
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceToward asymmetric aziridination with an iron complex supported by a D2-symmetric tetra-NHC†
Kevin M.
Blatchford
,
Carson J.
Mize
 ,
Sharani
Roy
and
David M.
Jenkins
*
,
Sharani
Roy
and
David M.
Jenkins
*
Department of Chemistry, University of Tennessee, Knoxville, Tennessee 37996, USA. E-mail: jenkins@ion.chem.utk.edu
First published on 5th April 2022
Abstract
A neutral D2-symmetric macrocyclic tetra-N-heterocyclic carbene ligand was synthesized. The macrocycle was ligated to iron(II) via transmetalation from an isolated silver complex that has two conformers. The iron complex catalyzed the first stereospecific aziridination between aryl azides and aliphatic alkenes, albeit with low ee's.
Catalytic C2 + N1 aziridination has undergone a revival in the last decade with the development of multiple approaches to form a three membered ring from a nitrene source (N1) and an alkene (C2).1–7 This research is driven by two competing dynamics: aziridines are effective reagents in organic synthesis due to their strained ring,8,9 but, unlike epoxides, there are no effective commercial aziridination catalysts across a variety of alkenes.
Most alkenes are prochiral, and thus either one or two chiral carbons are set during the catalytic reaction, yet even with some recent advances, catalytic synthesis of asymmetric aziridines remains highly limited.4,6,7 Researchers including Zhang, Schomaker, Katsuki, and Uchida have developed catalysts that produce single enantiomers, but with limitations such as performing only with very specific reagents like styrene and its derivatives for the alkene, or only working with (tosyliminoiodo)benzene as the nitrene source.2,10–14 This lack of stereo control is a pity, since many medicinal or biological applications benefit from forming a single enantiomer.15,16
Our group has developed macrocyclic tetra-NHC complexes on both iron and chromium that are effective for catalytic aziridination with organic azides and aliphatic alkenes.17–19 Recently, we developed a synthesis of the first D2-symmetric chiral macrocyclic NHC by linking C2-symmetric chiral diimidazoles bound on a cyclohexyl ring.20 We hypothesized that a similar, less sterically hindered, D2-symmetric macrocycle could produce an iron complex that would favour one enantiomer in a catalytic aziridination reaction.
In this manuscript, we report the synthesis of the first catalyst that gives enantiomeric excess of chiral aziridines prepared from aryl azides and aliphatic alkenes. Our approach was to prepare a new, more flexible, D2-symmetric tetra-imidazolium macrocycle that was then complexed to silver forming two distinct stereoisomeric dimers, both of which were effective in NHC transmetalation to iron. The iron complex was modestly effective as an aziridination catalyst and, in cases with ortho substituents on the aryl ring of the organic azide, measurable ee's for the aziridines were obtained. Computational results suggest that this approach is feasible with a limited scope of organic azides and provides suggestions about how to develop an improved future catalyst.
The ability to synthesize the desired D2-symmetric tetra-imidazolium macrocycle requires the synthesis of C2-symmetric diimidazoles. We had previously reported a two-step synthesis of (1S,2S)-1,2-di(imidazole)cyclohexane from its commercially available chiral diamine20 and a similar strategy proved successful here (Scheme 1A). Addition of 1,1-diethoxy-2-isothiocyanatoethane to (1S,2S)-(−)-1,2-diphenylethylenediamine in refluxing dry acetonitrile followed by addition of 1 M HCl led to the formation of 1,1′-((1S,2S)-1,2-diphenylethane)bis(1,3-dihydro-2H-imidazole-2-thione), 1. Compound 1 was isolated by precipitation at −20 °C and then washed with water to produce the pure product in 94% yield. Thione 1 was reduced to the diimidazole by addition of HNO3 and NaNO2 at 0 °C. After the reaction was completed, the product was extracted with CH2Cl2 from the aqueous layer and 1,1′-[(1S,2S)-1,2-diphenyl-1,2-ethanediyl]bis-imidazole (2) was purified with gradient column chromatography on silica gel (MeOH/CH2Cl2) giving a 45% yield. Notably, diimidazole 2 can be synthesized on a 5+ gram scale.
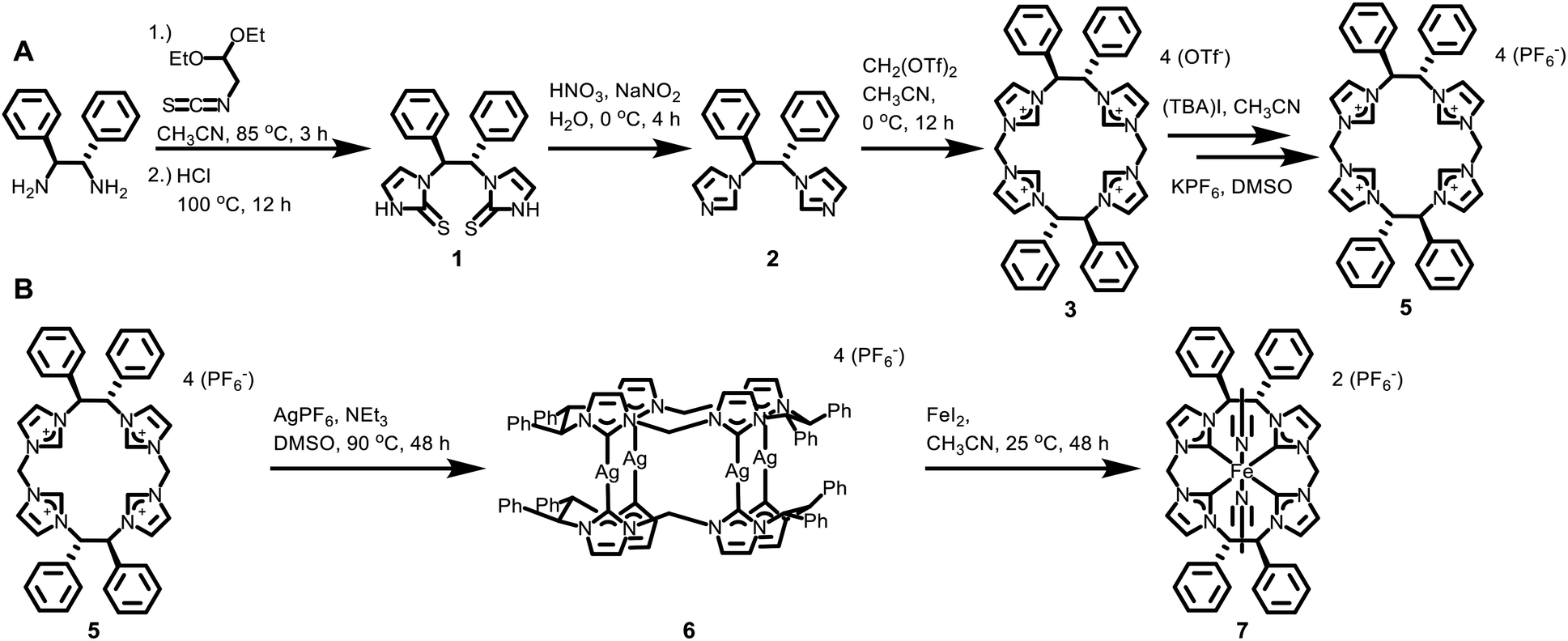 | ||
| Scheme 1 (A) Synthesis of D2-symmetric macrocyclic tetra-imidazolium. (B) Synthesis of chiral iron catalyst (7). Complex 6 is a mixture of conformers; 6a is shown. | ||
The chiral macrocycle ((S,S)-1,2-Ph2-Et,MeTCH)(OTf)4, 3, was formed by reaction of 2 with ditriflatomethane21 in dry acetonitrile at 0 °C for 12 hours (Scheme 1A). The solvent was removed under reduced pressure and the collected solid was washed with THF to yield pure 3 in 45% yield. To prevent anion confusion, we completed two ion exchange steps, via ((S,S)-1,2-Ph2-Et,MeTCH)(I)4 (4), to replace the triflate anion with PF6, giving ((S,S)-1,2-Ph2-Et,MeTCH)(PF6)4 (5) (see ESI for details†).
Crystallographic and spectroscopic characterization of 5 involved several notable features. First, single crystal X-ray diffraction of 5 confirms the absolute stereochemistry as (S,S,S,S) (Fig. 1A). Second, the 1H NMR and HSQC spectra for 5 shows the stereogenic protons at 6.31 ppm (Fig. S17 and S19†) suggesting that they are highly acidic compared to our original macrocycle, (Et,MeTCH)(OTf)4, which shows resonances for the ethyl bridging protons at 4.68 ppm.22
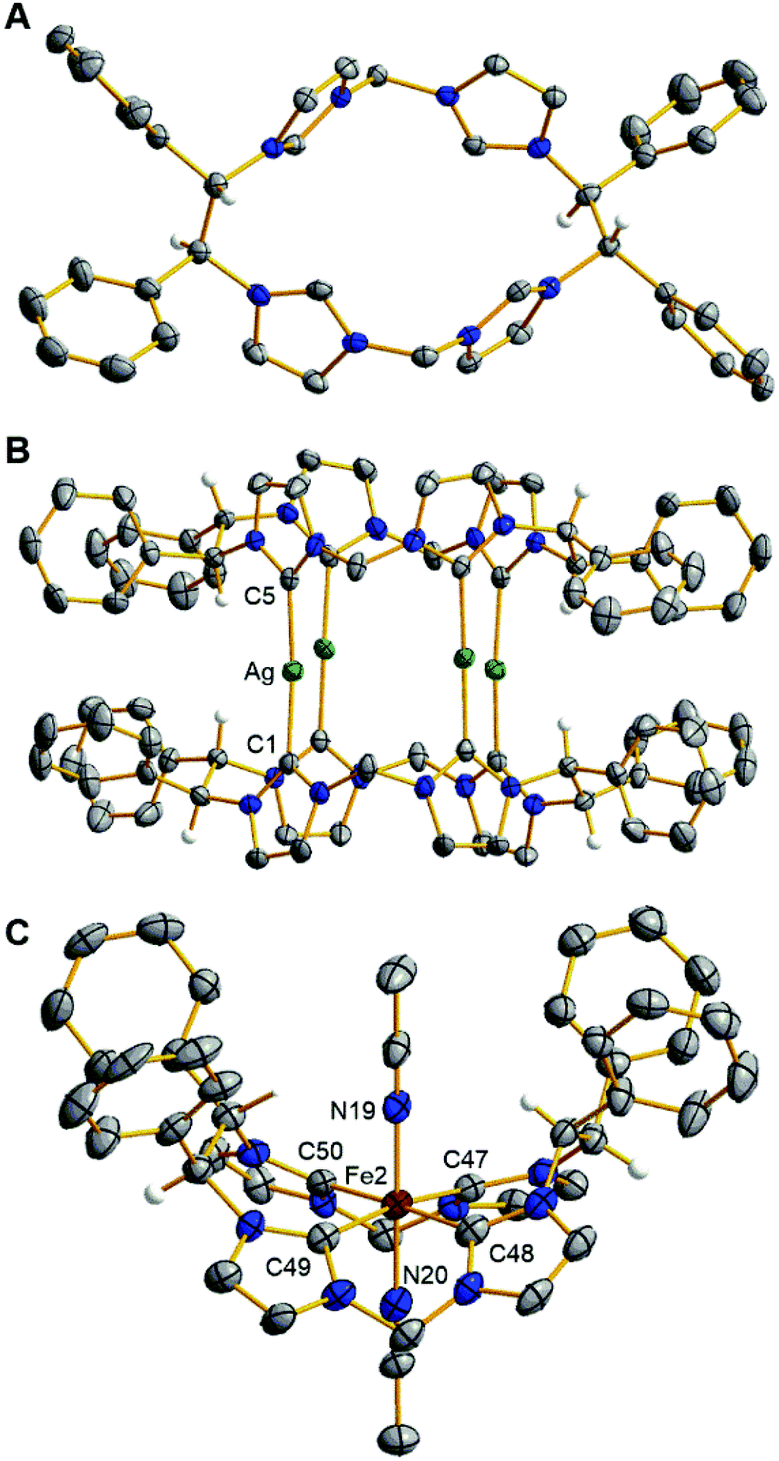 | ||
| Fig. 1 Solid state structures of (A) ((S,S)-1,2-Ph2-Et,MeTCH)(PF6)4, 5, (B) [((S,S)-1,2-Ph2-Et,MeTCH)2Ag4](PF6)4, 6a, and (C) [((S,S)-1,2-Ph2-Et,MeTCH)Fe(NCCH3)2](PF6)2, 7. Green, burgundy, blue, grey, and white ellipsoids (50% probability) represent Ag, Fe, N, C, and H atoms, respectively. Solvent molecules and H-atoms on non-stereogenic atoms are omitted for clarity; Selected bond lengths (Å) and angles (°) are as followed for 6a: Ag–C1, 2.107(3); Ag–C5, 2.093(3); C1–Ag–C5, 168.5(1) and 7: Fe2–C47, 2.001(6); Fe2–C48, 1.937(7); Fe2–C49, 1.992(6); Fe2–C50, 1.923(6); C47–Fe2–C49, 170.2(2); C48–Fe2–C50, 177.2(3); N19–Fe2–N20, 178.7(2). | ||
The relatively similar pKas for the stereogenic protons on the ethyl bridge and for the imidazolium protons prevented us from employing a direct deprotonation strategy for the macrocyclic ligand.18,21,23 Instead, we synthesized a silver complex to act as a transmetalation reagent, a strategy we previously employed with (Et,MeTCH)(PF6)4.22,23 Combining 5, AgPF6, and NEt3 in DMSO at 90 °C for 48 hours gave a brown solution. The product, [((S,S)-1,2-Ph2-Et,MeTCH)2Ag4](PF6)4 (6), was precipitated by addition of excess DI water to give a 45% yield. The initial analysis of the 1H NMR of the product (Fig. S23†) was highly perplexing until we realized that two separate conformers were formed. The conformers were separated by selective solvent extraction and confirmed by single crystal X-ray diffraction. The solid-state structure of the eclipsed conformer, designated as 6a, is shown in Fig. 1B, while the staggered conformer, 6b, where the macrocycles are rotated 90° versus each other is shown in Fig. S57.† Both conformers, 6a and 6b, retain the all-S chiral configuration.
The iron complex, [((S,S)-1,2-Ph2-Et,MeTCH)Fe(NCCH3)2](PF6)2 (7), was synthesized directly from 6 without the need to separate the conformers. Complex 6 and FeI2 were mixed in acetonitrile for 48 hours at 25 °C, which gave 7 in 95% yield after crystallization by vapor diffusion. Complex 7 is diamagnetic and the 1H NMR shows that it is rigid, giving diastereotopic protons on the methyl bridges at 6.24 and 5.79 ppm (Fig. S33†). The proton resonances off the stereogenic carbons in 7 are 6.88 and 6.79 ppm and are confirmed by HSQC (Fig. S35†). The X-ray crystal structure of 7 confirms the C2-symmetry of the iron complex (Fig. 1C). Two separate iron complexes were identified in the asymmetric unit, but both are structurally similar and have the same all-S chirality. Like similar iron complexes ligated to neutral NHC macrocycles, the Fe–C bond distances are between 1.92 and 2.00 Å, and acetonitrile ligands bind to the axial positions in the octahedral geometry.18,21,24,25
Since our group has been developing aziridination catalysis with aryl azides and aliphatic alkenes, we decided to focus on straight chain alkenes for initial tests as the resulting aziridines would have only a single chiral carbon (Scheme 2). Our initial test reaction with p-tolyl azide, 1-decene and 7 as the catalyst yielded 2-octyl-1-(p-tolyl)aziridine, 8, in 55% isolated yield after column chromatography on silica gel (Table 1, entry 1). Unlike epoxides, where there are many standard examples for determining effective chiral catalysts, there are so few examples for similar aziridines that we did not have a standard protocol for chiral separation of the enantiomers. We were able to separate the enantiomers consistently for the aziridines with ultra-high performance liquid chromatography (UHPLC) with a Phenomenex Lux 3 μm Cellulose-1 column with a mixture of acidified acetonitrile and water (see ESI for complete details†). Unfortunately, the UHPLC showed no enantiomeric excess for 8 (Fig. S43†). Substituting to a shorter alkene, 1-octene, had no significant difference in the result (Table 1, entry 2).
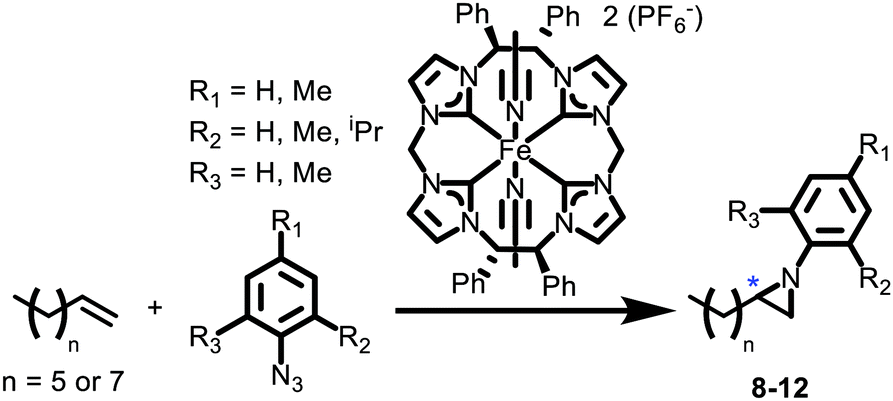 | ||
| Scheme 2 Catalytic aziridination reactions with 7. * Denotes chiral carbon. | ||
| Entrya | Aziridine | n | R1 | R2 | R3 | Isolated yield (%) | ee by UHPLCb (%) |
|---|---|---|---|---|---|---|---|
| a Between 50–70 mg of each azide was used in every reaction. Catalyst loading was 2% for each reaction. b A measured ee needed to be greater than 2% to be reported, even if integrations were not exactly equivalent. | |||||||
| 1 | 8 | 7 | Me | H | H | 55 | < 2 |
| 2 | 9 | 5 | Me | H | H | 65 | < 2 |
| 3 | 10 | 5 | H | Me | H | 64 | 3 |
| 4 | 11 | 5 | H | iPr | H | 15 | 4 |
| 5 | 12 | 5 | Me | Me | Me | — | — |
Switching to an ortho-substituted aryl azide, on the other hand, had significant impact on the catalytic reaction. A reaction with o-tolyl azide, 1-octene and 7 as the catalyst yielded 2-hexyl-1-(o-tolyl)aziridine, 10, in 64% isolated yield after column chromatography (Table 1, entry 3). Notably, UHPLC showed a clear, but small, differentiation between the enantiomers in this case with a 3% ee based on integration of the peaks (Fig. S50†). Increasing the steric bulk in the ortho position to isopropyl gave similar results for ee but with a reduced yield (Table 1, entry 4). Nevertheless, formation of 2-hexyl-1-(2-isopropylphenyl)aziridine, 11, confirmed that the enantioselective catalysis is compatible with multiple R-groups. At present, we have not been able to determine which enantiomer of the aziridines are favoured, since 10 and 11 are liquids and similar chiral aziridines have not been reported in the literature allowing for comparison by optical rotation.
Two additional organic azides were attempted which also showed the limitations of catalyst 7. First, a reaction with mesityl azide showed no reaction with the iron complex (Table 1, entry 5). Steric bulk in both ortho positions of the aryl ring shuts down catalysis. Second, complex 7 does not react with alkyl azides, such as octyl azide, which is consistent with our previous reports on catalysts supported by neutral NHC-macrocycles.18,19
To help elucidate these experimental results, we investigated some of these reaction profiles by DFT (see ESI for details†). In our previous publication on the mechanism of aziridination with iron catalysts, we noted that there are two key transition states.26 First, the addition of organic azide to iron complex forms an imide. Second, the addition of alkene to the iron imide gives a short-lived metalloradical intermediate, which subsequently quickly ring closes over a small energy barrier to form the aziridine. Adding steric bulk to form the C2-symmetric pocket about iron increases the activation free energy of the initial imide-forming step. For mesityl azide, the calculated free-energy barrier is 27.9 kcal mol−1, whereas for p-tolyl azide and o-tolyl azide, the free-energy barriers are 20.7 kcal mol−1 and 21.2 kcal mol−1, respectively. This result explains why we saw no reactivity with mesityl azide and 7 (Fig. S58 and Table S1†) and also explains the high temperatures required for the catalytic reaction.
More promisingly, DFT calculations do show a difference between the R and S enantiomers of the aziridine. The reaction profile for p-tolyl azide, 1-decene and 7 shows a ΔΔG‡ of 1.8 kcal mol−1 difference between the free energies of activation for formation of the R and S enantiomers of 8 (Fig. 2) during the ring closing step. The energy gap must increase sufficiently to lead to a measurable ee for ortho substituted aryl rings.
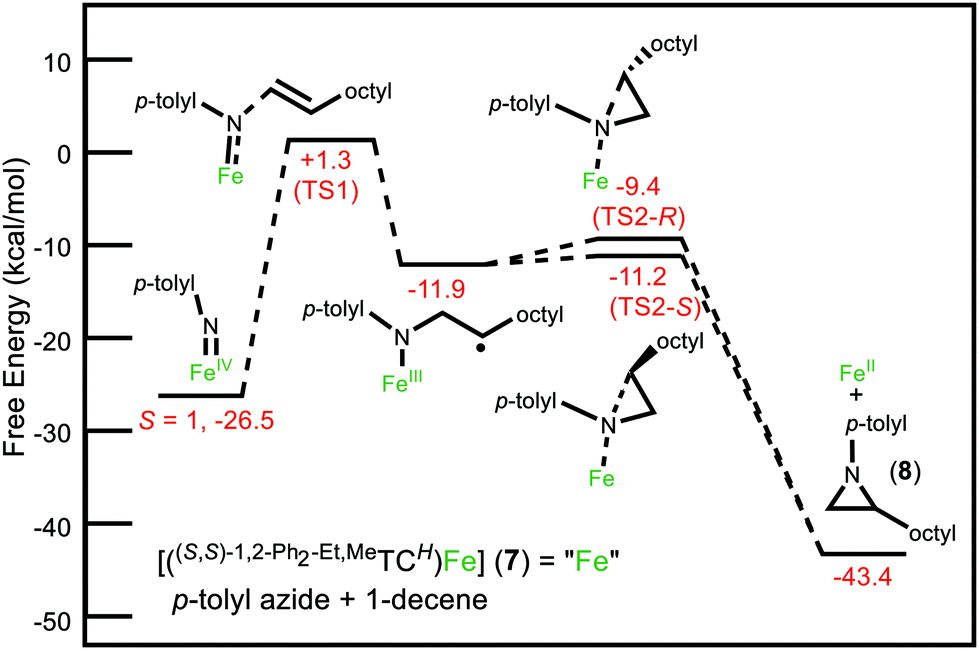 | ||
| Fig. 2 DFT-computed free-energy pathway for formation of aziridine from p-tolyl imide and 1-decene using catalyst 7. All species are in S = 1 (triplet) spin state, excluding the final aziridine product. TS designates a transition state. Free energies (ΔG) are given in kcal mol−1. | ||
A final consideration for developing improved catalysts for aziridination also includes considering the relatively high activation energies. While organic azides are excellent choices from an atom economy perspective,1 they can also form aziridines when heated in the presence of alkenes without a catalyst (often in low to modest yield).27,28 For example, 8 is formed in 32% yield without a catalyst (see ESI†). To improve the ee's of subsequent catalysts, it will be necessary to reduce the TS energies significantly so that little to no background reaction is occurring.
In conclusion, we have synthesized a new D2-symmetric tetra-NHC macrocycle and its subsequent silver and iron complexes. The absolute stereochemistry (S,S,S,S) of the macrocycle and complexes was confirmed by single crystal X-ray diffraction. The iron complex demonstrated the proof of principle that these C2-symmetric tetra-NHC complexes can favour one enantiomer over the other during the formation of aziridines from aryl azides and aliphatic alkenes, which, to our knowledge, has never been previously demonstrated. Finally, DFT calculations suggest that lowering the energy of the TS1 in Fig. 2 will be necessary to improve the ee's of the reactions by making the catalysis even more favourable than the background reaction, which is crucial for forming a single enantiomer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Xian Carroll for helpful discussions on X-ray crystallography. We thank Lindsay Brown from the Biological and Small Molecule Mass Spectrometry Core Facility for assistance on measurement of the MS of the aziridines. K.M.B. and D.M.J. are grateful to the National Institute of General Medical Sciences (R15GM117494-02) for financial support. C.J.M., S.R., and D.M.J. are grateful to the NIH (R15GM117494-01A) for financial support.Notes and references
- D. M. Jenkins, Synlett, 2012, 1267–1270 CrossRef CAS
.
- L.-M. Jin, X. Xu, H. Lu, X. Cui, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2013, 52, 5309–5313 CrossRef CAS PubMed
- K.-H. Chan, X. Guan, V. K.-Y. Lo and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 2982–2987 CrossRef CAS PubMed
- M. Ju and J. M. Schomaker, Nat. Rev. Chem., 2021, 5, 580–594 CrossRef CAS
- C. Damiano, S. Gadolini, D. Intrieri, L. Lay, C. Colombo and E. Gallo, Eur. J. Inorg. Chem., 2019, 2019, 4412–4420 CrossRef CAS
- H. Pellissier, Adv. Synth. Catal., 2014, 356, 1899–1935 CrossRef CAS
- L. Degennaro, P. Trinchera and R. Luisi, Chem. Rev., 2014, 114, 7881–7929 CrossRef CAS PubMed
- D. Tanner, Angew. Chem., Int. Ed. Engl., 1994, 33, 599–619 CrossRef
- R. Akhtar, S. A. R. Naqvi, A. F. Zahoor and S. Saleem, Mol. Diversity, 2018, 22, 447–501 CrossRef CAS PubMed
- V. Subbarayan, J. V. Ruppel, S. Zhu, J. A. Perman and X. P. Zhang, Chem. Commun., 2009, 4266–4268 RSC
- M. Ju, C. D. Weatherly, I. A. Guzei and J. M. Schomaker, Angew. Chem., Int. Ed., 2017, 56, 9944–9948 CrossRef CAS PubMed
- H. Nishikori and T. Katsuki, Tetrahedron Lett., 1996, 37, 9245–9248 CrossRef CAS
- K. Noda, N. Hosoya, R. Irie, Y. Ito and T. Katsuki, Synlett, 1993, 469–471 CrossRef CAS
- K. Omura, T. Uchida, R. Irie and T. Katsuki, Chem. Commun., 2004, 2060–2061 RSC
- T. J. Hodgkinson and M. Shipman, Tetrahedron, 2001, 57, 4467–4488 CrossRef CAS
- M. Kasai and M. Kono, Synlett, 1992, 778–790 CAS
- C. L. Keller, J. L. Kern, B. D. Terry, S. Roy and D. M. Jenkins, Chem. Commun., 2018, 54, 1429–1432 RSC
- S. A. Cramer and D. M. Jenkins, J. Am. Chem. Soc., 2011, 133, 19342–19345 CrossRef CAS PubMed
- P. P. Chandrachud, H. M. Bass and D. M. Jenkins, Organometallics, 2016, 35, 1652–1657 CrossRef CAS
- J. F. DeJesus and D. M. Jenkins, Chem. – Eur. J., 2020, 26, 1429–1435 CrossRef CAS PubMed
- M. R. Anneser, S. Haslinger, A. Pothig, M. Cokoja, J. M. Basset and F. E. Kuhn, Inorg. Chem., 2015, 54, 3797–3804 CrossRef CAS PubMed
- H. M. Bass, S. A. Cramer, J. L. Price and D. M. Jenkins, Organometallics, 2010, 29, 3235–3238 CrossRef CAS
- Z. Lu, S. A. Cramer and D. M. Jenkins, Chem. Sci., 2012, 3, 3081–3087 RSC
- A. A. Massie, C. Schremmer, I. Rüter, S. Dechert, I. Siewert and F. Meyer, ACS Catal., 2021, 11, 3257–3267 CrossRef CAS
- M. A. Bernd, F. Dyckhoff, B. J. Hofmann, A. D. Böth, J. F. Schlagintweit, J. Oberkofler, R. M. Reich and F. E. Kühn, J. Catal., 2020, 391, 548–561 CrossRef CAS
- S. B. Isbill, P. P. Chandrachud, J. L. Kern, D. M. Jenkins and S. Roy, ACS Catal., 2019, 9, 6223–6233 CrossRef CAS PubMed
- P. Scheiner, Tetrahedron, 1968, 24, 349–356 CrossRef
- J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247–258 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental details, NMR, IR, MS, and DFT calculations. CCDC 2152673–2152677. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00772j |
| This journal is © The Royal Society of Chemistry 2022 |