
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
M-CPOnes: transition metal complexes with cyclopropenone-based ligands for light-triggered carbon monoxide release†
Marc
Lehr
a,
Tjorge
Neumann
a,
Christian
Näther
 b and
Anna J.
McConnell
*a
b and
Anna J.
McConnell
*a
aOtto Diels Institute of Organic Chemistry, Christian-Albrechts-Universität zu Kiel, Otto-Hahn-Platz 4, Kiel 24098, Germany. E-mail: amcconnell@oc.uni-kiel.de
bInstitute of Inorganic Chemistry, Christian-Albrechts-Universität zu Kiel, Max-Eyth-Straße 2, Kiel 24118, Germany
First published on 21st April 2022
Abstract
A new class of CO-releasing molecules, M-CPOnes, was prepared combining cyclopropenone-based ligands for CO release with the modular scaffold of transition metal complexes. In proof-of-concept studies, M-CPOnes based on ZnII, FeII and CoII are stable in the dark but undergo light-triggered CO release with the cyclopropenone substituents and metal ions enabling tuning of the photophysical properties. Furthermore, the choice of metal allows the use of different spectroscopic methods to monitor photodecarbonylation from fluorescence spectroscopy to UV/vis spectroscopy and paramagnetic NMR spectroscopy. The modularity of M-CPOnes from the metal ion to the cyclopropenone substitution and potential for further functionalisation of the ligand make M-CPOnes appealing for tailored functionality in applications.
Introduction
Carbon monoxide has potential therapeutic applications,1e.g. for treatment of inflammation and vascular dysfunction,2–5 given the role of carbon monoxide in the regulation of ion channels6,7 and signalling pathways.8,9 Since the safe handling and delivery of gaseous carbon monoxide is problematic, carbon monoxide releasing molecules (CORMs)3 have been developed for the controlled release of carbon monoxide in response to stimuli. These include metal carbonyl complexes10 (such as tricarbonyldichlororuthenium(II) dimer (CORM-2),11 (tricarbonylchloro(glycinato)ruthenium (CORM-3)12), non-metallic/organic CORMs13 and carbon monoxide “prodrugs”.14,15 For applications, the CORM should have good stability but triggerable and quantitative CO release as well as tunable photophysical properties and synthetic accessibility.16Photo-CORMs17–20 take advantage of the spatial and temporal control of light to release carbon monoxide upon irradiation16,21 and in some cases, the loss of carbon monoxide could be tracked by fluorescence.22–24 While cyclopropenones25,26 undergo efficient photolysis27,28 upon irradiation with light resulting in carbon monoxide release and alkyne formation, they have been largely overlooked as CORMs.16 Instead, cyclopropenones have found use across different fields from organic chemistry (as substrates in annulations,29–33 metal-catalysed ring-opening reactions,34 cycloaddition reactions35,36 and allene syntheses37,38 as well as catalysts for nucleophilic substitutions39,40) to material science18,41,42 and biorthogonal chemistry.43–48
Cyclopropenones are appealing as prospective CORMs not only for their efficient photodecarbonylation but since they can be prepared via various synthetic methods (e.g. Friedel–Crafts reaction,49–53 [2 + 1] cycloaddition and hydrolysis,37,54–57 Favorskii rearrangement29,39,58,59 and substitution of cyclopropenone acetals60–63). In addition, they are stable in aqueous media and cellular environments45,47,64 and there is the potential to exploit multi-photon induced decarbonylation for carbon monoxide release at higher wavelengths (800–950 nm).65,66
We envisaged the combination of cyclopropenones with transition metal complexes could lead to a new class of CO-releasing molecules, M-CPOnes (Scheme 1), as an alternative to metal carbonyl complexes; light could be exploited as a reagent-free trigger for CO release from the cyclopropenone while the transition metal complex could serve as a modular scaffold for not only introducing multiple cyclopropenone moieties but also tuning properties such as the stability, solubility and photodecarbonylation wavelength through the choice of metal and ligands.
 | ||
| Scheme 1 Proof-of-concept of M-CPOnes: light triggered CO release from transition metal complexes with cyclopropenone-based ligands. The complexes form as a mixture of the fac and mer isomers but only the fac isomer is depicted for clarity. | ||
We report cyclopropenone ligands based on a 2,2′-bipyridine coordination motif are now synthetically accessible via a cycloaddition and subsequent hydrolysis and in addition, their photophysical properties are tunable through the R substituent. The M-CPOnes resulting from complexation with ZnII, FeII and CoII metal ions are stable in the dark but undergo photodecarbonylation upon irradiation with 365 nm light (Scheme 1). Thus, we demonstrate the proof-of-concept of M-CPOnes as a new class of CO-releasing molecules and this lays the groundwork for future investigation of their application as CORMs.
Results and discussion
Given diphenylcyclopropenone is one of the simplest and most well-studied diaryl-substituted cyclopropenones,67–69 the initial target ligand was 1a (Scheme 2) where one of the phenyl groups was replaced with a simple 2,2′-bipyridine as the coordination motif for the development of this new class of CO-releasing molecules, M-CPOnes. Furthermore, ligands 1b and 1c would also be prepared where the second substituent was varied from a phenyl group to an electron rich thienyl and second N-heterocyclic substituent, respectively, to study the electronic effects of the aryl substituents on the stability and photophysical properties of the cyclopropenones and their corresponding M-CPOnes (Scheme 1). | ||
| Scheme 2 Synthesis of N-heterocyclic cyclopropenones 1a-cvia a [2 + 1] cycloaddition of :CF2 with alkyne derivatives 2a-c and subsequent hydrolysis of the corresponding difluorocyclopropenes 3a-c. | ||
While there are numerous examples of alkyl- and carbocyclic aryl-substituted cyclopropenones,25–29,39,41,45,47,58,59,62,63,65,66,70–72 heterocyclic-based cyclopropenones are relatively rare73 and typically based on more electron-rich 5-membered heterocycles.37,74,75 Due to the incompatibility of Lewis acid reagents like AlCl3 with the 2,2′-bipyridine coordination motif in the commonly employed synthesis of cyclopropenones via a Friedel–Crafts reaction,76 we focused on an alternative synthetic strategy involving a [2 + 1] cycloaddition between a dihalocarbene77,78 and acetylene derivative followed by hydrolysis of the dihalocyclopropene (Scheme 2).
Even though the synthesis of difluorocyclopropenes via a cycloaddition is, in general, well established,78–80 there are a scarcity of examples of electron-deficient N-heterocyclic difluorocyclopropenes. We expected the cycloaddition to be challenging given the more electron-deficient nature of the substrates and since Lewis bases such as pyridine have been proposed to coordinate to the :CF2 during the cycloaddition causing decomposition or difluoromethylation.77,78,81–84 We hypothesised the reduced basicity of 2,2′-bipyridine and 6-quinoline relative to pyridine might limit these side-reactions. Therefore, the feasibility of the [2 + 1] cycloaddition reaction with 2,2′-bipyridine-based alkyne derivatives 2a-c (prepared according to Scheme S1†) was investigated in NMR scale experiments.
Derivatives 2a-c, 1.5 eq. TMSCF2Br and 3 mol% of the initiator TBABr were heated in toluene-d8 adapting literature procedures for related aryl-substituted difluorocyclopropenes.79,80 The reaction mixture was monitored by 1H and 19F NMR spectroscopy before and directly after a reaction time of 2 h (ESI, section 3†). For all three derivatives, at least one new set of signals appeared in the 1H NMR spectra (Fig. S50, S52 and S54†) and the 19F NMR spectra showed the consumption of TMSCF2Br as well as the presence of a new fluorine signal between −110 and −114 ppm (Fig. S51, S53 and S55†), which is consistent with the expected chemical shift of a difluorocyclopropene.79,80,84,85
The relative ratio of the unreacted alkyne and proposed difluorocyclopropene was determined by integrating the proton Hj signal (Scheme 2) due to its characteristic chemical shift and separation from other signals (Fig. S50, S52 and S54†). Derivative 2b showed the highest product/starting material ratio (93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7, Table 1) and furthermore, the 19F NMR spectrum showed nearly complete consumption of TMSCF2Br (Fig. S53†). In contrast, the amount of the product significantly decreased for 2a and even more so for 2c with a second N-heterocyclic substituent (Table 1). Thus, better conversion to the difluorocyclopropene is observed in cycloadditions with substrates containing more electron-rich R substituents, as expected.
:7, Table 1) and furthermore, the 19F NMR spectrum showed nearly complete consumption of TMSCF2Br (Fig. S53†). In contrast, the amount of the product significantly decreased for 2a and even more so for 2c with a second N-heterocyclic substituent (Table 1). Thus, better conversion to the difluorocyclopropene is observed in cycloadditions with substrates containing more electron-rich R substituents, as expected.
| Substrate |
3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2 ratioa :2 ratioa
|
Isolatedb yield (%) |
|---|---|---|
| a Determined from NMR scale experiments (0.1 mmol of the respective alkyne substrate) and integration of the proton Hj of the difluorocyclopropene 3a-c and alkyne 2a-c. b Following column chromatography in larger scale syntheses. | ||
| 2a | 52:48 |
32 |
| 2b | 93:7 |
50 |
| 2c | 18:82 |
21 |
Encouraged by the observed conversion in the NMR scale experiments, large scale syntheses were carried out in a pressure tube and difluorocyclopropenes 3a-c were isolated in moderate to good yields following column chromatography (Table 1 and ESI section 2†). In addition, the X-ray crystal structure of 3a was obtained showing the expected difluorocyclopropene structure (ESI section 2.1.2.1 and Fig. S12†). The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond length (1.33 Å) and C–F bond lengths (1.38 Å) in the cyclopropene ring are similar to those in related difluorocyclopropenes.86,87
C bond length (1.33 Å) and C–F bond lengths (1.38 Å) in the cyclopropene ring are similar to those in related difluorocyclopropenes.86,87
Although synthetic access to the desired difluorocyclopropenes was initially envisaged to be challenging, the cycloaddition strategy was tolerant of both the 2,2′-bipyridine coordination motif and a second heterocyclic 6-quinoline substituent. Furthermore, the difluorocyclopropene derivatives were stable during purification by column chromatography on silica gel. This was surprising since other difluorocyclopropenes and related difluorocyclopropanes are susceptible to hydrolysis under relatively mild conditions,38,56,74 including on silica gel,56,81,86,88 giving the corresponding cyclopropenones and cyclopropanones, respectively.
Indeed, attempted hydrolysis of 3a-c to cyclopropenones 1a-c on a small scale under a variety of mild conditions (wet CDCl3, Amberlyst® 15 or silica gel at room temperature for 24 h)38,56,74,80 revealed the stability of the difluorocyclopropenes (ESI, section 4†); while there was no hydrolysis of 3c under these conditions (Fig. S58A–C†), derivatives 3a-b showed partial hydrolysis using Amberlyst® 15 and silica gel (Fig. S56 and S57B, C†) and no hydrolysis in CDCl3 (Fig. S56 and S57A†). In contrast, complete hydrolysis was observed using 6 M HCl since the cyclopropenone was obtained following neutralisation and extraction with CDCl3 (Fig. S56 and S57D†). Despite the complete consumption of quinoline derivative 3c, it was necessary to shorten the reaction time to 1 h to prevent the formation of a by-product (Fig. S58D and D.1†).
Further optimisation of this hydrolysis method with HCl enabled the preparative isolation of N-heterocyclic cyclopropenones in good yields (65–91%) with a reaction time of 15 min for 1a and 1b and 1 h for derivative 1c. Evidence for the formation of 1a-c was given by the distinct shift of the carbonyl carbon39,50,89 around 150–156 ppm in the 13C NMR spectra (Fig. S14, S30 and S46†), observation of the molecular ion peak in the ESI mass spectra as well as the characteristic carbonyl stretches and ring vibrations25,28,39,90 in the ranges of 1835–1850 cm−1 and 1560–1625 cm−1 in the IR spectra (ESI sections 2.1.3, 2.2.3, 2.3.3†).
With the successful synthesis of the series of 2,2′-bipyridine-based ligands 1a-c, the stability of this new class of cyclopropenones was investigated regarding storage and handling. No decomposition of solid samples was observed over at least a month at room temperature under ambient light. However, solutions of the cyclopropenones in CDCl3 under ambient light showed evidence of decarbonylation over a period of one week (ESI section 5 and Fig. S62–64†) As a result, all cyclopropenones in solution were protected from light as a precaution.
Irradiation studies were carried out using UV/vis and NMR spectroscopy to investigate the influence of the R substituent on photodecarbonylation (Scheme 3 and ESI section 6†). Since the synthesis and study of the M-CPOnes was carried out in acetonitrile, the UV/vis spectra of ligands 1a-c as well as reference alkyne derivatives 2a-c were measured in acetonitrile. However, due to the limited solubility of the ligands at typical NMR concentrations in acetonitrile, the NMR studies with the ligands were performed in CDCl3.
 | ||
| Scheme 3 Photodecarbonylation of cyclopropenone ligands 1a-c to 2a-c. | ||
The UV/vis spectra of cyclopropenones 1a-c show broad absorption bands and as expected based on studies of related cyclopropenones with alkyl and aryl substituents,50 the R substituent tunes the absorption maximum from 321 nm (1a) to 333 nm (1c) and 341 nm (1b) (Fig. 1a). Photodecarbonylation of related cyclopropenones has been reported upon irradiation of the absorption bands around 240–325 nm and 360 nm assigned to the π–π* and n–π* transitions, respectively.50,91–95 Irradiation of the π–π* absorption band is proposed to produce the corresponding alkyne in the excited state in contrast to the ground state following irradiation of the n–π* absorption band.94 A wavelength of 365 nm was chosen to trigger the photodissociation in these studies for this reason and since the absorbance of the alkynes are minimal at this wavelength; in related systems, the alkyne photoproducts are reported to undergo subsequent photoreactions.50
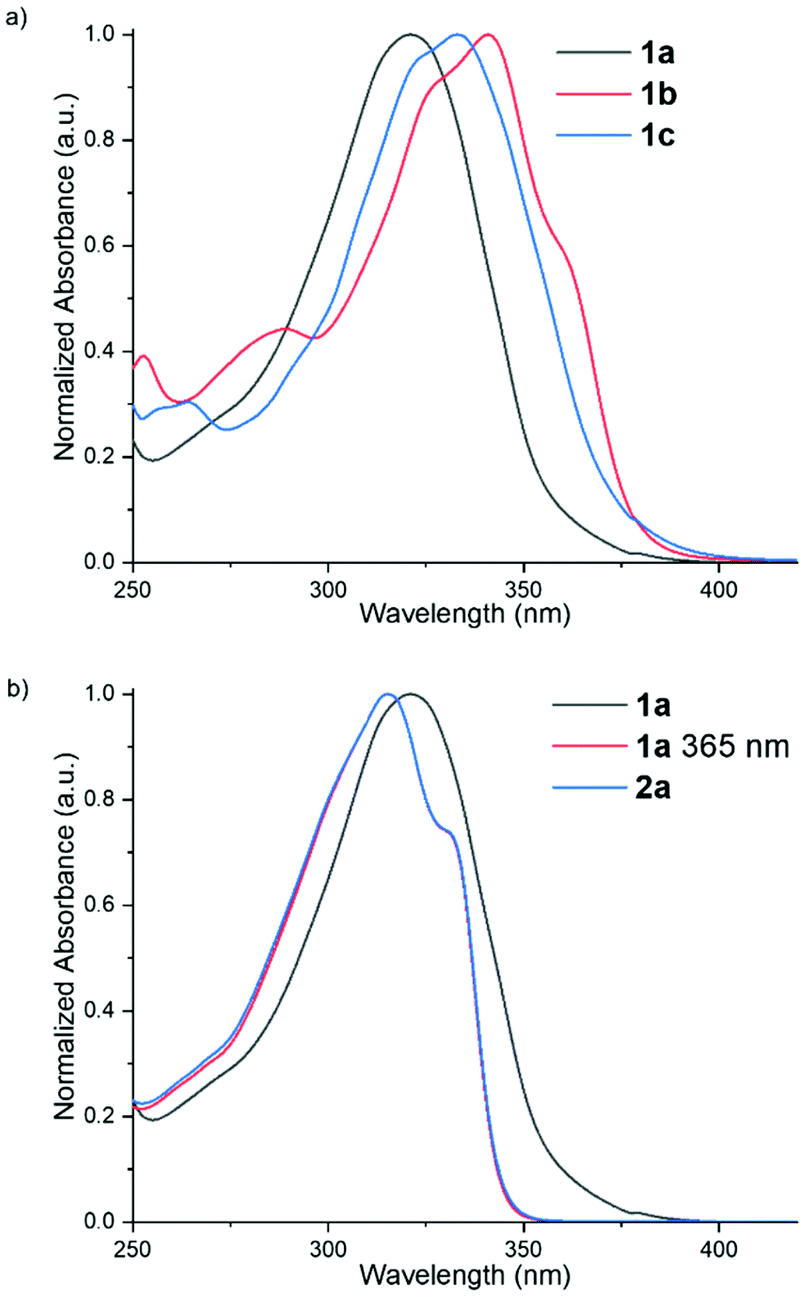 | ||
| Fig. 1 Comparison of the normalized UV/vis spectra (CH3CN, 298 K) of: (a) cyclopropenone ligands 1a-c; (b) cyclopropenone ligand 1a before (black) and after (red) irradiation with 365 nm for 1 min as well as the reference compound 2a (blue). | ||
Irradiation for 1 min with all cyclopropenones showed complete photodecarbonylation to the corresponding alkyne derivatives (Fig. 1b). The 1H and 13C NMR spectra as well as the UV/vis spectra were consistent with the reference alkyne spectra (Fig. S65–S73†). Furthermore, no by-products were observed in the NMR spectra. Complete photodecarbonylation was also observed by UV/visible spectroscopy following irradiation of 1a for 10 s (Fig. S67†), demonstrating that the longer irradiation time of 1 min does not lead to by-product formation and suggesting efficient photodecarbonylation. The quantum yields for the photodecarbonylation of related cyclopropenones are high (Φ 0.2–1.0).50,93
The scope of this study is demonstration of the proof of concept of light-triggered CO release from M-CPOnes. Therefore, we chose to focus on M-CPOnes based on kinetically labile FeII, CoII and ZnII complexes for these initial investigations to establish the compatibility of the cyclopropenone moiety with transition metal complexes regarding stability and photodecarbonylation. M-CPOnes-1a-c were prepared by mixing the respective metal salt (Fe(BF4)2·6H2O, Zn(OTf)2 or Co(NTf2)2) and three equivalents of the ligand in CD3CN and were characterised by NMR, UV/Vis, IR spectroscopy and ESI spectrometry either in situ or after precipitation with diethyl ether. Since these non-symmetric ligands could form a mixture of meridional (mer) and facial (fac) coordination isomers, up to four sets of NMR signals were to be expected.
In addition, the corresponding alkyne complexes M-2a-c (M = FeII, ZnII and CoII) were synthesised for comparison since they are formed upon photodecarbonylation of the M-CPOnes (Scheme 1). Although the nitrogen donor of the quinoline substituent in ligand 2c could, in principle, also coordinate to the metal leading to ill-defined complex mixtures, the NMR spectra for complexes Fe-2c and Co-2c were consistent with metal binding to the 2,2′-bipyridine motif only when a 1:3 metal/ligand ratio was used (Fig. S162–166 and S168–173†).
Complexation of cyclopropenone ligands 1a and 1b with ZnII and FeII resulted in the formation of diamagnetic complexes (Fig. S74, S75, S80, S81, S95, S96, S101 and S102†) with different 1H and 13C NMR spectra to the alkyne complexes Zn-2a,b and Fe-2a,b (Fig. S125, S126, S131, S132, S145, S146, S151, and 152†). However, complete assignment of the spectra was not possible for the Zn-CPOnes-1a,b due to broad linewidths and overlapping signals, although ESI mass spectra were consistent with the formation of ZnL3 complexes (Fig. S84 and S106†). Characterisation of Fe-CPOnes-1a,b was also challenging given the number of overlapping signals, however, the signals for Hj were distinct and four sets were observed (Fig. S74 and S95†). This along with the observation of four sets of signals for each carbon in the 13C NMR spectra suggests the formation of a mixture of fac and mer isomers (Fig. S75 and S96†). In contrast to the challenging NMR analysis with the ZnII and FeII complexes due to signal overlap, the formation of paramagnetic complexes with CoII resulted in greater signal dispersion due to the large paramagnetic shifts. Characterisation using our recently reported paramagnetic NMR toolbox96 revealed four ligand environments consistent with a mixture of the fac and mer isomers (Fig. S87–S93 and S107–S112†).
Evidence for the cyclopropenone moiety being intact following complexation comes from observation of the carbonyl carbon signals in the 13C NMR spectra of the diamagnetic FeII complexes at similar shifts to the free ligands as well as the distinctive carbonyl stretches and ring vibrations in the IR spectra of M-CPOne-1a,b (M = FeII, ZnII, CoII). Furthermore, ESI mass spectra confirmed the formation of M-CPOne-1a,b (M = FeII, ZnII, CoII) through the observation of the [ML3]2+ ion peaks as well as [ML3 − (CO)n]2+ (n = 1–3) ion peaks resulting from decarbonylation under the ionisation conditions (Fig. S79, S84, S94, S100, S106 and S113†).
The photophysical properties of the cyclopropenone in M-CPOne-1a,b (M = FeII, ZnII, CoII) were similar to those of the free cyclopropenone ligand 1a and 1b with small bathochromic shifts of the absorption maxima in the UV/vis spectra in acetonitrile (Fig. S176, S179, S181, S184, S189 and S191†). As with the free ligands, the R substituent tunes the absorption maximum with a 30 nm difference between Fe-CPOne-1a and Fe-CPOne-1b (Fig. 2a, inset).
 | ||
| Fig. 2 Comparison of the normalized UV/vis spectra (CH3CN, 298 K) of: (a) Fe-CPOnes-1a-c; (b) Fe-CPOne-1a before (black) and after (red) irradiation with 365 nm for 1 min as well as the reference complex Fe-2a (blue). The insets show an expansion of the spectra between 250 and 400 nm for comparison of the absorption maxima. | ||
Irradiation experiments were carried out by UV/vis and NMR spectroscopy using 365 nm light given the similarity of the absorption maxima to those of the free ligands (Scheme 1 and Fig. 1a, 2a). Following irradiation of M-CPOne-1a,b (M = FeII, ZnII, CoII) for 1 min, UV/vis spectra consistent with the corresponding alkyne complexes M-2a,b were obtained, suggesting complete photodecarbonylation of M-CPOne-1a,b (ESI section 9†). Further studies using NMR spectroscopy supported photodecarbonylation to the corresponding alkyne complexes M-2a,b. However, longer irradiation times of 4 min and 20 min were required for photodecarbonylation of Co-CPOne-1a,b and Fe-CPOne-1a,b, respectively. This was attributed to the increased concentration for the NMR studies (mM vs. μM for NMR and UV/vis studies, respectively) and competing absorbance of the alkyne complexes at 365 nm, although competing metal-based transitions could also contribute to the longer irradiation times. There was no evidence of by-product formation, for example from subsequent photoreactions of the alkyne complexes, with the longer irradiation times.
In addition to these photodecarbonylation properties, the M-CPOnes have tunable properties due to the presence of the metal. The fluorescent properties of Zn-CPOne-1a and its corresponding alkyne complex Zn-2a were also investigated. Upon excitation at 312 nm Zn-2a showed emission at 362 nm and a similar emission spectrum was obtained for Zn-CPOne-1a, although the fluorescence was weaker (Fig. S85, S86 and S177†). Similar spectra were obtained for the corresponding ligands 1a and 2a (Fig. S85 and S86†). We propose that Zn-CPOne-1a shows no/weak fluorescence and some photodecarbonylation takes place at this excitation wavelength, as observed by Popik and co-workers for related cyclopropenones.50 Thus, the fluorescence from the photodecarbonylated products of Zn-CPOnes could be exploited as another indicator of CO release. For Fe-CPOne-1a,b CO release could be followed through the hypsochromic shift of the MLCT band around 570 nm upon irradiation (Fig. 2b and S181, S184†), resulting in a colour change from purple to red (Fig. S182†). Finally, photodecarbonylation of Co-CPOne-1a,b resulted in shifts of up to 5 ppm for the bipyridine coordination motif signals in the paramagnetic NMR spectrum (Fig. S188 and S190†).
Over the course of our studies, we found the quinoline-containing ligand 1c showed different coordination properties to the corresponding alkyne ligand 2c and cyclopropenone ligands 1a–1b. Given the characterisation difficulty with Zn-CPOnes due to the broadness of the NMR spectra, we focussed on the preparation of M-CPOnes-1c (M = FeII, CoII) using the same procedure as previously described. While 1H NMR and mass spectra consistent with the formation of Fe-CPOne-1c and Co-CPOne-1c were observed immediately after preparation (Fig. S114, S116–S119 and S121–124†), the signals in the NMR spectra decreased in intensity over time (Fig. S115 and S120†). There was also a colour change as well as partial precipitation. We propose the observed spectral changes for Fe-CPOne-1c and Co-CPOne-1c are due to the rearrangement of kinetically formed metastable M-CPOnes-1c (M = FeII, CoII) to a dynamic combinatorial library of multiple interconverting species. This is attributed to the presence of an additional coordination motif, the 6-quinoline, and its orientation in the bent cyclopropenone ligand given complexes M-2c did not rearrange.
Indeed, irradiation experiments with the proposed dynamic combinatorial library samples as well as freshly prepared M-CPOnes-1c (M = FeII, CoII) using NMR spectroscopy showed similar NMR spectra to the respective alkyne complexes M-1c following irradiation at 365 nm for 4 min (M = CoII) or 20 min (M = FeII). In addition, the freshly prepared M-CPOne-1c complexes showed spectral changes consistent with photodecarbonylation (Fig. S185, S186, S192 and S193†). This suggests that the change of the NMR spectra over time was not due to decomposition of the cyclopropenone motif but due to additional coordination events since photodecarbonylation was still possible following rearrangement to the proposed dynamic combinatorial library.
Conclusions
A family of M-CPOnes was successfully prepared as a new class of CO-releasing molecules combining the light-triggered decarbonylation of cyclopropenones with the tunability of transition metal complexes. Firstly, we demonstrated N-heterocyclic cyclopropenones based on electron-deficient 2,2′-bipyridine coordination motifs are synthetically accessible via a cycloaddition and subsequent hydrolysis of the difluorocyclopropenes. Furthermore, the ligands are stable to coordination with ZnII, FeII and CoII forming the M-CPOnes. Special handling and storage of the ligands and their corresponding M-CPOnes was not necessary, other than exclusion of ambient light for samples in solution. Upon irradiation with a suitable wavelength, photodecarbonylation of M-CPOne to the corresponding alkyne complex was observed, thus demonstrating the proof-of-concept. More detailed photophysical studies of cyclopropenone-based ligands and their corresponding M-CPOnes will be the subject of future work.The advantage of M-CPOnes is their modularity since up to 3 CO-releasing ligands can be incorporated within one complex and their properties can be readily tuned via the metal ion as well as the cyclopropenone substituents. For example, the absorbance maximum of the cyclopropenones in the M-CPOnes and their corresponding ligands was modulated by the R substituent with a bathochromic shift of up to almost 30 nm for the thienyl relative to the phenyl substituent.
The presence of the metal ion confers additional properties so that the photodecarbonylation of the Zn-CPOnes could be detected by fluorescence, the Fe-CPOnes by the shift of the MLCT band and the Co-CPOnes by paramagnetic NMR spectroscopy due to the greater dispersion of the signals, particularly for the bipyridine coordination motif given its proximity to the CoII centre. The ability to track photodecarbonylation by a variety of spectroscopic methods dependent on the metal ion will be beneficial for future applications of M-CPOnes.
Furthermore, M-CPOnes are compatible with incorporation of a second coordination site, 6-quinoline, and the initially formed M-CPOnes-1c (M = CoII, FeII) were proposed to rearrange into dynamic combinatorial libraries that still undergo photodecarbonylation to the corresponding M-2c complexes. This opens up avenues for supramolecular M-CPOnes and will be the subject of future investigations as well as broadening the scope of the metals within M-CPOnes.
Author contributions
M. L.: conceptualisation (equal), data curation (lead), formal analysis (lead), investigation (lead), resources (lead), validation (lead), visualisation (lead), writing-original draft (lead), writing-review and editing (equal). T. N.: conceptualisation (equal), formal analysis (supporting), investigation (supporting), resources (supporting), validation (supporting), visualisation (supporting), writing-original draft (supporting), writing-review and editing (equal). C. N.: formal analysis (supporting), investigation (supporting), resources (supporting), writing-review and editing (supporting). A. J. M.: conceptualisation (lead), formal analysis (supporting), funding acquisition (lead), project administration (lead), resources (supporting), supervision (lead), visualisation (supporting), writing-original draft (supporting), writing-review and editing (equal).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG, project number 413396832) for financial support. We thank the spectroscopy department, Dr Claus Bier and Johanna Baum for NMR and mass spectral data collection. We also thank Marvin Grünhagen for preliminary studies.Notes and references
- S. H. Heinemann, T. Hoshi, M. Westerhausen and A. Schiller, Chem. Commun., 2014, 50, 3644–3660 RSC.
- L. Y. Wu and R. Wang, Pharmacol. Rev., 2005, 57, 585–630 CrossRef CAS PubMed.
- R. Motterlini, B. E. Mann and R. Foresti, Expert Opin. Invest. Drugs, 2005, 14, 1305–1318 CrossRef CAS PubMed.
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed.
- C. P. Hopper, L. Meinel, C. Steiger and L. E. Otterbein, Curr. Pharm. Des., 2018, 24, 2264–2282 CrossRef CAS PubMed.
- A. Walewska, A. Szewczyk and P. Koprowski, Int. J. Mol. Sci., 2018, 19, 3227 CrossRef PubMed.
- H. Bae, T. Kim and I. Lim, Korean J. Physiol. Pharmacol., 2021, 25, 227–237 CrossRef CAS PubMed.
- A. Verma, D. J. Hirsch, C. E. Glatt, G. V. Ronnett and S. H. Snyder, Science, 1993, 259, 381–384 CrossRef CAS PubMed.
- D. Stucki, J. Steinhausen, P. Westhoff, H. Krahl, D. Brilhaus, A. Massenberg, A. P. M. Weber, A. S. Reichert, P. Brenneisen and W. Stahl, Antioxidants, 2020, 9, 251–260 CrossRef PubMed.
- B. E. Mann, Organometallics, 2012, 31, 5728–5735 CrossRef CAS.
- R. Motterlini, J. E. Clark, R. Foresti, P. Sarathchandra, B. E. Mann and C. J. Green, Circ. Res., 2002, 90, E17–E24 CrossRef CAS PubMed.
- R. Foresti, J. Hammad, J. E. Clark, T. R. Johnson, B. E. Mann, A. Friebe, C. J. Green and R. Motterlini, Br. J. Pharmacol., 2004, 142, 453–460 CrossRef CAS PubMed.
- N. Abeyrathna, K. Washington, C. Bashur and Y. Liao, Org. Biomol. Chem., 2017, 15, 8692–8699 RSC.
- X. Y. Ji and B. H. Wang, Acc. Chem. Res., 2018, 51, 1377–1385 CrossRef CAS PubMed.
- J. T. B. Kueh, N. J. Stanley, R. J. Hewitt, L. M. Woods, L. Larsen, J. C. Harrison, D. Rennison, M. A. Brimble, I. A. Sammut and D. S. Larsen, Chem. Sci., 2017, 8, 5454–5459 RSC.
- T. Slanina and P. Sebej, Photochem. Photobiol. Sci., 2018, 17, 692–710 CrossRef CAS PubMed.
- U. Schatzschneider, Inorg. Chim. Acta, 2011, 374, 19–23 CrossRef CAS.
- L. S. Lazarus, A. D. Benninghoff and L. M. Berreau, Acc. Chem. Res., 2020, 53, 2273–2285 CrossRef CAS PubMed.
- J. Cheng, G. H. Gan, Z. Q. Shen, L. Gao, G. Y. Zhang and J. M. Hu, Angew. Chem., Int. Ed., 2021, 60, 13513–13520 CrossRef CAS PubMed.
- R. N. Pickens, G. L. Judd and J. K. White, Chem. Commun., 2021, 57, 7713–7716 RSC.
- W. Feng, S. Feng and G. Feng, Chem. Commun., 2019, 55, 8987–8990 RSC.
- P. Peng, C. M. Wang, Z. Shi, V. K. Johns, L. Y. Ma, J. Oyer, A. Copik, R. Igarashi and Y. Liao, Org. Biomol. Chem., 2013, 11, 6671–6674 RSC.
- L. K. C. De La Cruz, S. L. Benoit, Z. X. Pan, B. C. Yu, R. J. Maier, X. Y. Ji and B. H. Wang, Org. Lett., 2018, 20, 897–900 CrossRef CAS PubMed.
- Y. Q. Zheng, X. Y. Ji, B. C. Yu, K. L. Ji, D. Gallo, E. Csizmadia, M. Y. Zhu, M. R. Choudhury, L. K. C. De La Cruz, V. Chittavong, Z. X. Pan, Z. N. Yuan, L. E. Otterbein and B. H. Wang, Nat. Chem., 2018, 10, 787–794 CrossRef CAS PubMed.
- K. T. Potts and J. S. Baum, Chem. Rev., 1974, 74, 189–213 CrossRef CAS.
- K. Komatsu and T. Kitagawa, Chem. Rev., 2003, 103, 1371–1428 CrossRef CAS PubMed.
- N. K. Urdabayev, A. Poloukhtine and V. V. Popik, Chem. Commun., 2006, 454–456 RSC.
- A. Poloukhtine and V. V. Popik, J. Phys. Chem. A, 2006, 110, 1749–1757 CrossRef CAS PubMed.
- X. Li, C. Han, H. Yao and A. Lin, Org. Lett., 2017, 19, 778–781 CrossRef CAS PubMed.
- T. Matsuda, K. Yamanaka, Y. Tabata and T. Shiomi, Tetrahedron Lett., 2018, 59, 1458–1460 CrossRef CAS.
- T. Matsuda, Y. Tabata and H. Suzuki, New J. Chem., 2018, 42, 19178–19182 RSC.
- H. M. Xing, J. Chen, Y. S. Shi, T. L. Huang, L. Hai and Y. Wu, Org. Chem. Front., 2020, 7, 672–677 RSC.
- Y. S. Shi, H. M. Xing, T. L. Huang, X. X. Liu, J. Chen, X. Y. Guo, G. B. Li and Y. Wu, Chem. Commun., 2020, 56, 1585–1588 RSC.
- J. L. Xu, H. Tian, J. H. Kang, W. X. Kang, W. Sun, R. Sun, Y. M. Li and M. Sun, Org. Lett., 2020, 22, 6739–6743 CrossRef CAS PubMed.
- S. W. Liu, C. Yuan, X. F. Jiang, X. X. Wang and H. L. Cui, Asian J. Org. Chem., 2020, 9, 82–85 CrossRef CAS.
- J. Wu, W. X. Gao, X. B. Huang, Y. B. Zhou, M. C. Liu and H. Y. Wu, Org. Lett., 2020, 22, 5555–5560 CrossRef CAS PubMed.
- Y. L. Yang, Z. Zhang, X. N. Zhang, D. Wang, Y. Wei and M. Shi, Chem. Commun., 2014, 50, 115–117 RSC.
- A. V. Kuzmin and V. V. Popik, Chem. Commun., 2009, 5707–5709 RSC.
- C. M. Vanos and T. H. Lambert, Angew. Chem., Int. Ed., 2011, 50, 12222–12226 CrossRef CAS PubMed.
- E. D. Nacsa and T. H. Lambert, Org. Lett., 2013, 15, 38–41 CrossRef CAS PubMed.
- C. C. Shao, H. Y. Duan, Y. Q. Min and X. H. Zhang, Chin. Chem. Lett., 2020, 31, 299–302 CrossRef CAS.
- M. Denis, D. Gindre and F. X. Felpin, J. Mater. Sci., 2021, 56, 5006–5014 CrossRef CAS.
- D. A. Sutton and V. V. Popik, J. Org. Chem., 2016, 81, 8850–8857 CrossRef CAS PubMed.
- D. A. Sutton, S. H. Yu, R. Steet and V. V. Popik, Chem. Commun., 2016, 52, 553–556 RSC.
- R. D. Row, H. W. Shih, A. T. Alexander, R. A. Mehl and J. A. Prescher, J. Am. Chem. Soc., 2017, 139, 7370–7375 CrossRef CAS PubMed.
- W. Luo, P. Gobbo, C. D. McNitt, D. A. Sutton, V. V. Popik and M. S. Workentin, Chem. – Eur. J., 2017, 23, 1052–1059 CrossRef CAS PubMed.
- R. D. Row, S. S. Nguyen, A. J. Ferreira and J. A. Prescher, Chem. Commun., 2020, 56, 10883–10886 RSC.
- L. Y. Sun, Y. K. Gai, C. D. McNitt, J. Sun, X. H. Zhang, W. Xing, Z. H. Li, V. V. Popik and D. X. Zeng, J. Org. Chem., 2020, 85, 5771–5777 CrossRef CAS PubMed.
- J. L. Benham, R. West and J. A. T. Norman, J. Am. Chem. Soc., 1980, 102, 5047–5053 CrossRef CAS.
- A. P. Vladimir and V. V. Popik, J. Org. Chem., 2003, 68, 7833–7840 CrossRef PubMed.
- R. Chotima, T. Dale, M. Green, T. W. Hey, C. L. McMullin, A. Nunns, A. G. Orpen, I. V. Shishkov, D. F. Wass and R. L. Wingad, Dalton Trans., 2011, 40, 5316–5323 RSC.
- E. I. Klimova, E. A. V. Lopez, M. F. Alamo, L. A. Ortiz-Frade, G. Hernandez-Sanchez, V. H. S. Dominguez and M. M. Garcia, J. Heterocycl. Chem., 2012, 49, 1156–1162 CrossRef CAS.
- J. J. S. Garcia, R. S. Joo-Cisneros, D. Garcia-Bassoco, M. Flores-Alamo, J. M. M. Stivalet, J. Garcia-Valdes and E. I. Klimova, J. Organomet. Chem., 2021, 944, 121809 CrossRef.
- K. A. Netland, L. L. Gundersen and F. Rise, Synth. Commun., 2000, 30, 1767–1777 CrossRef CAS.
- O. Korner, R. Gleiter and F. Rominger, Synthesis, 2009, 3259–3262 Search PubMed.
- P. Rulliere, P. Cyr and A. B. Charette, Org. Lett., 2016, 18, 1988–1991 CrossRef CAS PubMed.
- T. Nanda, P. Biswal, B. V. Pati, S. K. Banjare and P. C. Ravikumar, J. Org. Chem., 2021, 86, 2682–2695 CrossRef CAS PubMed.
- R. Breslow, L. J. Altman, A. Krebs, E. Mohacsi, I. Murata, R. A. Peterson and J. Posner, J. Am. Chem. Soc., 1965, 87, 1326–1331 CrossRef CAS.
- K. Matsumoto, A. Okada, T. Girek, Y. Ikemi, J. C. Kim, N. Hayashi, H. Yoshida and A. Kakehi, Heterocycl. Commun., 2002, 8, 325–328 CAS.
- M. Isaka, R. Ando, Y. Morinaka and E. Nakamura, Tetrahedron Lett., 1991, 32, 1339–1342 CrossRef CAS.
- M. Isaka, S. Ejiri and E. Nakamura, Tetrahedron, 1992, 48, 2045–2057 CrossRef CAS.
- M. Nakamura, H. Isobe and E. Nakamura, Chem. Rev., 2003, 103, 1295–1326 CrossRef CAS PubMed.
- M. Cohen, U. Bretler and A. Albeck, Protein Sci., 2013, 22, 788–799 CrossRef CAS PubMed.
- A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G. J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769–15776 CrossRef CAS PubMed.
- N. K. Urdabayev, A. Poloukhtine and V. V. Popik, Chem. Commun., 2006, 454–456 RSC.
- C. D. McNitt, H. Cheng, S. Ullrich, V. V. Popik and M. Bjerknes, J. Am. Chem. Soc., 2017, 139, 14029–14032 CrossRef CAS PubMed.
- M. G. Wilkerson, J. Henkin and J. K. Wilkin, J. Am. Acad. Dermatol., 1984, 11, 802–807 CrossRef CAS PubMed.
- T. Wasylyszyn and K. Borowska, Acta Pol. Pharm., 2016, 73, 1455–1460 Search PubMed.
- K. Borowska and T. Wasylyszyn, Acta Pol. Pharm., 2017, 74, 459–464 CAS.
- E. I. Klimova, T. K. Berestneva, L. R. Ramirez, A. Cinquantini, M. Corsini, P. Zanello, S. Hernandez-Ortega and M. M. Garcia, Eur. J. Org. Chem., 2003, 4265–4272 CrossRef CAS.
- T. Klimova, E. I. Klimova, M. M. Garcia, C. A. Toledano and R. A. Toscano, J. Organomet. Chem., 2003, 665, 23–28 CrossRef CAS.
- Z. M. Strater, M. Rauch, S. Jockusch and T. H. Lambert, Angew. Chem., Int. Ed., 2019, 58, 8049–8052 CrossRef CAS PubMed.
- G. Ernouf, J. L. Brayer, B. Folleas, J. P. Demoute, C. Meyer and J. Cossy, J. Org. Chem., 2017, 82, 3965–3975 CrossRef CAS PubMed.
- S. S. Nguyen, A. J. Ferreira, Z. G. Long, T. K. Heiss, R. S. Dorn, R. D. Row and J. A. Prescher, Org. Lett., 2019, 21, 8695–8699 CrossRef CAS PubMed.
- Y. V. Skornyakov, N. A. Lozinskaya, M. V. Proskurnina and N. S. Zefirov, Russ. J. Org. Chem., 2005, 41, 689–693 CrossRef CAS.
- R. Sunke, S. B. Nallapati, J. S. Kumar, K. S. Kumar and M. Pal, Org. Biomol. Chem., 2017, 15, 4042–4057 RSC.
- D. L. S. Brahms and W. P. Dailey, Chem. Rev., 1996, 96, 1585–1632 CrossRef CAS PubMed.
- W. R. Dolbier and M. A. Battiste, Chem. Rev., 2003, 103, 1071–1098 CrossRef CAS PubMed.
- F. Wang, W. Zhang, J. M. Zhu, H. F. Li, K. W. Huang and J. B. Hu, Chem. Commun., 2011, 47, 2411–2413 RSC.
- L. C. Li, F. Wang, C. F. Ni and J. B. Hu, Angew. Chem., Int. Ed., 2013, 52, 12390–12394 CrossRef CAS PubMed.
- F. Wang, T. Luo, J. B. Hu, Y. Wang, H. S. Krishnan, P. V. Jog, S. K. Ganesh, G. K. S. Prakash and G. A. Olah, Angew. Chem., Int. Ed., 2011, 50, 7153–7157 CrossRef CAS PubMed.
- C. F. Ni and J. B. Hu, Synthesis, 2014, 46, 842–863 CrossRef.
- G. Tran, D. G. Pardo, T. Tsuchiya, S. Hillebrand, J. P. Vors and J. Cossy, Org. Lett., 2015, 17, 3414–3417 CrossRef CAS PubMed.
- R. Eujen and B. Hoge, J. Organomet. Chem., 1995, 503, C51–C54 CrossRef CAS.
- X. Y. Deng, J. H. Lin, J. Zheng and J. C. Xiao, Chem. Commun., 2015, 51, 8805–8808 RSC.
- A. K. Brisdon, I. R. Crossley, K. R. Flower, R. G. Pritchard and J. E. Warren, Angew. Chem., Int. Ed., 2003, 42, 2399–2401 CrossRef CAS PubMed.
- Z. L. Cheng and Q. Y. Chen, Synlett, 2006, 478–480 CrossRef CAS.
- T. Nihei, T. Hoshino and T. Konno, Org. Lett., 2014, 16, 4170–4173 CrossRef CAS PubMed.
- G. Kuzmanich, M. N. Gard and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2009, 131, 11606–11614 CrossRef CAS PubMed.
- S. Staicu, I. G. Dinulescu, F. Chiraleu and M. Avram, J. Organomet. Chem., 1976, 113, C69–C72 CrossRef CAS.
- G. Quinkert, K. Opitz, W. W. Wiersdorff and J. Weinlich, Tetrahedron Lett., 1963, 27, 1863–1868 CrossRef.
- Y. Hirata and N. Mataga, Chem. Phys. Lett., 1992, 193, 287–291 CrossRef CAS.
- R. W. Fessenden, P. M. Carton, H. Shimamori and J. C. Scalano, J. Phys. Chem., 1982, 86, 3803–3811 CrossRef CAS.
- Y. Hirata, T. Okada, N. Mataga and T. Nomoto, J. Phys. Chem., 1992, 96, 6559–6563 CrossRef CAS.
- S. Takeuchi and T. Tahara, J. Chem. Phys., 2004, 120, 4768–4776 CrossRef CAS PubMed.
- M. Lehr, T. Paschelke, E. Trumpf, A. M. Vogt, C. Näther, F. D. Sönnichsen and A. J. McConnell, Angew. Chem., Int. Ed., 2020, 59, 19344–19351 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2142653. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00835a |
| This journal is © The Royal Society of Chemistry 2022 |