
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and reactivity of an iridium complex based on a tridentate aminophosphano ligand†
Marco
Palmese
,
Jesús J.
Pérez-Torrente
 and
Vincenzo
Passarelli
*
and
Vincenzo
Passarelli
*
Departamento de Química Inorgánica, Instituto de Síntesis Química y Catalisis Homogenea (ISQCH), Universidad de Zaragoza-CSIC, C/Pedro Cerbuna 12, ES-50007 Zaragoza, Spain. E-mail: passarel@unizar.es
First published on 21st April 2022
Abstract
The iridium(III) hydride compound [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (1PF6) was obtained by reaction of [Ir(SiNP)(cod)][PF6] with CNtBu as the result of the intramolecular oxidative addition of the SiCH2–H bond to iridium(I) [SiNP = Si(CH3)2{N(4-tolyl)PPh2}2, SiNP–H = CH2Si(CH3){N(4-tolyl)PPh2}2]. The mechanism of the reaction was investigated by NMR spectroscopy and DFT calculations showing that the pentacoordinated intermediate [Ir(SiNP)(cod)(CNtBu)][PF6] (2PF6) forms in the first place and that further reacts with CNtBu, affording the square planar intermediate [Ir(SiNP)(CNtBu)2][PF6] (3PF6) that finally undergoes the intramolecular oxidative addition of the SiCH2–H bond. The reactivity of 1PF6 was investigated. On one hand, the reaction of 1PF6 with N-chlorosuccinimide or N-bromosuccinimide provides the haloderivatives [IrX{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (X = Cl, 4PF6; Br, 5PF6), and the reaction of 5PF6 with AgPF6 in the presence of acetonitrile affords the solvato species [Ir{κ3C,P,P′-(SiNP–H)}(CH3CN)(CNtBu)2]2+ (62+) isolated as the hexafluorophosphate salt. On the other hand, the reaction of 1PF6 with HBF4 gives the iridium(III) compound [IrH(CH2SiF2CH3)(HNP)2(CNtBu)2][BF4] (7BF4) as the result of the formal addition of hydrogen fluoride to the Si–N bonds of 1+ [HNP = HN(4-tolyl)PPh2]. A similar outcome was observed in the reaction of 1PF6 with CF3COOH rendering 7PO2F2. In this case the intermediate [IrH{κ2C,P-CH2SiMeFN(4-tolyl)PPh2}(HNP)(CNtBu)2]+ (8+) was observed and characterised in situ by NMR spectroscopy. DFT calculations suggests that the reaction goes through the sequential protonation of the nitrogen atom of the Si–N–P moiety followed by the formal addition of fluoride ion to silicon. Also, the crystal structures of SiNP, 1PF6, 4PF6 and 7BF4 have been determined by X-ray diffraction measurements.
Introduction
Aminophosphanes are easily accessible ligands and both nitrogen and phosphorus substituents can be easily varied thanks to the ample diversity of commercially available precursors. So far, aminophosphano ligands of general formula NHRPR2 have been used to prepare mononuclear,1 di- or oligonuclear species2 supported by 1κN,2κP aminophosphanes (Fig. 1, top). Alternatively, bidentate aminophosphano ligands have been employed to prepare mononuclear complexes (Fig. 1, bottom).3–6 In addition, the aminophosphano functionality has been successfully used to decorate ligating functionalities,7 in some cases as elusive as the silylene7a,i and germylene7a groups. Remarkably, the applications of these complexes are varied and span catalysis, bond activation, metaloenzyme mimics, drugs, and redox-active multimetallic systems, among others. | ||
| Fig. 1 Selected amino-phosphane scaffolds and related metal–ligand structures. | ||
Relevant to this paper, in 2001 Woolins reported5 the synthesis of SiMe2{N(2-pyridyl)PPh2}2 (SiNpyP) and of its palladium and platinum κ2P,P′-derivatives and thereafter we reported on the preparation of SiMe2{N(4-tolyl)PPh2}2 (SiNP) and its rhodium6a and iridium6b,c complexes. Notably, beside the expected κ2P,P′ coordination of SiNP, we reported two unprecedented examples6b,c of a κ3C,P,P′ coordination of SiNP as a result of the intramolecular SiCH2–H oxidative addition to iridium(I), triggered by π-acceptor ligands such as carbon monoxide or trimethyl phosphite on [Ir(SiNP)(cod)]+ (Scheme 1).
 | ||
| Scheme 1 Reactivity of [Ir(SiNP)(cod)]+ towards CO or P(OMe)3. | ||
On this background, aiming at further expand the family of metal complexes containing aminophosphano ligands, we decided to assess the capability of tert-butyl isocyanide, isoelectronic with CO, to promote the intramolecular SiCH2–H oxidative addition to iridium(I) and eventually explore the reactivity of the resulting complex. So, herein we report on the synthesis of a novel iridium(III) complex of formula [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2]+ as well as the detailed theoretical and experimental study of the course of its formation. In addition, a reactivity study was carried out on the above mentioned hydrido derivative, including the hydride-halide exchange and the following halide abstraction as well as protonation reactions rendering the unexpected fragmentation of the aminophosphano ligand.
Results and discussion
Synthesis of [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (1PF6)
The reaction of [Ir(SiNP)(cod)][PF6] with tert-butyl isocyanide (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio) yields the hydrido iridium(III) derivative [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (1PF6) (room temperature, 24 h) as a result of the intramolecular SiCH2–H oxidative addition to the metal centre along with the substitution of the cod ligand with two tert-butyl isocyanide ligands (SiNP–H = CH2SiMe{N(4-tolyl)PPh2}2) (Scheme 2).
:2 molar ratio) yields the hydrido iridium(III) derivative [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (1PF6) (room temperature, 24 h) as a result of the intramolecular SiCH2–H oxidative addition to the metal centre along with the substitution of the cod ligand with two tert-butyl isocyanide ligands (SiNP–H = CH2SiMe{N(4-tolyl)PPh2}2) (Scheme 2).
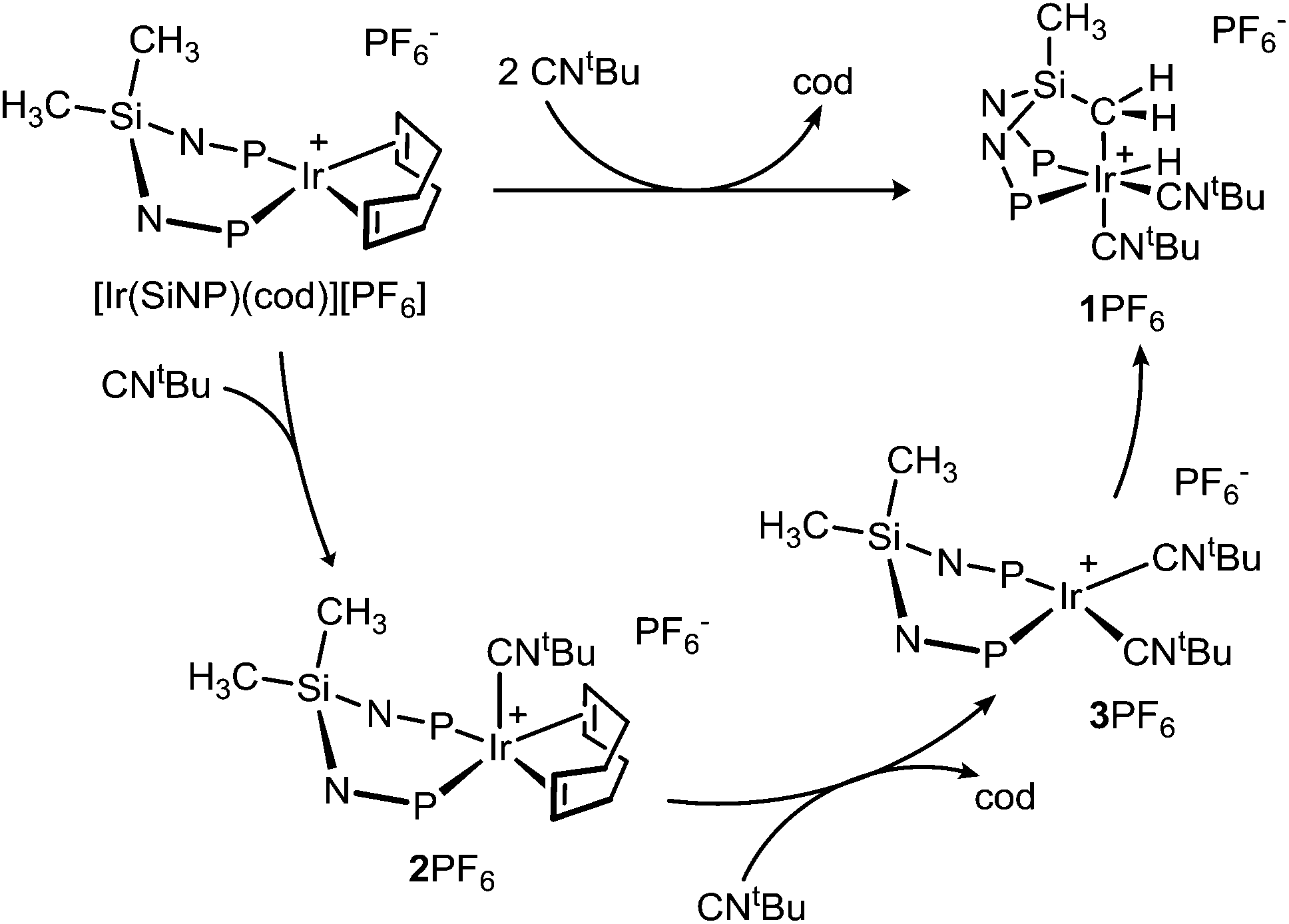 | ||
| Scheme 2 Reaction of [Ir(SiNP)(cod)]+ with CNtBu showing the observed intermediates. | ||
The crystal structure of 1PF6 was determined by single crystal X-ray diffraction measurements, and Fig. 2-top shows the ORTEP plot of the cation [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2]+ (1+). For the sake of comparison, the crystal structure of SiNP was also determined (Fig. 2-bottom). The metal centre of 1+ exhibits an octahedral environment, the metalated κ3C,P,P′-(SiNP–H) ligand occupying three mutually cis coordination sites [C1–Ir–P2 82.79(9)°, P1–Ir–P2 97.00(3)°, C1–Ir–P1 84.83(9)°]. The hydrido ligand lies cis to P1 and trans to P2 [P1–Ir–H 87.6(16)°, P2–Ir–H 169.2(16)°]. The remaining coordination sites are occupied by two mutually cis isocyanide ligands [C47–Ir–C41 86.18(13)°], one trans to P1 [C47–Ir–P1 169.51(10)°] and the other trans to the metallated carbon atom C1 [C41–Ir–C1 175.56(13)°]. Reasonably as a consequence of the metalation, the C1–Si–C2 angle of 1+ [C1–Si–C2 124.65(15)°] is wider than the C1–Si–C2 angle of SiNP [C1–Si–C2 111.48(10)°]. Also, the formation of two fused five member metalacycles in 1+ should account for the smaller Si–N–P angles of 1+ [P2–N2–Si 114.31(15)°, P1–N1–Si 112.48(15)°] when compared with SiNP [P1–N1–Si 121.40(9)°, P2–N2–Si 120.94(9)°]. In addition, it is also remarkable that the nitrogen atoms N1 and N2 of both 1+ and SiNP exhibit a planar geometry, i.e. the fragments N1–Si–P1–C15 and N2–Si–P2–C34 are almost planar in both 1+ and SiNP suggesting that a p–d(π) backdonation could imply nitrogen and phosphorus and/or silicon. In this connection, it is worth a mention that the tolyl rings attached to N1 and that attached to N2 lie almost perpendicular to the corresponding N1–Si–P1–C15 and N2–Si–P2–C34 planes in 1+ as well as SiNP (1+, N1 78.5°; N2 85.3°; SiNP, N1 71.5°, N2 72.2°), which rules out the delocalization of the nitrogen lone pair on the aromatic ring as the cause of the above mentioned planarity of the fragments N1–Si–P1–C15 and N2–Si–P2–C34.
 | ||
| Fig. 2 ORTEP plots of [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2]+ (1+) (top) and of SiNP (bottom). For clarity, most hydrogen are omitted and the tolyl and phenyl rings are represented in a wireframe style for [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2]+. Selected bond lengths (Å) and angles (°) are given in ESI.† | ||
As a confirmation, a QTAIM analysis was carried out on both the crystal and the calculated structures of 1+ and SiNP showing that the delocalization index DI (aka fuzzy bond orders, FBO) of the bonds at the nitrogen atoms are 1.08–1.11 (N–C), 1.28–1.32 (N–P) and 1.08–1.15 (N–Si) (Fig. 3) pointing at that some p–d(π) backdonation actually should exist mainly between nitrogen and phosphorus and that therefore it should be responsible for the planar geometry of the nitrogen atoms.
 | ||
| Fig. 3 Selected delocalization indexes (DI) for SiNP and 1+ (normal type, values for the X-ray structure; italic type, values for the DFT-calculated structure). | ||
The crystal structure of 1+ should be preserved in solution. Indeed, its 31P{1H} NMR spectrum shows two doublets at 40.3 and 34.4 ppm with a 2JPP coupling constant of 20.4 Hz, in agreement with a cis arrangement of the two phosphorus atoms. Also, two 1H singlets at 1.37 and 1.20 ppm are indicative of two non-equivalent tert-butyl isocyanide ligands. As for the HIrCH2Si moiety, one 13C doublet of doublets at −29.7 ppm (2JCP = 2.1, 3.7 Hz) and one 1H multiplet (vide infra) at −10.58 ppm have been observed. In addition, similar to the related trimethyl phosphito derivative [IrH{κ3C,P,P′-(SiNP–H)}{P(OCH3)3}2]+,6b the coupling pattern (see nJXY in Fig. 4) of the IrHCH2 moiety suggests that the conformation observed in the solid state is maintained in solution.‡
 | ||
| Fig. 4 Selected 1H and 31P NMR data for the IrHCH2 moiety of 1+. | ||
The formation of the hydrido iridium(III) derivative 1+ was observed to be stepwise (Scheme 2). As a matter of fact, the formation of 1+ was monitored by NMR spectroscopy at −80 °C showing that [Ir(SiNP)(cod)(CNtBu)]+ (2+) forms in the first place and further reacts with CNtBu upon raising the temperature, rendering the square planar intermediate [Ir(SiNP)(CNtBu)2]+ (3+, vide infra) which eventually evolves to 1+. As a confirmation, [Ir(SiNP)(cod)(CNtBu)][PF6] (2PF6) could be prepared in high yield upon reacting [Ir(SiNP)(cod)][PF6] with tert-butyl isocyanide (1:1 molar ratio) at −80 °C. Furthermore, the reaction of 2PF6 with CNtBu cleanly yielded 1PF6 through 3PF6. Remarkably, also in this case, 3+ formed along with 1+, which indicates that the formation of 3+ and its conversion into 1+ should exhibit similar activation barriers. 2PF6 was fully characterised in solution by means of multinuclear NMR spectroscopy. A 31P{1H} singlet is observed at 41.4 ppm along with one 1H singlet at 2.04 ppm for the two methyl moieties of the tolyl groups, suggesting that the two Si-Ntol-PPh2 arms of 2+ are equivalent. On the contrary, two 1H singlets at 0.56 and −0.21 ppm are observed for the two SiCH3 groups of 2+, which indicates that they are non-equivalent reasonably as a consequence of the coordination of the isocyanide ligand to iridium in [Ir(SiNP)(cod)]+ rendering a distorted square pyramidal geometry at the metal centre (vide infra for the DFT calculated structure). As for the cod ligand, broad 1H signals are observed even at −60 °C for the olefinic (3.46 ppm) and aliphatic hydrogen atoms (1.76 ppm), respectively, suggesting that even at that temperature the putatively non-equivalent olefinic CH moieties as well as the methylene hydrogen atoms are exchanging and their signals are averaged.
As far as the intermediate 3+ is concerned, it could be spectroscopically identified§in situ (1H, 31P NMR). Indeed, a 31P{1H} singlet at 53.7 ppm was assigned to its equivalent phosphorus atoms. Accordingly two equivalent tolyl groups as well as two equivalent tBu and two equivalent SiMe groups were observed.¶
DFT calculations nicely underpinned the proposed pathway for the formation of 1PF6. Fig. 5 shows the simplified Gibbs free energy profile for the reaction Ir(SiNP)(cod) + 2 CNtBu → 1+ + cod, including the calculated structure of the detected intermediates 2+ and 3+ as well as the transition state TS_3+-1+ of the oxidative addition of SiCH2–H to iridium. The first step is the exoergonic formation of the distorted square pyramidal complex [Ir(SiNP)(cod)(CNtBu)]+ (2+). In the following, [Ir(SiNP)(CNtBu)2]+ (3+) is obtained by reaction of 1+ with CNtBu (ΔGr = −13.3 kcal mol−1). Remarkably 3+ exhibits a boat conformation of the six member ring Ir–P–N–Si–N–P which allows one of the SiCH3 group, namely the flagpole one, to approach the metal centre (CH⋯Ir 2.683 Å, Fig. 5) and eventually add oxidatively to it (TS_3+-1+, Fig. 5).
 | ||
| Fig. 5 (left) Relative Gibbs free energy profile (kcal mol−1) of the reaction Ir(SiNP)(cod) + 2 CNtBu → 1+ + cod (M06/def2tzvp//B3PW91-GD3BJ/def2svp, 298 K, 1 atm); (right) view of the calculated structures of 2+, 3+ and TS_3+-1+ with selected interatomic distances (Å) and angles (°) (for clarity, most hydrogen atoms are omitted and only the ipso carbon atoms of the phenyl and tolyl moieties are shown). | ||
Reactivity of [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (1PF6)
In order to assess the applicability of 1PF6 as a catalyst in the functionalization of multiple carbon–carbon bonds, a preliminary reactivity study was undertaken. We observed that 1PF6 does not react either with alkynes – phenylacetylene or 1-hexyne – or alkenes – styrene or 1-hexene – even after prolonged reaction times (up to 48 h) and heating (70 °C in THF). Reasonably the stable κ3C,P,P′ coordination of the SiNP–H ligand along with the substitutional inertness of the CNtBu ligands hampered the reactivity of 1PF6. Thus, anticipating that the abstraction of a halido ligand could trigger some reactivity at the Ir{κ3C,P,P′-(SiNP–H)} platform, we decided to exchange the hydrido ligand with a halido ligand. Thus, the iridium haloderivatives of formula [IrX{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (X = Cl, 4PF6; Br, 5PF6) were prepared by reaction of 1PF6 with N-chlorosuccinimide or N-bromosuccinimide (Scheme 3). | ||
| Scheme 3 Hydride-halide exchange on [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (1PF6). | ||
The crystal structure of 4PF6 was determined by means of X-ray diffraction measurements and it exhibits an octahedral environment of the metal centre with a κ3C,P,P′-(SiNP–H) ligand along with the chlorido ligand and two tert-butyl isocyanide ligands (Fig. 6). The Ir{κ3C,P,P′-(SiNP–H)} moiety of 4+ and 1+ are virtually superimposable, and by the same token no significant differences are observed between the isocyanide ligands when comparing 1+ and 4+.
 | ||
| Fig. 6 ORTEP plot of [IrCl{κ3C,P,P′-(SiNP–H)}(CNtBu)2]+ (4+). For clarity, most hydrogen are omitted and the tolyl and phenyl rings are represented in a wireframe style. Selected bond lengths (Å) and angles (°) are given in ESI.† | ||
The solution structure of 4+ and 5+ should be similar to that of 4+ in the solid state. Indeed, the 31P{1H} NMR spectrum shows two doublets at 28.7 and 26.3 ppm (4+), and at 25.7 and 25.1 ppm (5+) with a coupling constant indicating a mutually cis disposition of the phosphorus atoms (2JPP = 18.2 Hz, 4+; 17.4 Hz, 5+). Also, the 1H NMR spectra contains two singlets at 1.45 and 1.25 (4+), and 1.48 and 1.29 (5+) ppm for the tert-butyl isocyanide ligands, two multiplets at 1.30 and 1.21 (4+), and 1.37 and 1.24 (5+) for the IrCH2 moiety, and one singlet at 0.15 ppm (4+) and 0.17 ppm (5+) for the SiCH3 group.
The bromido ligand of 5+ was easily abstracted by reaction with AgPF6 but a clean product, namely [Ir{κ3C,P,P′-(SiNP–H)}(CH3CN)(CNtBu)2][PF6]2 (6[PF6]2), could be isolated only in the presence of acetonitrile (Scheme 4), whereas intractable mixtures of products were obtained with styrene or phenylacetylene.
 | ||
| Scheme 4 Bromide abstraction from [IrBr{κ3C,P,P′-(SiNP–H)}(CNtBu)2]+ (5+). | ||
The κ3C,P,P′ coordination of SiNP–H is preserved in 62+ as judged by the 31P{1H} doublets observed at 25.1 and 18.0 ppm (2JPP = 19.4 Hz) and the 1H doublet of doublets at 1.44 and 1.30 ppm assigned to the IrCH2 moiety as well as the 1H singlet at 0.34 ppm for the SiCH3 group. The 1H singlets at 2.09, 1.48 and 1.29 ppm confirm the presence of one CH3CN and two CNtBu ligands, respectively. Unfortunately no reaction of 62+ with either styrene or phenylacetylene was observed indicating that neither CH3CN nor CNtBu ligands in 62+ are labile.
Protonation of [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (1PF6)
On another note, the reaction of 1PF6 with Brønsted acid was explored envisioning that the hydrido moiety could undergo protonation rendering dihydrogen and eventually an accessible coordination vacant. As a matter of fact, 1+ does react with Brønsted acids but with an unexpected outcome (Scheme 5). Indeed the reaction of 1+ with HBF4 (1:2 molar ratio) renders the hydrido iridium(III) derivative [IrH(CH2SiF2CH3)(HNP)2(CNtBu)2][BF4] (7BF4) [HNP = NH(4-tolyl)PPh2] as a result of the formal addition of two hydrogen fluoride molecules to the κ3C,P,P′-(SiNP–H) ligand along with the counterion exchange (Scheme 5). In addition, formally PF5 and BF3 should also result from the reaction, but unfortunately neither they nor any chemically related species could be identified in the course of the reaction.
 | ||
| Scheme 5 Reaction of [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] with HBF4. | ||
The crystal structure of 7BF4 was determined by means of single crystal X-ray diffraction measurements (Fig. 7). The metal centre exhibits an octahedral environment in which the newly formed ligands NH(4-tolyl)PPh2 and CH2SiF2CH3 are mutually cis, rendering an arrangement at the metal centre which is reminiscent of the κ3C,P,P′ coordination of the SiNP–H ligand of 1+ [P(1)–Ir–P(2) 98.52(2)°, C(1)–Ir–P(2) 89.12(7)°, C(1)–Ir–P(1) 92.46(7)°]. The hydrido ligand lies cis to C1 and P1 and trans to P2 and the remaining cis coordination sites are occupied by the tert-butyl isocyanide ligands. Remarkably the N1–H1N group is involved in an intramolecular NH⋯F hydrogen bond to F1 [N1–H1N, 0.940(19); H1N⋯F1 2.11(2); N1⋯F1 3.001(3), N1–H1N–F1 158(3)°]. The crystal structure of 7BF4 should be preserved in solution. Indeed two 31P doublets are observed at 11.4 and 3.1 ppm with a coupling constant 2JPP of 20.5 Hz pointing at a cis disposition of the two HNP ligands. The 1H NMR spectrum shows two singlets at 1.25 and 1.21 ppm, assigned to the tert-butyl isocyanide ligands, and a doublet of doublets of triplets at −11.86 ppm for the hydrido ligand as a result of the scalar coupling of the IrH hydrogen to the trans phosphorus (2JHP = 152.9 Hz), the cis phosphorus (2JHP = 17.4 Hz), and fluorine (4JHF = 2.2 Hz). As for the CH2SiF2CH3 moiety, two 19F signals at −127.5 and −129.0 ppm (2JFF = 20.6 Hz), and two 1H multiplets at 0.01 and −0.42 ppm for the IrCH2Si hydrogen atoms are observed (cf. Experimental section), which is indicative of two non-equivalent fluorine atoms and two non-equivalent hydrogen atoms, respectively. Also, while the 19F{1H} signal at −129.0 ppm is a doublet, the 19F{1H} signal at −127.5 ppm is a doublet of doublets due to the above mentioned fluorine–fluorine coupling and to a fluorine–phosphorus coupling (4JFP = 2.7 Hz, vide infra for this assignment).
 | ||
| Fig. 7 ORTEP plot of [IrH(CH2SiF2Me)(HNP)2(CNtBu)2]+ (7+). For clarity, most hydrogen are omitted and the tolyl and phenyl rings are represented in a wireframe style. Selected bond lengths (Å) and angles (°) are given in ESI.† | ||
Remarkably the NH⋯F hydrogen bond observed in the solid state is maintained in solution. For the sake of clarity, the numbering scheme of the crystal structure given in Fig. 7 will be used in the following discussion of the NMR data. While a 1H doublet (2JHP = 15.9 ppm) at 4.21 ppm is observed for the N2–H2N moiety, a 1H doublet of doublets at 5.33 ppm is observed for the N1–H1N group as a consequence of the scalar coupling of hydrogen to the phosphorus atom P1 (2JHP = 15.9 Hz) and to the fluorine atom F1 (JHF = 3.9 Hz). As a confirmation of the NH⋯F hydrogen bond and the consequent hampered rotation around the Ir–CH2Si bond, NOE cross peaks are observed in the 1H–1H NOESY spectrum between H1B (−0.42 ppm) and H1N (5.35 ppm) and between H2N (4.21 ppm) and both H1A (0.01 ppm) and H1B (−0.42 ppm). Finally for the sake of confirmation, selected NMR data were calculated by DFT methods (mPW1PW91/def2TZVP) confirming the proposed assignment.||
Aiming at investigating the influence of the acid – more specifically of its conjugated base – on the outcome of the reaction, 1PF6 was treated with different Brønsted acids, namely HPF6 in water (54% w/w) and CF3COOH. Surprisingly no reaction between 1PF6 and HPF6 was observed even after 48 h at room temperature. On the other hand, the reaction of 1PF6 with CF3COOH is slower than that with HBF4 and completeness is reached after 4 days and in the presence of a moderate excess of CF3COOH (1:4) at room temperature, rendering 7+ and the anion PO2F2−. Notably when the reaction was monitored by 31P{1H} NMR spectroscopy, the anion PF6− is quantitatively converted into PO2F2− (δF = −84.1, δP = −19.5, 1JPF = 957 Hz) after 24 h. As a confirmation, the reaction of NBu4PF6 with CF3COOH (1:4 molar ratio, in CD2Cl2) has a similar outcome cleanly affording PO2F2−. On these grounds, reasonably the formation of PO2F2− should not be metal-assisted and might follow a route similar to the chlorination of carboxylic acids with PCl5 and POCl3.8
When the reaction of 1+ with CF3COOH was monitored by 1H, 19F and 31P NMR spectroscopy, [IrH{κ2C,P-CH2SiMeFN(4-tolyl)PPh2}(HNP)(CNtBu)2]+ (8+) was detected as an intermediate as a result of the formal addition of one hydrogen fluoride molecule to one Si–N bond (Scheme 6). Fig. 8 shows selected areas of the 1H, 19F and 31P NMR spectra with the proposed assignment.**
 | ||
| Scheme 6 Reaction of [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] with HPF6 or CF3COOH. | ||
 | ||
| Fig. 8 Selected areas of the 1H, 19F, 19F{1H} and 31P{1H} NMR spectra of the mixture resulting from the reaction of 1PF6 with CF3COOH in CD2Cl2 after 24 h with the proposed assignment. Inset A shows the experimental and simulated 19F signal at −125.8 ppm of 8+. Inset B shows the expanded view of the 19F{1H} signal at −127.5 ppm of 7+. * unassigned. | ||
In view of the 31P{1H} and 1H NMR spectra, the formal addition of hydrogen fluoride to 1+ affording 8+ is regiospecific since four products (I+–IV+, Fig. 9) might form depending on which nitrogen atom undergoes protonation (N1 or N2) and on the orientation of the formal addition of the fluoride ion to silicon (syn or anti with respect to the protonated nitrogen atom).
 | ||
| Fig. 9 Sequential addition of HF to 1+ showing the possible intermediates and their relative Gibbs free energies (M06/def2tzvp//B3PW91-GD3BJ/def2svp, 298 K, 1 atm). | ||
In order to shed light on the above mentioned regiospecifity, a thorough examination of the calculated structure of 1+ suggested that the steric hindrances at each nitrogen atoms are similar. Further, the NBO charges of the nitrogen atoms are virtually identical (−1.171, −1.185 a.u.), suggesting that no preferential attack of H+ to one of the two nitrogen atoms should be expected as a consequence of the atomic charges at the reacting sites. On these grounds, the observed selectivity in the formation of 8+ should rely on the thermodynamic stability of the intermediate itself. With this in mind, the proton affinities (PA) of 1+ were calculated along with the relative Gibbs free energy for the sequential addition of hydrogen fluoride to the two Si–N bonds 1+, namely Si–N1 and Si–N2 (Fig. 9). In agreement with the proposed structure of 8+, the most stable protonated species 1H2+ is that resulting from the protonation of N2, that is the nitrogen atom bonded to the phosphorus trans to the hydrido moiety (PA = 143.2, N2; 139.4 kcal mol−1, N1, Fig. 9). Accordingly, the most stable intermediate I+ results from the formal addition of hydrogen fluoride to the bond Si–N2 of 1+ with an anti orientation of the attack of fluoride to silicon 1+.
Conclusions
tert-Butyl isocyanide triggers the oxidative addition of the SiCH2–H bond to iridium(I) in [Ir(SiNP)(cod)][PF6] yielding the iridium(III) hydrido derivative [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (1PF6). Reasonably as a consequence of the stable κ3C,P,P′ coordination of the SiNP–H ligand along with the substitutional inertness of the CNtBu ligands, 1+ as well as the related haloderivatives [IrX{κ3C,P,P′-(SiNP–H)}(CNtBu)2]+ (X = Cl, 4+; Br, 5+) and solvato complex [Ir{κ3C,P,P′-(SiNP–H)}(CH3CN)(CNtBu)2]2+ (62+) do not react with unsaturated molecules such as olefins or alkynes.On the other hand, 1+ does react with Brønsted acids such as HBF4 and CF3COOH undergoing the unexpected fragmentation of the SiNP backbone. Indeed the formal addition of two molecules of hydrogen fluoride to the Si–N bonds affords the iridium(III) derivative [IrH(CH2SiCH3F2)(HNP)2(CNtBu)2] (7+) through the formation of the intermediate [IrH{κ2C,P-CH2SiMeFN(4-tolyl)PPh2}(HNP)(CNtBu)2]+ (8+), observed as the result of the regiospecific formal addition of HF to one Si–N bond. Accordingly DFT calculation suggests that a sequential protonation of the Si–N–P moieties takes place followed by the formal addition of fluoride ion to silicon as well as that the observed regiospecificity relies on the thermodynamic stability of the observed intermediate.
Experimental
General section
All the operations were carried out using standard Schlenk tube techniques under an atmosphere of pre-purified argon or in a Braun glove-box under dinitrogen or argon. Organic solvents were dried by standard procedures and distilled under argon or obtained oxygen- and water-free from a Solvent Purification System (Innovative Technologies). The compounds SiMe2{(N(4-tolyl)(PPh2)}2 (SiNP)6a and [Ir(SiNP)(cod)][PF6]6b were prepared according to the literature. NMR spectra were recorded with Bruker spectrometers (AV300 and AV400) and are referred to SiMe4 (1H, 13C) and H3PO4 (31P), CFCl3 (19F). The proposed 1H, 13C, and 31P assignment relies on the combined analysis of 1D [1H, 1H{31P}, 13C{1H}-apt, 31P{1H}] and 2D NMR spectra (1H–1H COSY, 1H–1H NOESY, 1H–13C HSQC, 1H–13C HMBC, 1H–31P HMBC). In compounds containing two non-equivalent phosphorus atoms, namely P1 and P2, P1 indicates the phosphorus atom trans to H (1+, 8+), Cl (4+), Br (5+), or CH3CN (62+) and superscript labels “tol-P1/2” and “PhP1/2” are used for hydrogen and carbon atoms belonging to the tolyl and phenyl groups attached/linked to the phosphorus atom P1/2. C, H, and N analyses were carried out on a PerkinElmer 2400 CHNS/O analyzer.Synthesis of [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (1PF6)
Method 1. A dichloromethane solution (15 mL) of [Ir(SiNP)(cod)][PF6] (199.80 mg, 0.184 mmol, 1084.15 g mol−1) was added with CNtBu (53.5 μL, 0.473 mmol, 83.13 g mol−1, 0.735 g mL−1). The yellow resulting solution was stirred for 24 h, partially evaporated up to 1 mL and added with hexane (5 mL), affording a pale yellow solid which was filtered off and washed with tetrahydrofuran/hexane (1:1, 5 mL), dried in vacuo and finally identified as [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (1PF6, 149 mg, 0.130 mmol, 71% yield).
Method 2. A dichloromethane solution (12 mL) of [Ir(SiNP)(cod)(CNtBu)][PF6] (2PF6, vide infra, 619 mg, 0.530 mmol, 1167.29 g mol−1) was added with CNtBu (60.0 μL, 0.530 mmol, 83.13 g mol−1, 0.735 g mL−1) at 313 K. The resulting yellow solution was stirred for 14 h, partially evaporated and added with diethyl ether/hexane (1:1, 20 mL), affording a pale yellow solid which was filtered off and washed with tetrahydrofuran/hexane (1:1, 5 mL), dried in vacuo and finally identified as [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (1PF6, 464 mg, 0.406 mmol, 77% yield). Found: C, 52.99; H, 5.07; N, 4.85. Calcd for C50H58F6IrN4P3Si (1142.24): C, 52.57; H, 5.12; N, 4.91. 1H NMR (CD2Cl2 298 K): δH 7.68–7.56 (6H tot; 4H, o-P1Ph, 2H, m-P1Ph), 7.55–7.43 (4H tot; 2H, m-P2Ph, 2H, p-P1Ph), 7.44–7.32 (6H tot; 2H, p-P2Ph, 2H, o-P2Ph, 2H, m-P1Ph), 7.00 (d, 2H, 3JHH = 8.3 Hz, C3Htol-P2), 6.92 (td, 2H, 3JHH = 7.9 Hz, 4JHP = 2.3 Hz, m-P2Ph), 6.76 (d, 2H, 3JHH = 8.3 Hz, C2Htol-P2), 6.74 (d, 2H, 3JHH = 8.3 Hz, C3Htol-P1), 6.61 (ddd, 2H, 3JHP = 11.5 Hz, 3JHH = 8.1 Hz, 5JHP = 1.1 Hz, o-P2Ph), 6.24 (d, 2H, 3JHH = 8.3 Hz, C2Htol-P1), 2.29 (s, 3H, CH3tol-P1), 2.15 (s, 3H, CH3tol-P2), 1.37 (s, 9H, CH3tBu), 1.20 (s, 9H, CH3tBu), 0.67 (dddd, 1H, 3JHP = 15.0 Hz, 2JHH = 12.6 Hz, 3JHH = 2.6 Hz, 3JHP = 1.2 Hz, SiCHaHbIr), 0.46 (ddd, 1H, 2JHH = 12.6 Hz, 3JHP = 9.0 Hz, 3JHP = 1.2 Hz, SiCHaHbIr), −0.21 (s, 3H, SiCH3,), −10.58 (ddd, 1H, 2JHP trans = 149.6 Hz, 2JHP cis = 17.9 Hz, 3JHH = 2.6 Hz, IrH). 13C{1H} NMR (CD2Cl2, 298 K): δC 140.2 (dd, 1JCP = 45.0 Hz, 3JCP = 1.9 Hz, C1, PhP), 139.0 (d, 2JCP = 10.1 Hz, C1, tol-P2), 138.5 (d, 2JCP = 9.6 Hz, C1, tol-P1), 136.0 (d, 5JCP = 1.6 Hz, C4, tol-P1), 135.5 (C4, tol-P2), 135.4 (d, 2JCP = 13.8 Hz, C2, PhP2), 134.64 (d, 1JCP = 60.2 Hz, C1, PhP), 134.60 (d, 2JCP = 12.0 Hz, 4JCP = 1.2 Hz C2, PhP1), 133.90 (dd, 1JCP = 63.4 Hz, 3JCP = 4.5 Hz, C1, PhP), 131.8 (d, 2JCP = 10.9 Hz, C2, PhP1), 131.7 (d, 4JCP = 2.2 Hz, C4, PhP2), 131.5 (d, 4JCP = 2.0 Hz, C4, PhP1), 131.1 (d, 4JCP = 2.4 Hz, C4, PhP2), 130.5 (d, 2JCP = 10.2 Hz, C2, PhP2), 130.3 (d, 4JCP = 2.0 Hz, C4, PhP1), 129.7 (d, 4JCP = 1.4 Hz, C3, tol-P2), 129.64 (d, 4JCP = 0.7 Hz, C3, tol-P1), 129.61 (d, 4JCP = 1.4 Hz, C2, tol-P1), 129.1 (d, 3JCP = 1.4 Hz, C2, tol-P2), 128.4 (d, 3JCP = 10.8 Hz, C3, PhP2), 128.3 (d, 3JCP = 9.7 Hz, C3, PhP1), 128.0 (d, 3JCP = 11.1 Hz, C3, PhP2), 127.4 (d, 3JCP = 10.8 Hz, C3, PhP1), 58.4 (CtBu2), 58.2 (CtBu1), 30.0 (CH3tBu1), 29.8 (CH3tBu2), 20.5 (CH3tol-P2), 20.4 (CH3tol-P1), −0.9 (t, 3JCP = 7.6 Hz, CH3Si), −29.7 (dd, 2JCP = 3.7, 2.1 Hz, CH2Si). 31P{1H} NMR (CD2Cl2, 298 K): δP 40.3 (d, 2JPP 20.4 Hz, P1, SiNP), 34.4 (d, 2JPP 20.4 Hz, P2, SiNP), −144.4 (hept, 1JPF = 710.2 Hz, PF6−).
Synthesis of [Ir(SiNP)(cod)(CNtBu)][PF6] (2PF6)
A dichloromethane solution (10 mL) of [Ir(SiNP)(cod)][PF6] (178 mg, 0.164 mmol, 1084.15 g mol−1) was added with CNtBu (37.4 μL, 0.331 mmol, 83.13 g mol−1, 0.735 g mL−1) at 253 K. The yellow resulting solution was stirred for 30 minutes, partially evaporated and added with hexane (10 mL) affording a light yellow solid which was filtered off, dried in vacuo and finally identified as [Ir(SiNP)(cod)(CNtBu)][PF6] (2PF6, 176 mg, 0.151 mmol, 92% yield). Found: C, 53.97; H, 5.31; N, 3.65. Calcd for C53H61F6IrN3P3Si (1167.29): C, 54.53; H, 5.27; N, 3.60. 1H NMR (C6D6 298 K), the labels “up” and “down” are used for the CH or CH3moieties pointing towards the CNtBu ligand and apart from it, respectively: δH 7.70–7.55 (8H, o-PPh), 7.43–7.24 (12H, m-PPh and p-PPh), 7.19 (d, 2H, 3JHH = 8.2 Hz, C2Htol down), 6.83 (d, 2H, 3JHH = 8.2 Hz, C3Htol down), 6.64 (d, 2H, 3JHH = 8.2 Hz, C3Htol up), 6.24 (d, 2H, 3JHH = 8.2 Hz, C2Htol up), 3.47 (br, 4H, Csp2Hcod), 2.03 (s, 6H, CH3tol), 1.97 (s, 9H, CH3tBu), 1.76 (br, 8H, Csp3Hcod), 0.56 (s, 3H, SiCH3up), −0.21 (s, 3H, SiCH3down). 13C{1H} NMR (C6D6, 298 K): δC 139.6 (C4, tol), 136.7 (C1, tol), 135.6 (C2, PhP), 132.6 (C2, PhP), 132.0–131.9 (C2, tol up and C2, tol down), 131.8 (C3, PhP), 130.5 (C4, PhP), 129.6 (C3, tol down), 129.1 (C3, tol up), 127.6 (C3, PhP), 78.4 (Csp2, cod), 59.9 (CtBu), 32.6 (Csp3, cod), 30.4 (CH3tBu) 20.5 (CH3tol), 3.4 (t, 3JCP = 2.2 Hz, CH3Sidown), 2.1 (CH3Siup). 31P{1H} NMR (C6D6, 298 K): δP 41.3 (s, PSiNP), −142.5 (hept, 1JPF = 708.8 Hz, PF6−).Synthesis of [IrCl{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (4PF6)
A dichloromethane solution (8 mL) of [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (1PF6, 87.3 mg, 0.0764 mmol, 1142.24 g mol−1) was added with N-chlorosuccinimide (10.1 mg, 0.0756 mmol, 133.53 g mol−1). The resulting colourless solution was stirred for 32 h, partially evaporated and added with hexane (5 mL), affording a colourless solid which was filtered off and washed with diethyl ether (3 × 5 mL), dried in vacuo and finally identified as [IrCl{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (4PF6, 81.5 mg, 0.0693 mmol, 92% yield). Found: C, 51.65; H, 4.78; N, 4.70. Calcd for C50H57ClF6IrN4P3Si (1176.68): C, 51.04; H, 4.88; N, 4.76. 1H NMR (CD2Cl2 298 K): δH 7.62–7.47 (10H tot: 4H, o-PPh, 4H, m-P2Ph, 2H, p-P2Ph), 7.45 (m, 2H, m-P1Ph), 7.43–7.31 (4H tot: 2H o-P1Ph, 2H p-P2Ph), 7.11 (td, 2H, 3JHH = 7.9 Hz, 3JHP = 3.0 Hz, m-P2Ph), 6.92 (d, 2H, 3JHH = 8.2 Hz, C3Htol-P2), 6.86 (ddd, 2H, 3JHP = 12.5 Hz, 3JHH = 8.2 Hz, 3JHP = 1.0 Hz, o-P2Ph), 6.81 (d, 2H, 3JHH = 8.2 Hz, C3Htol-P1), 6.52 (d, 2H, 3JHH = 8.2 Hz, C2Htol-P2), 6.41 (d, 2H, 3JHH = 8.2 Hz, C2Htol-P1), 2.24 (s, 3H, CH3tol-P1), 2.18 (s, 3H, CH3tol-P2), 1.45 (s, 9H, CH3tBu2), 1.30 (dd, 1H, 2JHH = 13.1 Hz, 3JHP = 6.6 Hz, SiCH2Ir), 1.25 (s, 9H, CH3tBu1), 1.21 (ddd, 1H, 2JHH = 13.1 Hz, 3JHP = 3.6 Hz, 3JHP = 1.3 Hz, SiCH2Ir), 0.15 (s, 3H, SiCH3). 13C{1H} NMR (CD2Cl2, 298 K): δC 138.3 (d, 1JCP = 67.5 Hz, C1, PPh), 137.6 (d, 2JCP = 9.6 Hz, C1, tol-P1), 137.2 (C1, tol-P2), 136.5 (d, 5JCP = 1.9 Hz, C4, tol-P2), 135.8 (d, 5JCP = 1.5 Hz, C4, tol-P1), 134.3 (d, 2JCP = 10.0 Hz, C2, PhP1), 134.2 (d, 2JCP = 10.6 Hz, C2, PhP2), 133.2 (d, 2JCP = 10.8 Hz, C2, PhP2), 132.4 (d, 4JCP = 2.8 Hz, C4, PhP2), 132.04 (d, 4JCP = 2.9 Hz, C4, PhP1), 131.99 (d, 4JCP = 2.8 Hz, C4, PhP2), 131.4 (d, 4JCP = 2.7 Hz, C4, PhP1), 131.1 (d, 2JCP = 10.2 Hz, C2, PhP1), 129.8 (d, 5JCP = 1.7 Hz, C3, tol-P2), 129.5 (C3, tol-P1), 129.2 (d, 4JCP = 3.4 Hz, C2, tol-P2), 128.8 (d, 3JCP = 11.5 Hz, C3, PhP1), 128.6 (dd, 3JCP = 6.6 Hz, 5JCP = 3.2 Hz, C2, tol-P1), 128.5 (d, 3JCP = 11.3 Hz, C3, PhP1), 127.9 (d, 3JCP = 11.7 Hz, C3, PhP2), 127.2 (d, 3JCP = 11.3 Hz, C3, PhP1), 59.3 (CtBu2), 59.0 (CtBu1), 29.8 (CH3tBu1), 29.7 (CH3tBu2), 20.5 (CH3tol-P2), 20.3 (CH3tol-P1), −1.2 (t, 3JCP = 7.0 Hz, CH3Si), −16.2 (CH2Si). 31P{1H} NMR (CD2Cl2, 298 K): δP 28.6 (d, 2JPP = 18.2 Hz, SiNP1), 26.3 (d, 2JPP = 18.2 Hz, SiNP2), −144.4 (hept, 1JPF = 710.2 Hz, PF6−).Synthesis of [IrBr{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (5PF6)
A dichloromethane solution (8 mL) of [IrH{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (1PF6, 175 mg, 0.153 mmol, 1142.24 g mol−1) was added with N-bromosuccinimide (27.3 mg, 0.153 mmol, 177.98 g mol−1). The resulting colourless solution was stirred for 30 min, partially evaporated and added with hexane (5 mL), affording a colorless solid which was filtered off and washed with diethyl ether (3 × 5 mL), dried in vacuo and finally identified as [IrBr{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (5PF6, 147 mg, 0.120 mmol, 79% yield). Found: C, 49.27; H, 4.72; N, 4.39. Calcd for C50H57BrF6IrN4P3Si (1221.13): C, 49.18; H, 4.70; H, 4.59. 1H NMR (CD2Cl2 298 K): δH 7.66–7.41 (12H tot: 4H o-PPh, 4H m-PPh, 4H p-PPh), 7.37–7.28 (4H tot: 2H o-P1Ph, 2H m-P1Ph), 7.15 (td, 2H, 3JHH = 7.9 Hz, 4JHP = 3.0 Hz, m-P2Ph), 6.94 (ddd, 2H, 3JHP = 12.3 Hz, 3JHH = 7.9 Hz, 5JHP = 1.1 Hz, o-P2Ph), 6.91 (d, 2H, 3JHH = 8.2 Hz, C3Htol-P2), 6.83 (d, 2H, 3JHH = 8.2 Hz, C3Htol-P1), 6.50 (d, 2H, 3JHH = 8.2 Hz, C2Htol-P2), 6.43 (d, 2H, 3JHH = 8.2 Hz, C2Htol-P1), 2.24 (s, 3H, CH3tol-P2), 2.19 (s, 3H, CH3tol-P1), 1.44 (s, 9H, CH3tBu1), 1.37 (ddd, 1H, 2JHH = 13.2 Hz, 3JHP = 3.3 Hz, 3JHP = 1.3 Hz, SiCH2Ir), 1.25 (s, 9H, CH3tBu2), 1.24 (dd, 1H, 2JHH = 13.2 Hz, 3JHP = 8.6 Hz, SiCH2Ir), 0.17 (s, 3H, SiCH3). 13C{1H} NMR (CD2Cl2, 298 K): δC 137.8 (dd, 1JCP = 65.3 Hz, 3JCP = 2.7 Hz, C1, PhP), 137.7 (d, 2JCP = 9.1 Hz, C1, tol-P1), 137.1 (d, 2JCP = 9.0 Hz, C1, tol-P2), 136.4 (d, 5JCP = 2.0 Hz, C4, tol-P2), 135.8 (d, 5JCP = 1.6 Hz, C4, tol-P1), 134.1 (d, 2JCP = 10.1 Hz, C2, PhP2), 134.0 (d, 2JCP = 10.0 Hz, C2, PhP1), 133.4 (d, 2JCP = 10.8 Hz, C2, PhP1), 132.5 (d, 4JCP = 2.7 Hz, C4, PhP2), 132.1 (d, 4JCP = 2.8 Hz, C4, PhP1), 132.0 (d, 4JCP = 2.5 Hz, C4, PhP2), 131.5 (dd, 1JCP = 62.7 Hz, 3JCP = 1.6 Hz, C1, PhP), 131.4 (d, 4JCP = 2.8 Hz, C4, PhP1), 131.3 (d, 3JCP = 10.1 Hz, C2, PhP2), 130.3 (dd, 1JCP = 57.1 Hz, 3JCP = 1.4 Hz, C1, PhP), 129.7 (d, 5JCP = 1.6 Hz, C3, tol-P2), 129.5 (d, 5JCP = 1.2 Hz, C3, tol-P1), 129.0 (d, 4JCP = 3.5 Hz, C2, tol-P2), 128.8 (d, 2JCP = 11.3 Hz, C3, PhP2), 128.6 (d, 4JCP = 3.3 Hz, C2, tol-P1), 128.40 (d, 3JCP = 10.8 Hz, C3, PhP1), 128.37 (dd, 1JCP = 66.5 Hz, 3JCP = 1.2 Hz, C1, PhP), 128.1 (d, 3JCP = 11.6 Hz, C3, PhP2), 127.2 (d, 3JCP = 11.2 Hz, C3, PhP1), 59.3 (CtBu2), 58.9 (CtBu1), 29.8 (CH3tBu1), 29.6 (CH3tBu2), 20.4 (CH3tol-P2), 20.4 (CH3tol-P1), −1.1 (t, 3JCP = 7.1 Hz, CH3Si), −18.9 (d, 2JCP = 3.8 Hz, CH2Si). 31P{1H} NMR (CD2Cl2, 298 K): δP 25.7 (d, 2JPP = 17.4 Hz, P1 SiNP), 25.1 (d, 2JPP = 17.4 Hz, P2 SiNP), −144.4 (hept, 1JPF = 710.2 Hz, PF6−).Synthesis of [Ir{κ3C,P,P′-(SiNP–H)}(CH3CN)(CNtBu)2][PF6]2 (6[PF6]2)
An acetonitrile solution (10 mL) of [IrBr{κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6] (5PF6, 233 mg, 0.191 mmol, 1221.13 g mol−1) was added with AgPF6 (53.2 mg, 0.210 mmol, 252.83 g mol−1). The resulting colorless suspension was stirred for 12 h and filtered. The resulting solution was partially evaporated and added with diethyl ether (5 mL), affording a colourless solid which was filtered and washed with diethyl ether (3 × 5 mL), dried in vacuo and finally identified as [Ir(CH3CN) {κ3C,P,P′-(SiNP–H)}(CNtBu)2][PF6]2 (6[PF6]2, 179 mg, 0.0135 mmol, 71% yield). Found: C, 46.81; H, 4.69; N, 5.32. Calcd for: C52H60F12IrN5P4Si (1327.24): C, 47.06; H, 4.56; N, 5.28. 1H NMR (CD2Cl2 298 K): δH 7.73–7.46 (12H tot: 4H m-PPh, 4H p-PPh, 4H o-PPh), 7.46–7.31 (4H tot: 2H o-P2Ph, 2H m-P2Ph), 7.18 (td, 2H, 3JHH = 7.8 Hz, 3JHP = 3.2 Hz, m-P1Ph), 6.95 (m, 2H, m-P2Ph), 6.90 (br d, 4H, 3JHH = 8.0 Hz, C3Htol-P1 and C3Htol-P2), 6.50 (d, 2H, 3JHH = 8.0 Hz C2Htol-P1), 6.41 (d, 2H, 3JHH = 8.0 Hz, C2Htol-P2), 2.23 (br s, 6H, CH3tol-P1 and CH3tol-P2), 2.09 (s, 3H, CH3CN), 1.48 (s, 9H, CH3tBu1), 1.44 (dd, 1H, 2JHH = 13.0 Hz, 3JHP = 7.5 Hz, SiCH2Ir), 1.30 (dd, 1H, 2JHH = 13.0 Hz, 3JHP = 2.7 Hz, SiCH2Ir), 1.29 (s, 9H, CH3tBu2), 0.34 (s, 3H, SiCH3). 13C{1H} NMR (CD2Cl2, 298 K): δC 137.0 (d, 5JCP = 2.1 Hz, C4, tol-P2), 136.7 (d, 2JCP = 8.7 Hz, C1, tol1 or C1, tol2), 136.5 (d, 5JCP = 1.5 Hz, C4, tol-P1), 135.81 (d, 2JCP = 8.0 Hz, C1, tol1 or C1, tol2), 135.79 (d, 2JCP = 70.6 Hz, C2, PhP), 133.9 (d, 2JCP = 10.7 Hz, C2, PhP2), 133.7 (d, 2JCP = 11.1 Hz, C2, PhP1), 133.2 (d, 4JCP = 2.8 Hz, C4, PhP2), 133.0 (d, 4JCP = 1.9 Hz, C4, PhP1), 132.9 (d, 4JCP = 1.8 Hz, C4, PhP2), 132.5 (d, 4JCP = 2.8 Hz, C4, PhP1), 132.3 (d, 3JCP = 10.1 Hz, C2, PhP2), 131.4 (d, 3JCP = 10.2 Hz, C2, PhP1), 130.8 (d, 1JCP = 60.7 Hz, C1, PhP), 130.0 (d, 4JCP = 1.5 Hz, C3, tol-P2), 129.9 (d, 4JCP = 1.4 Hz, C3, tol-P1), 129.4 (d, 3JCP = 11.7 Hz, C2, PhP2), 129.4 (d, 3JCP = 11.2 Hz, C2, PhP1), 129.1 (d, 3JCP = 11.7 Hz, C3, PhP2), 128.9 (d, 4JCP = 3.7 Hz, C2, tol-P2), 128.6 (d, 3JCP = 12.0 Hz, C3, PhP1), 127.8 (d, 4JCP = 3.8 Hz, C2, tol-P1), 127.7 (d, 1JCP = 62.0 Hz, C1, PhP), 126.8 (d, 1JCP = 70.9 Hz, C1, PhP), 121.9 (d, 3JCP = 18.6 Hz, NCCH3), 61.1 (CtBu2), 60.7 (CtBu1), 29.4 (CH3tBu1), 29.3 (CH3tBu2), 20.4 (CH3tol1 or tol2), 20.3 (CH3tol1 or tol2), 2.6 (CH3CN), −1.5 (t, 3JCP = 6.8 Hz, CH3Si), −18.7 (t, 2JCP = 3.6 Hz, CH2Si). 31P{1H} NMR (CD2Cl2, 298 K): δP 25.1 (d, 2JPP 19.4 Hz, P1 SiNP), 18.0 (d, 2JPP 19.4 Hz, P2 SiNP), −144.4 (hept, 1JPF = 710.2 Hz, PF6−).Synthesis of [IrH(CH2SiF2CH3)(HNP)2(CNtBu)2][BF4] (7BF4)
A dichloromethane solution (5 mL) of [IrH(SiNP–H)(CNtBu)2][PF6] (1PF6, 116 mg, 0.102 mmol, 1142.24 g mol−1) was added with HBF4·Et2O (27.8 μL, 0.203 mmol, 161.93 g mol−1, 1.18 g mL−1) at 193 K. The resulting colourless solution was stirred for 30 min, allowed to warm up at room temperature, partially evaporated and added with hexane (10 mL), affording a colourless solid which was filtered off and washed with diethyl ether/hexane (1:10) (12 mL), dried in vacuo and finally identified as [IrH(CH2SiF2CH3)(HNP)2(CNtBu)2][BF4] (7BF4, 64.4 mg, 0.0573 mmol, 56% yield). Found: C, 53.29; H, 5.47; N, 5.09. Calcd for C50H60BF6IrN4P2Si (1124.09): C, 53.42; H, 5.38; N, 4.98. 1H NMR (CD2Cl2 298 K): δH 7.74–7.62 (4H tot: 2H p-P2Ph and 2H o-P2Ph), 7.62–7.45 (12H tot: 2H o-P1Ph, 2H o-P2Ph, 4H m-P1Ph, 2H m-P2Ph and 2H p-P1Ph,), 7.40 (td, 2H, 3JHH = 7.7 Hz, 4JHP = 2.8 Hz, m-P2Ph), 7.31 (m, 2H, o-P1Ph), 6.77 (d, 2H, 3JHH = 8.4 Hz, C3Htol-P1), 6.73 (d, 3JHH = 8.4 Hz, C3Htol-P2), 6.11 (d, 2H, 3JHH = 8.4 Hz, C2Htol-P1), 5.94 (d, 2H, 3JHH = 8.4 Hz, C2Htol-P2), 5.33 (dd, 1H, 2JHP = 15.9 Hz, 1JHF = 3.9 Hz, NHP1), 4.21 (d, 1H, 2JHP = 15.9 Hz, NHP2), 2.16 (s, 3H, CH3tol-P1), 2.15 (s, 3H, CH3tol-P2), 1.25 (s, 9H, CH3tBu1), 1.21 (s, 9H, CH3tBu2), 0.33 (t, 3H, 3JHF = 6.3 Hz, SiCH3), 0.01 (ddddd, 1H, 2JHH = 13.4 Hz, 3JHF = 13.4 Hz, 3JHP = 10.5 Hz, 3JHP = 4.7 Hz, 3JHF = 2.4 Hz, SiCHaHbIr), −0.42 (ddddd, 1H, 2JHH = 13.4 Hz, 3JHF = 13.4 Hz, 3JHP = 11.6 Hz, 3JHP = 3.6 Hz, 3JHF = 3.5 Hz, SiCHaHbIr), −11.86 (ddt, 1H, 2JHP trans = 152.9 Hz, 2JHP cis = 17.4 Hz, 4JHF = 2.2 Hz, IrH). 13C{1H} NMR (CD2Cl2, 298 K): δC 138.9 (d, 2JCP = 9.0 Hz, C1, tol-P2), 138.0 (d, 2JCP = 11.1 Hz, C1, tol-P1), 132.2 (d, 2JCP = 10.2 Hz, C2, PhP1), 131.9 (d, 4JCP = 2.3 Hz, C4, PhP1), 131.73 (d, 3JCP = 10.6 Hz, C2, PhP2), 131.67 (d, 3JCP = 11.0 Hz, C2, PhP2), 131.5 (dd, 3JCP = 10.0, 5JCP = 2.8 Hz, C2, PhP1), 130.8 (C4, tol-P1 and C4, tol-P2), 130.7 (d, 3JCP = 11.2 Hz, C3, PhP1), 129.1 (d, 3JCP = 11.2 Hz, C3, tol-P1), 129.0 (d, 4JCP = 7.6 Hz, C3, tol-P2), 128.9 (d, 4JCP = 1.6 Hz, C4, PhP2), 128.6 (d, 3JCP = 11.2 Hz, C3, PhP2), 118.4 (d, 3JCP = 5.7 Hz, C2, tol-P1), 117.9 (d, 3JCP = 5.1 Hz, C2, tol-P1), 58.9 (CtBu2), 58.8 (CtBu1), 29.7 (CH3tBu1), 29.4 (CH3tBu2), 20.0 (CH3tol-P1), 20.0 (CH3tol-P2), −3.6 (t, 2JCF = 19.3 Hz, CH3Si), −34.9 (CH2Si). 19F{1H} NMR (CD2Cl2, 298 K): δF −127.5 (dd, 1F, 2JFF = 20.6 Hz, 4JFP = 2.7 Hz, SiFaFb), −129.0 (d, 1F, 2JFF = 20.6 Hz, SiFaFb). 31P{1H} NMR (CD2Cl2, 298 K): δP 11.4 (d, 2JPP 20.5 Hz, P1), 3.1 (d, 2JPP 20.5 Hz, P2).
DFT calculations
Molecular structure optimizations and frequencies calculations were carried out with the Gaussian09 program (revision D.01)9 using the method B3PW91,10 including the D3 dispersion correction scheme by Grimme with Becke–Johnson damping.11 The def2-SVP12 basis and pseudo potential were used for all atoms and the “ultrafine” grid was employed in all calculations. Stationary points were characterized by vibrational analysis. The structures were optimized in dichloromethane (298 K, 1 atm) using the PCM method.13 In order to improve the accuracy of the calculated energies, single point energy calculations were carried out on the optimized structures of intermediates and transition states using the method M06,14 the def2-TZVP12 basis and pseudo potentials, where appropriate, and the SMD model15 for the solvent (dichloromethane). Finally a correction of +1.89 kcal mol−1 to Gibbs free energy was also applied for the change of the standard state from gas phase (1 atm) to solution (1 M) at 298 K.16 Delocalization indexes (DI) were calculated using Multiwfn.17Crystal structure determination
Single crystals of SiNP and 7BF4 were obtained by slow evaporation of dicholoromethane solutions of the compounds; single crystals of 1PF6 and 4PF6 were grown by slow diffusion of hexane into a THF (4PF6) or dichloromethane solution (1PF6) of the compounds. X-ray diffraction data were collected at 100(2) K on a Bruker APEX SMART (1PF6, 4PF6, 7BF4) or APEX DUO (SiNP) diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) using 0.6° ω rotations. Intensities were integrated and corrected for absorption effects with SAINT–PLUS18 and SADABS19 programs, both included in APEX2 package. The structures were solved by the Patterson method with SHELXS-9720 and refined by full matrix least-squares on F2 with SHELXL-201421 under WinGX.22Crystal data for SiNP
C40H40N2P2Si, 638.77 g mol−1, monoclinic, P21/c, a = 23.093(3) Å, b = 9.0864(13) Å, c = 17.039(2) Å, β = 107.176(2)°, V = 3415.9(8) Å3, Z = 4, reflections collected/independent 65969/6980 [R(int) = 0.0406], R1 = 0.0435 [I > 2σ(I)], wR2 = 0.1380 (all data). CCDC deposit number 2155640.†
Crystal data for 1PF6
2C50H58F6IrN4P3Si·CH2Cl2·C6H14, 2455.50 g mol−1, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.9615(7) Å, b = 12.7205(8) Å, c = 20.4705(12) Å, α = 86.1880(10)°, β = 77.4030(10)°, γ = 85.9510(10)°, V = 2774.8(3) Å3, Z = 1, reflections collected/independent 34093/11291 [R(int) = 0.0280], R1 = 0.0293 [I > 2σ(I)], wR2 = 0.0762 (all data). CCDC deposit number 2155643.†
, a = 10.9615(7) Å, b = 12.7205(8) Å, c = 20.4705(12) Å, α = 86.1880(10)°, β = 77.4030(10)°, γ = 85.9510(10)°, V = 2774.8(3) Å3, Z = 1, reflections collected/independent 34093/11291 [R(int) = 0.0280], R1 = 0.0293 [I > 2σ(I)], wR2 = 0.0762 (all data). CCDC deposit number 2155643.†
Crystal data for 4PF6
4C50H57ClF6IrN4P4Si·3C4H8O, 4922.88 g mol−1, monoclinic, C2/c, a = 46.824(5) Å, b = 10.9773(11) Å, c = 25.668(3) Å, β = 121.1930(10)°, V = 11286(2) Å3, Z = 2, reflections collected/independent 58271/12421 [R(int) = 0.0495], R1 = 0.0364 [I > 2σ(I)], wR2 = 0.0815 (all data). CCDC deposit number 2155642.†
Crystal data for 7BF4
C50H60BF6IrN4P2Si·CH2Cl2, 1208.98 g mol−1, monoclinic, P21/c, a = 16.3517(10) Å, b = 11.6078(7) Å, c = 28.9040(17) Å, β = 93.8810(10)°, V = 5473.6(6) Å3, Z = 4, reflections collected/independent 61703/11193 [R(int) = 0.0364], R1 = 0.0231 [I > 2σ(I)], wR2 = 0.0525 (all data). CCDC deposit number 2155641.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministerio de Ciencia e Innovación MCIN/AEI/10.13039/501100011033, under the Project PID2019-103965GB-I00, and the Departamento de Ciencia, Universidad y Sociedad del Conocimiento del Gobierno de Aragón (group E42_20R) is gratefully acknowledged.Notes and references
- V. Passarelli, J. J. Pérez-Torrente and L. A. Oro, Dalton Trans., 2016, 45, 951–962 RSC.
- (a) B. S. Mitchell, W. Kaminsky and A. Velian, Inorg. Chem., 2021, 60, 6135–6139 CrossRef CAS PubMed; (b) A. Aloisi, É. Crochet, E. Nicolas, J.-C. Berthet, C. Lescot, P. Thuéry and T. Cantat, Organometallics, 2021, 40, 2064–2069 CrossRef CAS; (c) J. A. Kephart, A. C. Boggiano, W. Kaminsky and A. Velian, Dalton Trans., 2020, 49, 16464–16473 RSC; (d) H. Zhang, G. P. Hatzis, C. E. Moore, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2019, 141, 9516–9520 CrossRef CAS PubMed; (e) J. A. Kephart, B. S. Mitchell, A. Chirila, K. J. Anderton, D. Rogers, W. Kaminsky and A. Velian, J. Am. Chem. Soc., 2019, 141, 19605–19610 CAS; (f) K. M. Gramigna, D. A. Dickie, B. M. Foxman and C. M. Thomas, ACS Catal., 2019, 9, 3153–3164 CrossRef CAS; (g) M. L. Bin Ismail and C.-W. So, Chem. Commun., 2019, 55, 2074–2077 RSC; (h) B. A. Barden, G. Culcu, J. P. Krogman, M. W. Bezpalko, G. P. Hatzis, D. A. Dickie, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2019, 58, 821–833 CrossRef CAS PubMed; (i) A. J. Ayres, A. J. Wooles, M. Zegke, F. Tuna and S. T. Liddle, Inorg. Chem., 2019, 58, 13077–13089 CrossRef CAS PubMed; (j) H. Zhang, B. Wu, S. L. Marquard, E. D. Litle, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Organometallics, 2017, 36, 3498–3507 CrossRef CAS; (k) G. Culcu, D. A. Iovan, J. P. Krogman, M. J. T. Wilding, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2017, 139, 9627–9636 CrossRef CAS PubMed; (l) F. Völcker and P. W. Roesky, Dalton Trans., 2016, 45, 9429–9435 RSC; (m) R. W. Carlsen and D. H. Ess, Dalton Trans., 2016, 45, 9835–9840 RSC; (n) W. K. Walker, B. M. Kay, S. A. Michaelis, D. L. Anderson, S. J. Smith, D. H. Ess and D. J. Michaelis, J. Am. Chem. Soc., 2015, 137, 7371–7378 CrossRef CAS PubMed; (o) W. K. Walker, D. L. Anderson, R. W. Stokes, S. J. Smith and D. J. Michaelis, Org. Lett., 2015, 17, 752–755 CrossRef CAS PubMed; (p) F. Völcker, F. M. Mück, K. D. Vogiatzis, K. Fink and P. W. Roesky, Chem. Commun., 2015, 51, 11761–11764 RSC; (q) J. P. Krogman, B. M. Foxman and C. M. Thomas, Organometallics, 2015, 34, 3159–3166 CrossRef CAS.
- Selected references for PNNP: (a) A. Prades, S. Núñez-Pertíñez, A. Riera and X. Verdaguer, Chem. Commun., 2017, 53, 4605–4608 RSC; (b) F. Trentin, A. M. Chapman, A. Scarso, P. Sgarbossa, R. A. Michelin, G. Strukul and D. F. Wass, Adv. Synth. Catal., 2012, 354, 1095–1104 CrossRef CAS; (c) S. W. Hunt, V. Nesterov and M. G. Richmond, J. Mol. Struct., 2012, 1010, 91–97 CrossRef CAS; (d) L. E. Bowen, M. Charernsuk, T. W. Hey, C. L. McMullin, A. G. Orpen and D. F. Wass, Dalton Trans., 2010, 39, 560–567 RSC; (e) B. R. Aluri, N. Peulecke, B. H. Müller, S. Peitz, A. Spannenberg, M. Hapke and U. Rosenthal, Organometallics, 2010, 29, 226–231 CrossRef CAS; (f) P. Fonteh and D. Meyer, Metallomics, 2009, 1, 427–433 CrossRef CAS PubMed; (g) J. Bravo, J. Castro, S. García-Fontán, M. C. Rodríguez-Martínez, G. Albertin, S. Antoniutti and A. Manera, J. Organomet. Chem., 2007, 692, 5481–5491 CrossRef CAS.
- Selected references of PNP: (a) M. Rodriguez-Zubiri, V. Gallo, J. Rosé, R. Welter and P. Braunstein, Chem. Commun., 2008, 64–66 RSC; (b) L.-C. Song, X.-F. Han, W. Chen, J.-P. Li and X.-Y. Wang, Dalton Trans., 2017, 46, 10003–10013 RSC; (c) L.-C. Song, L.-D. Zhang, W.-W. Zhang and B.-B. Liu, Organometallics, 2018, 37, 1948–1957 CrossRef CAS; (d) S. A. Bartlett, J. Moulin, M. Tromp, G. Reid, A. J. Dent, G. Cibin, D. S. McGuinness and J. Evans, Catal. Sci. Technol., 2016, 6, 6237–6246 RSC; (e) A. M. Lifschitz, N. A. Hirscher, H. B. Lee, J. A. Buss and T. Agapie, Organometallics, 2017, 36, 1640–1648 CrossRef CAS; (f) S. Orgué, T. León, A. Riera and X. Verdaguer, Org. Lett., 2015, 17, 250–253 CrossRef PubMed; (g) C. Mu, J. He, S. Lü, J. Yang, Y. Xie, K. Hu, P. Yan and Y.-L. Li, Polyhedron, 2021, 200, 115087 CrossRef CAS; (h) S. Todisco, V. Gallo, P. Mastrorilli, M. Latronico, N. Re, F. Creati and P. Braunstein, Inorg. Chem., 2012, 51, 11549–11561 CrossRef CAS PubMed; (i) S. Naik, J. T. Mague and M. S. Balakrishna, Inorg. Chim. Acta, 2013, 407, 139–144 CrossRef CAS.
- (a) M. L. Clarke, A. M. Z. Slawin and J. D. Woolins, Phosphorus, Sulfur Silicon Relat. Elem., 2001, 168–169, 329–332 CAS; (b) S. M. Aucott, M. L. Clarke, A. M. Z. Slawin and J. D. Woolins, J. Chem. Soc., Dalton Trans., 2001, 972–976 RSC.
- (a) V. Passarelli and F. Benetollo, Inorg. Chem., 2011, 50, 9958–9967 CrossRef CAS PubMed; (b) V. Passarelli, J. J. Pérez-Torrente and L. A. Oro, Dalton Trans., 2015, 44, 18596–18606 RSC; (c) V. Passarelli, J. J. Pérez-Torrente and L. A. Oro, Inorg. Chem., 2014, 53, 972–980 CrossRef CAS PubMed.
- (a) M. Zhong, J. Wei, W.-X. Zhang and Z. Xi, Organometallics, 2021, 40, 310–313 CrossRef CAS; (b) X. Xin and C. Zhu, Dalton Trans., 2020, 49, 603–607 RSC; (c) L.-C. Song, X.-Y. Yang, X.-Y. Gao and M. Cao, Inorg. Chem., 2019, 58, 39–42 CrossRef CAS PubMed; (d) G. Feng, M. Zhang, P. Wang, S. Wang, L. Maron and C. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17654–17658 CrossRef CAS PubMed; (e) J. Ji, F. Wu, L.-M. Shi, A.-Q. Jia and Q.-F. Zhang, J. Organomet. Chem., 2019, 885, 1–6 CrossRef CAS; (f) A. A. Kassie, P. Duan, E. T. McClure, K. Schmidt-Rohr, P. M. Woodward and C. R. Wade, Inorg. Chem., 2019, 58, 3227–3236 CrossRef CAS PubMed; (g) A. A. Kassie, P. Duan, M. B. Gray, K. Schmidt-Rohr, P. M. Woodward and C. R. Wade, Organometallics, 2019, 38, 3419–3428 CrossRef CAS; (h) S. Khan, S. Pal, N. Kathewad, I. Purushothaman, S. De and P. Parameswaran, Chem. Commun., 2016, 52, 3880–3882 RSC; (i) A. Alzamly, S. Gambarotta and I. Korobkov, Organometallics, 2013, 32, 7107–7115 CrossRef CAS; (j) S. Zhang, R. Pattacini and P. Braunstein, Dalton Trans., 2011, 40, 5711 RSC; (k) S. Zhang, R. Pattacini and P. Braunstein, Organometallics, 2010, 29, 6660–6667 CrossRef CAS.
- J. Clayden, Organic chemistry, Oxford University Press, Oxford, 2005. ISBN 978-0-19-850346-0 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, C. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- J. P. Perdew, in Electronic Structure of Solids '91, Ed. P. Ziesche and H. Eschrig, Akademie Verlag, Berlin, 1991 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- V. S. Bryantsev, M. S. Diallo and W. A. Goddard III, J. Phys. Chem. B, 2008, 112, 9709–9719 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- SAINT+: Area-Detector Integration Software, version 6.01, Bruker AXS, Madison, WI, 2001 Search PubMed.
- G. M. Sheldrick, SADABS program, University of Göttingen, Göttingen, Germany, 1999 Search PubMed.
- G. M. Sheldrick, SHELXS 97, Program for the Solution of Crystal Structure, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystal data, NMR spectra, and atomic coordinates of calculated structures. CCDC 2155640–2155643. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00794k |
| ‡ The hydrido signal was observed as a 1H{31P} doublet due to the scalar coupling of the IrH hydrogen atom to one of the IrCH2 hydrogen atoms (3JHH = 2.6 Hz). Accordingly, the 1H{31P} signals at 0.46 and 0.67 ppm, assigned to the IrCH2 moiety, are a doublet (2JHH = 12.6 Hz) and a doublet of doublets (2JHH = 12.6, 3JHH = 2.6 Hz), respectively. As already discussed for the related hydrido derivative [IrH{κ3C,P,P′-(SiNP–H)}{P(OCH3)3}2]+,6b this pattern is the consequence of the dependence of the 3JHH constant on the H–X–Y–H dihedral angle (cf. M. J. Minch, Concepts Magn. Reson., 1994, 6, 41–56). |
| § It is worth a mention that in the course of the related reaction of [Ir(SiNP)(cod)][PF6] with P(OCH3)3 the pentacoordinated derivative [Ir(SiNP)(cod){P(OCH3)3}][PF6], analogous to 2+, could not be isolated and was characterised in situ whereas the putative square planar complex [Ir(SiNP){P(OCH3)3}2]+, analogous to 3+, could not even be observed and was proposed only based on DFT calculations (see ref. 6b). |
| ¶ Selected 1H NMR data for 3+ (CD2Cl2, 298 K) are: δH 7.14 (d, 3JHH = 8.1 Hz, 2H, C2Htol), 6.87 (d, 3JHH = 8.1 Hz, 2H, C3Htol), 2.54 (s, 6H, CH3tol), 1.41 ppm (s, 18H, CH3tBu), 0.86 ppm (s, 6H, SiCH3). |
| || The calculated heteronuclear spin–spin constants JXY are negligible in all the cases except for F2 and P2 (6.5 Hz, calc.), thus the observed fluorine-phosphorus coupling is the consequence of the conformation adopted by the F–Si–C–Ir–P fragment rather than of the NH⋯F hydrogen bond. On the other hand, when it comes to the NH⋯F hydrogen bond, the calculated heteronuclear spin–spin constants are negligible in all the cases except for H1N and F1 (23.2 Hz, calc.), nicely matching the proposed assignment. |
| ** The 31P{1H} doublets at 43.0 and 11.9 ppm (2JPP = 19.0 Hz) of 8+ are indicative of two mutually cis phosphorus atoms. The 19F doublet of quartets of doublets at −125.8 ppm results from the scalar coupling of the fluorine atom to the SiCH3 moiety (3JHF = 6.4 Hz) and the SiCH2 non-equivalent hydrogen atoms (3JHF = 16.4 Hz, 2.0 Hz). Accordingly, the 1H doublet at 0.22 ppm (3JHF = 6.4 Hz) was assigned to the SiCH3 group (δC = −1.3 ppm, doublet, 2JCF = 6.7 Hz) and the 1H multiplets at 0.20 and −0.24 ppm were assigned to the two non-equivalent SiCH2 hydrogen atoms. The 1H signal of the hydrido ligand was observed at −10.8 ppm as a doublet of doublets (2JHP, trans = 146.1 Hz, 2JHP, cis = 26.3 Hz). |
| This journal is © The Royal Society of Chemistry 2022 |