
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular ruby: exploring the excited state landscape†
Winald Robert
Kitzmann
 a,
Charusheela
Ramanan
bc,
Robert
Naumann
a and
Katja
Heinze
*a
a,
Charusheela
Ramanan
bc,
Robert
Naumann
a and
Katja
Heinze
*a
aDepartment of Chemistry, Johannes Gutenberg University of Mainz, Mainz, Germany. E-mail: katja.heinze@uni-mainz.de
bDepartment of Physics and Astronomy, Vrije Universiteit Amsterdam, Amsterdam, Netherlands
cMax-Planck-Institute for Polymer Research, Mainz, Germany
First published on 5th April 2022
Abstract
The discovery of the highly NIR-luminescent molecular ruby [Cr(ddpd)2]3+ (ddpd = N,N′-dimethyl-N,N′-dipyridin-2-ylpyridine-2,6-diamine) has been a milestone in the development of earth-abundant luminophors and has led to important new impulses in the field of spin–flip emitters. Its favourable optical properties such as a high photoluminescence quantum yield and long excited state lifetime are traced back to a remarkable excited state landscape which has been investigated in great detail. This article summarises the results of these studies with the aim to create a coherent picture of the excited state ordering and the ultrafast as well as long-timescale dynamics. Additional experimental data is provided to fill in gaps left by previous reports.
Introduction
In metal-to-ligand charge transfer (MLCT) emitters such as [Ru(bpy)3]2+ an excited state ordering of 1GS, 3MLCT, 1MLCT and 3MC is commonly found and exploited for different applications like photocatalysis (GS = ground state, MC = metal-centered state, bpy = 2,2′-bipyridine).1In contrast, spin–flip emitters with the prototype ruby (Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cr3+) possess a richer excited state landscape in the relevant energy region.2,3 For a d3 electron configuration in Oh symmetry as found in octahedral CrIII compounds, the metal-centered 4A2, 2E/2T1, 2T2 and 4T2 states are important (Fig. 1). Additionally, charge transfer states can play a role in excitation and decay processes.4,5
:Cr3+) possess a richer excited state landscape in the relevant energy region.2,3 For a d3 electron configuration in Oh symmetry as found in octahedral CrIII compounds, the metal-centered 4A2, 2E/2T1, 2T2 and 4T2 states are important (Fig. 1). Additionally, charge transfer states can play a role in excitation and decay processes.4,5
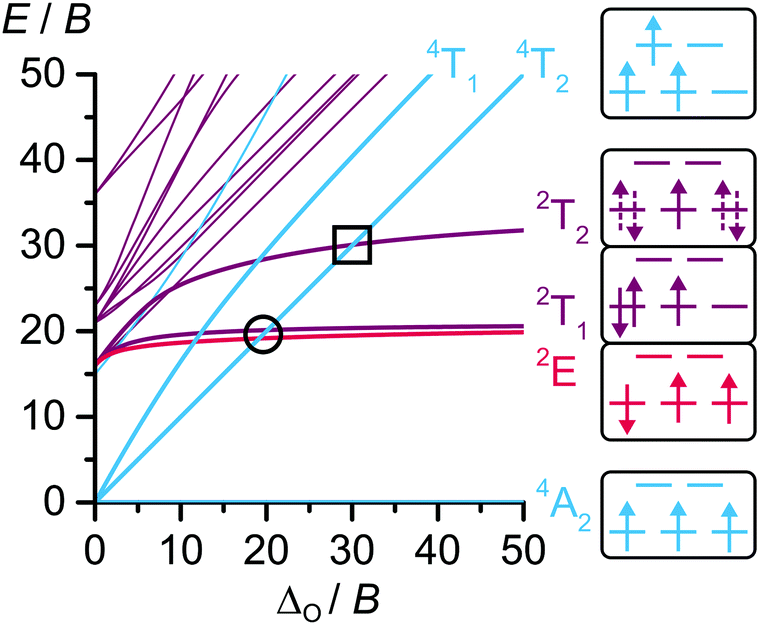 | ||
| Fig. 1 Tanabe–Sugano (TS) diagram of the d3 electron configuration in an octahedral ligand field with C/B = 4 and exemplary microstates.4,14 The two important excited state quartet/doublet crossing points are highlighted with a circle and a square, respectively. Dotted lines in the microstates indicate strong electronic mixing. | ||
In d3 spin–flip emitters, excitation to the 4T2 state is followed by intersystem crossing (ISC) to the doublet states 2E/2T1, from which phosphorescence to the quartet GS 4A2 occurs.6 As the term “spin–flip emitter” suggests, this transition merely involves the flipping of a single electron spin. The 2E state features only unpaired electrons, while two electrons are paired in 2T1 and 2T2 states (Fig. 1).4 For spin–flip emission, the 2E/2T1 states need to be the lowest excited states. Additionally, a large energy gap to the 4T2 state is necessary to prevent 2E/2T1 → 4T2 back-ISC (bISC) and fast relaxation via4T2 → 4A2 fluorescence, internal conversion (IC) or ligand dissociation.7 This is achieved via a large ligand field splitting Δ0, since the energy of the interconfigurational 4T2 state is linearly dependent on Δ0, while the energies of the intraconfigurational states 2E, 2T1 and 2T2 are essentially independent of Δ0 and instead vary with interelectronic repulsion quantified by the Racah parameters B and C (Fig. 1).8 In general, CrIII ions possess a small instrinsic ligand field splitting due to the primogenic effect.9
In ruby, the six oxido ligands exert a strong ligand field on the Cr3+ ions because of the short Cr–O distance imposed by the corundum host lattice. This results in a 4T2 absorption band at 551 nm and strong phosphorescence at 694.3 (R1 line) as well as 692.9 nm (R2 line) via the slightly split 2E → 4A2 transitions.2,10 This emission with a lifetime of 4268 ± 6 μs and a photoluminescence quantum yield of 90 ± 5% was the cornerstone of the first laser in 1960.2,3,11 In contrast, molecular CrIII complexes like [Cr(bpy)3]3+ (Φ = 0.089%)12 or [Cr(tpy)2]3+ (Φ < 0.001%)13 suffer from poor quantum yields due to the small Δ0 facilitating bISC and other deactivation pathways (tpy = 2,2′:6′,2′′-terpyridine).4
Ground and excited states of the molecular ruby
In 2015, a breakthrough was achieved with the discovery of the strongly near infrared (NIR) luminescent complex [Cr(ddpd)2]3+ (Fig. 2, ddpd = N,N′-dimethyl-N,N′-dipyridin-2-ylpyridine-2,6-diamine). Irradiation in the 4A2 → 4T2 and ligand-to-metal charge transfer (LMCT) absorption band at 435 nm yields a very intense dual phosphorescence at 738 and 775 nm (2T1/2E → 4A2, purple area in Fig. 3) with a lifetime of 1122 μs and quantum yields of 11% in aqueous and 13.7% in acetonitrile solution at room temperature.7,15 The excitation spectra observed at 775 nm closely follow the absorption spectra in the region 300–500 nm demonstrating that all states efficiently evolve to the emissive states. Indeed, only a weak fluorescence (4T2 → 4A2, blue area in Fig. 3) at 500 nm was found with a lifetime7 of 3 ns and a quantum yield of approximately 0.01% (see ESI†). Overall, [Cr(ddpd)2]3+ vastly outperforms classical chromium(III) complexes and hence was called molecular ruby.16 Detailed follow-up studies revealed several aspects underlying the reasons for its success.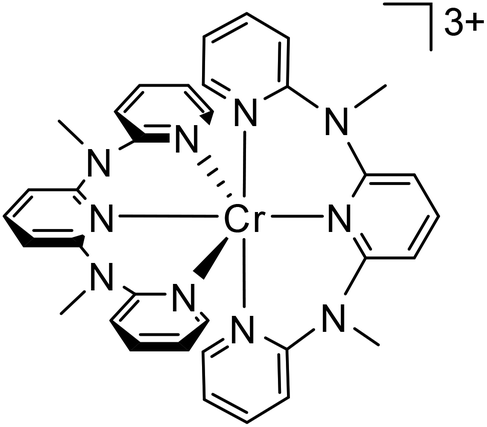 | ||
| Fig. 2 Molecular structure of [Cr(ddpd)2]3+. | ||
 | ||
| Fig. 3 Absorption spectra (H2O: black line, CD3CN: red line, see ESI†), MCD spectrum (H2O/glycerol, 7 K, 7 T) and separately normalized fluorescence (blue area) and phosphorescence spectra (purple area, aerated H2O) of [Cr(ddpd)2]3+ and TD-DFT (B3LYP/Def2-TZVPP) calculated difference densities of the 4A2 → 4T2 transitions with an isosurface value of 0.005 a.u.7,17 | ||
The 4A2 GS of [Cr(ddpd)2]3+ exhibits an almost spherical spin distribution as expected for a d3 configuration (Fig. 4). A small zero-field splitting of D = +0.18 cm−1 and E = −0.06 cm−1 was found through Q-band electron paramagnetic resonance (EPR) spectroscopy at 5 K.17 The [CrN6] σ-skeleton is close to octahedral, but the π-bonding lowers the actual symmetry (point group D2). Thus, all E and T terms found in the TS diagram (Fig. 1) should be split in [Cr(ddpd)2]3+.
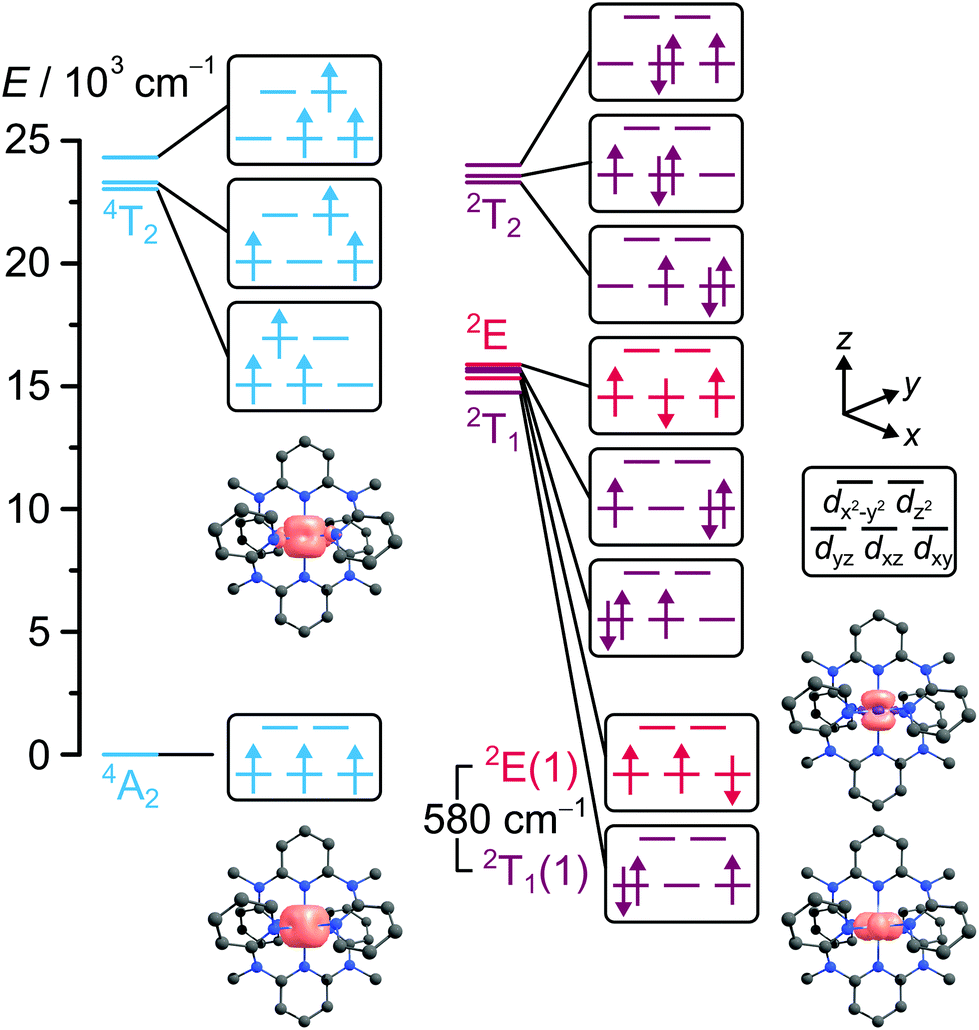 | ||
| Fig. 4 CASSCF(7,12)-NEVPT2 derived state diagram at the GS geometry of [Cr(ddpd)2]3+ with dominating electron configurations in the microstates and calculated spin-densities at an isosurface value of 0.05 a.u.20 | ||
How large is the ligand field splitting Δ0 in [Cr(ddpd)2]3+, in other words, where is [Cr(ddpd)2]3+ located in the TS diagram (Fig. 1)? Investigation of the absorption bands including the 4A2 → 4T2 transition at 435 nm with magnetic circular dichroism (MCD) spectroscopy at 7 K and 7 T in frozen solution and DFT calculations revealed Δ0 ≫ 20 B (Fig. 3).17 Thus, the large 4T2–2T1/2E energy gap effectively suppresses bISC to the 4T2 states as relaxation pathway. The large Δ0 was rationalized with the strongly σ-donating nature of the electron-rich ligand ddpd and its six-membered chelate rings leading to an almost perfectly octahedral [CrN6] coordination geometry and a large metal–ligand σ-orbital overlap.7
Multireference calculations with perturbation correction and ab initio ligand field analysis (CASSCF(3,5)-NEVPT2-AILFT) yielded Δ0 = 23000 cm−1, B = 964 cm−1 and C/B = 3.08 as ligand field parameters (B(free Cr3+ ion) = 918 cm−1)18 and split E and T states.19 A larger active space in a CASSCF(7,12)-NEVPT2 calculation was used to generate the state diagram in Fig. 4.20 According to this calculation, Δ0 of [Cr(ddpd)2]3+ is located close to the second quartet-doublet crossing point 4T2–2T2 in the TS diagram (black square in Fig. 1). Although the absolute calculated values are too high, trends can be well reproduced.19 In contrast to perfectly octahedral complexes such as [Cr(CN)6]3−, CASSCF(7,12)-NEVPT2 calculations of [Cr(ddpd)2]3+ place a spin-paired 2T1-derived microstate below a 2E-derived true spin–flip microstate (2T1(1) and 2E(1) in Fig. 4).19,20 Thus the energy level ordering calculated for [Cr(ddpd)2]3+ (Fig. 4) differs from the TS diagram (Fig. 1).
The sharp phosphorescence bands (purple area in Fig. 3) suggest that the emissive excited doublet states 2T1(1) and 2E(1) are essentially nested, i.e. only weakly distorted relative to the 4A2 GS, as expected for the unchanged (t2g)3 electron configuration in the doublet states compared to the GS (Fig. 4).
The CASSCF calculated energy difference between the lowest doublet states ΔE amounts to 580 cm−1.20 Experimentally, values of 650 cm−1 and 700 cm−1 were derived from the emission band energy difference and the slopes of Boltzmann plots obtained from temperature dependent emission spectra in various solvents (Fig. 5), respectively.21
 | ||
| Fig. 5 Phosphorescence spectra of [Cr(ddpd)2][BF4]3 in H2O at variable temperature. The inset shows the Boltzmann plot ln(I738/I775) vs. T−1/K−1. The slope corresponds to −ΔE/kB with the energy difference of the emissive states ΔE and the Boltzmann constant kB.21 | ||
The 2E(1) and 2T1(1) states share the same lifetime as well as a similar geometry as the 4A2 ground state suggesting only a very small barrier for internal conversion (IC) between the two lowest doublet states. The two lowest emissive states are in thermal equilibrium (Fig. 5).21 Using ΔE = 700 cm−1 a Boltzmann factor N738/N775 of 0.032 is calculated at 293 K.
The area ratio A740/A776 of the emission bands in MeCN fitted with two Voigt functions amounts to 0.091 (see ESI, Fig. S2 and Table S2†) indicating that the radiative transition 2E(1) → 4A2 at 738 nm is approximately 0.091/0.032 = 2.8 times more allowed than the 2T1(1) → 4A2 radiative transition at 775 nm. The spin-forbidden 4A2 → 2T1/2E absorption bands (red line in Fig. 3) with extinction coefficients of <0.3 M−1 cm−1 in a concentrated acetonitrile solution were fitted with three Voigt functions and give a similar area ratio of A736/A770 = 2.96 (see ESI, Fig. S1 and Table S1†). This also indicates that the 4A2 → 2E(1) transition is ca. 3 times more allowed than the 4A2 → 2T1(1) transition. Based on the calculations presented in Fig. 4 these bands were assigned to 4A2 → 2T1(1), 4A2 → 2E(1) and 4A2 → 2T1(2) transitions, respectively (see ESI, Table S1† for details). The area ratio of the 736 and 770 nm absorption bands of 3.0 agrees with the value derived from the emission data (see above). The small Stokes shifts of 37 and 83 cm−1 for the high-energy and low-energy emission bands, respectively, support the proposed nested nature of the doublet excited states. The data also suggest a slightly more pronounced geometric distortion of the 2T1 state compared to the 2E state, which may facilitate the radiative transitions between the GS and 2E(1) state.
Below ca. 130 K (KBr pellet), the high-energy emission band vanishes and only the lowest 2T1 derived microstate is populated (Fig. 6). Furthermore, the 2T1(1) → 4A2 emission band shows vibrational progression at 795 and 811 nm (Fig. 6).22 The doublet state population 2E(1)/2T1(1) was modulated in Fourier-transform infrared (FTIR) pump–pump–probe(IR) and pump–dump–probe(IR) experiments at 290 and 20 K.22 Step-scan FTIR spectroscopy differentiated 2E and 2T1 derived microstates and suggested that [Cr(ddpd)2]3+ in the 2E(1) state nearly perfectly resembles the 4A2 geometry, while the 2T1(1) derived microstate is slightly distorted giving rise to an excited state IR spectrum which differs from the GS IR spectrum.22 These experiments show the effect of pairing two electrons in a t2g-orbital. However, the exact (Jahn–Teller) distortional mode is not fully elucidated, yet.
 | ||
| Fig. 6 Phosphorescence spectra of [Cr(ddpd)2][BF4]3 as KBr pellet between 290 and 10 K after excitation at 355 nm. The inset shows the region of the transition at about 739 nm, where the low-energy emission intensity at each temperature is normalized to 1 for better visibility of the decrease of the high-energy band at lower temperature.22 | ||
Pressure shifts both emissive doublet states to lower energies similar to ruby (−0.8 cm−1 kbar−1)23 but with a much stronger effect, i.e. 13–15 cm−1 kbar−1 for the low-energy 2T1(1) and ca. 8 cm−1 kbar−1 for the high-energy 2E(1) emission (Fig. 7).20 These energy shifts result from small geometric changes under pressure likely involving N–Cr–N angles which is much more pronounced in the molecular ruby than in the oxidic lattice of ruby.20
 | ||
| Fig. 7 Normalized pressure-dependent phosphorescence spectra of [Cr(ddpd)2][BF4]3 between 0.001 and 45.469 kbar in nujol together with a ruby crystal (right).20,23 | ||
Ultrafast dynamics and dark excited states
According to CASSCF-NEVPT2 calculations, the 4T2 states are approximately degenerate with the higher-energy 2T2 states at the Franck Condon (FC) geometry (Fig. 4). This implies, that Δ0 of [Cr(ddpd)2]3+ is well-separated from the first excited state crossing point 2T1/2E/4T2 (black circle in Fig. 1) but very close to the second crossing point 2T2/4T2 in the TS diagram (in the FC state at GS geometry, black square in Fig. 1). Consequently, when exciting [Cr(ddpd)2]3+ to the 4T2 states, a high density of 2T2 states with their vibrational levels is present in this FC region. Additionally, the CASSCF calculations yielded significant spin–orbit couplings (SOCs) of 42, 97 and 45 cm−1 for the pathways from 4T2(1) to 2T2(2), 2E(1) and 2T1(2), respectively.19 This is in agreement with El-Sayed's rule that states that SOC between two states is large when a change in multiplicity (here: quartet → doublet) is accompanied by a change in orbital angular momentum (here: eg* → t2g).24 Overall, the high density of states, relatively large SOC and small 4T2–2T2 energy difference at FC geometry could facilitate 4T2 → 2T2 ISC in [Cr(ddpd)2]3+.25In fact, the fully relaxed, emissive doublet states 2T1/2E of [Cr(ddpd)2]3+ are reached within τ = 3.5 ps after excitation to 4T2 states.26 Very likely, the ISC process is even faster (τISC < 200 fs) and the picosecond time constant rather applies to vibrational cooling (VC) and IC between doublet states. The fluorescence quantum yield ΦFl ≈ 0.01% allows to derive an upper limit for the ISC quantum yield of ΦISC < 99.99%. In the structurally similar [Cr(bpmp)2]3+ (bpmp = 2,6-bis(2-pyridylmethyl)pyridine) a lower limit for ΦISC of 92 ± 5% was determined via doublet–triplet energy transfer and laser flash photolysis experiments.19
Interestingly, in the classical organic sensitizer benzophenone Ph2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O the predicted relaxation cascade S1 → T2 → T1 → S0 with almost isoenergetic T2 and S1 states27 is analogous to the one proposed for [Cr(ddpd)2]3+ with 4T2 → 2T2 → 2T1/2E → 4A2.
O the predicted relaxation cascade S1 → T2 → T1 → S0 with almost isoenergetic T2 and S1 states27 is analogous to the one proposed for [Cr(ddpd)2]3+ with 4T2 → 2T2 → 2T1/2E → 4A2.
Femtosecond transient absorption spectra covering the Vis-NIR range after excitation with 435 nm pulses show broad excited state absorptions (ESA) covering the 500–900 nm spectral range (ESA1: 14000 cm−1 and ESA2: 12300 cm−1, Fig. 8). These evolve with a common time constant of ca. 3.2 ps to ESAs at 534 nm (ESA3: 18700 cm−1) and 1389 nm (ESA4: 7200 cm−1, see ESI, Fig. S4 and S5†).26 ESA3 and ESA4 are assigned to 2T1/2E → 2LMCT and 2T1/2E → 2T2 transitions, respectively. Addition of ESA3 to the 2T1 energy yields an energy of 31600 cm−1 (316 nm) for the 2LMCT state, which corresponds to the respective 4LMCT absorptions found in the GS absorption spectra (Fig. 3). ESA4 in the NIR region helps to experimentally estimate the energy of the dark 2T2 states by summation of the emission energy (775 nm, 12900 cm−1) and the ESA4 energy (1389 nm, 7200 cm−1) giving 20100 cm−1 (498 nm). This energy is indeed slightly below the CASSCF calculated energy of 4T2 states at the GS geometry.
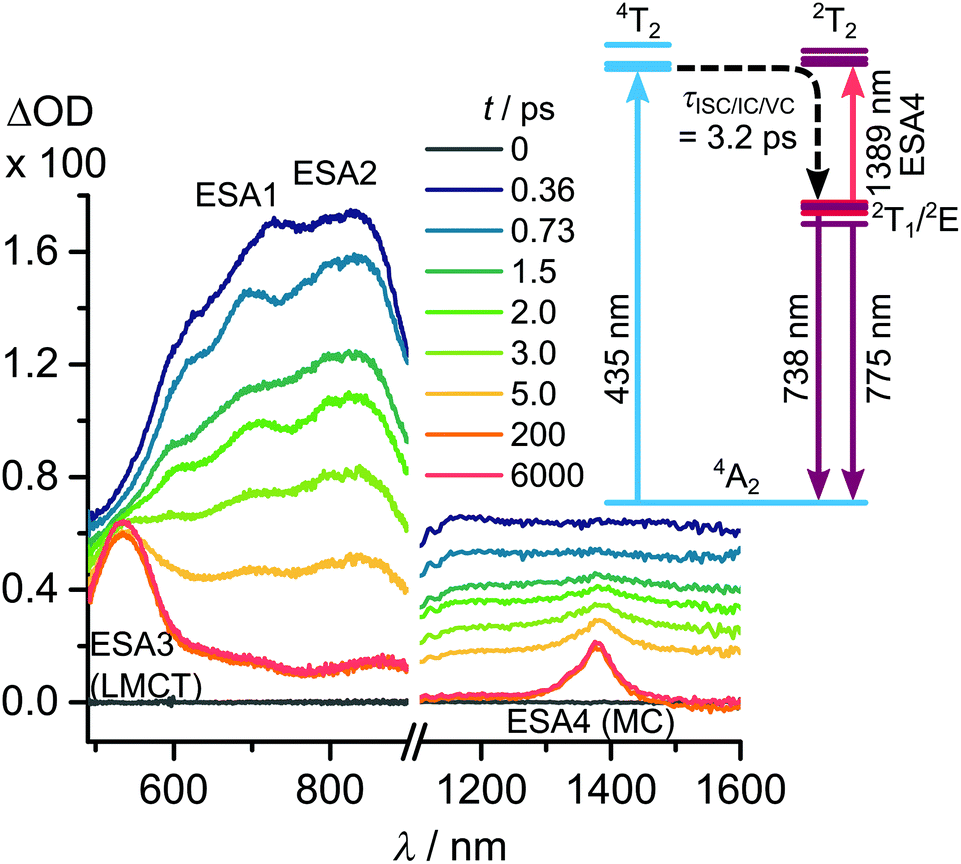 | ||
| Fig. 8 Fs-Transient absorption spectra of [Cr(ddpd)2]3+ in CH3CN after excitation at 435 nm and excited state diagram with CASSCF(7,12)-NEVPT2 calculated energies (see ESI† for details, ESA = excited state absorption).26 | ||
The 4T2 states should be Jahn–Teller distorted due to the singly occupied eg* orbitals and the doubly occupied t2g orbitals. Indeed DFT calculations reveal, that the optimised lowest 4T2 microstate displays elongated Cr–N bond lengths of terminal pyridines in a single ddpd ligand (2.08 Å → 2.42 Å).7 As the energy of this quartet state drops dramatically with increasing Cr–N distances, it is essential for high stability and strong emission that the ISC is fast and preferably in the FC region before vibrational relaxation in the 4T2 state takes place.
With this interpretation, [Cr(ddpd)2]3+ hits a sweet spot in the TS diagram which enables a fast ISC pathway 4T2(1) → 2T2(2) to the doublet states at the FC geometry via the dark 2T2 states in addition to the direct 4T2(1) → 2E(1) and 4T2(1) → 2T1(2) pathways with a higher energy difference. The vibrational relaxation within the quartet states along the Cr–N Jahn–Teller modes (in x- and y-direction)28 might be comparably slow due to a low potential energy gradient at the FC geometry. In any case, the fast ISC avoids vibrational relaxation within the quartet states and thus suppresses undesired fluorescence from the 4T2 state, non-radiative decay by 4T2 surface crossing with the GS, and even Cr–N bond dissociation. In fact, [Cr(ddpd)2]3+ is particularly photostable, even at low pH in contrast to e.g. [Cr(bpy)3]3+.6,7
Long-lived spin–flip states
The dynamics of the long-lived excited states depend on the presence of high-energy oscillators29 and quenchers,26,30 the excited state distortion22 and the temperature.21,22The doublet states can relax non-radiatively via energy transfer (EnT) to vibrational overtones of nearby C–H oscillators (multiphonon relaxation).31 [Cr(ddpd)2]3+ has a comparably small spectral overlap integral (SOI) with the ν4 and the ν5 C–H overtones associated with the pyridine rings (Fig. 9). This results in a small rate constant for non-radiative decay knr and thus a high quantum yield.29
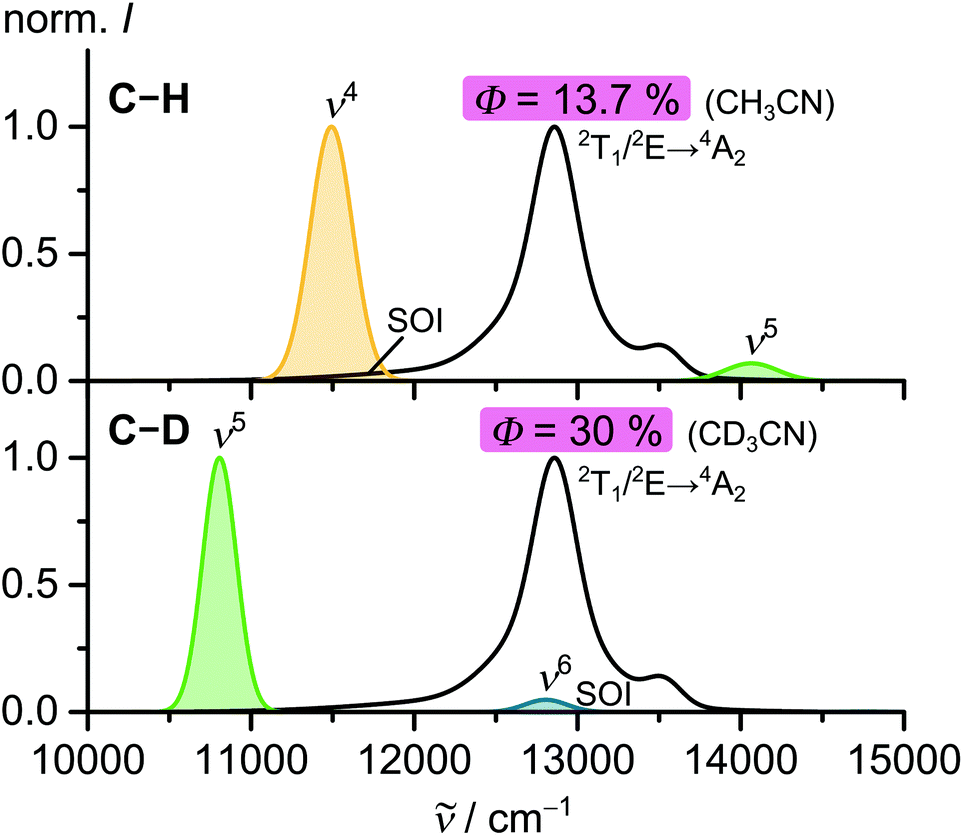 | ||
| Fig. 9 C–H and C–D vibrational overtone spectra of model pyridine ligands and their overlap with the phosphorescence bands of [Cr(ddpd)2]3+ (SOI = spectral overlap integral).29 | ||
After deuteration of the ligand, there is only a significant SOI with a higher C–D overtone (ν6) with an extinction coefficient which is roughly two orders of magnitude smaller than ν5(C–H).29 Thus, deuterated [Cr(ddpd)2]3+ shows an even smaller knr and a record quantum yield of 30%. The limited increase from 13.7 to 30% might in part be associated with the near degeneracy of the fifth C–D overtone (ν6) and the 2T1(1) → 4A2 emission band (Fig. 9),29 yet direct 2T1 → 4A2 ISC plays a role as well, irrespective of the deuteration level.
As stated above, at high enough thermal energy the 2E and 2T1 states equilibrate leading to dual emission. Below ca. 130 K, the 2E(1) band vanishes, the 2T1(1) emission band sharpens and shifts to lower energy (785 nm in KBr, Fig. 6) showing that the 2T1(1) state is stabilized (vertical displacement of the 2T1(1) potential energy well). The phosphorescence quantum yield increases dramatically between 70 and 10 K (Fig. 6).22
The increase in phosphorescence quantum yield at very low temperatures might be associated with the sharpening of the 2T1(1) emission band, which reduces the SOI with the third C–H vibrational overtone (ν4) of the ligand. Additional explanations for the higher phosphorescence quantum yield at low T might be a reduction of other thermally activated non-radiative decay pathways and the enhancement of the radiative decay via larger distortion of the 2T1(1) state relative to the ground state lifting Laporte's rule.32 In fact, the radiative rate kr decreases dramatically from pseudooctahedral [Cr(ddpd)2]3+ (kr = Φ/τ = 122 s−1 in CH3CN)15 to the electronically similar but perfectly centrosymmetric CrIII complex [Cr(tpe)2]3+ (kr = 18.2 s−1, tpe = 1,1,1-tris(pyrid-2-yl)ethane).33
Triplet oxygen quenches the long-lived phosphorescence of [Cr(ddpd)2]3+ induced by 435 nm excitation via Dexter energy transfer to form the ground state complex and 1O2 with a quantum yield of 61% in acetonitrile.26 The typical 1O2 phosphorescence at 1274 nm appears even after direct spin-forbidden excitation of [Cr(ddpd)2]3+ to its 2T1/2E states at 771 and 735 nm, respectively (see ESI, Fig. S3†), confirming that the doublet states of [Cr(ddpd)2]3+ sensitize 1O2.
The excited state landscape of [Cr(ddpd)2]3+ is sketched in Fig. 10 along with experimentally determined energies, rate constants and quantum yields, summarising excitation, excited state dynamics to the emissive doublet states, excited state absorptions, non-radiative relaxation in particular multiphonon relaxation as well as radiative transitions.
 | ||
| Fig. 10 Experimentally7,21,26,29 determined excited state energy landscape of the molecular ruby [Cr(ddpd)2]3+ in CH3CN and time constants determined so far with exemplary microstates for the MC states derived from CASSCF(7,12)-NEVPT calculations,20 the rate constant of non-radiative decay knr and the rate constant for energy transfer to vibrational overtones kEnT (which is included in knr). The Jahn–Teller coordinate refers to the 4T2 state while the 4/2LMCT states likely display other distortions from the GS geometry. | ||
Future outlook
Following the report of [Cr(ddpd)2]3+ the underlying design principles gave rise to a large number of highly luminescent CrIII complexes.19,33–35,36 Current efforts are directed to shifting the emission energy of molecular ruby derivatives to lower or higher energy by ligand modifications19,37 and by modification of the central metal and electron configuration from d3-CrIII to d2-VIII.38 Variation of the counter ions of [Cr(ddpd)2]3+ and surrounding matrix as well as shielding of the metal center by sterically demanding groups proved to be fruitful strategies to enhance quantum yields and lower oxygen sensitivity.15 Applications of molecular ruby derivatives in upconversion,39 circularly polarized emission,34,35,40 sensing,20,21,41 photosensitization for organic synthesis26 or photodynamic therapy30 and others had already been demonstrated or are currently explored.Author contributions
W. R. K. provided the resources (synthesis of [Cr(ddpd)2]3+), performed the investigation (measurement of ΦFl), visualized the data and wrote the original drafts. C. R. performed the fs-transient absorption spectroscopy. R. N. performed the investigation of the phosphorescence lifetimes, the spin-forbidden absorption and 1O2 sensitization experiments with NIR excitation. K. H. conceptualized the article, acquired the funding, supervised the project and wrote the original drafts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Johannes Hunger for his support in this project and Dr Christoph Förster for constructive criticism of the manuscript. Financial support from the Deutsche Forschungsgemeinschaft [DFG, Priority Program SPP 2102 “Light-controlled reactivity of metal complexes” (HE 2778/13-1)] is gratefully acknowledged. We thank the DFG for grant INST 247/1018-1 FUGG to K. H.; W. R. K. is grateful for a Kekulé scholarship of the Fonds der Chemischen Industrie.Notes and references
- (a) F. Teplý, Phys. Sci. Rev., 2020, 5, 20170171 Search PubMed; (b) O. S. Wenger, Chem. – Eur. J., 2019, 25, 6043–6052 CrossRef CAS PubMed.
- T. H. Maiman, Nature, 1960, 187, 493–494 CrossRef.
- C. Degli Esposti and L. Bizzocchi, J. Chem. Educ., 2007, 84, 1316 CrossRef CAS.
- W. R. Kitzmann, J. Moll and K. Heinze, Photochem. Photobiol. Sci., 2022 DOI:10.1007/s43630-022-00186-3 , in press.
- R. D. Dill, R. I. Portillo, S. G. Shepard, M. P. Shores, A. K. Rappé and N. H. Damrauer, Inorg. Chem., 2020, 59, 14706–14715 CrossRef CAS PubMed.
- S. Otto, M. Dorn, C. Förster, M. Bauer, M. Seitz and K. Heinze, Coord. Chem. Rev., 2018, 359, 102–111 CrossRef CAS.
- S. Otto, M. Grabolle, C. Förster, C. Kreitner, U. Resch-Genger and K. Heinze, Angew. Chem., Int. Ed., 2015, 54, 11572–11576 CrossRef CAS PubMed.
- D. Zare, B. Doistau, H. Nozary, C. Besnard, L. Guénée, Y. Suffren, A.-L. Pelé, A. Hauser and C. Piguet, Dalton Trans., 2017, 46, 8992–9009 RSC.
- (a) Y. Tang, S. Zhao, B. Long, J.-C. Liu and J. Li, J. Phys. Chem. C, 2016, 120, 17514–17526 CrossRef CAS; (b) J. K. McCusker, Science, 2019, 363, 484–488 CrossRef CAS PubMed.
- D. D. Ragan, R. Gustavsen and D. Schiferl, J. Appl. Phys., 1992, 72, 5539–5544 CrossRef CAS.
- R. S. Quimby and W. M. Yen, J. Appl. Phys., 1980, 51, 1780–1782 CrossRef CAS.
- A. D. Kirk and G. B. Porter, J. Phys. Chem., 1980, 84, 887–891 CrossRef CAS.
- J.-R. Jiménez, B. Doistau, C. Besnard and C. Piguet, Chem. Commun., 2018, 54, 13228–13231 RSC.
- (a) Y. Tanabe and S. Sugano, J. Phys. Soc. Jpn., 1954, 9, 753–766 CrossRef CAS; (b) Y. Tanabe and S. Sugano, J. Phys. Soc. Jpn., 1954, 9, 766–779 CrossRef CAS; (c) J. Oppenheim and J. Miller, Tanabe-Sugano for Mathematica, 2019, available at: https://github.com/JulesOpp/Tanabe-Sugano, accessed 7 December 2021 Search PubMed.
- C. Wang, W. R. Kitzmann, F. Weigert, C. Förster, X. Wang, K. Heinze and U. Resch-Genger, ChemPhotoChem, 2022, e202100296 Search PubMed.
- (a) P. A. Scattergood, in Organometallic Chemistry, ed. N. J. Patmore and P. I. P. Elliott, Royal Society of Chemistry, Cambridge, 2020, vol. 43, pp. 1–34 Search PubMed; (b) M. Dorn, N. R. East, C. Förster, W. R. Kitzmann, J. Moll, F. Reichenauer, T. Reuter, L. Stein and K. Heinze, in Comprehensive Inorganic Chemistry III, ed. J. Reedijk, Elsevier, San Diego, 2022 Search PubMed.
- S. Lenz, H. Bamberger, P. P. Hallmen, Y. Thiebes, S. Otto, K. Heinze and J. van Slageren, Phys. Chem. Chem. Phys., 2019, 21, 6976–6983 RSC.
- A. B. P. Lever, Inorganic electronic spectroscopy, Elsevier, Amsterdam, 1st edn, 1968 Search PubMed.
- F. Reichenauer, C. Wang, C. Förster, P. Boden, N. Ugur, R. Báez-Cruz, J. Kalmbach, L. M. Carrella, E. Rentschler, C. Ramanan, G. Niedner-Schatteburg, M. Gerhards, M. Seitz, U. Resch-Genger and K. Heinze, J. Am. Chem. Soc., 2021, 143, 11843–11855 CrossRef CAS PubMed.
- S. Otto, J. P. Harris, K. Heinze and C. Reber, Angew. Chem., Int. Ed., 2018, 57, 11069–11073 CrossRef CAS PubMed.
- S. Otto, N. Scholz, T. Behnke, U. Resch-Genger and K. Heinze, Chem. – Eur. J., 2017, 23, 12131–12135 CrossRef CAS PubMed.
- P. Boden, P. Di Martino-Fumo, G. Niedner-Schatteburg, W. Seidel, K. Heinze and M. Gerhards, Phys. Chem. Chem. Phys., 2021, 23, 13808–13818 RSC.
- R. A. Forman, G. J. Piermarini, J. D. Barnett and S. Block, Science, 1972, 176, 284–285 CrossRef CAS.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- S. Otto, A. M. Nauth, E. Ermilov, N. Scholz, A. Friedrich, U. Resch-Genger, S. Lochbrunner, T. Opatz and K. Heinze, ChemPhotoChem, 2017, 1, 344–349 CrossRef CAS.
- (a) K. Shizu and H. Kaji, J. Phys. Chem. A, 2021, 125, 9000–9010 CrossRef CAS; (b) S. Yabumoto, S. Sato and H. Hamaguchi, Chem. Phys. Lett., 2005, 416, 100–103 CrossRef CAS.
- K. Mack, A. Wünsche von Leupoldt, C. Förster, M. Ezhevskaya, D. Hinderberger, K. W. Klinkhammer and K. Heinze, Inorg. Chem., 2012, 51, 7851–7858 CrossRef CAS PubMed.
- C. Wang, S. Otto, M. Dorn, E. Kreidt, J. Lebon, L. Sršan, P. Di Martino-Fumo, M. Gerhards, U. Resch-Genger, M. Seitz and K. Heinze, Angew. Chem., Int. Ed., 2018, 57, 1112–1116 CrossRef CAS PubMed.
- U. Basu, S. Otto, K. Heinze and G. Gasser, Eur. J. Inorg. Chem., 2019, 37–41 CrossRef CAS.
- E. Kreidt, C. Kruck and M. Seitz, in Handbook on the physics and chemistry of rare earths. Including actinides, ed. J.-C. G. Bünzli, V. K. Pecharsky, G.-Y. Adachi, K. A. Gschneidner and E. LeRoy, North-Holland, Amsterdam, Oxford, 2018, vol. 53, pp. 35–79 Search PubMed.
- O. Laporte and W. F. Meggers, J. Opt. Soc. Am., 1925, 11, 459 CrossRef CAS.
- S. Treiling, C. Wang, C. Förster, F. Reichenauer, J. Kalmbach, P. Boden, J. P. Harris, L. M. Carrella, E. Rentschler, U. Resch-Genger, C. Reber, M. Seitz, M. Gerhards and K. Heinze, Angew. Chem., Int. Ed., 2019, 58, 18075–18085 CrossRef CAS PubMed.
- J.-R. Jiménez, B. Doistau, C. M. Cruz, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, J. Am. Chem. Soc., 2019, 141, 13244–13252 CrossRef PubMed.
- J.-R. Jiménez, M. Poncet, S. Míguez-Lago, S. Grass, J. Lacour, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, Angew. Chem., Int. Ed., 2021, 60, 10095–10102 CrossRef.
- (a) J.-R. Jiménez, M. Poncet, B. Doistau, C. Besnard and C. Piguet, Dalton Trans., 2020, 49, 13528–13532 RSC; (b) S. Otto, C. Förster, C. Wang, U. Resch-Genger and K. Heinze, Chem. – Eur. J., 2018, 24, 12555–12563 CrossRef CAS PubMed.
- (a) N. Sinha, J.-R. Jiménez, B. Pfund, A. Prescimone, C. Piguet and O. S. Wenger, Angew. Chem., Int. Ed., 2021, 60, 23722–23728 CrossRef CAS PubMed; (b) L. Stein, P. Boden, R. Naumann, C. Förster, G. Niedner Schatteburg and K. Heinze, Chem. Commun., 2022, 58, 3701–3704 RSC.
- (a) M. Dorn, J. Kalmbach, P. Boden, A. Kruse, C. Dab, C. Reber, G. Niedner-Schatteburg, S. Lochbrunner, M. Gerhards, M. Seitz and K. Heinze, Chem. Sci., 2021, 12, 10780–10790 RSC; (b) M. Dorn, K. Mack, L. M. Carrella, E. Rentschler, C. Förster and K. Heinze, Z. Anorg. Allg. Chem., 2018, 644, 706–712 CrossRef CAS; (c) J. P. Zobel, T. Knoll and L. González, Chem. Sci., 2021, 12, 10791–10801 RSC; (d) M. S. Fataftah, S. L. Bayliss, D. W. Laorenza, X. Wang, B. T. Phelan, C. B. Wilson, P. J. Mintun, B. D. Kovos, M. R. Wasielewski, S. Han, M. S. Sherwin, D. D. Awschalom and D. E. Freedman, J. Am. Chem. Soc., 2020, 142, 20400–20408 CrossRef CAS.
- (a) J. Kalmbach, C. Wang, Y. You, C. Förster, H. Schubert, K. Heinze, U. Resch-Genger and M. Seitz, Angew. Chem., Int. Ed., 2020, 59, 18804–18808 CrossRef CAS PubMed; (b) C. Wang, F. Reichenauer, W. R. Kitzmann, C. Kerzig, K. Heinze and U. Resch-Genger, Angew. Chem., Int. Ed., 2022, 61, e202202238 Search PubMed.
- (a) C. Dee, F. Zinna, W. R. Kitzmann, G. Pescitelli, K. Heinze, L. Di Bari and M. Seitz, Chem. Commun., 2019, 55, 13078–13081 RSC; (b) M. Poncet, A. Benchohra, J.-R. Jiménez and C. Piguet, ChemPhotoChem, 2021, 5, 880–892 CrossRef CAS.
- C. Wang, S. Otto, M. Dorn, K. Heinze and U. Resch-Genger, Anal. Chem., 2019, 91, 2337–2344 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt00569g |
| This journal is © The Royal Society of Chemistry 2022 |