
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the role of flux, pressure and temperature on polymorphism in ThB2O5†
Yucheng
Hao
 a,
Gabriel L.
Murphy
*b,
Philip
Kegler
b,
Yan
Li
c,
Piotr M.
Kowalski
de,
Simon
Blouin
fg,
Yang
Zhang
a,
Shuao
Wang
h,
Lars
Robben
ij,
Thorsten M.
Gesing
ij and
Evgeny V.
Alekseev
*k
a,
Gabriel L.
Murphy
*b,
Philip
Kegler
b,
Yan
Li
c,
Piotr M.
Kowalski
de,
Simon
Blouin
fg,
Yang
Zhang
a,
Shuao
Wang
h,
Lars
Robben
ij,
Thorsten M.
Gesing
ij and
Evgeny V.
Alekseev
*k
aSchool of Energy Materials and Chemical Engineering, Hefei University, Hefei 230000, China. E-mail: haoyco@hfuu.edu.cn
bInstitute of Energy and Climate Research (IEK-6), Forschungszentrum Jülich GmbH, 52428 Jülich, Germany. E-mail: murphy@fz-juelich.de
cState Key Laboratory of Optoelectronic Materials and Technologies, School of Materials Science and Engineering, Sun Yat-sen (Zhongshan) University, Guangzhou, 510275, PR China
dInstitute of Energy and Climate Research (IEK-13), Forschungszentrum Jülich GmbH, 52428 Jülich, Germany
eJARA Energy & Center for Simulation and Data Science (CSD), Jülich, Germany
fDepartment de Physique, University of Montreal, Montreal, QC H3C 3J7, Canada
gDepartment of Physics and Astronomy, University of Victoria, Victoria BC V8P 5C2, Canada
hSchool for Radiological and Interdisciplinary Sciences (RAD-X) and Collaborative Innovation Center of Radiation Medicine of Jiangsu Higher Education Institutions, Soochow University, Suzhou 215123, China
iUniversity of Bremen, Institute of Inorganic Chemistry and Crystallography, D-28359 Bremen, Germany
jUniversity of Bremen, MAPEX Center for Materials and Processes, D-28359 Bremen, Germany
kInstitute of Energy and Climate Research (IEK-9), Forschungszentrum Jülich GmbH, 52428 Jülich, Germany. E-mail: e.alekseev@fz-juelich.de
First published on 19th August 2022
Abstract
A novel polymorph of ThB2O5, denoted as β-ThB2O5, was synthesised under high-temperature high-pressure (HT/HP) conditions. Via single crystal X-ray diffraction measurements, β-ThB2O5 was found to form a three-dimensional (3D) framework structure where thorium atoms are ten-fold oxygen coordinated forming tetra-capped trigonal prisms. The only other known polymorph of ThB2O5, denoted α, synthesised herein using a known borax, B2O3–Na2B4O7, high temperature solid method, was found to transform to the β polymorph when exposed to conditions of 4 GPa and ∼900 °C. Compared to the α polymorph, β-ThB2O5 has smaller molar volume by approximately 12%. Exposing a mixture of the α and β polymorphs to HT/HP conditions ex situ further demonstrated the preferred higher-pressure phase being β, with no α phase material being observed via Rietveld refinements against laboratory X-ray powder diffraction (PXRD) measurements. In situ heating PXRD measurements on α-ThB2O5 from RT to 1030 °C indicated that α-ThB2O5 transforms to the β variant at approximately 900 °C via a 1st order mechanism. β-ThB2O5 was found to exist only over a narrow temperature range, decomposing above 1050 °C. Ab initio calculations using density functional theory (DFT) with the Hubbard U parameter indicated, consistent with experimental observations, that β is both the preferred phase at higher temperatures and high pressures. Interestingly, it was found by switching from B2O3–Na2B4O7 to H3BO3–Li2CO3 flux using consistent high temperature solid state conditions for the synthesis of the α variant, β-ThB2O5 could be generated. Comparison of their single crystal measurements showed this was identical to that obtained from HT/HP conditions.
1. Introduction
Traditional interest in thorium chemistry has been somewhat overshadowed by its f-block sister uranium,2,3 however contemporary studies have unravelled significant applications and properties in the context of advanced functional material development, reinvigorating focus in the most naturally abundant actinide element.4–6 Examples of such successes can be seen in nuclear waste separation technology,7a radiation detection7b and also MOF development.7c In these, the chemical advantages derived from the typically tetravalent Th cation, as opposed to its higher actinide counterparts (U, Np, Pu), relate to its ability to adopt a relatively more spherical chemical environment whilst retaining somewhat f-block character.8 This allows for the construction of unique higher dimensional structures than those typically found for instance in uranyl compounds. Accordingly, together with property measurements and applications, inorganic chemists have shifted their attention towards deriving facile synthesis routes to unlock novel thorium phases and study their structural properties.Since the exceptional Tc ion-exchangeable thorium borate, NDTB-1 (ThB5O6(OH)6 BO(OH)2·2.5H2O) was first reported in 2010, interest in thorium borate synthesis has grown.9 However, surprisingly only a few thorium borates without additional countercations have been reported.1,9,10 Prior to NDTB-1, Cousson and Gasperin in 1991 reported the first example of a thorium borate, ThB2O5 (denoted hereon as α-phase), which was obtained through high-temperature flux synthesis.1 It consists of a 3D thorium borate framework structure, in which thorium–thorium stacking sequences exist with a diamond packing motif forming a 3D thorium-network. B2O5 dimers edge share with thorium polyhedra and fill the voids of the Th-cation framework along the [−101] direction. NDTB-1 was originally synthesised using a low-temperature boric acid flux method in which this thorium borate is constructed from crown-like B10O24 groups and twelve-fold coordinated thorium ions. These groups form a porous super-tetrahedral 3D thorium-borate framework. The super-tetrahedra cationic framework in NDTB-1 contributes to its superb anion exchange properties. Hinteregger et al. prepared the third member of the thorium borates system, ThB4O8, which was synthesised under extreme conditions (5.5 GPa/1100 °C) through a high temperature/high pressure (HT/HP) method.10 Its structure is based on 2D polyborate layers composed of corner-sharing BO4 tetrahedra where the ten-fold coordinated thorium cations are located in the interlayer space. Currently, there are no further reports of other thorium borates than the three described.
With the motivation of understanding the structural chemistry and behavior of thorium borates under variable conditions a part of our general focus on exploring chemistry of actinide compounds,2–9 we have examined the Th–B–O system towards expanding knowledge of it and ascertaining fundamental chemical insight. Herein, we report a new polymorph in the ThB2O5 family, β-ThB2O5, from HT/HP conditions and examine the temperature and pressure relationship it has to the only other reported polymorph,1 α, using experimental and theoretical methods. Interestingly although β-ThB2O5 is found to be the preferred higher pressure and temperature phase, relative to α-ThB2O5, it can also be accessed through careful choice of flux under ambient pressure solid state high temperature conditions. The synthetic conditions, structural chemistry and topology, phase relationship and phase decomposition process, thermodynamics stability, theoretical analysis as well as Raman and IR spectra of both polymorphs are discussed.
2. Experiment section
2.1 Materials and methods
Thorium nitrate Th(NO3)4·5H2O (International Bioanalytical Industries, Inc.), Lithium carbonate Li2CO3 (Alfa-Aesar, 99.9%), Boric acid H3BO3 (Alfa-Aesar, 99.9%), Sodium Tetraborate Na2B4O7 (Alfa-Aesar, 99.5%), Boron Oxide B2O3 (Alfa-Aesar, 99.5%).2.2 Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.2 at ∼780 °C for 36 hours. The samples were then washed with boiled water and ethanol several times to eliminate impurities.
:B = 1:6 were used. All the reagents were thoroughly ground in an agate mortar and then transferred to a small platinum crucible of 4 mm diameter and 7 mm height which was sealed. A pressure of 4.0 GPa was applied within 0.5 hours before heating (cold piston in). The reaction mixtures were kept at 4.0 GPa for the whole experimental run. The temperature was increased up to 1000 °C in 0.5 hours. The temperature was held at 1000 °C for 6 hours then slowly cooled down to 700 °C with a rate of 5 °C h−1. The following next step cooled the assembly down to 450 °C at a rate of 10 °C h−1 followed by quenching to room temperature and subsequent pressure release. For extracting the resulting products, the platinum crucible was broken. Colorless pallet shaped crystals were isolated.
:2.2 at ∼780 °C for 36 hours. The samples were then washed with boiled water and ethanol several times to eliminate impurities.
:B = 1:6 were used. All the reagents were thoroughly ground in an agate mortar and then transferred to a small platinum crucible of 4 mm diameter and 7 mm height which was sealed. A pressure of 4.0 GPa was applied within 0.5 hours before heating (cold piston in). The reaction mixtures were kept at 4.0 GPa for the whole experimental run. The temperature was increased up to 1000 °C in 0.5 hours. The temperature was held at 1000 °C for 6 hours then slowly cooled down to 700 °C with a rate of 5 °C h−1. The following next step cooled the assembly down to 450 °C at a rate of 10 °C h−1 followed by quenching to room temperature and subsequent pressure release. For extracting the resulting products, the platinum crucible was broken. Colorless pallet shaped crystals were isolated.
A nearly pure polycrystalline sample of β-ThB2O5 was prepared quantitatively by reacting a mixture of Th(NO3)4·5H2O(0.306 g, 0.54 mmol) and H3BO3 (0.090 g, 1.45 mmol) with a molar ratio of 3:8 and exposed to conditions of 4 GPa/900 °C for 36 hours. The samples were then washed with boiling water several times to eliminate impurities. Energy dispersive X-ray spectroscopy (EDS) elemental analysis on several single crystals gave an average molar ratio of Th:O = 1:5.35 for β-ThB2O5, which is in good agreement with those obtained from single crystal X-ray diffraction studies (Fig. S1†).
2.3. Ex situ and in situ experimentation on α and β phases
2.4 Analysis crystallographic study and X-ray powder diffraction
| a R 1 = Σ||Fo| − |Fc||/Σ|Fo| b wR2 = {Σw[(Fo)2 − (Fc)2]2/Σw [(Fo)2]2}1/2 | ||
|---|---|---|
| Compound | β-ThB2O5 | α-ThB2O51 |
| FW | 333.66 | 333.66 |
| Space group | P21/m | C2/c |
| a (Å) | 4.2535(2) | 11.545(3) |
| b (Å) | 6.8733(3) | 6.937(2) |
| c (Å) | 6.3242(3) | 10.263(3) |
| α (°) | 90 | 90 |
| β (°) | 106.316(5) | 101.5(3) |
| γ (°) | 90 | 90 |
| V (Å3) | 177.445(14) | 805.44 |
| Z | 2 | 8 |
| λ (Å) | 0.71073 | |
| F(000) | 280 | |
| D c (g cm−3) | 6.245 | 5.500 |
| GOF on F2 | 0.851 | |
| R 1 | 0.0261 | |
| wR2b | 0.0609 | |
θ = 10–80° with 5 s/step and a step width of 0.02°. The aperture of the fixed divergence slit was set to 0.2 mm and the receiving slit with 8.0 mm, respectively. The discriminator of the detector was set to an interval of [0.16–0.25 V]. Collected data for the α-ThB2O5/β-ThB2O5 mix pre- and post-HT/HP treatment were analysed using the Rietveld refinement method using the program GSAS-II13 Starting models were used derived from SC-XRD structural solutions where the peak shapes were modelled using a pseudo-Voigt function and the background was estimated using a 6 term shifted Chebyschev function. The scale factor, detector, zero point, lattice parameters, atomic coordinates and atomic displacement parameters for cations and phase fractions were refined together with the peak profile parameters.
2.5 Scanning electron microscopy (SEM)/energy-dispersive X-ray spectroscopy (EDS) analysis
Scanning electron microscopy image and energy-dispersive X-ray spectroscopy (SEM/EDS) was collected on a FEI Quanta 200F Environment Scanning Electron Microscope with a low-vacuum mode at 0.6 mbar. SEM/EDS results were given as ESI (Fig. S1†).2.6 Raman and IR spectroscopy
Unpolarised Raman spectra were measured with a Horiba LabRAM HR spectrometer using a Peltier cooled multichannel CCD detector. An objective lens with a 50× magnification was linked to the spectrometer, allowing the analysis of samples as small as 2 μm in diameter. The samples were in the form of single crystals. The incident radiation was produced by a He–Ne laser line at a power of 17 mW (λ = 632.8 nm). The focal length of the spectrometer was 800 mm, and an 1800 gr mm−1 grating was used. The spectral resolution was approximately 1 cm−1 with a slit of 100 μm. The spectrum was recorded in the range of 100–4000 cm−1. Infrared (IR) spectrum was measured using polycrystalline samples mixed with KBr powder, with FT-IR spectrometer Bruker Equinox 55 at room temperature.2.7 Bond-valence analysis
As a common method in coordination chemistry to evaluate the oxidation states of atoms, BVS of all atoms in the thorium borate phase were calculated and agree well with corresponding formal values of oxidation states. The bond-valence parameters for Th(IV)–O and B(III)–O were used according to Brese and O'Keeffe.142.8 Ab initio calculations
The computational studies of the ThB2O5 system were performed with density functional theory (DFT) method using Quantum-ESPRESSO15 simulation package. Because we are interested in good computations of structures of considered materials we applied PBEsol exchange/correlation functional,16 which was successfully applied in the previous studies of an-bearing systems.8,17 The core electrons of the constituent atoms were modeled with ultrasoft pseudopotentials18 and the 6s26p66d27s2 electrons of thorium atom were computed explicitly. Based on previous experience with computing Th-bearing systems, we selected 50Ryd as plane-wave energy cutoff.19The α- and β-ThB2O5 structures were modeled with 16 and 64 atoms contained supercells and the calculations were performed on the 6 × 4 × 4 and 2 × 4 × 3 Methfessel–Paxton k-points grids.20 The calculations of phonons and vibrational entropy contributions to Gibbs free energies were performed with the density functional perturbation theory using PHONON and DYNMAT packages of the Quantum-ESPRESSO code.
3.1 Results and discussion
3.1 Syntheses
β-ThB2O5 was successfully synthesised via a HT/HP method with conditions of 4 GPa and temperature of 1000 °C. It was further found that β-ThB2O5 could be successfully synthesised through a high temperature flux method with H3BO3–Li2CO3. It was found that by switching the fluxes from H3BO3–Li2CO3 to B2O3–Na2B4O7 the known α polymorph1 could be obtained. It is pertinent that we used different initial reactants (Th(NO3)4·5H2O/Li2CO3/H3BO3) for preparation of β-ThB2O5 compared to (ThO2/Na2B4O7/B2O3) the synthesis of α-ThB2O5. It was found that by heating a sample α-ThB2O5 between 800 to 1200 °C, β-ThB2O5 could be obtained indicating that α transforms to β with increasing temperature. If a mixture of α-ThB2O5 and β-ThB2O5 or a single sample of α-ThB2O5 was exposed to HT/HP conditions of 4 GPa, ∼900 °C, only the β polymorph with some additional minor ThO2 was obtained. This indicates that a pressure induced phase transition occurs from α to β-ThB2O5 with increasing pressure which is logical because β-phase has significantly higher density. A difference in volume of ∼12 Å3, at P = 4 GPa, corresponds to ΔVP = 29 kJ mol−1, which is larger than the difference in the free energy calculated between α and β phases (Table 2) from ab initio calculations. Hinteregger et al. reported the synthesis of ThB4O8, under similar HT/HP conditions to that of the present investigation (5.5 GPa, 1000 °C).10 The density of ThB4O8 (5.970 g cm−3) is larger than that of α-ThB2O5 (5.50 g cm−3) and smaller compared to that of β-ThB2O5 (6.245 g cm−3).| ΔU (0 K) | ΔU + ZPE (0 K) | ΔG (300 K) | ΔG (1000 K) | ΔG (1200 K) | ΔG (1500 K) | |
|---|---|---|---|---|---|---|
| ΔG = G(β) − G(α) | 34.4 | 31.8 | 28.1 | 12.6 | 8.1 | 1.5 |
3.2 Structural description of β-ThB2O5 and comparison to α-ThB2O5
Structural refinements against single crystal X-ray diffraction data reveal that novel β-ThB2O5 crystallises in the monoclinic space group P21/m. It consists of one crystallographically independent Th, two B and four O atoms. The 3D framework of β-ThB2O5 is based on 1D B chains and 2D Th layers (see Fig. 1). In the structure of β-ThB2O5, B(1) atoms are existing as B(1)O4 tetrahedra and B(2) atoms are forming B(2)O3 planar triangles. B(1)O4 tetrahedra and B(2)O3 triangles are connected with each other through corner sharing, forming a 1D zigzag B–O chain along the a-axis (see Fig. 1b). The oxo-borate fragments are polymerising into 2D layers in the structure of the phases from the family of closely related compounds AB2O5 (A = Zr, Hf).21,22 B atoms occur only as BO4 tetrahedra within the structures of AB2O5 (A = Zr, Hf). | ||
| Fig. 1 Polyhedral representation of β-ThB2O5 structure. The 3D thorium borate structure along the a-axis (a), a 1D boron–oxygen chain along the a-axis (b), a 2D thorium layer on ab-plane (c). Thorium polyhedra, BO3 triangles and BO4 tetrahedra are shown in yellow and green, oxygen atoms are shown in red, respectively. | ||
In β-ThB2O5, two BO4 tetrahedra are corner sharing with two ThO10 polyhedra, forming four-membered ring channels along the a-axis. B–O bond lengths in B(1)O4 tetrahedra are found to range from 1.455(7) Å to 1.472(11) Å. The O–B(1)–O bond angles are in the range of 105.8(7)°–113.1(5)°. The B–O bond distances in B(2)O3 are in the range from 1.356(11) Å to 1.390(11) Å. The O–B(2)–O bond angles are in the range of 114.2(7)°–127.4(8)°, which are consistent with the borates reported previously (see Table S2†).23
The Th atoms in β-ThB2O5 are coordinated by eight shorter and two longer Th–O bonds giving a ten-fold oxygen coordination, resulting in ThO8+2 tetra-caped trigonal prisms (Fig. 2 and Fig. S6). The shorter eight Th–O bonds range in length from 2.302(5) Å to 2.583(6) Å, whereas the additional longer two Th-O bonds are 3.067(6) Å. These ThO8+2 polyhedra are edge and face sharing with each other forming 2D sheets parallel to the ab-plain (see Fig. 1c). The zigzag B–O chains further link the 2D Th-based sheets, through corner (BO4), edge (BO3), and face (BO4) sharing, and form the 3D Th borate framework (see Fig. S7†). In the 3D thorium borate framework, a four-membered ring channel can be observed along the a-axis. The size of this channel is ∼4.5 Å × 3.5 Å (distances are between two opposite O–O atoms) (see Fig. S8†). BVS calculations indicate that Th cations are tetravalent with a value of 3.97. The BVS values suggest that the B cation is trivalent with B(1) and B(2) BVS of ∼3.07 and ∼2.97, respectively.14
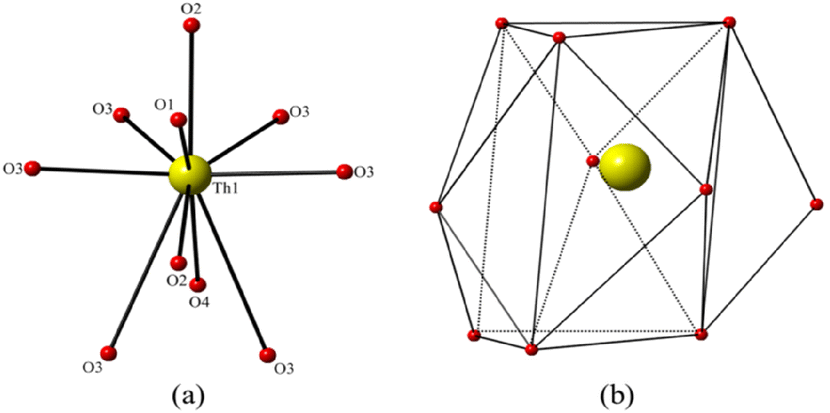 | ||
| Fig. 2 (a) The oxygen coordination environment of thorium cation with ball and stick mode, (b) the four caped trigonal prismatic shape of thorium polyhedra for β-ThB2O5. Th and O atoms are shown as yellow and red, respectively. | ||
Contrasting the structures of α-ThB2O5 and β-ThB2O5, both possess 3D framework structures, which are composed of two independent B and one Th atoms per unit cell. However, B atoms are only presented as BO3 triangles in the α phase.
In the structure of α-ThB2O5, two BO3 triangles are corner sharing linked forming isolated B2O5 dimers. Each BO3 triangle is corner sharing with two and edge sharing with one Th polyhedra, forming BTh3 fragments with linear geometry (see Fig. S9†). In the structure of β-ThB2O5, as described above, B(1)O4 tetrahedra and B(2)O3 triangles are corner sharing and form 1D zigzag chains. Within the 1D B–O chains, the two shortest B–B distances are 2.489(3) Å and 2.594(1) Å, respectively. For comparison, the shortest B(1)–B(2) distance in the structure of α-ThB2O5 is 2.445(1) Å. Th cations are eight-fold coordinated in the structure of α-ThB2O5 and exist as distorted one-caped pentagonal bipyramids. These ThO8 distorted one-caped pentagonal bipyramids are edge sharing with each other, forming a 3D Th–O framework. Each Th cation is connected to four other Th cations in a diamond packing mode in the structure of α-ThB2O5. The four shortest Th–Th distances are ranging from 4.071(1) Å to 4.131(3) Å in the structure of α-ThB2O5. There are seven neighboring Th cations with Th-Th distances ranging from 3.965(5) Å to 4.411(2) Å in the structure of β-ThB2O5 (see Fig. 3a and b). We suspect that changing of packing modes between α- and β-ThB2O5 is necessary for the large molar volume change (∼12%), where there is a similar density increase α to β phase. This is comparable with to the ThMo2O8 polymorphs, where a large volume change (∼20%) for the HT/HP–ThMo2O8 modification is described compared with its other ambient pressure polymorphs.19
 | ||
| Fig. 3 The adjacent Th–Th bond distances in two polymorphs for α-ThB2O5 (a) and β-ThB2O5 (b). | ||
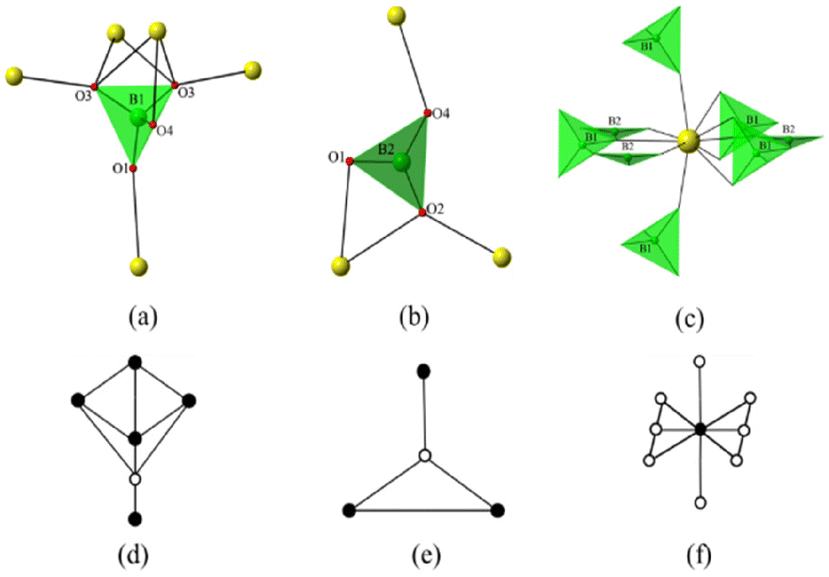 | ||
| Fig. 4 For β-ThB2O5; the coordination environment of a B(1)O4 tetrahedron (a), coordination environment of a B(2)O3 triangle (b), coordination environment of ThO8+2 polyhedra (c) and the corresponding schematic topological representation of square pyramid geometry (d), schematic topological representation of a trigonal geometry (e), schematic topological representation of a hexagonal bipyramidal shape of Th coordination (f). | ||
3.3 Temperature and pressure relationship between α- and β-ThB2O5
To understand the temperature and pressure dependence of ThB2O5 polymorphs, both were examined using in situ powder X-ray diffraction, and under ex situ HT/HP conditions (post PXRD examination). | ||
| Fig. 5 Temperature dependence of a portion of the PXRD profiles for α-ThB2O5 heated from 0 to 1030 °C illustrating the transition from the α to β phase between 807 °C and 927 °C. | ||
Interestingly, continual heating above 840 towards 927 °C further reflections can be observed to occur as can be seen in Fig. 5 but also particularly highlighted in Fig. 6. That these reflections occur independently with respect to the main reflections of β-ThB2O5 indicates that the sample is undergoing a discontinuous 1st order phase transformation. This corresponds to the formation of β-ThB2O5. The phase transformation motif is further supported when the refined lattice volumes are compared (Fig. S4†). β-ThB2O5 is found to occur only over a narrow temperature range as further heating above 1020 °C results in its decomposition. This is supported by TGA measurements performed on α-ThB2O5 to 1200 °C (see section below).
 | ||
| Fig. 6 Portion of PXRD data between 13 and 24° for ThB2O5 heated from 807 to 1007 °C. The occurrence of additional reflections above 887 °C (most noticeable at 14.9, 19.5 and 22° as shown by the arrows) indicates a 1st order phase transformation is occurring resulting in β-ThB2O5. | ||
 | ||
Fig. 7 Rietveld refinements profiles for the α-ThB2O5/β-ThB2O5 mix pre- (top) and post- (bottom) HT/HP treatment. For top; β-ThB2O5 was refined in a monoclinic structure (SG = P21/m), where a = 4.2400(16) Å, b = 6.8504(7) Å, c = 6.302(3) Å, and β = 106.300(7)°, α-ThB2O5 was refined in a monoclinic structure (SG = C2/c), where a = 11.548(7) Å, b = 6.9360(20) Å, c = 10.272(6) Å, and β = 101.56(2)°, ThO2 was refined in a cubic structure (SG = Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), where a = 5.597(1) Å and where wR = 15.58%. For bottom; β-ThB2O5 was refined in a monoclinic structure (SG = P21/m), where a = 4.2417(8) Å, b = 6.8549(5) Å, c = 6.3047(19) Å, and β = 106.305(4)°, ThO2 was refined in a cubic structure (SG = Fmm), where a = 5.5304(8) Å, and where wR = 17.60%. m), where a = 5.597(1) Å and where wR = 15.58%. For bottom; β-ThB2O5 was refined in a monoclinic structure (SG = P21/m), where a = 4.2417(8) Å, b = 6.8549(5) Å, c = 6.3047(19) Å, and β = 106.305(4)°, ThO2 was refined in a cubic structure (SG = Fmm), where a = 5.5304(8) Å, and where wR = 17.60%. | ||
For the initial (pre-HT/HP) α-ThB2O5/β-ThB2O5 mixture monoclinic models in space groups C2/c and P21/m could be refined against the PXRD data consistent with α-ThB2O5 and β-ThB2O5 respectively. An additional ThO2 impurity phase was also identified, speculated to arise from the initial as synthesised ThB2O5. Phase analysis indicated the sample to be 85.6(15), 12.9(6) and 1.48(2) wt% for α-ThB2O5, β-ThB2O5 and ThO2 respectively largely consistent with initial experimental input amounts. For the post-HT/HP α-ThB2O5/β-ThB2O5 mixture, monoclinic and cubic models in space group P21/m and Fmm respectively consistent with β-ThB2O5 and ThO2 could be successfully refined against the collected PXRD data. Phase analysis indicates the sample to be 97(1) and 2.3(3) wt% for β-ThB2O5 and ThO2 respectively. From these ex situ measurements and analysis, it indicates that the application of HT/HP conditions to the α-ThB2O5/β-ThB2O5 mix results in the conversion of α-ThB2O5 to β-ThB2O5, such that β-ThB2O5 is the preferred phase under HT/HP conditions. The occurrence of a slightly increased relative amount of ThO2 is speculated to be related to partial sample decomposition occurring.
3.4 Vibrational spectroscopy
Raman spectrum of β-ThB2O5 was measured in the region of 100–1500 cm−1. As shown in Fig. S13,† the modes in low-frequency region from 187–389 cm−1 are raised from the lattice vibrations. The Raman band with a very strong peak around 526 cm−1 can be attributed to the bending character ν4 mode of BO4 tetrahedra. Raman bands with weak peaks around 800 cm−1 are due to the symmetric stretching ν1 mode of BO4 units. The Raman band at around 966 cm−1 were assigned to the B–O–B symmetrical stretching ν3 mode in BO3 triangles. The peaks around 1362 cm−1 can be attributed to the doubly degenerate asymmetrical stretching ν4 mode of the B–O bonds in the trigonal BO3 units. For α-ThB2O5, Raman band with a strong peak at 472 cm−1 are attributed to the doubly degenerate in-plane O–B–O bending ν1 mode from the planar trigonal BO3 groups. The weak peaks at 700 cm−1 and 803 cm−1 can be assigned to the B–O bending ν2 mode in the BO3 triangles. The symmetrical stretching ν3 modes of BO3 groups have caused the peaks at Raman bands of 918 cm−1 and 1000 cm−1. The Raman band with a weak peak at 1416 cm−1 is attributed to the doubly degenerate asymmetrical stretching ν4 mode of BO3 units.26,27IR spectra of both phases were measured in the range of 4000–400 cm−1 (see Fig. 8 black curve). The IR spectra of both phases show very high transmittance in the range of 4000–1600 cm−1 (2.50–6.25 μm). The absorption bands between 1027 cm−1 and 1438 cm−1 in both curves can be assigned to the antisymmetric stretching vibrations of the BO3 groups in the two compounds. The peaks associated with the BO4 symmetric stretch vibrations appeared in the range of 928–735 cm−1 for β-ThB2O5. The absorption peaks at 529–652 cm−1 can be assigned to bending vibrations of BO3 and BO4 groups for α-ThB2O5 and BO3 groups for β-ThB2O5. These assignments are consistent with the borates reported previously.22
 | ||
| Fig. 8 IR spectra experimental (black) and calculated (red) curves of α-ThB2O5 and β-ThB2O5. | ||
3.5 DFT investigations of α- and β-polymorphs
To understand the thermodynamic stability and spectroscopic properties of studied polymorphic modifications we performed a DFT study of both structures. The computed lattice parameters for the α-ThB2O5 and β-ThB2O5 phases are reported in Table S3.† The lattice parameters of β-ThB2O5 phase are reproduced with surprisingly good accuracy with relative error much smaller than 1% and the volume being off from the experimental value by just 0.6%. On the other hand, the lattice parameters of α-ThB2O5 are off the experimental values by up to 1.5% with the volume being overestimated by 2.5%, although this is still reasonably good performance of DFT. Despite these offsets, we notice that both structures are well reproduced, and the DFT-based geometry relaxation did not result in significant deformation of the experimental structures, which were taken as starting configurations. In Table 2 we present the values of relative internal energies (enthalpies) and free energies computed via adding vibrational entropy and zero point energy contributions for temperatures of 300K (ambient conditions), 1000 K, 1200 K and 1500 K. The Gibbs free energies are computed as:| G = U + ZPE − ST |
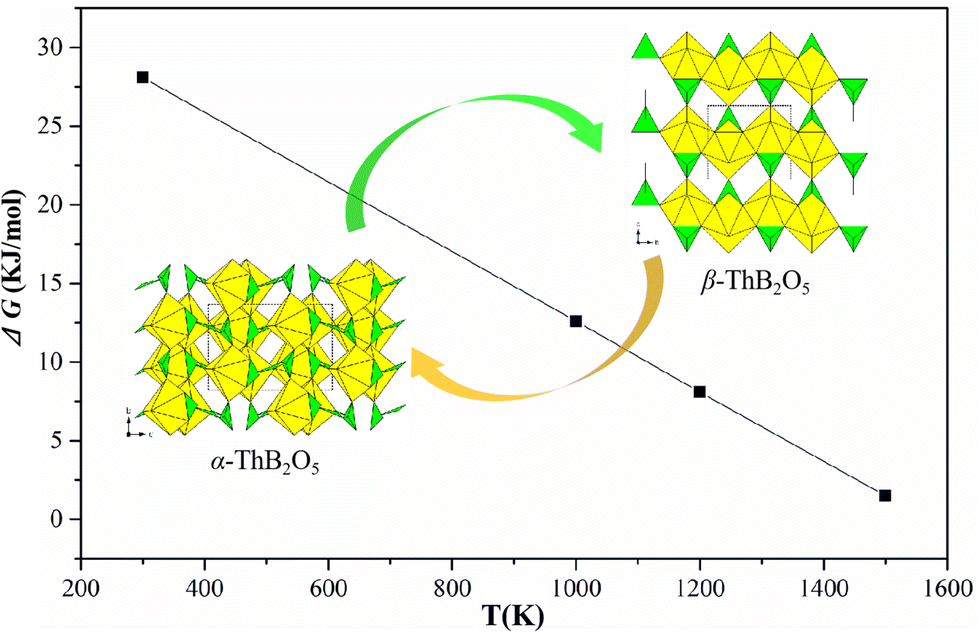 | ||
| Fig. 9 The computed difference in Gibbs free energy between α-ThB2O5 and β-ThB2O5 phases. ΔG = G(β-ThB2O5) − G(α-ThB2O5). | ||
The calculations show that α-ThB2O5 phase is the more stable phase at low temperature with the internal energy difference at 0 K of 34.4 kJ mol−1. This is consistent with experimental observation which have been discussed in thermal and in situ PXRD analyses section. Increasing the temperature, the free energy difference decreases reaching zero at temperature just above 1500 K.
4. Conclusions
Only the second known polymorph of ThB2O5, β-ThB2O5, was synthesised from HT/HP conditions and characterised using X-ray single crystal, powder diffraction and spectroscopic methods. The structure of β-ThB2O5 is starkly different in comparison to the known α-phase1 and to the wider family of AB2O5 compounds (A = Zr, Hf).21,22 The β-modification features a dense 3D thorium borate framework, which is composed of [36] topological 2D thorium layers on the ab-plane and 1D boron–oxygen zigzag chains along the a-axis. The structure further possesses tetra-capped trigonal prisms of ThO10 polyhedra that are apparently the first observed in inorganic thorium compounds. The previously known α variant with regard to the β, was found to be the lower temperature and lower pressure preferred phase as shown by HT/HP synthesis experiments, in situ heating PXRD measurements and supported by ab initio calculations. From in situ heating PXRD measurements the α polymorph was observed to transform to the β form at approximately 900 °C via a 1st order discontinuous phase transformation. β-ThB2O5 was found to exist only over a narrow temperature range, decomposing above 1050 °C. Interestingly it was found that by switching fluxes from borax, used to synthesis the α variant under high temperature conditions, to carbonates, the β variant could be synthesised, which was identical to that obtained from HT/HP synthesis. This study demonstrates how subtle changes to synthesis conditions, including temperature, pressure but also and pertinently choice of flux, can dramatically influence different results in structure formation of ThB2O5 polymorphs and potentially maybe applicable in synthesis design of other inorganic compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors are grateful to the Natural Science Foundation of the Education Department of Anhui Province (KJ2019A0831), Hefei University Talent Funding (18-19RC14), Natural Science Foundation of Anhui Province (2008085QB56), HFUU Bilingual Course Construction Project (2021Yyykc02) and Teaching and Research Project of Hefei 2019hfjyxm09. E. V. A. is grateful for DFG for the funding within the AL1527/3-1 project.References
- A. Cousson and M. Gasperin, Synthesis and Structure of Thorium Borate: ThB2O5, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, C47, 10 CrossRef CAS.
- J. Arnold, T. L. Gianetti and Y. Kashtan, Thorium lends a fiery hand, Nat. Chem., 2014, 6, 554 CrossRef CAS PubMed.
- (a) D. L. Clark, M. P. Neu, W. Runde and D. W. Keogh, Kirk-Othmer Encyclopedia of Chemical Technology, 2006 Search PubMed; (b) G. L. Murphy, Y. Wang, P. Kegler, Y. Wang, S. Wang and E. V. Alekseev, The first actinide polyiodate: a complex multifunctional compound with promising X-ray luminescence properties and proton conductivity, Chem. Commun., 2021, 57, 496 RSC.
- (a) R. M. Hazen, R. C. Ewing and D. A. Sverjensky, Evolution of uranium and thorium minerals, Am. Mineral., 2009, 94, 1293 CrossRef CAS; (b) G. L. Murphy, P. Kegler, M. Klinkenberg, A. Wilden, M. Henkes, D. Schneider and E. V. Alekseev, Incorporation of iodine into uranium oxyhydroxide phases, Dalton Trans., 2021, 50, 17257 RSC; (c) G. L. Murphy, B. J. Kennedy, J. A. Kimpton, Q. Gu, B. Johannessen, G. Beridze, P. M. Kowalski, D. Bosbach, M. Avdeev and Z. Zhang, Nonstoichiometry in Strontium Uranium Oxide: Understanding the Rhombohedral–Orthorhombic Transition in SrUO4, Inorg. Chem., 2016, 55, 9329 CrossRef CAS PubMed.
- (a) W. Stoll, Ullmann's Encyclopedia of Industrial Chemistry, 2000 Search PubMed; (b) G. L. Murphy, C.-H. Wang, G. Beridze, Z. Zhang, J. A. Kimpton, M. Avdeev, P. M. Kowalski and B. J. Kennedy, Unexpected crystallographic phase transformation in nonstoichiometric SrUO4−x: Reversible oxygen defect ordering and symmetry lowering with increasing temperature, Inorg. Chem., 2018, 57, 5948 CrossRef CAS PubMed.
- (a) P. C. Burns, The Crystal Chemistry of Uranium in Uranium: Mineralogy Geochemistry and The Environment, in Reviews in Mineralogy and Geochemistry, ed. P. C. Burns and R. Finch, Mineralogical Society of America, Washington, D. C., 1999, vol. 38, pp. 23 CrossRef; (b) G. L. Murphy, Z. Zhang, R. Tesch, P. M. Kowalski, M. Avdeev, E. Y. Kuo, D. J. Gregg, P. Kegler, E. V. Alekseev and B. J. Kennedy, Tilting and Distortion in Rutile-Related Mixed Metal Ternary Uranium Oxides: A Structural, Spectroscopic, and Theoretical Investigation, Inorg. Chem., 2021, 60, 2246 CrossRef CAS PubMed; (c) G. L. Murphy, B. J. Kennedy, Z. Zhang, M. Avdeev, H. E. A. Brand, P. Kegler and E. V. Alekseev, Structure and phase transition in BaThO3: A combined neutron and synchrotron X-ray diffraction study, J. Alloys Compd., 2017, 727, 1044 CrossRef CAS.
- (a) C. Xiao, A. Khayambashi and S. Wang, Separation and Remediation of 99TcO4− from Aqueous Solutions, Chem. Mater., 2019, 31, 3863 CrossRef CAS; (b) L. Jian, K. T. Praveen and S. Denis, Metal–Organic Frameworks for Removal of Xe and Kr from Nuclear Fuel Reprocessing Plants, Langmuir, 2012, 28, 11584 CrossRef PubMed; (c) S. L. Griffin and N. R. Champness, A periodic table of metal-organic frameworks, Coord. Chem. Rev., 2020, 414, 213295 CrossRef CAS.
- B. Xiao, J. Dellen, H. Schlenz, D. Bosbach, E. V. Suleimanov and E. V. Alekseev, Unexpected Structural Complexity in Cesium Thorium Molybdates, Cryst. Growth Des., 2014, 14, 2677 CrossRef CAS.
- S. Wang, E. V. Alekseev, J. Diwu, W. H. Casey, B. L. Phillips, W. Depmeier and T. E. Albrecht Schmitt, NDTB-1: A Supertetrahedral Cationic Framework that Removes TcO4− from Solution, Angew. Chem., Int. Ed., 2010, 49, 1057 CrossRef CAS PubMed.
- E. Hinteregger, T. S. Hofer, G. Heymann, L. Perfler, F. Kraus and H. Huppertz, High-Pressure Synthesis and Characterization of New Actinide Borates, AnB4O8 (An = Th, U), Chem. – Eur. J., 2013, 19, 15985 CrossRef CAS PubMed.
- (a) CrystalClear, version 1.3.5, Rigaku Corp., Woodlands, TX, 1999 Search PubMed; (b) G. M. Sheldrick, SHELXTL, Crystallographic Software Package, version 5.1, Bruker-AXS, Madison, WI, 1998 Search PubMed; (c) A. L. Spek, PLATON, Utrecht University, Utrecht, The Netherlands, 2001 Search PubMed; (d) V. A. Blatov and V. N. Serezhkin, Stereoatomic Model of the Structure of Inorganic and Coordination Compounds, Russ. J. Inorg. Chem., 2000, 45, S105 Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock and P. A. Wood, Mercury CSD 2.0-new features for the visualization and investigation of crystal structures, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, GSAS-II: the genesis of a modern open-source all purpose crystallography software package, J. Appl. Crystallogr., 2013, 46, 544 CrossRef CAS.
- (a) I. D. Brown and D. Altermatt, Bond-valence parameters obtained from a systematic analysis of the inorganic crystal structure database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1985, B41, 244 CrossRef CAS; (b) N. E. Brese and M. O'Keeffe, Bond-valence parameters for solids, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1991, B47, 192 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. F. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.: Condens. Matter, 2009, 21, 5502 CrossRef PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Restoring the Density-gradient Expansion for Exchange in Solids and Surfaces, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- G. Murphy, B. Kennedy, J. Kimpton, Q.-F. Gu, B. Johannesen, G. Beridze, P. M. Kowalski, D. Bosbach, M. Avdeev and Z. Zhang, Nonstoichiometry in Strontium Uranium Oxide: Understanding the Rhombohedral–Orthorhombic Transition in SrUO4, Inorg. Chem., 2016, 55, 9329 CrossRef CAS PubMed.
- D. Vanderbilt, Soft Self-consistent Pseudopotentials in a Generalized Eigenvalue Formalism, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892 CrossRef PubMed.
- X. Bin, K. Philip, M. G. Thorsten, R. Lars, A. Kowalski, P. Li, Y. Klepov, V. V. Bosbach, D. Alekseev and E. V, Giant Volume Change and Topological Gaps in Temperature–and Pressure–Induced Phase Transitions: Experimental and Computational Study of ThMo2O8, Chem. – Eur. J., 2016, 22, 946 CrossRef PubMed.
- M. Methfessel and A. T. Paxton, High-precision Sampling for Brillouin-zone Integration in Metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616 CrossRef CAS PubMed.
- J. S. Knyrim and H. Huppertz, High-Pressure Synthesis, Crystal Structure, and, Properties of the First Ternary Hafniumborate β-HfB2O5, J. Solid State Chem., 2007, 180, 742 CrossRef CAS.
- J. S. Knyrim and H. Huppertz, High-pressure Synthesis, Crystal Structure, and, Properties of the First Ternary Zirconium Borate β-ZrB2O5, Z. Naturforsch., B, 2008, 63, 707 CrossRef CAS.
- (a) S. Wang, E. V. Alekseev, J. T. Stritzinger, G. Liu, W. Depmeier and T. E. Albrecht-Schmitt, Structure-Property Relationships in Lithium, Silver, and Cesium Uranyl Borates, Chem. Mater., 2010, 22, 5983 CrossRef CAS; (b) Y. Hao, V. V. Klepov, G. L. Murphy, G. Modolo, D. Bosbach, T. E. Albrecht-Schmitt, B. J. Kennedy, S. Wang and E. V. Alekseev, Influence of Synthetic Conditions on Chemistry and Structural Properties of Alkaline Earth Uranyl Borates, Cryst. Growth Des., 2016, 16, 5923 CrossRef CAS; (c) Y. Hao, P. Kegler, D. Bosbach, T. E. Albrecht-Schmitt, S. Wang and E. V. Alekseev, Divergent Structural Chemistry of Uranyl Borates Obtained from Solid State and Hydrothermal Conditions, Cryst. Growth Des., 2017, 17, 5898 CrossRef CAS; (d) Y. Hao, G. L. Murphy, D. Bosbach, G. Modolo, T. E. Albrecht-Schmitt and E. V. Alekseev, Porous Uranyl Borophosphates with Unique three dimensional Open Framework Structure, Inorg. Chem., 2017, 56, 9311 CrossRef CAS PubMed.
- Y. Li, Z. Weng, Y. Wang, L. Chen, D. Sheng, Y. Liu, J. Diwu, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Centrosymmetric and chiral porous thorium organic frameworks exhibiting uncommon thorium coordination environments, Dalton Trans., 2015, 44, 20867 RSC.
- (a) Y. Li, Z. Weng, Y. Wang, L. Chen, D. Sheng, J. Diwu, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Surprising Coordination for Low-valent Actinides Resembling Uranyl (VI) in Thorium(IV) Organic Hybrid Layered and Framework Structures Based on a Graphene-like (6, 3) Sheet Topology, Dalton Trans., 2016, 45, 918 RSC; (b) Y. Hao and E. V. Alekseev, Giant Volume Change and Topological Gaps in Temperature–and Pressure–Induced Phase Transitions: Experimental and Computational Study of ThMo2O8, Chem. – Eur. J., 2016, 22, 946 CrossRef PubMed.
- (a) Y. C. Hao, C. L. Hu, X. Xu, F. Kong and J. G. Mao, SrGe2B2O8 and Sr3Ge2B6O16: Novel Strontium Borogermanates with Three-Dimensional and Layered Anionic Architectures, Inorg. Chem., 2013, 52, 13644 CrossRef CAS PubMed; (b) E. Smith and G. Dent, Modern Raman spectroscopy: a practical approach, John Wiley & Sons, 2013, 298. K. Nakamoto, A. Part John Wiley & Sons, Inc., 2009; Search PubMed; (c) Y. Hao, L. He, G. Ge, Q. Zhang, N. Luo, S. Huang and E. V. Alekseev, Mg3Pt(BO3)2O2: The First Platinum Borate from the Flux Technique, J. Solid State Chem., 2020, 281, 121046 CrossRef CAS.
- (a) Y. C. Hao, X. Xu, F. Kong, J. L. Song and J. G. Mao, PbCd2B6O12 and EuZnB5O10: syntheses, crystal structures and characterizations of two new mixed metal borates, CrystEngComm, 2014, 16, 7689 RSC; (b) G. Barros, E. N. Silva, A. P. Ayala, I. Guedes, C. K. Loong and J. Y. Wang, Raman spectroscopic characterization of RECa4O(BO3)(RE=La and Gd) crystals, Vib. Spectrosc., 2008, 46, 100 CrossRef CAS; (c) Y. Hao, V. V. Klepov, P. Kegler, G. Modolo, D. Bosbach, T. E. Albrecht-Schmitt, S. Wang and E. V. Alekseev, Synthesis and Study of the First Zeolitic Uranium Borate, Cryst. Growth Des., 2017, 18, 498 CrossRef; (d) S. Wang, E. M. Villa, J. Diwu, E. V. Alekseev, W. Depmeier and T. E. Albrecht-Schmitt, Role of Anions and Reaction Conditions in the Preparation of Uranium(VI), Neptunium(VI), and Plutonium(VI) Borates, Inorg. Chem., 2011, 50, 2527 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files in CIF format, SEM and EDS measurement, selected bond distances and angles, TG-DSC curves, and vis-IR spectroscopy. CCDC 2106445. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt01049f |
| This journal is © The Royal Society of Chemistry 2022 |