
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the reactivity of homoleptic organozincs towards SO2: synthesis and structure of a homologous series of organozinc sulfinates†
Adam
Tulewicz
 *a,
Vadim
Szejko
b,
Iwona
Justyniak
a,
Małgorzata
Wolska-Pietkiewicz
b and
Janusz
Lewiński
*ab
*a,
Vadim
Szejko
b,
Iwona
Justyniak
a,
Małgorzata
Wolska-Pietkiewicz
b and
Janusz
Lewiński
*ab
aInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: atulewicz@ichf.edu.pl; lewin@ch.pw.edu.pl
bDepartment of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland
First published on 11th April 2022
Abstract
Studies on the reactivity of zinc alkyl compounds towards SO2 are relatively less explored than either oxygenation or hydrolysis reactions. We report on the environmentally friendly and efficient syntheses of a homologous series of [(RSO2)ZnR]n complexes from reactions involving homoleptic R2Zn (R = Me, tBu, Ph) compounds and SO2. Diffusion ordered spectroscopy experiments indicate that the resulting compounds predominately occur as solvated dimers, [(RSO2)ZnR(THF)]2, in THF solution irrespective of the character of the group bonded to the zinc centres. In turn, these organozinc sulfinates exhibit structurally diversified molecular and supramolecular arrangements in the solid state, as evidenced by single-crystal X-ray diffraction studies. The methyl compound crystallises as a one-dimensional polymer, [(MeSO2)ZnMe]n, and the use of tBu2Zn and Ph2Zn leads to molecular aggregates, a tetramer [(tBuSO2)ZntBu]4, and a solvated [(PhSO2)ZnPh]2·2THF dimer, respectively. In addition, new theoretical insights have been gained by modelling the direct trapping of homoleptic organozinc compounds with SO2 using DFT calculations.
Introduction
Studies on the reactivities of zinc alkyl compounds towards small inorganic molecules lie at the heart of organometallic chemistry. Primary attempts to react homoleptic organozinc compounds with small inorganic reagents can be traced back to the middle of the XIXth century.1,2 Armoured with experimental creativity, it was Edward Frankland who synthesised ‘zincethyl’, known nowadays as diethylzinc, and reacted it with molecular dioxygen and water. The legacy of Frankland has continued ever since. Modern experiments performed in our group on the activation of dioxygen on Zn–C reactive species led to the isolation of the first alkylzinc alkylperoxides and provided a new look at the reactivities of zinc alkyls towards dioxygen.3,4 Strikingly, the first structurally well-defined product derived from the hydrolysis of a dialkylzinc compound was reported only in 2011.5 Since then, a set of compelling results regarding the hydrolysis of homo- and heteroleptic zinc alkyls including stable zinc hydroxide and zincoxane molecular clusters6,7 or even ZnO quantum dots have been reported.8–11 Furthermore, it was demonstrated that reactions between selected alkylzinc hydroxides and carbon dioxide could efficiently provide novel clusters and nanomaterials based on zinc carbonates with unique physicochemical properties.12–14 In turn, studies on the reactivities of homoleptic and heteroleptic organozinc compounds towards SO2 have been explored to a lesser extent, which may seem surprising, for example, in view of the potential use of heteroleptic zinc sulfinates as catalysts for the copolymerisation of CO2 with epoxides15–18 or zinc bis(alkanesulfinate)s as general reagents for the formation of radical species in organic synthesis.19–22 The latter have been usually generated from the corresponding sulfonyl chlorides by treatment with zinc dust, and the requirement of a large excess of Zn metal has restricted their employment under in situ conditions.19 Thus, the development of efficient synthetic procedures for zinc organosufinates is highly desirable.To provide sufficient breadth and depth of literature coverage on the direct trapping of organometallic compounds with SO2, we note that the research in the field started many decades ago and represents efficient methods for the preparation of a vast array of metal sulfinates,19,23–28 featuring exceptionally rich coordination chemistry.29–32 The renewed interest has been rekindled by reactions of frustrated Lewis pairs or related compounds with SO2.32–37 Remarkably, to our knowledge, there are only two reports where the crystal structures of the products of SO2 insertion into homoleptic organozincs have been investigated. Roesky et al.38 showed that the reaction between SO2 and zincocens affords a double insertion product, [Zn(O2SCp*)2(tmeda)], as well as [Zn4(O2SCp*)6O] clusters that contain a Zn4O structural motif. Moreover, thermal decomposition of [Zn(O2SCp*)2(tmeda)] resulted in the formation of [Zn2(μ-SO3)(μ-S2O4)(tmeda)2]. The latter result carries valuable information on the thermodynamic stability of the O2SCp*− anion, in particular, regarding the possibility of its transformations into other ions, i.e., sulfuroxo anions. More recently, our group initiated studies on the Et2Zn/SO2 system.39 We showed that various multidimensional networks bearing either (EtSO2)ZnEt or (EtSO2)2Zn motifs could be obtained with respect to (i) the reaction stoichiometry, (ii) the presence of a certain Lewis base and (iii) the type of solvent chosen for performing the reaction.
We and others have well documented that the character of Zn-bonded alkyl groups dramatically affects the reactivity of organozincs towards small molecules and the composition and structure of the resulting products.40–46 Driven by both curiosity to explore new aspects of reactivities of organozinc systems towards SO2 and the search for new (L)ZnR-type precursors or building blocks of functional materials, herein we turn our attention to the studies of equimolar reactions between selected homoleptic organozinc compounds with SO2. In particular, we report on reactions between R2Zn (R = Me, tBu, Ph) and SO2 in THF, which allowed for the synthesis of a homologous series of organozinc sulfinates in the form of solvated [(RSO2)ZnR(THF)]2 dimers in a THF solution, and the isolation of structurally diversified molecular and supramolecular arrangements in the solid state, i.e., a one-dimensional coordination polymer [(MeSO2)ZnMe]n (1), a tetrameric cluster [(tBuSO2)ZntBu]4 (2) to a solvated dimer of the formula [(PhSO2)ZnPh]2·2THF (3), respectively (Scheme 1).
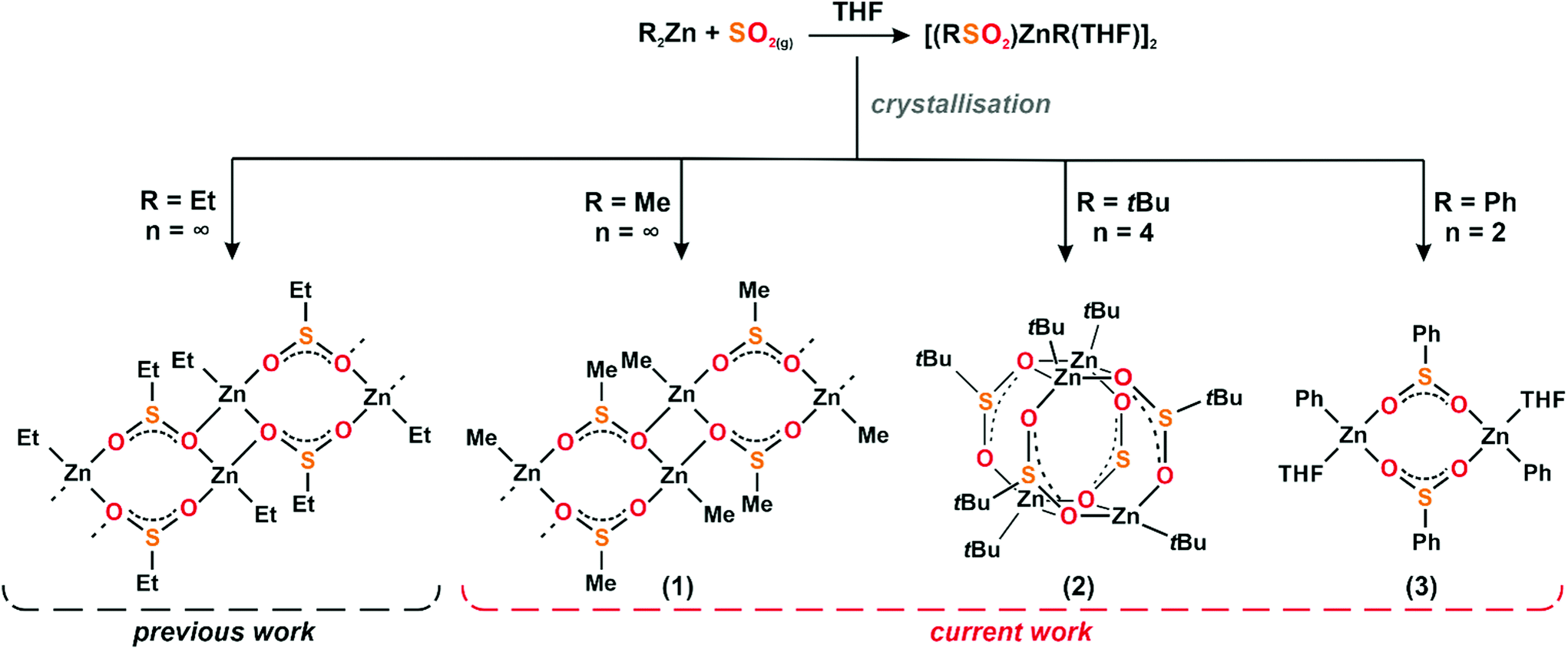 | ||
| Scheme 1 Reactivity of Me2Zn, tBu2Zn and Ph2Zn towards SO2 leading to (MeSO2)ZnMe (1), (tBuSO2)ZntBu (2) and (PhSO2)ZnPh (3). | ||
Results and discussion
Synthesis and spectroscopic characterisation
Immediate reactions between R2Zn (R = Me, tBu, Ph) and gaseous SO2 in the molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were carried out according to Scheme 1. Colourless cubic crystals of either 1, 2 or 3 were isolated in high yield after standard workup (for details, see the Experimental section). Compounds 1–3 were fully characterised using elemental analysis, spectroscopic and single-crystal X-ray diffraction techniques. The FTIR spectra of compounds 1–3 comprise intense to medium bands in the range 876–1043 cm−1, assigned to the sulfur–oxygen vibrations,35 which confirms the presence of the sulfinate anion. The 1H NMR spectrum of 1 shows two well-resolved singlets at 2.38 ppm and −0.91 ppm, derived from the protons of MeSO2 and MeZn groups, respectively (see the ESI, Fig. S1†), while the 13C NMR spectrum of 1 shows two signals at 49.33 ppm and −16.9 ppm from the carbon atoms of the methylsulfinate anion and the Zn-Me group, respectively (see the ESI, Fig. S2†). Similarly, inspection of the 1H NMR spectrum of 2 reveals two separated sharp singlets at 1.07 ppm and 1.01 ppm with equal intensities, assigned to the methyl protons of tBuSO2 and tBuZn groups, respectively (see the ESI, Fig. S4†). The 13C NMR spectrum of 2 shows two intensive sharp singlets of the primary carbons of the tBuSO2 and tBuZn groups at 32.89 ppm and 20.77 ppm, respectively. Less intensive signals from the quaternary carbons were found at 55.42 ppm and 19.72 ppm, respectively (see the ESI, Fig. S5†). The 1H NMR spectrum of 3 comprises an overlapping multiplet from phenyl protons that fall within a range between 6.98 ppm and 7.79 ppm. In addition, the characteristic multiplets of THF protons were recorded at 1.77 ppm and 3.62 ppm, with the intensities indicative of the partial desolvation of compound 3 (see the ESI, Fig. S7†). The 13C NMR spectrum of 3 features signals from the phenyl groups at 153.11 and 152.38 ppm, and in the range of 139.59–125.62 ppm. Signals assigned to the secondary and primary carbons of THF were found at 26.37 ppm and 68.25 ppm, respectively. Moreover, the DOSY experiments indicate that compounds 1–3 occur as solvated dimers [(RSO2)ZnR(THF-d8)]2 in THF-d8 solution irrespective of the character of the alkyl group bonded to zinc centres (for details, see the ESI†).
:1 were carried out according to Scheme 1. Colourless cubic crystals of either 1, 2 or 3 were isolated in high yield after standard workup (for details, see the Experimental section). Compounds 1–3 were fully characterised using elemental analysis, spectroscopic and single-crystal X-ray diffraction techniques. The FTIR spectra of compounds 1–3 comprise intense to medium bands in the range 876–1043 cm−1, assigned to the sulfur–oxygen vibrations,35 which confirms the presence of the sulfinate anion. The 1H NMR spectrum of 1 shows two well-resolved singlets at 2.38 ppm and −0.91 ppm, derived from the protons of MeSO2 and MeZn groups, respectively (see the ESI, Fig. S1†), while the 13C NMR spectrum of 1 shows two signals at 49.33 ppm and −16.9 ppm from the carbon atoms of the methylsulfinate anion and the Zn-Me group, respectively (see the ESI, Fig. S2†). Similarly, inspection of the 1H NMR spectrum of 2 reveals two separated sharp singlets at 1.07 ppm and 1.01 ppm with equal intensities, assigned to the methyl protons of tBuSO2 and tBuZn groups, respectively (see the ESI, Fig. S4†). The 13C NMR spectrum of 2 shows two intensive sharp singlets of the primary carbons of the tBuSO2 and tBuZn groups at 32.89 ppm and 20.77 ppm, respectively. Less intensive signals from the quaternary carbons were found at 55.42 ppm and 19.72 ppm, respectively (see the ESI, Fig. S5†). The 1H NMR spectrum of 3 comprises an overlapping multiplet from phenyl protons that fall within a range between 6.98 ppm and 7.79 ppm. In addition, the characteristic multiplets of THF protons were recorded at 1.77 ppm and 3.62 ppm, with the intensities indicative of the partial desolvation of compound 3 (see the ESI, Fig. S7†). The 13C NMR spectrum of 3 features signals from the phenyl groups at 153.11 and 152.38 ppm, and in the range of 139.59–125.62 ppm. Signals assigned to the secondary and primary carbons of THF were found at 26.37 ppm and 68.25 ppm, respectively. Moreover, the DOSY experiments indicate that compounds 1–3 occur as solvated dimers [(RSO2)ZnR(THF-d8)]2 in THF-d8 solution irrespective of the character of the alkyl group bonded to zinc centres (for details, see the ESI†).
Furthermore, some combinations of heteroleptic (L)ZnR-type complexes with homoleptic R2Zn compounds act as arguably the most important organozinc catalytic systems in organic synthesis47,48 or precursors of various functional materials.14,49 Nevertheless, the chemistry of (L)ZnR/R2Zn systems remains both relatively poorly understood and a subject of constant debate.50,51 Thus, we performed controlled experiments on the reactivity of selected [(RSO2)ZnR]/R′2Zn systems. In this regard, we prepared THF solutions of 1 and 2 that were subsequently treated with equimolar amounts of tBu2Zn and Me2Zn, respectively (Scheme 2). Strikingly, we found that in the first case, (MeSO2)ZnMe/tBu2Zn, the zinc-bonded methyl group was easily substituted by the tert-butyl group (see the ESI, Fig. S10 and 11†). Conversely, in the case of the (tBuSO2)ZntBu/Me2Zn system no exchange was observed as indicated by the NMR studies (see the ESI, Fig. S12 and 13†). The reasons for such different reactivities are unclear and must be further investigated.
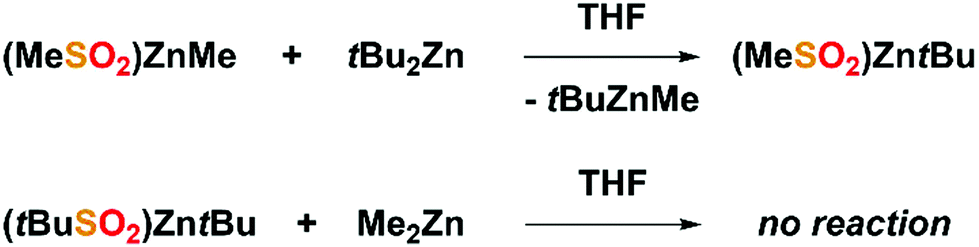 | ||
| Scheme 2 Controlled experiments on the reactivities of [(RSO2)ZnR] species towards homoleptic organozincs. | ||
Solid-state structures
Remarkably, the nature of the zinc-bonded alkyl substituents has a profound effect on the solid-state structure of the title organozinc sulfinates. Compound 1 crystallises in triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) as a 1D coordination polymer constructed of dinuclear [(MeSO2)ZnMe]2 units incorporating sulfinato-O,O′-type ligands (Fig. 1) and interconnected through the [Zn2(μ2-O2)] rings to form an extended chain along the a axis. The structural constitution of 1 reflects the polymeric structure of the previously reported ethylzinc ethylsulfinate derivative,38 [(EtSO2)ZnEt]n. The Zn–O bond distances within the molecular [(MeSO2)ZnMe]2 unit fall in the range of 1.988–2.096 Å, and the Zn–O bonds connecting these units are of a similar length of 2.075 Å. The S–C bond length is 1.772 Å, and the S–O bonds of length 1.517–1.551 Å, along with O–S–O angles (106.9°), are similar to values observed for other metal sulfinate complexes.30,38,39
as a 1D coordination polymer constructed of dinuclear [(MeSO2)ZnMe]2 units incorporating sulfinato-O,O′-type ligands (Fig. 1) and interconnected through the [Zn2(μ2-O2)] rings to form an extended chain along the a axis. The structural constitution of 1 reflects the polymeric structure of the previously reported ethylzinc ethylsulfinate derivative,38 [(EtSO2)ZnEt]n. The Zn–O bond distances within the molecular [(MeSO2)ZnMe]2 unit fall in the range of 1.988–2.096 Å, and the Zn–O bonds connecting these units are of a similar length of 2.075 Å. The S–C bond length is 1.772 Å, and the S–O bonds of length 1.517–1.551 Å, along with O–S–O angles (106.9°), are similar to values observed for other metal sulfinate complexes.30,38,39
 | ||
| Fig. 1 Molecular structure and single polymeric chain (view along the crystallographic a axis) of 1. Hydrogen atoms have been omitted for clarity. | ||
Compound 2 crystallises in the P triclinic space group. An inspection of the structure of 2 revealed that metal centres adopt a tetrahedral geometry, and four tBuZn moieties are stabilised by four tert-butylsulfinate anions (Fig. 2a). As a result, the molecular structure of 2 comprises two sulfinato-O,O′-type [(tBuSO2)ZntBu]2 heterocycles joined within two [Zn2(μ2-O2)] moieties forming tetranuclear cluster [(tBuSO2)ZntBu]4. Interestingly, the core of compound 2 is isostructural to quasi-cube-shaped tert-buthylzinc diorganophosphate [tBuZn(O2P(OR)2)] (R = Me, Ph) complexes previously reported by us.52 The Zn–O bond lengths in 2 were found to be in the range between 2.005 and 2.121 Å and the S–O distances between 1.510 and 1.555 Å, similar to the bond values found in the case of compound 1 and related alkylzinc sulfinates.39 Compound 3 crystallises in triclinic space group P. An introduction of the aromatic group provided dinuclear solvate [(PhSO2)ZnPh(THF)]2 (Fig. 2b). Two metal centres of 3 are connected by the PhSO2-μ2-bridging moieties forming an octanuclear macrocycle that adopts a chair conformation similar to that observed for compound 1, with the phenyl groups located trans to each other across the ring. However, contrary to 1 and 2, the tetrahedral coordination sphere located on each metal centre of 3 is completed by a single THF molecule. That results in a monomeric structural motif, preferable over either the 1D coordination polymer or tetranuclear cluster, likely due to the steric hindering introduced by phenyl rings. The Zn–Osulfinate distances of 3 are observed to be in the range of 1.987–2.088 Å, and the Zn–OTHF bonds to outer solvent molecules are longer and are of length 2.088 Å. The S–O bonds are similar within the molecule and their lengths are between 1.527 and 1.528 Å. The O–S–O conical angles (107.37°) together with the aforementioned parameters are similar to that observed for other zinc sulfinate compounds.30,38,39
 | ||
| Fig. 2 Molecular structure of 2 (a) and 3 (b). Hydrogen atoms have been omitted for clarity. | ||
DFT studies
DFT calculations were subsequently performed to explore the thermodynamic landscape associated with insertions of SO2 into the studied systems as well as to deeply investigate the studies on the reactivity of SO2 towards various alkylzincs (Fig. 3). In all the considered cases, the reaction between R2Zn and SO2 starts with the formation of van der Waals complexes, where the oxygen atom from SO2 is attracted by the metal centre, similarly to our previous studies with Et2Zn/SO2 systems. In the next step, the formation of the transition state occurs. In the case of Ph2Zn/SO2, the geometry of the transition state remains only slightly distorted, in contrast to the geometries of transition states found within the Me2Zn/SO2 and tBu2Zn/SO2 systems. This may explain to some extent the relatively low barrier of reaction found for the insertion of SO2 into Ph2Zn. Note that the elongation of the Zn–C bond, associated with the R2Zn/SO2 transition state, is energetically highly unfavoured, in particular for the small alkyls. This effect may explain the higher value of the energy barrier of +14.9 kcal mol−1, found for the insertion of SO2 into Me2Zn. Finally, the overall energetic effect of the R2Zn/SO2 reactions remains highly negative and similar to the results obtained within related studies,24,25 and suggests that reactions between R2Zn and SO2 should be driven in a one way direction. | ||
| Fig. 3 The comparison of reaction pathways of SO2 insertion into Me2Zn (a; blue line), tBu2Zn (b; red line) and Ph2Zn (c; violet line). The values of ΔG have been referenced to the Gibbs free energies of the van der Waals complexes. | ||
Experimental section
General remarks
All reactions and post-synthetic manipulations were conducted under an argon or nitrogen atmosphere using standard Schlenk and glovebox techniques (MBraun UniLab Plus; <0.1 ppm O2, <0.1 ppm H2O). All solvents were purified by passage through activated aluminium oxide (MBraun SPS) and stored over 3 Å molecular sieves. Prior to use, deuterated solvents were dried over Na/K, distilled under an argon atmosphere before use, and stored over molecular sieves. Di-tert-butylzinc was prepared according to the literature procedure.53 All reagents used in this study were of commercial grade and obtained from Sigma-Aldrich Co or abcr GmbH. Stoichiometric amounts of SO2 (determined by using the Clausius–Clapeyron equation) were added to the reaction mixtures through a syringe. NMR spectra were acquired with a Varian Mercury 400 MHz spectrometer at 298 K unless otherwise specified and were referenced to the residual 1H or 13C signals of the deuterated solvents. FTIR spectra were recorded with a Bruker Vertex 80V spectrometer equipped with a diamond ATR (attenuated total reflection) unit. Elemental analysis was performed by using a UNICUBE (Elementar Analysensysteme GmbH).X-Ray structure determination
The X-ray data for complexes 1, 2, and 3 were collected at 100(2) K on a SuperNova Agilent diffractometer using graphite monochromated MoKα radiation (λ = 0.71073 Å). The data were processed with CrysAlisPro.54 The structure was solved by direct methods using the SHELXS-97 program and was refined by full matrix least-squares on F2 using the program SHELXL.55 All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were added to the structure model at geometrically idealised coordinates and refined as riding atoms. (no. 2), a = 4.5135(8) Å, b = 7.7984(11) Å, c = 7.8400(13) Å, α = 89.141(12)°, β = 80.887(14)°, γ = 84.090(13)°, U = 271.02(8) Å3, Z = 2, F(000) = 160, Dc = 1.954 g cm−3, μ(Mo-Kα) = 4.785 mm−1, θmax = 26.500°, 1117 unique reflections. Refinement converged at R1 = 0.0701, wR2 = 0.1688 for all data (R1 = 0.0632, wR2 = 0.1609 for 977 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal to 1.088.
(no. 2), a = 11.9492(3) Å, b = 12.8183(7) Å, c = 16.1223(5) Å, α = 86.157(3)°, β = 78.812(2)°, γ = 75.515(3)°, U = 2345.05(16) Å3, Z = 2, F(000) = 1024, Dc = 1.380 g cm−3, μ(Mo-Kα) = 2.238 mm−1, θmax = 26.997°, 10221 unique reflections. Refinement converged at R1 = 0.0611, wR2 = 0.0720 for all data (R1 = 0.0374, wR2 = 0.0646 for 7731 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal to 1.019.
(no. 2), a = 8.792(5) Å, b = 9.326(5) Å, c = 10.669(5) Å, α = 78.722(5)°, β = 74.655(5)° γ = 67.772(5)°, U = 776.4(7) Å3, Z = 1, F(000) = 368, Dc = 1.522 g cm−3, μ(Mo-Kα) = 1.722 mm−1, θmax = 30.163°, 3931 unique reflections. Refinement converged at R1 = 0.0449, wR2 = 0.0872 for all data (R1 = 0.0361, wR2 = 0.0826 for 3412 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal to 1.050.
Theoretical details
The geometries of the molecules chosen as model systems were optimised by using density functional theory (DFT) with the B3LYP56,57 functional in the 6-31++G(2d,2p) basis set with the D3(BJ) correction58,59 added in order to improve the description of dispersion forces. Frequency analysis was performed in order to confirm the nature of minima and maxima. Intrinsic reaction coordinate calculations (IRC)60 were additionally done to check the quality of the transition states. All the calculations were done with the Gaussian suite of codes.61 Figures have been prepared with the CYLview visualisation software.62Conclusions
Finding new potential (L)ZnR-type reagents for organic synthesis or precursors of nanomaterials that can be efficiently synthesised from some air pollutants may represent an important step toward a better future. Simultaneously, explorations of reactivity of homoleptic dialkylzincs towards sulfur dioxide still leave many opportunities for scientific discoveries. In this vein, we performed the first systematic studies on the effect of the nature of Zn-bonded alkyl groups on both the SO2 insertion process and structure of the resulting products in the solid state and solution. The direct trapping of homoleptic R2Zn compounds with SO2 allowed for the isolation and characterisation of a homologous series of organozinc sulfinate [(RSO2)ZnR] species that are hardly accessible with alternative methods. Moreover, we showed that the resulting organozinc sulfinates occur as solvated dimers [(RSO2)ZnR(THF)]2 in THF solution irrespective of the character of the substituent. In turn, the identity of the Zn-R group determines whether the [(RSO2)ZnR]n product acquires a dimeric, tetrameric or 1D-polymeric constitution in the solid state. Interestingly, the DFT calculations performed on the set of equimolar R2Zn/SO2 systems confirm that the SO2 insertion into the Zn–C bond can be characterised by a moderate value of the energy barriers and the highly exothermic character of the SO2 insertion.Finally, the reported investigations fill a missing gap in the realm of (L)ZnR complexes as new precursors of functional materials and further studies on the application of organozinc sulfinates as efficient precursors of surface-modified ZnO nanostructures are in progress.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the National Science Centre, Poland (grant OPUS 2017/25/B/ST5/02484 and grant MINIATURA 2021/05/X/ST4/00520) for financial support. Moreover, the ICM University of Warsaw Computer Center is acknowledged for facilities and computer time allocation within the grant G70-21. The authors would also like to thank Dr Piotr Bernatowicz for assistance with DOSY experiments.Notes and references
- E. von Frankland, Justus Liebigs Ann. Chem., 1849, 71, 171–213 CrossRef.
- D. Seyferth, Organometallics, 2001, 20, 2940–2955 CrossRef CAS.
- J. Lewiński, W. Śliwiński, M. Dranka, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2006, 45, 4826–4829 CrossRef PubMed.
- T. Pietrzak, I. Justyniak, M. Kubisiak, E. Bojarski and J. Lewiński, Angew. Chem., Int. Ed., 2019, 58, 8526–8530 CrossRef CAS PubMed.
- W. Bury, E. Krajewska, M. Dutkiewicz, K. Sokołowski, I. Justyniak, Z. Kaszkur, K. J. Kurzydłowski, T. Płociński and J. Lewiński, Chem. Commun., 2011, 27, 5467–5469 RSC.
- D. Prochowicz, K. Sokołowski and J. Lewiński, Coord. Chem. Rev., 2014, 270–271, 112–126 CrossRef CAS.
- K. Sokołowski, I. Justyniak, W. Śliwiński, K. Sołtys, A. Tulewicz, A. Kornowicz, R. Moszyński, J. Lipkowski and J. Lewiński, Chem. – Eur. J., 2012, 18, 5637–5645 CrossRef PubMed.
- D. Lee, M. Wolska-Pietkiewicz, S. Badoni, A. Grala, J. Lewiński and G. De Paëpe, Angew. Chem., Int. Ed., 2019, 58, 17163–17168 CrossRef CAS PubMed.
- M. Terlecki, S. Badoni, M. K. Leszczyński, S. Gierlotka, I. Justyniak, H. Okuno, M. Wolska-Pietkiewicz, D. Lee, G. De Paëpe and J. Lewiński, Adv. Funct. Mater., 2021, 2105318 CrossRef CAS.
- Z. Zhao, Y. Wang, C. Delmas, C. Mingotaud, J. D. Marty and M. L. Kahn, Nanoscale Adv., 2021, 3, 6696–6703 RSC.
- P. Krupiński, A. Grala, M. Wolska-Pietkiewicz, W. Danowski, I. Justyniak and J. Lewiński, ACS Sustainable Chem. Eng., 2021, 9, 1540–1549 CrossRef.
- K. Sokołowski, W. Bury, I. Justyniak, D. Fairen-Jimenez, K. Sołtys, D. Prochowicz, S. Wang, M. Schröder and J. Lewiński, Angew. Chem., Int. Ed., 2013, 52, 13414–13418 CrossRef PubMed.
- K. Sokołowski, W. Bury, I. Justyniak, A. M. Cieślak, M. Wolska, K. Sołtys, I. Dzięcielewski and J. Lewiński, Chem. Commun., 2013, 49, 5271 RSC.
- K. Sokołowski, W. Bury, A. Tulewicz, A. M. Cieślak, I. Justyniak, D. Kubicki, E. Krajewska, A. Milet, R. Moszyński and J. Lewiński, Chem. – Eur. J., 2015, 21, 5496–5503 CrossRef PubMed.
- R. Eberhardt, M. Allmendinger, M. Zintl, C. Troll, G. A. Luinstra and B. Rieger, Macromol. Chem. Phys., 2004, 205, 42–47 CrossRef CAS.
- B. Y. Lee, H. Y. Kwon, S. Y. Lee, S. J. Na, S. Han, H. Yun, H. Lee and Y. Park, J. Am. Chem. Soc., 2005, 127, 3031–3037 CrossRef CAS PubMed.
- M. F. Pilz, C. Limberg, B. B. Lazarov, K. C. Hultzsch and B. Ziemer, Organometallics, 2007, 26, 3668–3676 CrossRef CAS.
- R. Eberhardt, M. Allmendinger, G. A. Luinstra and B. Rieger, Organometallics, 2008, 22, 211–214 CrossRef.
- F. O'Hara, R. D. Baxter, A. G. O'Brien, M. R. Collins, J. A. Dixon, Y. Fujiwara, Y. Ishihara and P. S. Baran, Nat. Protoc., 2013, 8, 1042–1047 CrossRef PubMed.
- D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed.
- S. Liang, K. Hofman, M. Friedrich and G. Manolikakes, Eur. J. Org. Chem., 2020, 4664–4676 CrossRef CAS.
- J. L. Nova-Fernández, M. J. García, L. Mollari, G. Pascual-Coca, S. Cabrera and J. Alemán, Chem. Commun., 2022, 58, 4611–4614 RSC.
- A. Wojcicki, Acc. Chem. Res., 1971, 598, 344–352 CrossRef.
- A. Wojcicki, in Advances in Organometallic Chemistry, Elsevier, 1974, pp. 31–81 Search PubMed.
- R. G. Severson and A. Wojcicki, J. Am. Chem. Soc., 1979, 5, 877–883 CrossRef.
- C. E. Kefalidis, C. Jones and L. Maron, Dalton Trans., 2016, 45, 14789–14800 RSC.
- E. Louyriac, P. W. Roesky and L. Maron, Dalton Trans., 2017, 46, 7660–7663 RSC.
- J. M. Smith, J. A. Dixon, J. N. Degruyter and P. S. Baran, J. Med. Chem., 2019, 62, 2256–2264 CrossRef CAS PubMed.
- W. A. Schenk, Angew. Chem., Int. Ed. Engl., 1987, 26, 98–109 CrossRef.
- W. A. Schenk, Dalton Trans., 2011, 40, 1209–1219 RSC.
- A. J. Boutland, I. Pernik, A. Stasch and C. Jones, Chem. – Eur. J., 2015, 21, 15749–15758 CrossRef CAS PubMed.
- S. V. Klementyeva, N. Arleth, K. Meyer, S. N. Konchenko and P. W. Roesky, New J. Chem., 2015, 39, 7589–7594 RSC.
- M. Sajid, A. Klose, B. Birkmann, L. Liang, B. Schirmer, T. Wiegand, H. Eckert, A. J. Lough, R. Fröhlich, C. G. Daniliuc, S. Grimme, D. W. Stephan, G. Kehr and G. Erker, Chem. Sci., 2013, 4, 213–219 RSC.
- D. W. Stephan and G. Erker, Chem. Sci., 2014, 5, 2625–2641 RSC.
- K. Y. Ye, M. Bursch, Z. W. Qu, C. G. Daniliuc, S. Grimme, G. Kehr and G. Erker, Chem. Commun., 2017, 53, 633–635 RSC.
- N. Aders, L. Keweloh, D. Pleschka, A. Hepp, M. Layh, F. Rogel and W. Uhl, Organometallics, 2019, 38, 2839–2852 CrossRef CAS.
- N. Szynkiewicz, J. Chojnacki and R. Grubba, Inorg. Chem., 2020, 59, 6332–6337 CrossRef CAS PubMed.
- R. P. Kelly, N. Kazeminejad, C. A. Lamsfus, L. Maron and P. W. Roesky, Chem. Commun., 2016, 52, 13090–13093 RSC.
- A. Tulewicz, M. Wolska-Pietkiewicz, M. Jędrzejewska, T. Ratajczyk, I. Justyniak and J. Lewiński, Chem. – Eur. J., 2019, 25, 14072–14080 CrossRef CAS PubMed.
- S. Jana, R. J. F. Berger, R. Fröhlich, T. Pape and N. W. Mitzel, Inorg. Chem., 2007, 46, 4293–4297 CrossRef CAS PubMed.
- I. Dranka, M. Kubisiak, I. Justyniak, M. Lesiuk, D. Kubicki and J. Lewiński, Chem. – Eur. J., 2011, 17, 12713–12721 CrossRef CAS PubMed.
- M. Kubisiak, K. Zelga, W. Bury, I. Justyniak, K. Budny-Godlewski, Z. Ochal and J. Lewiński, Chem. Sci., 2015, 6, 3102–3108 RSC.
- T. Pietrzak, M. D. Korzyński, I. Justyniak, K. Zelga, A. Kornowicz, Z. Ochal and J. Lewiński, Chem. – Eur. J., 2017, 23, 7997–8005 CrossRef CAS PubMed.
- Z. Wrobiel, T. Pietrzak, I. Justyniak and J. Lewiński, Chem. Commun., 2017, 53, 10808–10811 RSC.
- K. Budny-Godlewski, I. Justyniak, M. K. Leszczyński and J. Lewiński, Chem. Sci., 2019, 10, 7149–7155 RSC.
- Ł. Mąkolski, V. Szejko, K. Zelga, A. Tulewicz, P. Bernatowicz, I. Justyniak and J. Lewiński, Chem. – Eur. J., 2021, 27, 5666–5674 CrossRef PubMed.
- L. Pu and H. Yu, Chem. Rev., 2001, 101, 757–824 CrossRef CAS PubMed.
- K. Soai, T. Kawasaki and A. Matsumoto, Acc. Chem. Res., 2014, 47, 3643–3654 CrossRef CAS PubMed.
- K. L. Orchard, M. S. P. Shaffer and C. K. Williams, Chem. Mater., 2012, 24, 2443–2448 CrossRef CAS.
- J. Lewiński, W. Marciniak, Z. Ochal, J. Lipkowski and I. Justyniak, Eur. J. Inorg. Chem., 2003, 43, 2753–2755 CrossRef.
- S. V. Athavale, A. Simon, K. N. Houk and S. E. Denmark, Nat. Chem., 2020, 12, 412–423 CrossRef CAS PubMed.
- M. Wolska-Pietkiewicz, A. Świerkosz, I. Justyniak, A. Grala, K. Sokołowski and J. Lewiński, Dalton Trans., 2016, 45, 18813–18816 RSC.
- J. Lewiński, M. Dranka, W. Bury, W. Śliwiński, I. Justyniak and J. Lipkowski, J. Am. Chem. Soc., 2007, 129, 3096–3098 CrossRef PubMed.
- CrysAlisPro, Data Collection and Processing Software for Agilent X-ray Diffractometers, ver. 1.171.35.21b, Agilent Technologies, 2012 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 15410 CrossRef PubMed.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009 (https://www.cylview.org) Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic and crystallographic data, and computation details. CCDC 2116783–2116785. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00577h |
| This journal is © The Royal Society of Chemistry 2022 |