
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium containing molecular electrocatalyst on glassy carbon for electrochemical water splitting†
Lin
Li‡
 *abc,
Biswanath
Das‡
*b,
Ahibur
Rahaman
b,
Andrey
Shatskiy
d,
Fei
Ye
e,
Peihong
Cheng
a,
Chunze
Yuan
ac,
Zhiqi
Yang
a,
Oscar
Verho
*b,
Markus D.
Kärkäs
d,
Joydeep
Dutta
e,
Tsu-Chien
Weng
ac and
Björn
Åkermark
*b
*abc,
Biswanath
Das‡
*b,
Ahibur
Rahaman
b,
Andrey
Shatskiy
d,
Fei
Ye
e,
Peihong
Cheng
a,
Chunze
Yuan
ac,
Zhiqi
Yang
a,
Oscar
Verho
*b,
Markus D.
Kärkäs
d,
Joydeep
Dutta
e,
Tsu-Chien
Weng
ac and
Björn
Åkermark
*b
aSchool of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China. E-mail: lilin1@shanghaitech.edu.cn
bDepartment of Organic Chemistry, Arrhenius Laboratory Stockholm University, Svante Arrhenius v-g 16C, 10691 Stockholm, Sweden. E-mail: bjorn.akermark@su.se; oscar.verho@ilk.uu.se; Das.Biswanath85@gmail.com
cCenter for Transformative Science, ShanghaiTech University, Shanghai 201210, China
dDepartment of Chemistry, KTH Royal Institute of Technology, Stockholm SE-100 44, Sweden
eDepartment of Applied Physics, Functional Materials, KTH Royal Institute of Technology, 106 91 Stockholm, Sweden
First published on 6th May 2022
Abstract
Electrochemical water splitting constitutes one of the most promising strategies for converting water into hydrogen-based fuels, and this technology is predicted to play a key role in the transition towards a carbon-neutral energy economy. To enable the design of cost-effective electrolysis cells based on this technology, new and more efficient anodes with augmented water splitting activity and stability will be required. Herein, we report an active molecular Ru-based catalyst for electrochemically-driven water oxidation (overpotential of ∼395 mV at pH 7 phosphate buffer) and two simple methods for preparing anodes by attaching this catalyst onto glassy carbon through multi-walled carbon nanotubes to improve stability as well as reactivity. The anodes modified with the molecular catalyst were characterized by a broad toolbox of microscopy and spectroscopy techniques, and interestingly no RuO2 formation was detected during electrocatalysis over 4 h. These results demonstrate that the herein presented strategy can be used to prepare anodes that rival the performance of state-of-the-art metal oxide anodes.
Introduction
The conversion of solar energy into hydrogen-based fuels is considered to be one of the most promising alternatives to fossil fuels.1 Water is an inexpensive and readily available natural source of hydrogen, which has made electrochemical water splitting an intensively pursued research field in recent decades.2 However, the anodic reaction that leads to the oxidation of water to molecular oxygen is plagued by a high activation barrier, which has steered research towards the development of active and durable water oxidation catalysts (WOCs).3 So far, the most intensively studied type of catalysts is the molecular ones, which have the advantage of straightforward structural modification and electronic control.4 However, molecular catalysts generally suffer from limited stability, which in many cases can be improved through the attachment of the catalysts onto conductive surfaces, where bimolecular decomposition pathways are impeded by restricted translational mobility.5Catalysts based on 1st row transition metals are considered to be more attractive for large-scale industrial applications compared to their 2nd and 3rd row counterparts due to their low cost and environmentally benign character.6 However, in practice, higher stability and catalytic efficiency have been observed for ruthenium (Ru) and iridium (Ir)-based catalysts, which have made them the most widely studied.7 Earlier, we reported a highly active Ru-based water oxidation catalyst, [Ru(mcbp)(py)2] (mcbp2− = 2,6-bis(1-methyl-4-(carboxylate)-benzimidazol-2-yl)pyridine, py = pyridine, 1 in Fig. 1).8 This catalyst shows an impressive maximum turnover frequency (TOFmax) of approximately 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1 (calculated from the foot-of-the-wave analysis) at pH 9.0,9 which shares the world record in efficiency with a related catalyst developed by Llobet and co-workers.10 Given the remarkable performance of this catalyst, we became interested in immobilizing it on an electrode for practical applications in electrocatalytic water oxidation.
000 s−1 (calculated from the foot-of-the-wave analysis) at pH 9.0,9 which shares the world record in efficiency with a related catalyst developed by Llobet and co-workers.10 Given the remarkable performance of this catalyst, we became interested in immobilizing it on an electrode for practical applications in electrocatalytic water oxidation.
 | ||
| Fig. 1 Chemical structure of the previously reported Ru-based catalyst 1,8 the structure of Ru-based catalyst 2 (this work), and optimized by unrestricted DFT calculations structure of 2 (for cartesian coordinates, see the ESI†) in aqueous medium (SMD model).16 Blue, red, grey, and turquoise colors represent nitrogen, oxygen, carbon, and ruthenium atoms, respectively. | ||
There exists a wide range of strategies for attaching molecular catalysts onto metal oxide electrodes.11 One of the most common approaches involves the utilization of either phosphonate or carboxylate anchoring groups.12 However, a major limitation of these systems is their instability during electrochemical oxidation in alkaline aqueous electrolytes.13
Multi–walled carbon nanotubes (MWCNTs) have also been studied as support for molecular catalysts, and they have shown great promise for improving catalytic performance thanks to their high conductivity and large electrochemically active surface area.12 Here, previous approaches to anchor water oxidation catalysts onto MWCNTs have involved modified ligands serving as linkers,13 containing mostly aromatic or polyaromatic moieties14 that can interact non-covalently with the surface of carbon nanotubes via π-stacking and electrostatic interactions.15 However, equipping water oxidation catalysts with aromatic or polyaromatic moieties is relatively complicated, which hinders large-scale synthesis and the application of such catalysts.
Herein, we report the synthesis of catalyst [Ru(mcbp)(4,4′-bpy)2] (2) (Fig. 1) and two methods for conveniently attaching it onto a glassy carbon surface using MWCNTs as conductive support. [Ru(mcbp)(4,4′-bpy)2] (bpy = bipyridine) (2) represents a structural modification of the catalyst [Ru(mcbp)(py)2] (1), in which the two axial pyridine ligands are replaced by 4,4′-bipyridines. This structural modification was shown to greatly improve the interaction between catalyst 2 and the MWCNTs, while not altering the catalytic features of the catalyst significantly. 4,4′-Bipyridine linkers have previously been reported to serve as bridges between a series of metal centers to give coordination polymers.17 In an analogous fashion, we prepared the corresponding coordination polymer of 2 and directly anchored it onto the MWCNTs. In this work, we describe the synthesis of the new catalyst 2 and its characterization under homogeneous and heterogeneous (after attachment to MWCNTs) conditions. Furthermore, the structural features of catalyst 2 on MWCNTs before and after electrolysis on the electrodes were investigated.
Results and discussion
Catalyst 2 was synthesized following a step-wise procedure (for details, see the ESI†), initiated by refluxing a mixture of H2mcbp, [Ru(DMSO)4Cl2], and triethylamine in ethanol–water (1:1), followed by the addition of two equivalents of 4,4′-bpy. Continuous reflux of the reaction mixture at 80 °C over 5 days under N2 atmosphere resulted in precipitate of the major product (Scheme S1 and Fig. S1, S2†). The selectivity of the reaction towards the formation of the monomeric complex 2 could be improved by following an alternative step-wise procedure, in which H2mcbp and [Ru(DMSO)4Cl2] were refluxed in ethanol–water (1:1) for 4 h, followed by the addition of triethylamine and two equivalents of 4,4′-bpy and continued reflux for 3 days. The monomeric complex 2 was characterized by Nuclear Magnetic Resonance (NMR), High Resolution Mass Spectrometry (HRMS), and Ultraviolet–Visible (UV–Vis) spectroscopy (for details, see the ESI†). The 1H NMR spectrum of 2 shows a singlet at δ 4.48 ppm integrating to 6H belonging to the two methyl groups, and all aromatic protons are present within the 7.48–8.73 ppm range (Fig. S1†). The presence of the two bipyridine units in 2 was further confirmed by HRMS, demonstrating a singly-charged Ru-complex at m/z 862.1424 (Fig. S3†) with its characteristic isotope pattern. Several attempts to obtain single crystals of 2 suitable for X-ray diffraction analysis were unsuccessful.
A DFT optimized [B97D3/def2tzvp-6-31G(d,p), aqueous medium (smd model), unrestricted] structure of 2 (Fig. 1) reveals that the neutral molecule has one free carboxylate and one ruthenium bonded carboxylate unit (for the cartesian coordinates, see the ESI†). The Ru–N (benzimidazole) bond distance is significantly shorter at the side where the carboxylate unit is coordinated (∼1.93 Å) compared to the side where it remains as a free carboxylate (∼2.15 Å) unit (Fig. 1 and S11†). The other three Ru–N (to the central pyridine and two axial bipyridine ligands) bond distances are between 1.95 and 2.08 Å. Attempts to optimize the structure of [RuII(mcbp)(4,4′-bpy)2] (2) with both carboxylates coordinated to the ruthenium center did not result in a feasible structure. The UV-Vis spectra of complexes 1 and 2 in aqueous phosphate buffer solutions at pH 2.0 and 7.0 displayed metal-to-ligand charge-transfer (MLCT) bands (λmax ∼490–510 nm) that are typical for RuII polypyridine complexes (Fig. S4†).
The cyclic voltammograms (CV) of both 1 and 2 showed two quasi-reversible waves between 0.60 to 1.45 V vs. NHE in pH 2.0 phosphate buffer (Fig. 2A), which can be attributed to the RuIII/RuII and RuIV/RuIII redox couples. Differential pulse voltammetry (DPV) demonstrated the redox potentials for these couples at 0.72 and 1.31 V vs. NHE for 1 and 0.81 and 1.33 V vs. NHE for 2. These redox waves are followed by an irreversible anodic wave characteristic of water oxidation. The similar CV behavior for 1 and 2 indicates that the change of ligand from pyridine to 4,4′-bipyridine did not cause any significant differences in the electrochemical behavior, especially for the RuIV/III couple at pH 2.0. At pH 7, 1 showed slightly lower (around 70 mV) overpotential for water oxidation in the first scan and the catalytic current remained relatively unchanged during multiple CV scans (Fig. 2B). Interestingly, in the case of 2, the electrocatalytic wave continuously increased upon sequential CV cycling, while the onset potential gradually shifted towards more negative potentials. After ten consecutive scans, the catalytic current almost doubled and considerable water oxidation beginning at ca. 1.21 V vs. NHE could be observed, i.e., showing an overpotential of ∼395 mV at pH 7 phosphate buffer.§ Under this condition only RuIII/II reversible redox wave was visible for 2 at around 0.69 V and RuIV/III redox couple gets hidden under the WO wave (Fig. S5†). After ten scans at pH 7, both of the electrodes were removed, washed with water, and dried. These electrodes were then tested as working electrodes in phosphate buffer solutions at pH 7.0 in absence of any additional catalysts (Fig. S6†). The modified electrodes generated during the electrochemical experiment with 2 showed more than ten times higher catalytic current in comparison to the electrode generated with 1, suggesting a greater accumulation of the catalyst on the electrode surface during the CV experiment with 2 (Fig. S6†) and a better interaction of the bipyridine units of 2 with the electrode surface. The molecularity of 2 on glassy carbon electrode was confirmed by characteristic RuIII/II (at 0.47 V) and RuIV/III (at 0.68 V) waves (Fig. S6† inset). These results suggest that 2 represents a better candidate for heterogeneous water splitting. Upon continuous CV cycling, the catalytic current using both electrodes steadily decrease, indicating detachment of the catalysts from the surface.
 | ||
| Fig. 2 CV and DPV measurements (inset A) of 0.1 mM solutions of complexes 1 and 2 at pH 2.0 (A) and pH 7.0 (B). Inset B shows the first and the tenth CV cycles. The scan rate for CV measurements was 100 mV s−1. A glassy carbon disk, a Pt disk, and Ag/AgCl electrodes were used as working, counter, and reference electrodes, respectively. | ||
To prevent detachment and increase the loading of catalyst, MWCNTs were added as supporting material onto the glassy carbon (GC) electrodes. Two types of electrodes (type A and B) were prepared as illustrated in Scheme 1. In short, electrodes of type A were prepared by drop-casting MWCNTs onto the GC electrodes, followed by dipping them into an H2O/DMSO-solution of 2 for 24 h. Type B electrodes were prepared by first attaching 2 onto the MWCNTs in THF solution, followed by drop casting them onto a GC electrode. Both type A and B electrodes were thoroughly washed with EtOH and H2O and dried under a high flow of N2 prior to their further use as described in ESI.†
 | ||
| Scheme 1 Illustration of the preparation procedure for electrodes A and B. | ||
Fig. 3A shows the cyclic voltammetric features of electrodes A and B at pH 7.0. The red trace, which is the type A electrode, prepared from 1, shows almost no catalytic activity. In contrast, the black trace which comes from the same type (A) of electrode prepared from 2, shows appreciable catalytic current starting at around 1.25 V vs. NHE. This result indicates that 2 was attached to the MWCNTs electrodes under these reaction conditions while 1 was not. The reason could be the improved electrostatic interaction and π-stacking of 2 as compared to 1 with the MWCNTs. Electrode B (blue trace) shows a very similar onset potential for the electrocatalytic current associated with water oxidation at around 1.25 V vs. NHE, but a slightly lower electrocatalytic response compared to electrode A. However, the current density of electrode B is significantly higher than that of electrode A in the region of 0.2 to 1 V vs. NHE, which indicates higher catalyst loading for the electrode B.
 | ||
| Fig. 3 (A) CVs of electrodes A or B as the working electrodes (WE) in a pH 7.0 phosphate buffer solution. The scan rate was 100 mV s−1. A Pt disk and a Ag/AgCl were used as counter electrode and reference electrode, respectively. (B) Bulk electrolysis using type A electrode with catalyst 2 as working electrode (black) and without catalyst 2 (red) in a phosphate-buffered solution at pH 7.0 at 1.4 V vs. NHE. | ||
The catalytic efficiency of the best performing electrode (type A, loaded with 2) was further investigated by bulk electrolysis experiments at an applied potential of 1.4 V vs. NHE for over 4 h (Fig. 3B). The current steadily decreased to close to 0 after 4.5 h of continuous electrolysis, which is similar to other related state-of-the-art electrocatalytic systems.11a,13 Looking at the current densities of electrodes A and B with 2 (Fig. 3B and Fig. S7B†), it is quite clear that electrode A starts oxidizing water with much higher current density, but deactivates faster than electrode B. Nearly a two-fold increase of the current density was observed during the first 5000 seconds, followed by a slight decrease (Fig. S7B†) before reaching a constant current for electrode B with 2. This result points towards possible structural changes of the catalyst or the surface morphology under the oxidative conditions of bulk electrolysis. Altogether, it can be inferred that although the loading is much higher for electrode B, the morphology of the catalyst on MWCNTs is not suitable for higher current density.
In order to characterize the electrodes further, a series of spectroscopic studies were performed, which included scanning electron microscopy (SEM), transmission electron microscopy (TEM), Fourier transform infrared spectroscopy (FTIR), Raman spectroscopy, and X-ray photoelectron spectroscopy (XPS).
To correlate the electrochemical behavior of electrodes A and B with the preparation procedure, the surface morphology of the two electrodes as well as an electrode prepared from pure MWCNTs were examined by SEM and TEM (Fig. 4). The SEM image of MWCNTs (Fig. 4A) shows that the carbon nanotubes have a diameter around 20 nm and a length close to 200 nm. For electrode A, a smooth and uniform covering layer of 2 on the MWCNTs is observed (Fig. 4B). The thickness of the MWCNTs is around 30 nm, reflecting an excellent attachment of 2 onto the surface of the MWCNTs. In contrast, electrode B (Fig. 4C) with 2 displays an irregular coating of around 30 nm on the MWCNTs that results in agglomeration. These indicate that the catalyst could evenly cover the MWCNTs on the surface of electrode A, while catalyst-coated MWCNTs are randomly interconnected on the surface of electrode B. Further visualization of individual carbon nanotubes was provided by high-resolution TEM micrographs. From these, it can be seen that both electrodes A and B show the presence of hollow channels with the orientation of graphene layers, which is the typical appearance of the MWCNTs with a diameter of approximately 30 nm. It can also be noticed that the outside of the crystalline lattice of the MWCNTs is covered by amorphous materials for electrode B (Fig. 4E), while this cannot be seen for electrode A (Fig. 4D). On the other hand, the aggregation of catalyst 2 can rarely be seen for electrode A. These observations suggest that the 4,4′-bipyridine unit facilitates the absorption of catalyst 2 onto the surface of MWCNTs, most likely via improved π-stacking and electrostatic interactions.15,20
 | ||
| Fig. 4 Scanning electron microscopy (SEM) images of (A) MWCNTs, (B) electrode type A with 2, and (C) electrode type B with 2. Transmission electron microscopy (TEM) images of (D) electrode type A with 2 and (E) electrode type B with 2. | ||
In order to further verify the catalyst's coverage on the MWCNTs, TEM combined with EDX was used to explore the elemental composition of the electrodes. The elemental mappings of C, N, O, and Ru in electrode B are presented in Fig. 5. From this result, it can be seen that the atomic ratio between N and Ru is around 7:1, which agrees with the expected composition of catalyst 2. Unfortunately, we were unable to estimate the elemental composition of electrode A, due to the low loading of catalyst 2 (Fig. S8†).
 | ||
| Fig. 5 Scanning transmission electron microscopy (STEM) image of electrode type B with 2 and corresponding elemental mappings using energy dispersive X-ray spectroscopy (EDS). | ||
FTIR and Raman spectroscopy confirmed the molecular level attachment of the complexes to the MWCNTs as observed by electron microscopy. The FTIR spectra of electrode A and MWCNTs are shown in Fig. 6A. Both MWCNTs and catalyst-coated MWCNTs on electrodes show the characteristic C–H stretching (2955–2840 cm−1) and C–H bending (1465–1360 cm−1) bands. A relatively stronger intensity of the stretching and bending bands of C–H from the complex coating on the electrode implies the cumulative signals from the C–H moieties of MWCNTs and the catalyst 2. In addition, the band at 1227 cm−1 can be assigned to the C–N stretching of pyridine, further emphasizing the successful attachment of 2 onto the MWCNTs. A very weak band centered at 1730 cm−1 is observed for both MWCNTs and the catalyst-coated electrode, which could correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching from surface groups on the MWCNTs and from 2 that coats the electrode. The Raman spectra of electrode A before and after electrolysis were compared (Fig. 6B). The major peaks are from the G and D bands of MWCNTs, which are associated with in-plane vibration modes. This can be explained by the low loading of the catalyst.
O stretching from surface groups on the MWCNTs and from 2 that coats the electrode. The Raman spectra of electrode A before and after electrolysis were compared (Fig. 6B). The major peaks are from the G and D bands of MWCNTs, which are associated with in-plane vibration modes. This can be explained by the low loading of the catalyst.
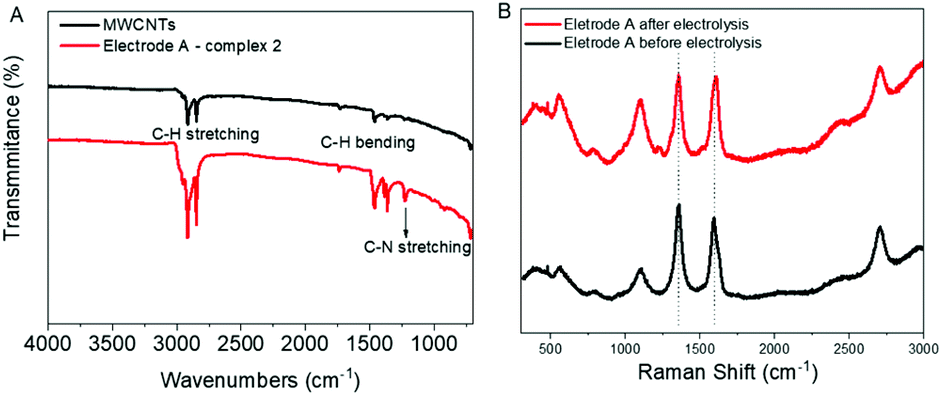 | ||
| Fig. 6 (A) FTIR spectra of electrode A and MWCNTs. (B) Raman spectral analysis of MWCNTs and electrode A with 2 before and after electrolysis. See Fig. S9† for Raman spectra of MWCNTs before immobilizing catalysts. | ||
The G band at 1510 cm−1 and D band at 1350 cm−1 are characteristic Raman signals for amorphous carbon.18 They are reported to overlap sometimes and are difficult to predict. The D band is generally derived from the defective structures and an increasing value for ID/IG ratio is an indicator of the defect amounts in CNTs. In the case of electrode type A with 2 this D band is significant (Fig. 6B), whereas, for electrode B with 2, it is almost negligible (Fig. S9†).
A slight rise of the signal around 1106 cm−1 after electrolysis for electrode type A indicates possible small structural changes of the catalyst. No such changes were observed for electrode type B (Fig. S9†), most probably due to the lower WO activity. No characteristic Raman spectral features of RuO2, such as a sharp peak located around 515 to 528 cm−1 (due to the Eg band of RuO2) could be observed for both type A and B electrodes.19 Thus, there is no evidence of the formation of RuO2 during the electrolysis based on the Raman spectral analysis.
Furthermore, XPS was used to investigate the elemental composition and chemical state of the electrode material before and after the electrocatalysis (Fig. 7 and Fig. S10†). The full XPS spectra of electrodes A and B before and after electrolysis show the existence of C, N, Ru, and O (Fig. 7A). High-resolution XPS spectra for C1s and Ru3d are shown in Fig. 7B and C, wherein the C 1s peak at 284.6 eV (for electrode A) comes from the C–C of graphite overlapping with Ru 3d3/2 at 286.2 eV. The peak at 280.8 eV corresponds to Ru 3d5/2. Curve fitting of the Ru 3d and C 1s core level peaks was performed to isolate the contribution of the different species. More than three types of carbon were found from the peaks centred around 284.6, 285.3, and 286.1 eV, which are proposed to be due to the C–C of the graphite peak (284.6 eV) and the C–C, C–N and CO bonds of the catalytic material 2, respectively. There were no significant changes regarding the positions of the C-peaks except for the relative intensity of the different species after electrolysis. In other words, the positions of the peaks before and after electrolysis are consistent, which indicates that no new or structurally very different species are being formed upon electrolysis. The change in the proportion of the species before and after electrolysis might be due to the activation of materials during the water oxidation reaction. For instance, more Ru2+ species were converted to Ru3+ species. It can also be noticed that the intensity of the peak at 281 eV (Ru 3d5/2) is weaker for the electrodes after electrolysis. The weaker signal indicates lower quantities of ruthenium-based materials, which might be due to a slight loss of the catalytic material during the electrolysis process. Therefore, the slow detachment of 2 from the electrode might be the reason for lower current density when continuous electrolysis was carried out as shown in Fig. 3. The peak intensities of both Ru 3d3/2 and Ru 3d5/2 are much higher for electrode B (Fig. 7C) than for electrode A (Fig. 7B). This can again be attributed to the higher loading of the catalyst in electrode B. Fig. 7D and E show the high-resolution scan for the N 1s peak. All the spectra show clear N 1s peaks, which indicate the existence of ligands containing N in both types of electrodes before and after the electrolysis process. New peaks at higher binding energy were found after the electrolysis that indicates the presence of a more highly oxidized environment around the N. This is most likely due to a higher oxidation state of the metal center of the molecular catalysts. Fig. 7F and G show the high-resolution scan for the O 1s peak. All the spectra show similar peak positions, although different electrodes show different proportions of the two species. Also, here the peak positions are more or less identical for the electrode before and after electrolysis, which indicates the presence of molecular catalysts rather than ruthenium oxide before and after the electrolysis.
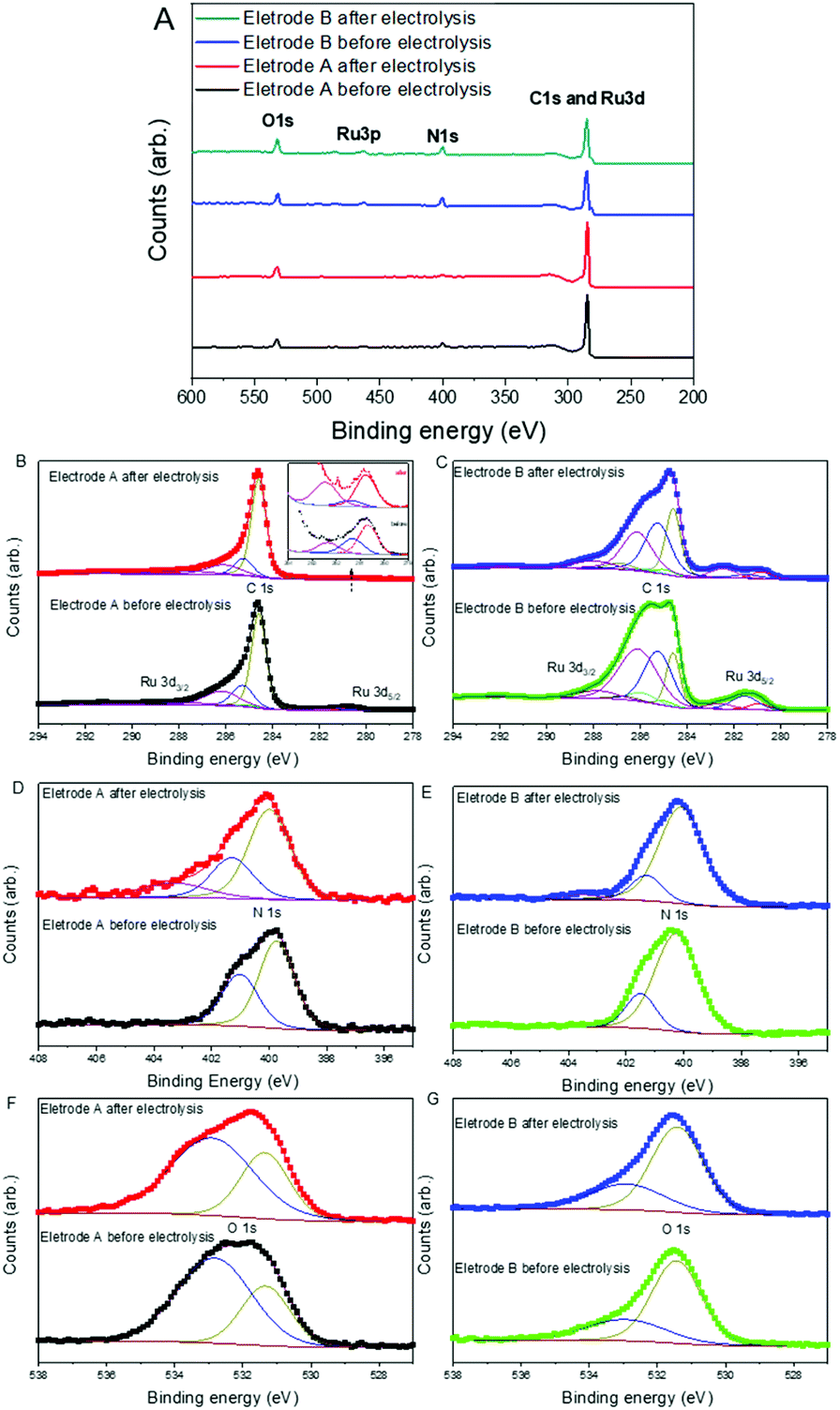 | ||
| Fig. 7 (A) Full XPS spectra of electrode A and electrode B before and after electrolysis. The high-resolution scan for C 1s and Ru 3d ((B) and (C)), N 1s ((D) and (E)), O 1s ((F) and (G)) of electrode A and electrode B before and after electrolysis. | ||
Conclusion
In summary, we report on a molecular Ru catalyst, which was modified from our previously published highly active ruthenium-based water oxidation catalyst ([Ru(mcbp)(py)2]) by replacing the pyridines with 4,4′-bipyridine units. This catalyst was anchored onto glassy carbon through MWCNTs using two different methods, which both form highly active electrodes for electrochemically driven water oxidation. These electrodes were characterized by SEM, TEM, EDX, FTIR, Raman, and XPS before and after electrolysis, and interestingly no sign of RuO2 formation was detected during the electrocatalysis, demonstrating the high stability as well as low overpotential (for WO) of the molecular catalyst on MWCNTs. In 2020, Llobet and co-workers reported another ruthenium-based metal complex which had a similar efficiency and stability as reported in this study.20 However, it is important to point out that the catalyst reported in the current work operates at an even lower potential than that one prepared by Llobet and co-workers. This demonstrates the positive influence of the non-innocent imidazole ligand in electro-catalytic water oxidation. Furthermore, it shows that proper utilization of the imidazole unit in water oxidation electrocatalysts and the manufacture of anodes for electrolysis cells can be a viable strategy for the future.Experimental section
Materials
All reagents and solvents were commercially available and used as received unless otherwise noted. Reagent grade organic solvents were used for the purification and HPLC grade solvents were used for synthesis and other operations. Multi-walled carbon nanotubes (MWCNTs, 10–20 nm (diam), 5–15 μm (length), product number is C2150) were purchased from Tokyo chemical industry co. LTD. MilliQ water (18.2 MΩ cm) was used for all electrochemical experiments. H2mcbp (mcbp2− = 2,6-bis(1-methyl-4-(carboxylate)-benzimidazol-2-yl) pyridine),8 and [Ru(DMSO)4Cl2] were synthesized according to the previous reports.21 Phosphate buffered solutions of pH 2.0, and 7.0 (0.1 ionic strength) were prepared using the standard procedure. For pH 2.0 phosphate buffer (0.1 ionic strength), 3.012 g of NaH2PO4 and 1.80 mL of 85 wt% H3PO4 were mixed and diluted with water to 250 mL. For pH 7.0 phosphate buffer (0.1 ionic strength) 997 mg of Na2HPO4 and 489 mg of NaH2PO4 were mixed and diluted with water to 250 mL, and the pH was adjusted to 7.0 with the previously prepared pH 2.0 phosphate buffer solution.Synthetic procedures
:1). This mixture was then purged with nitrogen for 10 min 3.5 mL of the mixture was transferred to the reaction vessel under the protection of N2, resulting an homogeneous yellow solution. The reaction mixture was then stirred at 80 °C under nitrogen, under which it gradually turned to a dark reddish-brown color. After 5 days the dark-brown reaction mixture was allowed to cool gradually to room temperature and was stored in the fridge overnight, which resulted in the formation of a dark-brown precipitate. The precipitate was collected by filtration, washed with ice-cold water (3 × 5 mL) and ethanol (2 × 5 mL), and dried overnight under vacuum, resulting in the title compound as a dark-brown solid of 2 (Fig. S1 and S2†). Upon heating for an extended period (from 5 days to 7 days), the formation of a higher quantity of the oligomers and polymers could be identified by their lower solubility and the NMR spectroscopy features (Fig. S1b†).
DFT calculations
The structure of 2 was optimized by unrestricted DFT without any restriction.16 The standalone functional B97D3 (as implemented in the Gaussian09 package) was used. The def2-TZVP basis set (and associated effective core potential) for the ruthenium atom and 6-31G(d,p) basis set for C, H, N and O atoms were used for the optimization.22 Solvation effects were evaluated using the SMD model on the optimized geometries at the same level of theory as the geometry optimizations with water as the surrounding solvent. Cartesian coordinates can be found in the ESI.†Characterization
NMR spectra were recorded on Bruker Ascend or Bruker Avance spectrometers and internally calibrated against the residual undeuterated solvent peaks (DMSO-d6: δ 2.50 for 1H NMR). Chemical shifts (δ) are reported in ppm and peak multiplicity is designated as s (singlet), d (doublet), t (triplet), m (multiplet), dd (doublet of doublets), dt (doublet of triplets), and tt (triplet of triplets). Aqueous samples were recorded on an NMR spectrometer equipped with a cryoprobe. ESI-HRMS measurements were performed using the Bruker Daltonics micrOTOF mass spectrometer (direct injection, positive mode, all samples in MeOH). UV-vis spectra were recorded on a Varian Cary 50 Bio UV-vis spectrometer. Fourier transform infrared spectra between 4000 cm−1 to 650 cm−1 were recorded using a Bruker Vertex V70 apparatus with a minimum of 32 scans. Raman spectra were obtained using a Shamrock SR-500I-D2-1F1 500mm focal length, motorized Czerny-Turner Spectrograph. XPS (Thermo Scientific ESCALAB 250Xi) was performed using a monochromatic Al Kα source (hν = 1486.6 eV). All of the XPS spectra were corrected in regards to C 1s peak at a binding energy of 284.6 eV. The Thermo Scientific™ Avantage software was used for data analysis and curve fitting. Scanning Electron Microscopy (SEM, JEOL JSM-7800F) operating at 15 kV was used to observe the morphology of the samples. High-resolution transmission electron microscopy (HRTEM) images were captured using a JEOL JEM-2100 Plus apparatus.Electrode preparation
:1. This mixture was purged with nitrogen for 10 min 0.8 mL of the mixture was transferred to the reaction vessel under the protection of N2. The reaction vessel was sonicated for 5 minutes, then the reaction mixture was stirred at 80 °C under nitrogen. After 5 days the obtained dark reaction mixture was allowed to cool gradually to room temperature and was stored in the fridge overnight. The precipitate was collected by centrifugation, washed with water, and evacuated overnight.
Electrochemical measurements
All electrochemical measurements were carried out using a Biologic SP-240 potentiostat. The measurements were performed in a standard electrochemical cell fitted with glassy carbon disk (d is 0.3 cm and area are 0.07 cm2) as the working electrode, Ag/AgCl as the reference electrode, and platinum disk as the counter electrode. Phosphate buffered solutions of pH 2.0 and 7.0 (0.1 ionic strength) were used as electrolyte and 0.1 mM of catalysts were dissolved in the electrolyte. The scan rate for cyclic voltammetry (CV) was 100 mV s−1. Differential pulse voltammetry (DPV) was recorded with the following acquisition parameters: potential increment 0.004 V, pulse height 0.05 V, pulse width 0.05 s, sample width 0.0167 s, pulse period 0.5 s. Potentials vs. Ag/AgCl were converted to vs. NHE by using Ru(bpy)3Cl2 as a reference. The redox potential of Ru2+/3+ couple was obtained at 1.095 vs. Ag/AgCl in our experiments, which should be 1.26 V vs. NHE. Therefore, all obtained potentials vs. Ag/AgCl was obtained by adding 0.165 V to convert to vs. NHE.The Electrochemically active surface area (ECSA) was estimated by the double-layer capacitance according to eqn (1).6
 | (1) |
C S is the specific capacitance of the sample and CDL is the electrochemical double layer capacitance. Here, we use general specific capacitances of CS = 0.04 mF cm−2 based on the typical reported values.
C DL was obtained by measuring the double-layer charging current (ic) from the scan-rate dependence of cyclic voltammograms (CVs) in a typical 0.1 V potential window centered at the open-circuit potential (OCP), where no apparent faradaic processes occurred and therefore all measured current is assumed to be due to double-layer charging. The double-layer charging current (ic) is the product of the electrochemical double-layer capacitance (CDL) times the scan rate (v), as given by eqn (2).
| ic = CDL × v | (2) |
Thus, the electrochemical double layer capacitance (CDL) is equal to the slope of the straight line of a plot of ic as a function of v. The CDL measured from the scan-rate dependent CVs for the electrode A is 0.01 mF. Thus, ECSA for the electrode A is estimated to be 0.25 cm2, which is very similar with the pristine MWCNTs electrode. For electrode B, these values are CDL = 0.02 mF, ECSA = 0.50 cm2. These results are obtained based on the average of three repeated experiments.
Author contributions
Lin Li: Conceptualization, investigation, writing – original draft, writing – review & editing. Biswanath Das: Investigation, methodology, writing – original draft, writing – review & editing. Ahibur Rahaman, Andrey Shatskiy, Fei Ye, Peihong Cheng, Chunze Yuan, Zhiqi Yang: Investigation. Oscar Verho: Supervision, investigation, writing – review & editing. Markus D. Kärkäs: Supervision, investigation. Joydeep Dutta: Supervision, investigation. Tsu-Chien Weng: Supervision. Björn Åkermark: Supervision, conceptualization, investigation, writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We all thank Futura Foundation for the financial support. L. L., P. C., Z. Y. and T. W. thank National Natural Science Foundation of China for the financial support (NSFC-21727801) and are grateful for the support from the Analytical Instrumentation Center (#SPST-AIC 10112914), SPST, ShanghaiTech University. B. D. and B. Å. acknowledge the computational facilities from Swedish National Infrastructure for Computing [(SNIC 2020/13-64) awarded to B. D.]. We appreciate Lars Eriksson and Erik Svensson Grape at the department of materials and environmental chemistry at Stockholm University, Sweden for the trials with our samples for the single-crystal X-ray diffraction. We thank Jonas Ståhle at the department of organic chemistry at Stockholm University, Sweden for the help with the ESI-HRMS measurements.References
- A. Heller, Science, 1984, 223, 1141 CrossRef CAS PubMed.
- (a) M. Yamamoto and K. Tanaka, ChemPlusChem, 2016, 81, 1028 CrossRef CAS PubMed; (b) B. Das, A. Rahaman, A. Shatskiy, O. Verho, M. D. Kärkäs and B. Åkermark, Acc. Chem. Res., 2021, 54(17), 3326 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- (a) M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed; (b) B. Das, A. Thapper, S. Ott and S. B. Colbran, Sustainable Energy Fuels, 2019, 3, 2159 RSC.
- (a) D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef CAS; (b) C. Jia, K. Ching, P. V. Kumar, C. Zhao, N. Kumar, X. Chen and B. Das, ACS Appl. Mater. Interfaces, 2020, 12, 41288 CrossRef CAS PubMed; (c) B. Das, C. Jia, K. Ching, M. Bhadbhade, X. Chen, G. E. Ball, S. B. Colbran and C. Zhao, ChemCatChem, 2020, 12, 1292 CrossRef CAS; (d) C. Casadevall, Water, 2022, 14, 371 CrossRef CAS.
- (a) J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974 CrossRef CAS PubMed; (b) B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120 CrossRef CAS; (c) C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed.
- (a) S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC; (b) P. Garrido-Barros, C. Gimbert-Suriñach, R. Matheu, X. Sala and A. Llobet, Chem. Soc. Rev., 2017, 46, 6088 RSC; (c) N. Mamaca, E. Mayousse, S. Arrii-Clacens, T. W. Napporn, K. Servat, N. Guillet and K. B. Kokoh, Appl. Catal., B, 2012, 111–112, 376 CrossRef CAS; (d) B. Das, L. Ezzedinloo, M. Bhadbhade, M. P. Bucknall and S. B. Colbran, Chem. Commun., 2017, 53, 10006 RSC; (e) F. L. Huber, A. M. Wernbacher, D. Perleth, D. Nauroozi, L. Gonza'lez and S. Rau, Inorg. Chem., 2021, 60(17), 13299 CrossRef CAS.
- A. Shatskiy, A. A. Bardin, M. Oschmann, R. Matheu, J. Benet-Buchholz, L. Eriksson, M. D. Kärkäs, E. V. Johnston, C. Gimbert-Suriñach, A. Llobet and B. Åkermark, ChemSusChem, 2019, 12, 2251 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235 CrossRef CAS.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786 CrossRef CAS PubMed.
- (a) L. Wang, K. Fan, H. Chen, Q. Daniel, B. Philippe, H. Rensmo and L. Sun, Catal. Today, 2017, 290, 73 CrossRef CAS; (b) F. Li, H. Yang, W. Li and L. Sun, Joule, 2018, 2, 36 CrossRef CAS.
- N. Karousis, N. Tagmatarchis and D. Tasis, Chem. Rev., 2010, 110, 5366 CrossRef CAS PubMed.
- J. Creus, R. Matheu, I. Peñafiel, D. Moonshiram, P. Blondeau, J. Benet-Buchholz, J. García-Antón, X. Sala, C. Godard and A. Llobet, Angew. Chem., Int. Ed., 2016, 55, 15382 CrossRef CAS PubMed.
- F. Li, L. Li, L. Tong, Q. Daniel, M. Göthelid and L. Sun, Chem. Commun., 2014, 50, 13948 RSC.
- (a) A. Maurin and M. Robert, J. Am. Chem. Soc., 2016, 138, 2492 CrossRef CAS; (b) F. Li, B. Zhang, X. Li, Y. Jiang, L. Chen, Y. Li and L. Sun, Angew. Chem., Int. Ed., 2011, 50, 12276 CrossRef CAS PubMed; (c) D. Schindler, M. Gil-Sepulcre, J. O. Lindner, V. Stepanenko, D. Moonshiram, A. Llobet and F. Würthner, Adv. Energy Mater., 2020, 10, 2002329 CrossRef CAS; (d) E. Cohen, H. Dodiuk, A. Ophir, S. Kenig, C. Barry and J. Mead, Compos. Sci. Technol., 2013, 79, 133 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- K. Biradha, M. Sarkar and L. Rajput, Chem. Commun., 2006, 4169 RSC.
- A. Orlando, F. Franceschini, C. Muscas, S. Pidkova, M. Bartoli, M. Rovere and A. Tagliaferro, Chemosensors, 2021, 9, 1–28 CrossRef.
- S. Y. Mar, C. S. Chen, Y. S. Huang and K. K. Tiong, Appl. Surf. Sci., 1995, 90, 497 CrossRef CAS.
- M. A. Hoque, M. Gil-Sepulcre, A. de Aguirre, J. A. A. W. Elemans, D. Moonshiram, R. Matheu, Y. Shi, J. Benet-Buchholz, X. Sala, M. Malfois, E. Solano, J. Lim, A. Garzón-Manjón, C. Scheu, M. Lanza, F. Maseras, C. Gimbert-Suriñach and A. Llobet, Nat. Chem., 2020, 12, 1060 CrossRef CAS PubMed.
- E. Dulière, M. Devillers and J. Marchand-Brynaert, Organometallics, 2003, 22, 804 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt00824f |
| ‡ Contributed equally to the project. |
| § The overpotential for water oxidation is defined as EWox − (1.23 − (0.059 pH)), EWox is the onset potential where WO presumably starts. |
| This journal is © The Royal Society of Chemistry 2022 |