
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall compound – big colors: synthesis and structural investigation of brightly colored alkaline earth metal 1,3-dimethylviolurates†‡
Volker
Lorenz
a,
Phil
Liebing
 ab,
Matthias
Müller
c,
Liane
Hilfert
a,
Martin
Feneberg
d,
Elias
Kluth
d,
Marcel
Kühling
a,
Magnus R.
Buchner
c,
Rüdiger
Goldhahn
d and
Frank T.
Edelmann
*d
ab,
Matthias
Müller
c,
Liane
Hilfert
a,
Martin
Feneberg
d,
Elias
Kluth
d,
Marcel
Kühling
a,
Magnus R.
Buchner
c,
Rüdiger
Goldhahn
d and
Frank T.
Edelmann
*d
aChemisches Institut, Otto-von-Guericke-Universität Magdeburg, Universitätsplatz 2, 39106 Magdeburg, Germany
bInstitut für Anorganische und Analytische Chemie, Humboldtstr. 8, 07743 Jena, Germany
cFachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Straße 4, 35032 Marburg, Germany
dInstitut für Physik, Otto-von-Guericke-Universität Magdeburg, Universitätsplatz 2, 39106 Magdeburg, Germany. E-mail: frank.edelmann@ovgu.de
First published on 4th May 2022
Abstract
A series of brightly colored alkaline earth metal 1,3-dimethylviolurates M(Me2Vio)2 have been prepared and fully characterized. The title compounds AE(Me2Vio)2·nH2O (AE = Mg, n = 6 (3); AE = Ca, n = 8 (4), AE = Sr, n = 6 (5); AE = Ba, n = 4 (6)) were obtained by neutralizing 1,3-dimethylvioluric acid monohydrate (=H(Me2Vio)·H2O; 2) with 0.5 equiv. of the corresponding metal dihydroxides AE(OH)2. The hair-like appearance of the Sr derivative 5 prevented the growth of single-crystals. This problem could be solved by crystallizing the crown ether derivative Sr(Me2Vio)2(18-crown-6) (5a). The isolated salts exhibit intense colors ranging from red to purple. Various attempts to prepare the beryllium derivative Be(Me2Vio)2 failed. Instead, work-up of the reaction mixtures provided pink crystals of a new modification of 2 formulated as [H3O][Me2Vio] (2b) as shown by an X-ray diffraction study. An unexpected oxidation reaction of the barium salt Ba(Me2Vio)2 led to formation of the novel mixed-anion salt Ba(Me2Vio)(Me2NO2Barb)·2H2O (8, Me2NO2Barb− = 1,3-dimethyl-5-nitrobarbiturate anion). Compound 8 could also be synthesized deliberately by treatment of Ba(OH)2 with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 2 and 1,3-dimethyl-5-nitrobarbituric acid (7, =H(Me2NO2Barb)·H2O). All new compounds were fully characterized by their IR, Raman, NMR (1H, 13C{1H}) and UV-vis spectra as well as elemental analyses. Single-crystal X-ray diffraction studies revealed that the solid-state structures of compounds 3, 4, 5a and 6 are governed by the typical coordination behavior of the alkaline-earth metals, i.e. increasing coordination numbers and a decreasing degree of hydration when going from Mg to Ba. The dimensions of the structures range from hydrogen-bonded ions (3) over monomeric, neutral complex molecules (4, 5a), to polymeric networks (6). The successful isolation of the mixed-anion barium salt 8 adds a new facet to the coordination chemistry of violurate and related ligands.
:1 mixture of 2 and 1,3-dimethyl-5-nitrobarbituric acid (7, =H(Me2NO2Barb)·H2O). All new compounds were fully characterized by their IR, Raman, NMR (1H, 13C{1H}) and UV-vis spectra as well as elemental analyses. Single-crystal X-ray diffraction studies revealed that the solid-state structures of compounds 3, 4, 5a and 6 are governed by the typical coordination behavior of the alkaline-earth metals, i.e. increasing coordination numbers and a decreasing degree of hydration when going from Mg to Ba. The dimensions of the structures range from hydrogen-bonded ions (3) over monomeric, neutral complex molecules (4, 5a), to polymeric networks (6). The successful isolation of the mixed-anion barium salt 8 adds a new facet to the coordination chemistry of violurate and related ligands.
Introduction
Chromic phenomena involving stimulated color changes play an important role in science and technology.1 These phenomena include (but are not limited to) well-known “chromisms” such as photochromism, solvatochromism, thermochromism, electrochromism, and mechanochromism, while typical applications are found e.g. in visual displays, thermochromic paints and thermal papers, optical data storage, self-dimming smart windows, and photochromic eyeglass lenses.1–3 A rather spectacular chromic phenomenon is displayed by the small molecule violuric acid (= H3Vio, compound 1 in Scheme 1).§ Violuric acid (1) is commercially available in the form of its monohydrate, which forms colorless crystals. What makes the compound 1 particularly fascinating is its ability to form brightly colored (e.g. red, purple, and blue) salts with colorless cations, including alkali and alkaline earth metal ions, NH4+ as well as various organoammonium cations.4–13 This effect, known as pantochromism, results from an n → π* transition of the violurate anion in these salts.12–14 During his famous studies on uric acid more than 150 years ago, Adolf Baeyer first reported the preparation of violuric acid by the reaction of barbituric acid with nitrous acid.15–19 Another convenient way to prepare violuric acid involves treatment of alloxan with hydroxylamine hydrochloride.20,21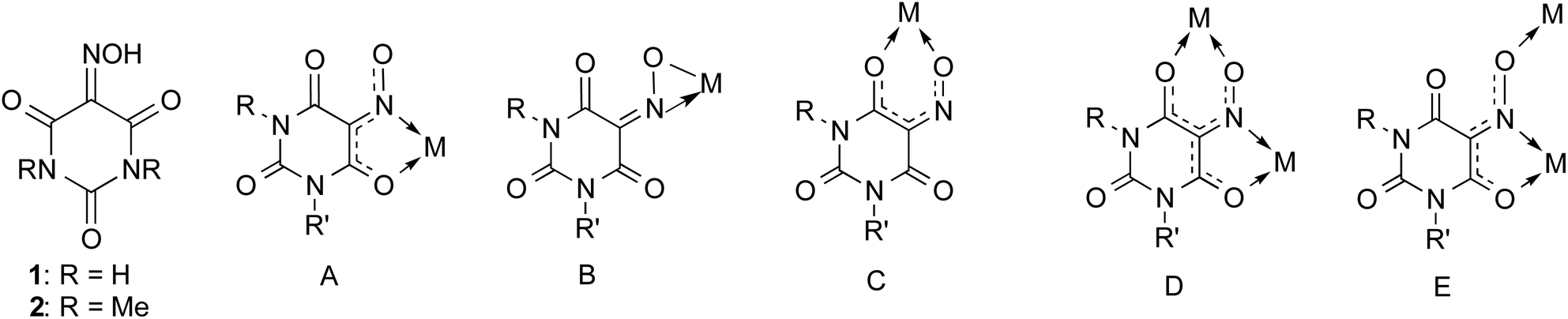 | ||
| Scheme 1 Schematic representation of violuric acids and structurally characterized chelating coordination modes of violurate anions. (A) N,O-Bidentate, κN,O; (B) η2-NO; (C) O,O-bidentate, κO,O′; (D) bis(bidentate)-bridging, κN,O:κO′,O′′; (E) tridentate bridging, κN,O:κO′′; R, R′ = H, Me, and Ph. | ||
Various related classes of organic compounds also show the phenomenon of pantochromism, for example imidovioluric acid derivatives (one C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O unit formally replaced by CNH),22–24 4-thiovioluric acid,25,26 nitrosomalonamide,27 oximidoketones,28 and oximido-oxazolones.29–31 Frequently studied organo-substituted violuric acid derivatives include e.g. 1-methylvioluric acid,32 1,3-dimethylvioluric acid (2)33–35 and 1,3-diphenylvioluric acid.34
O unit formally replaced by CNH),22–24 4-thiovioluric acid,25,26 nitrosomalonamide,27 oximidoketones,28 and oximido-oxazolones.29–31 Frequently studied organo-substituted violuric acid derivatives include e.g. 1-methylvioluric acid,32 1,3-dimethylvioluric acid (2)33–35 and 1,3-diphenylvioluric acid.34
Violurate anions not only form a variety of pantochromic metal salts, but they are also versatile ligands for main-group, transition metal and f-element complexes. Quite often hydrogen-bonding is observed in such coordination compounds which can lead to 1D, 2D and 3D supramolecular self-assembly in the crystalline state.36 In 2019 we published the first comprehensive review article summarizing the coordination chemistry of violurate ligands.37 Due to their multifunctional character, violurate anions usually act either as chelating (bi- or tridentate) or bridging ligands. Structurally characterized coordination modes are illustrated in Scheme 1. Most commonly observed is the κN,O-chelating mode A in which a five-membered chelate ring is formed through coordination of the oxime N atom and a neighboring carbonyl oxygen to the metal atom. Examples for coordination modes B–E have also been reported in the literature, but are more rare.36
1,3-Dimethylvioluric acid (2) and various salt-like 1,3-dimethylviolurates have already been investigated more than 120 years ago.33,38,39 All previously reported salts derived from 2 were found to show pantochromic behavior.7,32 With the only exception of the free acid, none of these derivatives is colorless. Recently we reported the X-ray structural characterization of the full series of alkali metal 1,3-dimethylviolurates M(Me2Vio) (M = Li–Cs).40 It was found that these salts exist as one- (Li, Na), two- (K, Cs), or three-dimensional (Rb) coordination polymers in the crystalline state. In view of this rather diverse structural chemistry of the alkali metal 1,3-dimethylviolurate salts we now investigated the similar series of alkaline earth metal salts AE(Me2Vio)2 (AE = Mg–Ba). All possible alkaline earth metal 1,3-dimethylviolurate salts AE(Me2Vio)2 (AE = Be–Ba) were already mentioned in the early literature. In 1894 Techow first described the barium salt Ba(Me2Vio)2·H2O which was obtained as a “heavy red precipitate” from a reaction of BaCl2 with 2 equiv. of Na(Me2Vio).38 Only one year later (1895) Andreasch reported the preparation of the Mg, Sr, and Ba salts by reaction of 2 equiv. of the free acid with either MgO, SrCO3, or BaCO3.39 In 1909 Hantzsch and Isherwood listed all possible alkaline earth metal 1,3-dimethylviolurate salts ranging from beryllium to barium. Besides the red-brown Be derivative, all other salts were described as red crystalline solids.7
Characterization of the alkaline earth metal 1,3-dimethylviolurate salts described in the early literature was mainly limited to elemental analysis. Thus in the course of the present study we set out to isolate the entire series and fully characterize the compounds through analytical and spectroscopic methods as well as single-crystal X-ray diffraction studies. As in the case of the recently investigated alkali metal 1,3-dimethylviolurates M(Me2Vio) (M = Li–Cs)40 we expected the formation of interesting supramolecular crystal structures. Here we report the results of this study, including failed attempts to prepare the beryllium compound Be(Me2Vio)2 and an unexpected oxidation reaction of Ba(Me2Vio)2 under formation of the novel mixed-anion salt Ba(Me2Vio)(Me2NO2Barb)·2H2O (8, Me2NO2Barb− = 1,3-dimethyl-5-nitrobarbiturate anion).
Results and discussion
Preparation and properties
The starting material 1,3-dimethylvioluric acid 2 (isolated as monohydrate H(Me2Vio)·H2O) was prepared according to the literature procedure by reaction of commercially available 1,3-dimethylbarbituric acid with in situ generated HNO2.41 The preparative results obtained in the present study are illustrated in Scheme 2. | ||
| Scheme 2 Summary of the preparative results reported in this study. | ||
We then set out trying to repeat the original work by Hantzsch and Isherwood.7 For this purpose, commercially available basic beryllium acetate, Be4O(CH3COO)6, was treated with 6 or 8 equiv. of 1,3-dimethylvioluric acid monohydrate (2) in water or ethanol solutions, typically for 3 d at room temperature. In all cases, a pink solution containing a small amount of white precipitate (assumedly BeO or BeCO3) was obtained. Alternatively, we studied reactions of BeSO4·H2O with Ba(Me2Vio)2·H2O in a molar ratio of 1:1. Heating of the two starting materials in water (2 h at 70 °C) resulted in dissolution of the bright red barium salt and (as expected) formation of a heavy white precipitate of BaSO4. In both cases filtration of the reaction mixtures yielded pale pink solutions, from which orange (!), needle-like crystals deposited upon standing at r.t. (denoted here as 2a). Despite the distinct orange color, X-ray diffraction studies of 2a revealed the presence of the starting material 1,3-dimethylvioluric acid monohydrate 2. A rather surprising result was obtained when the orange form of 2 was recrystallized from DMSO. This resulted in the formation of bright pink, needle-like crystals which where suitable for X-ray diffraction. X-Ray analysis revealed the presence of a new form of 1,3-dimethylvioluric acid monohydrate which can be formulated as hydronium 1,3-dimethylviolurate, [H3O][Me2Vio] (2b). Fig. 1 illustrates the markedly different colors of compound 2, 2a, and 2b.
 | ||
| Fig. 1 Two forms of 1,3-dimethylvioluric acid monohydrate (=HMe2Vio·H2O; 2 and 2a) and a sample of hydronium 1,3-dimethylviolurate, [H3O][Me2Vio] (2b). | ||
Compounds 2, 2a and 2b were characterized through IR, 1H NMR and 13C{1H} NMR spectroscopy. The 1H NMR spectra for compounds 2–2b, measured in DMSO-d6, showed two signals for the chemically inequivalent methyl groups bound to nitrogen for 2 at 3.17 and 3.11, for 2a at 3.30 and 3.04 and for 2b at 3.21 and 3.03 ppm. The chemical shifts in the NMR of 2a and 2b are almost the same because 2b was synthesized by recrystallization of 2a in DMSO. The most significant differences between 2 and 2a/2b were found in the 13C{1H} NMR. The signals for the CN moiety were observed for 2 at 135.6 ppm and for 2a/2b the signal is shifted to 142.4/143.1 ppm.
After all, our findings cast serious doubts on the existence of the previously reported compound beryllium 1,3-dimethylviolurate. However, they produced the exciting result that a new form of 1,3-dimethylvioluric acid monohydrate (2b) can be isolated and structurally characterized.
:1 in methanol/water mixtures according to Scheme 2. In all cases the reaction mixtures quickly turned red or purple, and the crystalline alkaline earth 1,3-dimethylviolurates were readily isolated as precipitates. Isolated yields were in the range of 71% (5) to 83% (6).
In the case of the Mg (3), Ca (4), and Ba (6) salts, single crystals suitable for X-ray diffraction formed upon standing of the filtered mother liquid at room temperature for a few days. A notable exception is the Sr derivative 5. As early as 1895, Andreasch reported that this salt formed a matted, hair-like mass, which could be taken out of the reaction mixture by wrapping it around a glass rod.39 This behavior was fully confirmed in the course of the present study. The hair-like appearance of compound 5 made it impossible to grow single-crystals of reasonable size which were suitable for X-ray diffraction. For example, crystallization from DMSO and diglyme or mixtures of DMSO and diglyme resulted again in formation of the hair-like pink microcrystals which were unsuitable for X-ray studies. A solution to this problem was finally found in the complexation of the strontium ion with 18-crown-6. Addition of 1 equiv. of 18-crown-6 to a solution of 5 in DMSO resulted in crystallization of the new salt Sr(Me2Vio)2(18-crown-6) (5a) in the form of small, dark pink crystals. These crystals were again needle-like, but individual single-crystals were significantly more compact than those of 5, so that an X-ray diffraction study of 5a became possible. Fig. 2 illustrates the colors of the isolated title compounds 3–6.
 | ||
| Fig. 2 Characteristic solid-state colors of the title compounds 3–6. | ||
All new 1,3-dimethylviolurate salts 3–6 were fully characterized through elemental analyses as well as IR, Raman, 1H NMR and 13C{1H} NMR spectroscopy. The most prominent IR bands could be readily assigned (cf. Experimental section).45 Raman spectra of 3–6 show broad unstructured bands around 1400 cm−1 whose width hampers any further interpretation. Most remarkably, sharp peaks at around 750 cm−1 are observed for all compounds investigated and additionally around 1050 cm−1 for 5–6. Both sets of sharp Raman signals systematically shift to higher wavenumbers with increasing AE metal atomic mass. For compounds 5 and 5a, both peaks are found at the identical wavenumber, indicating that these peaks have a metal-related origin. However, it has to be mentioned that compound 1 as a reference also shows a Raman peak around 700 cm−1 though none around 1050 cm−1. A mode at 644 cm−1 was convincingly assigned to a ring breathing mode in violuric acid in an earlier study,46 in our spectra this mode seems shifted to higher wavenumbers. Due to the strong luminescence background of all colored salts, we tried to reproduce these Raman signals in the ultraviolet. Unfortunately, these attempts were unsuccessful; all compounds yielded very similar spectra without the sharp peaks observed under green laser excitation.
1H NMR spectra of compound 3–6, measured in DMSO-d6, showed the typical signals for the two inequivalent methyl groups which were observed around 3.23 and 3.00 ppm. The 1H NMR of compound 5a showed the signal of the CH2 moiety of the crown ether. The 13C{1H} NMR spectra are also very similar in the series of compounds 3–6. The additional signal of the CH2 unit of the crown ether in complex 5a was detected at δ = 69.9 ppm.
Previous studies had shown that the different colors of violurate and 1,3-dimethylviolurate salts (e.g. red, purple, blue) are primarily a solid-state phenomenon.9,12–14,40 Even the bright blue rubidium and cesium violurates and dimethylviolurates dissolve in water or DMSO with the same pink-violet color of the free violurate anions. Thus it did not come as a surprise that the UV-vis spectra of the title compounds revealed the same effect. Fig. 3 displays the UV-vis spectra of free 1,3-dimethylvioluric acid (2) and the alkaline earth metal salts 3–6 in aqueous solutions. Solvatochromism as reported for thyraminium violurate13 was not observed.
 | ||
| Fig. 3 UV-vis spectra of compounds 2–6. | ||
:1:1. Heating of the reaction mixture to 75 °C for 1 h led to formation of a clear, pink-violet solution which upon cooling to r.t. deposited red-violet plate-like crystals of Ba(Me2Vio)(Me2NO2Barb)·2H2O (8) in 71% yield. IR and NMR spectra of 8 differed only slightly from those of the original barium violurate salt 6. A broad IR band at 1436 cm−1 could be assigned to the ν(NO) vibrations. The 1H NMR spectrum of 8 differed from that of 6 in that it showed three signals for the two inequivalent methyl groups of the Me2Vio ligand at 3.24 and 3.03 ppm plus a new resonance with the double relative intensity at 3.08 ppm which was due to the chemically equivalent methyl groups of the Me2NO2Barb− ligand. The coordinated water molecules gave rise to a singlet at 3.37 ppm. In the 13C{1H} NMR spectrum of 8 the 1,3-dimethyl-5-nitrobarbiturate anion of 8 gave rise to additional signals at δ = 157.5 (CO), 151.0 (CO), 113.8 (C–NO2) and 27.3 (CH3) ppm.
Molecular and crystal structures
Crystallization of the title compounds 2b, 3, 4, 5a, 6 and 8 is outlined in the Experimental section. Details of the crystal structure analyses are summarized in Table 1. Fig. 4 depicts the asymmetric unit in the crystal structure of the pink, orthorhombic modification of 1,3-dimethylvioluric acid monohydrate (2b), containing two formula units. Structure refinement disclosed that all the acidic hydrogen atoms are most likely located around the water oxygen atoms, and consequently, compound 2b can formally be denoted as “hydronium 1,3-dimethylviolurate”. By contrast, the previously reported colorless, monoclinic modification of HMe2Vio·H2O (2) contains neutral NOH moieties and H2O molecules.47 However, it must be pointed out that the validity of hydrogen positions based on X-ray data are limited and neutron diffraction experiments would be necessary to obtain more reliable data.
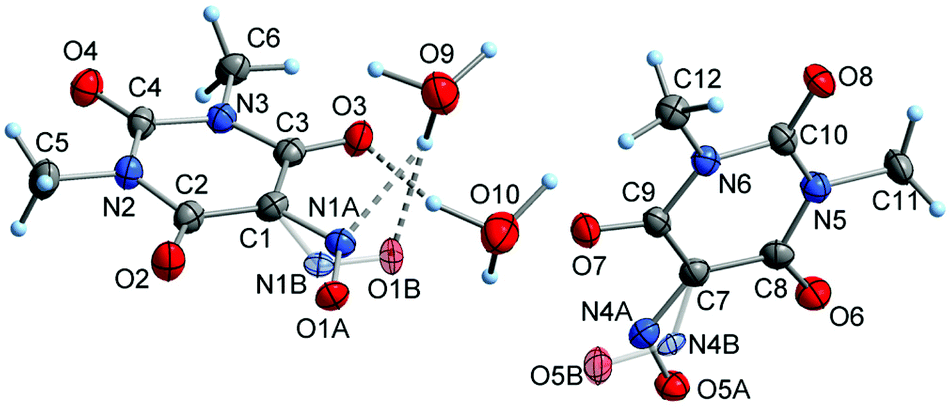 | ||
| Fig. 4 Asymmetric unit in the crystal structure of the orthorhombic modification of 1,3-dimethylvioluric acid – monohydrate (formally denoted as “hydronium 1,3-dimethylviolurate”; 2b), showing the atom numbering scheme and disorder of the NO groups. Displacement ellipsoids are drawn with 50% probability and hydrogen atoms as spheres of arbitrary size. | ||
| a Due to the absence of heavy atoms, the Flack test results are meaningless. | ||||||
|---|---|---|---|---|---|---|
| Compound | 2b | 3 | 4 | 5a | 6 | 8 |
| CCDC deposition number | 2111883 | 2111884 | 2111885 | 2111886 | 2111887 | 1865464 |
| Molecular formula sum | C6H9N3O5 | C12H24MgN6O14 | C12H28CaN6O16 | C24H36N6O14Sr | C12H20BaN6O12 | C12H16BaN6O11 |
| Formula weight/g mol−1 | 203.16 | 500.68 | 552.48 | 720.21 | 577.68 | 557.65 |
| Crystal system | Orthorhombic | Triclinic | Monoclinic | Orthorhombic | Triclinic | Triclinic |
| Space group | Pna21 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c | Pbca |
P |
P |
| Cell metric a/Å | 14.9362(5) | 7.1355(7) | 15.1065(5) | 7.2969(2) | 8.2100(3) | 7.0946(3) |
| b/Å | 4.5201(1) | 8.2367(8) | 12.2974(4) | 15.7231(7) | 8.3910(3) | 8.3205(4) |
| c/Å | 25.0667(7) | 9.7745(9) | 24.2005(7) | 24.7319(7) | 14.5232(5) | 15.0306(7) |
| α/deg. | 90 | 93.769(7) | 90 | 90 | 82.535(3) | 85.940(4) |
| β/deg. | 90 | 102.940(7) | 92.938(2) | 90 | 73.457(3) | 84.382(4) |
| γ/deg. | 90 | 112.708(7) | 90 | 90 | 86.207(3) | 83.417(3) |
| Cell volume/Å3 | 1692.33(8) | 509.03(9) | 4489.8(2) | 2837.5(2) | 950.51(6) | 875.59(7) |
| Molecules per cell z | 8 | 1 | 8 | 4 | 2 | 2 |
| Electrons per cell F000 | 848 | 262 | 2320 | 1488 | 572 | 548 |
| Calcd density, ρ/g cm−3 | 1.595 | 1.633 | 1.635 | 1.686 | 2.018 | 2.115 |
| μ/mm−1 (Mo-Kα) | 0.140 | 0.175 | 0.371 | 1.984 | 2.169 | 2.347 |
| Crystal shape and color | Pink plate | Pink plate | Red prism | Pink rod | Pink plate | Purple |
| Crystal size/mm | 0.62 × 0.14 × 0.06 | 0.51 × 0.33 × 0.10 | 0.40 × 0.25 × 0.22 | 0.26 × 0.10 × 0.06 | 0.17 × 0.17 × 0.05 | 0.25 × 0.22 × 0.04 |
| T/K | 133(2) | 133(2) | 133(2) | 133(2) | 133(2) | 153(2) |
| θ range/deg. | 2.727–25.999 | 2.169–25.993 | 2.271–26.996 | 2.591–25.993 | 2.449–29.158 | 2.469–26.996 |
| Reflections collected | 14231 |
4579 | 14608 |
24932 |
13063 |
8147 |
| Reflections unique | 3245 | 1994 | 4900 | 2779 | 5084 | 3800 |
| Reflections with I > 2σ(I) | 2805 | 1833 | 3993 | 2066 | 4672 | 3466 |
| Completeness of dataset | 99.9% | 99.7% | 99.7% | 99.9% | 99.1% | 99.7% |
| Absorption correction | None | None | None | Numerical58 | Numerical58 | Numerical58 |
| R int | 0.0741 | 0.0468 | 0.0584 | 0.0956 | 0.0446 | 0.0387 |
| Parameters; restraints | 213; 31 | 172; 6 | 383; 20 | 207; 0 | 308; 8 | 292; 4 |
| R 1 (all data, I > 2σ(I)) | 0.0608; 0.0486 | 0.0451; 0.0422 | 0.0522; 0.0393 | 0.0703; 0.0464 | 0.0372; 0.0324 | 0.0268; 0.0225 |
| wR2 (all data, I > 2σ(I)) | 0.1319; 0.1192 | 0.1192; 0.1148 | 0.1152; 0.1051 | 0.1230; 0.1028 | 0.0806; 0.0781 | 0.0535; 0.0525 |
| GooF (F2) | 1.084 | 1.049 | 1.069 | 1.143 | 1.094 | 1.036 |
| Max. residual peaks | −0.357; 0.328 | −0.335; 0.331 | −0.399; 0.308 | −0.655; 1.357 | −0.938; 2.009 | −0.827; 0.529 |
| Extinction coefficient | — | 0.09(2) | — | — | — | 0.0034(6) |
| Flack parameter | −0.3(1)a | — | — | — | — | — |
The crystal structure of magnesium 1,3-dimethylviolurate – hexahydrate (3) contains separated [Mg(H2O)6]2+ ions (Fig. 5). The two symmetry-equivalent HMe2Vio− anions do not contribute to metal coordination and are fixed by O–H⋯O and O–H⋯N hydrogen bonds with NO and CO acceptor groups, resulting in the formation of a three-dimensional, supramolecular network structure (Fig. 6). The [Mg(H2O)6]2+ ion, which displays a typical octahedral metal coordination, has been reported for numerous hydrated magnesium salts with other anions deposited in the Cambridge Structural Database (CSD).48
 | ||
| Fig. 5 A pair of Me2Vio− and [Mg(H2O)6]2+ ions observed in the crystal structure of magnesium 1,3-dimethylviolurate – hexahydrate (3), showing the atom numbering scheme. Displacement ellipsoids are drawn with 50% probability and hydrogen atoms as spheres of arbitrary size. The cation exhibits crystallographically exposed inversion symmetry with the inversion center () being located on the Mg atom. | ||
 | ||
| Fig. 6 Illustration of the coordination sphere around the Mg atom and adjacent hydrogen-bonded Me2Vio− ions in the crystal structure of [Mg(H2O)6](Me2Vio)2 (3). | ||
The calcium salt Ca(Me2Vio)2·4H2O (4) crystallizes with one formula unit in the asymmetric unit (Fig. 7). Different from the magnesium salt 3, the two 1,3-dimethylviolurate anions are coordinated to the metal ion in a chelating κ2O,O′-mode involving the NO moiety and an adjacent carbonyl group. All four H2O molecules in the asymmetric unit are terminally coordinated to calcium. The resulting eight-coordination of the central metal atom is remarkably symmetric and best described as square-antiprismatic, with the two Me2Vio− ligands defining one base area and the four H2O ligands the other (angle between planes: 2.68(4)°). The Ca–O(Me2Vio) and the Ca–OH2 bonds are very similar in length and cover ranges of 239.2(1)–247.3(1) pm and 236.8(2)–248.2(2) pm, respectively. A very similar coordination has been observed in the crystal structure of the related hydrated calcium violurate (Ca–O(H2Vio) 241.9(1)–243.3(1) pm, Ca–OH2 242.2(1)–247.6(1) pm).49 Comparable Ca–O distances as in compound 4 have been observed for other hydrated calcium salts with oxygen donor anions deposited in the CSD, where the coordination number of calcium is larger than six.48
 | ||
| Fig. 7 Molecular structure of calcium 1,3-dimethylviolurate – tetrahydrate (4), showing the atom numbering scheme. Displacement ellipsoids are drawn with 50% probability and hydrogen atoms as spheres of arbitrary size. | ||
The crystal structure of the crown-ether-complexed strontium salt Sr(Me2Vio)2(18-crown-6) (5a) contains distinct monomeric complex molecules having crystallographically imposed inversion symmetry (Fig. 8). The Sr atom is situated in the center of the hexadentate 18-crown-6 ring (Sr–O 270.0(3)–274.0(3) pm) and is sterically saturated by the NO donor groups of two symmetry-equivalent 1,3-dimethylviolurate ligands. The Sr–O(Me2Vio) bond lengths are 251.6(3) pm and therefore comparable with rather strong bonds of strontium to other oxygen anions, e.g. carboxylates (which are typically in a range of ca. 250–280 pm).48 Notably, the NO group is coordinated to strontium in a tilted orientation, resulting in a relatively short Sr–N contact of 283.2(4) pm. It can be regarded as a comparatively weak coordinative Sr–N contact, as Sr–N bonds cover a range of ca. 240–300 pm in the CSD.48 That is, the coordination of the NO group can be described as an intermediate between η1(O)- and η2(N,O)-coordination. The η2-mode is rare for metal violurates and has previously only been seen with cesium 1,3-dimethylviolurate to the best of our knowledge.40
 | ||
| Fig. 8 Molecular structure of the complex strontium salt 5a, showing the atom numbering scheme. Displacement ellipsoids are drawn with 50% probability and hydrogen atoms as spheres of arbitrary size. The molecule exhibits crystallographically exposed inversion symmetry with the inversion center () being located on the Sr atom. | ||
Similarly to the calcium salt 4, barium 1,3-dimethylviolurate crystallizes as a tetrahydrate (6) with one formula moiety in the asymmetric unit (Fig. 9). The bonding environments of the two crystallographically inequivalent Me2Vio− anions are very different. One of the 1,3-dimethylviolurate moieties displays a mixture of chelating and bridging coordination involving two symmetry-equivalent Ba atoms (κ3O,O′:O′′), while the other 1,3-dimethylviolurate anion is not coordinated to barium and only fixed by O–H⋯O hydrogen bonding with H2O. The H2O ligands also display different coordination patterns and only one of them is coordinated in a terminal mode. The other three H2O ligands are in a μ-bridging coordination in order to achieve steric saturation of the large barium ion. As a result of the bridging coordination of 1,3-dimethylviolurate and water, a two-dimensional polymeric structure is formed in the crystal. The Ba atom adopts a rather irregular ten-coordination with Ba–O separations of 279.0(3)–281.9(2) pm (Ba–O(Me2Vio)), 270.7(3) pm (Ba–OH2, terminal), and 279.9(3)–299.9(2) pm (Ba–(μ-OH2)). In the previous literature, a mixed-anion salt of barium with violurate (H2Vio−) has been reported.50 In this compound, the Ba atom adopts a nine-coordination with the same coordination mode and very similar bond lengths as observed in 6 (κ3O,O′:O′′; Ba–O(H2Vio) 279.4(5)–286.7(6) pm).
 | ||
| Fig. 9 The asymmetric unit in the crystal structure barium 1,3-dimethylviolurate – tetrahydrate (6), showing the atom numbering scheme. Displacement ellipsoids are drawn with 50% probability and hydrogen atoms as spheres of arbitrary size. Bonds to symmetry-related atoms are indicated by semi-transparent lines. | ||
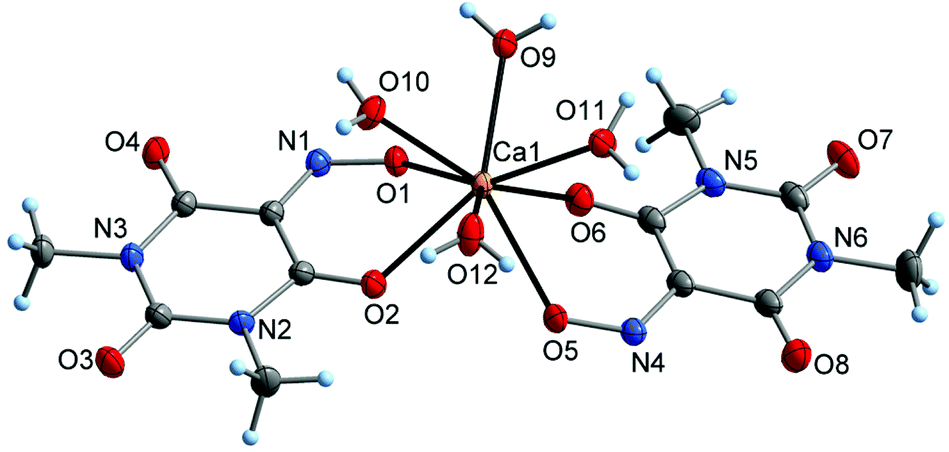
X-Ray diffraction studies confirmed the identity of the by-product obtained during the synthesis of 6 and the deliberately synthesized mixed-anion salt Ba(Me2Vio)(Me2NO2Barb)·2H2O (8; Me2NO2Barb− = 1,3-dimethyl-5-nitrobarbiturate). In the crystal structure of 8, the Ba atom adopts an irregular ten-coordination by oxygen atoms of the Me2Vio−, Me2NO2Barb−, and H2O ligands (Fig. 10). The Me2Vio− anion displays the same coordination mode as in the binary barium salt 6, κ3O,O′:O′′. The corresponding Ba–O bond lengths are 271.8(2)–285.4(2) pm and therefore similar to those observed for 6. The Me2NO2Barb− ligand is in a dual-chelating κ3O,O′:O,O′′ coordination mode, involving a nitro group (Ba–O 283.6(2)–301.4(2) pm) and a carbonyl group (Ba–O 273.5(2) pm). While the latter bond is similar in lengths as the related Ba–O(Me2Vio) bonds in 6, the Ba–O2N bonds are relatively long. This finding can possibly be addressed to the electron-poor nature of the nitro group. Crystallographically characterized salts with the 1,3-dimethyl-5-nitrobarbiturate anion are not known to the best of our knowledge, but the previous literature contains several examples of salts with the unsubstituted 5-nitrobarbiturate anion, e.g. with [AE(H2O)6]2+ (AE = Mg, Ca),51 and with different ammonium cations.52,53
 | ||
| Fig. 10 The asymmetric unit in the crystal structure of the mixed-anion barium salt Ba(Me2Vio)(Me2NO2Barb)·2H2O (7), showing the atom numbering scheme. Displacement ellipsoids are drawn with 50% probability and hydrogen atoms as spheres of arbitrary size. Bonds to symmetry-related atoms are indicated by semi-transparent lines. | ||
Conclusions
In summarizing the results reported in this contribution, a series of brightly colored alkaline earth metal 1,3-dimethylviolurates AE(Me2Vio)2(H2O)n (AE = Mg, n = 6 (3); AE = Ca, n = 8 (4), AE = Sr, n = 6 (5); AE = Ba, n = 4 (6)) as well as Sr(Me2Vio)2(18-crown-6) (5a) and Ba(Me2Vio)(Me2NO2Barb)·2H2O (8) have been fully characterized by their IR, Raman and NMR (1H, 13C{1H}) spectra, elemental analyses, and single-crystal X-ray diffraction. The colors of the crystalline salts range from red to purple and show less diversity than the corresponding alkali metal 1,3-dimethylviolurates where the salts of the heavier elements K, Rb, and Cs are bright blue.40 In contrast, the colors of the crystalline title compounds are mainly red to red-violet. Various attempts to synthesize the corresponding beryllium derivative failed, although a red-brown material of the composition Be(Me2Vio)2 had been mentioned in the early literature. Instead, work-up of the aqueous reaction mixtures yielded a new form of 1,3-dimethylvioluric acid monohydrate which can be formulated as [H3O][Me2Vio] (2b). While single crystals of the Mg, Ca, and Ba salts (3, 4, 6) could be readily obtained from the concentrated mother liquids of the original reactions mixture, crystallization of the Sr derivative 5 proved impossible due to the formation of “mats” of hair-like microcrystals. However, single-crystal X-ray diffraction could be carried out using the 18-crown-6 derivative Sr(Me2Vio)2(18-crown-6) (5a). The solid-state structures of compounds 3, 4, 5a and 6 are governed by the typical coordination behavior of the alkaline-earth metals, comprising increasing coordination numbers and a decreasing degree of hydration in going from Mg to Ba. The dimensions of the structures range from hydrogen-bonded ions (3) over monomeric, neutral complex molecules (4, 5a), to polymeric networks (6). Finally, the successful isolation and characterization of the mixed-anion barium salt 8 adds a new facet to the coordination chemistry of violurate and related ligands. Crystal structure elucidation of 8 showed that the 1,3-dimethylviolurate anion is readily replaced by its oxidation product 1,3-dimethyl-5-nitrobarbiturate.Experimental
General and materials
All operations were performed under ambient conditions without exclusion of air unless stated otherwise. The starting materials 1,3-dimethylbarbituric acid (Aldrich), 18-crown-6 (Merck) and AE(OH)2 or their hydrates (AE = Mg–Ba, 97% purity, Aldrich, Grüssing, Merck) were obtained from commercial suppliers and used as received. BeCl2 and BeBr2 were synthesized according to the literature,54 while BeCO3, BeSO4 (Alfa Aesar) and Be4O(CH3COO)6 (Carl Roth) were obtained from commercial suppliers. 1,3-Dimethylvioluric acid (=H(Me2Vio)·H2O; 2)41 and 1,3-dimethyl-5-nitrobarbituric acid (=H(Me2NO2Barb)·H2O; 7)38 were prepared according to the published procedures. 1H and 13C{1H} NMR spectra were recorded in DMSO-d6 solutions on a Bruker-Biospin AVIII 400 MHz spectrometer at 25 °C, while the beryllium-containing compounds were measured on a Bruker-Biospin HD300 spectrometer. Chemical shifts are referenced to tetramethylsilane. IR spectra were measured with a Bruker-Optics VERTEX 70v spectrometer equipped with a diamond ATR unit between 4000 cm−1 and 50 cm−1. Unpolarized Raman spectra were measured in back scattering geometry under an optical microscope. The Raman signal was dispersed by a grating monochromator and recorded by a Peltier cooled charge coupled device camera. As excitation a frequency doubled YAG:Nd laser was used having a wavelength of 532 nm. UV/Vis spectra were measured with aqueous solutions using a Cary 4E UV/Vis spectrometer at wavelengths of 250–800 nm. All spectra were recorded without using a polarization analyzer. Microanalysis (C, H, N) of the compounds were performed using a “vario EL cube” apparatus from Elementar Analysensysteme GmbH. Melting/decomposition points were measured on a Büchi Melting Point B-540 apparatus. The single-crystal X-ray intensity data were collected on a STOE IPDS 2T diffractometer equipped with a 34 cm image-plate detector, using Mo-Kα radiation. The crystal structures were solved with SHELXT-2018/355 and refined by full matrix least-squares methods on F2 with SHELXL-2018/3,56 using the Olex 1.2 environment.57 CCDC 1865464 and 2111883–2111887‡ contain the supplementary crystallographic data for this paper (see Table 1).Orange form of H(Me2Vio)2·H2O (2a)
(a) 1.63 g (8.0 mmol) of 2 dissolved in ca. 75 mL of ethanol were added to a solution of 0.41 g (1.0 mmol) Be4O(CH3COO)6 in 50 ml of hot water. The color of the suspension changed to pink. The mixture was stirred for 3 d at room temperature. A white precipitate was filtered off, the resulting clear pink solution was allowed to evaporate. During two weeks the volume of the solution was reduced to one half and orange, needle-like crystals of H(Me2Vio)2·H2O (2a) were formed. Yield: 1.40 g (86%). Mp 128 °C (dec.). C6H9N3O5 (203.15): calc. C 35.47, H 4.47, N 20.68; found: C 35.28, H 4.65, N 20.93%.(b) 2.88 g (5.0 mmol) Ba(Me2Vio)2·4H2O and 0.9 g (5.1 mmol) BeSO4·4H2O were suspended in 100 ml water. The suspension was stirred and heated for 2 h to a temperature of ca. 70 °C. During that time a heavy white precipitate was formed. After filtration the clear pink solution was allowed to evaporate. During one week the volume of the solution was reduced to 30 ml, and orange, needle-like crystals of H(Me2Vio)2·H2O (2a) were formed. Yield: 0.85 g (84%). Mp 129 °C (dec.). C6H9N3O5 (203.15): calc. C 35.47, H 4.47, N 20.68; found: C 35.20, H 4.75, N 21.02%. 1H NMR (400.1 MHz, DMSO-d6, 20 °C): δ = 3.60 (s, 16H, H2O), 3.21 (s, 3H, CH3), 3.04 (s, 3H, CH3) ppm. 13C{1H} NMR (100.6 MHz, DMSO-d6, 21 °C): δ = 162.1 (NNCO), 151.3 (CO), 151.1 (CO), 142.4 (CN), 27.6 (CH3), 26.7 (CH3) ppm. IR (ATR): ν = 3505 m(br) ν(H2O), 3460 m(br) ν(H2O), 2957 w ν(C–H), 1726 w, 1652 s ν(CO), 1505 w, 1453 m, 1416 m ν(NO), 1369 m, 1279 m, 1209 m, 1044 m, 983 m, 961 m, 809 m, 789 m, 749 m, 716 m, 682 m, 634 m, 592 m, 550 m, 496 m, 403 m, 363 s, 271 m, 261 m, 247 m, 118 s, 103 s, 66 m cm−1.
[H3O][Me2Vio] (2b)
0.25 g of 2a were dissolved in 10 ml DMSO. The pink-violet solution was placed in a vial, 5 ml Et2O were added to form a second layer, the vial was sealed and placed for one week at room temperature then for 24 hours at ca. 5 °C and again for about one month at room temperature to form thin pink, needle-like crystals which were suitable for X-ray crystal structure measurement. Yield: 0.13 g (52%). Mp 131 °C (dec.). C6H9N3O5 (203.15): calc. C 35.47, H 4.47, N 20.68; found: C 35.56, H 4.56, N 20.89%. 1H NMR (400.1 MHz, DMSO-d6, 20 °C): δ = 3.21 (s, 3H, CH3), 3.03 (s, 3H, CH3) ppm. 13C{1H} NMR (100.6 MHz, DMSO-d6, 21 °C): δ = 162.6 (NNCO), 151.4 (CO), 151.0 (CO), 143.1 (CN), 27.6 (CH3), 26.6 (CH3) ppm. IR (ATR): ν = 3475 m(br) ν(H–O), 3188 m(br) ν(H–O), 3090 m(br) ν(H–O), 2964 w ν(C–H), 1653 s ν(CO), 1622 vs ν(CO), 1516 w, 1415 s ν(NO), 1384 s, 1363 s, 1317 m, 1263 s, 1219 s, 1054 m, 974 m, 926 m, 807 m, 784 m, 754 s, 732 m, 641 m, 502 s, 479 m, 412 s, 386 m, 370 m, 348 m, 288 m, 191 s, 167 s, 110 s, 85 s cm−1.
Non-coordinating solvents. BeCl2 or BeBr2 (0.04 mol) and 14.8 mg (0.08 mmol) of anhydrous 1,3-dimethylvioluric acid or 16.2 mg of 2 (0.08 mmol) were weighed into a J. Young NMR tube in an argon filled glove box. Subsequently, 0.45 ml of CD2Cl2 were added with an adjustable air displacement pipette. This led to the instantaneous formation of large amount of precipitate. No NMR signals of the ligand or a formed complex could be detected.
Coordinating solvents. BeCl2 or BeBr2 (0.04 mol) and 14.8 mg (0.08 mmol) of anhydrous 1,3-dimethylvioluric acid were weighed into a J. Young NMR tube in an argon filled glove box. Subsequently, 0.45 ml of THF or DMF were added with an adjustable air displacement pipette. The solvent of the obtained colorless solutions was reduced in vacuo. This led to the precipitation of the known solvates BeCl2(THF)2, [Be(DMF)4]Cl2 and [Be(DMF)4]Br2, respectively. Also addition of an auxiliary base like pyridine or NEt3, did not produce beryllium-1,3-dimethylviolurates.
Forcing conditions. BeCl2, BeBr2 or BeCO3 (0.04 mol) and 14.8 mg (0.08 mmol) of anhydrous 1,3-dimethylvioluric acid were weighed into a J. Young NMR tube in an argon filled glove box. Subsequently 0.45 ml of dimethoxyethane were added with an adjustable air displacement pipette and the reaction mixtures heated to 100 °C for 6 weeks. Upon cooling to ambient temperature colorless crystals of MeHN(CO)CONHMe deposited on the glass wall.
[Mg(H2O)6](Me2Vio)2 (3)
2.03 g (10.0 mmol) of 2 dissolved in ca. 100 mL of hot methanol were added to a suspension of 0.29 g (5.0 mmol) Mg(OH)2 in 50 ml of hot water. Immediately the color of the suspension turned to red. The mixture was stirred for 3 d, and the red precipitate was filtered off, washed with methanol and dried at r.t. Pink plate-like single crystals of [Mg(H2O)6](Me2Vio)2 (3) were formed in the mother liquor after standing for 1 week at 20 °C. Yield: 1.8 g (72%). Mp 124 °C (dec.). C12H24MgN6O14 (M = 500.66): calcd C 28.79, H 4.83, N 16.79; found: C 28.45, H 4.98, N 15.34%. 1H NMR (400.1 MHz, DMSO-d6, 20 °C): δ = 3.60 (s, 16H, H2O), 3.23 (s, 6H, CH3), 3.05 (s, 6H, CH3) ppm. 13C{1H} NMR (100.6 MHz, DMSO-d6, 21 °C): δ = 162.4 (NNCO), 153.1 (CO), 151.0 (CO), 143.8 (CN), 27.7 (CH3), 26.9 (CH3) ppm. IR (ATR): ν = 3500 m(br) ν(H2O), 3436 m(br) ν(H2O), 3308 w ν(H2O), 3028 w, 2969 w ν(C–H), 1688 m ν(CO), 1652 m ν(CO), 1616 vs ν(CO), 1523 w, 1481 m, 1431 s ν(NO), 1397 m, 1328 w, 1287 s, 1261 s, 1233 s, 1081 m, 1055 m, 972 w, 940 w, 807 m, 777 m, 727 m, 690 s, 633 s, 596 m, 509 s, 478 m, 447 w, 415 s, 396 m, 339 m, 350 m, 289 m, 261 w, 241 w, 198 m, 177 m, 162 m, 121 s, 100 s, 66 m cm−1. Raman (532 nm): ν = 718 w, 1422 w cm−1.
Ca(Me2Vio)2·8H2O (4)
2.03 g (10.0 mmol) of 2 dissolved in ca. 100 ml hot methanol were added to a suspension of 0.37 g (5.0 mmol) Ca(OH)2 in 50 ml hot water. The resulting mixture was stirred over night and the red precipitate was filtered off, washed with methanol and dried at r.t. Red-orange prisms of Ca(Me2Vio)2·8H2O (4) were formed in the mother liquor after standing for 2 weeks at 20 °C. Yield: 2.1 g (76%). Mp 185 °C (dec.). C12H28CaN6O16 (M = 552.48): calcd C 26.09, H 5.11, N 15.21; found: C 26.95, H 4.92, N 15.74%. 1H NMR (400.1 MHz, DMSO-d6, 20 °C): δ = 3.60 (s, 16H, H2O), 3.23 (s, 6H, CH3), 3.05 (s, 6H, CH3) ppm. 13C{1H} NMR (100.6 MHz, DMSO-d6, 21 °C): δ = 163.7 (NNCO), 151.6 (CO), 151.5 (CO), 145.2 (CN), 27.8 (CH3), 26.8 (CH3) ppm. IR (ATR): ν = 3496 m(br), 2957 w ν(C–H), 1725 m ν(CO), 1621 vs ν(CO), 1514 w, 1440 s ν(NO), 1401 m, 1276 s, 1231 s, 1080 s, 1056 m, 974 m, 945 m, 809 m, 784 s, 752 s, 729 m, 694 m, 632 w, 612 m, 602 w, 594 w, 505 vs, 448 m, 409 vs, 339 m, 285 m, 208 s, 199 s, 141 m, 123 m, 68 m cm−1. Raman (532 nm): ν = 715 w cm−1.
Sr(Me2Vio)2·6H2O (5) and Sr(Me2Vio)2(18-crown-6) (5a)
2.03 g (10.0 mmol) of 2 dissolved in ca. 100 ml hot methanol were added to a suspension of 1.33 g (5.0 mmol) Sr(OH)2·8H2O in 50 ml hot water. Immediately the color of the suspension turned to red-violet. The mixture was stirred for 12 h and the pink-violet precipitate was filtered off, washed with methanol and dried at r.t. Pink-violet hair-like microcrystals of Sr(Me2Vio)2·6H2O (5) were formed in the mother liquor after standing for 1 week at 20 °C. The dry solid compound was recrystallized from DMSO and diglyme or mixtures of DMSO and diglyme resulting again in mats of hair-like pink microcrystals. A DMSO solution of 5 was treated with 18-crown-6 leading to the formation of needle-like, dark pink crystals of 5a suitable for single-crystal structure determination.Sr(Me2Vio)2·6H2O (5)
Yield 2.0 g (71%). Mp 110 °C (dec.). C12H24N6O14Sr (M = 564.04): calcd C 25.56, H 4.29, N 14.90; found: C 25.85, H 4.62, N 14.77%. 1H NMR (400.1 MHz, DMSO-d6, 20 °C): δ = 3.62 (s, 12 H, H2O), 3.19 (s, 6 H, CH3), 2.98 (s, 6 H, CH3) ppm. 13C{1H} NMR (100.6 MHz, DMSO-d6, 21 °C): δ = 164.0 (NNCO), 151.4 (CO), 151.4 (CO), 145.3 (CN), 27.8 (CH3), 26.7 (CH3) ppm. IR (ATR): ν = 3485 m(br) ν(H2O), 2958 w ν(C–H), 1727 m, 1646 s ν(CO), 1632 vs ν(CO), 1513 w, 1441 s ν(NO), 1399 m, 1340 m, 1302 s, 1233 s, 1085 m, 1051 m, 973 w, 947 w, 809 w, 781 m, 752 m, 691 w, 618 m, 603 m, 550 w, 499 m, 441 m, 407 s, 337 m, 299 m, 264 w, 182 m, 135 s, 81 m, 68 m cm−1. Raman (532 nm): ν = 697 w, 1063 m cm−1; (266 nm): ν = 666 m, 1389 m, 1481 m, 1612 s cm−1.
Sr(Me2Vio)2(18-crown-6) (5a)
Yield 0.3 g (8%). Mp 117 °C (dec.). C24H36N6O14Sr (M = 720.21): calcd C 40.03, H 5.04, N 11.67; found: C 39.74, H 5.12, N 11.37%. 1H NMR (400.1 MHz, DMSO-d6, 20 °C): δ = 3.50 (s, 24 H, CH2, 18-crown-6), 3.23 (s, 6H, CH3), 3.03 (s, 6H, CH3) ppm. 13C{1H} NMR (100.6 MHz, DMSO-d6, 21 °C): δ = 163.8 (NNCO), 151.5 (CO), 150.6 (CO), 145.7 (CN), 69.8 (CH2, 18-crown-6), 27.6 (CH3), 26.6 (CH3) ppm. IR (ATR): ν = 2933 w ν(C–H), 2885 w ν(C–H), 2836 w ν(C–H), 1713 m ν(CO), 1662 vs ν(CO), 1511 w, 1456 m ν(NO), 1418 s, 1382 s, 1347 m, 1321 m, 1257 s, 1221 s, 1129 w, 1116 w, 1089 vs, 1071 vs, 1046 vs, 967 vs, 942 m, 838 m, 827 m, 803 m, 781 m, 749 s, 681 w, 609 m, 583 w, 535 w, 505 m, 471 w, 434 w, 409 m, 400 s, 358 m, 344 m, 332 w, 293 m, 286 m, 270 m, 242 m, 177 s, 163 s, 131 vs, 94 m, 76 m, 60 m cm−1. Raman (532 nm): ν = 697 w, 1063 s cm−1.
Ba(Me2Vio)2·4H2O (6)
2.03 g (10.0 mmol) 2 dissolved in ca. 100 ml of hot water were added to a suspension of 1.58 g (5.0 mmol) Ba(OH)2·8H2O in 50 ml hot water. Immediately the color of the suspension turned to violet-red. After stirring for 12 h the pink-violet precipitate was filtered off, washed with methanol and dried at r.t. Red-violet plates of Ba(Me2Vio)2·4H2O (6) suitable for single crystal diffraction measurements were formed in the mother liquor after standing for 1 week at 20 °C. Combined yield 2.4 g (83%). Mp 106 °C (dec.). C12H20N6O12Ba (M = 577.65): calcd C 24.95, H 3.49, N 14.55; found: C 24.74, H 3.52, N 14.87%. 1H NMR (400.1 MHz, DMSO-d6, 20 °C): δ = 3.40 (s, 8 H, H2O), 3.21 (s, 6 H, CH3), 2.98 (s, 6 H, CH3) ppm. 13C{1H} NMR (100.6 MHz, DMSO-d6, 21 °C): δ = 163.9 (NNCO), 151.4 (CO), 150.3 (CO), 145.9 (CN), 27.5 (CH3), 26.4 (CH3) ppm. IR (ATR): ν = 3519 m(br) ν(H2O), 3441 m(br) ν(H2O), 3381 m(br) ν(H2O), 3005 w, 2964 w, 1712 m ν(CO), 1628 vs ν(CO), 1513 w, 1460 m ν(NO), 1432 s, 1393 m, 1334 m, 1281 s, 1269 s, 1225 s, 1075 s, 1055 m, 973 w, 943 w, 808 m, 785 m, 754 s, 737 w, 644 w, 618 m, 598 m, 561 w, 508 s, 476 s, 435 m, 414 s, 399 s, 345 m, 281 w, 258 m, 187 m, 174 m, 150 m, 100 m, 86 m, 70 s, 56 m cm−1. Raman (532 nm): ν = 690 m, 719 w, 1052 s, 1320 broad m, 1555 broad m cm−1.
Ba(Me2Vio)(Me2NO2Barb)·2H2O (7)
0.203 g (1.00 mmol) HMe2Vio·H2O (2) and 0.219 g (1.00 mmol) H(NO2Barb)·H2O (7) dissolved in ca. 30 ml hot water were added to a suspension of 0.315 g (1.0 mmol) Ba(OH)2·8H2O in 30 ml hot water. Immediately the color of the suspension turned to violet-red. After stirring for 1 h at 75 °C the pink-violet solution was cooled to room temperature. Pale red-violet plates of Ba(Me2Vio)(NO2Barb)·2H2O (8) were formed in the mother liquor after standing for 2 h at 20 °C. Combined yield 0.4 g (71%). Mp 154 °C (dec.). C12H16N6O11Ba (M = 557.65): calcd C 25.85, H 2.89, N 15.07; found: C 25.54, H 3.11, N 14.73%. 1H NMR (400.1 MHz, DMSO-d6, 20 °C): δ = 3.37 (s, 4 H, H2O), 3.24 (s, 3 H, CH3, Me2Vio), 3.08 (s, 6 H, CH3, NO2Barb), 3.03 (s, 3 H, CH3, Me2Vio) ppm. 13C{1H} NMR (100.6 MHz, DMSO-d6, 21 °C): δ = 163.9 (NNCO, V), 157.5 (CO, NB), 151.4 (CO, V), 151.0 (NNCO, NB), 150.3 (CO, V), 145.9 (CN, V), 113.8 (C–NO2, NB) 27.5 (CH3, V), 27.3 (CH3, NB), 26.4 (CH3, V) ppm [V = Me2Vio, NB = NO2Barb]. IR (ATR): ν = 3522 w ν(H2O), 3415 m ν(H2O), 3268 m(br) ν(H2O), 1721 m ν(CO), 1615 s ν(CO), 1520 w, 1508 w, 1436 m ν(NO), 1406 m, 1395 m, 1339 m, 1285 s, 1271 m, 1225 s, 1079 m, 157 m, 976 w, 947 w, 806 m, 793 m, 779 m, 751 m, 723 m, 690 m, 673 m, 628 m, 601 w, 557 w, 542 w, 509 m, 484 m, 453 m, 435 w, 403 m, 372 m, 335 w, 280 m, 248 w, 195 m, 142 m, 98 vs, 69 vs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
General financial support of this work by the Otto-von-Guericke-Universität Magdeburg is gratefully acknowledged.Notes and references
- P. Bamfield and M. Hutchings, Chromic Phenomena; Technological Applications of Colour Chemistry, RSC Publishing, Cambridge, 3rd edn, 2018 Search PubMed.
- K. M. Hutchins, S. Gutta, B. P. Loren and L. R. MacGillivray, Chem. Mater., 2014, 26, 3042–3044 CrossRef CAS.
- K. Bašnec, L. S. Perše, B. Šumiga, M. Huskić, A. Meden, A. Hladnik, B. B. Podgornik and M. K. Gunde, Sci. Rep., 2018, 8, 2–11 CrossRef PubMed.
- G. Magnanini, Z. Phys. Chem., 1893, 12, 56–62 CrossRef.
- H. Euler, Ber. Dtsch. Chem. Ges., 1906, 39, 1607–1613 CrossRef CAS.
- A. Hantzsch, Ber. Dtsch. Chem. Ges., 1909, 42, 966–983 CrossRef CAS.
- A. Hantzsch and P. C. C. Isherwood, Ber. Dtsch. Chem. Ges., 1909, 42, 986–1000 CrossRef CAS.
- A. Hantzsch and B. Issaias, Ber. Dtsch. Chem. Ges., 1909, 42, 1000–1007 CrossRef CAS.
- P. Liebing, F. Stein, L. Hilfert, V. Lorenz, K. Oliynyk and F. T. Edelmann, Z. Anorg. Allg. Chem., 2019, 645, 36–43 CrossRef CAS.
- J. Bonacin, A. L. B. Formiga, V. de Melo and H. E. Toma, Vib. Spectrosc., 2007, 44, 133–141 CrossRef CAS.
- G. S. Nichol and W. Clegg, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, o609–o612 CrossRef CAS PubMed.
- T. Kolev, B. B. Koleva, R. W. Seidel, M. Spiteller and W. S. Sheldrick, Cryst. Growth Des., 2009, 9, 3348–3352 CrossRef CAS.
- M. Gryl, A. Rydz, J. Wojnarska, A. Krawczuk, M. Kozieł, T. Seidler, K. Ostrowsky, M. Marzec and K. M. Stadnicka, IUCrJ, 2019, 6, 226–237 CrossRef CAS PubMed.
- R. M. Awadallah, A. A. M. Belal, R. M. Issa and R. D. Peacock, Spectrochim. Acta, Part A, 1991, 47, 1541–1546 CrossRef.
- A. Baeyer, Justus Liebigs Ann. Chem., 1863, 127, 199–236 CrossRef.
- A. Baeyer, Justus Liebigs Ann. Chem., 1864, 129, 129–175 CrossRef.
- A. Baeyer, Justus Liebigs Ann. Chem., 1864, 131, 291–302 CrossRef.
- J. Wagner, Z. Phys. Chem., 1893, 12, 314–321 CrossRef.
- F. G. Donnan, Z. Phys. Chem., 1896, 19, 465–488 CrossRef CAS.
- M. Ceresole, Ber. Dtsch. Chem. Ges., 1883, 16, 1133–1135 CrossRef.
- J. Guinchard, Ber. Dtsch. Chem. Ges., 1899, 32, 1723–1741 CrossRef CAS.
- J. Lifschitz and L. Kritzmann, Ber. Dtsch. Chem. Ges., 1917, 50, 1719–1738 CrossRef.
- J. Lifschitz, Ber. Dtsch. Chem. Ges., 1922, 55, 1619–1631 CrossRef.
- N. A. Illán-Cabeza, A. R. García-García and M. N. Moreno-Carretero, Inorg. Chim. Acta, 2011, 366, 262–267 CrossRef.
- R. Robinson and M. L. Tomlinson, J. Chem. Soc., 1935, 467–470 RSC.
- H. von Hahn, B. Prijs and H. Erlenmeyer, Helv. Chim. Acta, 1956, 39, 341–347 CrossRef.
- L. Cambi and A. Cagnasso, Gazz. Chim. Ital., 1933, 63, 767–778 CAS.
- A. Hantzsch, Ber. Dtsch. Chem. Ges., 1910, 43, 82–91 CrossRef CAS.
- A. Hantzsch and J. Heilbron, Ber. Dtsch. Chem. Ges., 1910, 43, 68–82 CrossRef CAS.
- J. Lifschitz, Ber. Dtsch. Chem. Ges., 1913, 46, 3233–3250 CrossRef.
- J. Lifschitz, Ber. Dtsch. Chem. Ges., 1914, 47, 1068–1076 CrossRef CAS.
- R. Andreasch, Monatsh. Chem., 1900, 21, 281–300 CrossRef CAS.
- E. Fischer and L. Ach, Ber. Dtsch. Chem. Ges., 1895, 28, 3135–3143 CrossRef CAS.
- A. Hantzsch and R. Robison, Ber. Dtsch. Chem. Ges., 1910, 43, 45–68 CrossRef CAS.
- M. E. Taylor and R. J. Robison, Anal. Chem., 1962, 34, 533–536 CrossRef CAS.
- H. C. Garcia, R. Diniz, N. L. Speziali and L. F. C. de Oliveira, J. Mol. Struct., 2014, 1070, 117–124 CrossRef CAS.
- V. Lorenz, P. Liebing, F. Engelhardt, F. Stein, M. Kühling, L. Schröder and F. T. Edelmann, J. Coord. Chem., 2019, 72, 1–34 CrossRef CAS , and references cited therein.
- W. Techow, Ber. Dtsch. Chem. Ges., 1894, 27, 3082–3089 CrossRef CAS.
- R. Andreasch, Monatsh. Chem., 1895, 16, 17–33 CrossRef CAS.
- V. Lorenz, P. Liebing, L. Hilfert, L. Schröder and F. T. Edelmann, Z. Anorg. Allg. Chem., 2020, 646, 1854–1860 CrossRef CAS.
- P. A. Leermakers and W. A. Hoffman, J. Am. Chem. Soc., 1958, 80, 5663–5667 CrossRef CAS.
- M. R. Buchner and Z. Naturforsch, B., 2020, 75, 405–412 CAS.
- V. K. Bel'skii, N. R. Strel'tsova, B. M. Bulychev, L. V. Ivakina and P. A. Storozhenko, Zh. Strukt. Khim., 1987, 28, 148–149 Search PubMed.
- M. Müller and M. R. Buchner, Inorg. Chem., 2019, 58, 13276–13248 CrossRef PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B, Wiley-Interscience Publication, 5th edn, 1998 Search PubMed.
- L. F. C. de Oliveira, P. S. Santos and J. C. Rubim, J. Raman Spectrosc., 1991, 22, 197–203 CrossRef CAS.
- R. Banik, S. Roy, A. Bauza, A. Frontera and S. Das, RSC Adv., 2015, 5, 10826–10836 RSC.
- C. R. Groom and F. H. Allen, Angew. Chem., Int. Ed., 2014, 53, 662–671 CrossRef CAS PubMed.
- W. Clegg and K. Guille, CSD Communication, Ref. code UTIWUS, 2021.
- W. Clegg and K. Guille, CSD Communication, Ref. code AZABEL, 2021.
- O. Simonsen, Acta Chem. Scand., 1997, 51, 861–864 CrossRef CAS.
- O. Simonsen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, C41, 1258–1260 CrossRef CAS.
- J. Han, L. Zhao, C.-W. Yau and T. C. W. Mak, Cryst. Growth Des., 2009, 9, 308–319 CrossRef CAS.
- M. Müller, F. Pielnhofer and M. R. Buchner, Dalton Trans., 2018, 47, 12506–12510 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- Stoe & Cie, X-Area and X-Red, Darmstadt, Germany, 2002.
Footnotes |
| † Dedicated to Professor Wolfgang Beck on the occasion of his 90th birthday. |
| ‡ Electronic supplementary information (ESI) available: IR, NMR (1H and 13C), Raman, and UV-vis spectra. CCDC 1865464 and 2111883–2111887. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00606e |
| § Other names: 2,4,5,6(1H,3H)-pyrimidinetetrone-5-oxime, alloxan-5-oxime, 5-(hydroxyimino)barbituric acid, 5-isonitrosobarbituric acid. |
| This journal is © The Royal Society of Chemistry 2022 |