
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Use of heterometallic alkali metal–magnesium aryloxides in ring-opening polymerization of cyclic esters†
Rafał
Petrus
 *a,
Tadeusz
Lis
b and
Adrian
Kowaliński
a
*a,
Tadeusz
Lis
b and
Adrian
Kowaliński
a
aFaculty of Chemistry, Wrocław University of Science and Technology, 23 Smoluchowskiego, 50-370 Wrocław, Poland. E-mail: rafal.petrus@pwr.edu.pl; Tel: +48 71 320 32 08
bFaculty of Chemistry, University of Wrocław, 14 F. Joliot-Curie, 50-383 Wrocław, Poland
First published on 30th May 2022
Abstract
In this work, alkali metal–magnesium aryloxides [Mg2Li2(MesalO)6] (1), [Mg2Na2(MesalO)6(THF)x] for x = 2 or 4 (2), and [Mg2K2(MesalO)6(THF)4] (3) derived from the reaction of MgnBu2 and nBuLi, metallic Na or K with methyl salicylate (MesalOH) were used as molecular platforms for the synthesis of new heterometallic compounds [Mg2Li2(EtsalO)6] (4), [MgK(EtsalO)3]n (5), [Mg6Na4Al(MesalO)13(OH)6(MesalOH)(THF)0.5(H2O)0.5] (6), and [Mg4Na2(MesalO)6(SalO)2(THF)4] (7) (EtsalOH = ethyl salicylate and SalOH2 = salicylic acid) by the reaction with EtOH, exposure to atmospheric moisture or addition of stoichiometric quantities of water. Compounds 4 and 5 were synthesized by transesterification of 1 and 3. Cluster 6 was formed haphazardly by exposing a THF solution of 2 derived using MgnBu2 stabilized with 1 wt% AlEt3 to atmospheric moisture. Compound 7 was synthesized by partial hydrolysis of 2. Homometallic magnesium aryloxide [Mg4(MesalO)4(OMe)4(HOMe)4] (8) was obtained by reaction of MgnBu2 and MesalOH in a methanol solution. The catalytic activity of 1–3 and 6–8 was investigated in the ring-opening polymerization (ROP) of L-lactide (L-LA) or benzaldehyde Tishchenko reaction.
Introduction
Over the last 40 years, s-block metal reagents have attracted special attention in sustainable synthesis, polymerization, and a wide range of organic transformations. The mixed-metal organometallic compounds of the 1 and 2 groups were particularly important.1 These reagents are powerful bases for the deprotonative metallation of organic substrates, metal halogen exchange, or C–C bond formation. Combining a group 1 metal center with a less electropositive s-block metal center such as Mg provides reactivity enhancement and better chemo- and regioselectivities than monometallic bases.2,3 These heterobimetallic systems displayed high reactivity associated with group 1 compounds as well as the regioselective control and functional group tolerance characteristic of magnesium derivatives. Synergic behavior arising from the chemical cooperativity of two different metal species allows transformations to be performed that would be unachievable using conventional monometallic reagents. Most reported systems are based on alkyl/aryl,4,5 or amido6 alkali metal magnesiates, which were applicable for magnesium–halogen exchange of aryl substrates,7 enantioselective alkylation of aldehydes,8 cyclization of alkynols,9 transfer hydrogenation of alkenes,10 or hydroamination of alkynes and alkenes.11 In organic synthesis, very important synergistic heterobimetallic reagents were alkyl or amido alkali metal–magnesium halides, so-called turbo-Grignard or turbo-Hauser bases.12,13 Recently, MgR2 treatment with M′(OR) for M′ = Li, Na, K was investigated as an attractive economic way for the synthesis of alkyl(alkoxy)alkali metal or tetra(alkyl)lithium magnesiates [MgM′R2(OR)] and [MgLi2R4] for magnesium–halogen exchanges.14,15 Incorporating chiral alkoxide/aryloxide ligands into alkyl lithium magnesiates leads to the formation of alkylmagnesium–lithium alkoxides/aryloxides and provides facile access to asymmetric synthesis.16–21 Despite the above mentioned examples of heteroleptic alkali metal magnesiates, their homoleptic alkoxide or aryloxide derivatives are uncommon. The structures of only 18 such compounds have been deposited in the Cambridge Structural Database.22 Most of these alkali metal–magnesium alkoxides/aryloxides have shown excellent potential in polymerization catalysis, including lactone ring-opening polymerizations (ROP), as well as CO2/epoxide or anhydride/epoxide copolymerizations. However, the heterometallic cooperativity of these compounds was very limited.23 One of the few examples is [Mg2Li2(OAr)6] (where ArO = 1-(phenyldiazenyl)-2-naphtholato), which showed better stereoselectivity and polymerization control than its mononuclear counterparts in the ROP of rac-LA (Pr = 0.75, while for Li or Mg initiators Pr = 0.67 or 0.46).24 The synergistic effect of the magnesium and lithium centers in [nBu2MgLi(OR)]2 and [nBuMgLi3(O)(OR)2]2 (for RO = 1-({[2-(dimethylamino)ethyl](methyl)amino}methyl)cyclohexan-1-olato) was observed in the Meerwein–Ponndorf–Verley reaction of benzaldehyde and isopropanol. Both catalysts provide the 90–93% conversion of benzaldehyde to benzyl alcohol in 6 h at 110 °C, and these values were 30% higher than those obtained with [Mg(OR)2]2.25n-Butylmagnesium–lithium or sodium aryloxides of [nBuMgM′(EDBP)(Et2O)]2 (for M′ = Li, Na; and EDBP = 2,2′-ethylidene-bis(4,6-di-t-butylphenolato)) were efficient initiators for highly isoselective polymerization of methyl methacrylate at −30 °C in 1 h, leading to polymers with high isotacticity ([mm] = 95.4–96.1%), molecular weights (Mn) of 58.9–86.3 kDa and narrow dispersity (Đ) of 1.35–1.58.26 Hetero-bimetallic [nBuMgLi(OAr)] (for ArO = 6,6′-methylene-bis-(2,4-di-t-butyl-phenolato)) combined with 1 equiv. of neopentyl alcohol was a highly active and stereoselective initiator for the polymerization of rac-LA to heterotactic PLA. For this system, the catalytic activity and stereocontrol of polymerization depended on the solvent used, which determined the aggregation of active species in the solution (Pr = 0.54 for THF, 0.64 for toluene, and 0.87 for THF/toluene).27 Recently, the Williams group reported a highly active magnesium–sodium copolymerization catalyst, which was able to selectively switch the polymerization course from cyclohexene oxide/phthalic anhydride ring-opening copolymerization to cyclohexene oxide ring-opening polymerization, allowing for controlled polyether block formation.28 The same group also established that the copolymerization course is influenced by appropriate metal selections, different coordination modes, metal–metal separation, ligand electronics, and complex stability.29There are also several examples in which the formation of heterometallic clusters led to a decrease in reactivity compared to that of the homometallic counterparts. Magnesium–lithium bisphenoxide or trisphenoxide intitiators [MgLi2(TBBP)2(THF)4] and [Mg2Li2(PBTP)2(THF)4] (where TBBP = 3,3′,5,5′-tetra-t-butylbiphenyl-2,2′-diolato and PBTP = 2,2′-((5-t-butyl-2-oxy-1,3-phenylene)bis(methylene))bis(4,6-di-t-butylphenolato)) show low catalytic activity and convert (90–92%) 150 equiv. of L-LA in 90 or 110 °C in 24 or 36 h, respectively.30,31 Another example was the magnesium–lithium 1,3-dipropoxy-p-tertbutylcalix[4]arene complex, which converted (9%) 100 equiv. of rac-LA in the presence of 1 equiv. MeOH in toluene in 1 h, while for the magnesium initiator an almost seven-times greater conversion was achieved in 2 h.32
Heterobimetallic alkali metal–magnesium alkoxides are also attractive single-source molecular precursors for the preparation of alkali metal-doped MgO nanoparticles,33 which have found applications as catalysts in the oxidative coupling of methane34 or semiconducting materials.35
Here, we report the preparation and characterization of homometallic and heterometallic magnesium aryloxides, namely [Mg2Li2(EtsalO)6] (4), [MgK(EtsalO)3]n (5), [Mg6Na4Al(MesalO)13(OH)6(MesalOH)(THF)0.5(H2O)0.5] (6), [Mg4Na2(MesalO)6(SalO)2(THF)4] (7), and [Mg4(MesalO)4(OMe)4(HOMe)4] (8) (where MesalOH = methyl salicylate, EtsalOH = ethyl salicylate, and SalOH2 = salicylic acid).
Compounds 4–7 were prepared by reaction of [Mg2M′2(MesalO)6(THF)x] for M′ = Li(1), Na(2), or K(3) with EtOH, exposure to atmospheric moisture, or deliberate addition of water. We found that 6 was formed haphazardly using MgnBu2 stabilized with 1 wt% of AlEt3. We investigated the catalytic activities of 1–3 and 6–8 in the ROP of the L-lactide (L-LA) or benzaldehyde Tishchenko reaction to determine the cooperativity effect between the metal sites. The effects of the initiator structure on the catalytic L-LA polymerization process and the physicochemical properties of the polyester products were investigated.
Results and discussion
In our previous work, a series of heterometallic alkali metal–magnesium aryloxides of the general formula [Mg2M′(MesalO)6(THF)x] (for M′ = Li, Na, K, and x = 0, 2, 4) were synthesized by reaction of MgnBu2 and nBuLi, metallic Na or K with the ligand precursor in a THF solution at stoichiometries of Mg/M′/MesalOH = 1/1/3 to give [Mg2Li2(MesalO)6] (1, 66%), [Mg2Na2(MesalO)6(THF)x], for x = 2 or 4 (2, 57%) and [Mg2K2(MesalO)6(THF)4] (3, 66%) as summarized in Scheme 1. Compounds 1–3 were used as efficient catalysts to recycle high consistency silicone rubber waste by alcoholysis to alkoxysilane derivatives.36 Detailed descriptions of the structural chemistry of 1–3 were discussed in this work because of their impact on the performed spectroscopic or thermal analysis studies, synthesis of high-nuclearity heterometallic clusters, or catalytic applications. Compounds 1–3 are centrosymmetric tetranuclear clusters based on a double-open dicubane core structure with two missing vertices (Scheme 1, ESI, Fig. S1–S3†). The metal ions in 1–3 are held together by four μ- and two μ3-O(aryloxo) bridges. The vertices of the common face of the central core in 1 are occupied by Mg1 and Mg1i ions, and the external vertices are Li1 and Li1i ions (Scheme 1). For compounds 2–3, the reverse arrangements of the metal atoms in the tetranuclear units are observed with peripheral Mg1 and Mg1i atoms, and Na1 and Na1i or K1 and K1i occupy the vertices of the common face (Scheme 1). Continuous-shape measurements (CShM)37 of the coordination environment around magnesium centers in 1–3 revealed the presence of an insignificant departure from the ideal octahedral geometry with the metric shape parameters S(Oh) = 0.684–0.979. The CShM parameters define the distance between the real and ideal polyhedra and are used to gauge the similarity between both structures (S(Oh) = 0 for the ideal octahedron; <1 indicates minor distortions; >1 reveals more severe deviations from the reference shape). For the six-fold coordinated Na1 and K1 atoms, the corresponding metric parameters of 1.742 and 3.976 suggested the presence of more significant geometrical distortions.38 Four-fold coordinated Li1 and Li1i atoms in 1 surrounded by one carbonyl oxygen donor and three aryloxo oxygen donors adopt axially vacant trigonal bipyramidal geometries (Scheme 1).39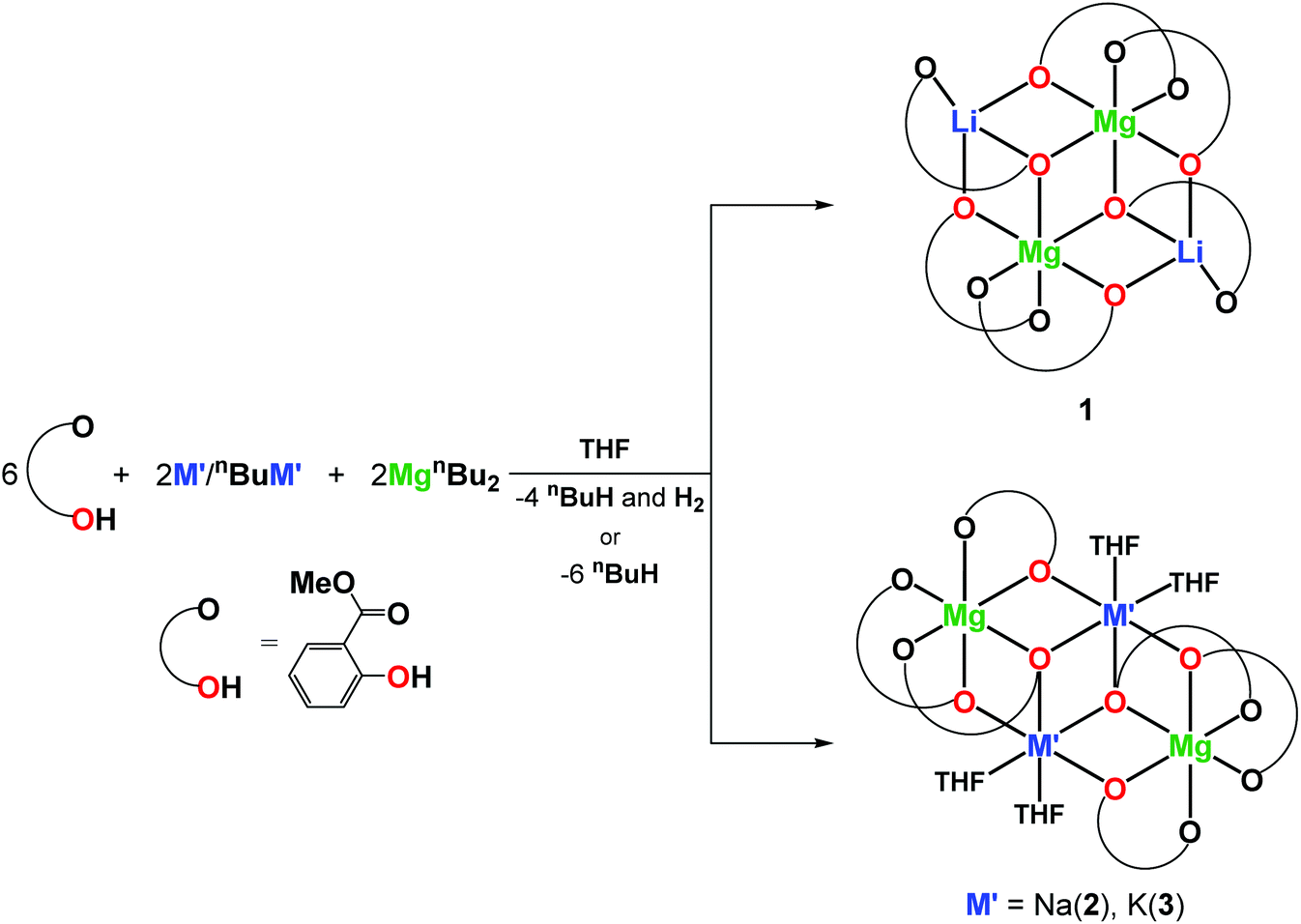 | ||
| Scheme 1 Synthesis of 1–3. | ||
The decomposition behavior of 1–3 from 25 to 1000 °C with a heating rate of 10 °C min−1 under N2 was investigated by thermogravimetry and differential thermal analysis (TGA-DTA). Thermograms presented in Fig. 1 indicate that compounds 1–3 underwent multistep thermal decomposition. Compound 1 was stable up to 238 °C with a mass loss not exceeding 2.5%. For 2 and 3, the decomposition starts above 90 °C with the removal of the THF ligand. The aromatic ligands begin to decompose and are removed at 238 °C for 1, 219 °C for 2, and 228 °C for 3. The total mass losses of 84.5% for 1, 85.1% for 2, and 84.8% for 3 correspond well to estimated values of 84.1%, 84.7%, and 83.4% for the mixture of MgO and M′2CO3 (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
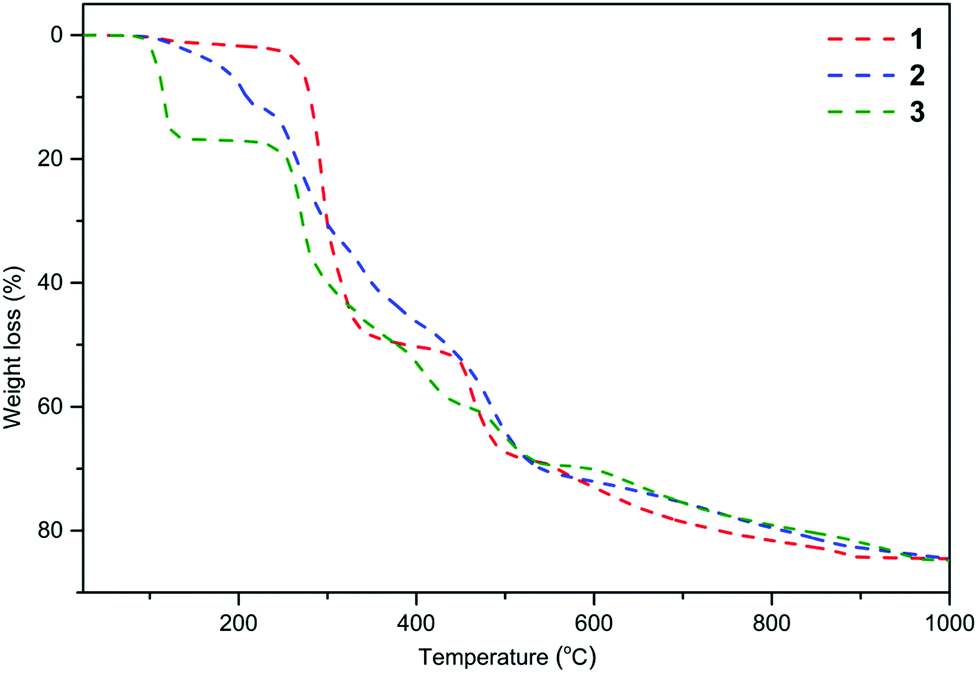 | ||
| Fig. 1 TGA curves (dashed-dotted lines) for 1–3 measure at a heating rate of 10 °C min−1 under a nitrogen atmosphere over the temperature range of 25 to 1000 °C. | ||
The PXRD diffractograms of the resulting powders (Fig. 2) confirmed the formation of MgO (periclase) as the main phase. However, a wide range of metal carbonates was also detected, i.e., MgCO3 (magnesite), Li2CO3 (zabuyelite), Na2CO3, K2CO3, Na2Mg(CO3)2 (eitelite), and K2Mg(CO3)2·4H2O (baylissite). Furthermore, the diffractograms also contain small intensity diffraction peaks corresponding to various magnesium or alkali metal silicates, i.e., Li4SiO4, Na4SiO4, K4SiO4, MgSiO3, Na2Si2O5, K2Si2O5, K2Si4O9, Li2MgSiO4, Na2MgSiO4, K2MgSiO4, and K2MgSi5O12 probably formed as the result of silicone grease contaminations.
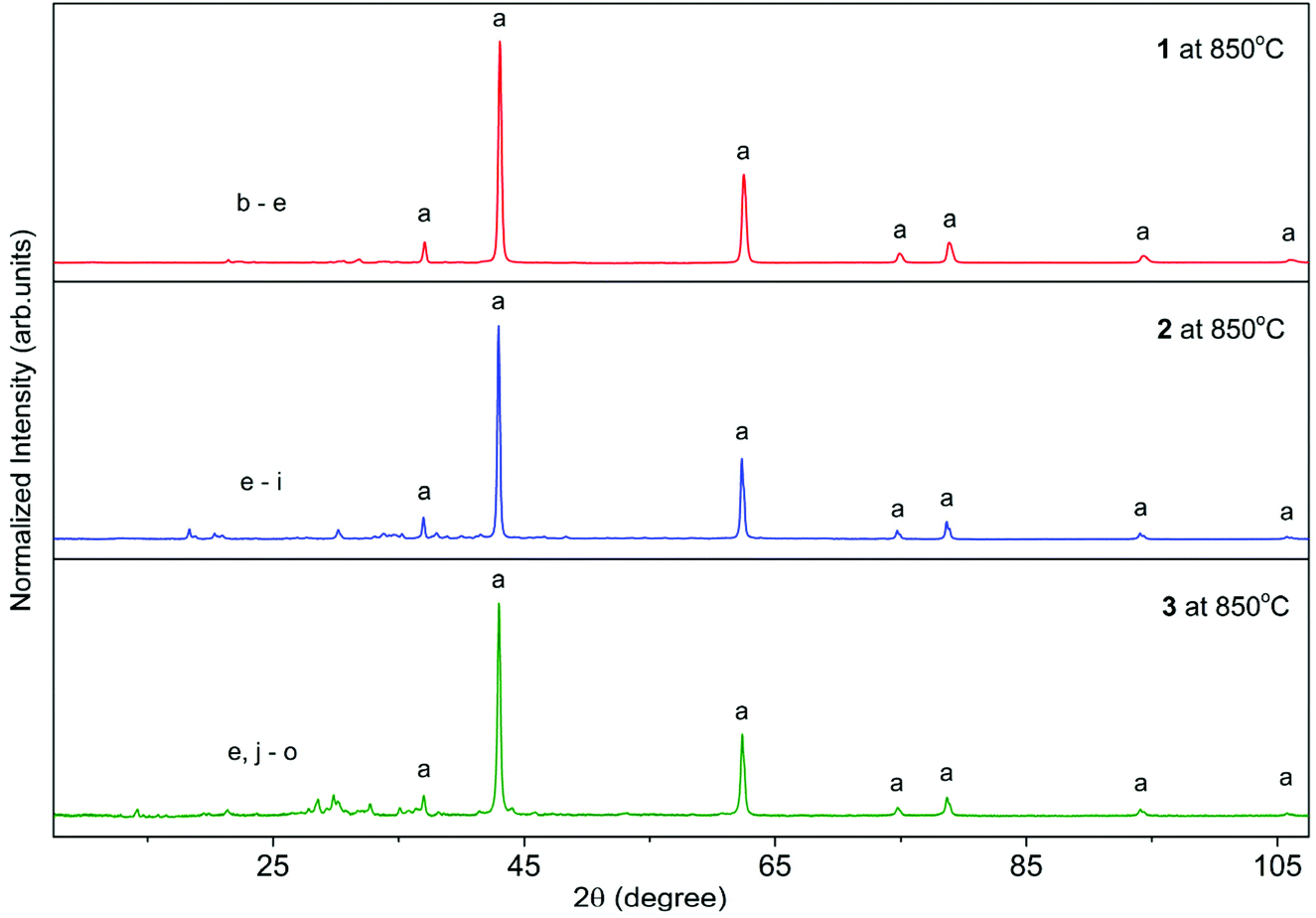 | ||
| Fig. 2 PXRD patterns of materials prepared by calcination of 1–3 at 850 °C. [Crystallography Open Database (COD)]: MgO 969006806 (a), Li2CO3 969008284 (b), Li4SiO4 961539514 (c), Li2MgSiO4 961537454 (d), MgCO3 969002811 (e), Na2CO3 962106298 (f), Na4SiO4 961527120 (g), Na2MgSiO4 962101699 (h), Na2Mg(CO3)2 961011094 (i), K2Mg(CO3)2·4H2O 969012597 (j), K2Si2O5 962003027 (k), K4SiO4 961539724 (l), K2MgSi5O12 962101101 (m), K2Si4O9 969000878 (n), K2CO3 962107220 (o). | ||
In this work, we decided to investigate the reactivity of 1–3 with EtOH or H2O to find an undemanding synthesis route into high-nuclearity heterometallic clusters. When 1 was reacted with excess EtOH in a toluene solution, the isostructural compound [Mg2Li2(EtsalO)6] (4, 62%) supported by the ethyl salicylate ligand (EtsalO) was obtained, as shown in Scheme 2. The X-ray diffraction structure of 4 presented in Fig. 3 is the molecular model that visualizes EtsalO ligand formation by transesterification of MesalO chelates. The structural and geometric parameters of 4 are similar to those reported for 1. When the same reaction was carried out with 3, the one-dimensional magnesium–potassium coordination polymer [MgK(EtsalO)3]n (5, 72%) was received, as shown in Scheme 2. In this reaction, the rearrangement of the aryloxo-bridged tetranuclear dicubane core structure of 3 into the aryloxo and carbonyl group bridged coordination polymer network of 5 was found. The structure of 5 presented in Fig. 4 is important in the field of structural chemistry because there are no known examples of alkali metal–magnesium coordination polymers with alkoxide/aryloxide ligands.
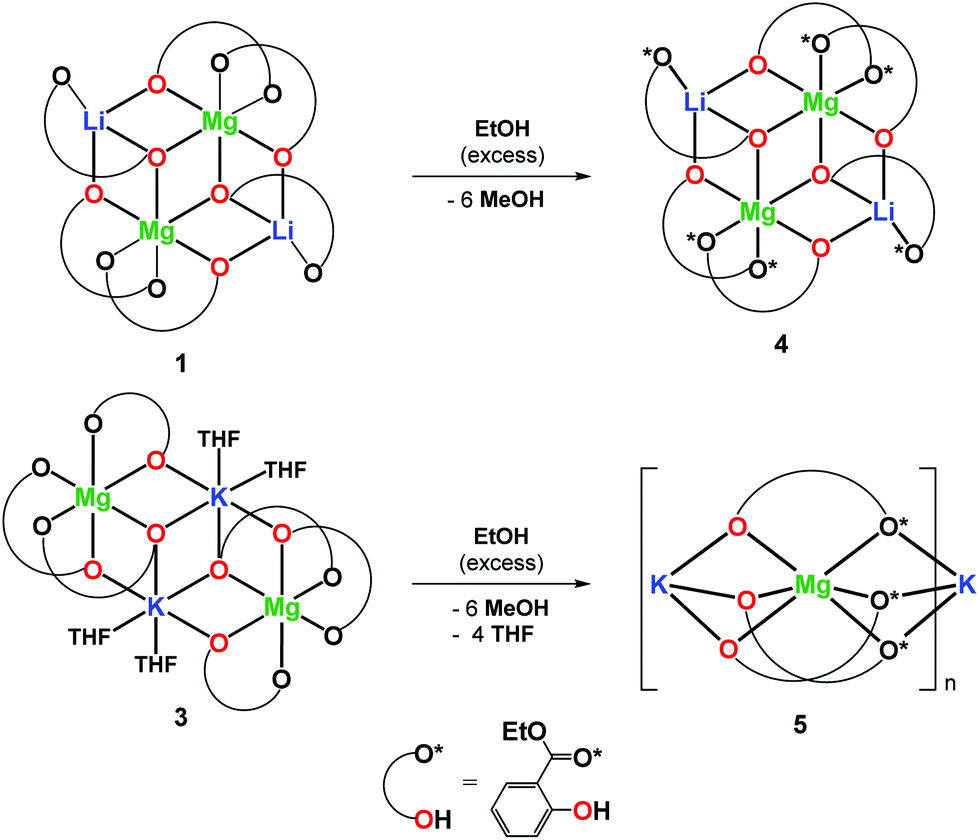 | ||
| Scheme 2 Synthesis of 4 and 5. | ||
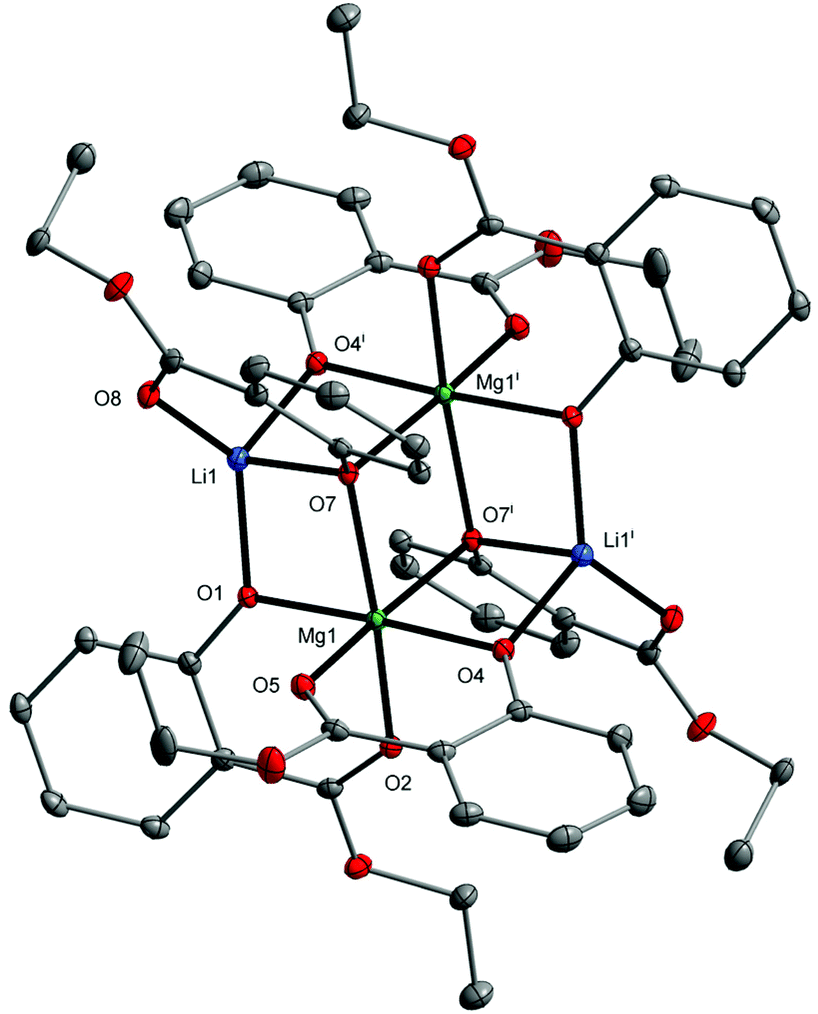 | ||
| Fig. 3 Molecular structure of [Mg2Li2(EtsalO)6] (4) with displacement ellipsoids drawn at the 25% probability level. Hydrogen atoms are omitted for the sake of clarity [symmetry code: (i) −x + 1, −y + 1, −z + 1]. | ||
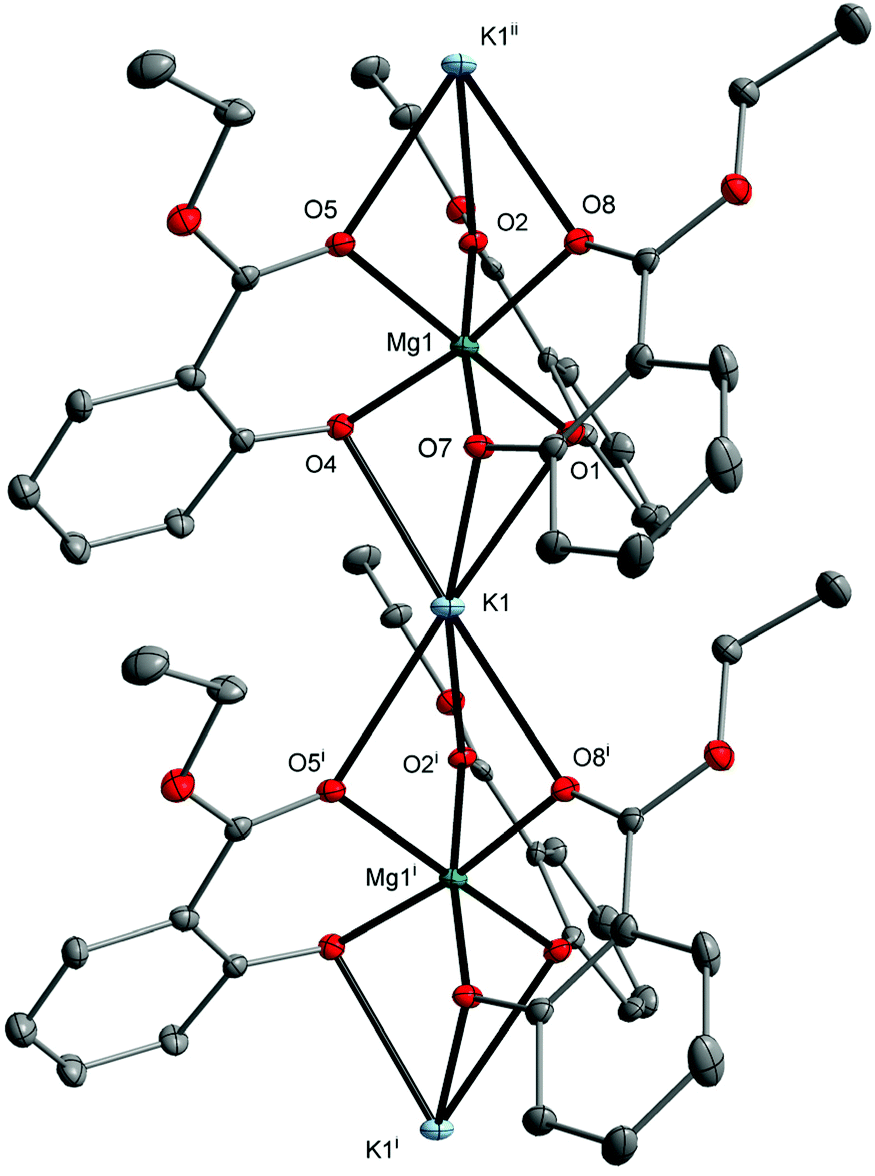 | ||
| Fig. 4 Molecular structure of [MgK(EtsalO)3]n (5) with displacement ellipsoids drawn at the 25% probability level. Hydrogen atoms are omitted for the sake of clarity [symmetry code: (i) x + 1, y, z; (ii) x − 1, y, z]. | ||
The formation of an infinite heterometallic coordination network was usually observed in the presence of carboxylate, diketonate or cyanurate ligands, i.e., [Mg2Li(BTB)(HBTB)(DMF)2]n (H3BTB = benzene-1,3,5-tribenzoic acid),40 [Mg2Na2(C2O4)3(H2O)2]n,41 [MgNa(NTA)2(H2O)]n (H3NTA = nitrilotriacetic acid),42 [MgCs2(C2O4)2(H2O)4]n,43 [MgK2(μ-H2C3N3O3)4(μ-H2O)4]n, [MgCs2(μ-H2C3N3O3)4(μ-H2O)8(H2O)2]n,44 [Mg2Rb2(CF3COO)6(CF3COOH)2(H2O)3]n, [Mg4Rb4(CF3COO)12(H2O)2]n,45 and [Mg2Cs2(L)3(MeCN)2] (H2L = 1,2-bis(pentoxy-2,4-dione)benzene).46 The metal atoms in 5 are octahedrally coordinated with six oxygen donor atoms from three chelating MesalO ligands for Mg1, three aryloxo oxygen atoms, and three carbonyl oxygen atoms from six MesalO ligands for K1. The Mg1 and K1 atoms are connected by three μ-O(aryloxo) bridges, while the K1 and Mg1i atoms are held together by three μ-O(carbonyl) bridges. For alkyl salicylate derivative ligands, the presence of metal bridging carbonyl groups was observed only in the structure of [Zn2Na4(MesalO)8]n.36 The CShM calculations revealed an insignificant departure from the ideal octahedral geometry around Mg1 atom with an S(Oh) parameter of 0.424 compared to a highly distorted octahedron formed by K1 atom with an S(Oh) parameter of 8.521. Compound 5 does not retain its structure in THF solution and transforms into a tetranuclear cluster similar to those reported for 3.
In coordination, chemistry is many examples of metal compounds formed haphazardly by the action of impurities or additives (stabilizers, antioxidants, reductants) present in solvents, organic reagents, and a wide range of various metal precursors.47–49 For example, MgnBu2, a convenient reagent used in organic synthesis or for the preparation of various magnesium compounds, contains up to 1 wt% of AlEt3 as a viscosity reducer. The presence of AlEt3 in the heptane solution of MgnBu2 is generally imperceptible. However, when we exposed the Schlenk flask containing the crude reaction mixture of 2 for contact with atmospheric moisture after four weeks, the formation of plate-like crystals of an undecanuclear magnesium–sodium–aluminum cluster [Mg6Na4Al(MesalO)13(OH)6(MesalOH)(THF)0.5(H2O)0.5] (6, 45%) was observed (Scheme 3). For the first time, the isolation of the aluminum compound [nBu3Al(IPr)] (IPr = 1,3-bis-(2,6-diisopropylphenyl)imidazol-2-ylidene) using commercially purchased MgnBu2 was reported during the synthesis of the bisalkyl magnesium carbene adduct [(nBu2Mg)4(IPr)2].50
 | ||
| Scheme 3 Synthesis of 6 and 7. | ||
The combination of metal ions Mg(II)/Na(I)/Al(III) presented in 6 is uncommon and was previously observed only in the natural mineral Zhemchuzhnikovite {[Mg(H2O)6][NaAl0.55Fe0.45(C2O4)3]·2H2O}n or its synthetic isostructural analog {[Mg(H2O)6][NaAl(C2O4)3]·3H2O}n.51,52
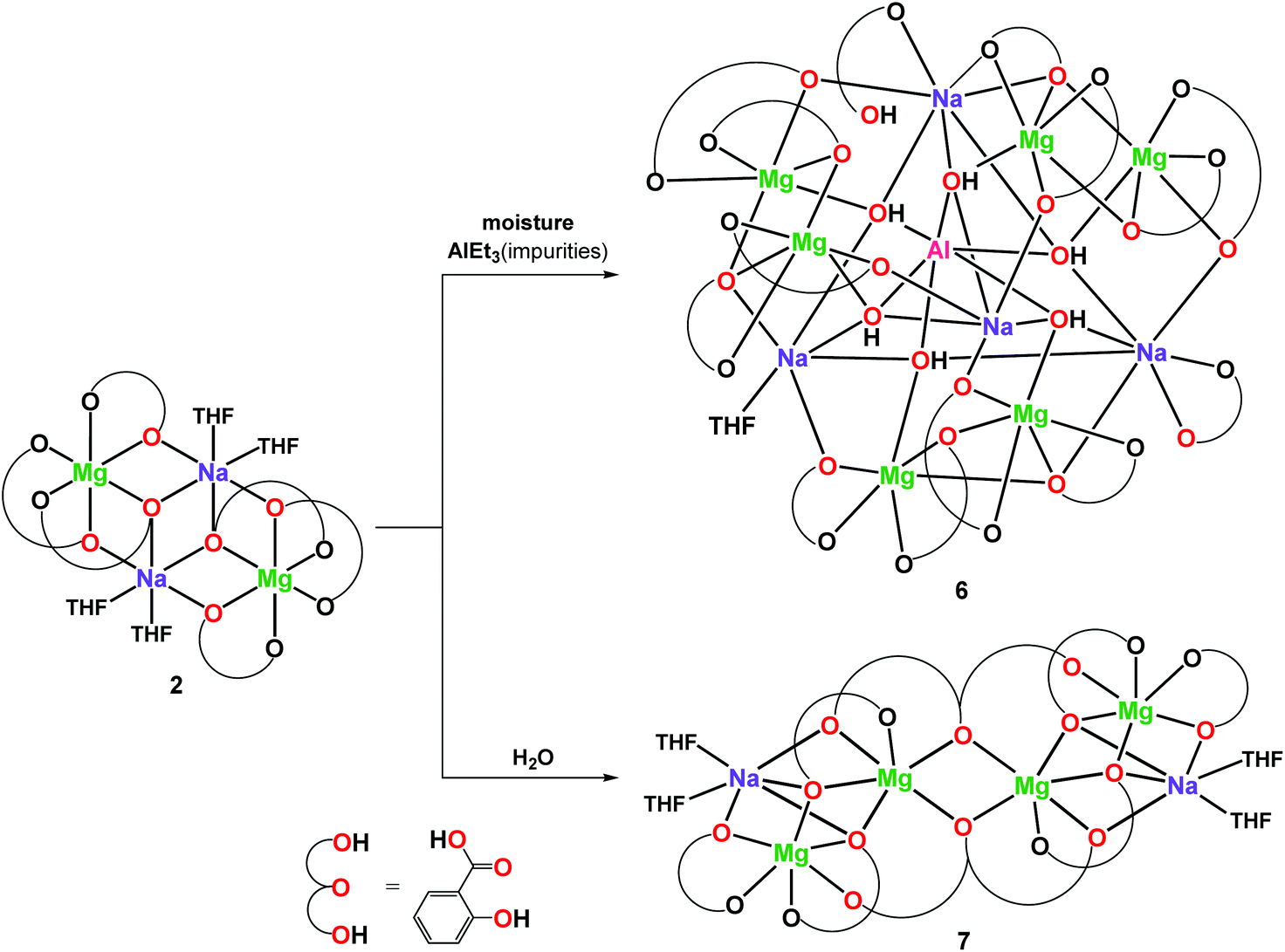
The XRD structure of 6 presented in Fig. 5 shows that Al(OH)63− was capped inside the central decametalate motif formed by six Mg and four Na atoms. Each OH group of the Al(OH)63− octahedron acts as a μ4-O bridge and binds the Al(III) ion with one Mg(II) and two Na(I) ions. The six-coordinated metal atoms adopted an octahedral (Al1, Mg1–Mg6, Na4) or a trigonal prismatic (Na1) geometry. Na2 and Na3 atoms surrounded by seven oxygen donor atoms form capped trigonal prisms.
 | ||
| Fig. 5 Molecular structure of [Mg6Na4Al(MesalO)13(OH)6(MesalOH)(THF)0.5(H2O)0.5] (6) with displacement ellipsoids drawn at the probability level of 20% (left); hydrogen atoms are omitted for clarity. Polyhedral representation of the central core structure Mg6Na4AlO34 with the removed Na1 polyhedron for clarity (right). | ||
Then we decided to broaden our research and perform partial hydrolysis of 2 in THF solution in a 2:H2O ratio of 1:2. However, instead of the heterometallic cluster similar to 6, the formation of a hexanuclear compound [Mg4Na2(MesalO)6(SalO)2(THF)4] (7, 56%) was observed. The molecular structure of 7 presented in Fig. 6 shows that the direct addition of H2O to the THF solution of 2 leads to the formation of salicylato ligands (SalO) by hydrolysis of the ester group MesalO.
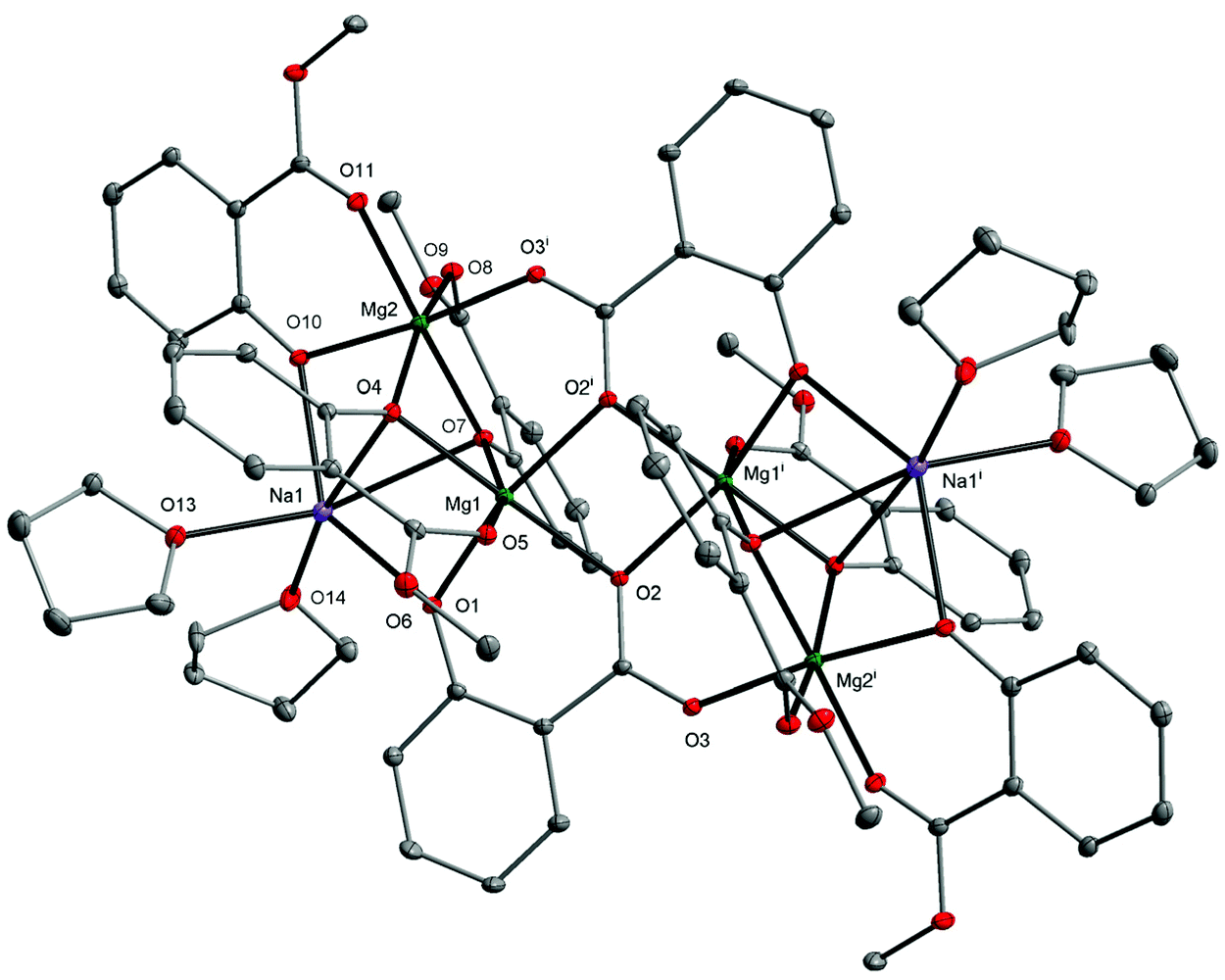 | ||
| Fig. 6 Molecular structure of [Mg4Na2(MesalO)6(SalO)2(THF)4] (7) with displacement ellipsoids drawn at the 25% probability level. Hydrogen atoms are omitted for clarity [symmetry code: (i) −x + 1, −y + 1, −z + 1]. | ||
Each SalO ligand binds to three Mg atoms and one Na atom in a tridentate bridging chelate mode. The carboxylate group binds to the Mg1i and Mg2i atoms through O2 and O3 oxygen donors. The O2 and O1 atoms act as μ-O bridges and link Mg1 and Mg1i or Mg1 and Na1, respectively. To date, such a coordination mode has previously been observed only in {[Na2Mn4(SalO)4(pyca)4(MeOH)2]·2H2O}n (pyca = pyridine-2-carboxylate).53 All metal ions in 7, surrounded by O6 donor sets, adopt distorted octahedral geometries as verified by the metric shape parameters S(Oh) equal to 3.925 for Na1, 1.898 for Mg1, and 0.918 for Mg2. The structural analysis of 7 indicated that it was formed by incorporating the species [Mg2(SalO)2] into 2, which was realized by scission of four Na–OMg bonds and rearrangement of the tetranuclear unit, leading to the external vertices occupied by Na(I) ions.
Recently, we also established that the direct reaction of two equiv. of MesalOH with MgnBu2 in the THF/EtOH solution leads to dinuclear [Mg2(EtsalO)4(EtOH)2] as shown in Scheme 4. When we performed the same reaction with MeOH, heteroleptic tetranuclear [Mg4(OMe)4(MesalO)4(HOMe)4] (8, 82%) was received. Compound 8 was obtained as crystalline, but the resulting material was poorly soluble in classic organic solvents. The tetranuclear structure of 8 presented in Fig. 7 is similar to that found in [Mg4(OMe)8(HOMe)8],54 [Mg4(OMe)4(tmhd)4(HOMe)4] (tmhdH = 2,2,6,6-tetramethylheptane-3,5-dione),55 [Mg4(OMe)4(mhp)4(HOMe)8] (mhpH = 6-methyl-2-pyridone).56 The structures of compounds 4–8 were confirmed by elemental analysis, 1H and 13C nuclear magnetic resonance (NMR) spectroscopy, and Fourier-transform infrared attenuated total reflectance (FTIR-ATR) spectroscopy (ESI, Fig., S4–S22†).
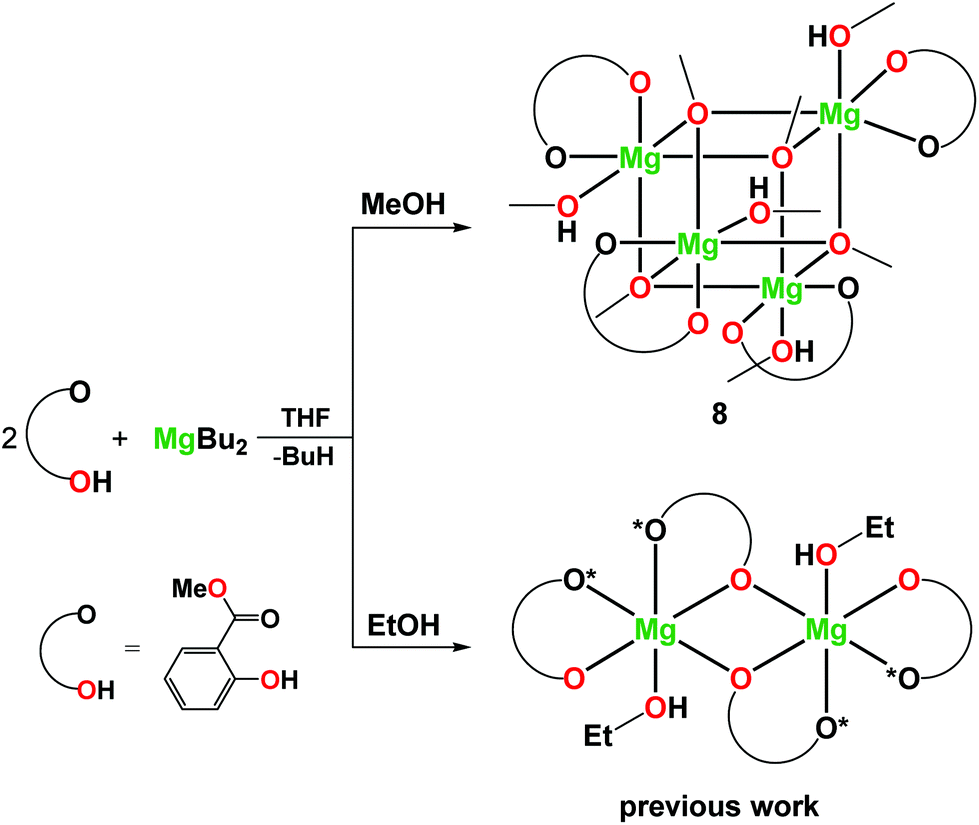 | ||
| Scheme 4 Synthesis of 8 and [Mg2(EtsalO)4(EtOH)2].36 | ||
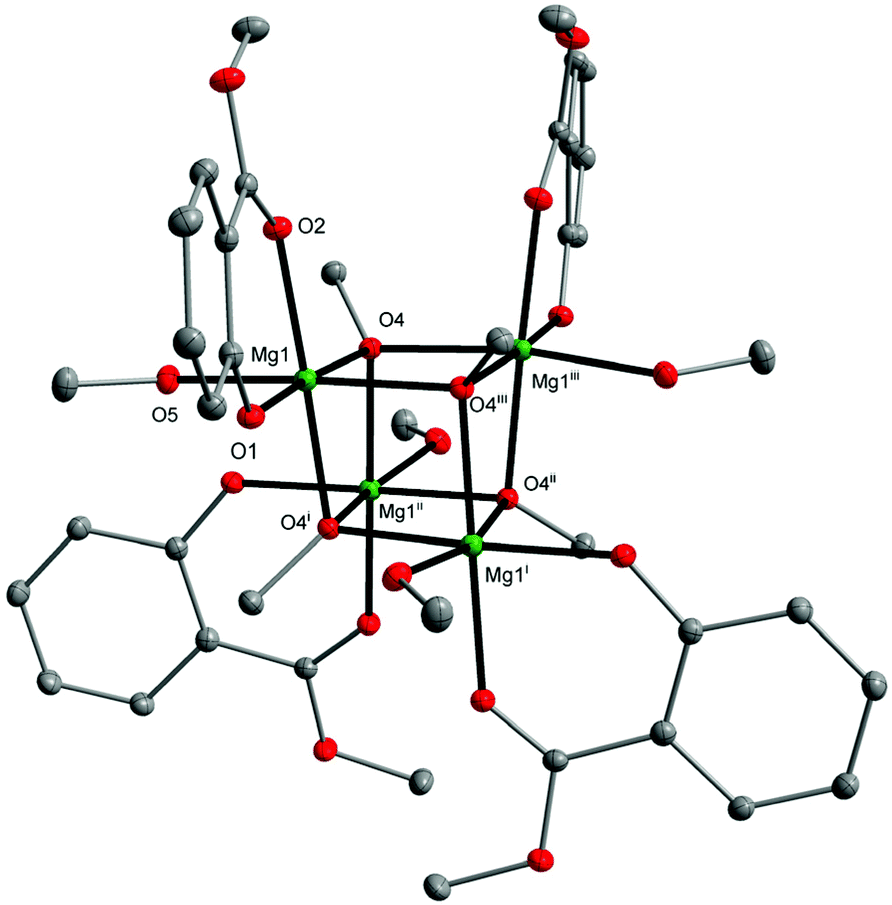 | ||
| Fig. 7 Molecular structure of [Mg4(MesalO)4(OMe)4(HOMe)4] (8) with displacement ellipsoids drawn at the 25% probability level. Hydrogen atoms are omitted for clarity [symmetry code: (i) −x + 2, −y + 1/2, z; (ii) y + 3/4, −x + 1.25, −z + 1.25; (iii) y + 3/4, −x + 1.25, −z + 1.25; (iv) −y + 1.25, x − 3/4, −z + 1.25]. | ||
Ring-opening polymerization of L-lactide
The ROP of L-LA with compounds 1–3, 6–8, and cetyl alcohol (ROH) was used as a model system to investigate the effect of heterometallic cooperativity. The amount of compound used in each reaction was calculated on the basis of the number of metal centers (M) in 1–3 and 6–8. Representative results for the L-LA polymerizations performed are summarized in Table 1. The number average molecular weights (Mn) and dispersity indices (Đ) of the polymers were determined by size-exclusion chromatography (SEC) with a multiangle laser-light scattering (MALLS) detector. The bulk ROP of L-LA was performed in a [L-LA]/[M]/[ROH] ratio of 100/1/1 at 110 °C. The SEC analysis of isolated poly(L-lactide)s (PLLAs) shows a monomodal weight distribution, with narrow Đ values ranging from 1.13 to 1.53 (Table 1, entries 1–8). Within the [Mg2M′2(MesalO)6(THF)x] series, the most active was the magnesium–sodium cluster 2, then the magnesium–potassium 3, and the least active was the magnesium–lithium 1. The Mn values of the PLLAs synthesized using 1–3 were comparable and ranged from 5.1 to 5.8 kDa with Đ indices of 1.34 to 1.43 (Table 1, entries 1–3), suggesting that the observed differences in catalytic activity are mainly affected by the central core structure and the ionic radius of M′ ions. The higher activity of 2 compared to 3 deviates from the activity trend commonly observed for the group 1 ROP initiators: Li(I) < Na(I) < K(I),57 where larger metal ions usually enhance monomer coordination and polymerization activity. The steric coverage of the K(I) coordination sphere due to the presence of metal–π interactions with the neighboring phenyl ring (3.215(2) Å) makes the coordination of the monomer more difficult than for Na(I).| Entry | Initiator | [L-LA]/[M]/[ROH] | t (min) | C (%) |
M
n
(kDa) |
Đ |
|---|---|---|---|---|---|---|
| a Polymerization conditions: L-LA (0.7 g, 4.86 mmol), cetyl alcohol (0.012 g, 0.0486 mmol), and an appropriate amount of initiator calculated based on the number of metal centers M (0.0486 mmol); carried out in bulk at 110 °C, in a nitrogen atmosphere. b Obtained from 1H NMR spectroscopic analysis. c Obtained from SEC analysis with MALLS detector. | ||||||
| 1 | 1 | 100/1/1 | 39 | 99 | 5.8 | 1.34 |
| 2 | 2 | 100/1/1 | 20 | 99 | 5.1 | 1.43 |
| 3 | 3 | 100/1/1 | 29 | 99 | 5.8 | 1.38 |
| 4 | Na(MesalO) | 100/1/1 | 90 | 96 | 6.3 | 1.32 |
| 5 | K(MesalO) | 100/1/1 | 90 | 94 | 6.2 | 1.13 |
| 6 | 8 | 100/1/1 | 37 | 99 | 3.2 | 1.53 |
| 7 | 6 | 100/1/1 | 47 | 99 | 6.3 | 1.43 |
| 8 | 7 | 100/1/1 | 49 | 98 | 6.6 | 1.43 |
The 1H-DOSY NMR investigations of reactions 1–3 with 4 equiv. of ROH in THF-d8 revealed the presence of two different sets of diffusion coefficients with logD of −9.08 m2 s−1 for uncoordinated ROH and from −9.21 to −9.32 m2 s−1 for 1–3 (Fig. 8 and Fig. S23–S24 in ESI†). The performed analysis did not reveal the formation of alcoholysis products, i.e., no free MesalOH and no OR bridged metal species were detected. The disproportionation of heterometallic clusters to the mixture of homometallic counterparts has also been excluded (Fig. 8 and Fig. S23 and S24 in ESI†).
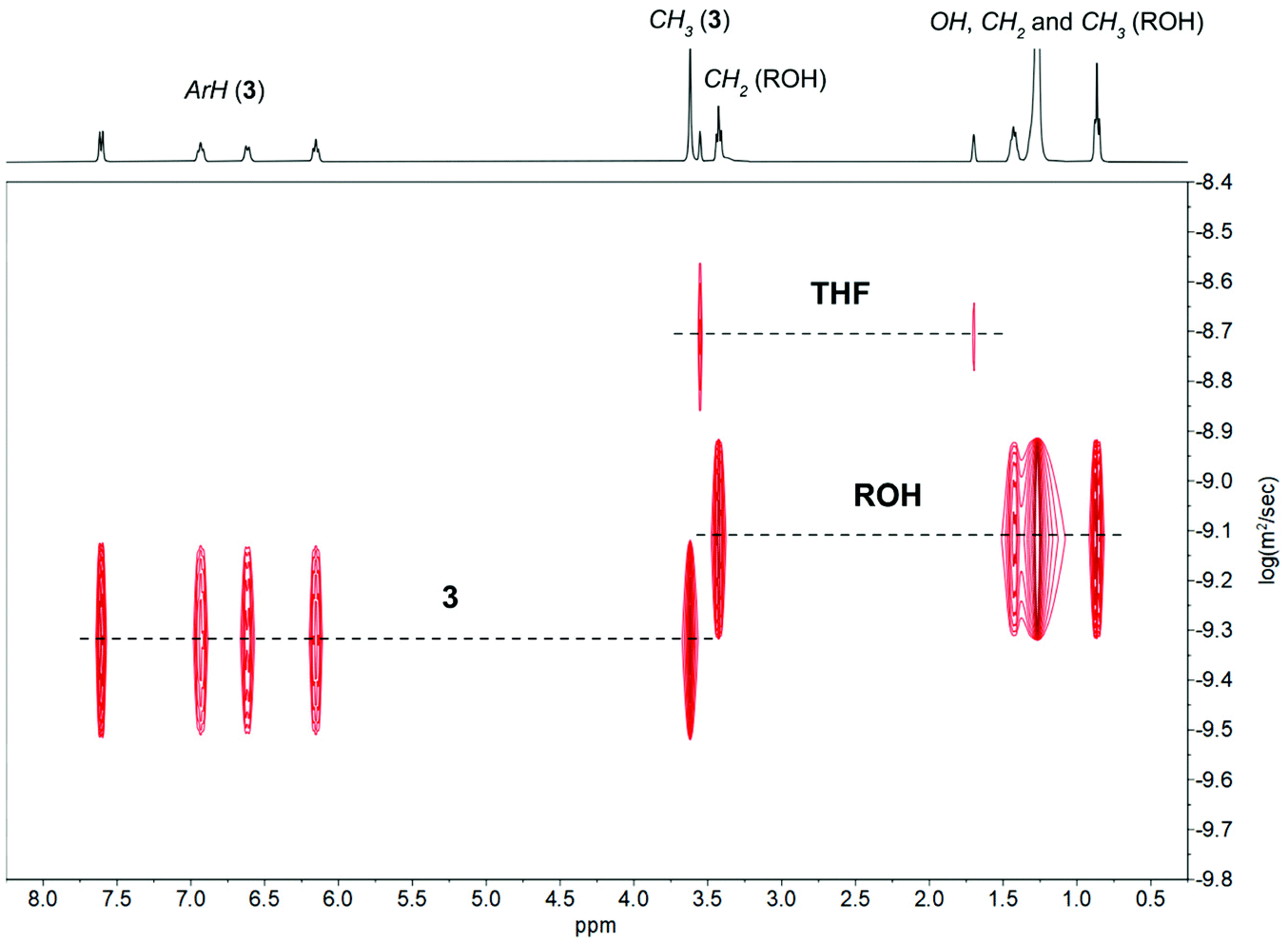 | ||
| Fig. 8 1H-DOSY NMR spectrum of the mixture of 3 and 4 equiv. of ROH in THF-d8. | ||
The formula weights and the hydrodynamic radii values estimate for 1–3 according to the Stokes–Einstein Gierer–Wirtz method (Fw = 944 (1), 1214 (2), 1354 (3) g mol−1 and rH = 8.42 (1), 9.16 (2), 9.49 (3) Å) suggested the presence of tetranuclear species in solution. We additionally verified this assumption using known internal references and correlating their molecular weights with relative diffusion coefficients via linear regression plots of the logarithms of D against the reference Fws. The estimated molecular weights of 1156 for 1, 1273 for 2, and 1516 g mol−1 for 3 correlate well with the values calculated from the Stokes–Einstein Gierer–Wirtz method and those derived from X-ray structures (ESI, Tables S2, S3 and Fig. S25–S30†).
The relative stability of tetranuclear clusters 1–3 towards ROH suggested that they operate via an activated-monomer mechanism. We verified this hypothesis by adding the excess of L-LA to the samples analyzed before and performing NMR-scale reactions at reagent stoichiometry [L-LA]/[M]/[ROH] = 10/1/1. The coordination of ROH to tetranuclear clusters 1–3 and the formation of oligolactides capped by the hexadecyl ester group provide clear evidence that the reaction proceeds through an activated monomer mechanism (Fig. 9 and Fig. S31 and S32 in ESI†).
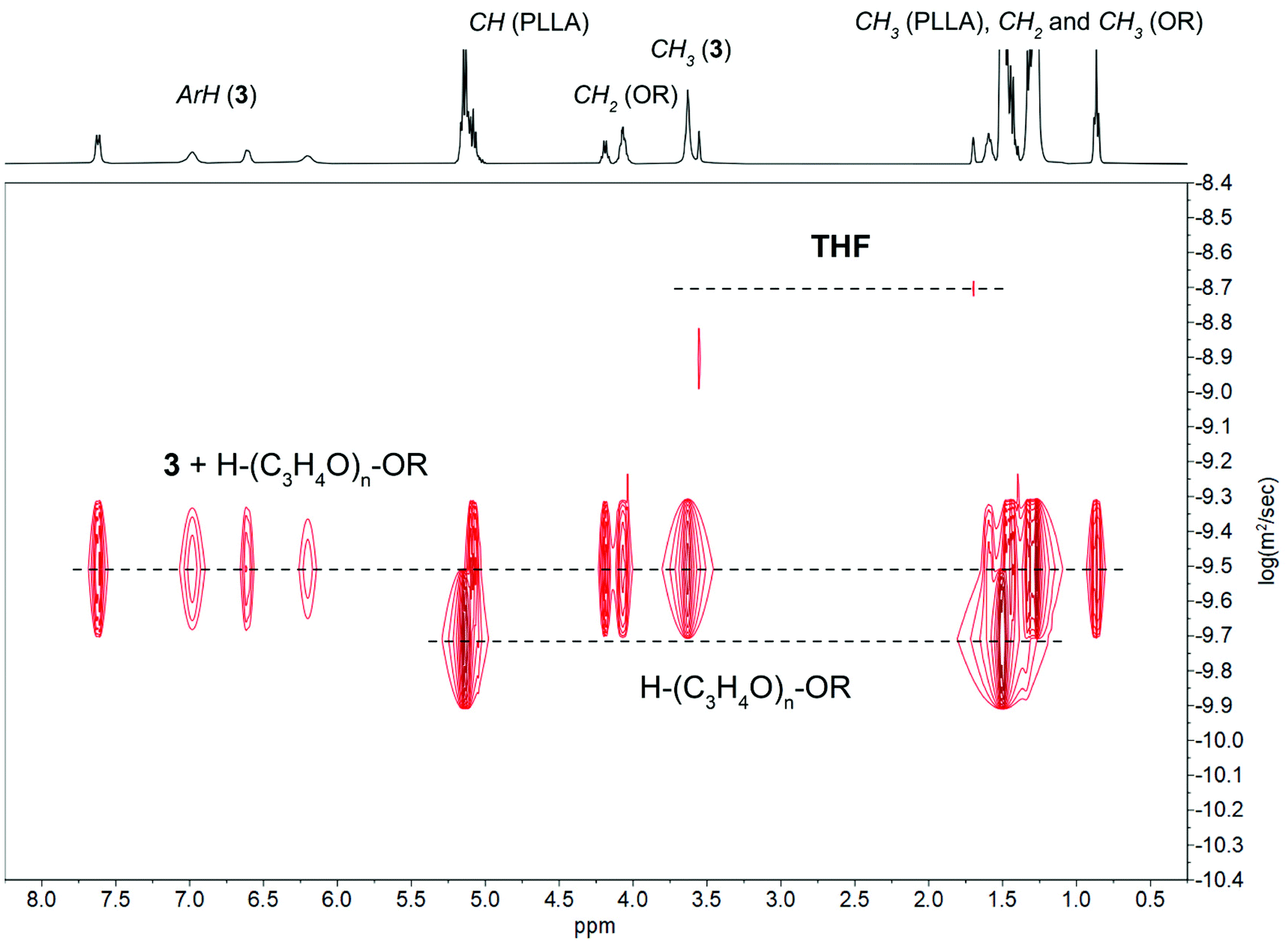 | ||
| Fig. 9
1H-DOSY NMR spectrum in THF-d8 of PLLA synthesized using 3 and ROH (1:4). | ||
Moreover, based on the molecular structures of 2 and 3 that contain Na(I) and K(I) ions coordinated by THF molecules, it is proposed that monomer coordination occurs on larger Na(I)/K(I) with greater availability of coordination sites than Mg(II). The more electronegative Mg(II) centers mediated ROH activation through Mg–O(phenolate)⋯H–OR hydrogen bonds formation, which facilitates the nucleophilic attack of ROH towards the L-LA carbonyl group.
When we used sodium, and potassium aryloxides, for the synthesis of PLLAs, their catalytic activity was much lower than that observed for 2 and 3. The 94–96% monomer conversion was achieved after 90 min (Table 1, entries 4 and 5). When homometallic magnesium aryloxide 8 was applied as the initiator for comparison with the above results, 99% of the L-LA conversion was obtained with a reaction time similar to that observed for 1–3. However, the Mn value of the resulting PLLA was almost half that of the polymers obtained with 1–3 due to the presence of magnesium bounded OMe and MeOH groups, which participate in the ROP of L-LA (Table 1, entry 6). These results suggest that heterometallic cooperation within tetranuclear clusters 2 and 3 arises from the differentiated roles of both metal centers. The large and Lewis acidic Na(I) and K(I) ions activate the L-LA carbonyl group toward the nucleophilic attack of ROH, accelerated by Mg(II).
The hexanuclear and undecanuclear compounds 6 and 7 within 47–49 min led to PLLAs with Mn of 6.3–6.6 and Đ ∼ 1.43 at [L-LA]/[M]/[ROH] ratio of 100/1/1 (Table 1, entries 7 and 8). In these examples, the increased aggregation state of heterometallic magnesium clusters reduces the kinetic mobility of the catalyst species. The non-bonding Mg⋯Na distances of 3.150(3)–3.421(3) Å for 6 and 3.130(2)–3.133(2) Å for 7 compared to 2 (3.323(2)–3.392(2) Å) revealed decreases the steric accessibility of the metal center. The presence of μ-OH or salicylato bridges that engage some of the Mg(II) and Na(I) ions to perform only structural functions could also affect the slightly higher Mn. However, based on the results of 1H DOSY NMR studies of 6–7 (ESI, Table S3,† and Fig. S33–S36†), it was not possible to exclude the possibility of a polymerization initiated by disaggregated active species. From a structural chemistry point of view, the most efficient in the ROP of L-LA were aryloxides 2 and 3, which contain Mg(II) ions on the external and Na(I)/K(I) ions on the common vertices of the tetranuclear unit. The presence of two Na(I) ions externally located within the central core structure of 7 does not lead to its higher catalytic activity in the ROP of L-LA compared to 2. The central Mg2M′2O6 motif discussed here is crucial for optimal cooperative interactions of both metal centers. It plays an important role in the ROP of cyclic esters and the alcoholysis of high consistency silicone rubber.36
The representative 1H NMR spectrum in C6D6 of the resulting PLLA showed resonance signals at 4.26, 2.58, and 1.39 ppm from the CH, OH, and CH3 groups of hydroxyl end units, the protons of the CH and CH3 groups of the main chain at 5.06 and 1.48 ppm, and resonances for the alkyl protons of the hexadecyl ester group at 4.01, 1.15 and 0.77 ppm (Fig. 10). The ESI-MS spectrum presented in Fig. 11 confirmed the formation of PLLA terminated by the hexadecyl ester group.
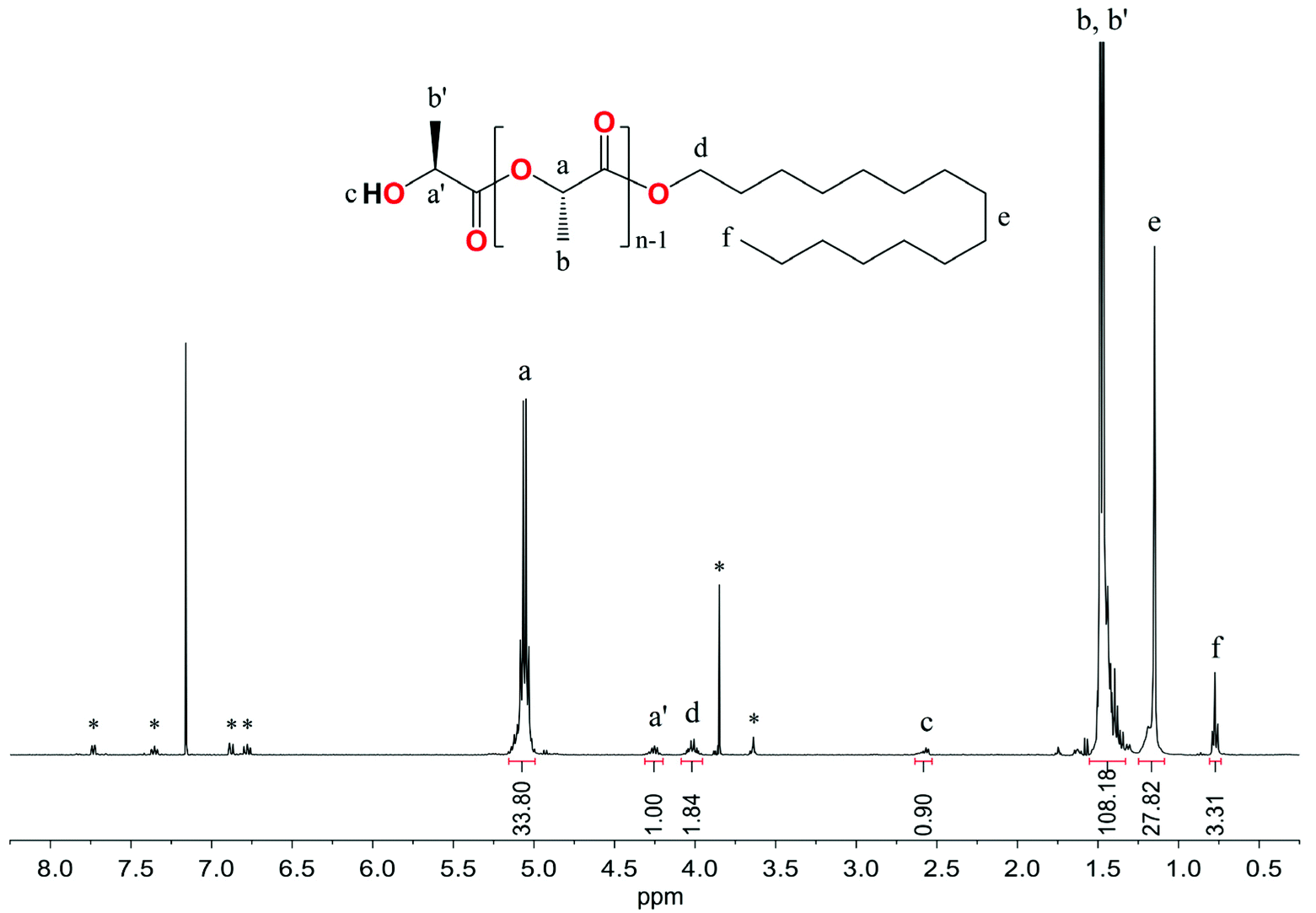 | ||
| Fig. 10 1H NMR spectrum in C6D6 of the resulting PLLA. * assigned to the signal of the MesalO ligand. | ||
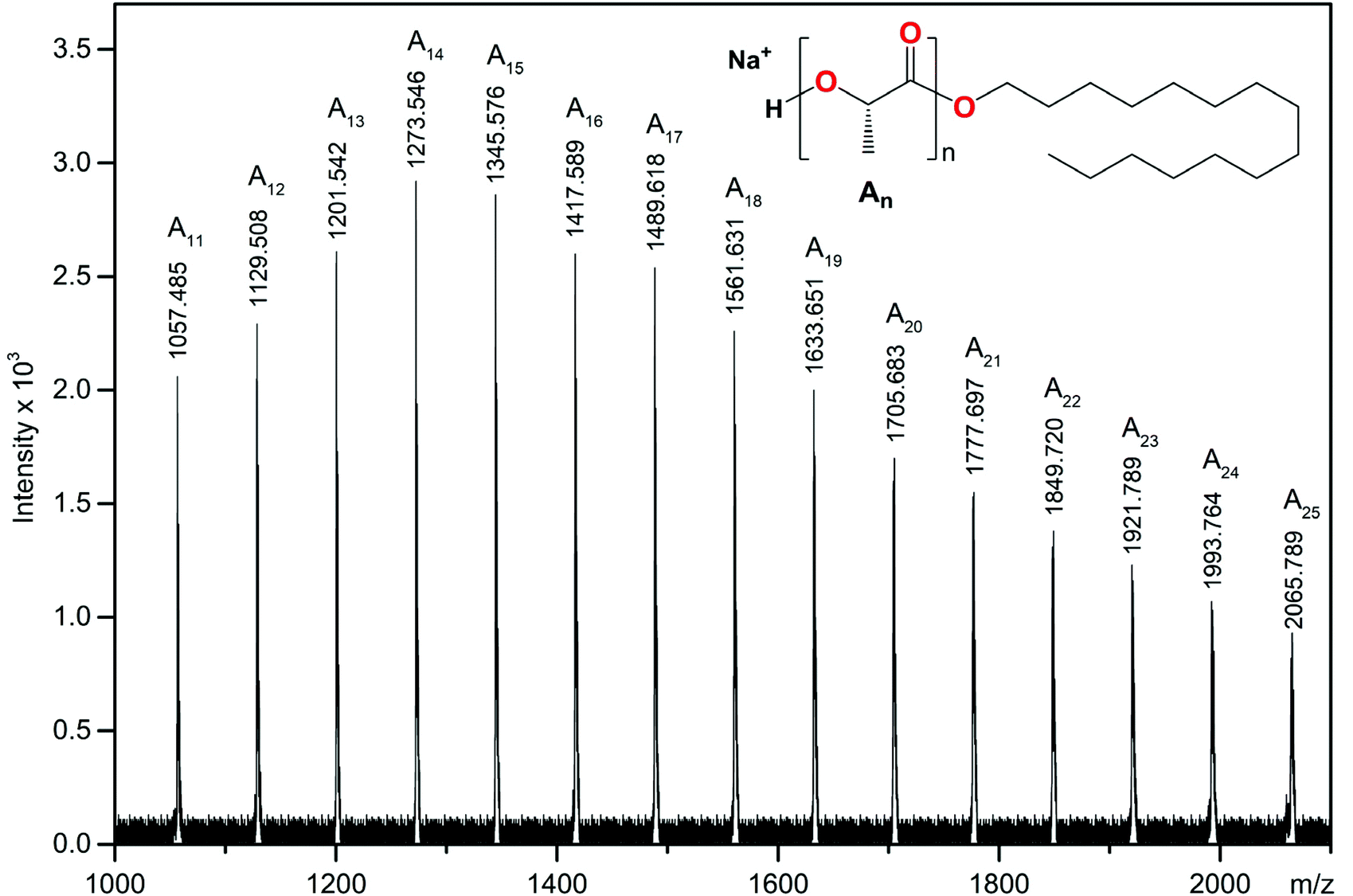 | ||
| Fig. 11 ESI-MS spectrum of the resulting PLLA. | ||
Synthesis of benzyl benzoate
The catalytic activity of 1–3 and 6–8 was also investigated in the Tishchenko reaction using benzaldehyde (PhCHO) in a PhCHO/M ratio of 200/1. Initially, compounds 1–3 were used as catalysts under mild reaction conditions, i.e., at 25–70 °C temperature using THF, toluene, CH3CN, and DMF as solvents, but no reactivity was observed within 120 h. Then we decide to perform the Tishchenko reaction using benzaldehyde as both the solvent and reactant at 120 °C in the presence of 1–3 and 6–8. The 1H NMR monitoring of the reactions performed showed that the highest catalytic activity revealed 8, which led to benzyl benzoate with an 88% conversion yield in 45 min (Fig. 12). In the presence of heterometallic clusters 1–3 and 6–7 after 48 h, the conversion of PhCHO does not exceed 8%. When under similar conditions, the catalytic activity of the bis-aryloxide magnesium compound [Mg2(EtsalO)2(EtOH)2] was tested only a trace of benzyl benzoate was detected after 48 h. The weak reactivity of homoleptic aryloxides 1–3 and 6–7 in benzaldehyde dimerization could be explained by previous studies of the Coles group, which revealed that the active species in the Tishchenko reaction is the alkoxide, (L)Mg(OCH2Ph) for LH = 1,4,6-triazabicyclo[3.3.0]oct-4-ene, 1,5,7-triazabicyclo[4.4.0]dec-5-ene.58,59 The formation of active catalytic species using sterically crowded aryloxides 1–3 and 6–7 is not thermodynamically favored. We established that the best performance and highest yields in the dimerization of benzaldehyde were achieved using compound 8, which contains active alkoxide groups Mg–OMe that undergo further transformation to Mg–OCH2Ph species.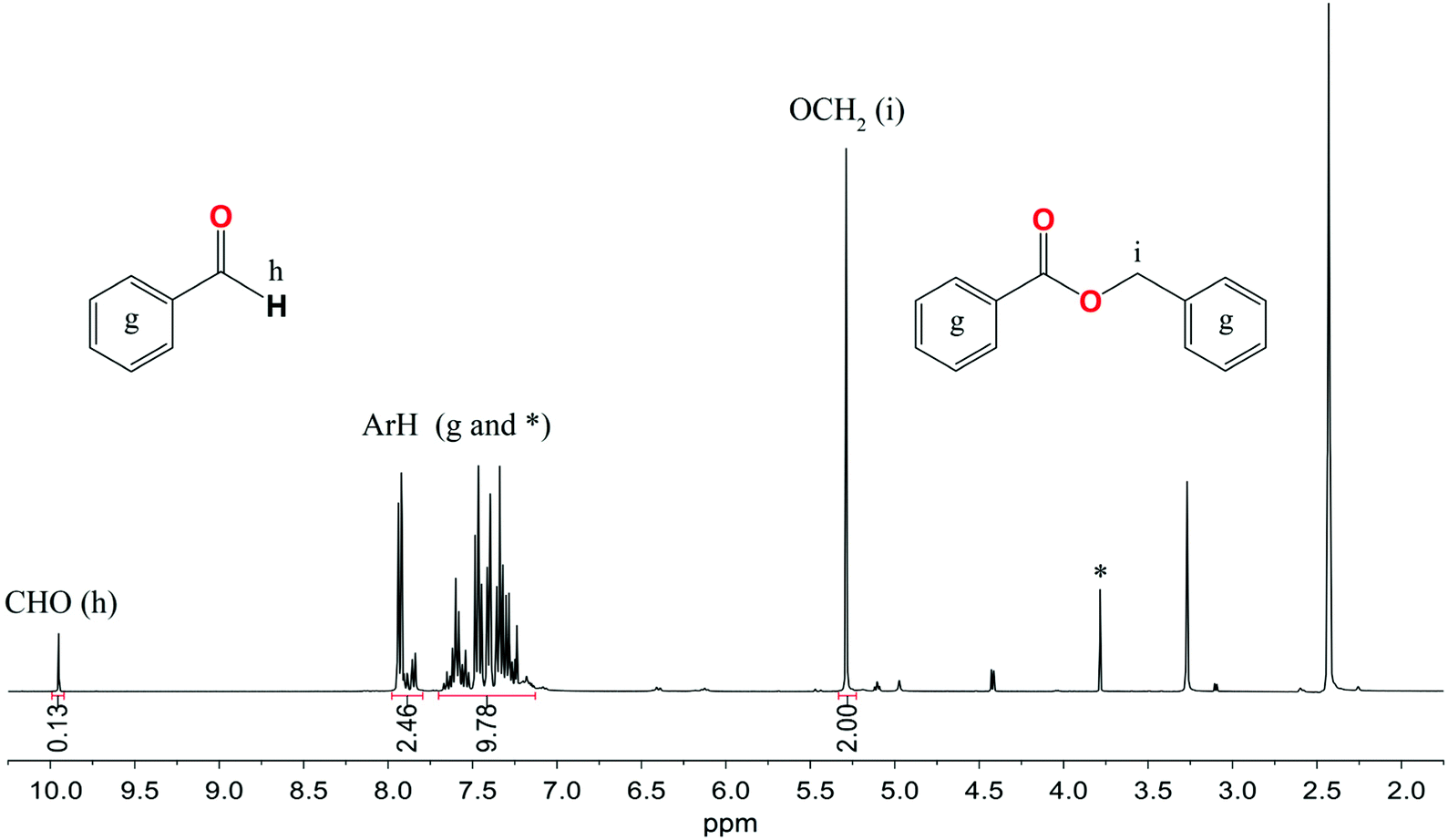 | ||
| Fig. 12 1H NMR spectrum in DMSO-d6 of the Tishchenko reaction with benzaldehyde using 8 as a catalyst. * assigned to the signal of the MesalO ligand. | ||
Conclusion
In this work, a series of alkali metal–magnesium aryloxides of the general formula [Mg2M′(MesalO)6(THF)x] (for M′ = Li (1), Na (2), K(3), and x = 0, 2, 4) were used as precursors for the synthesis of new heterometallic clusters by reaction with EtOH, exposure to atmospheric moisture or deliberate addition of stoichiometric quantities of water. We determined the structure of the undecanuclear cluster [Mg6Na4Al(MesalO)13(OH)6(MesalOH)(THF)0.5(H2O)0.5] (6) with an uncommon combination of metal ions Mg(II)/Na(I)/Al(III) previously observed only in the natural mineral Zhemchuzhnikovite. We found that the source of Al(III) ions in 6 was MgnBu2 stabilized with AlEt3. Transesterification or partial hydrolysis of 1–3 were used for the synthesis of [Mg2Li2(EtsalO)6] (4), [MgK(EtsalO)3]n (5) or [Mg4Na2(MesalO)6(SalO)2(THF)4] (7). Homometallic magnesium aryloxide [Mg4(MesalO)4(OMe)4(HOMe)4] (8) was obtained by reaction of MgnBu2 and MesalOH in a methanol solution. The catalytic activity of compounds 1–3 and 6–8 was investigated in ring-opening polymerization (ROP) of the L-lactide (L-LA) or benzaldehyde Tishchenko reaction. It was determined that the structure of the initiators and catalysts used strongly influenced both investigated reactions. We show that the most active in the ROP of L-LA were heterometallic aryloxides 2 and 3. For these initiators, valuable synergic effects between Mg(II) and Na(I)/K(I) ions were observed in the ROP of L-LA. Their catalytic activity was much better than that of the corresponding homometallic aryloxides. It was proposed that heterometallic cooperation within tetranuclear clusters 2 and 3 arises from differentiated roles of both metal centers: the Lewis acidic Na(I) and K(I) ions activate the L-LA carbonyl group towards the nucleophilic attack of ROH accelerated by the Mg(II). We established that the best performance and highest yields in the Tischenko benzaldehyde reaction revealed homometallic 8, which contains an ancillary aryloxide ligand combined with an Mg–OMe species.Overall, these results will be helpful in the development of industrially important chemicals, i.e., benzyl benzoate or biodegradable polyesters using homometallic and heterometallic magnesium catalysts with different central core structures. These findings should also help guide future polymerization catalyst designs, especially those with improved heterometallic cooperation.
Experimental section
Materials and methods
All syntheses were performed under a dry N2 atmosphere and using Schlenk techniques. Standard methods purified the reagents: toluene, hexane, and THF were distilled over Na, and EtOH was distilled over Mg. All chemical reagents were purchased from commercial sources: methyl salicylate, metallic sodium, metallic potassium, MgnBu2 1.0 M in heptane, nBuLi solution 1.6 M in hexane, L-lactide, benzaldehyde (Sigma-Aldrich, St Louis, MO, USA); toluene, hexane, THF, CH2Cl2 and C2H5OH (Chempur); DMSO-d6, THF-d8 (Eurisotop). L-Lactide was recrystallized twice from toluene, sublimed, and kept over P2O5. The 1H, 7Li, and 13C{1H} NMR spectra were recorded at room temperature on JEOL JNM-ECZ 400 MHz and Bruker Avance 600 MHz spectrometers. Chemical shifts were reported in parts per million and referenced the residual protons in deuterated solvents. The 7Li spectra were referenced to a 0.1 M solution of LiNO3 in D2O. The calculation of formula weights (Fws) and the hydrodynamic radii (rH) of 1–3 and 5 were performed using the Stokes–Einstein Gierer–Wirtz estimation method. The diffusion coefficients (D) of magnesium clusters were first normalized using as reference Dref of residual THF in the deuterated solvent. Then the Fw and rH estimation was performed using the SEGWE D/MW calculator according to the procedure described by Evans and coworkers.60 The temperature value from 21.5 to 22.6 °C was applied for the calculations performed. The results of these studies are summarized in Table S2 in ESI.† FTIR-ATR spectra were recorded on a Bruker Vertex 70 vacuum spectrometer. Elemental analyzes were performed on a PerkinElmer 2400 CHN elemental analyzer. Metal ion concentrations were determined by ICP-OES using a Thermo Scientific iCAP 7400 Duo spectrometer. The thermal decomposition of the metal aryloxide precursors was performed using an NT 1313 furnace (Neotherm) equipped with a KXP4 thermostat. The resulting metal oxide materials were investigated by powder X-ray diffraction with an Empyrean (PANalytical) diffractometer. The samples were analyzed using the PDF-4+ and COD powder diffraction database. The SEC traces of PLLAs were recorded at 30 °C with a system consisting of an Agilent 1100 Series isocratic pump, a degasser, an autosampler, a thermostatic box for columns, and a set of TSK Gel columns (2 × PLGel 5 μm MIXED-C). A Wyatt Optilab rEX interferometric refractometer and a MALLS DAWN EOS laser photometer (Wyatt Technology Corp., USA) were used as detectors. The eluent was CH2Cl2 with a flow rate of 0.8 ml min−1. The specific refractive index increment (dn/dc) was 0.1620 ml g−1. The Mn and Đ values were calculated from the experimental traces using the Wyatt ASTRA v 4.90.07 program. TGA/DTA was performed in a nitrogen atmosphere with a TGA/DSC 3+ system (Mettler-Toledo) at a heating rate of 10 °C min−1.Single-crystal X-ray diffraction studies
Single-crystal XRD data were collected using an Xcalibur Ruby, PX Xcalibure, or Agilent SuperNova Dual Atlas diffractometer at 100 K for 4–8. Experimental details and crystal data are given in Table S1.† Structures were solved by direct methods and refined by the full-matrix least-squares method on F2, using the SHELXTL package.61 Non-hydrogen atoms were refined with anisotropic thermal parameters. All hydrogen atoms were positioned geometrically and added to the structure factor calculations but were not refined. Molecular graphics for the resulting structures were created using Diamond (version 3.1e).62 CCDC 2123619–2123623† contains the supplementary crystallographic data for this article.Synthesis of [Mg2Li2(MesalO)6] (1), [Mg2Na2(MesalO)6(THF)x] for x = 2 or 4 (2), [Mg2K2(MesalO)6(THF)4] (3)
Structural, spectroscopic, and analytical data for 1–3 have been published in ref. 36. For catalytic studies, compounds 1–3 were synthesized according to the previously published procedure.Synthesis of [Mg2Li2(EtsalO)6] (4)
Compound 1 (0.75 g, 0.77 mmol) was added to the mixture of 50 ml of EtOH and 10 ml of toluene and heated under reflux to complete dissolution. The reaction mixture was then cooled and left for crystallization at room temperature. After four weeks of crystallization, the resulting block-like colorless crystals were filtered off, washed with hexane (3 × 10 ml), and dried under vacuum. Yield: 0.51 g (62%). Anal. calc. for C54H54O18Li2Mg2: C, 61.56; H, 5.17; Mg, 4.61; Li 1.32. Found: C, 61.61; H, 5.19; Mg, 4.69; Li 1.29. 1H NMR (600 MHz, THF-d8): δ 7.80–7.31 (6H, m, ArH), 7.28–6.58 (12H, m, ArH), 6.28 (6H, m, ArH), 4.60–3.40 (12H, m, CH2), 1.44–1.03 (18H, m, CH3). 13C NMR (151 MHz, THF-d8): δ 172.0 (12C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O), 135.9, 135.4 (6C, ArH), 131.8 (6C, ArH), 124.9, 120.0 (6C, ArH), 114.8, 113.7 (6C, ArH), 112.9, 112.7 (6C, Ar), 61.6, 61.1 (6C, CH2), 14.7, 14.3 (6C, CH3). 7Li NMR (THF-d8, 155 MHz): δ 5.37, 4.14. 1H NMR (400 MHz, THF-d8, 50 °C): δ 7.61 (6H, m, ArH), 7.03 (6H, m, ArH), 6.72 (6H, m, ArH), 6.30 (6H, m, ArH), 4.15, 3.72 (12H, m, CH2), 1.11, 1.01 (18H, m, CH3). 13C NMR (101 MHz, THF-d8, 50 °C): δ 172.1 (12C, CO, C–O), 135.4 (6C, ArH), 131.5 (6C, ArH), 124.5 (6C, ArH), 114.8 (6C, ArH), 113.01 (6C, Ar), 61.4, 61.1 (6C, CH2), 14.4, 14.3 (6C, CH3). FTIR-ATR: 3049 (vw), 3028 (vw), 2978 (w), 2933 (w), 2870 (vw), 2701 (vw), 2162 (vw), 2113 (vw), 1980 (vw), 1953 (vw), 1919 (vw), 1808 (vw), 1685 (m), 1672 (m), 1649 (s), 1601 (m), 1557 (w), 1543 (m), 1467 (s), 1444 (s), 1396 (w); 1371 (m), 1351 (m), 1324 (m), 1296 (m), 1261 (m), 1221 (vs), 1186 (m), 1176 (m), 1154 (s), 1141 (m), 1113 (w), 1082 (s), 1044 (w), 1033 (m), 1003 (w), 981 (vw), 951 (w), 891 (w), 870 (w), 856 (w), 827 (m), 798 (w), 759 (vs), 709 (s), 661 (m), 583 (s), 552 (vw), 533 (m), 492 (w), 473 (w), 440 (w), 413 (vw).
O, C–O), 135.9, 135.4 (6C, ArH), 131.8 (6C, ArH), 124.9, 120.0 (6C, ArH), 114.8, 113.7 (6C, ArH), 112.9, 112.7 (6C, Ar), 61.6, 61.1 (6C, CH2), 14.7, 14.3 (6C, CH3). 7Li NMR (THF-d8, 155 MHz): δ 5.37, 4.14. 1H NMR (400 MHz, THF-d8, 50 °C): δ 7.61 (6H, m, ArH), 7.03 (6H, m, ArH), 6.72 (6H, m, ArH), 6.30 (6H, m, ArH), 4.15, 3.72 (12H, m, CH2), 1.11, 1.01 (18H, m, CH3). 13C NMR (101 MHz, THF-d8, 50 °C): δ 172.1 (12C, CO, C–O), 135.4 (6C, ArH), 131.5 (6C, ArH), 124.5 (6C, ArH), 114.8 (6C, ArH), 113.01 (6C, Ar), 61.4, 61.1 (6C, CH2), 14.4, 14.3 (6C, CH3). FTIR-ATR: 3049 (vw), 3028 (vw), 2978 (w), 2933 (w), 2870 (vw), 2701 (vw), 2162 (vw), 2113 (vw), 1980 (vw), 1953 (vw), 1919 (vw), 1808 (vw), 1685 (m), 1672 (m), 1649 (s), 1601 (m), 1557 (w), 1543 (m), 1467 (s), 1444 (s), 1396 (w); 1371 (m), 1351 (m), 1324 (m), 1296 (m), 1261 (m), 1221 (vs), 1186 (m), 1176 (m), 1154 (s), 1141 (m), 1113 (w), 1082 (s), 1044 (w), 1033 (m), 1003 (w), 981 (vw), 951 (w), 891 (w), 870 (w), 856 (w), 827 (m), 798 (w), 759 (vs), 709 (s), 661 (m), 583 (s), 552 (vw), 533 (m), 492 (w), 473 (w), 440 (w), 413 (vw).
Synthesis of [MgK(EtsalO)3]n (5)
Compound 3 (0.75 g, 0.57 mmol) was added to the mixture of 50 ml of EtOH and 20 ml of toluene and heated under reflux to complete dissolution. The reaction mixture was then cooled, concentrated under vacuum to half the volume, and left for crystallization at room temperature. After several weeks of crystallization, the resulting colorless block-like crystals were filtered, washed with hexane (3 × 10 ml) and dried under vacuum. Yield: 0.46 g (72%). Anal. calc. for C27H27O9KMg: C, 58.02; H, 4.87; Mg, 4.35; K 7.00. Found: C, 58.09; H, 4.89; Mg, 4.40; K 6.97. 1H NMR (400 MHz, THF-d8): δ 7.66 (3H, dd, J = 8.0, 1.8 Hz, ArH), 6.94 (3H, m, ArH), 6.66 (3H, m, ArH), 6.17 (3H, m, ArH), 4.13 (6H, q, J = 7.0 Hz, CH2), 1.24 (16H, t, J = 7.0 Hz, CH3). 13C NMR (101 MHz, THF-d8): δ 174.0 (3C, CO), 171.9 (3C, C–O), 135.4 (3C, ArH), 131.6 (3C, ArH), 125.0 (3C, ArH), 114.2 (3C, Ar), 111.6 (3C, ArH), 60.7 (3C, CH2), 14.7 (3C, CH2). FTIR-ATR: 3047 (vw), 3027 (vw), 2978 (w), 2935 (w), 2903 (w), 2868 (vw), 2691 (vw), 2588 (vw), 2324 (vw), 2162 (vw), 1919 (vw), 1822 (vw), 1808 (vw), 1748 (vw), 1670 (w), 1624 (s), 1532 (m), 1507 (w), 1464 (s), 1450 (s), 1396 (m); 1374 (m), 1357 (m), 1326 (m), 1255 (s), 1218 (vs), 1184 (m), 1176 (m), 1157 (s), 1143 (m), 1119 (w), 1088 (s), 1031 (m), 1018 (m), 981 (vw), 951 (w), 895 (m), 872 (vw), 854 (w), 825 (m), 799 (w), 758 (vs), 708 (s), 659 (m), 589 (s), 562 (m), 531 (m), 458 (w), 436 (m), 422 (m).
Synthesis of [Mg6Na4Al(MesalO)13(OH)6(MesalOH)(THF)0.5(H2O)0.5] (6)
Metallic Na (0.092 g, 4 mmol) was added to a solution of MesalOH (1.56 ml, 12 mmol) in 80 ml of THF, and the reaction was stirred at room temperature for 5 h. Then, MgnBu2 (4 ml, 4 mmol) was added dropwise and the reaction was carried out for an additional 12 h. The solution was evaporated under vacuum to half of the volume, and the Schlenk flask was open for contact with atmospheric moisture. After four weeks, colorless plate-like crystals were received. The 4 ml of MgnBu2 used for synthesis contains ca. 1 wt% of AlEt3 (0.0285 g, 0.25 mmol) as a viscosity reducer. The reaction yield was calculated using AlEt3 as the limiting reagent. Yield: 0.29 g (45%). Anal. calc. for C116H113O49AlNa4Mg6: C, 54.51; H, 4.46; Mg, 5.71; Na 3.60; Al, 1.06. Found: C, 54.56; H, 4.49; Mg, 5.74; Na 3.90; Al, 1.02. 1H NMR (400 MHz, THF-d8): δ 10.80 (1H, s, OH), 7.61 (14H, dd, J = 8.1, 1.5 Hz, ArH), 7.02 (14H, m, ArH), 6.66 (14H, m, ArH), 6.19 (14H, m, ArH), 3.65 (39H, s, CH3), 3.62 (4H, m, THF), 2.88–2.60 (6H, s, OH), 1.77 (4H, m, THF). 13C NMR (101 MHz, THF-d8): δ 173.3 (14C, CO), 172.0 (14C, C–O), 134.9 (14C, ArH), 131.4 (14C, ArH), 124.8 (14C, ArH), 114.2 (14C, Ar), 111.8 (14C, ArH), 68.0 (2C, CH2), 51.3 (14C, CH3), 26.2 (2C, CH2). FTIR-ATR (cm−1): 3449 (w), 3020 (vw), 2953 (w), 2851 (vw), 2658 (vw), 2323 (vw), 2150 (vw), 2050 (vw), 1919 (vw), 1811 (vw), 1719 (w), 1689 (m), 1677 (m), 1656 (s), 1600 (m), 1574 (w), 1551 (w), 1539 (m), 1468 (s), 1437 (s), 1379 (w), 1337 (m), 1316 (m), 1260 (m), 1224 (vs), 1193 (s), 1155 (m), 1144 (m), 1084 (s), 1032 (m), 963 (w), 860 (m), 822 (w), 800 (w), 757 (s), 709 (m), 661 (w), 579 (m), 564 (m), 534 (m), 499 (w), 447 (w), 426 (w).
Synthesis of [Mg4Na2(MesalO)6(SalO)2(THF)4] (7)
Compound 2 (0.75 g, 0.62 mmol) was dissolved in 50 ml of THF and 0.022 ml (1.23 mmol) of H2O was added. The reaction mixture was stirred under reflux for 12 h and left for crystallization at room temperature. After several weeks of crystallization, the resulting colorless block-like crystals were filtered, washed with hexane (3 × 10 ml) and dried under vacuum. Yield: 0.28 g (56%). Anal. calc. for C78H82O28Na2Mg4: C, 58.16; H, 5.13; Mg, 6.04; Na 2.85. Found: C, 58.19; H, 5.15; Mg, 6.17; Na 2.89. 1H NMR (400 MHz, THF-d8): δ 8.18 (2H, dd, J = 8.0, 2.0 Hz, ArH), 7.87 (2H, dd, J = 8.3, 1.8 Hz, ArH), 7.57 (2H, dd, J = 8.0, 2.0 Hz, ArH), 7.41 (2H, m, ArH), 7.27 (2H, m, ArH), 7.13 (2H, ddd, J = 8.7, 6.9, 2.0 Hz, ArH), 7.01 (4H, m, ArH), 6.88 (4H, m, ArH), 6.47 (2H, m, ArH), 6.31 (8H, m, ArH), 6.20 (2H, m, ArH), 4.12 (6H, s, CH3), 3.62 (16H, m, CH2), 3.41 (6H, s, CH3), 2.64 (6H, s, CH3), 1.78 (16H, m, CH2). 13C NMR (101 MHz, THF-d8): δ 176.1 (2C, CO), 174.2 (2C, CO), 173.7 (2C, CO), 171.9 (2C, CO), 170.9 (2C, C–O), 170.7 (2C, C–O), 169.6 (2C, C–O), 168.9 (2C, C–O), 135.9 (2C, ArH), 135.4 (2C, ArH), 134.9 (2C, ArH), 134.8 (2C, ArH), 133.1 (2C, ArH), 131.9 (2C, ArH), 131.3 (2C, ArH), 130.7 (2C, ArH), 126.2 (2C, ArH), 124.5 (2C, ArH), 124.0 (2C, ArH), 123.4 (2C, ArH), 120.6 (2C, Ar), 115.5 (2C, Ar), 114.8 (2C, ArH), 114.7 (2C, Ar), 114.6 (2C, ArH), 113.8 (2C, Ar), 112.2 (2C, ArH), 111.6 (2C, ArH), 68.0 (4C, CH2), 67.8 (4C, CH2), 52.6 (2C, CH3), 51.7 (2C, CH3), 51.2 (2C, CH3), 26.4 (4C, CH2), 26.2 (4C, CH2). FTIR-ATR (cm−1): 3057 (vw), 3025 (vw), 2953 (w), 2871 (vw), 2855 (vw), 2594 (vw), 2325 (vw), 2164 (vw), 2053 (vw), 2040 (vw), 1945 (vw), 1676 (m), 1653 (s), 1602 (s), 1572 (m), 1551 (m), 1541 (m), 1469 (s), 1451 (s), 1439 (s), 1402 (w), 1378 (m), 1323 (s), 1260 (m), 1230 (vs), 1195 (m), 1157 (m), 1142 (m), 1089 (m), 1039 (m), 963 (w), 884 (w), 866 (m), 825 (m), 799 (w), 756 (vs), 709 (s), 663 (m), 585 (m), 537 (w), 454 (w).
Synthesis of [Mg4(MesalO)4(OMe)4(HOMe)4] (8)
MgnBu2 (2 ml, 2 mmol) was added dropwise to a solution of MesalOH (0.52 ml, 4 mmol) in 100 ml of MeOH. The mixture was stirred at room temperature for 6 h. The cloudy solution obtained was left overnight. The colorless cuboid-like crystals were crystalized after 24 h on the walls of the Schlenk flask. The resulting crystals were filtered off, washed with hexane (3 × 10 ml), and dried under vacuum. Yield: 1.56 g (82%). Anal. calc. for C40H56O20Mg4: C, 50.36; H, 5.92. Found: C, 50.40; H, 5.94. 1H NMR (400 MHz, THF-d8): δ 8.07–6.84 (8H, m, ArH), 6.84–5.87 (8H, m, ArH), 4.43–3.29 (40H, m, CH3, OH). 13C NMR (101 MHz, THF-d8): δ 173.5–172.3 (4C, CO), 172.1–170.9 (4C, C–O), 135.9–134.8 (4C, ArH), 131.9–139.5 (4C, ArH), 126.8–123.6 (4C, ArH), 115.8–112.8 (4C, Ar; 4C, ArH), 51.8 (4C, CH3) 49.7 (4C, CH3OH). FTIR-ATR (cm−1): 3142 (w), 2952 (w), 2823 (w), 1662 (vs), 1602 (w), 1540 (m), 1468 (m) 1434 (m), 1330 (m), 1261 (m), 1223 (vs), 1193 (m), 1158 (m), 1143 (m), 1083 (m), 1054 (s), 959 (w), 866 (m), 824 (w), 798 (w), 758 (s), 709 (m), 661 (w), 587 (m), 563 (w), 536 (w), 436 (m), 411 (m).
Polymerization procedure
The typical cyclic esters polymerization procedure for PLLA synthesis was as follows. A mixture of cetyl alcohol (0.012 g, 0.0486 mmol) and an appropriate amount of 1–3 or 6–8 calculated based on the number of metal centers M (0.0486 mmol) was introduced into the melted monomer (0.70 g, 4.86 mmol) at 110 °C with a stoichiometry of [L-LA]/[M]/[ROH] = 100/1/1 (Table 1, entries 1–6). The reaction mixtures were stirred for the prescribed time and stopped when the polymer became solid. The resulting materials were cooled and manually ground. A small portion of these materials was dissolved in C6D6 to determine monomer conversion by 1H NMR spectroscopy. The solid PLLAs were washed with hexane to remove the monomer residues. The PLLAs were then dissolved in CH2Cl2, and the resulting solutions were concentrated under a vacuum. Polymers were precipitated from CH2Cl2 solutions using cooled MeOH (−30 °C). Then, the polymers were filtered off and dried under a vacuum.General procedure for the synthesis of benzyl benzoate
To 1 ml of benzaldehyde (9.80 mmol) in a 10 ml Schlenk tube, an appropriate amount of catalysts 1–3 or 6–8 was added at a stoichiometry of [PhCHO]/[M] = 200/1. Reactions were carried out at 120 °C and monitored by 1H NMR from 0.5 to 48 h. The conversion yield of benzyl benzoate was determined from 1H NMR spectra in DMSO-d6 by integration of the signals at 5.29 ppm (s, OCH2, benzyl benzoate) and 9.95 (s, CHO, benzaldehyde).Author contributions
Rafał Petrus: involved in conceptualization, investigation, project administration, funding acquisition, and original draft writing and editing. Tadeusz Lis: single-crystal X-ray diffraction measurements of 4–7. Adrian Kowaliński: technical support.All authors have read and approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Polish National Science Center for financial support, grant number 2017/26/D/ST5/01123. The authors thank Dr J. Utko and Prof. P. Sobota for their helpful comments on the synthesis method examined in this paper and their substantive support.Notes and references
- J. M. Gil-Negrete and E. Hevia, Chem. Sci., 2021, 12, 1982–1992 RSC.
- A. J. Martínez-Martínez, S. Justice, B. J. Fleming, A. R. Kennedy, I. D. H. Oswald and C. T. O'Hara, Sci. Adv., 2017, 3, 1–9 Search PubMed.
- A. J. Martínez-Martínez, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Science, 2014, 346, 834–837 CrossRef PubMed.
- A. J. Martínez-Martínez and C. T. O'Hara, Adv. Organomet. Chem., 2016, 65, 1–46 CrossRef.
- J. Francos, S. Zaragoza-Calero and C. T. O'Hara, Dalton Trans., 2014, 43, 1408–1412 RSC.
- P. E. Eaton, C. H. Lee and Y. Xiong, J. Am. Chem. Soc., 1989, 111, 8016–8018 CrossRef CAS.
- C. Yeardley, A. R. Kennedy, P. C. Gros, S. Touchet, M. Fairley, R. McLellan, A. J. Martínez-Martínez and C. T. O'Hara, Dalton Trans., 2020, 49, 5257–5263 RSC.
- R. Noyori, S. Suga, K. Kawai, S. Okada and M. Kitamura, Pure Appl. Chem., 1988, 60, 1597–1606 CrossRef CAS.
- M. Fairley, L. Davin, A. Hernán-Gómez, J. García-Álvarez, C. T. O'Hara and E. Hevia, Chem. Sci., 2019, 10, 5821–5831 RSC.
- T. X. Gentner, A. R. Kennedy, E. Hevia and R. E. Mulvey, ChemCatChem, 2021, 13, 2371–2378 CrossRef CAS.
- L. Davin, A. Hernán-Gómez, C. McLaughlin, A. R. Kennedy, R. McLellan and E. Hevia, Dalton Trans., 2019, 48, 8122–8130 RSC.
- S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef CAS PubMed.
- R. L.-Y. Bao, R. Zhao and L. Shi, Chem. Commun., 2015, 51, 6884–6900 RSC.
- L. J. Bole, N. R. Judge and E. Hevia, Angew. Chem., Int. Ed., 2021, 60, 7626–7631 CrossRef CAS PubMed.
- N. R. Judge, L. J. Bole and E. Hevia, Chem. – Eur. J., 2021, e202104164 Search PubMed.
- S. Touchet, S. S. R. Kommidi and P. C. Gros, ChemistrySelect, 2018, 3, 3939–3942 CrossRef CAS.
- D. Catel, F. Chevallier, F. Mongin and P. C. Gros, Eur. J. Org. Chem., 2012, 53–57 CrossRef CAS.
- J. Francos, P. C. Gros, A. R. Kennedy and C. T. O'Hara, Organometallics, 2015, 34, 2550–2557 CrossRef CAS.
- J. Farkas, S. J. Stoudt, E. M. Hanawalt, A. D. Pajerski and H. G. Richey, Organometallics, 2004, 23, 423–427 CrossRef CAS.
- R. Noyori, S. Suga, K. Kawai, S. Okada and M. Kitamura, Pure Appl. Chem., 1988, 60, 1597–1606 CrossRef CAS.
- J. E. Chubb and H. G. Richey, Organometallics, 2002, 21, 3661–3666 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., 2016, 72, 171–179 CrossRef CAS PubMed.
- W. Gruszka and J. A. Garden, Advances in heterometallic ring-opening (co)polymerisation catalysis, Nat. Commun., 2021, 12, 3252 CrossRef CAS.
- C. Gallegos, V. Tabernero, F. M. García-Valle, M. E. G. Mosquera, T. Cuenca and J. Cano, Organometallics, 2013, 32, 6624–6627 CrossRef CAS.
- Y. Hua, Z. Guo, H. Han and X. Wei, Organometallics, 2017, 36, 877–883 CrossRef CAS.
- M.-L. Hsueh, B.-T. Ko, T. Athar, C.-C. Lin, T.-M. Wu and S.-F. Hsu, Organometallics, 2006, 25, 4144–4149 CrossRef CAS.
- J. Char, E. Brulé, P. C. Gros, M.-N. Rager, V. Guérineau and C. M. Thomas, J. Organomet. Chem., 2015, 796, 47–52 CrossRef CAS.
- A. J. Plajer and C. K. Williams, ACS Catal., 2021, 11, 14819–14828 CrossRef CAS.
- A. C. Deacy, C. B. Durr, R. W. F. Kerr and C. K. Williams, Catal. Sci. Technol., 2021, 11, 3109–3118 RSC.
- L. Wang, J. Zhang, L. Yao, N. Tang and J. Wu, Inorg. Chem. Commun., 2011, 14, 859–862 CrossRef CAS.
- Y. Sun, L. Wang, D. Yu, N. Tang and J. Wu, J. Mol. Catal. A: Chem., 2014, 393, 175–181 CrossRef CAS.
- M. J. Walton, S. J. Lancaster and C. Redshaw, ChemCatChem, 2014, 6, 1892–1898 CrossRef CAS.
- S. Heitz, J. D. Epping, Y. Aksu and M. Driess, Chem. Mater., 2010, 22, 4563–4571 CrossRef CAS.
- K. Kwapien, J. Paier, J. Sauer, M. Geske, U. Zavyalova, R. Horn, P. Schwach, A. Trunschke and R. Schlögl, Angew. Chem., Int. Ed., 2014, 53, 8774–8778 CrossRef CAS PubMed.
- M. M. Tardío, R. Ramírez, R. González and Y. Chen, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 1–8 CrossRef.
- R. Petrus, J. Utko, R. Gniłka, M. G. Fleszar, T. Lis and P. Sobota, Macromolecules, 2021, 54, 2449–2465 CrossRef CAS.
- M. Pinsky and D. Avnir, Inorg. Chem., 1998, 37, 5575–5582 CrossRef CAS PubMed.
- S. Alvarez, Chem. Rev., 2015, 115, 13447–13483 CrossRef CAS PubMed.
- S. Alvarez, P. Alemany, D. Casanova, J. Cirera, M. Llunell and D. Avnir, Coord. Chem. Rev., 2005, 249, 1693–1708 CrossRef CAS.
- P. J. Calderone, D. Banerjee, L. A. Borkowski and J. B. Parise, Inorg. Chem. Commun., 2011, 14, 741–744 CrossRef CAS.
- X. A. Chen, F. P. Song, X. A. Chang, H. G. Zang and W. Q. Xiao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m983 CrossRef CAS.
- S. Q. Ma, D. Zhao and F. F. Li, Z. Kristallogr. – New Cryst. Struct., 2015, 230, 367–368 CAS.
- U. Kolitsch, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, m129–m133 CrossRef PubMed.
- X. Meng, F. Liang, K. Kang, J. Tang, Q. Huang, W. Yin, Z. Lin and M. Xia, Dalton Trans., 2019, 48, 9048–9052 RSC.
- R. G. Szlag, L. Suescun, B. D. Dhanapala and F. A. Rabuffetti, Inorg. Chem., 2019, 58, 3041–3049 CrossRef CAS PubMed.
- R. W. Saalfrank, N. Mooren, A. Scheurer, H. Maid, F. W. Heinemann, F. Hampel and W. Bauer, Eur. J. Inorg. Chem., 2007, 4815–4822 CrossRef CAS.
- R. Petrus, J. Utko, T. Lis and P. Sobota, Inorg. Chem., 2017, 56, 3324–3334 CrossRef CAS PubMed.
- L. C. Pop and M. Saito, Coord. Chem. Rev., 2016, 314, 64–70 CrossRef CAS.
- V. Nahrstedt, A. Raauf, C. Hegemann, V. Brune, J. Schläfer and S. Mathur, Z. Anorg. Allg. Chem., 2021, 647, 1102–1109 CrossRef CAS.
- A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Dalton Trans., 2010, 39, 9091–9099 RSC.
- I. Huskić, I. V. Pekov, S. V. Krivovichev and T. Friščić, Sci. Adv., 2016, 2, 1–8 Search PubMed.
- H. Riesen and A. D. Rae, Dalton Trans., 2008, 4717–4722 RSC.
- X. S. Tan, Z. S. Ma, N. C. Shi, D. G. Fu, J. Chen and W. X. Tang, J. Chem. Soc., Dalton Trans., 1996, 13, 2735–2738 RSC.
- S. Wuttke, A. Lehmann, G. Scholz, M. Feist, A. Dimitrov, S. I. Troyanov and E. Kemnitz, Dalton Trans., 2009, 4729–4734 RSC.
- H. O. Davies, A. C. Jones, T. J. Leedham, M. J. Crosbie, P. J. Wright, N. M. Boag and J. R. Thompson, Chem. Vap. Deposition, 2000, 6, 71–75 CrossRef CAS.
- G. S. Nichol and W. Clegg, Inorg. Chim. Acta, 2006, 359, 3474–3480 CrossRef CAS.
- F. M. García-Valle, R. Estivill, C. Gallegos, T. Cuenca, M. E. G. Mosquera, V. Tabernero and J. Cano, Organometallics, 2015, 34, 477–487 CrossRef.
- B. M. Day, W. Knowelden and M. P. Coles, Dalton Trans., 2012, 41, 10930–10933 RSC.
- B. M. Day, N. E. Mansfield, M. P. Coles and P. B. Hitchcock, Chem. Commun., 2011, 47, 4995–4997 RSC.
- R. Evans, G. Dal Poggetto, M. Nilsson and G. A. Morris, Anal. Chem., 2018, 90(6), 3987–3994 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef.
- K. Brandenburg, “DIAMOND” Crystal Impact GbR, Bonn, Germany, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data for 4–8 (CIF); NMR, IR, and crystallographic data for 1–8 (PDF). CCDC 2123619–2123623. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00731b |
| This journal is © The Royal Society of Chemistry 2022 |