintered samples were ground, annealed, and for electrical measurements electroded with gold. ε(T) and polarization–electric field hysteresis loops were determined using an HP 4192A impedance analyzer and an AixACCT TF2000 ferroelectric measurement setup, respectively. Before XRD analysis, the ground samples were annealed at 400 °C for 2 h. X-ray powder diffractograms were then recorded by using a Bruker D8 Advance diffractometer with CuKα radiation. The analysis of the full patterns was performed using the Jana2006 package.42 The 207Pb solid-state NMR spectra were recorded using a Bruker Avance III spectrometer operating with a 7.1 T magnet. The sintered ceramic pellets were cut to dimensions of approximately 1.2 × 3 × 6.2 mm, packed in 4 mm zirconia rotors with γ-Al2O3 as a packing powder, and spun at the magic angle at a rate of 8 kHz. In addition, a small piece of sintered PBZ06 was crushed and annealed at 400 °C for 2 h to alleviate mechanical stresses and then the NMR spectra were recorded at 8 kHz MAS. 207Pb 2D-PASS experiments were carried out with five π pulses of 5.8 μs following the initial π/2 pulse of 2.6 μs, and a relaxation delay of 10 s. A modified version of the sequence with a shifted echo was used to accommodate for the free induction decays of PBZ dying out in less than 150 μs,40 with a duration of one rotor period added to the delay before and after the last π pulse. Sixteen pitch increments were used to separate the spinning sidebands by following the timings given by Antzutkin et al.41 The phase of each of the π pulses was cycled independently in 120° steps, yielding the 243-step scheme given in therein. 972 or 1215 scans were averaged on each pitch. 207Pb chemical shifts were referenced to the PbII signal of commercial PbZrO3 at −1363 ppm following the data published by Zhao et al.35 Line shape simulations of CSA patterns were carried out with the program DMFit.43Results & discussion
The low-angle segments of the XRD patterns of PbZrO3 (PZ), Pb0.94Ba0.06ZrO3 (PBZ06), and Pb0.88Ba0.12ZrO3 (PBZ12) are depicted in Fig. 1a. While the diffractograms of PZ and PBZ06 are qualitatively similar, PBZ12 lacks several superlattice reflections and additional splittings in the range of 20–60°. This difference is confirmed by our Rietveld analysis (Fig. S1†), which allows refinement with orthorhombic (space group no. 55, Pbam) symmetry for PZ and PBZ06, but indicates a predominantly rhombohedral (no. 161, R3c) symmetry in the case of PBZ12, in accordance with previously published data.27 This confirms the composition-induced AFE → FE phase transition when the amount of barium at the perovskite A-site is increased from x = 0.06 to 0.12. However, we note that a minor amount of the orthorhombic phase was also detected in PBZ12. In addition, the different states of our XRD samples (ground and annealed powders) in comparison with our NMR samples (sintered pellets) should be kept in mind.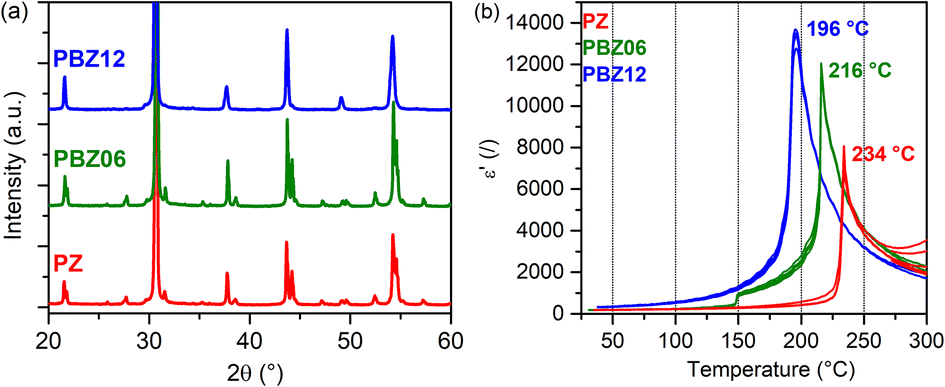 | ||
| Fig. 1 (a) XRD patterns of PZ, PBZ06, and PBZ12. (b) ε′(T) measurements upon heating for PZ, PBZ06, and PBZ12. | ||
The FE nature of PBZ12 is also supported by the electrical measurements. The polarization–electric field hysteresis loop is shown in the ESI (Fig. S2†). While it has been initially reported that the AFE state in PBZ persists up until x = 0.175 at room temperature,25,26 the exact location of this phase boundary remains unclear and the transition behavior is complex.8 Other experimental studies have reported FE-like polarization hysteresis for x = 0.1 and more.22 Measurements of the dielectric permittivity ε′ as a function of temperature (Fig. 1b) can provide additional information on the phase transitions between the paraelectric (PE), FE, and AFE states. Upon heating, PBZ06 exhibits two anomalies in ε′ at around 150 °C and at 216 °C. They are related to two temperature-induced phase transitions and attributed to the AFE → FE and FE → PE transitions, respectively. While the AFE → FE transition is subject to a large thermal hysteresis of around 50 °C, which is expected for a first-order transition,45,46 the FE → PE transition has a small hysteresis of 8 K. This behavior is similar to that of pure PZ, although here the first anomaly is only observable upon cooling (Fig. S3†). In contrast, PBZ12 exhibits only a single transition at 196 °C (heating), further supporting our conclusion on its room-temperature FE state.
The structure of orthorhombic (Pbam) PZ9 with the crystallographic sites PbI and PbII is depicted in Fig. 2a. Its 207Pb NMR spectrum at room temperature is known to exhibit two signals corresponding to the two inequivalent sites,47 the full CSA tensors of which have been determined from powder line shapes.48 PbI and PbII differ not only with respect to the volume of the PbO12 coordination polyhedra, but also their displacements from the centers thereof which are characteristic of the AFE state.
 | ||
| Fig. 2 (a) Structure of orthorhombic (Pbam) PZ9 with crystallographic sites PbI and PbII visualized with the program VESTA.44 The two sites feature differently sized coordination spheres and different displacement magnitudes. In 207Pb NMR spectra, the site with the shortest Pb–O distance is assigned to the signal with the less negative isotropic CS. (b) 207Pb 2D-PASS spectrum of ceramic PZ and visualization of the sample geometry. The two diagonals correspond to the isotropic signals with SSB manifolds of PbI and PbII. After a shearing operation, the purely isotropic signal displayed in the top projection can be obtained. | ||
As shown in the inlay, both sites feature an antiparallel arrangement of displacements. However, the displacement magnitudes vary between the two sites. In NMR, the signal with the less negative isotropic CS and larger CSA has previously been assigned to the lead site with the shortest Pb–O distance.35,39 With the 2D-PASS pulse sequence (Fig. 2b), the two signals can be separated along the diagonals of the spectrum, and simulations of the intensity patterns of the extracted SSB manifolds yield δPbI = −1026 ppm, CSAPbI = −865 ± 20 ppm and δPbII = −1368 ppm, CSAPbII = −550 ± 10 ppm. The NMR parameters are qualitatively in line with the previously reported values for PZ (Table S1†), and we suggest that any deviations of our data can be attributed to temperature gradients in the sample configuration, which can result in frictional heating.49 At the same time, we have not observed significant differences in line shape between pellet and powder samples (Fig. S4†). The chemical shift of PZ has been reported to be temperature-dependent between 0 and 30 °C,39 which is why we refrain from assigning too much importance to minor chemical shift changes in this work.
The 2D-PASS spectrum of PBZ06 depicted in Fig. 3a still features two distinct sites but is broadened compared to the spectrum of pure PZ. In a Hahn-Echo spectrum of the same ceramic pellet, this additional line width is significant enough to prevent any meaningful extraction of NMR parameters due to the heavy overlap of broadened SSB (Fig. S5†). 2D-PASS, in contrast, still allows for the discrimination between the two SSB manifolds. After shearing the individual rows of the spectrum with multiples of the MAS rate (Fig. S6†), a sum projection of the rows only consists of the isotropic contributions to the CS.41,47 As shown in Fig. 3a, this “purely isotropic” projection features two distinct signals with maxima at δPbI = −1025 ppm and δPbII = −1355 ppm for PBZ06.
 | ||
| Fig. 3 (a) 207Pb 2D-PASS spectra and sum projections after shear (purely isotropic projections) of (a) a PBZ06 pellet and (b) a PBZ12 pellet recorded at 8 kHz MAS. The diagonal positions of the two sites in PZ have been included as a guide to the eye. | ||
A superposition of the purely isotropic projections is depicted in Fig. 4. The increase of Gaussian line width between PZ and PBZ06 from ΔPbI = 39 ppm and ΔPbII = 45 ppm to ΔPbI = 187 ppm and ΔPbII = 217 ppm (Fig. S7 and Table S2†) is due to a dispersion of isotropic CS. This indicates the presence of a distribution of varying local environments, each of which contributes a slightly different CS value to the signal. The FWHM of the purely isotropic NMR lines is thus a measure of the underlying parameter distributions. We suggest that, in principle, two different effects might cause the increase of the width of the chemical shift distributions upon the addition of barium. Firstly, the presence of next-nearest neighboring (NNN) barium atoms could cause binary changes in the direct local environment of surrounding Pb2+ cations (compositional disorder). Secondly, the addition of barium to the lattice could cause continuous variations in the Pb–O bond length distributions (displacive disorder).
 | ||
| Fig. 4 Comparison of purely isotropic projections taken from the 207Pb 2D-PASS spectra of the ceramic pellets of PZ, PBZ06, and PBZ12 shown in Fig. 2 and 3. | ||
Assuming a random distribution of A-site cations, the probability of a Pb2+ cation in PBZ06 to have at least one next-nearest Ba2+ neighbor is 31% (Fig. S8†). Hence, any compositional contribution to the disorder would only be attributed to a small fraction of Pb2+ cations. In addition, such an effect is expected to result in a bimodal line shape reflecting the presence of two distinct lead environments for every crystallographic lead site in the structure, which is not supported by our experiments.
On the other hand, Zhou et al.36 have previously correlated the isotropic CS of 207Pb with effective Pb–O bond lengths: when Pb2+ cations are displaced from the center of their coordination spheres, the Pb–O distances split up, causing the CS to reflect a bond length that is dominated by the shortest value. In the same sense, an observed increase of line width can be attributed to a significant increase of Pb–O bond length disorder.34 They were able to demonstrate that a unimodal Gaussian-like line shape like the one observed in our work could thus be well explained by displacive disorder in a single direction, i.e. disorder of displacement magnitudes. Thus, we argue here that the distribution of isotropic CS observed in barium-modified PZ is mainly due to this kind of displacive disorder; however, we cannot exclude a combined effect.
Applying the relationships established by Fayon et al.,36,37 we can estimate that the distributions of effective bond lengths increase from σ(PbI–O) = σ(PbII–O) = 0.002 Å for PZ to σ(PbI–O) = 0.009 Å and σ(PbII–O) = 0.011 Å for PBZ06. Thus, it increases by one order of magnitude as a result of the addition of 6% barium. The relative peak areas of the two signals are found to be 38% and 62% in PBZ06 compared to 37% and 63% in pure PZ. It should be noted that in both cases, they deviate from the expected 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 intensity ratio. We attribute this to the fact that the r.f. pulse was in resonance with the PbII signal and therefore had a higher excitation efficiency for this signal.
:1 intensity ratio. We attribute this to the fact that the r.f. pulse was in resonance with the PbII signal and therefore had a higher excitation efficiency for this signal.
The spectrum of PBZ12 only exhibits one diagonal (Fig. 3b) corresponding to one broad signal with a maximum at −975 ppm in the purely isotropic projection (Fig. 4). The disappearance of one signal between PBZ06 and PBZ12 is also evident in the superposition in Fig. 4. In the context of our XRD and dielectric measurements, we can conclude that while the AFE state of PBZ features the two lead sites known from PZ, the FE state has only one distinct site. This result is consistent with the FE state of PBZ being an R3c perovskite with only one crystallographic A-site (Fig. S9†).50 Considering the relationship between lead displacements and effective Pb–O bond lengths, we can also interpret the disappearance of one signal as the collapse of two different displacement magnitudes into one.
The site that prevails in PBZ12 is the one with the more positive isotropic CS and larger CSA. Hence, the coordination sphere and the magnitude of lead displacements in PBZ12 are similar to that of the PbI site in PZ and PBZ06. PbI features larger displacements and the shorter shortest Pb–O distance, but longer average bonds. As depicted in Fig. 5, our Rietveld refinements find that the lattice volume of the reduced perovskite unit cell significantly increases during the AFE → FE transition from 71.47 Å3 (PBZ06) to 72.38 Å3 (PBZ12). This is mostly due to an abrupt increase in the reduced c parameter. A lattice volume increase with barium content is well in line with the larger ionic radii (r(Ba2+) = 1.61 Å, r(Pb2+) = 1.49 Å),51 and has also been reported by Pokharel et al.46 We suggest that the global lattice expansion translates into lattice expansions around the barium atoms on a local scale, which is in agreement with a larger, PbI-like coordination sphere being favored by the local structure. At the same time, the PbI sites also feature larger lead displacements, indicating that the lead ions prefer off-center positions as the cell expands. A similar effect has been reported by Zhou et al.36 for (1 − x)PMN-xPSN solid solutions, albeit for B-site substitution. The resemblance of the local lead environment in PBZ12 to the PbI site in PZ and PBZ06 can invite speculation on whether their local displacement directions are the same despite their long-range structures indicating average displacements along different directions. However, the Gaussian-style broadening of the NMR signals only allows us to infer that displacements occur in a single, unique direction33,36 which may differ from the average polarization direction.19,52 For more in-depth conclusions, a study of the local structure dynamics would be required.
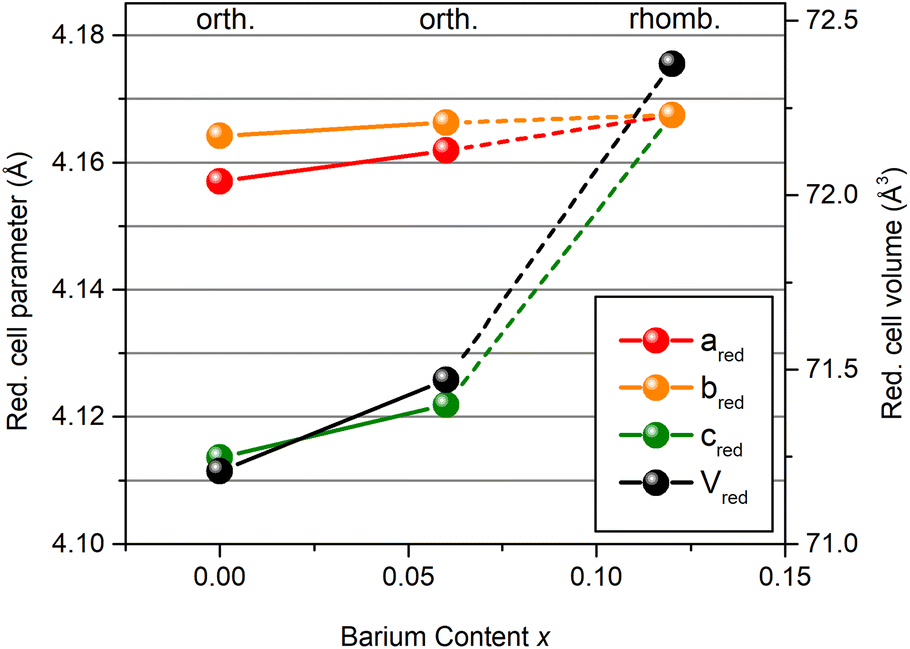 | ||
| Fig. 5 Reduced cell parameters and cell volumes obtained from Rietveld refinements of the XRD data. | ||
Analyzing the spectra in more detail, slight differences with respect to the spectral parameters can be observed. The line width increases from 188 ppm to 201 ppm between PBZ06 and PBZ12, indicating a further increase of the Pb–O bond length disorder and corresponding to an effective distribution of σ(PbI–O) = 0.010 Å. This change is small compared to the strong increase from PZ to PBZ06. Since in PBZ12, the probability of any Pb2+ cation to have at least one Ba2+ NNN can be estimated at 55%, part of the increase of the line width from PBZ06 to PBZ12 can in fact be attributed to an increase in compositional disorder.
The isotropic chemical shift of the PbI site increases by 64 ppm from PBZ06 to PBZ12, reflecting a shortening of effective PbI–O distances. This can be well explained by lead ions moving further off-center in the larger PbO12 cavities of the FE structure. It is known from the XRD data that the PbI site in pure PZ favors larger displacements,9 and previous studies have shown that Pb2+ cations tend to move off-center from high-symmetry positions to accommodate their lone pair, which can help explain this observation.52 Applying the formula by Fayon et al.,37 a decrease of effective PbI–O distances of roughly 0.007 Å can be estimated. In comparison, the difference between PbI and PbII in PZ is 0.03 Å for the average Pb–O distances and 0.04 Å for the shortest distances.9 Thus, the change induced in PBZ12 is not on the same order of magnitude as the difference between the two sites.
A possible explanation for the observed local structural changes can be found in the cubic structure and larger lattice volume of BaZrO3 compared to PZ.9,53 As mentioned above, introducing barium to the lattice might cause a local-scale expansion around the Ba2+ cations, facilitating larger Pb2+ displacements. As more barium is added to the lattice, these displacements are ultimately correlated as ferroelectricity during the structure-induced AFE → FE phase transition. The experimental data based on the 207Pb NMR spectra clearly demonstrate a larger distribution of the shortest Pb–O bond lengths with increasing barium content and a more covalent Pb environment in the barium-rich FE phase. However, the origin of the observed effect, explained in terms of the larger size and lower polarizability of Ba2+, and how this affects the local environment of lead sites in the direct neighbourhood of substituents and beyond, remains an open hypothesis to be tested in future studies.
In addition, the different polarizabilities of barium compared to lead54 might influence the bonding interactions, a change which should reflect in the correlation of isotropic CS and CSA.36 The 2D-PASS spectra contain information about both the isotropic CS and the CSA. Anisotropic projections (Fig. 6) can be obtained as vertical slices at the positions of the signal maxima in the sheared data. While the slices are essentially point spectra and therefore cannot be equated with full experimental SSB manifolds, their widths and intensity patterns are still characteristic of the value of the CSA and can be reproduced with a fit. However, the determination of an accurate value of the asymmetry parameter η exceeds the estimated accuracy of the line shape simulation. As the Pb2+ cations have their greatest displacements along one axis of symmetry (〈110〉PC in the orthorhombic structure9 and 〈111〉PC in the rhombohedral structure50), axial symmetry with an asymmetry parameter of η ≈ 0 is assumed instead.
 | ||
| Fig. 6 Comparison of the anisotropic projections taken from the 207Pb 2D-PASS spectra of PZ, PBZ06, and PBZ12 shown in Fig. 2 and 3. In (a) and (c), the intensity scale is halved for better visibility. | ||
Between PZ and PBZ06, no changes of the CSA outside of the estimated margins of error are determined. The chemical shift tensor values fall in the order of magnitude described by Zhou et al.36 as midway between covalent and ionic. However, for PBZ12, the CSA increases by 50 ppm (Table 1). While the increase is small compared to the estimated maximal margins of error of ≤30 ppm, the increase of SSB intensity between 500 and −500 ppm is still evident in the intensity patterns. It is possible that this change, in combination with the increase of the isotropic CS, reflects a transition to a slightly more covalent/less ionic bonding environment.36 This contrasts the notion that Pb-based compositions tend to be more covalent than Ba-based compositions,55i.e. the Pb2+ environment becomes more covalent instead of being less covalent after Ba2+ addition. One can speculate that the lower polarizability of Ba2+ compared to that of Pb2+ could potentially lead to weaker Ba–O bonds, in turn facilitating the formation of stronger Pb–O bonds in their vicinity. Further information on the structural changes could be obtained in the future by neutron diffraction, EXAFS or HRTEM experiments, which are beyond the scope of the current study.
| PbI | PbII | |||
|---|---|---|---|---|
| δ iso/ppm | CSA/ppm | δ iso/ppm | CSA/ppm | |
| PZ | −1026 ± 2 | −865 ± 20 | −1368 ± 2 | −550 ± 10 |
| PBZ06 | −1025 ± 10 | −858 ± 18 | −1355 ± 10 | −510 ± 15 |
| PBZ12 | −975 ± 5 | −905 ± 30 | ||
After applying an electric field to the PBZ12 sample, the spectrum remains unchanged (Fig. S9 and S10†), further corroborating the interpretation that it is already in the FE state. The lack of changes in the NMR spectra indicates that unpoled and poled FE states for this compound merely differ by microstructural texturing of the sample. Studying PZ and PBZ06 post-electric fields by 207Pb NMR spectroscopy could give insights into the reversibility of the AFE → FE transitions in these compositions. However, the electric fields necessary to induce the phase transitions exceeded the dielectric breakdown strengths at room temperature in the case of our samples, rendering such analysis inaccessible in our study.
Conclusions
In this work, changes in the local environment of lead upon substitution with barium were analyzed in the AFE model system (Pb1−xBax)ZrO3 by 207Pb NMR spectroscopy. 207Pb isotropic chemical shifts reflect effective Pb–O distances and therefore, displacements of Pb2+ ions within their oxygen cages. The electric field-induced AFE → FE phase transition in ceramic PbZrO3 is irreversible; for x = 0.06, the AFE state is expected to be destabilized. 207Pb NMR revealed an increase of isotropic line widths, indicating that the substitution with barium induces Pb–O bond length disorder with a distribution width of approx. 0.01 Å. Meanwhile, the isotropic chemical shift, the relative integrals of the PbI:PbII signals, and the CSA remained unchanged between the two compounds, reflecting that the global structure is not strongly influenced by the introduction of barium.Upon crossing a threshold value for x, barium induces a macroscopic phase transition to a FE state, which was observed by XRD and, on a local scale, the collapse of two lead environments into one. For x = 0.12, lead environments and displacements are of similar nature to those of the PbI site in pure PZ. The stabilization of the larger volume site is in accordance with a lattice expansion as determined by Rietveld refinements. Meanwhile, the favoring of shorter shortest Pb–O distances reflects a tendency for larger Pb2+ displacements. We suggest that the introduction of Ba2+ ions to the lattice can cause local-scale lattice expansions in their vicinity, facilitating the displacements. Compared to the PbI site in PbZrO3, a further off-center shift of Pb2+ ions in the larger PbO12 cavities of the FE structure is observed. An accompanying increase of the CSA gives an indication of a more covalent bonding environment in PBZ12, which could potentially be related to the lower polarizability of Ba2+ facilitating the formation of stronger Pb–O bonds in their vicinity. From the local structural point of view, the composition-induced AFE → FE phase transition can therefore potentially be described as an increasing correlation of larger lead displacements in larger oxygen cavities as more barium is added to the lattice.
The dielectric breakdown did not allow for an ex situ study of the (Pb1−xBax)ZrO3 post-electric field for x = 0, 0.06 in this study. For x = 0.12, no changes were observed (Fig. S10 and 11†), indicating that the poled ferroelectric state for this compound only differs with respect to the microstructural texturing of the sample.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Hessian State Ministry for Higher Education, Research and the Arts under the LOEWE collaborative project FLAME (Fermi level engineering of anti-ferroelectric materials for energy storage and insulation systems). B. M. acknowledges the financial support by the Slovenian Research Agency (core funding P2-0105). G. B. acknowledges the financial support by the German Research Foundation under the contract Bu-911/28-1. P. G. acknowledges the financial support by the Dutch Research Council (NWO) for the ECCM Tenure Track funding under the project number ECCM.006.References
- Z. Liu, T. Lu, J. Ye, G. Wang, X. Dong, R. Withers and Y. Liu, Antiferroelectrics for Energy Storage Applications: A Review, Adv. Mater. Technol., 2018, 3, 1800111 CrossRef.
- A. Mischenko, Q. Zhang, J. F. Scott, R. W. Whatmore and N. D. Mathur, Giant Electrocaloric Effect in Thin Film PbZr0.95Ti0.05O3, Science, 2006, 311, 1270–1271 CrossRef CAS PubMed.
- N. Novak, F. Weyland, S. Patel, H. Guo, X. Tan, J. Rödel and J. Koruza, Interplay of Conventional with Inverse Electrocaloric Response in (Pb,Nb)(Zr,Sn,Ti)O3 Antiferroelectric Materials, Phys. Rev. B, 2018, 97, 094113 CrossRef CAS.
- E. Sawaguchi, G. Shirane and Y. Takagi, Phase Transition in Lead Zirconate, J. Phys. Soc. Jpn., 1951, 6, 333–339 CrossRef CAS.
- G. Shirane, E. Sawaguchi and Y. Takagi, Dielectric Properties of Lead Zirconate, Phys. Rev., 1951, 84, 476–481 CrossRef CAS.
- G. Shirane, Ferroelectricity and Antiferroelectricity in Ceramic PbZrO3 Containing Ba or Sr, Phys. Rev., 1952, 86, 219–227 CrossRef CAS.
- P.-H. Chien, K. J. Griffith, H. Liu, Z. Gan and Y.-Y. Hu, Recent Advances in Solid-State Nuclear Magnetic Resonance Techniques for Materials Research, Annu. Rev. Mater. Res., 2020, 50, 493–520 CrossRef CAS.
- H. Liu and B. Dkhil, A Brief Review on the Model Antiferroelectric PbZrO3 Perovskite-Like Material, Z. Kristallogr., 2011, 226, 163–170 CrossRef CAS.
- D. L. Corker, A. M. Glazer, J. Dec, K. Roleder and R. W. Whatmore, A Re-Investigation of the Crystal Structure of the Perovskite PbZrO3 by X-ray and Neutron Diffraction, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 135–142 CrossRef.
- X. Tan, C. Ma, J. Frederick, S. Beckman and K. G. Webber, The Antiferroelectric ↔ Ferroelectric Phase Transition in Lead-Containing and Lead-Free Perovskite Ceramics, J. Am. Ceram. Soc., 2011, 94, 4091–4107 CrossRef CAS.
- F. Jona, G. Shirane, F. Mazzi and R. Pepinsky, X-Ray and Neutron Diffraction Study of Antiferroelectric Lead Zirconate, PbZrO3, Phys. Rev., 1957, 105, 849–856 CrossRef CAS.
- E. Sawaguchi, H. Maniwa and S. Hoshino, Antiferroelectric Structure of Lead Zirconate, Phys. Rev., 1951, 85, 1078 CrossRef.
- A. M. Glazer, Structure and Disorder in Single-Crystal Lead Zirconate, PbZrO3, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 846–852 CrossRef.
- B. Xu, O. Hellman and L. Bellaiche, Order-Disorder Transition in the Prototypical Antiferroelectric PbZrO3, Phys. Rev. B, 2019, 100, 20102 CrossRef CAS.
- J. Íñiguez, M. Stengel, S. Prosandeev and L. Bellaiche, First-Principles Study of the Multimode Antiferroelectric Transition in PbZrO3, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 220103 CrossRef.
- A. K. Tagantsev, K. Vaideeswaran, S. B. Vakhrushev, A. V. Filimonov, R. G. Burkovsky, A. Shaganov, D. Andronikova, A. I. Rudskoy, A. Q. R. Baron, H. Uchiyama, D. Chernyshov, A. Bosak, Z. Ujma, K. Roleder, A. Majchrowski, J.-H. Ko and N. Setter, The Origin of Antiferroelectricity in PbZrO3, Nat. Commun., 2013, 4, 2229 CrossRef CAS PubMed.
- P. Vales-Castro, K. Roleder, L. Zhao, J.-F. Li, D. Kajewski and G. Catalan, Flexoelectricity in Antiferroelectrics, Appl. Phys. Lett., 2018, 113, 132903 CrossRef.
- H. Liu, Origin of the Intermediate Phase in Lead Zirconate, PbZrO3, J. Am. Ceram. Soc., 2018, 101, 5281–5286 CrossRef CAS.
- S. Teslic, T. Egami and D. Viehland, Structural Instabilities in PZT, Ferroelectrics, 1997, 194, 271–285 CrossRef CAS.
- O. E. Fesenko, R. V. Kolesova and Y. G. Sindeyev, The Structural Phase Transitions in Lead Zirconate in Super-High Electric Fields, Ferroelectrics, 1978, 20, 177–178 CrossRef CAS.
- X. Hao, J. Zhai, L. B. Kong and Z. Xu, A Comprehensive Review on the Progress of Lead Zirconate-Based Antiferroelectric Materials, Prog. Mater. Sci., 2014, 63, 1–57 CrossRef CAS.
- K. H. Yoon and S. C. Hwang, Dielectric and Field-Induced Strain Behaviour of (Pb1−xBax)ZrO3 Ceramics, J. Mater. Sci., 1997, 32, 17–21 CrossRef CAS.
- S. Roberts, Dielectric Properties of Lead Zirconate and Barium-Lead Zirconate, J. Am. Ceram. Soc., 1950, 33, 63–66 CrossRef CAS.
- G. Shirane and S. Hoshino, X-Ray Study of Phase Transitions in PbZrO3 Containing Ba or Sr, Acta Crystallogr., 1954, 7, 203–210 CrossRef CAS.
- I. El-Harrad, A. Ridah, C. Carabatos-Nédelec, P. Becker, J. Handerek, Z. Ujma and D. Dmytrow, Raman Scattering Investigation with Temperature of Phase Transitions in (Pb1−xBax)ZrO3 Ceramics at Critical Compositions x = 0.175 and 0.35, J. Raman Spectrosc., 1998, 29, 123–129 CrossRef CAS.
- B. P. Pokharel and D. Pandey, Effect of Ba2+ Substitution on the Stability of the Antiferroelectric and Ferroelectric Phases in (Pb1−xBax)ZrO3: Phenomenological Theory Considerations, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 214108 CrossRef.
- B. P. Pokharel, R. Ranjan, D. Pandey, V. Siruguri and S. K. Paranjpe, Rhombohedral Superlattice Structure and Relaxor Ferroelectric Behavior of (Pb0.70Ba0.30)ZrO3 Ceramics, Appl. Phys. Lett., 1999, 74, 756–758 CrossRef CAS.
- B. P. Pokharel and D. Pandey, Irreversibility of the Antiferroelectric to Ferroelectric Phase Transition in (Pb0.90Ba0.10)ZrO3 ceramics, J. Appl. Phys., 1999, 86, 3327–3332 CrossRef CAS.
- J. Rödel and J.-F. Li, Lead-Free Piezoceramics: Status and Perspectives, MRS Bull., 2018, 43, 576–580 CrossRef.
- M.-H. Zhang, L. Fulanović, S. Egert, H. Ding, P. B. Groszewicz, H.-J. Kleebe, L. Molina-Luna and J. Koruza, Electric-Field-Induced Antiferroelectric to Ferroelectric Phase Transition in Polycrystalline NaNbO3, Acta Mater., 2020, 200, 127–135 CrossRef CAS.
- M.-H. Zhang, N. Hadaeghi, S. Egert, H. Ding, H. Zhang, P. B. Groszewicz, G. Buntkowsky, A. Klein and J. Koruza, Design of Lead-Free Antiferroelectric (1−x)NaNbO3−xSrSnO3 Compositions Guided by First-Principles Calculations, Chem. Mater., 2021, 33, 266–274 CrossRef CAS.
- P. B. Groszewicz, NMR Spectroscopy of Electroceramics – Applications to Lead-Free Perovskite Oxides, Open Ceram., 2021, 5, 100083 CrossRef CAS.
- C. E. Avalos, B. J. Walder and L. Emsley, Lead–Oxygen Bond Length Distributions of the Relaxor Ferroelectric 0.67PbMg1/3Nb2/3O3–0.33PbTiO3 from 207Pb Nuclear Magnetic Resonance, J. Phys. Chem. C, 2019, 123, 15744–15750 CrossRef CAS.
- A. Baldwin, P. A. Thomas and R. Dupree, A Multi-Nuclear NMR Study of the Local Structure of Lead Zirconate Titanate, PbZr1−xTixO3, J. Phys.: Condens. Matter, 2005, 17, 7159–7168 CrossRef CAS.
- P. Zhao, S. Prasad, J. Huang, J. J. Fitzgerald and J. S. Shore, Lead-207 NMR Spectroscopic Study of Lead-Based Electronic Materials and Related Lead Oxides, J. Phys. Chem. B, 1999, 103, 10617–10626 CrossRef CAS.
- D. H. Zhou, G. L. Hoatson, R. L. Vold and F. Fayon, Local Structure in Perovskite Relaxor Ferroelectrics by 207Pb NMR, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 305 Search PubMed.
- F. Fayon, I. Farnan, C. Bessada, D. Massiot and J. P. Coutures, Empirical Correlations Between 207Pb NMR Chemical Shifts and Structure in Solids, J. Am. Chem. Soc., 1997, 119, 6837–6843 CrossRef CAS.
- C. Dybowski and G. Neue, Solid State 207Pb NMR Spectroscopy, Prog. Nucl. Magn. Reson. Spectrosc., 2002, 41, 153–170 CrossRef CAS.
- P. Rossi, M. R. Dvorak and G. S. Harbison, 207Pb NMR of PbZrO3 and PbZr1−xTixO3 Solid Solutions, MRS Online Proc. Libr., 2000, 658, 351 Search PubMed.
- F. Fayon, C. Bessada, A. Douy and D. Massiot, Chemical Bonding of Lead in Glasses Through Isotropic vs Anisotropic Correlation: PASS Shifted Echo, J. Magn. Reson., 1999, 137, 116–121 CrossRef CAS PubMed.
- O. N. Antzutkin, S. C. Shekar and M. H. Levitt, Two-Dimensional Sideband Separation in Magic-Angle-Spinning NMR, J. Magn. Reson., Ser. A, 1995, 115, 7–19 CrossRef CAS.
- V. Petříček, M. Dušek and L. Palatinus, Crystallographic Computing System JANA2006: General Features, Z. Kristallogr., 2014, 229, 345–352 Search PubMed.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Modelling One- and Two-Dimensional Solid-State NMR Spectra, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- K. Momma and F. Izumi, VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- B. P. Pokharel and D. Pandey, Dielectric Studies of Phase Transitions in (Pb1−xBax)ZrO3, J. Appl. Phys., 2000, 88, 5364–5373 CrossRef CAS.
- B. P. Pokharel and D. Pandey, High Temperature X-Ray Diffraction Studies on Antiferroelectric and Ferroelectric Phase Transitions in (Pb1−xBax)ZrO3 (x = 0.05, 0.10), J. Appl. Phys., 2001, 90, 2985–2994 CrossRef CAS.
- F. G. Vogt, J. M. Gibson, D. J. Aurentz, K. T. Mueller and A. J. Benesi, Multiple-Rotor-Cycle 2D PASS Experiments with Applications to 207Pb NMR Spectroscopy, J. Magn. Reson., 2000, 143, 153–160 CrossRef CAS PubMed.
- G. Neue, C. Dybowski, M. L. Smith, M. A. Hepp and D. L. Perry, Determination of 207Pb2+ Chemical Shift Tensors from Precise Powder Lineshape Analysis, Solid State Nucl. Magn. Reson., 1996, 6, 241–250 CrossRef CAS PubMed.
- A. Bielecki and D. P. Burum, Temperature Dependence of 207Pb MAS Spectra of Solid Lead Nitrate. An Accurate, Sensitive Thermometer for Variable-Temperature MAS, J. Magn. Reson., Ser. A, 1995, 116, 215–220 CrossRef CAS.
- H. Yokota, N. Zhang, A. E. Taylor, P. A. Thomas and A. M. Glazer, Crystal Structure of the Rhombohedral Phase of PbZr1−xTixO3 Ceramics at Room Temperature, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 104109 CrossRef.
- R. D. Shannon, Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- W. Dmowski, M. K. Akbas, P. K. Davies and T. Egami, Local Structure of Pb(Sc1/2,Ta1/2)O3 and Related Compounds, J. Phys. Chem. Solids, 2000, 61, 229–237 CrossRef CAS.
- A. Perrichon, E. Jedvik Granhed, G. Romanelli, A. Piovano, A. Lindman, P. Hyldgaard, G. Wahnström and M. Karlsson, Unraveling the Ground-State Structure of BaZrO3 by Neutron Scattering Experiments and First-Principles Calculations, Chem. Mater., 2020, 32, 2824–2835 CrossRef CAS.
- P. Schwerdtfeger and J. K. Nagle, 2018 Table of Static Dipole Polarizabilities of the Neutral Elements in the Periodic Table, Mol. Phys., 2019, 117, 1200–1225 CrossRef CAS.
- Z. Wu and H. Krakauer, Charge-Transfer Electrostatic Model of Compositional Order in Perovskite Alloys, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 184113 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD patterns, polarization–electric field (P–E) loops, temperature dependence of dielectric permittivity, 207Pb Hahn-Echo spectra, sheared 207Pb 2D-PASS spectra, NMR line shape simulations, 207Pb 2D-PASS spectra of the Pb0.88Ba0.12ZrO3 post-electric-field, and statistical evaluation of next-nearest neighbour configurations. See DOI: https://doi.org/10.1039/d2dt01302a |
| This journal is © The Royal Society of Chemistry 2022 |