
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectroscopic investigation of the covalence of An(III) complexes with tetraethylcarboxamidopyridine†
Thomas
Sittel
 *ab,
Patrik
Weßling
ab,
Dennis
Großmann
b,
Eliane
Engels
b,
Andreas
Geist
a and
Petra J.
Panak
ab
*ab,
Patrik
Weßling
ab,
Dennis
Großmann
b,
Eliane
Engels
b,
Andreas
Geist
a and
Petra J.
Panak
ab
aKarlsruhe Institute of Technology (KIT), Institute for Nuclear Waste Disposal (INE), P.O. Box 3640, 76021 Karlsruhe, Germany. E-mail: thomas.sittel@kit.edu; Tel: +49 (0)721 60824652
bHeidelberg University, Institute for Physical Chemistry, Im Neuenheimer Feld 234, 69120 Heidelberg, Germany
First published on 25th April 2022
Abstract
In this work, we report a combined NMR spectroscopic and time-resolved laser fluorescence spectroscopic (TRLFS) study of the complexation of N,N,N′,N′-tetraethyl-2,6-carboxamidopyridine (Et-Pic) with Ln(III) (La, Sm, Eu, and Lu), Y(III) and An(III) (Am and Cm). The focal point of this study was the metal–ligand interaction in the [M(Et-Pic)3]3+ (M = An and Ln) complexes. The NMR analyses found slight differences between the An(III)–N and Ln(III)–N interactions in contrast to the similar properties of the Am(III)–O and Ln(III)–O interactions. These results were supported by TRLFS which shows that the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 Cm(III) complex is by one order of magnitude more stable than the respective Eu(III) complex. Thus, the ligand's selectivity lies in between those of pure N- and O-donor ligands. The selectivity results from a small partial covalent bonding between the An(III) ions and Et-Pic.
:3 Cm(III) complex is by one order of magnitude more stable than the respective Eu(III) complex. Thus, the ligand's selectivity lies in between those of pure N- and O-donor ligands. The selectivity results from a small partial covalent bonding between the An(III) ions and Et-Pic.
Introduction
Soft N-donor ligands such as bis(triazinyl)pyridine (BTP) or bis(triazinyl)bipyridine (BTBP) are known for their selectivity towards trivalent actinide ions, An(III), over their lanthanide counterparts, Ln(III).1,2 The molecular origin of this selectivity remains a topic of fundamental scientific interest. Generally speaking, the interaction of organic ligands with An(III) and Ln(III) is mainly driven by electrostatic forces. However, a slightly larger covalent contribution to the An(III)–N interaction compared to the Ln(III)–N interaction has been confirmed by NMR studies.3–5 As a result, An(III) forms stronger complexes with N-donor ligands than Ln(III).6–8 For example, the formation constant of the [Cm(BTP)3]3+ complex is approximately 2.5 orders of magnitude greater than that of the respective [Eu(BTP)3]3+.7,9 This is in conformity with the separation factor for Cm(III) over Eu(III), SFCm/Eu ≈ 230, as determined by solvent extraction.10 The selectivity for Am(III) over Eu(III) is SFAm/Eu ≈ 140.10In contrast to N-donor ligands, pure O-donor ligands do not differentiate between An(III) and Ln(III). In fact, their affinity for a given An(III) is similar to that of Ln(III) with a similar ionic radius.11,12 This is also the case for diglycolamides;13–17 the Am(III) and Cm(III) distribution ratios obtained by solvent extraction experiments fall within those of light Ln(III). For example, TODGA (N,N,N,N-tetraoctyl diglycolamide) extracts Am(III) and Cm(III) slightly less well than Eu(III), SFEu/Am ≈ 8 and SFEu/Cm ≈ 5. Accordingly, the stability constants of Cm(III) and Eu(III) complexes formed with such diglycolamides have values in the same order of magnitude.13
It is to be expected that N,O-donor ligands act intermediately. Indeed, picolindiamides show separation factors in the range of SFAm/Eu = 1.4–6.18,19 Well studied representatives of this ligand type are tetraethyl-2,6-dicarboxamidpyridine (Et-Pic),20–24N,N′-diethyl-N,N′-diphenyl-2,6-dicarboxamidpyridine (Et2–Ph2–Pic)21,25–27 and N,N′-diethyl-N,N′-bis(4-methylphenyl)-2,6-dicarboxamidpyridine (Et2–MePh2–Pic).19,28–30 Although their extraction properties are well understood, spectroscopic studies on the complexation of picolindiamides, including studies on bonding properties, are scarce.24 To further improve the knowledge of the bonding properties in Ln(III) and An(III) complexes, we present a first approach to study the bonding properties of Ln(III) (La, Sm, Eu, and Lu), Y(III) and An(III) (Am and Cm) complexes with the N,O-donor ligand Et-Pic (Fig. 1) using a combined approach of NMR spectroscopy and TRLFS.
 | ||
| Fig. 1 Structure of tetraethyl-2,6-dicarboxamidpyridine (Et-Pic). | ||
Experimental section
Methods and materials
All chemicals were used as purchased without further purification. Et-Pic was synthesized according to the literature procedure.31TRLFS experiments were performed at 298 K with a Nd:YAG (Surelite II laser, Continuum) pumped dye laser system (NarrowScan D-R; Radiant Dyes Laser Accessories GmbH). A wavelength of 394.0 nm (Eu(III)) or 396.6 nm (Cm(III)) was used to excite the metal ions. Spectral decomposition was performed with a spectrograph (Shamrock 303i, ANDOR) with 300, 1199 or 2400 lines per mm gratings. The fluorescence was detected by an ICCD Camera (iStar Gen III, ANDOR). A delay of 1 μs was used to discriminate short-lived organic fluorescence and light scattering.
NMR spectra were recorded using a Bruker Avance III 400 spectrometer operating at 400.13 MHz for 1H, 100.63 MHz for 13C and 40.58 MHz for 15N at 300 K. The spectrometer was equipped with a broadband observe probe (BBFOplus) with direct x-magnetization detection for proton and heteronuclear detection experiments. Chemical shifts are referenced internally to TMS (δ(TMS) = 0 ppm) for 1H and 13C and to 15NH4Cl with δ(15NH4Cl) = 0 ppm for 15N. For all spectra, standard Bruker pulse sequences were used. The 1D spectra of 1H and 13C were recorded with 32k data points and are zero filled to 64k data points. The 15N data at natural abundance were obtained from high resolution 1H,15N-HMQC spectra with a resolution of 4k data points in the indirect dimension. Signal multiplicity was determined as s (singlet), d (doublet), t (triplet), q (quartet), quin (quintet), sex (sextet), sept (septet), m (multiplet) and br. s (broad signal). Mass spectra using ESI ionization methods were recorded using a Bruker ApexQe FT-ICR instrument.
Sample preparation
TRLFS samples were prepared by adding 4.7 μL of a Cm(III) stock solution (2.12 × 10−5 mol L−1 Cm(ClO4)3 in 0.01 mol L−1 HClO4: 248Cm: 89.7%, 246Cm: 9.4%, 243Cm: 0.4%, 244Cm: 0.3%, 245Cm: 0.1%, 247Cm: 0.1%) or 9.4 μL of a Eu(III) stock solution (1.07 × 10−3 mol L−1 Eu(ClO4)3 in 0.01 mol L−1 HClO4) to 900 μL of acetonitrile and 95.3 μL (Cm(III)) or 90.6 μL (Eu(III)) H2O, respectively, resulting in initial concentrations of 10−7 mol L−1 Cm(III) or 10−5 mol L−1 Eu(III).
NMR sample preparation
Results and discussion
Complexation of Ln(III) and Am(III) with Et-Pic
:L 1:0.5 and Lu:L 1:1.5, the signals of a first complex species are observed. At Lu:L 1:1, a second complex species forms. The intensity share of this species increases significantly up to Lu:L 1:2.0. Increasing the ligand concentration further leads to the formation of a third complex species. This represents the only complex species at Lu:L 1:3 and beyond. At a surplus of ligand, the signals of the free ligand are observed, indicating that complexation is completed and that the inner coordination sphere of Lu(III) is saturated. In conclusion, Fig. 2 shows the stepwise formation of the 1:1, 1:2 and 1:3 complexes of Lu(III) with Et-Pic. The results are consistent with reports in the literature as well as with similar experiments conducted with Sm(III), La(III) and Y(III). ESI-MS fragmentation at a M:L ratio of 1:3 (M = La, Sm, Y, and Lu) further confirmed the formation of the 1:3 complexes for all these ions (see the ESI†). Besides the speciation, 1H NMR delivers information on the complex structure and insights into the bonding properties between the ligand and metal ion. Based on the weak shifts of the aromatic protons of the pyridine ring, the impact of complexation on the electron density distribution of the pyridine ring is similar for the 1:1, 1:2 and 1:3 complexes. In conclusion, the M–Npyr interaction is rather similar in all complex species. Thus, the M–Npyr bond length only changes slightly between the 1:1 and 1:3 complexes. In contrast to the aromatic proton signals, the NMR signals of the CH2 groups change significantly with speciation. This is caused by the restriction of the free rotation of the ethyl moieties with an increasing number of coordinated ligands. As a result, the protons of each CH2 group become diastereotopic which leads to signal splitting. This can be clearly seen in the 1:3 and, to some extent, in the 1:2 complexes.
 | ||
| Fig. 2 1H NMR spectra (400.18 MHz, 300 K) of the Lu(III)–Et-Pic complexes ([Lu] = 17 mmol L−1) depending on the Et-Pic(L)/metal ion ratio in CD3CN (* δ(H2O)). | ||
:3 complexes of Am(NO3)3, La(III), Lu(III), Sm(III) and Y(III) (as OTf− salts) were prepared and characterized by 1D and 2D NMR methods.
The general strategy is to derive chemical shift trends from the 13C and 15N NMR data in order to abstract conclusions about the metal–ligand interactions. For the M(III)–Npyr interaction, this seems to be rather simple by gathering the 15N shift of the coordinated nitrogen atom. In addition, carbon NMR shifts provide further insights into the M(III)–Npyr interaction due to the conjugated nature of the aromatic pyridine ring.
In comparison to the M(III)–Npyr interaction, probing the Ln(III)–O or An(III)–O interaction by NMR is rather difficult as the NMR active nucleus 17O (I = 5/2) has a low natural abundance of 0.04%. However, using the electronic properties of the amide group provides an indirect approach to probe the M(III)–O interaction. The amide group consists of a conjugated system that involves the C–O double bond and the lone pair of electrons of the neighboring nitrogen atom. Due to this conjugation, changes in the electron density distribution at the oxygen will also affect the amide carbon and nitrogen effectively. Thus, essential insights into the bonding properties between oxygen and metal ion can be derived from the 13C and 15N NMR shifts.
For C-1 no significant disparities in the chemical shift are observed between all ions. The chemical shift δ(C-1) varies between 141.2 ppm and 142.3 ppm. Thus, all complexes show a small downfield shift by +3.0–4.1 ppm in comparison to the free ligand. In the case of the Ln(III) complexes, C-2 is also deshielded which leads to 13C shifts up to +5.1 ppm. On the contrary, C-2 is shifted slightly upfield of about −1.0 ppm in the Am(III) complex and therefore is additionally shielded compared to the free ligand. For C-3, all complexes show upfield shifts. However, this shift is less pronounced in the Am(III) complex compared to the Ln(III) complexes (−0.9 ppm vs. −3.4 to −6.4 ppm).
All in all, the chemical shifts of the aromatic carbon atoms show slightly different trends between Am(III) and Ln(III). This indicates that the Am(III)–Npyr and the Ln(III)–Npyr bonds do not share the same properties. This is further proved by comparing our observed trends to the already reported NMR data of [Am(nPr-BTP)3]3+/[Ln(nPr-BTP)3]3+ by Adam et al.5 The observed trends in the present work, especially for the Ln(III) ions, are quite consistent with the literature data (for comparison, see the ESI†). This indicates clearly that the bonding properties of Am(III)–Npyr and Ln(III)–Npyr are dissimilar. Due to the nature of the amide bond, the carbon shifts of C-4 provide a well-suited tool to investigate the M(III)–O bonding properties. Overall, all C-4 carbon atoms show a downfield shift compared to the free ligand. The smallest shifts of about +0.4 ppm to 1.3 ppm are observed for the diamagnetic ions La(III), Lu(III) and Y(III). The weak paramagnetic ions, Am(III) and Sm(III), show the strongest influence on δ(C-4) with shifts of +4.5 ppm and 3.2 ppm, respectively. Overall, δ(C-4) shows similar trends across all the metal ions studied. All signals show a downfield shift, with Am(III) and Sm(III) being shifted the most. This indicates the similar bonding properties of the Ln(III)–O and Am(III)–O bonds.
In contrast to Npyr, the NMR shifts of the amide nitrogen atom Namide were collected for all complexes, including Am(III) and Sm(III) (see Table 2). Intriguingly, the data for Ln(III) and Am(III) differ slightly. The Ln(III) complexes show 15N shifts between 13–16 ppm, independent of the ionic radius or paramagnetic properties. The observed shifts suggest a stronger Ln(III)–O interaction compared to the Ln(III)–Npyr interaction. This is in line with the expectation that hard Ln(III) ions prefer the coordination of a hard donor atom such as oxygen. In the case of the Am(III) complex, the Namide atom shows a downfield shift of 8 ppm. Considering the consistency of the chemical shift of Namide in the Ln(III) complexes, the Am(III)–O interaction seems to differ slightly. However, the differences are too small to project different bonding modes for Am(III)–O and Ln(III)–O.
Overall, the NMR data suggest similar bonding mechanisms in Ln(III)–O and Am(III)–O—mainly driven by electrostatic interaction—and slight differences in the Am(III)–Npyr and Ln(III)–Npyr interactions. For the latter, a higher fraction of covalence in the Am(III)–N bond is expected which is in accordance with previous NMR studies on similar N-donor complexes.
Complexation of Cm(III) and Eu(III) with Et-Pic
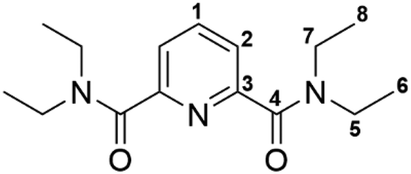
 | ||
| Fig. 3 Normalized Cm(III) emission spectra for the complexation of Cm(III) with Et-Pic in acetonitrile containing 10 vol% H2O as a function of Et-Pic concentration. [Cm(III)]ini = 10−7 mol L−1. | ||
In the absence of Et-Pic the Cm(III) emission spectrum is characterized by an emission band at 594.6 nm. The emission band is slightly shifted compared to the Cm(III) emission spectrum in water (λmax = 593.8 nm)32 due to the replacement of water molecules in the first coordination sphere of Cm(III) by acetonitrile. The lifetime of the solvent species is τ = 71 + 4 μs (cf. Fig. S21†). Both emission spectrum and fluorescence lifetime are in good agreement with the literature.33
The addition of Et-Pic results in a bathochromic shift of the Cm(III) emission band with respect to the solvent species. This is due to the increased ligand field splitting of the 6D′7/2 state upon complexation of Cm(III) with Et-Pic. With increasing Et-Pic concentration, three new emission bands at 600.0 nm, 606.3 nm and 611.7 nm can be observed. These emission bands are attributed to the [Cm(Et-Pic)n]3+ complexes (n = 1–3). For ligand concentrations greater than 0.201 mol L−1 Et-Pic no further changes in the Cm(III) emission spectrum are detected. The fluorescence lifetime at this Et-Pic concentration is τ = 559 ± 28 μs (cf. Fig. S.2†). Using the Kimura equation,32 this corresponds to 0.3 ± 0.5 molecules of water in the first coordination sphere suggesting the formation of the 1:3 complex. The emission bands at 600.0 nm and 606.3 nm are assigned to the 1:1 and 1:2 complexes accordingly.
The single component spectra for the 1:1, 1:2, 1:3 complexes and the Cm(III) solvent species are shown in Fig. 4. Peak deconvolution is employed to derive the area of each species contributing to an experimental spectrum. An example is given in the ESI (Fig. S20†). As can be seen from Fig. S23† the total fluorescence intensity (normalized to the intensity of the solvent species) increases upon the addition of Et-Pic. This is described by the fluorescence intensity factor (FI). The fluorescence intensity factor denotes the intensity of a given species in relation to the solvent species. The FI factors are used to convert the areas determined by peak deconvolution into molecular fractions (χi). χi as a function of the free ligand concentration, which is calculated according to eqn (1), is plotted in Fig. 5.
| [Et-Pic]free = [Et-Pic]ini − [Cm(III)]·(χ1 + 2χ2 + 3χ3) | (1) |
 | ||
| Fig. 4 Single component spectra of the Cm(III) solvent species and the [Cm(Et-Pic)n]3+ complexes (n = 1–3). | ||
 | ||
| Fig. 5 Relative fractions of the Cm(III) solvent species and the [Cm(Et-Pic)n]3+ complexes (n = 1–3) as a function of free Et-Pic concentration. Dots denote the experimental data. Lines are calculated using logβ1 = 4.3, logβ2 = 7.2 and logβ3 = 7.6. | ||
As can be seen in Fig. 5, the complexation of Cm(III) with Et-Pic starts at concentrations higher than 10−6 mol L−1. The 1:2 and 1:3 complexes start forming at concentrations of 3 × 10−5 mol L−1 and 10−3 mol L−1 Et-Pic.
To verify the complex stoichiometry, slope analyses are performed on the basis of a consecutive complexation model (eqn (2)).
| [Cm(Et-Pic)n−1]3+ + Et-Pic ⇌ [Cm(Et-Pic)n]3+ | (2) |
Applying the mass action law gives
 | (3) |
The logarithmic form of eqn (3) is used for the slope analyses (Fig. 6).
 | (4) |
 | ||
| Fig. 6 Double logarithmic plot of the concentration ratios of [Cm(Et-Pic)n]3+/[Cm(Et-Pic)n−1]3+ (n = 1–3) as a function of free Et-Pic concentration. | ||
The derived slopes of m1 = 0.93 ± 0.03, m2 = 1.11 ± 0.03 and m3 = 0.94 ± 0.05 verify the single component spectra. In the following, stability constants are calculated according to eqn (5).
 | (5) |
The stability constants for the complexation of Cm(III) with Et-Pic in acetonitrile containing 10 vol% H2O are logβ1′ = 4.3 ± 0.3, logβ2′ = 7.2 ± 0.3 and logβ3′ = 7.6 ± 0.4.
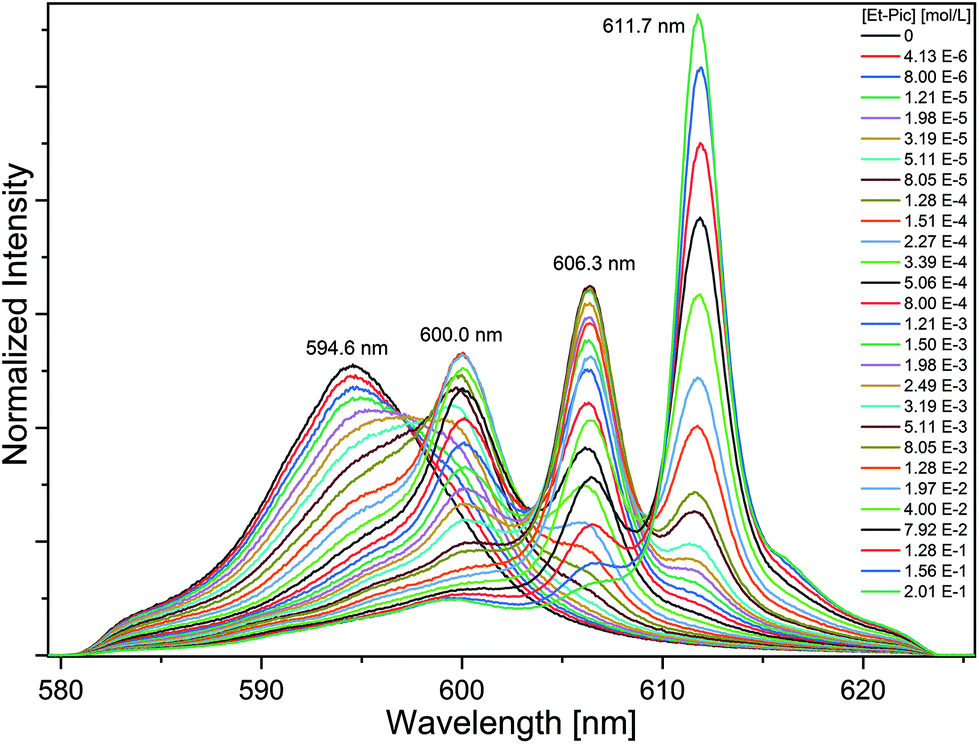
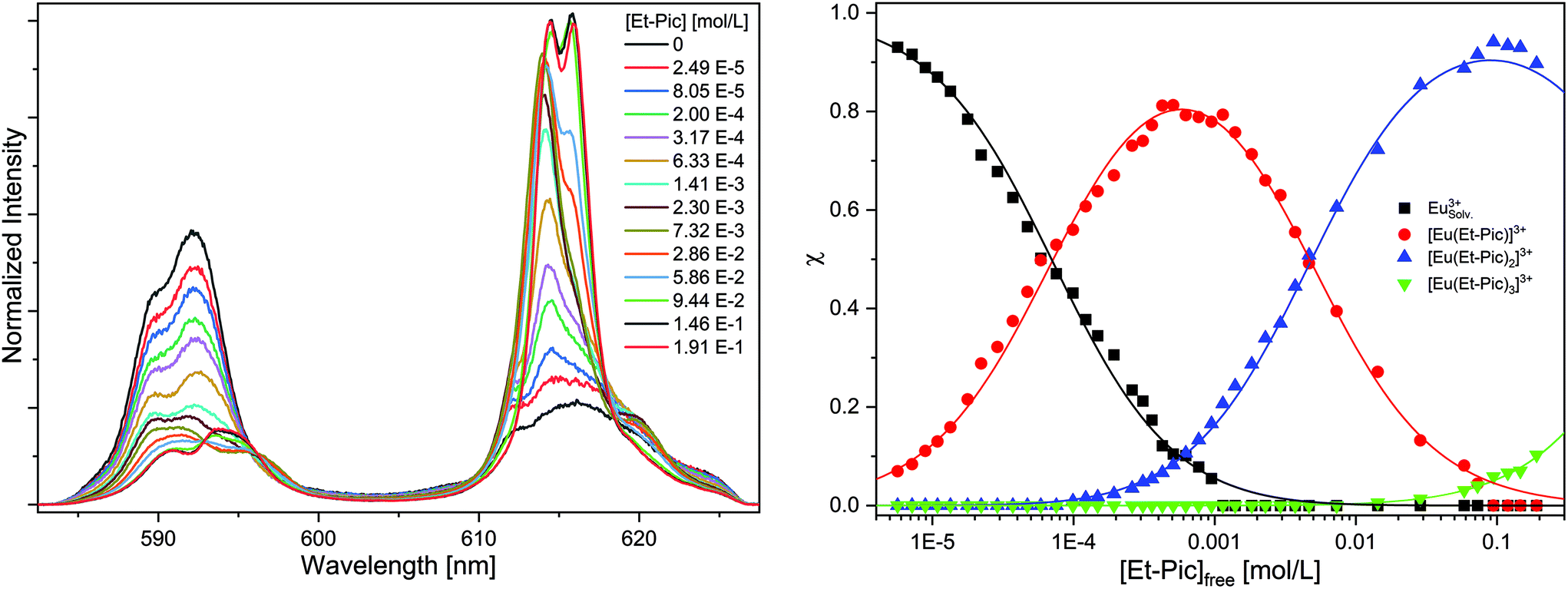 | ||
| Fig. 7 Left: Normalized Eu(III) emission spectra resulting from the 5D0 → 7Fn (n = 1, 2) transition for the complexation of Eu(III) with Et-Pic in acetonitrile containing 10 vol% H2O as a function of Et-Pic concentration. [Eu(III)ini = 10−5 mol L−1]. Right: Relative fractions of the Eu(III) solvent species and the [Eu(Et-Pic)n]3+ complexes (n = 1–3) as a function of free Et-Pic concentration. Dots denote experimental data. Lines are calculated using logβ1 = 4.2, logβ2 = 6.5 and logβ3 = 6.3. | ||
Without the addition of Et-Pic, the Eu(III) spectrum in acetonitrile containing 10 vol% of H2O is characterized by an intensive 7F1 emission band at 592.2 nm and a shoulder band at 589.7 nm. In comparison, the 7F2 emission band (λmax = 616.0 nm) is of weak intensity (intensity(7F2)/intensity(7F1) = 0.65). The fluorescence lifetime of the Eu(III) solvent species is τ = 116 ± 6 μs (cf. Fig. S.1†). Upon the addition of Et-Pic, the 7F2 emission band gains intensity. With increasing Et-Pic concentration, an emission band at 614.5 nm, and, with further progress, an emission band at 615.9 nm appears. The spectrum at the highest Et-Pic concentration is characterized by double split 7F1 (λmax = 590.7 nm and 593.9 nm) and 7F2 emission bands (λmax = 614.5 nm and 615.9 nm). The ratio of the intensity of the 7F2 and 7F1 emission bands is 4.05 indicating the formation of the 1:3 complex. The fluorescence lifetime is τ = 1740 ± 87 μs (cf. Fig. S22†) which corresponds to zero water molecules in the first coordination sphere of Eu(III).
Peak deconvolution is performed to determine the speciation which is shown in Fig. 7 (right). The single component spectra of the [Eu(Et-Pic)n]3+ complexes, an exemplary peak deconvolution, the evolution of the fluorescence intensity and slope analyses are given in the ESI in Fig. S24–S27.†
The formation of the 1:1 complex starts at Et-Pic concentrations greater 10−5 mol L−1 while the 1:2 and 1:3 complexes are observed at concentrations greater than 10−4 mol L−1 and 3 × 10−2 mol L−1. Using eqn (5) the stability constants for the [Eu(Et-Pic)n]3+ complexes (n = 1–3) are logβ1′ = 4.2 ± 0.3, logβ2′ = 6.5 ± 0.3 and logβ3′ = 6.3 ± 0.3.
Nephelauxetic effect of the 7F0 shift on Eu(III)–Et-Pic complexes
Although 4f elements are mainly known for their electrostatic interactions, the interaction between Ln(III) and the Et-Pic ligand is also covalent to a certain degree. A partially covalent bonding character of the Eu(III)–ligand bond has been reported in the literature for different O-34,35 and N-donor ligands36 using the shift of the 5D0 → 7F0 transition. Since the total angular momentum of both energy states is zero, no ligand field splitting is observed, resulting in only one emission band for each species present in the system. The position of the 7F0 band depends directly on the Eu(III) ligand field and complexation induces a bathochromic shift. The shift is due to the electronic changes caused by the Eu(III)–ligand bond. This shift—called the nephelauxetic effect—has been used in the literature to measure covalence in an Eu(III)–ligand bond.35–37Choppin et al. studied a variety of different O-donor ligands and found an empirical correlation between the shift Δν of the 7F0 emission band and the number of coordinated donor atoms CNL (eqn (6)).38 The shift Δν is given in wavenumbers and with respect to the corresponding Eu(III) solvent complex.
| CNL = 0.237·Δν + 0.628 | (6) |
| Δν = ν(Eu(solv.)) − ν(Eu(complex)) in [cm−1] |
Wagner et al. investigated the complexation of Eu(III) with different softer N-donor ligands which exhibit a significantly higher covalence in the Eu(III)–ligand bond than in complexes with hard O-donors.36 Since the nephelauxetic effect is closely related to the covalence of the Eu–ligand bond, stronger shifts of the 7F0 emission band were observed. Consequently, eqn (6) overestimates the number of donor atoms. Therefore, a new linear empirical correlation between the shift Δν of the 7F0 emission band and the number of coordinated donor atoms CNL had to be established (eqn (7)).
| CNL = 0.109·Δν + 0.134 | (7) |
To study the covalence in Eu(III) complexes with the mixed N,O donor ligand Et-Pic, the Eu(III) emission spectra of the 5D0 → 7F0 transition are recorded in acetonitrile containing 10 vol% H2O as a function of the Et-Pic concentration (Fig. 8).
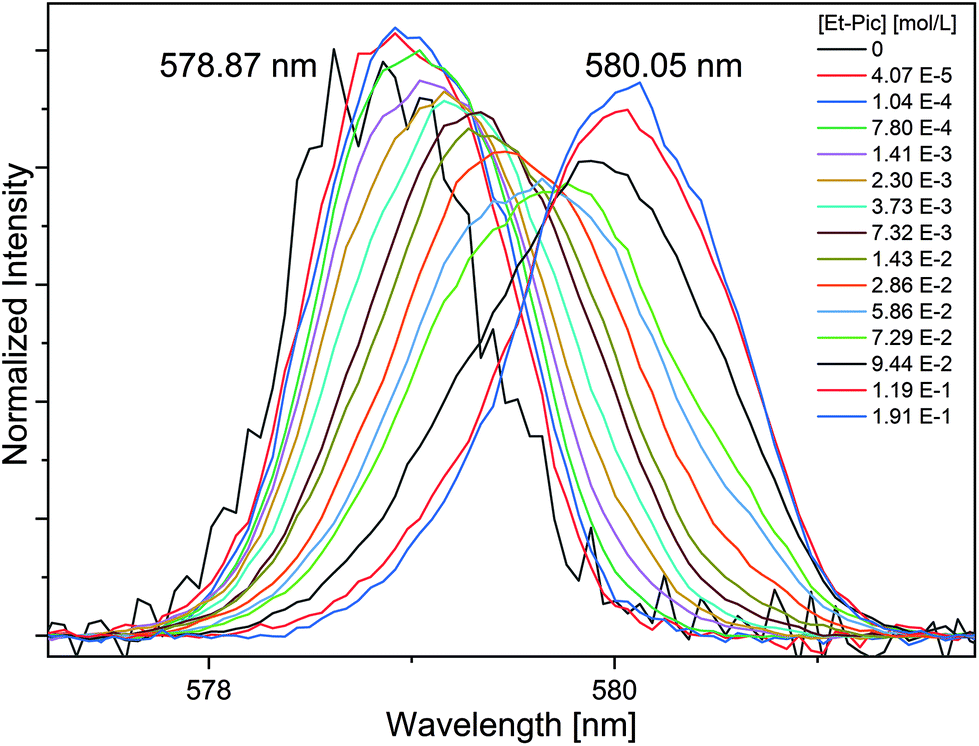 | ||
| Fig. 8 Normalized Eu(III) emission spectra resulting from the 5D0 → 7F0 transition for the complexation of Eu(III) with Et-Pic in acetonitrile containing 10 vol% H2O as a function of the Et-Pic concentration. [Eu(III)]ini = 10−5 mol L−1. | ||
The 7F0 emission band of Eu(III) dissolved in acetonitrile containing 10 vol% H2O is observed at 578.87 nm. The shift is similar to that of Eu(III) dissolved in water (578.8 nm).38 Upon the addition of Et-Pic, a bathochromic shift is observed and at the highest ligand concentration a symmetrical emission band at 580.05 nm is detected. Under these conditions, 90% of [Eu(Et-Pic)2]3+ and 10% [Eu(Et-Pic)3]3+ are present in the system. The 1:3 complex is a highly symmetric species with D3 symmetry. Thus, the intensity of the 5D0 → 7F0 transition for this species strongly decreases as the transition is prohibited due to its inversion symmetry. Therefore, the observed emission band only corresponds to the 1:2 complex with six donor atoms. According to eqn (6) or eqn (7), six donor atoms would induce a shift of Δν = 22.7 cm−1 and Δν = 53.8 cm−1, respectively. The shift of the [Eu(Et-Pic)2]3+ complex is Δν = 35.1 cm−1 and lies between the expected values from eqn (6) and (7). Consequently, the covalence of the Eu(III) complex with the mixed N,O-donor is greater than with pure O-donor and smaller than with pure N-donor ligands, which is in good agreement with our NMR and TRLFS findings.
Comparison and discussion of the stability constants of the Cm(III) and Eu(III) Et-Pic complexes
Table 3 gives an overview of the stability constants of the 1:3 complexes of Cm(III) and Eu(III) with TODGA,39 a pure O-donor ligand, nPr-BTP,8 a pure N-donor ligand and Et-Pic, a mixed N,O-donor ligand. Although the stability constants determined by TRLFS cannot be compared directly as they were measured in different solvents, important trends can be derived.
β and separation factors (determined by TRLFS SFTRLFSCm/Eu or solvent extraction SFExtr.Cm/Eu) of the 1:3 Cm(III) and Eu(III) complexes with the O-donor ligand TODGA, the N-donor ligand nPr-BTP and the mixed N,O-donor ligand Et-Pic
The O-donor ligand TODGA forms stronger complexes with Eu(III) (Δlogβ3,Eu–Cm = 0.8) while the N-donor ligand nPr-BTP forms stronger complexes with Cm(III) (Δlogβ3,Cm–Eu = 2.5). In accordance with the theory of hard and soft acids and bases (HSAB principle),41 trivalent actinides have a higher affinity towards soft donor ligands compared to trivalent lanthanide ions. The mixed N,O-donor ligand Et-Pic forms slightly stronger complexes with Cm(III) compared to Eu(III) (Δlogβ3,Cm–Eu = 1.3). The selectivity of Et-Pic lies between that of TODGA and nPr-BTP. Our NMR study reveals no significant differences in the Ln(III)–O and Am(III)–O-bond. Therefore, the enhanced selectivity of Et-Pic is due to the higher covalent bond character in the Cm(III)–N-bond, which is consistent with the 13C shifts of the pyridine rings in the NMR experiments.
Moreover, Table 3 compares separation factors determined by TRLFS (SFTRLFSCm/Eu) and solvent extraction (SFExtr.Cm/Eu). TODGA exhibits a small selectivity for Eu(III) over Cm(III). Separation factors determined by TRLFS and solvent extraction are consistent. In contrast, nPr-BTP is a highly selective ligand designed for the separation of An(III) from Ln(III) which reflects the separation factors of SFTRLFSCm/Eu = 3008 and SFExtr.Cm/Eu = 230.10 In the case of Et-Pic, the separation factor determined by TRFLS is SFTRLFSCm/Eu = 20. Extraction studies with similar picolindiamides (Bu–Pic and Et2–MePh2–Pic) report separation factors between 1.4–6 for the separation of Am(III) from Eu(III).18,19 The separation factor for Et-Pic is in the same range as the separation factors for structurally similar picolindiamides. This illustrates that the selectivity of the N,O donor ligand lies in between that of pure O- and N-donor ligands.
Conclusions
Using a combined approach of NMR spectroscopy and TRLFS, this study provides novel insights into the M(III)–N and M(III)–O interaction in [M(Et-Pic)3]3+ complexes (M = An and Ln). The carbon and nitrogen NMR data confirm the assumption of an almost identical metal–ligand interaction between Am(III)–O and Ln(III)–O. In contrast, the NMR data suggest different bonding modes for the Am(III)–Npyr and Ln(III)–Npyr interactions. The acquired NMR data are consistent with the TRLFS data presented in this work. The TRLFS results show that Cm(III) forms a stronger 1:3 complex by one order of magnitude compared to the respective Eu(III) complex. Therefore, Et-Pic exhibits a minor selectivity towards An(III) ions which matches the known extraction data of picolindiamides given in the literature. The data suggest that the observed selectivity results from an increased partially covalent interaction in the An(III)–N bond.
Although an increased knowledge of the M(III)–ligand interaction in f-element complexes has been achieved in recent years by using different spectroscopic techniques, many questions are yet to be answered. In this respect, the present study serves as a valuable reference for future systematic studies of metal–ligand interactions in lanthanide/actinide complexes using NMR spectroscopy and TRLFS.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the German Federal Ministry for Research and Education (grant agreement no. 02NUK059A and 02NUK059C).Notes and references
- A. Geist and P. J. Panak, Solvent Extr. Ion Exch., 2021, 39, 128–151 CrossRef CAS.
- P. J. Panak and A. Geist, Chem. Rev., 2013, 113, 1199–1236 CrossRef CAS PubMed.
- C. Adam, V. Rohde, U. Müllich, P. Kaden, A. Geist, P. J. Panak and H. Geckeis, Procedia Chem., 2016, 21, 38–45 CrossRef.
- C. Adam, B. B. Beele, A. Geist, U. Müllich, P. Kaden and P. J. Panak, Chem. Sci., 2015, 6, 1548–1561 RSC.
- C. Adam, P. Kaden, B. B. Beele, U. Müllich, S. Trumm, A. Geist, P. J. Panak and M. A. Denecke, Dalton Trans., 2013, 42, 14068–14074 RSC.
- S. Trumm, G. Lieser, M. R. S. Foreman, P. J. Panak, A. Geist and T. Fanghanel, Dalton Trans., 2010, 39, 923–929 RSC.
- S. Trumm, P. J. Panak, A. Geist and T. Fanghänel, Eur. J. Inorg. Chem., 2010, 2010, 3022–3028 CrossRef.
- S. Trumm, G. Lieser, M. R. Foreman, P. J. Panak, A. Geist and T. Fanghanel, Dalton Trans., 2010, 39, 923–929 RSC.
- A. Bremer, U. Müllich, A. Geist and P. J. Panak, New J. Chem., 2015, 39, 1330–1338 RSC.
- N. L. Banik, M. A. Denecke, A. Geist, G. Modolo, P. J. Panak and J. Rothe, Inorg. Chem. Commun., 2013, 29, 172–174 CrossRef CAS.
- D. F. Peppard, G. W. Mason, J. L. Maier and W. J. Driscoll, J. Inorg. Nucl. Chem., 1957, 4, 334–343 CrossRef CAS.
- A. Geist, L. Berthon, M.-C. Charbonnel and U. Müllich, Solvent Extr. Ion Exch., 2020, 38, 681–702 CrossRef CAS.
- A. Wilden, G. Modolo, S. Lange, F. Sadowski, B. B. Beele, A. Skerencak-Frech, P. J. Panak, M. Iqbal, W. Verboom, A. Geist and D. Bosbach, Solvent Extr. Ion Exch., 2014, 32, 119–137 CrossRef CAS.
- S. A. Ansari, P. Pathak, P. K. Mohapatra and V. K. Manchanda, Chem. Rev., 2012, 112, 1751–1772 CrossRef CAS PubMed.
- D. Whittaker, A. Geist, G. Modolo, R. Taylor, M. Sarsfield and A. Wilden, Solvent Extr. Ion Exch., 2018, 36, 223–256 CrossRef CAS.
- Y. Sasaki, Y. Sugo, S. Suzuki and S. Tachimori, Solvent Extr. Ion Exch., 2001, 19, 91–103 CrossRef CAS.
- D. Magnusson, A. Geist, R. Malmbeck, G. Modolo and A. Wilden, Procedia Chem., 2012, 7, 245–250 CrossRef CAS.
- L. Nigond, C. Musikas and C. Cuillerdier, Solvent Extr. Ion Exch., 1994, 12, 261–296 CrossRef CAS.
- A. Paulenova, M. Y. Alyapyshev, V. A. Babain, R. S. Herbst and J. D. Law, Sep. Sci. Technol., 2008, 43, 2606–2618 CrossRef CAS.
- E. Makrlík, P. Vaňura, P. Selucký, V. A. Babain and I. V. Smirnov, J. Radioanal. Nucl. Chem., 2010, 284, 629–633 CrossRef.
- M. Y. Alyapyshev, V. A. Babain and I. V. Smirnov, Radiochemistry, 2004, 46, 270–271 CrossRef CAS.
- E. Makrlík, P. Vaňura, P. Selucký, V. A. Babain and I. V. Smirnov, J. Radioanal. Nucl. Chem., 2009, 279, 743–747 CrossRef.
- V. N. Romanovskiy, V. A. Babain, M. Y. Alyapyshev, I. V. Smirnov, R. S. Herbst, J. D. Law and T. A. Todd, Sep. Sci. Technol., 2006, 41, 2111–2127 CrossRef CAS.
- F. Renaud, C. Piguet, G. Bernardinelli, J.-C. G. Bünzli and G. Hopfgartner, Chem. – Eur. J., 1997, 3, 1646–1659 CrossRef CAS.
- V. A. Babain, M. Y. Alyapyshev and R. N. Kiseleva, Radiochim. Acta, 2007, 95, 217–223 CrossRef CAS.
- E. Makrlík, P. Vaňura, P. Selucký, V. A. Babain and I. V. Smirnov, J. Radioanal. Nucl. Chem., 2010, 283, 839–844 CrossRef.
- Y. A. Ustynyuk, I. P. Gloriozov, S. N. Kalmykov, A. A. Mitrofanov, V. A. Babain, M. Y. Alyapyshev and N. A. Ustynyuk, Solvent Extr. Ion Exch., 2014, 32, 508–528 CrossRef CAS.
- M. Bubeníková, J. Rais, P. Selucký and J. Kvíčala, Radiochim. Acta, 2013, 101, 753–759 CrossRef.
- M. Y. Alyapyshev, V. A. Babain, L. I. Tkachenko, I. I. Eliseev, A. V. Didenko and M. L. Petrov, Solvent Extr. Ion Exch., 2011, 29, 619–636 CrossRef CAS.
- J. L. Lapka, A. Paulenova, M. Y. Alyapyshev, V. A. Babain, R. S. Herbst and J. D. Law, Radiochim. Acta, 2009, 97, 291–296 CAS.
- P. V. Ivchenko, I. E. Nifant'ev and I. V. Buslov, Tetrahedron Lett., 2013, 54, 217–219 CrossRef CAS.
- T. Kimura and G. R. Choppin, J. Alloys Compd., 1994, 213, 313–317 CrossRef.
- S. Trumm, G. Lieser and P. J. Panak, Radiochim. Acta, 2011, 99, 783–790 CrossRef CAS.
- O. L. Malta, H. J. Batista and L. D. Carlos, Chem. Phys., 2002, 282, 21–30 CrossRef CAS.
- L. D. Carlos, O. L. Malta and R. Q. Albuquerque, Chem. Phys. Lett., 2005, 415, 238–242 CrossRef CAS.
- C. Wagner, C. Ruff, U. Müllich, A. Geist and P. J. Panak, New J. Chem., 2016, 40, 9232–9237 RSC.
- M. Latva and J. Kankare, J. Coord. Chem., 1998, 43, 121–142 CrossRef CAS.
- G. R. Choppin and Z. M. Wang, Inorg. Chem., 1997, 36, 249–252 CrossRef CAS.
- A. Wilden, G. Modolo, S. Lange, F. Sadowski, B. B. Beele, A. Skerencak-Frech, P. J. Panak, M. Iqbal, W. Verboom, A. Geist and D. Bosbach, Solvent Extr. Ion Exch., 2014, 32, 119–137 CrossRef CAS.
- A. Geist, U. Mullich, D. Magnusson, P. Kaden, G. Modolo, A. Wilden and T. Zevaco, Solvent Extr. Ion Exch., 2012, 30, 433–444 CrossRef CAS.
- R. G. Pearson, Inorg. Chim. Acta, 1995, 240, 93–98 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data and spectra, ESI-MS data and additional TRLFS data. See DOI: https://doi.org/10.1039/d2dt00757f |
| This journal is © The Royal Society of Chemistry 2022 |