
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalo complexes of gold(I) containing glycoconjugate carbene ligands: synthesis, characterization, cytotoxicity and interaction with proteins and DNA model systems†
Alfonso
Annunziata
 ab,
Giarita
Ferraro
a,
Maria Elena
Cucciolito
ab,
Paola
Imbimbo
a,
Angela
Tuzi
a,
Daria Maria
Monti
*a,
Antonello
Merlino
*a and
Francesco
Ruffo
*ab
ab,
Giarita
Ferraro
a,
Maria Elena
Cucciolito
ab,
Paola
Imbimbo
a,
Angela
Tuzi
a,
Daria Maria
Monti
*a,
Antonello
Merlino
*a and
Francesco
Ruffo
*ab
aDipartimento di Scienze Chimiche, Università di Napoli Federico II, Complesso Universitario di Monte Sant'Angelo, Via Cintia 21, 80126, Napoli, Italy. E-mail: dariamaria.monti@unina.it; antonello.merlino@unina.it; ruffo@unina.it
bConsorzio Interuniversitario di Reattività Chimica e Catalisi (CIRCC), Via Celso Ulpiani 27, 70126, Bari, Italy
First published on 1st June 2022
Abstract
New neutral carbene complexes of gold(I) [Au(Im-Me)X] (X = Cl, Au1; X = Br, Au2; X = I, Au3) have been synthesized and fully characterized by different techniques, including NMR and UV-vis absorption spectroscopy and single crystal X-ray diffraction. The carbene ligand Im-Me is decorated with a glucoside fragment via a triazole linker, obtainable through a click chemistry reaction. The compounds retain the Au–NHC fragment in aqueous solvents, and an equilibrium between the neutral halo- and the cationic di-carbene form [Au(Im-Me)2]+ is observed, whose extent follows the trend Au1 < Au2 < Au3. Cytotoxicity studies on two cancer and two non-tumorigenic cell lines reflect the solution behavior, as a certain difference among the complexes was disclosed, with the iodo complex Au3 being more active and selective. The compounds interact with both DNA and protein model systems. The X-ray structure of the adduct formed upon the reaction of Au1 with bovine pancreatic ribonuclease (RNase A) reveals Au binding at the side chain of His105 of both protein molecules A and B of the asymmetric unit. The binding of gold atoms at both the nitrogen atoms of the imidazole ring of His15 and at the N-terminal tail has been found in the adduct formed with hen egg white lysozyme.
Introduction
Gold(I) N-heterocyclic carbene (NHC) complexes are promising candidates in the pool of anticancer metallodrugs.1–3 Indeed, the design of new Au-based complexes requires the presence of stabilizing ligands, due to the marked oxidizing character of gold species which can easily undergo reduction to Au(0) in biological aqueous systems, with consequent deactivation and uncontrolled damage to biological tissues.4 NHC ligands give rise to strong carbon-to-metal bonds, resulting in highly robust complexes in physiological conditions.5–7 Moreover, pro-carbene scaffolds are easily prepared to allow the exploration of unlimited variations on the theme.8,9The interest in AuI(I) NHC complexes began in the early 2000s when Berners-Price et al. studied the biological activity of cationic mono- and di-nuclear Au(I) bis-NHC complexes aiming to target mitochondria.10–12 They explored a consistent panel of complexes with different structural motifs and correlated the lipophilicity of the compounds with their anti-mitochondrial activity. Moreover, they identified the mitochondrial enzyme thioredoxin reductase (TrxR) as the potential target for this class of metallodrugs. TrxR is an enzyme belonging to the thioredoxin system (Trx), a multi-protein assembly involved in maintaining the redox balance in the cell, and it plays a key role in cancer cell growth and proliferation.13 TrxR is a seleno-protein, containing selenocysteine in the C-terminal domain.14 Such a residue is a highly reactive nucleophilic site, crucial for the activity of the enzyme and a suitable target for gold-based drugs.15 Following Berners-Price works, several Au(I) NHC complexes have been studied,16–31 involving their in-solution stability, reactivity with model biomolecules, inhibition of TrxR, and cytotoxic properties.
Structure–activity relationships were studied by the Ott research group,32–35 who investigated the impact of the other ligands coordinated to the linear gold–NHC scaffold. TrxR inhibition was correlated with the bond dissociation energy of the ancillary ligand,34 with the chloro complexes ([Au(NHC)Cl]) showing higher inhibitory activity with respect to the cationic bis-carbene and phosphine complexes ([Au(NHC)2]+ and [Au(NHC)PR3]+). In their comprehensive studies,33 they also observed that the most lipophilic agent, i.e. a triphenylphosphine Au–NHC complex, was the most cytotoxic agent among those investigated, probably due to its more efficient internalization in cells. Such a complex was found to efficiently inhibit TrxR and the zinc-finger protein PARP-1. More extensive investigations35 disclosed that the same triphenylphosphine complex causes an alteration in the redox balance of the cell and affects mitochondria by irreversible inhibition of their respiratory activity. Finally, signaling associated with DNA damage was detected.
The interaction between AuNHC complexes and DNA was intensively studied by Casini et al.36 A cationic caffeine derivative bis-carbene complex (AuTMX2) was found to selectively interact with G-quadruplex DNA structures.4,37 Quadruplexes have a fundamental role in several cellular processes38–40 and are considered suitable targets for anticancer metallodrugs.41 Further studies42 evidenced the structural parameters that explain the preferential binding of AuTMX2 with G-quadruplexes over double helices. The binding mode was studied43 and the data revealed that the interaction seems to occur by π–π and weak electrostatic interactions. Recently, proteomic studies disclosed the binding to several proteins in human ovarian cancer cells (A2780), confirming a variegated mechanism of action of AuNHC complexes.44
All these data suggest a highly versatile multi-modal effect of gold NHC complexes for triggering their anticancer activity.
The conjugation of metal complexes to bio-derived fragments is a widely explored strategy to improve the pharmacological properties of the resulting metallodrugs.45,46 Sugar containing complexes have enhanced the biocompatibility, water solubility and selectivity due to the increased uptake of nutrients in cancer cells with respect to non-cancer ones (Warburg effect).47 Such a strategy has been successfully applied to several metal scaffolds for anticancer therapy and diagnostics.48 Our research group successfully applied glycoconjugation to organometallic platinum complexes with NHC49–51 and N-heterocyclic ligands.52–55 The concept was also extended to gold(I) NHC complexes. In 2017, D'Amora et al. described the synthesis, characterization and anticancer activity of a series of neutral and cationic Au(I) complexes with a glycoconjugate NHC ligand derived from methylimidazole.56 A cationic phosphine derivative displayed anticancer activity comparable to cisplatin while neutral chloro species were inactive. Recently, a series of analogous di-nuclear Au(I) carbene complexes were designed and studied for their anticancer properties.57
To further investigate the properties of glycoconjugate anticancer Au(I) NHC complexes, new neutral halo complexes have been synthesized and fully characterized (Au1–Au3 in Scheme 1). The carbene ligand Im-Me is decorated with a glucoside fragment via a triazole linker, easily obtainable through a click chemistry reaction. The in-solution behaviour, the reactivity with model biomolecules and the biological activity of the new complexes have been studied and discussed.
 | ||
| Scheme 1 Synthesis of the gold(I) NHC complexes Au1–Au3. | ||
Results and discussion
Synthesis and characterization
The pro-carbene ligand Im-Me was obtained by reacting the glycoconjugate imidazole Im, reported in previous works,52,55 with an excess of iodomethane in chloroform for 24 h. The alkylated product was isolated by removing the volatiles under vacuum (Scheme 2). | ||
| Scheme 2 Synthesis of the pro-carbene ligand Im-Me. | ||
The 1H NMR spectra (Fig. S1†) and elemental analysis confirmed the positive outcome of the reaction. The formation of the cationic quaternary nitrogen atom resulted in a general downfield shift of the NMR proton signals with respect to the parent Im. The NCHN atom shifted from 7.51 ppm in Im to 9.91 ppm in Im-Me, while the singlet of the triazole and the doublet of the backbone imidazole protons shifted from 7.91 to 8.88 ppm and from 7.30 to 7.95 ppm, respectively. The two non-equivalent methyl groups were found at 4.10 and 4.15 ppm.
The gold complexes Au1–Au3 were synthesized according to Scheme 1. The chloro-derivative Au1 was prepared by treating the precursor [Au(tht)Cl] (tht = tetrahydrothiophene) with the silver carbene intermediate, formed in situ upon the reaction between Im-Me and Ag2O in dichloromethane. The trans-metalation occurred with selective precipitation of AgI, which avoided the formation of mixed chloro/iodo species. The other halo-complexes were obtained through a halide exchange reaction in acetone, given the major affinity of gold(I) for the softer halides.
All the compounds were characterized by 1H and 13C NMR spectroscopy, as well as by elemental analysis. The formation of the carbene complexes was evident upon comparing their spectra with that of the pro-carbene Im-Me (Fig. S2†). One evidence was the disappearance of the singlet at 9.87 ppm belonging to the NCHN proton. Moreover, the coordination to the metal center resulted in a low-frequency shift of the imidazole proton. The signals of the sugar protons were found at the expected values of chemical shifts, with the coupling patterns for glucose in β-configuration.
The proton spectra of the three complexes were perfectly overlapping, as the halide did not affect the signals of the carbene moiety. Conversely, the effect of the X ligand was observable in the 13C NMR spectra, as shown in Fig. 1. The Ccarbene nucleus resonates progressively at higher frequencies from the chloro- to the iodo-derivative.58 This trend reflects the progressive increase of the halide donor strength along the sequence I > Br > Cl and has been explained considering the intrinsic electronic structure of NHC ligands that acquire increasing ‘free NHC ligand’ character as the trans-influence of the halide increases.59
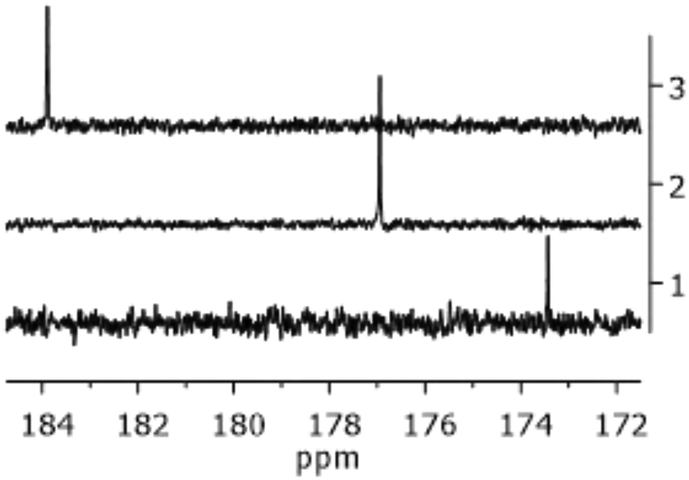 | ||
| Fig. 1 Portion of the 13C NMR spectra of Au1 (trace 1), Au2 (trace 2), and Au3 (trace 3) in CDCl3 at 400 MHz. | ||
Single crystals suitable for XRD were obtained by slow evaporation of a toluene/acetonitrile solution at room temperature. The X-ray diffraction analysis confirmed the identity of Au1 as a neutral mononuclear Au(I)–NHC complex (Fig. 2). The complex crystallizes in the orthorhombic P212121 space group with one molecule and one disordered solvate toluene in the independent unit. The crystal data and structure details are reported in the ESI (Tables S1–S4 and Fig. S3–S5†).
 | ||
| Fig. 2 ORTEP view of the Au1 molecular structure with thermal ellipsoids drawn at the 30% probability level. The solvent toluene molecule is not drawn for clarity. Selected bond lengths and angles: Au1–C1 = 1.970(8), Au1–Cl1 = 2.296(2), C1–N1 = 1.360(10), C1–N2 = 1.347(10), C2–C3 = 1.338(11), C3–C6 = 1.456(11), N3–N4 = 1.304(9), N4–N5 = 1.340(8) Å; N1–C1–N2 = 105.2(7), C1–Au1–Cl1 = 179.6(3)°. | ||
The Au–Cl and Au–C bond distances are comparable to those of analogous complexes.56,60 A linear environment of the two-coordinated Au atom is observed (C1–Au1–Cl1 = 179.6(3)°). In the complex, the imidazole and the triazole rings are significantly tilted to each other (angle of 32.5(5)° between the mean planes), at variance with similar compounds where a coplanarity of the two rings is observed.60,61 Such deviations from coplanarity may be related to the crystal packing weak interactions and indicate a quite great extent of conformational flexibility in the ligand. In the molecule, the encumbered pendant pyranose ring adopts the usual chair conformation and disposes almost perpendicular to the imidazole ring plane to favour van der Waals interactions in the crystal packing. The presence of the lattice solvent prevents the possibility of intermolecular C–H⋯Au contacts or aurophilic Au⋯Au interactions in the crystal and the crystal packing is stabilized by weak C–H⋯N and C–H⋯O interactions.
In-solution stability of the complexes
The in-solution stability of complexes Au1–Au3 was first evaluated by 1H NMR spectroscopy in acetone-d6 and DMSO-d6. In both solvents, the complexes gave rise to spectral profiles that did not change over 24 h (Fig. S6† shows the spectra in DMSO-d6).Besides the signals attributable to the NHC/halo complexes, another minor species was clearly detectable for Au2 and Au3. These less abundant species have been hypothesized as the bis-carbene complexes [Au(Im-Me)2]+[AuX2]− (Au2′–Au3′) formed in solution through ligand scrambling, as previously described for [Au(NHC)X] complexes.5–7,62
The 13C NMR spectrum of complex Au3 in DMSO-d6 (Fig. 3) confirms this hypothesis by disclosing the presence of two Ccarbene signals at 185 ppm (less intense) and 181 ppm. According to previously reported data,63 the former one can be attributed to the di-carbene species Au3′ that is expected to resonate at higher frequencies than the iodo-derivative Au3. The relative abundances of the second species were quantified at ∼3% and ∼12% for Au2 and Au3, respectively.
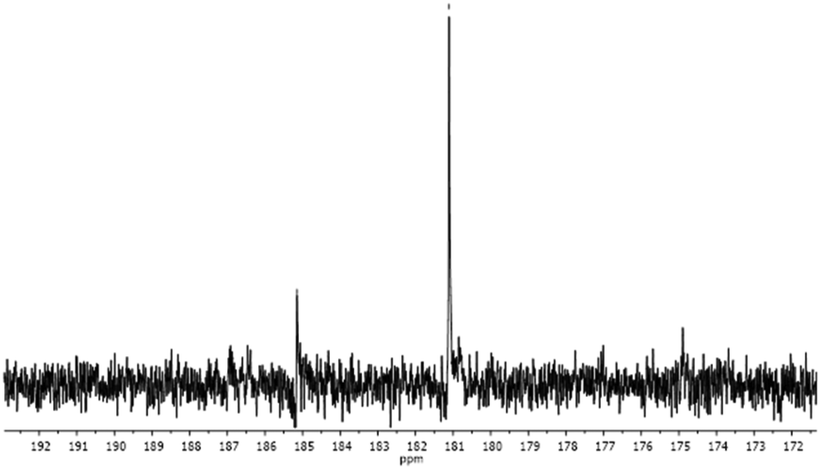 | ||
| Fig. 3 Portion of the 13C NMR spectrum of complex Au3 in DMSO-d6. | ||
The stability of the three gold complexes in pure DMSO has been also evaluated by UV-vis absorption spectroscopy. The UV-vis spectra of Au1, Au2 and Au3 were recorded for 3 h and then after 24 h at room temperature. The superimposition of the spectra reveals that the three compounds are stable over 24 h, showing a single peak in the region between 260–265 nm (Fig. 4). These results are consistent with NMR data, confirming that the gold complexes retain their structure in pure DMSO.
 | ||
| Fig. 4 UV-vis spectra of Au1 (top), Au2 (middle), and Au3 (bottom) in 100% DMSO as a function of time. Compound concentration: 100 μM. | ||
Mixed D2O–acetone-d6 and D2O–DMSO-d6 mixtures were then used to investigate the stability in aqueous systems. The maximum percentage of D2O used was 50%, due to precipitation of the compounds in the presence of higher amounts of water. The spectral profiles resembled those observed in pure organic solvents, although the equilibrium between the mono- and di-carbene species reached more slowly.
Fig. S7† shows the spectra of the bromo-derivative Au2 recorded in 50%–50% D2O–DMSO-d6 over time. After 24 h the abundance of Au2′ was almost 10% of the starting complex with respect to 3% recorded in pure DMSO-d6.
Similar trends were observed for Au1 (5% of Au1′ after 24 h) and for Au3 (almost 25% of Au3′ after 24 h). Such behaviour reflected the polarity of the solvent mixture, which increased in the presence of water.64 The reactivity observed in aqueous solvent mixtures was perfectly in line with the results reported for gold(I) complexes.5–7,65
In summary, compounds Au1–Au3 are stable in aqueous solvent mixtures with the retention of the NHC fragment. The equilibrium between the neutral halo- and the cationic di-carbene forms is dependent on the halide and the presence of water. This observation may play a role in the biological activity of the compounds because cationic bis-carbene species are usually more active than their neutral counterparts,66,67 and therefore, the speciation of complexes Au1–Au3 in biological environments could govern the cytotoxic effect as well as the propensity to react with biomolecules. These considerations encouraged the testing of the biological activity of the compounds on a cell-based model.
Evaluation of the biological activity of Au1–Au3 complexes
The biological activity of the NHC gold(I) complexes Au1–Au3 was assessed on two cancer cell lines, human epidermoid carcinoma cells (A431) and murine fibroblast BALB/c-3T3 transformed with SV40 virus (SVT2) and, on two non-cancer cell lines, immortalized human keratinocytes (HaCaT) and immortalized murine fibroblasts (BALB/c-3T3). Cells were incubated with increasing concentrations of Au1–Au3. After 48 h incubation, the cytotoxicity was evaluated by MTT assay and cell survival was expressed as the percentage of viable cells in the presence of Au complexes compared with that of untreated cells (Fig. S8†). The IC50 values (the concentration of the complex able to reduce to 50% of the cell viability) and the selectivity index (SI) of the compounds (the ratio between the IC50 values of the non-cancer cell line and its cancer counterpart) are reported in Table 1.| IC50 (μM) | ||||||
|---|---|---|---|---|---|---|
| HaCaT | A431 | SI | BALB/c-3T3 | SVT2 | SI | |
| Au1 | >150 | 91 ± 2 | N.D. | 76 ± 1 | 84 ± 6 | 0.90 |
| Au2 | >100 | >100 | N.D. | 82 ± 4 | 92 ± 4 | 0.87 |
| Au3 | 92 ± 0.1 | 83 ± 5 | 1.1 | 83 ± 8 | 35 ± 6 | 2.4 |
The complexes displayed IC50 values in the high micromolar range, lower than those obtained for the related glycoconjugated dinuclear Au(I) complexes, thus demonstrating an increased cytotoxicity.57 Some distinctions by changing the halide ligand could be observed. The chloro derivative Au1 exerted a cytotoxic effect on A431 cells, whereas no IC50 value was reached on HaCaT cells, stating some selectivity of this complex towards cancer cells. Similarly, the iodide complex Au3 was almost 2-times more toxic on SVT2 when compared to BALB/c-3T3. A comparison of the IC50 values obtained with CDDP for the HaCaT and A431 cell lines showed that CDDP was more toxic than the newly synthesized agents but the SI (0.17) indicated the complete absence of selectivity in the case of CDDP.50 On the other hand, the IC50 values obtained for the BALB/c-3T3 and SVT2 cells were much lower in the case of Au complexes with respect to CDDP, with Au3 having the highest SI (1.23 for CDDP and 2.4 for Au3). Generally speaking, some carbene-containing platinum complexes analyzed in previous studies revealed a higher toxicity but the total absence of selectivity for the analyzed cell lines.50
Binding to DNA
Prompted by the results of the biological experiments, we studied whether the investigated gold(I) complexes might interact with DNA, which is commonly believed to represent the major target of anticancer metallodrugs.68 The DNA binding properties of complexes Au1–Au3 were tested in vitro by circular dichroism (CD) and fluorescence spectroscopy. In particular, the conformational changes of calf thymus DNA (ct-DNA) induced by the three gold complexes were examined by CD: the far UV-CD spectra of ct-DNA were recorded in the absence and in the presence of increasing amounts of Au1–Au3 after 24 h incubation. The CD spectrum of Au-free ct-DNA contains a positive band at 278 nm due to base-stacking and a negative band at 245 nm due to the right-handed helicity (Fig. 5). Both bands are quite sensitive to the interaction with small molecules.69 | ||
Fig. 5 CD spectra of ct-DNA recorded after 24 h of incubation with Au1 (top), Au2 (middle), and Au3 (bottom) in the molar ratios of DNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au 1:0.5 (pink line) and 1:1 (blue line). The spectrum of native DNA is also reported as the control (black line). :Au 1:0.5 (pink line) and 1:1 (blue line). The spectrum of native DNA is also reported as the control (black line). | ||
Intercalation into the DNA double helix typically leads to an increase in the elliptical signal associated with helicity (245 nm) and base pair stacking interactions (278 nm). In contrast, minor groove binding agents typically cause a decrease in the elliptical signal associated with hydrogen bonding interactions.68 The superposition of the CD spectra of ct-DNA with those of the double helix in the presence of the gold compounds shows that the presence of Au(NHC) complexes induces a variation in the DNA conformation. The intensity of the CD signal increases for the positive band and decreases for the negative band when the concentration of gold complexes increases. These data suggest that the gold(I) complexes may influence both the base-stacking and the helicity of DNA by intercalation. Moreover, a red shift of the negative band occurs, from 245 to 248 nm, in the presence of all the complexes. This shift can be ascribed to a change in the DNA conformation.70 The induced variations are more pronounced in the presence of Au3, suggesting a stronger interaction of this complex with the double helix. It could be speculated that this stronger interaction might be the reason for the higher biological activity of Au3 when compared to the other two complexes.
These findings are further supported by the results of the ethidium bromide (EtBr) displacement assay. In this experiment, the DNA intercalation ability of the studied gold(I) complexes was evaluated following the variation in the fluorescence emission of EtBr upon excitation at 540 nm as a function of Au concentration. Data, reported in Fig. 6, show that the fluorescence intensity of EtBr in complex with DNA decreases when the concentration of gold complexes increases, suggesting that metal compounds can bind ct-DNA by replacing EtBr.
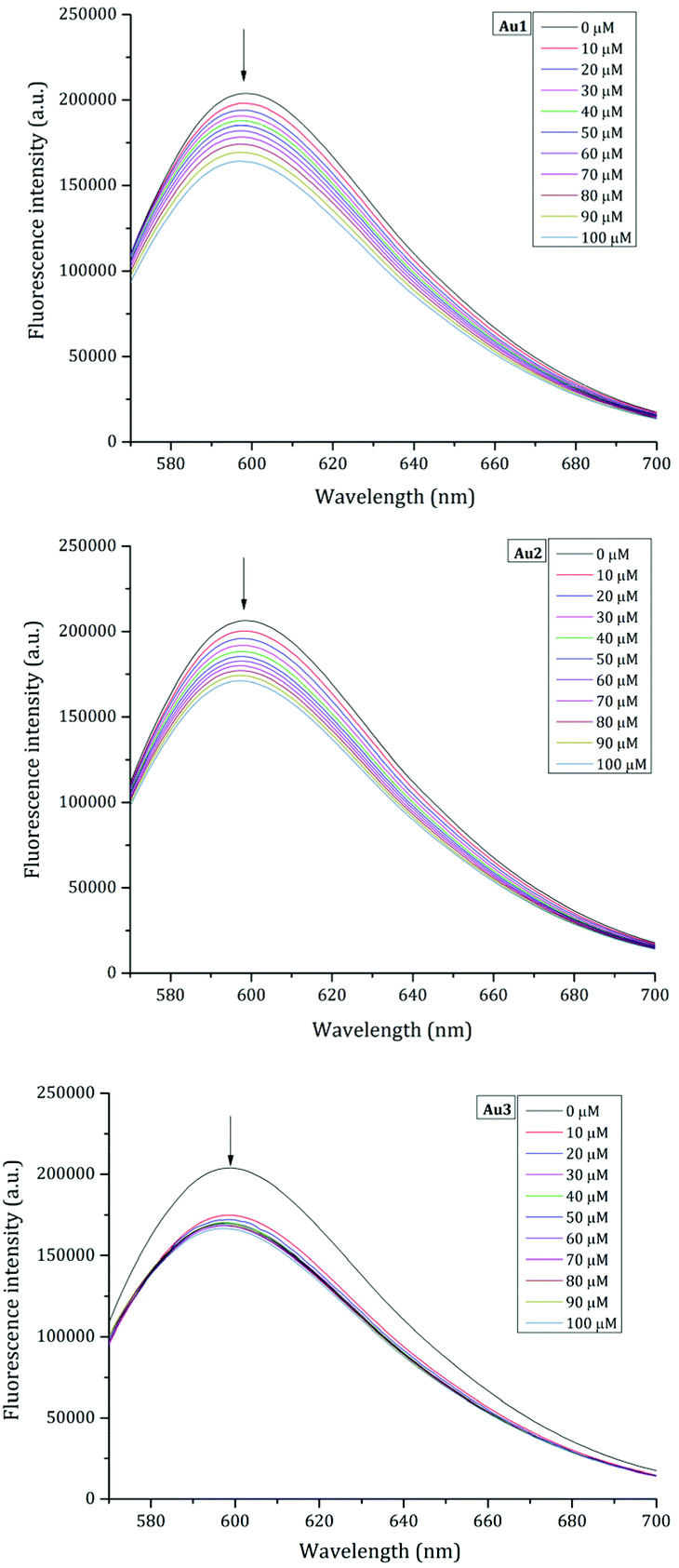 | ||
| Fig. 6 Fluorescence emission spectra of the EtBr/DNA complex upon titration with a solution of 30 mM of Au1 (top), Au2 (middle), and Au3 (bottom). | ||
Binding to proteins
With the aim to obtain further information on the possible ability of the synthesized Au(NHC) compounds to recognize biological macromolecules, the interaction with proteins, that are often considered the biological targets of Au compounds, has been evaluated.71 Thus, the X-ray structures of the potential adducts of Au1 and Au2 with bovine pancreatic ribonuclease (RNase A) and hen egg white lysozyme (lysozyme), respectively, have been solved (Fig. 7–9). Both these proteins have been frequently used as model systems for metalation studies,72,73 including structural studies on the interaction with metal(NHC) compounds74,75 | ||
| Fig. 7 Overall structures of the adducts of Au with (A) lysozyme (violet) and (B) RNase A (molecules A and B of the asymmetric unit are reported in light green and grey, respectively) obtained upon treating protein crystals with Au2 and Au1, respectively. Au binding sites are highlighted. | ||
 | ||
| Fig. 8 Details of the Au binding sites in the structure of the Au/RNase A adduct formed upon the treatment of monoclinic protein crystals with 2 molecules in the asymmetric unit with Au1. The 2Fo − Fc electron density maps are contoured at 1σ (blue) while the anomalous difference maps are contoured at 3σ (magenta). A second peak in the anomalous difference electron density map is found close to Au bound to His105 of molecule A, suggesting a possible alternative position of the Au center at this site. | ||
 | ||
| Fig. 9 Details of the Au binding sites in the structure of the Au/lysozyme adduct formed upon the treatment of lysozyme crystals with Au2. The 2Fo − Fc electron density maps are contoured at 1σ (blue) while the anomalous difference maps are contoured at 3σ (magenta). | ||
To verify whether the crystallization conditions affect the stability of Au1 and Au2, the UV-vis spectra of Au1 in 22% PEG4K and 10 mM sodium citrate buffer at pH 5.1 and of Au2 in 2.0 M sodium formate and 0.1 M HEPES buffer at pH 7.5 have been recorded as a function of time (Fig. S9†). Au1 appears to be stable over time under the analyzed experimental conditions. In fact, no change in the spectral profile can be detected in 24 h. On the other hand, the spectrum of Au2 shows a profile that changes with time. The spectrum of the compound presents a peak at 260 nm and a shoulder around 300 nm that remains stable for 3 h. After 24 h, a decrease in the intensity is observed, which could be attributed to the precipitation of the compound.
The structure of the Au/lysozyme adduct obtained upon the reaction of the protein crystals with Au2 has been solved at 1.1 Å resolution. Upon refinement, the structure of the Au/lysozyme adduct converges to Rfactor/Rfree values of 0.132/0.170. A comparison of this structure to the metal-free protein (PDB CODE 193L)76 showed no major structural variations. This is supported by the root-mean-square deviations between CA atoms (RMSD) that are as low as 0.25 Å. In the structure, clear binding of gold atoms at both the ND1 and NE2 atoms of His15 is observed (Fig. 9A).
A third Au-binding site is found close to the N-terminal tail (Fig. 9B). Notably, in all the gold binding sites, the metal has low occupancy (0.20) and the gold ligands cannot be identified in the electron density maps. In the structure, the average Au–N distance is 2.15 Å. His15 has been identified as the Au binding site in previous studies: for example, in the structures of the adduct formed upon the reaction of lysozyme with Auoxo3.77,78 The N-terminal tail has been previously found as the Pt binding site in the adduct of lysozyme with the platinum(II)–terpyridine complex79 and of thaumatin with cisplatin.80
The structure of the Au/RNase A adduct formed upon treatment of protein crystals with Au1 has been solved at 1.42 Å resolution and refined to Rfactor/Rfree values of 0.189–0.219.
Au binding to RNase A is observed at the His105 residue of both protein molecules A and B found in the asymmetric unit of the monoclinic protein crystal form (Fig. 8A and B). Also, for this structure the Au occupancy is low (0.30) and Au ligands have not been observed in the electron density maps.
Au metalation of RNase A has been already studied in previous works81 and His105 has been already identified as a gold binding site.82
Moreover, in the structure of the Au adduct with RNase A, the binding of the metal to the protein does not significantly alter the overall structure of the protein (RMSD = 0.29/0.51 Å).
Conclusions
This study exploited the wide versatility of the imidazole scaffold to prepare the new glycoconjugated carbene ligand Im-Me. This fragment was used to decorate three gold(I) complexes [Au(Im-Me)X] containing Cl−, Br− and I− as the anionic ligand X−, respectively. This offered the opportunity to disclose the effect of the halide on stability and activity. The carbene complexes were found to be stable in an aqueous environment, and this condition encouraged the study of cytotoxicity. Two data have emerged: the first is that the compounds demonstrate activity that grows along the series Cl− < Br− < I−, according to their tendency to form cationic dicarbene species of type [Au(Im-Me)2]+, that are acknowledged as valuable agents. Second, the more active complex [Au(Im-Me)I] was also somewhat selective, with a selectivity index of up to 2.4 in the case of the couple BALB/c-3T3/SVT2. CD and fluorescence spectroscopy indicated that Au(NHC) complexes can interact with DNA double helices by intercalation. Furthermore, the crystallographic data demonstrated the ability of the complexes to interact with proteins. Thus, the ability of these complexes to bind different biological targets, both proteins and DNA, makes them promising candidates as potential anticancer drugs.Experimental
General
Reagents and solvents were purchased from Sigma-Aldrich and were used without further purification. Human A431 epidermoid carcinoma, murine BALB/c-3T3 and SVT2 fibroblasts were from ATCC. Human HaCaT keratinocyte cells were from Innoprot. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Sigma-Aldrich, St Louis, MO, USA), supplemented with 10% foetal bovine serum (HyClone), 2 mM L-glutamine and antibiotics, all from Sigma-Aldrich, under a 5% CO2 humidified atmosphere at 37 °C. Au(tht)Cl was prepared according to the literature starting from H[AuCl4]. NMR spectra were acquired on a 400 Bruker Avance UltrashieldTM 400 and a 500 Varian Inova, located at the Dipartimento di Scienze Chimiche, Università di Napoli Federico II, Napoli (Italy). The solvents used were CDCl3 (CHCl3, δ 7.26, and 13CDCl3, δ 77.0, as the internal standards), acetone-d6 ((CD2H)2CO, δ 2.09, as the internal standard), D2O (HDO, δ 4.80 as the internal standard), and DMSO-d6 ((CD2H)2SO, δ 2.49, as the internal standard). The following abbreviations were used for describing NMR multiplicities: s, singlet; d, doublet; dd, double doublet; triplet; ddd, doublet of doublet of doublets; m, multiplet. NMR spectra are reported in Fig. S10–S15.†Synthesis of the pro-carbene Im-Me
The imidazole precursor Im (0.60 g, 1.2 mmol) was dissolved in 10 mL of chloroform and iodomethane (2 mL, 35 mmol) was added. The solution was stirred at RT for 24 h and then the solvent was removed under vacuum. The product was isolated as a brown oil which was treated with diethyl ether resulting in a white powder. Yield: 97% (0.73 g). 1H NMR (400 MHz, CDCl3) δ 9.91 (br, 1H, H2-Im), 8.88 (s, 1H, H-triazole), 7.95 (d, J = 1.6 Hz, 1H, H4-Im), 6.09 (d, JH1–H2 = 9.3 Hz, 1H, H1-glu), 5.62 (t, JH2–H3 = 9.4 Hz, 1H, H2-glu), 5.42 (t, JH3–H4 = 9.5 Hz, 1H, H3-glu), 5.29 (t, 1H, JH4–H5 = 9.6 Hz, H4-glu), 4.28 (dd, J = 13.0, 5.3 Hz, 1H, H6-glu), 4.21–4.02 (m, 2H, H5-glu and H6-glu), 4.15 (s, 3H, Me-Im), 4.10 (s, 3H, Me-Im), 2.03 (s, 3H, OAc), 2.02 (s, 3H, OAc), 1.98 (s, 3H, OAc), 1.84 (s, 3H, OAc). Anal. calcd (found) for C21H28IN5CO9: C, 40.59 (40.12), H, 4.54 (4.71), N, 11.27 (11.54).Synthesis of complex Au1
The pro-carbene Im-Me (0.30 g, 0.49 mmol) was dissolved in 5 mL of acetone and Ag2O (0.062 g, 0.26 mmol) was added. The mixture was stirred at RT for 3 h and then [Au(tht)Cl] (0.33 g, 0.45 mmol) was added. After stirring for 1.5 h the mixture was filtered through Celite and the solvent was removed. The crude material was dissolved in DCM and crystallized by adding diethyl ether. Yield: 78% (0.26 g). 1H NMR (400 MHz, CDCl3) δ 7.92 (s, 1H, H-triazole), 7.17 (s, 1H, H-Im), 5.86 (d, JH1–H2 = 9.0 Hz, 1H, H1-glu), 5.40 (t, JH2–H3 = JH3–H4 = 9.4 Hz, 1H, H3-glu), 5.33 (t, 1H, H2-glu), 5.20 (t, JH4–H5 = 9.6 Hz, 1H, H4-glu), 4.28 (dd, JH6–H6′ = 12.7, JH6–H5 = 5.0 Hz, 1H, H6-glu), 4.11 (d, JH6′–H5 = 1.4 Hz, 1H, H6′-glu), 3.99 (ddd, 1H, H5-glu), 3.92 (s, 1H, Me-Im), 3.81 (s, 3H, Me-Im), 2.02 (s, 3H, OAc), 2.02 (s, 3H, OAc), 1.98 (s, 3H, OAc), 1.84 (s, 3H, OAc). 13C NMR (101 MHz, CDCl3) δ 173.44, 170.42, 169.79, 169.37, 169.08, 136.46, 124.85, 120.81, 120.77, 86.06, 75.46, 72.20, 70.52, 67.62, 61.47, 38.52, 37.48, 20.72, 20.54, 20.51, 20.16. Anal. calcd (found) for C21H27AuIN5CO9: C, 34.75 (34.39), H, 3.75 (3.82), N, 9.65 (9.47).Synthesis of complexes Au2 and Au3
Complex Au1 (0.13 g, 0.17 mmol) was dissolved in acetone (4 mL) and the appropriate salt (LiBr or NaI, 2.0 mmol) was added. The mixture was stirred at RT for 12 h and then the solvent was removed. The crude was dissolved in dichloromethane (2 mL) and filtered through a pad of silica. The solvent was reduced in volume and the addition of diethyl ether resulted in the crystallization of the product. Yield: 80–85%. Au2: 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H, H-Im), 7.24 (s, 1H, H-triazole), 5.93 (d, JH1–H2 = 8.9 Hz, 1H), 5.43 (m, 2H, H2-glu and H3-glu), 5.27 (t, JH3–H4 = JH4–H5 = 9.3, 1H, H4-glu), 4.34 (dd, JH6–H6′ = 12.7, JH6–H5 = 5.0 Hz, 1H, H6-glu), 4.17 (dd, JH6′–H5 = 2.0 Hz, 1H, H6′-glu), 4.06 (ddd, 1H, H5-glu), 3.98 (s, 3H, Me-Im), 3.87 (s, 3H, Me-Im), 2.16 (s, 3H, OAc), 2.08 (s, 3H, OAc), 2.07 (s, 3H, OAc), 2.03 (s, 3H, OAc). 13C NMR (101 MHz, CDCl3) δ 176.94, 170.41, 169.78, 169.36, 169.08, 136.47, 124.79, 120.76, 120.73, 86.07, 75.47, 72.20, 70.51, 67.61, 61.47, 38.41, 37.37, 20.72, 20.54, 20.51, 20.17. Anal. calcd (found) for C21H27AuBrN5CO9: C, 32.74 (31.63), H, 3.53 (3.67), N, 9.09 (8.59). Au3: 1H NMR (400 MHz, CDCl3) δ 7.99 (s, 1H, H-Im), 7.23 (s, 1H, H-triazole), 5.92 (d, J = 9.0 Hz, 1H, H1-glu), 5.43 (m, 2H, H2-glu and H3-glu), 5.26 (t, JH4–H3 = 9.6 Hz, 1H, H4-glu), 4.34 (dd, JH6–H6′ = 12.6, JH6–H5 = 5.0 Hz, 1H, H6-glu), 4.16 (dd, JH6′–H5 = 1.6 Hz, 1H, H6′-glu), 4.06 (ddd, 1H, H5-glu), 4.00 (s, 3H, Me-Im), 3.87 (s, 3H, Me-Im), 2.09 (s, 3H, OAc), 2.08 (s, 3H, OAc), 2.04 (s, 3H, OAc), 1.90 (s, 3H, OAc). 13C NMR (101 MHz, CDCl3) δ 183.89, 170.40, 169.78, 169.37, 169.11, 136.52, 124.66, 120.65, 120.59, 86.09, 75.50, 72.19, 70.50, 67.61, 61.46, 38.12, 37.10, 20.72, 20.54, 20.51, 20.17. Anal. calcd (found) for C21H27AuIN5CO9: C, 30.86 (29.94), H, 3.35 (3.67), N, 8.15 (8.59).In-solution experiments
Stock solutions of each complex were prepared for each set of experiments. The appropriate complex (10 mM) was dissolved in 0.5 mL of DMSO-d6 or acetone-d6. The calculated volumes of the solution were diluted to 600 μL with the appropriate volumes of D2O or DMSO-d6/acetone-d6 to provide a final concentration of 3 mM of the gold complex with the appropriate v/v ratio of solvents.The UV–vis spectra were recorded using a Jasco V-750 UV-vis spectrophotometer and the following parameters: wavelength range 240–700 nm, data pitch 1 nm, scanning speed 400 nm min−1, a quartz cuvette with 1 cm path length, and compound concentration 100 μM. The spectra were collected over time at room temperature under the following experimental conditions: 100% DMSO, 2.0 M sodium formate and 10 mM Hepes buffer pH 7.5 (Au2), 22% PEG4 K and 10 mM sodium citrate buffer pH 5.1 (Au1).
Far UV-CD spectra were recorded on a Jasco J-715 spectropolarimeter equipped with a Peltier thermostatic cell holder (Model PTC-348WI) in the range of 220–320 nm, using a ct-DNA concentration of 300 μM in 10 mM ammonium acetate buffer pH 7.5 and a 0.1 cm path length quartz cell. ct-DNA was incubated with an increasing amount of each gold(I) complex (molar ratios = 1:0.5, 1:1). The spectra were collected after 24 h incubation and compared with the spectrum of the native DNA. Other experimental parameters were 1.0 nm data pitch, 2.0 nm bandwidth, 50 nm min−1 scanning speed, 2.0 s response time, and 25 °C. Each spectrum was obtained by averaging three scans.
The ethidium bromide (EtBr) displacement assay has been used to evaluate the DNA intercalation ability of the studied gold(I) complexes using the procedure already described in previous works.52,53 Briefly, the variation in the fluorescence emission of EtBr upon excitation at 540 nm was followed as a function of Au concentration. EtBr is a double helix intercalator that emits when it interacts with DNA. When it is displaced from the double helix and is free in solution, its emission is quenched. The fluorescence spectra were recorded using a Horiba Scientific Fluoromax-4 spectrofluorometer equipped with a thermostat bath, using quartz cuvettes with 1.0 cm path length. ct-DNA (2.0 μM in 0.050 M ammonium acetate buffer at pH 7.5) was incubated with EtBr in a molar ratio of 1:5 (ct-DNA:EtBr) for 30 minutes at room temperature in the dark. Then, fluorescence quenching was evaluated by adding to it increasing amounts of gold compounds (30 mM) dissolved in pure DMSO. Samples were equilibrated for 5 min before collecting each spectrum. Other experimental settings: 1.0 cm quartz cell, excitation/emission slit 5.0 nm, 560–750 nm range, 50 nm min−1 scanning speed. The data were obtained as the average of three independent measurements.
MTT assay
To test the cytotoxicity of Au1–Au3, cells were seeded at a density of 2.5 × 103 cells per well in 96-well plates. After 24 h, increasing concentrations of each compound, dissolved in DMSO, were added to the cells (0.1–150 μM). After 48 h incubation, cell viability was assessed by the MTT assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) as previously described.83 Cell survival was expressed as the percentage of viable cells in the presence of Au1–Au3 compared to the controls, represented by untreated cells and cells supplemented with identical volumes of DMSO. Each sample was tested in three independent analyses, each carried out in triplicate.Crystallization and X-ray diffraction data collection
The crystallization of both lysozyme and RNase A was undertaken at 295 K by the hanging drop vapor diffusion method. To obtain crystals of the Au/lysozyme adduct, crystals of lysozyme were grown using 2.0 M sodium formate and 0.10 M Hepes pH 7.5 and then soaked in a saturated solution of Au2 dissolved in DMSO. Crystals of RNase A were obtained using the procedure reported.84 Briefly, crystals were grown in 22% PEG4K and 0.010 M sodium citrate at pH 5.1. These crystals were then soaked in a solution of saturated Au1. For both protein crystals, a solution of the reservoir with 30% glycerol was used as the cryoprotectant. X-ray diffraction data from lysozyme and RNase A crystals treated with Au2 and Au1, respectively, were recorded at 100 K using the XRD2 beamline of Elettra synchrotron in Trieste using an X-ray wavelength of 1.0 Å. Data collection statistics are reported in Table S5.†Structure resolution and refinement
The crystal structures were solved by molecular replacement using Phaser85 and the structures from the PDB code 193L76 and 1JVT (chain A)86 for lysozyme and RNase A, respectively, as starting models. Then rigid body and restrained refinements were carried out using REFMAC587 in CCP4i. Model building and map visualization were carried out using WinCoot.88 Refinement statistics are reported in Table S1.† Assignment of gold atoms to residual peaks in the electron density maps has been verified by analyzing anomalous difference electron density maps. Metal occupancy has been evaluated by minimizing the peaks in correspondence to the gold centre in the residual Fo − Fc electron density maps. The structures have been deposited in the Protein Data Bank under the accession codes 7R1P (Au/RNase A) and 7R1Q (Au/lysozyme).X-ray crystallography of Au1
Single crystals of Au1 were obtained under slow evaporation of a toluene/acetonitrile solution at room temperature. Data were measured in flowing N2 at 173 K using a Bruker-Nonius Kappa CCD four-circle diffractometer equipped with Oxford Cryostream apparatus (graphite monochromated Mo Kα radiation, λ = 0.71073 Å, CCD rotation images, thick slices, φ and ω scans to fill asymmetric unit). The structure was solved by direct methods (SIR97 program89) and refined by the full-matrix least-squares method on F2 using the SHELXL-2018/3 program90 with the aid of the program WinGX.91 Anisotropic parameters were used for non-H atoms. All the H atoms were generated stereochemically and refined accordingly to the riding model with C–H distances in the range of 0.95–1.00 Å and Uiso(H) equal to 1.2Ueq of the carrier atom (1.5Ueq for Cmettyl).In the crystal toluene lattice solvent was found to be disordered at two positions (refined occupancy factors 0.55(2) and 0.45(2)), and some constraints were introduced in the last stage of refinement to regularize the geometry and the displacement parameters using DFIX and the ISOR commands of the SHELXL program. Details on the crystal data and refinement parameters are reported in Table S1 of the ESI.† The figures were generated using the ORTEP-391 and Mercury CSD 4.292 programs.
Author contributions
Alfonso Annunziata: data curation, formal analysis, investigation, and writing – original draft; Maria Elena Cucciolito: data curation and formal analysis; Giarita Ferraro: data curation, formal analysis, and investigation; Paola Imbimbo: data curation, formal analysis, and investigation; Angela Tuzi: data curation, formal analysis, investigation, and writing – original draft; Daria Maria Monti: funding acquisition, data curation, formal analysis, and writing – original draft; Antonello Merlino: data curation, formal analysis, and writing – original draft; Francesco Ruffo: conceptualization, funding acquisition, supervision, and writing – review & editing.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
DMM & FR thank Università di Napoli Federico II for financial support (grant 000023_ALTRI_CDA_75_2021_FRA_RUFFO).Notes and references
- B. Bertrand and A. Casini, Dalton Trans., 2014, 43, 4209–4219 RSC.
- I. Ott, Coord. Chem. Rev., 2009, 253, 1670–1681 CrossRef CAS.
- W. Liu and R. Gust, Coord. Chem. Rev., 2016, 329, 191–213 CrossRef CAS.
- B. Bertrand, L. Stefan, M. Pirrotta, D. Monchaud, E. Bodio, P. Richard, P. Le Gendre, E. Warmerdam, M. H. de Jager, G. M. M. Groothuis, M. Picquet and A. Casini, Inorg. Chem., 2014, 53, 2296–2303 CrossRef CAS PubMed.
- Ö. Karaca, V. Scalcon, S. M. Meier-Menches, R. Bonsignore, J. M. J. L. Brouwer, F. Tonolo, A. Folda, M. P. Rigobello, F. E. Kühn and A. Casini, Inorg. Chem., 2017, 56, 14237–14250 CrossRef PubMed.
- C. Schmidt, L. Albrecht, S. Balasupramaniam, R. Misgeld, B. Karge, M. Brönstrup, A. Prokop, K. Baumann, S. Reichl and I. Ott, Metallomics, 2019, 11, 533–545 CrossRef CAS PubMed.
- S. K. Goetzfried, C. M. Gallati, M. Cziferszky, R. A. Talmazan, K. Wurst, K. R. Liedl, M. Podewitz and R. Gust, Inorg. Chem., 2020, 59, 15312–15323 CrossRef CAS PubMed.
- A. Collado, A. Gómez-Suárez, A. R. Martin, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2013, 49, 5541 RSC.
- F. Nahra, N. V. Tzouras, A. Collado and S. P. Nolan, Nat. Protoc., 2021, 16, 1476–1493 CrossRef CAS.
- P. J. Barnard, M. V. Baker, S. J. Berners-Price, B. W. Skelton and A. H. White, Dalton Trans., 2004, 1038–1047 RSC.
- P. J. Barnard, M. V. Baker, S. J. Berners-Price and D. A. Day, J. Inorg. Biochem., 2004, 98, 1642–1647 CrossRef CAS PubMed.
- J. L. Hickey, R. A. Ruhayel, P. J. Barnard, M. V. Baker, S. J. Berners-Price and A. Filipovska, J. Am. Chem. Soc., 2008, 130, 12570–12571 CrossRef CAS PubMed.
- H. Ghareeb and N. Metanis, Chem. – Eur. J., 2020, 26, 10175–10184 CrossRef CAS PubMed.
- A. Holmgren, C. Johansson, C. Berndt, M. E. Lönn, C. Hudemann and C. H. Lillig, Biochem. Soc. Trans., 2005, 33, 1375–1377 CrossRef CAS PubMed.
- L. Zhong and A. Holmgren, J. Biol. Chem., 2000, 275, 18121–18128 CrossRef CAS PubMed.
- R. Rubbiani, I. Kitanovic, H. Alborzinia, S. Can, A. Kitanovic, L. A. Onambele, M. Stefanopoulou, Y. Geldmacher, W. S. Sheldrick, G. Wolber, A. Prokop, S. Wölfl and I. Ott, J. Med. Chem., 2010, 53, 8608–8618 CrossRef CAS PubMed.
- W. Liu, K. Bensdorf, M. Proetto, U. Abram, A. Hagenbach and R. Gust, J. Med. Chem., 2011, 54, 8605–8615 CrossRef CAS PubMed.
- E. Schuh, C. Pflüger, A. Citta, A. Folda, M. P. Rigobello, A. Bindoli, A. Casini and F. Mohr, J. Med. Chem., 2012, 55, 5518–5528 CrossRef CAS PubMed.
- L. Kaps, B. Biersack, H. Müller-Bunz, K. Mahal, J. Münzner, M. Tacke, T. Mueller and R. Schobert, J. Inorg. Biochem., 2012, 106, 52–58 CrossRef CAS.
- W. Liu, K. Bensdorf, M. Proetto, A. Hagenbach, U. Abram and R. Gust, J. Med. Chem., 2012, 55, 3713–3724 CrossRef CAS PubMed.
- F. Hackenberg, H. Müller-Bunz, R. Smith, W. Streciwilk, X. Zhu and M. Tacke, Organometallics, 2013, 32, 5551–5560 CrossRef CAS.
- R. Rubbiani, E. Schuh, A. Meyer, J. Lemke, J. Wimberg, N. Metzler-Nolte, F. Meyer, F. Mohr and I. Ott, MedChemComm, 2013, 4, 942 RSC.
- J. K. Muenzner, B. Biersack, H. Kalie, I. C. Andronache, L. Kaps, D. Schuppan, F. Sasse and R. Schobert, ChemMedChem, 2014, 9, 1195–1204 CrossRef CAS.
- T. Zou, C. T. Lum, C.-N. Lok, W.-P. To, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 5810–5814 CrossRef CAS PubMed.
- L. Messori, L. Marchetti, L. Massai, F. Scaletti, A. Guerri, I. Landini, S. Nobili, G. Perrone, E. Mini, P. Leoni, M. Pasquali and C. Gabbiani, Inorg. Chem., 2014, 53, 2396–2403 CrossRef CAS PubMed.
- B. Bertrand, A. Citta, I. L. Franken, M. Picquet, A. Folda, V. Scalcon, M. P. Rigobello, P. Le Gendre, A. Casini and E. Bodio, JBIC, J. Biol. Inorg. Chem., 2015, 20, 1005–1020 CrossRef CAS PubMed.
- W. Walther, O. Dada, I. Ott, A. Prochnicka, B. Büttner, X. Zhu and M. Tacke, Trends Cancer Res., 2018, 13, 63 CrossRef.
- M. J. Matos, C. Labão-Almeida, C. Sayers, O. Dada, M. Tacke and G. J. L. Bernardes, Chem. – Eur. J., 2018, 24, 12250–12253 CrossRef CAS PubMed.
- J. Oberkofler, B. Aikman, R. Bonsignore, A. Pöthig, J. Platts, A. Casini and F. E. Kühn, Eur. J. Inorg. Chem., 2020, 2020, 1040–1051 CrossRef CAS.
- M. Bian, R. Fan, G. Jiang, Y. Wang, Y. Lu and W. Liu, J. Med. Chem., 2020, 63, 9197–9211 CrossRef CAS PubMed.
- M. Safir Filho, T. Scattolin, P. Dao, N. V. Tzouras, R. Benhida, M. Saab, K. Van Hecke, P. Lippmann, A. R. Martin, I. Ott and S. P. Nolan, New J. Chem., 2021, 45, 9995–10001 RSC.
- X. Cheng, P. Holenya, S. Can, H. Alborzinia, R. Rubbiani, I. Ott and S. Wölfl, Mol. Cancer, 2014, 13, 221 CrossRef PubMed.
- R. Rubbiani, L. Salassa, A. de Almeida, A. Casini and I. Ott, ChemMedChem, 2014, 9, 1205–1210 CrossRef CAS PubMed.
- R. Rubbiani, S. Can, I. Kitanovic, H. Alborzinia, M. Stefanopoulou, M. Kokoschka, S. Mönchgesang, W. S. Sheldrick, S. Wölfl and I. Ott, J. Med. Chem., 2011, 54, 8646–8657 CrossRef CAS PubMed.
- P. Holenya, S. Can, R. Rubbiani, H. Alborzinia, A. Jünger, X. Cheng, I. Ott and S. Wölfl, Metallomics, 2014, 6, 1591–1601 CrossRef CAS PubMed.
- A. Casini and S. R. Thomas, Chem. Lett., 2021, 50, 1516–1522 CrossRef CAS.
- L. Stefan, B. Bertrand, P. Richard, P. Le Gendre, F. Denat, M. Picquet and D. Monchaud, ChemBioChem, 2012, 13, 1905–1912 CrossRef CAS PubMed.
- W. A. Harrell Jr., S. Neidle and S. Balasubramanian, Quadruplex Nucleic Acids, The Royal Society of Chemistry, 2006 Search PubMed.
- Y. Xu, Chem. Soc. Rev., 2011, 40, 2719–2740 RSC.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef CAS PubMed.
- S. Balasubramanian and S. Neidle, Curr. Opin. Chem. Biol., 2009, 13, 345–353 CrossRef CAS PubMed.
- C. Bazzicalupi, M. Ferraroni, F. Papi, L. Massai, B. Bertrand, L. Messori, P. Gratteri and A. Casini, Angew. Chem., 2016, 128, 4328–4331 CrossRef.
- Ö. Karaca, S. M. Meier-Menches, A. Casini and F. E. Kühn, Chem. Commun., 2017, 53, 8249–8260 RSC.
- S. M. Meier-Menches, B. Neuditschko, K. Zappe, M. Schaier, M. C. Gerner, K. G. Schmetterer, G. Del Favero, R. Bonsignore, M. Cichna-Markl, G. Koellensperger, A. Casini and C. Gerner, Chem. – Eur. J., 2020, 26, 15528–15537 CrossRef CAS PubMed.
- S. van Zutphen and J. Reedijk, Coord. Chem. Rev., 2005, 249, 2845–2853 CrossRef CAS.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- A. Pettenuzzo, R. Pigot and L. Ronconi, Metallodrugs, 2015, 1, 36–61 Search PubMed.
- G. Bononi, D. Iacopini, G. Cicio, S. Di Pietro, C. Granchi, V. Di Bussolo and F. Minutolo, ChemMedChem, 2021, 16, 30–64 CrossRef CAS PubMed.
- A. Annunziata, M. E. Cucciolito, R. Esposito, P. Imbimbo, G. Petruk, G. Ferraro, V. Pinto, A. Tuzi, D. M. Monti, A. Merlino and F. Ruffo, Dalton Trans., 2019, 48, 7794–7800 RSC.
- A. Annunziata, A. Amoresano, M. E. Cucciolito, R. Esposito, G. Ferraro, I. Iacobucci, P. Imbimbo, R. Lucignano, M. Melchiorre, M. Monti, C. Scognamiglio, A. Tuzi, D. M. Monti, A. Merlino and F. Ruffo, Inorg. Chem., 2020, 59, 4002–4014 CrossRef CAS PubMed.
- A. Annunziata, M. E. Cucciolito, R. Esposito, G. Ferraro, D. M. Monti, A. Merlino and F. Ruffo, Eur. J. Inorg. Chem., 2020, 2020, 918–929 CrossRef CAS.
- M. E. Cucciolito, A. D'Amora, G. De Feo, G. Ferraro, A. Giorgio, G. Petruk, D. M. Monti, A. Merlino and F. Ruffo, Inorg. Chem., 2018, 57, 3133–3143 CrossRef CAS PubMed.
- M. E. Cucciolito, F. De Luca Bossa, R. Esposito, G. Ferraro, A. Iadonisi, G. Petruk, L. D'Elia, C. Romanetti, S. Traboni, A. Tuzi, D. M. Monti, A. Merlino and F. Ruffo, Inorg. Chem. Front., 2018, 5, 2921–2933 RSC.
- A. Annunziata, M. E. Cucciolito, P. Imbimbo, A. Silipo and F. Ruffo, Inorg. Chim. Acta, 2021, 516, 120092 CrossRef CAS.
- A. Annunziata, D. Liberti, E. Bedini, M. E. Cucciolito, D. Loreto, D. M. Monti, A. Merlino and F. Ruffo, Int. J. Mol. Sci., 2021, 22, 8704 CrossRef CAS PubMed.
- M. E. Cucciolito, M. Trinchillo, R. Iannitti, R. Palumbo, D. Tesauro, A. Tuzi, F. Ruffo and A. D'Amora, Eur. J. Inorg. Chem., 2017, 2017, 4955–4961 CrossRef CAS.
- F. Tresin, V. Stoppa, M. Baron, A. Biffis, A. Annunziata, L. D'Elia, D. M. Monti, F. Ruffo, M. Roverso, P. Sgarbossa, S. Bogialli and C. Tubaro, Molecules, 2020, 25, 3850 CrossRef CAS PubMed.
- S. Guo, H. Sivaram, D. Yuan and H. V. Huynh, Organometallics, 2013, 32, 3685–3696 CrossRef CAS.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- M. Monticelli, S. Bellemin-Laponnaz, C. Tubaro and M. Rancan, Eur. J. Inorg. Chem., 2017, 2017, 2488–2495 CrossRef CAS.
- M. J. MacLeod and J. A. Johnson, J. Am. Chem. Soc., 2015, 137, 7974–7977 CrossRef CAS PubMed.
- H. Sivaram, R. Jothibasu and H. V. Huynh, Organometallics, 2012, 31, 1195–1203 CrossRef CAS.
- S. Guo, J. C. Bernhammer and H. V. Huynh, Dalton Trans., 2015, 44, 15157–15165 RSC.
- A. L. Hormann-Arendt and C. F. Shaw, Inorg. Chem., 1990, 29, 4683–4687 CrossRef CAS.
- S. K. Goetzfried, P. Kapitza, C. M. Gallati, A. Nindl, M. Cziferszky, M. Hermann, K. Wurst, B. Kircher and R. Gust, Dalton Trans., 2022, 51, 1395–1406 RSC.
- H. Sivaram, J. Tan and H. V. Huynh, Dalton Trans., 2013, 42, 12421–12428 RSC.
- I. Ott, in Advances in Inorganic Chemistry, Elsevier, 2020, vol. 75, pp. 121–148 Search PubMed.
- A. Kellett, Z. Molphy, C. Slator, V. McKee and N. P. Farrell, Chem. Soc. Rev., 2019, 48, 971–988 RSC.
- P. Uma Maheswari and M. Palaniandavar, J. Inorg. Biochem., 2004, 98, 219–230 CrossRef CAS PubMed.
- P. Shi, Q. Jiang, Y. Zhao, Y. Zhang, J. Lin, L. Lin, J. Ding and Z. Guo, JBIC, J. Biol. Inorg. Chem., 2006, 11, 745–752 CrossRef CAS PubMed.
- A. Giorgio and A. Merlino, Coord. Chem. Rev., 2020, 407, 213175 CrossRef CAS.
- L. Messori and A. Merlino, Coord. Chem. Rev., 2016, 315, 67–89 CrossRef CAS.
- A. Merlino, Chem. Commun., 2021, 57, 1295–1307 RSC.
- M. P. Sullivan, M. K. Nieuwoudt, G. A. Bowmaker, N. Y. S. Lam, D. Truong, D. C. Goldstone and C. G. Hartinger, Chem. Commun., 2018, 54, 6120–6123 RSC.
- G. Ferraro, C. Gabbiani and A. Merlino, Bioconjugate Chem., 2016, 27, 1584–1587 CrossRef CAS PubMed.
- M. C. Vaney, S. Maignan, M. Riès-Kautt and A. Ducruix, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1996, 52, 505–517 CrossRef CAS PubMed.
- I. Russo Krauss, L. Messori, M. A. Cinellu, D. Marasco, R. Sirignano and A. Merlino, Dalton Trans., 2014, 43, 17483–17488 RSC.
- L. Messori, F. Scaletti, L. Massai, M. A. Cinellu, C. Gabbiani, A. Vergara and A. Merlino, Chem. Commun., 2013, 49, 10100–10102 RSC.
- G. Ferraro, T. Marzo, T. Infrasca, A. Cilibrizzi, R. Vilar, L. Messori and A. Merlino, Dalton Trans., 2018, 47, 8716–8723 RSC.
- I. Russo Krauss, G. Ferraro and A. Merlino, Inorg. Chem., 2016, 55, 7814–7816 CrossRef CAS PubMed.
- L. Messori, F. Scaletti, L. Massai, M. A. Cinellu, I. Russo Krauss, G. di Martino, A. Vergara, L. Paduano and A. Merlino, Metallomics, 2014, 6, 233–236 CrossRef CAS PubMed.
- M. Serratrice, L. Maiore, A. Zucca, S. Stoccoro, I. Landini, E. Mini, L. Massai, G. Ferraro, A. Merlino, L. Messori and M. A. Cinellu, Dalton Trans., 2016, 45, 579–590 RSC.
- M. M. Rigano, A. Raiola, G. C. Tenore, D. M. Monti, R. Del Giudice, L. Frusciante and A. Barone, J. Agric. Food Chem., 2014, 62, 11519–11527 CrossRef CAS PubMed.
- L. Vitagliano, A. Merlino, A. Zagari and L. Mazzarella, Protein Sci., 2000, 9, 1217–1225 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- L. Vitagliano, A. Merlino, A. Zagari and L. Mazzarella, Proteins: Struct., Funct., Bioinf., 2002, 46, 97–104 CrossRef CAS PubMed.
- G. N. Murshudov, P. Skubák, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 355–367 CrossRef CAS PubMed.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2151285. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00423b |
| This journal is © The Royal Society of Chemistry 2022 |