
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anionic ordering in Pb2Ti4O9F2 revisited by nuclear magnetic resonance and density functional theory†‡
Kengo
Oka§
 *a,
Tom
Ichibha§
*b,
Daichi
Kato
c,
Yasuto
Noda
d,
Yusuke
Tominaga
d,
Kosei
Yamada
a,
Mitsunobu
Iwasaki
a,
Naoki
Noma
e,
Kenta
Hongo
f,
Ryo
Maezono
g and
Fernando A.
Reboredo
b
*a,
Tom
Ichibha§
*b,
Daichi
Kato
c,
Yasuto
Noda
d,
Yusuke
Tominaga
d,
Kosei
Yamada
a,
Mitsunobu
Iwasaki
a,
Naoki
Noma
e,
Kenta
Hongo
f,
Ryo
Maezono
g and
Fernando A.
Reboredo
b
aDepartment of Applied Chemistry, Faculty of Science and Engineering, Kindai University, Higashiosaka, Osaka 577-8502, Japan. E-mail: koka@apch.kindai.ac.jp
bMaterials Science and Technology Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: ichibha@icloud.com
cDepartment of Energy and Hydrocarbon Chemistry, Graduate School of Engineering, Kyoto University, Kyoto 615-8510, Japan
dDivision of Chemistry, Graduate School of Science, Kyoto University, 606-8502 Kyoto, Japan
eJoint Research Center, Kindai University, Higashiosaka, Osaka 577-8502, Japan
fResearch Center for Advanced Computing Infrastructure, JAIST, Asahidai 1-1, Nomi, Ishikawa 923-1292, Japan
gSchool of Information Science, JAIST, Asahidai 1-1, Nomi, Ishikawa 923-1292, Japan
First published on 23rd September 2022
Abstract
A combination of 19F magic angle spinning (MAS) nuclear magnetic resonance (NMR) and density functional theory (DFT) were used to study the ordering of F atoms in Pb2Ti4O9F2. This analysis revealed that F atoms predominantly occupy two of the six available inequivalent sites in a ratio of 73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :27. DFT-based calculations explained the preference of F occupation on these sites and quantitatively reproduced the experimental occupation ratio, independent of the choice of functional. We concluded that the Pb atom's 6s2 lone pair may play a role (∼0.1 eV per f.u.) in determining the majority and minority F occupation sites with partial density of states and crystal orbital Hamiltonian population analyses applied to the DFT wave functions.
:27. DFT-based calculations explained the preference of F occupation on these sites and quantitatively reproduced the experimental occupation ratio, independent of the choice of functional. We concluded that the Pb atom's 6s2 lone pair may play a role (∼0.1 eV per f.u.) in determining the majority and minority F occupation sites with partial density of states and crystal orbital Hamiltonian population analyses applied to the DFT wave functions.
1 Introduction
Understanding the mechanisms that produce ionic ordering in materials may lead to control of the self-assembly of ordered super lattices on an underlying crystal structure. Because ordered and disordered structures have remarkably different electronic and transport properties, controlling ordering is a key step in material design. For example, the properties of mixed-anion compounds strongly depend on the degree of order or disorder of the anions.1,2 Anionic ordering can cause heteroleptic coordinations or low-dimensional structures, which in turn modify electronic properties.1,2 This intriguing possibility to modify a material has attracted significant attention.3–6 Some ABX3 perovskites are typical examples. SrTaO2N and BaTaO2N have a high dielectric constant because of their O/N anionic ordering.7–9 In addition, an oxyhydride SrVO2H shows two-dimensional electron conduction and compression anisotropy because of the O/H anionic ordering.10In complex composite materials, multiple anions and cations are heteroleptically coordinated, and their concentrations typically obey valence charge neutrality conditions. When differences in ionic radii, electronegativity, or polarizability are large, some materials tend to exhibit ionic ordering.11,12 However, although O2− and F− are neighboring anions in the periodic table, the oxyfluorides nevertheless exhibit anionic ordering, depending on the structure. Whereas simple cubic perovskites (SrFeO2F,13 BaFeO2F,14 PbScO2F,15 BaScO2F,16 AgFeOF2,17 BaInO2F,18 and AgTiO2F19) adopt disordered configurations, a variety of Ruddlesden–Popper-type layered perovskites (Sr2CuO2F2,20 Sr2FeO3F,21,22 Ba2InO3F,23 Ba2ScO3F,24 Sr2MnO3F,25 Sr3Fe2O5−xFy21,26) exhibit ordered configurations of F−.27 For an ordered structure to be formed, migration energy barriers must be small enough to allow the minimum free energy configuration to be achieved via practical annealing temperatures and times. The lowest energy structure frequently can be found by considering Pauling's second rule,28 which explains that an ionic structure will be stable when the sum of the strength of the electrostatic bonds around an ion are equal to its charge. Thus, according to Pauling's second rule, F− should prefer a more open site compared with O2−, leading to O/F anionic ordering. Some nonlayered oxyfluorides with or akin to perovskite structure showing a complete or partial anionic ordering have been reported, such as Pb2Ti2O5.4F1.229 Pb2OF2,30 and Pb2Ti4O9F2.31 The anionic ordering of some nonlayered oxyfluorides is considered to be due to the Jahn–Teller distortion by the 6s2 lone pair.29,31
Among the nonlayered oxyfluorides with anionic ordering, Pb2Ti4O9F2 is especially attractive to study because this material uniquely has an isostructural oxide Bi2Ti4O11.31,32 Both Pb2+ and Bi3+ have the same electronic configuration and 6s2 lone pairs. Bi2Ti4O11 undergoes antiferroelectric-paraelectric transitions from C2/c to C2/m, whereas Pb2Ti4O9F2 does not.31,32 The high-temperature paraelectric phase of Bi2Ti4O11 (C2/m) adopts the same space group symmetry as Pb2Ti4O9F2.32 Because the Bi2Ti4O11-type structure is significantly low symmetric, an anionic ordering can exist in Pb2Ti4O9F2. Structural analyses based on the synchrotron X-ray diffraction (SXRD) patterns revealed the presence of anionic ordering.31 However, this conclusion is debatable because heavier Pb atoms in the system could hamper the identification of F occupation sites.
In this work, the anionic ordering in Pb2Ti4O9F2 was reexamined via a combination of 19F magic angle spinning (MAS) nuclear magnetic resonance (NMR) experiments and density functional theory (DFT) simulations. The 19F MAS NMR analysis revealed that F atoms randomly occupy two of the six sites in a ratio of 73:27, overturning the previous conclusion that F atoms selectively occupy a single site.31 DFT calculations identified the majority and minority F occupation sites and quantitatively reproduced the experimental occupation ratio, independent of the choice of functional. Partial density of states (PDOS) and crystal orbital Hamiltonian population (COHP) analyses were performed on the DFT results, showing that the 6s2 lone pairs may play a role (∼0.1 eV per f.u.) in determining the majority and minority F occupation sites. On the other hand, DFT calculations revealed that the low-symmetric anionic coordinates around the cations may barely be due to the steric effects of 6s2 lone pairs. This result goes against the current discussion in this class of materials and implies that the influence of 6s2 lone pairs on the structural distortion might be similarly not significant in some of the other Pb-based oxyfluorides such as Pb2Ti2O5.4F1.229 and Pb2OF2.30
2 Experimental details
The powder samples of Pb2Ti4O9F2 and Bi2Ti4O11 were prepared by solid-state reaction, as previously reported.31,32 The Pb2Ti4O9F2 was synthesized from a mixture of PbO (99.9%, Rare Metallic Co.), PbO2 (99.9%, Rare Metallic Co.), PbF2 (99.9%, Rare Metallic Co.), and TiO2 (rutile, 99.9%, Rare Metallic Co.) powders that were weighed to be 10 mol% F-rich to compensate for the loss of F during the reaction.33 The pelletized mixture was sealed in an evacuated Pyrex tube and treated at 823 K for 12 h in an electric furnace, followed by natural cooling to room temperature. The Bi2Ti4O11 was synthesized from a stoichiometric mixture of Bi2O3 (99.9%, Rare Metallic Co.) and TiO2 (99.9%, Rare Metallic Co.) powders. The pelletized mixture was treated at 1273 K for 12 h in air in an electric furnace, followed by natural cooling to room temperature.Production of a single phase for both Pb2Ti4O9F2 and Bi2Ti4O11 samples were confirmed via SXRD. SXRD patterns were collected with a large Debye-Scherrer camera installed at beamline BL02B2 of the SPring-8 synchrotron radiation facility using a glass capillary and a solid-state detector.34 The crystallographic parameters were refined by the Rietveld method using the RIETAN-FP program.35 The electron density distributions were estimated by the maximum entropy method (MEM) using the Dysnomia program.36
Solid-state NMR experiments were conducted on a homemade spectrometer with a 4 mm T3 probe (Varian) in a magnetic field of 4.7 T. All 19F NMR transients under MAS were accumulated using a background suppression method. The radio frequency field strength was 100 kHz, corresponding to 2.5 μs of π/2 pulse length. The longitudinal relaxation time (T1) was obtained by analyzing a build-up curve measured with a saturation recovery method. Rotational resonance experiments, which allow for solving whether the chemical-shift filter was set to half of the inverse of the difference between two signals and the MAS rate, was set to the inverse of the difference between two signals in rotational resonance experiments (further details given in ESI‡).
3 Calculation details
The DFT calculations were performed with Quantum Espresso.37 Perdew–Burke–Ernzerhof (PBE),38 Becke–Lee–Yang–Parr (BLYP),39,40 and Perdew–Wang 1991 (PW91)41,42 semi-local exchange–correlation functionals were employed. The valence orbitals were expanded with plane waves. The cutoff energy was 100 Ry, and the k-point mesh was 7 × 7 × 5 for a unit cell. With this choice of parameters, the energy difference between the first and second most stable anionic ordering patterns of Pb2Ti4O9F2 converged below 2 meV per f.u. These two ordering patterns are denoted as F-in-site6 and F-in-site5 in section 4.1. The core orbitals were described by the projector augmented wave (PAW) method.43 PAW pseudopotentials were taken from the pslibrary.44 Comparing the results of PAW and ultrasoft pseudopotentials in the pslibrary44 revealed that the relative total energies among the different anionic orderings are identical within 1 meV per f.u. Therefore, the errors from pseudopotential approximation would be negligible for this system. Ultrasoft pseudopotentials were used to calculate the electrostatic energies because PAW pseudopotentials cannot separately provide the electrostatic energies because the one-center term includes both electrostatic and exchange–correlation energies.45 The PDOS of Pb2Ti4O9F2 was obtained with the PBE functional. The LOBSTER code46 was used to perform the COHP analyses based on the PBE-DFT results with the pbeVaspFit2015 basis set.47,48 Every DFT calculation was performed for the energetically optimized structure.4 Results and discussion
4.1 Determination of anionic configurations
As shown in Fig. 1, Pb2Ti4O9F2 has six different anion sites, which are denoted by site 1–6. Previous structural analysis based on SXRD patterns has indicated that F atoms selectively occupy site 6, which is the closest site to the Pb atom.31 Here, that earlier conclusion is reexamined using NMR experiments and DFT simulations. | ||
| Fig. 1 The crystal structure of Pb2Ti4O9F2 at 300 K. The numbers indicate the numbers of anion sites. | ||
A 19F MAS NMR spectrum of Pb2Ti4O9F2 is shown together with a peak fitting result in Fig. 2. By fitting the spectrum with three Gaussians, the peak positions were obtained as −45 ppm, −58 ppm, and −63 ppm, with an area ratio of 26.1:1.8:72.1. The peak positions correspond to the 19F atoms in different distinct sites, and the area ratio indicates their occupancy ratio. Through-space correlation NMR experiments were conducted to confirm whether the 19F atoms showing in the two main peaks exist in the same crystal phase. When the MAS rate was matched to the resonant frequency difference of the two main peaks, both peak intensities varied periodically with mixing time. However, when the MAS rate was faster than the resonant frequency difference, neither peak intensity changed (details in ESI‡). This means that the dipole interaction that disappeared because of MAS was reintroduced by rotational resonance, and the magnetization was exchanged during the mixing time. Thus, the F sites showing the two main signals are nearby in the same crystalline phase, Pb2Ti4O9F2.
 | ||
| Fig. 2 A 19F MAS NMR spectrum of Pb2Ti4O9F2 (top) and peak fitting result (bottom). The asterisks indicate spinning sidebands. | ||
The position of the F occupation sites corresponding to the two main peaks in the 19F MAS NMR experiment must be determined using another method. Therefore, the energies of different F occupation patterns were evaluated with DFT to find the majority and minority F occupation sites corresponding to the two main peaks. In reality, numerous F occupation patterns are possible, and we cannot study all of them. Therefore, targets were restricted to be the cases in which F atoms selectively occupy each of the sites, and the F atom's stability in each site was evaluated. Table 1 lists the relative total energies given by PBE,38 BLYP,39,40 and PW9141,42 functionals. In the table, for example, F-in-site2 indicates the case in which F atoms selectively occupy site 2. The F stability at site 3 was evaluated as E (F-in-site3) ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) 2E (one F atom in site 3 and the other in site 6 in the unit cell) – E (F-in-site6) because the multiplicity of site 3 is one (multiplicity of the other sites is two). The three functionals qualitatively and quantitatively agree with each other. F-in-site6 and F-in-site5 give the first and second lowest energies, so the majority and minority F occupation sites are sites 6 and 5, respectively. The experimental third tiny peak at −58 ppm corresponds to the F occupancy in site 4. The percentages in the parentheses in Table 1 indicate the ratios of Boltzmann factors of the relative energies under the synthesis temperature, 823 K. These percentages correspond to the F occupation ratio of the anion sites. The ratio between the percentages of F-in-site6 and F-in-site5, approximately 70:30, closely agrees with that between the experimental F occupation ratios of the majority and minority sites, 73:27.
2E (one F atom in site 3 and the other in site 6 in the unit cell) – E (F-in-site6) because the multiplicity of site 3 is one (multiplicity of the other sites is two). The three functionals qualitatively and quantitatively agree with each other. F-in-site6 and F-in-site5 give the first and second lowest energies, so the majority and minority F occupation sites are sites 6 and 5, respectively. The experimental third tiny peak at −58 ppm corresponds to the F occupancy in site 4. The percentages in the parentheses in Table 1 indicate the ratios of Boltzmann factors of the relative energies under the synthesis temperature, 823 K. These percentages correspond to the F occupation ratio of the anion sites. The ratio between the percentages of F-in-site6 and F-in-site5, approximately 70:30, closely agrees with that between the experimental F occupation ratios of the majority and minority sites, 73:27.
| ΔE (PBE) | ΔE (BLYP) | ΔE (PW91) | |
|---|---|---|---|
| F-in-site1 | 1.019 (0.0%) | 0.965 (0.0%) | 1.018 (0.0%) |
| F-in-site2 | 0.935 (0.0%) | 0.889 (0.0%) | 0.937 (0.0%) |
| F-in-site3 | 0.513 (0.1%) | 0.514 (0.1%) | 0.543 (0.0%) |
| F-in-site4 | 0.321 (0.8%) | 0.308 (0.9%) | 0.322 (0.7%) |
| F-in-site5 | 0.061 (29.5%) | 0.066 (28.0%) | 0.058 (30.3%) |
| F-in-site6 | 0.000 (69.7%) | 0.000 (71.1%) | 0.000 (69.0%) |
4.2 Reason why sites 5 and 6 have a preference for F atoms
To understand the origin of the F-in-site6 and F-in-site5 stabilization, PBE,38 BLYP,39,40 and PW9141,42 functionals were used to compare the electrostatic energies with the total energies, as shown in Fig. 3. The results of the three functionals are qualitatively consistent with each other. The total energies in Fig. 3 are identical to those listed in Table 1. The total energies are roughly proportional to the electrostatic energies, consistent with a previous systematic study on NdNiO2F by simulations.49 However, F-in-site6, which had the lowest total energy, is an exception to Pauling's second rule28 because the lowest electrostatic energy was predicted for the F-in-site5.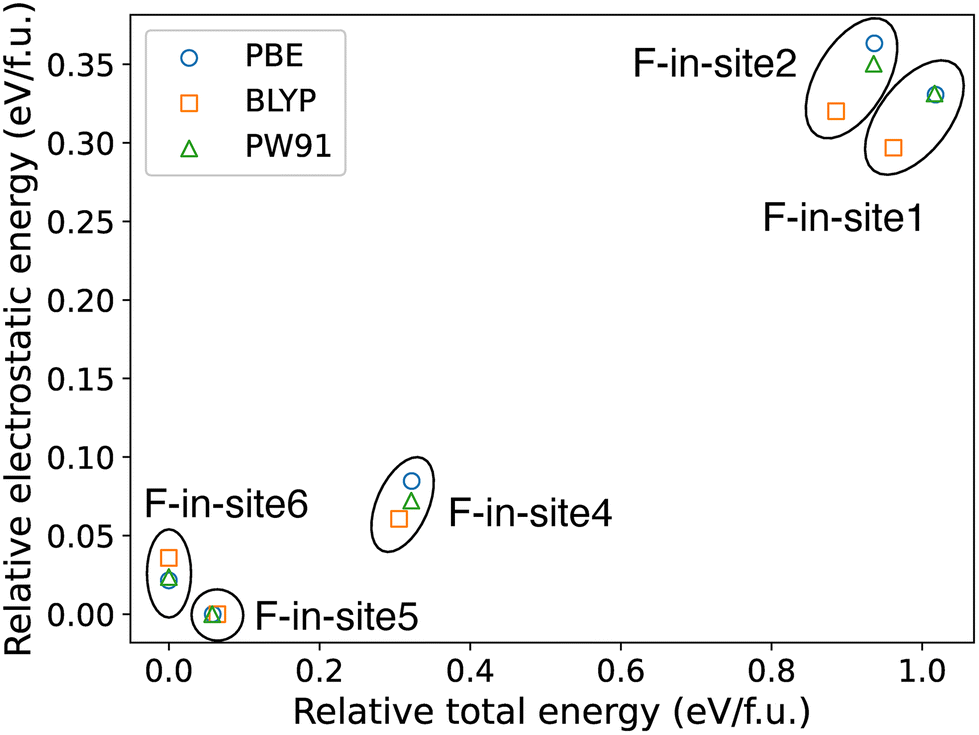 | ||
| Fig. 3 Comparison of electrostatic energies and total energies calculated by DFT, using PBE (blue circles), BLYP (orange squares), and PW91 (green triangles) functionals. The minimum electrostatic energy (i.e., F-in-site5) and total energy (i.e., F-in-site6) are set to be zero, and the others are relative to this for every functional. | ||
The reason of the small deviation (∼0.1 eV per f.u.) from Pauling's second rule28 is unclear. However, the steric effects introduced by the 6s2 lone pairs may explain this deviation. Fig. 4 shows the PDOS of each F ordering pattern in Pb2Ti4O9F2 given by the PBE functional. A peak of Pb-6s (indicated by an arrow) hybridized with O-2p exists at the valence band maximum (VBM). This peak is considered to be a signal of the orbital hybridization described by the revised lone-pair (RLP) model.50 This model proposes that the antibonding hybridized orbital of Pb-6s and O-2pz is further stabilized by hybridizing with the Pb-6p orbital. This antibonding orbital distributes opposite to the O50 and interrupts bonding with the other anions. The PDOS figures show that F-in-site6 has the smallest Pb-6s peak (i.e., the weakest RLP hybridization), presumably owing to the absence of O6 (i.e., an O ion in site 6) that is the closest to Pb and has the largest 2p density of states (DOS) at the VBM. Furthermore, the magnitudes of the hybridization between Pb-6s and O/F6-2p were evaluated for F-in-site5 and F-in-site6 in terms of integrated COHP (ICOHP) by the COHP analysis applied to the PBE–DFT results. Here, ICOHP indicates how much the hybridization contributes to the stabilization by the binding. The ICOHPs of Pb-O6 in F-in-site5 and Pb-F6 in F-in-site6 are −2.81 and −0.90 eV, so the contribution of the 6s2 lone pair is certainly smaller when F atoms occupy site 6. This explanation for the F-in-site6 stabilization is an exception to Pauling's second rule.28
 | ||
| Fig. 4 PDOS of different F occupation patterns obtained by DFT. The arrows indicate the peaks accompanied by the antibonding hybrid orbitals explained by the lone-pair model. For the F-in-site6 case, the O(site6)-2p distribution is not shown because site 6 is occupied by F atoms. | ||
4.3 Comparison between Pb2Ti4O9F2 and Bi2Ti4O11
Fig. 5 compares the experimental structures of Pb2Ti4O9F2 and Bi2Ti4O11, which were obtained by the Rietveld analysis of SXRD patterns. The SXRD patterns are provided in the ESI.‡ Comparing the isostructural Pb2Ti4O9F2 and Bi2Ti4O11, the anionic configurations around the cations are more symmetric in Pb2Ti4O9F2 than in Bi2Ti4O11. For example, the difference of the smallest (blue) and largest (red) interatomic distances are smaller in Pb2Ti4O9F2 than in Bi2Ti4O11. It is worth noting that this trend is the exact opposite of some Ruddlesden–Popper-type layered perovskites. The (B site metal)–O6 octahedra in layered perovskite oxides are often distorted by the Jahn–Teller effect.21,51 The F atoms selectively occupy the apical sites and lead to significantly longer (B site metal)–Fapical distance than (B site metal)–Oapical and Oequatorial distances.20–25 However, the oxyfluoride Pb2Ti4O9F2 is less distorted than the isostructural oxide Bi2Ti4O11, as shown in Fig. 5. | ||
| Fig. 5 Experimental geometries of Pb2Ti4O9F2 and Bi2Ti4O11 obtained via Rietveld refinement of SXRD patterns. The smallest and largest interatomic distances are shown with blue and red colors, respectively. | ||
Table 2 compares the experimental structure and theoretical F-in-site6 and F-in-site5 structures of Pb2Ti4O9F2. The theoretical values are given as the mean values of the three functionals. The errors indicate the unbiased deviations of the means. The F-in-site5 is slightly less symmetric than F-in-site6, but their differences are not significant, which indicates that the low-symmetric anionic configurations around the cations in Pb2Ti4O9F2 are barely due to the steric effects of 6s2 lone pairs: the structure is naively low-symmetric. The less symmetric structure of Bi2Ti4O11 than Pb2Ti4O9F2 can be simply explained by that Bi3+ cations are more positive than Pb2+ cations and prefer to be closer to anions. In the case of Bi2Ti4O11, the steric effects of 6s2 lone pairs may also contribute to the low-symmetric anionic configurations as discussed based on the MEM analyses in the ESI.‡ The finding from Table 2 demonstrates that the influence of 6s2 lone pairs on the structural distortion might be similarly not significant in some of the other Pb-based oxyfluorides such as Pb2Ti2O5.4F1.229 and Pb2OF2.30
| a | b | c | β (°) | Pb-O/F4 | Pb-O/F5 | Pb-O/F6shorter | Pb-O/F6longer | ||
|---|---|---|---|---|---|---|---|---|---|
| Expt. | Å | 14.63 | 3.83 | 10.75 | 135.58 | 2.56 | 2.95 | 2.42 | 2.56 |
| F-in-site6 | Å | 15.07(8) | 3.84(1) | 10.54(7) | 133.44(6) | 2.54(1) | 2.93(2) | 2.44(1) | 2.58(1) |
| % | +3.0(6) | +0.3(3) | −2.0(7) | −1.6(1) | −0.1(3) | −0.8(5) | +1.0(2) | +0.9(2) | |
| F-in-site5 | Å | 14.83(7) | 3.84(1) | 10.60(6) | 132.30(2) | 2.58(1) | 3.03(1) | 2.37(1) | 2.58(2) |
| % | +1.4(5) | +0.2(3) | −1.4(6) | −2.4(1) | +1.6(3) | +2.7(4) | −2.0(4) | +0.9 (6) | |
5 Conclusion
A combination of 19F MAS NMR experiments and DFT simulations were used to investigate the anionic ordering in Pb2Ti4O9F2. The 19F MAS NMR experiments showed that F atoms predominantly occupy two of the six distinct available sites in Pb2Ti4O9F2 in a ratio of 73:27. DFT calculations identified the majority and minority F occupation sites to be sites 6 and 5, respectively. The occupation ratios between sites 6 and 5 were quantitatively reproduced by theory, independent of the choice of functional. PDOS and COHP analyses revealed that the 6s2 lone pairs of Pb atom may play a role (∼0.1 eV per f.u.) in determining the site 6 (5) the majority (minority) site, against what is predicted by Pauling's second rule.28
The low-symmetric anionic coordinates around the cations in Pb2Ti4O9F2 have been considered to be the consequence of the steric effect by the 6s2 lone pairs, as well as Bi2Ti4O11,32 Pb2Ti2O5.4F1.2,29 and some Aurivillius oxides ABi2Nb(Ta)2O9.52 However, our DFT results indicate that the 6s2 lone pairs may barely induce a structural distortion in Pb2Ti4O9F2. This finding demonstrates that the influence of 6s2 lone pairs on the structural distortion might be similarly not significant in some of the other Pb-based oxyfluorides such as Pb2Ti2O5.4F1.229 and Pb2OF2.30
Author contributions
K.O., T.I., and D.K. conceived the ideas of this work. All the authors contributed to the discussion and writing of the paper. K.O. and K.Y. synthesized the Pb2Ti4O9F2 and Bi2Ti4O11 samples, analyzed the crystal structures, and performed MEM analyses. Y.N., Y.T., and N.N. performed the 19F MAS NMR measurements. T.I., D.K., and K.H. performed the ab initio calculations. M.I., R.M., and F.A.R. supervised the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Erica Heinrich for valuable help with manuscript preparation. The VESTA program53 was used to visualize the experimental and DFT crystal structures and electronic densities. The authors thank Prof. Sudo and Mr. Kitao of Kindai University for their help in the solid state NMR measurement. This work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Area “Mixed Anion (Project JP17H05489, JP19H04706)” (JSPS). The synchrotron radiation experiments were performed at the BL02B2 of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2016A1157 and 2018A1227). The ab initio calculations were performed with the computational resources of the Research Center for Advanced Computing Infrastructure (RCACI) at JAIST. The authors gratefully thank Division of Joint Research Center, Kindai University, for the solid-state NMR measurements. T. I. and F. A. R. acknowledge support from US Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division. K. O. is grateful for financial support from Kansai Research Foundation for technology promotion and a Grant-in-Aids for Scientific Research (C) (Project JP16K05731 and 21K04659). K. H. is grateful for financial support from MEXT-KAKENHI (JP19K05029, JP21K03400, JP21H01998, and JP22H02170), and the Air Force Office of Scientific Research (Award Numbers: FA2386-20-1-4036). R. M. is grateful for financial supports from MEXT-KAKENHI (22H05146, 21K03400 and 19H04692), from the Air Force Office of Scientific Research (AFOSR-AOARD/FA2386-17-1-4049;FA2386-19-1-4015), and from JSPS Bilateral Joint Projects (JPJSBP120197714).References
- H. Kageyama, T. Yajima, Y. Tsujimoto, T. Yamamoto, C. Tassel and Y. Kobayashi, Exploring Structures and Properties through Anion Chemistry, Bull. Chem. Soc. Jpn., 2019, 92, 1349–1357 CrossRef CAS.
- K. Maeda, F. Takeiri, G. Kobayashi, S. Matsuishi, H. Ogino, S. Ida, T. Mori, Y. Uchimoto, S. Tanabe, T. Hasegawa, N. Imanaka and H. Kageyama, Recent Progress on Mixed-Anion Materials for Energy Applications, Bull. Chem. Soc. Jpn., 2022, 95, 26–37 CrossRef CAS.
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Expanding frontiers in materials chemistry and physics with multiple anions, Nat. Commun., 2018, 9, 772 CrossRef PubMed.
- R. Kuriki, T. Ichibha, K. Hongo, D. Lu, R. Maezono, H. Kageyama, O. Ishitani, K. Oka and K. Maeda, A Stable, Narrow-Gap Oxyfluoride Photocatalyst for Visible-Light Hydrogen Evolution and Carbon Dioxide Reduction, J. Am. Chem. Soc., 2018, 140, 6648–6655 CrossRef CAS PubMed.
- T. Oshima, T. Ichibha, K. S. Qin, K. Muraoka, J. J. M. Vequizo, K. Hibino, R. Kuriki, S. Yamashita, K. Hongo, T. Uchiyama, K. Fujii, D. Lu, R. Maezono, A. Yamakata, H. Kato, K. Kimoto, M. Yashima, Y. Uchimoto, M. Kakihana, O. Ishitani, H. Kageyama and K. Maeda, Undoped Layered Perovskite Oxynitride Li2LaTa2O6N for Photocatalytic CO2 Reduction with Visible Light, Angew. Chem., Int. Ed., 2018, 57, 8154–8158 CrossRef CAS PubMed.
- T. Oshima, T. Ichibha, K. Oqmhula, K. Hibino, H. Mogi, S. Yamashita, K. Fujii, Y. Miseki, K. Hongo, D. Lu, R. Maezono, K. Sayama, M. Yashima, K. Kimoto, H. Kato, M. Kakihana, H. Kageyama and K. Maeda, Two-Dimensional Perovskite Oxynitride K2LaTa2O6N with an H+/K+ Exchangeability in Aqueous Solution Forming a Stable Photocatalyst for Visible-Light H2 Evolution, Angew. Chem., Int. Ed., 2020, 59, 9736–9743 CrossRef CAS PubMed.
- Y.-I. Kim, P. M. Woodward, K. Z. Baba-Kishi and C. W. Tai, Characterization of the Structural, Optical, and Dielectric Properties of Oxynitride Perovskites AMO2N (A = Ba, Sr, Ca; M = Ta, Nb), Chem. Mater., 2004, 16, 1267–1276 CrossRef CAS.
- K. Page, M. W. Stoltzfus, Y.-I. Kim, T. Proffen, P. M. Woodward, A. K. Cheetham and R. Seshadri, Local Atomic Ordering in BaTaO2N Studied by Neutron Pair Distribution Function Analysis and Density Functional Theory, Chem. Mater., 2007, 19, 4037–4042 CrossRef CAS.
- M. Yang, J. Oró-Solé, J. A. Rodgers, A. B. Jorge, A. Fuertes and J. P. Attfield, Anion order in perovskite oxynitrides, Nat. Chem., 2010, 3, 47–52 CrossRef PubMed.
- T. Yamamoto, D. Zeng, T. Kawakami, V. Arcisauskaite, K. Yata, M. A. Patino, N. Izumo, J. E. McGrady, H. Kageyama and M. A. Hayward, The role of π-blocking hydride ligands in a pressure-induced insulator-to-metal phase transition in SrVO2H, Nat. Commun., 2017, 8, 1217 CrossRef PubMed.
- J. P. Attfield, Principles and Applications of Anion Order in Solid Oxynitrides, Cryst. Growth Des., 2013, 13, 4623–4629 CrossRef CAS.
- A. Fuertes, Prediction of Anion Distributions Using Pauling's Second Rule, Inorg. Chem., 2006, 45, 9640–9642 CrossRef CAS PubMed.
- C. M. Thompson, C. K. Blakely, R. Flacau, J. E. Greedan and V. V. Poltavets, Structural and magnetic behavior of the cubic oxyfluoride SrFeO2F studied by neutron diffraction, J. Solid State Chem., 2014, 219, 173–178 CrossRef CAS.
- F. J. Berry, F. C. Coomer, C. Hancock, O. Helgason, E. A. Moore, P. R. Slater, A. J. Wright and M. F. Thomas, Structure and magnetic properties of the cubic oxide fluoride BaFeO2F, J. Solid State Chem., 2011, 184, 1361–1366 CrossRef CAS.
- T. Katsumata, M. Nakashima, H. Umemoto and Y. Inaguma, Synthesis of the novel perovskite-type oxyfluoride PbScO2F under high pressure and high temperature, J. Solid State Chem., 2008, 181, 2737–2740 CrossRef CAS.
- R. L. Needs and M. T. Weller, A New 2+/3+ Perovskite: The Synthesis and Structure of BaScO2F, J. Solid State Chem., 1998, 139, 422–423 CrossRef CAS.
- F. Takeiri, T. Yamamoto, N. Hayashi, S. Hosokawa, K. Arai, J. Kikkawa, K. Ikeda, T. Honda, T. Otomo, C. Tassel, K. Kimoto and H. Kageyama, AgFeOF2: A Fluorine-Rich Perovskite Oxyfluoride, Inorg. Chem., 2018, 57, 6686–6691 CrossRef CAS.
- T. Katsumata, R. Suzuki, N. Satoh, S. Suzuki, M. Nakashima, Y. Inaguma, D. Mori, A. Aimi and Y. Yoneda, Synthesis of new perovskite-type oxyfluorides, BaInO2F and comparison of the structure among perovskite-type oxyfluorides, J. Solid State Chem., 2019, 279, 120919 CrossRef CAS.
- Y. Inaguma, K. Sugimoto and K. Ueda, Synthesis of the perovskite-type oxyfluoride AgTiO2F: an approach adopting the HSAB principle, Dalton Trans., 2020, 49, 6957–6963 RSC.
- M. Ai-Mamouri, P. P. Edwards, C. Greaves and M. Slaski, Synthesis and superconducting properties of the strontium copper oxy-fluoride Sr2CuO2F2+δ, Nature, 1994, 369, 382–384 CrossRef.
- G. Simon Case, A. L. Hector, W. Levason, R. L. Needs, M. F. Thomas and M. T. Weller, Syntheses, powder neutron diffraction structures and Mössbauer studies of some complex iron oxyfluorides: Sr3Fe2O6F0.87, Sr2FeO3F and Ba2InFeO5F0.68, J. Mater. Chem., 1999, 9, 2821–2827 RSC.
- A. L. Hector, J. A. Hutchings, R. L. Needs, M. F. Thomas and M. T. Weller, Structural and Mössbauer study of Sr2FeO3X (X = F, Cl, Br) and the magnetic structure of Sr2FeO3F, J. Mater. Chem., 2001, 11, 527–532 RSC.
- R. L. Needs and M. T. Weller, Synthesis and structure of Ba2InO3F: oxide/fluoride ordering in a new K2NiF4 superstructure, J. Chem. Soc., Chem. Commun., 1995, 353–354 RSC.
- R. L. Needs, M. T. Weller, U. Scheler and R. K. Harris, Synthesis and structure of Ba2InO3X (X = F, Cl, Br) and Ba2ScO3F; oxide/halide ordering in K2NiF4-type structures, J. Mater. Chem., 1996, 6, 1219–1224 RSC.
- Y. Su, Y. Tsujimoto, Y. Matsushita, Y. Yuan, J. He and K. Yamaura, High-Pressure Synthesis, Crystal Structure, and Magnetic Properties of Sr2MnO3F: A New Member of Layered Perovskite Oxyfluorides, Inorg. Chem., 2016, 55, 2627–2633 CrossRef CAS PubMed.
- Y. Tsujimoto, K. Yamaura, N. Hayashi, K. Kodama, N. Igawa, Y. Matsushita, Y. Katsuya, Y. Shirako, M. Akaogi and E. Takayama-Muromachi, Topotactic Synthesis and Crystal Structure of a Highly Fluorinated Ruddlesden–Popper-Type Iron Oxide, Sr3Fe2O5+xF2−x(x∼0.44), Chem. Mater., 2011, 23, 3652–3658 CrossRef CAS.
- L.-S. Du, F. Wang and C. P. Grey, High-Resolution 19F MAS and 19F–113Cd REDOR NMR Study of Oxygen/Fluorine Ordering in Oxyfluorides, J. Solid State Chem., 1998, 140, 285–294 CrossRef CAS.
- L. Pauling, The nature of the chemical bond and the structure of molecules and crystals: an introduction to modern structural chemistry, Cornell University Press, Ithaca, N.Y., 1960, pp. 543–562 Search PubMed.
- K. Oka, H. Hojo, M. Azuma and K. Oh-ishi, Temperature-Independent, Large Dielectric Constant Induced by Vacancy and Partial Anion Order in the Oxyfluoride Pyrochlore Pb2Ti2O6−δF2δ, Chem. Mater., 2016, 28, 5554–5559 CrossRef CAS.
- Y. Inaguma, K. Ueda, T. Katsumata and Y. Noda, Low-temperature formation of Pb2OF2 with O/F anion ordering by solid state reaction, J. Solid State Chem., 2019, 277, 363–367 CrossRef CAS.
- K. Oka and K. Oh-ishi, Observation of Anion Order in Pb2Ti4O9F2, Inorg. Chem., 2015, 54, 10239–10242 CrossRef CAS PubMed.
- V. Kahlenberg and H. Böhm, The structures of α- and β-Bi2Ti4O11, Acta Crystallogr., Sect. B: Struct. Sci., 1995, 51, 11–18 CrossRef.
- T. Katsumata, M. Nakashima, Y. Inaguma and T. Tsurui, Synthesis of New Perovskite-Type Oxyfluoride, PbMnO2F, Bull. Chem. Soc. Jpn., 2012, 85, 397–399 CrossRef CAS.
- S. Kawaguchi, M. Takemoto, K. Osaka, E. Nishibori, C. Moriyoshi, Y. Kubota, Y. Kuroiwa and K. Sugimoto, High-throughput powder diffraction measurement system consisting of multiple MYTHEN detectors at beamline BL02B2 of SPring-8, Rev. Sci. Instrum., 2017, 88, 085111 CrossRef CAS PubMed.
- F. Izumi and K. Momma, Three-Dimensional Visualization in Powder Diffraction, Solid State Phenomena, 2007, 130, 15–20 CAS.
- K. Momma, T. Ikeda, A. A. Belik and F. Izumi, Dysnomia, a computer program for maximum-entropy method (MEM) analysis and its performance in the MEM-based pattern fitting, Powder Diffr., 2013, 28, 184–193 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- J. P. Perdew and Y. Wang, Accurate and simple analytic representation of the electron-gas correlation energy, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- J. P. Perdew and Y. Wang, Erratum: Accurate and simple analytic representation of the electron-gas correlation energy [Phys. Rev. B 45, 13244 (1992)], Phys. Rev. B, 2018, 98, 079904 CrossRef.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- A. Dal Corso, Pseudopotentials periodic table: From H to Pu, Comput. Mater. Sci., 2014, 95, 337–350 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- R. Nelson, C. Ertural, J. George, V. L. Deringer, G. Hautier and R. Dronskowski, LOBSTER: Local orbital projections, atomic charges, and chemical-bonding analysis from projector-augmented-wave-based density-functional theory, J. Comput. Chem., 2020, 41, 1931–1940 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids, J. Comput. Chem., 2013, 34, 2557–2567 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, LOBSTER: A tool to extract chemical bonding from plane-wave based DFT, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- Y. Kurauchi, T. Katayama, A. Chikamatsu and T. Hasegawa, Two-Dimensional Fluorine Distribution in a Heavily Distorted Perovskite Nickel Oxyfluoride Revealed by First-Principles Calculation, J. Phys. Chem. C, 2019, 123, 31190–31195 CrossRef CAS.
- A. Walsh, D. J. Payne, R. G. Egdell and G. W. Watson, Stereochemistry of post-transition metal oxides: revision of the classical lone pair model, Chem. Soc. Rev., 2011, 40, 4455–4463 RSC.
- D. Lee and H. N. Lee, Controlling Oxygen Mobility in Ruddlesden–Popper Oxides, Materials, 2017, 10, 368 CrossRef.
- Ismunandar and B. J. Kennedy, Gunawan and Marsongkohadi, Structure ofABi2Nb2O9 (A = Sr, Ba): Refinement of Powder Neutron Diffraction Data, J. Solid State Chem., 1996, 126, 135–141 CrossRef CAS.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
Footnotes |
| † This manuscript has been authored by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with the US Department of Energy (DOE). The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (https://energy.gov/downloads/doe-public-access-plan). |
| ‡ Electronic supplementary information (ESI) available. CCDC 2159898, 2159899 and 2191023. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00839d |
| § These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |