
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomimics of [FeFe]-hydrogenases incorporating redox-active ligands: synthesis, redox properties and spectroelectrochemistry of diiron-dithiolate complexes with ferrocenyl-diphosphines as Fe4S4 surrogates†
Georgia R. F.
Orton
a,
Shishir
Ghosh
ab,
Lucy
Alker
b,
Jagodish C.
Sarker
a,
David
Pugh
 a,
Michael G.
Richmond
c,
František
Hartl
d and
Graeme
Hogarth
*a
a,
Michael G.
Richmond
c,
František
Hartl
d and
Graeme
Hogarth
*a
aDepartment of Chemistry, King's College London, 7 Trinity Street, London, SE1 1DB, UK. E-mail: graeme.hogarth@kcl.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
cDepartment of Chemistry, University of North Texas, 1155 Union Circle, Box 305070, Denton, Texas 76203, USA
dDepartment of Chemistry, University of Reading, Whiteknights, Reading RG6 6DX, UK
First published on 6th June 2022
Abstract
[FeFe]-Ase biomimics containing a redox-active ferrocenyl diphosphine have been prepared and their ability to reduce protons and oxidise H2 studied, including 1,1′-bis(diphenylphosphino)ferrocene (dppf) complexes Fe2(CO)4(μ-dppf)(μ-S(CH2)nS) (n = 2, edt; n = 3, pdt) and Fe2(CO)4(μ-dppf)(μ-SAr)2 (Ar = Ph, p-tolyl, p-C6H4NH2), together with the more electron-rich 1,1′-bis(dicyclohexylphosphino)ferrocene (dcpf) complex Fe2(CO)4(μ-dcpf)(μ-pdt). Crystallographic characterisation of four of these show similar overall structures, the diphosphine spanning an elongated Fe–Fe bond (ca. 2.6 Å), lying trans to one sulfur and cis to the second. In solution the diphosphine is flexible, as shown by VT NMR studies, suggesting that Fe2⋯Fe distances of ca. 4.5–4.7 Å in the solid state vary in solution. Cyclic voltammetry, IR spectroelectrochemistry and DFT calculations have been used to develop a detailed picture of electronic and structural changes occurring upon oxidation. In MeCN, Fe2(CO)4(μ-dppf)(μ-pdt) shows two chemically reversible one-electron oxidations occurring sequentially at Fe2 and Fc sites respectively. For other dppf complexes, reversibility of the first oxidation is poor, consistent with an irreversible structural change upon removal of an electron from the Fe2 centre. In CH2Cl2, Fe2(CO)4(μ-dcpf)(μ-pdt) shows a quasi-reversible first oxidation together with subsequent oxidations suggesting that the generated cation has some stability but slowly rearranges. Both pdt complexes readily protonate upon addition of HBF4·Et2O to afford bridging-hydride cations, [Fe2(CO)4(μ-H)(μ-dcpf)(μ-pdt)]+, species which catalytically reduce protons to generate H2. In the presence of pyridine, [Fe2(CO)4(μ-dppf)(μ-pdt)]2+ catalytically oxidises H2 but none of the other complexes do this, probably resulting from the irreversible nature of their first oxidation. Mechanistic details of both proton reduction and H2 oxidation have been studied by DFT allowing speculative reaction schemes to be developed.
Introduction
The active site of [FeFe]-H2ases (a) (Chart 1) consists of diiron [2Fe]H and tetrairon [Fe4S4]H sub-units linked via a cysteine moiety. Strong electronic communication occurs between these subunits via redox potential levelling,1,2 which is essential if both electron transfer events required for the interconversion of protons and hydrogen are to occur with a similar driving force.3 Over the past 20+ years, significant effort has been made to prepare biomimics of this site,4 although the majority of these do not include a mimic of the Fe4S4 cluster, a number of strategies have also been adopted for the inclusion of a second redox centre.5–10 In 2005, Pickett and co-workers reported the [FeFe]-H2ase biomimetic (b) (Chart 1, L = 1,3,5-tris((4,6-dimethyl-3-mercaptophenyl)thio)-2,4,6-tris(p-tolyl-thio)benzene) containing both Fe2 and Fe4 centres, and showed that it functioned as an electrocatalyst for proton-reduction.6 Later, Camara and Rauchfuss prepared biomimetic (c) (Chart 1, Bn = CH2Ph) in which the 4Fe–4S cluster is replaced by a permethylated ferrocene linked to the diiron centre through an appended phosphine.7 The latter was chosen as the redox partner as the electron-releasing methyl groups give rise to a significantly lower oxidation potential. Importantly, (c) is oxidised at mild potentials allowing access to a dication in which both the ferrocene and diiron centres are oxidised, which cleaves dihydrogen. | ||
| Chart 1 | ||
An attractive Fe4S4 surrogate is commercially available, 1,1′-bis(diphenylphosphino)ferrocene (dppf), which shows well-defined Fe(II)/Fe(III) redox chemistry.11 In 2014 we reported our preliminary findings on the synthesis, electrochemistry and electrocatalysis of Fe2(CO)4(μ-dppf)(μ-pdt) (1).8 Herein we develop this chemistry. Our initial goal was to coordinate dppf in a chelating fashion to a single iron atom, viz. to prepare Fe2(CO)4(κ2-dppf)(μ-dithiolate), since unsymmetrically substituted chelate complexes have been predicted to be the best electrocatalysts.12 Indeed, in related studies with Ph2PN(R)PPh2-functionalised biomimics, we have shown experimentally that chelate isomers are more efficient electrocatalysts than bridged analogues.13 Unfortunately, we have not been able to realise this, but have successfully prepared a small number of other dppf-bridged diiron complexes with both dithiolate and bis(dithiolate) bridges and these together with detailed electrochemical and spectroelectrochemical (SEC) studies on their oxidation chemistry are described herein.
Results and discussion
Reactions of Fe2(CO)6(μ-dithiolate) with 1,1′-bis(diphenylphosphino)ferrocene (dppf) and 1,1′-bis(dicyclohexylphosphino)ferrocene (dcpf)
Our initial aim was to prepare chelate complexes Fe2(CO)4(κ2-dppf)(μ-pdt) and Fe2(CO)4(κ2-dcpf)(μ-pdt) as this framework closely mimics the active site of [FeFe]-H2ase.12 To achieve this, we first explored reaction of ca. equimolar amounts of dppf and Fe2(CO)6(μ-pdt) under a variety of experimental conditions. In MeCN with added Me3NO·2H2O, pentanuclear {Fe2(CO)5(μ-pdt)}2(μ-κ1,κ1-dppf)14 rapidly formed in high yields but heating at 70 °C for 6 h resulted in no significant change. In refluxing toluene, the reaction was slow, and after 24 h a 31P{1H} NMR spectrum showed a series of signals attributed to the presence of {Fe2(CO)5(μ-pdt)}2(μ-κ1,κ1-dppf) (55.4 ppm),14 dppf (−16.9 ppm), a signal at −17.4 ppm tentatively assigned to Fe2(CO)5(κ1-dppf)(μ-pdt) and Fe2(CO)4(μ-dppf)(μ-pdt) (1) (51.3 ppm). Refluxing the mixture for a further 4 d resulted in the disappearance of all signals apart from that associated with 1 and removal of volatiles followed by washing with hexanes gave 1 as a dark red solid in 52% yield (Scheme 1). In a separate experiment, we found that heating {Fe2(CO)5(μ-pdt)}2(μ-κ1,κ1-dppf) (X)14 with dppf in refluxing toluene also gave 1. The most expedient preparation of 1 was found to be from heating Fe2(CO)6(μ-pdt) and dppf in xylene over 1 d, although yields (48%) are slightly lower than from toluene. While this work was in progress, Li and co-workers independently reported the synthesis of Fe2(CO)4(μ-dppf)(μ-SCH2CHPhCH2S),15 from reaction of Fe2(CO)6(μ-SCH2CHPhCH2S) with dppf, its formation also proceeding via an initially formed pentanuclear intermediate. | ||
| Scheme 1 Synthesis of diphosphine-bridged complexes 1–3. | ||
Heating Fe2(CO)6(μ-pdt) and dcpf in refluxing toluene, even over prolonged periods, gave no evidence of Fe2(CO)4(μ-pdt)(μ-dcpf) (2) formation, but 2 could be isolated (55% yield) upon refluxing Fe2(CO)6(μ-pdt) and dcpf in xylene for 16 h. No intermediate(s) were observed, and the reaction was not affected by addition of Me3NO. It is not clear why this is the outome, especially given the facile formation of X, but it may be that the bulky dcpf ligand binds to Fe2(CO)6(μ-pdt) in a basal site which is pre-organised for bridge formation. In contrast, substitution of Fe2(CO)6(μ-pdt) and related diiron-dithiolate complexes by dppf occurs at an apical position and for bridge formation an apical-basal isomerisation must occur, which likely has a high activation energy. We also attempted analogous reactions of Fe2(CO)6(μ-pdt) with dipf (R = iPr) and dtbpf (R = tBu) but in both cases were unable to isolate a stable product (see Experimental section for details).
The synthesis of Fe2(CO)4(μ-dppf)(μ-edt) (3) has been claimed by Li and co-workers,16 but inspection of their spectroscopic data shows they prepared pentanuclear {Fe2(CO)5(μ-edt)}2(μ-κ1,κ1-dppf).17 We independently prepared this complex and heated it in the presence of added dppf but were unable to isolate the μ-dppf product. Similarly, heating Fe2(CO)5(κ1-dppf)(μ-edt), formed from Fe2(CO)6(μ-edt) and dppf in MeCN in the presence of the decarbonylation agent Me3NO,19 also resulted only in decomposition. We were able to isolate relatively small amounts of 3 (ca. 20% yield) upon heating Fe2(CO)6(μ-edt) and 2 equivalents of dppf in xylene for 24 h. Separation from excess unreacted dppf and other minor products was by thin layer chromatography (TLC), the second band being confirmed as 3.
IR spectra of 1–3 are characteristic of an Fe2(CO)4(μ-diphosphine)(μ-dithiolate) motif,18 the ν(CO) region consisting of four absorptions at 1986s, 1949vs, 1918s 1896w cm−1 for 1 and 1974s, 1934vs, 1903s, 1879w cm−1 for 2, the shift to lower wavenumbers of ca. 15 cm−1 being expected for the more electron-donating dcpf vs. dppf ligand. In the 1H NMR spectrum of 1, four broad singlet resonances of equal intensity between δ 4.93–4.01 are seen for the cyclopentadienyl protons and three broad multiplets between δ 2.60–2.13 for the methylene groups of the dithiolate. The 31P{1H} NMR spectrum of 1 shows a singlet at 51.3 ppm but for 2, no phosphorus signals were observed at room temperature and only broad, uninformative, signals were seen in the 1H NMR spectrum, both being dynamically involved with a fluxional process (see later). The edt complex 3 has a characteristic pattern of ν(CO) bands at 1989s, 1952vs, 1921s and 1900w cm−1, and a singlet is seen in the 31P{1H} NMR spectrum at 54.9 ppm.
Synthesis of Fe2(CO)4(μ-dppf)(μ-SC6H4-p-R)2
To further expand the range of dppf-bridged diiron complexes, we explored reactions of the bis(thiolate) complexes, Fe2(CO)6(μ-SC6H4-p-R)2 (R = H, Me, NH2), with dppf. In xylene at 120–145 °C for ca. 30–60 min Fe2(CO)4(μ-dppf)(μ-SC6H4-p-R)2 (4a–c) were formed in moderate yields after work up (Scheme 2). The ν(CO) pattern is again characteristic, and the 1H NMR spectrum of 4c shows a broad peak at δ 2.72 attributed to the amine while 4b shows two sharp methyl singlets assigned to the inequivalent thiolate ligands. The (relatively) rapid formation of 4a–c is in stark contrast to the slow formation of 1–2 suggesting these reaction reactions proceed via a different pathway, and no evidence of a dppf-linked pentanuclear intermediate was observed in the open-bridged complexes. | ||
| Scheme 2 Synthesis of dppf-bridged complexes 4a–c. | ||
Solid-state structures
Molecular structures of 1 (Fig. 1a),83 (Fig. 1b), 4a (Fig. 1c) and 4c (Fig. 1d) have been elucidated by X-ray crystallography and selected bond lengths and angles are given in Table 2. Each contains a diiron core spanned by dppf and dithiolate ligands. Iron–iron bond lengths of >2.61 Å are elongated as compared to related Fe2(CO)4(μ-diphosphine)(μ-dithiolate) complexes, which typically range between 2.46–2.53 Å3. They are similar to that of 2.6246(3) Å in Fe2(CO)4{μ-Ph2P(CH2)4PPh2}(μ-pdt)20 which also contains a highly flexible backbone. This highlights the soft nature of the Fe–Fe contact in these complexes, being easily distorted by the steric requirements of the bridging ligand(s). Diphosphine coordination results in the inequivalence of the two thiolate bridges, the dppf ligand lying trans to one and cis to the second. In 4c the aryl substituents adopt a relative anti-conformation as noted for other diphosphine complexes of this type such as Fe2(CO)4(μ-dppm)(μ-SAr2)2.21 The iron centre in the ferrocene is quite remote from the diiron centres with Fe2⋯Fc distances ranging from 4.5–4.7 Å. Molecular structures of 1–2 are similar to those reported for Fe2(CO)4(μ-dppf)(μ-SCH2CHPhCH2S)15 and Fe2(CO)4(μ-dppf){μ-SCH2N(R)CH2S} (R = CH2CH2OH, CH2CH2SC(O)Me).22 | ||
| Fig. 1 Molecular structures of (a) 1, (b) 3, (c) 4a and (d) 4c. Thermal ellipsoids are displayed at 50% probability and hydrogen atoms omitted for clarity. | ||
| Compound | 1·0.5CH2Cl2 [ref. 8] | 3 | 4a | 4c |
|---|---|---|---|---|
| Empirical formula | C41.50H35ClFe3O4P2S2 | C40H32Fe3O4P2S2 | C50H38Fe3O4P2S2 | C50H40Fe3N2O4P2S2 |
| Formula weight | 926.75 | 870.26 | 996.41 | 1026.45 |
| Temperature (K) | 150 | 100 | 150 | 150 |
| Crystal system | Triclinic | Monoclinic | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
P |
P |
| a (Å) | 9.7365(19) | 15.4691(8) | 11.7727(4) | 12.2875(6) |
| b (Å) | 13.149(3) | 11.0407(8) | 12.3881(3) | 12.6980(6) |
| c (Å) | 16.654(3) | 22.8109(10) | 18.5833(3) | 18.6853(9) |
| α (°) | 99.609(3) | 90 | 76.239(2) | 79.137(4) |
| β (°) | 94.376(3) | 106.043(4) | 79.887(2) | 74.698(4) |
| γ (°) | 111.343(3) | 90 | 82.837(3) | 83.481(4) |
| Volume (Å3) | 1936.1(7) | 3744.1(3) | 2581.76(12) | 2755.3(2) |
| Z | 2 | 4 | 2 | 2 |
| D calc. (Mg m−3) | 1.588 | 1.544 | 1.282 | 1.237 |
| μ (Mo Kα) (mm−1) | 1.411 | 11.381 | 1.013 | 0.953 |
| F(000) | 944 | 1776 | 1020 | 1052 |
| Crystal size (mm) | 0.38 × 0.32 × 0.16 | 0.15 × 0.08 × 0.02 | 0.23 × 0.19 × 0.12 | 0.22 × 0.19 × 0.15 |
| θ range (°) | 2.59–28.35 | 3.09–70.27 | 2.78–26.00 | 3.51–29.25 |
| Limiting indices | −12 ≤ h ≥ 12, −17 ≤ k ≥ 17, −21 ≤ l ≥ 21 | −18 ≤ h ≥ 17, −13 ≤ k ≥ 13, −22 ≤ l ≥ 27 | −14 ≤ h ≥ 12, −15 ≤ k ≥ 11, −22 ≤ l ≥ 17 | −16 ≤ h ≥ 16, −12 ≤ k ≥ 16, −24 ≤ l ≥ 22 |
| Reflections collected | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 800 |
21109 |
17867 |
18816 |
| Independent reflections (Rint) | 8886 (0.0333) | 6780 (0.0756) | 11779 (0.0239) |
12487 (0.0331) |
| Data/restraints/parameters | 8886/0/511 | 6780/0/460 | 11779/0/550 |
12487/0/569 |
| Goodness of fit on F2 | 1.049 | 1.029 | 1.024 | 1.039 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0345, wR2 = 0.0911 | R 1 = 0.0722, wR2 = 0.1740 | R 1 = 0.0349, wR2 = 0.0729 | R 1 = 0.0492, wR2 = 0.1129 |
| R indices (all data) | R 1 = 0.0374, wR2 = 0.0929 | R 1 = 0.0941, wR2 = 0.1864 | R 1 = 0.0474, wR2 = 0.0799 | R 1 = 0.0661, wR2 = 0.1228 |
| Largest difference in peak and hole (e Å−3) | 0.597 and −0.725 | 1.35 and −0.73 | 0.43 and −0.37 | 0.73 and −0.49 |
| 1 [ref. 8] | 3 | 4a | 4c | |
|---|---|---|---|---|
| Fe–Fe | 2.6133(6) | 2.6289(13) | 2.6267(4) | 2.6268(6) |
| Fe–P | 2.2256(6) | 2.2556(17) | 2.2434(6) | 2.2608(8) |
| 2.2679(6) | 2.2595(19) | 2.2584(5) | 2.2463(8) | |
| Fe–Scis | 2.2540(6) | 2.2491(19) | 2.2553(5) | 2.2744(8) |
| 2.2565(6) | 2.2354(16) | 2.2792(5) | 2.2495(7) | |
| Fe–Strans | 2.2410(6) | 2.2353(17) | 2.2552(6) | 2.2759(8) |
| 2.2508(6) | 2.2421(18) | 2.2792(5) | 2.2553(7) | |
| Fe⋯Fe | 4.613(1) | 4.493(1) | 4.579(1) | 4.581(1) |
| 4.581(1) | 4.618(1) | 4.586(1) | 4.605(1) | |
| Fe–Fe–P | 120.10(2) | 115.79(6) | 119.06(2) | 118.22(2) |
| 118.65(2) | 120.16(6) | 118.72(2) | 119.24(2) | |
| Fe–S(1)–Fe | 71.15(2) | 71.91(5) | 70.80(2) | 70.99(2) |
| Fe–S(2)–Fe | 70.81(2) | 71.78(5) | 70.99(2) | 70.86(2) |
| P–Fe–Fe–P | 14.12(4) | 22.28(8) | 13.41(8) | 14.70(5) |
Solution fluxionality
Dppf has a high degree of conformational flexibility which results from the tilting motion of the two cyclopentadienyl ring planes and a torsional rotation around the Cp–Fe–Cp axis. Consequently, many dppf complexes are fluxional.23,24 In the solid state, the two ends of the dppf ligand in 1 and 4c are chemically inequivalent, while in solution at room temperature a singlet is observed suggesting they are equivalent, being associated with the flexibility of the dppf ligand. For 2, in the 31P{1H} NMR spectrum at room temperature no signals were seen (Fig. 2), however, upon cooling to −30 °C two phosphorus signals appeared, their difference in linewidth suggesting that an additional fluxional process may still be occurring, being most probably “flipping” of the pdt bridgehead.25 Warming to 80 °C resulted in coalescence of these signals, as shown by the appearance of a sharp singlet. Non-equivalence of the two phosphorus environments renders the iron centres inequivalent.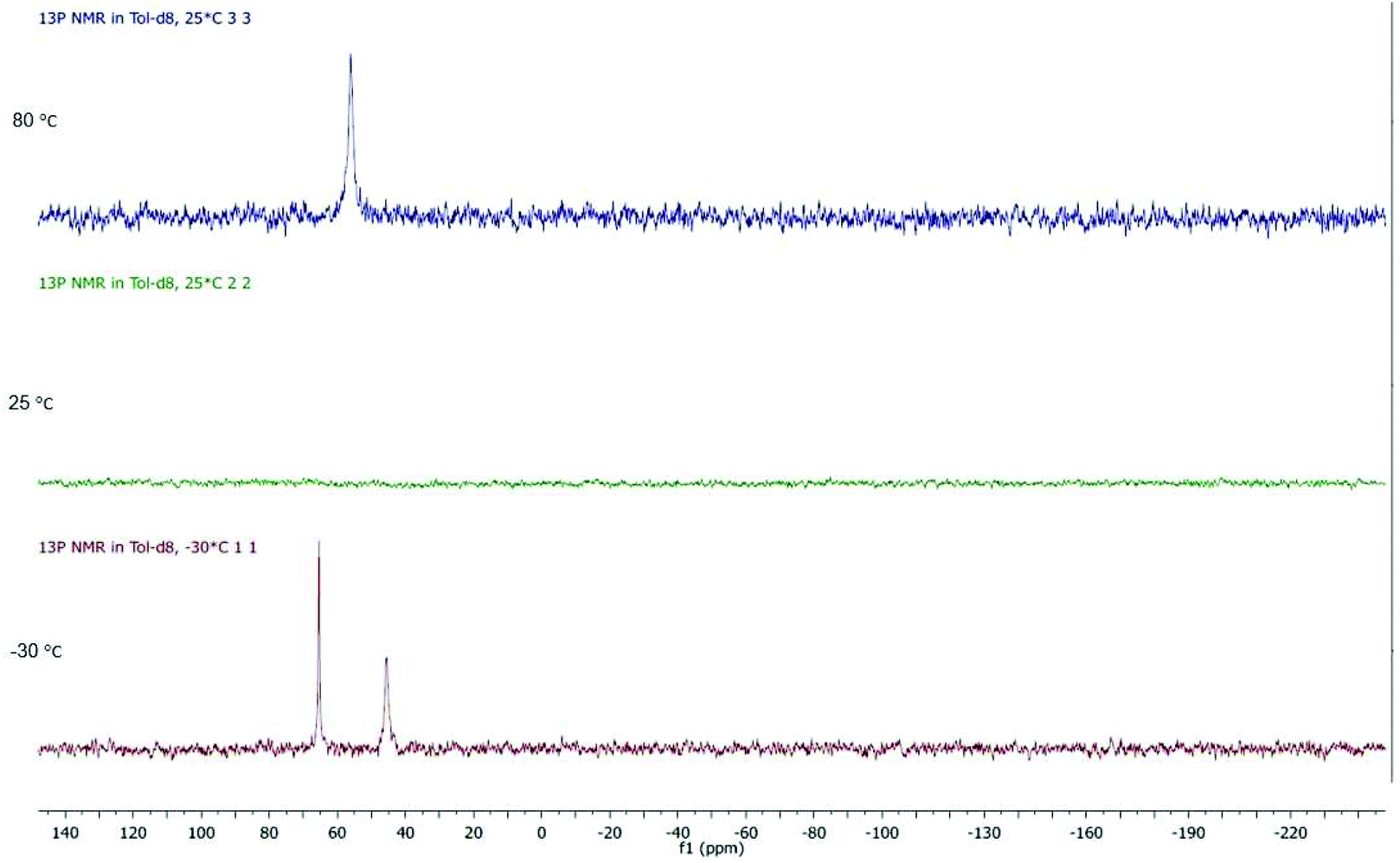 | ||
| Fig. 2 VT 31P{1H} NMR of 2 in toluene-d8 between −30 and 80 °C. | ||
Non-equivalence of the two phosphorus atoms and dibasal coordination of the diphosphine should lead to all eight ferrocenyl protons being inequivalent. In the room temperature 1H NMR spectrum of 2 (Fig. 3) all cyclohexyl and methylene protons appear below 2.8 ppm and the spectrum is difficult to interpret. A broad feature centred at 4.5 ppm is assigned to protons 1/1′ and 4/4′ which interconvert on the NMR timescale. Upon reducing the temperature to −30 °C, separate resonances for protons 1 and 4 are seen at 5.45 and 5.04 ppm respectively, while resonances for 1′ and 4′ overlap at 3.85 ppm. The larger Δδ between 1/1′ as compared to 4/4′ might be explained by the proximity of 1/1′ to the dithiolate bridge. At 80 °C, the other four ferrocenyl protons appear as a single peak, which splits into a ‘pseudo doublet’ upon cooling at 25 °C due to slowed wagging of the diphosphine (slower 2 → 2′ and 3 → 3′ conversions). The two peak maxima correspond to the overlapping signals of 2 and 3, and 2′ and 3′. Increased broadening of this peak at −30 °C indicates freezing out of these four very similar environments. In is worth noting that the two conformers of the dithiolate bridgehead are not frozen out at temperatures reached in this experiment.
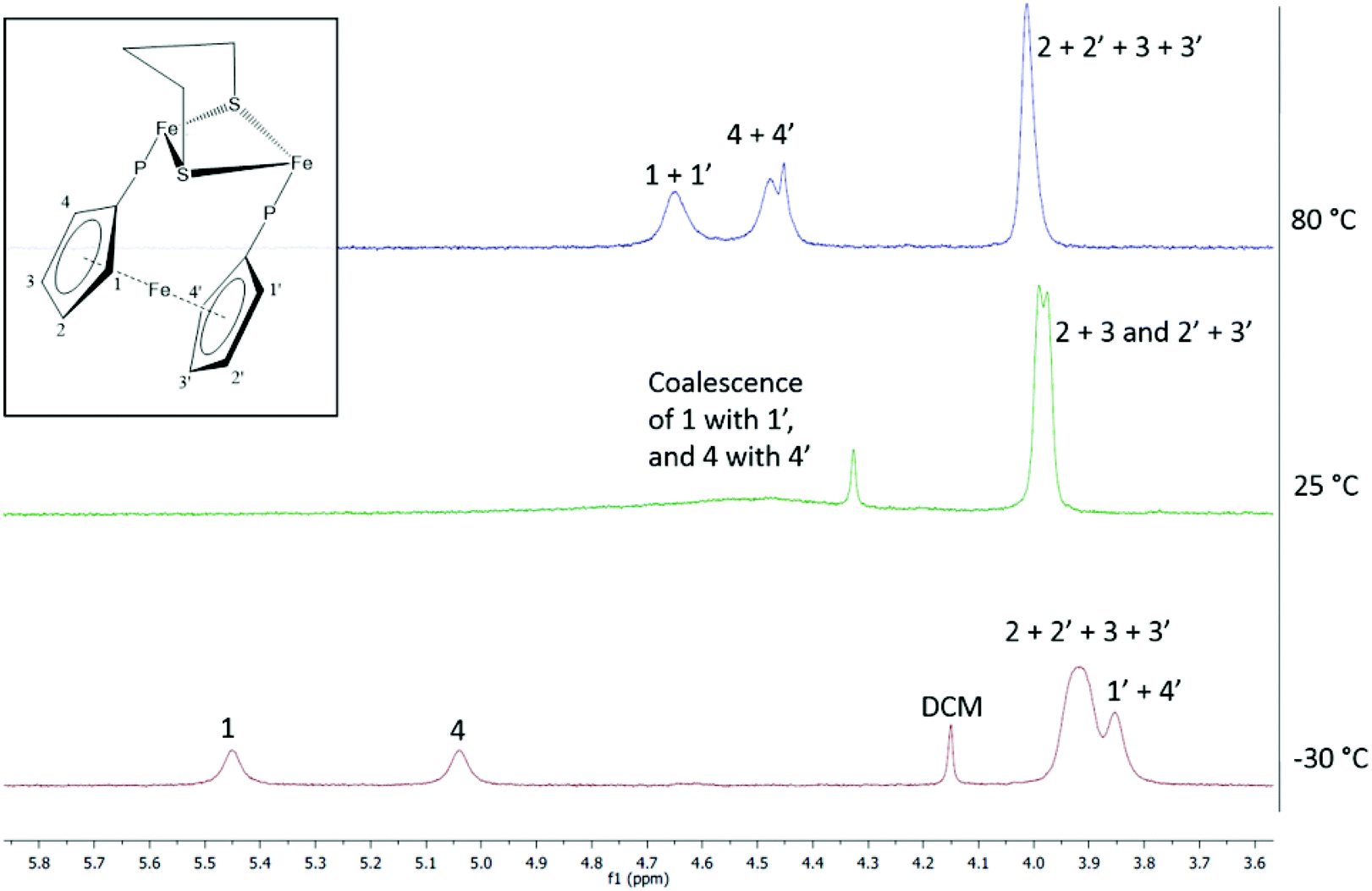 | ||
| Fig. 3 VT 1H NMR of 2 in toluene-d8 between −30 and 80 °C. | ||
We also carried out VT 1H NMR studies of 1 and 4b and see changes in the cyclopentadienyl region of the spectrum consistent with dppf twisting. Importantly, in 4b the two methyl resonances do not change with temperature, showing that the fluxional process is not associated with the movement of the diphosphine from one side of the molecule to the other, via a so-called double trigonal twist process, as has been observed for Fe2(CO)4(μ-dppm)(μ-dithiolate).26 Thus, in solution the dppf and dcpf ligands in these complexes are highly flexible.
DFT calculations on bridge and chelate isomers of Fe2(CO)4(μ-pdt)(Me2PC5H4FeC5H4PMe2)
In order to better understand why we were unable to isolate (or identify) chelate dppf complexes we have carried out a series of DFT calculations on isomers of Fe2(CO)4(μ-pdt)(Me2PC5H4FeC5H4PMe2), replacing phenyl with methyl groups for computational simplicity, while realising that this necessarily underestimates steric considerations. The calculated ground state structure of Fe2(CO)4(μ-pdt)(μ-Me2PC5H4FeC5H4PMe2) (A) (Fig. 4a) is in good accord with that found for 1. The Fe–Fe distance of 2.598 Å is slightly shorter than in 1 (2.6133(6) Å) and the Me2P groups are staggered somewhat from the normally observed eclipsed geometry in diiron hexacarbonyls, a feature also seen in 2, and attributed to the low temperature inequivalence of the phosphorus centres.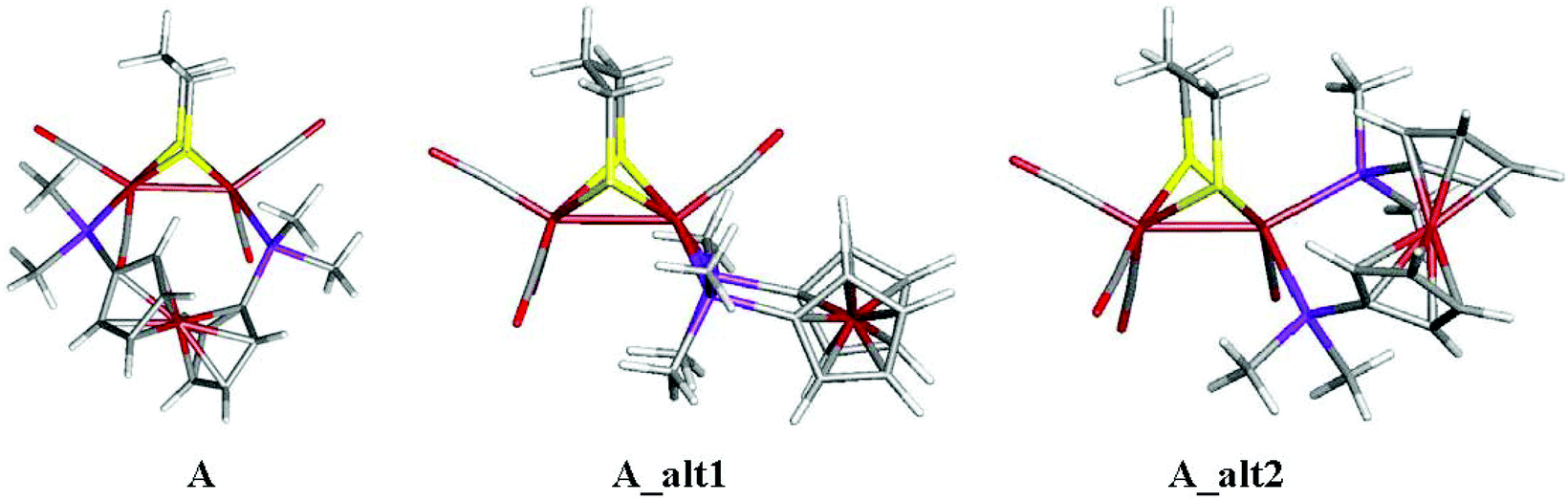 | ||
| Fig. 4 Ground-state structure of (a) Fe2(CO)4(μ-pdt)(μ-Me2PC5H4FeC5H4PMe2) (A) and chelate analogues (b) A_alt1 and (c) A_alt2. | ||
We also calculated structures for the unobserved dibasal and apical-basal chelate isomers, A_alt1 and A_alt2 respectively (Fig. 4b and c). The bridge isomer A is thermodynamically favoured, with the dibasal (2.5 kcal mol−1) and apical-basal (5.9 kcal mol−1) ones being higher in energy. Nevertheless, these small energy differences do not eliminate the formation of the chelate isomers solely on thermodynamic grounds.
For small bite-angle diphosphines such as Ph2PC(Me2)PPh227 and Ph2PN(R)PPh213 we have shown that chelate isomers are kinetic products of reactions with Fe2(CO)6(μ-pdt) and (slowly) convert to the thermodynamically stable bridge isomers upon heating. A similar chelate-bridge transformation has also been noted for Os3(CO)10(dppf), the chelate complex transforming to the bridge isomer upon heating at 110 °C for 3 h.23 In the reaction of Fe2(CO)6(μ-pdt) with dppf, initial formation of Fe2(CO)5(κ1-dppf)(μ-pdt) must be quickly followed by addition of a second equivalent of Fe2(CO)6(μ-pdt) to the free phosphorus centre, a low-energy process which is not available to small-angle diphosphine equivalents. Thus, it is this lack of stability of Fe2(CO)5(κ1-dppf)(μ-pdt) under the reaction conditions which precludes formation of chelate complexes.
Cyclic voltammetry (CV) of 1–4
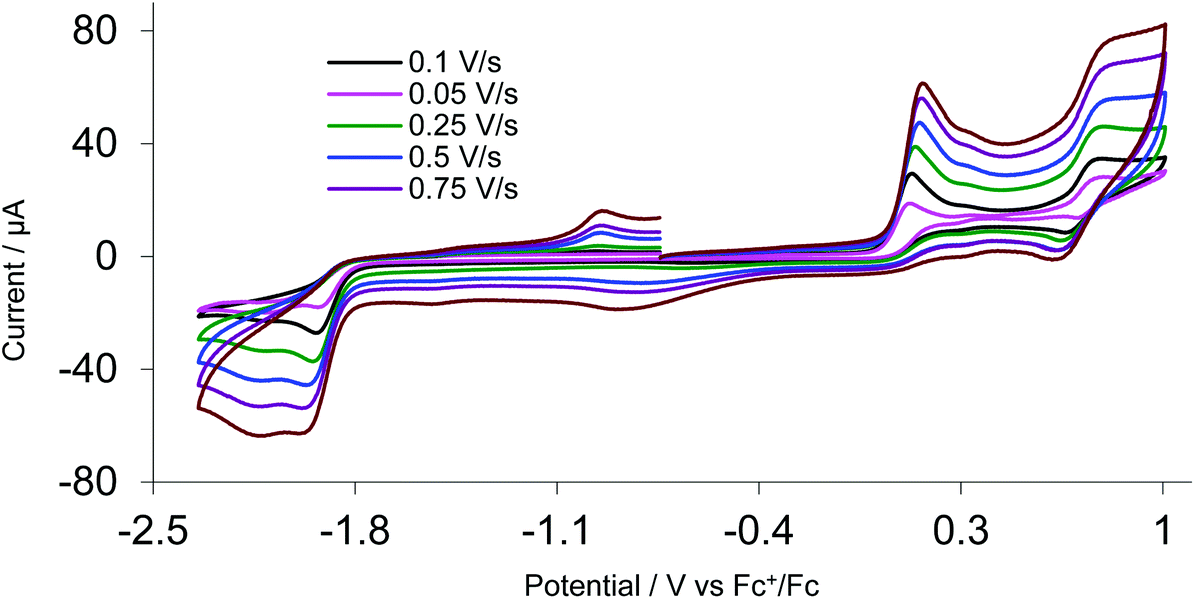
A key feature of [FeFe]-H2ases is the interaction of Fe2 and Fe4S4 redox centres in the H-cluster. To probe electronic communication between the two redox centres in 1–3 we initially used cyclic voltammetry (CV). Unlike the reversible oxidation–reduction of ferrocene, free dppf undergoes an oxidation at +0.20 V, which is not electrochemically reversible due to the rapid formation of dimeric [dppf2]2+.11 However, when coordinated to a metal centre, oxidation usually becomes chemically reversible and is shifted to more positive potentials.28We first measured CVs of 1 in MeCN at various scan rates (Fig. 5 and Fig. S1†). At 0.1 V s−1, a reversible oxidation at E1/2 = +0.05 V (ΔE = 60 mV) and a quasi-reversible oxidation at E1/2 = +0.685 V were observed, processes associated with oxidation of the diiron and ferrocene centres respectively. CVs also show two overlapping irreversible reduction peaks at Ep = –2.10 V and Ep = –2.19 V, which become separated at higher scan rates (≥0.25 V s−1), better separation being observed when the positive potential window was scanned first. Two small oxidation peaks at Ep = –1.80 V and Ep = –1.53 V on the return scan are associated with the product formed in the reductive process, while the small reduction peak at Ep = –0.35 V on the return scan is associated with the first oxidation product. The reversibility of both oxidative processes is maintained at all scan rates with both features originating from a diffusion-controlled one-electron process. This is verified by the scan rate (ν) dependence of the peak current which gives a linear ipvs. √ν plot for both oxidative processes (Fig. S2†). The solubility of 1 in MeCN is low and insufficient for spectroelectrochemical (IR-SEC) studies (see later), while 2 is completely insoluble in MeCN. We thus also carried CVs of 1 in CH2Cl2 (Fig. S3†) as 1 and 2 show good solubility in this solvent. At 0.1 V s−1 three irreversible oxidations are seen at Ep = +0.63 V, Ep = +1.16 V and Ep = +1.42 V and there is also an irreversible reduction at Ep = –1.91 V, with a shoulder at Ep = –1.72 V, the latter becoming prominent when the positive potential window is scanned first. The first oxidation shows reversibility when the potential is cycled below +1.30 V and it becomes fully chemically reversible (ipa/ipc ∼ 1) when the potential is cycled below +1 V. The enhanced reversibility of the oxidations in MeCN vs. CH2Cl2 probably represents some solvent coordination with the generated cationic centres.
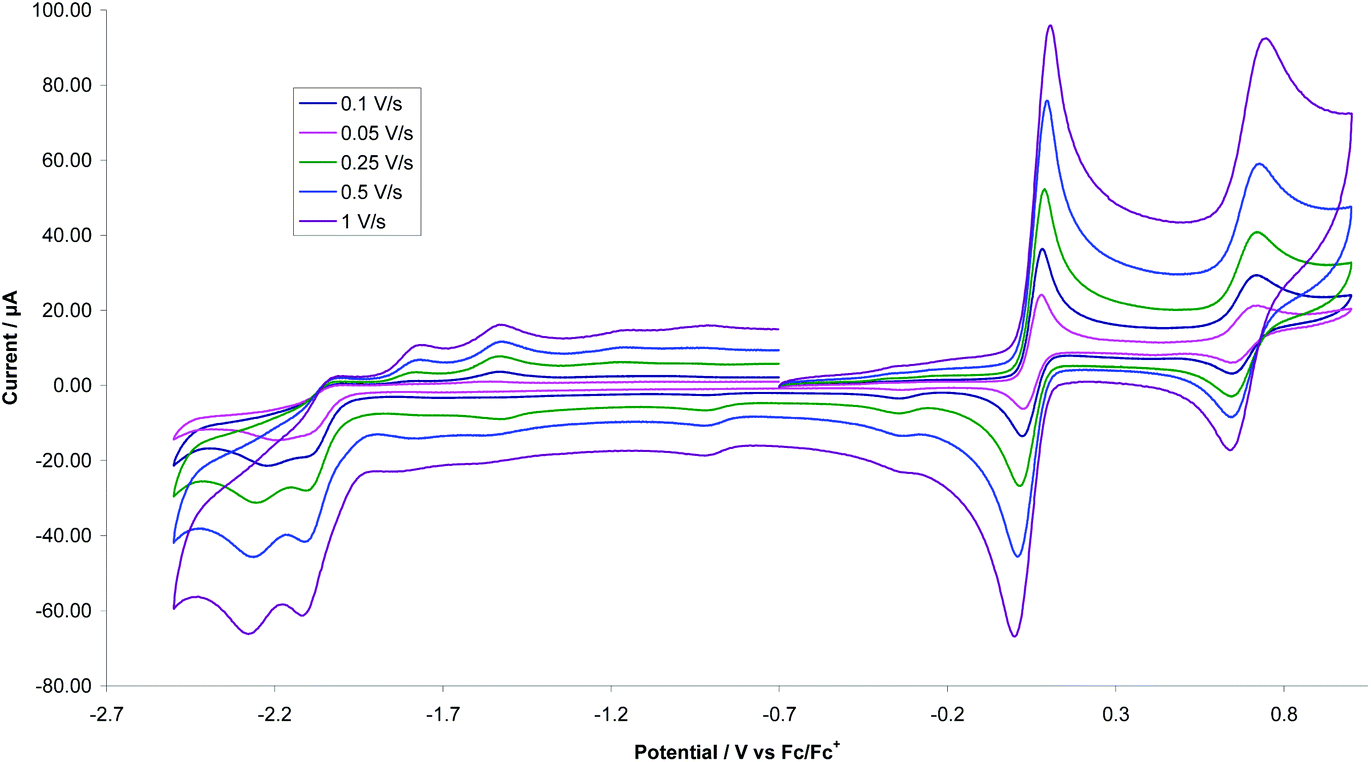 | ||
| Fig. 5 CVs of 1 at various scan rates in MeCN (1 mM solution, supporting electrolyte [NBu4][PF6], glassy carbon electrode, potential vs. Fc+/Fc) scanning positive potential first. | ||
CVs of the edt-complex 3 (in MeCN) are also quite different from those of 1. Thus, while 1 shows two reductive features, 3 shows only one. However, the most significant difference is that while the first oxidation of 1 is completely chemically reversible, the process in 3 is irreversible at all scan rates, showing only slight reversibility at higher scan rates when the CV is cycled between −0.8 V and 0.3 V (Fig. 6). Thus, the nature of the dithiolate bridge significantly alters the stability of the oxidised products. The more flexible pdt-bridge allows full reversibility of electron loss from the Fe2 centre, while in contrast the more rigid edt renders the initially oxidised species unstable, possibly resulting from Fe–S bond scission.
 | ||
| Fig. 6 CVs of 3 at various scan rates in MeCN (1 mM solution, supporting electrolyte [NBu4][PF6], glassy carbon electrode, potential vs. Fc+/Fc) scanning positive potential first. | ||
CVs of 2 were recorded in CH2Cl2 and decamethylferrocene was used as an internal reference, however, for ease of comparison with 1, data are reported vs. ferrocene (Fig. 7). Electrode passivation was a problem, and reproducible CVs were obtained only when the (glassy carbon) electrode was polished between each scan. At room temperature, 2 shows two quasi-reversible oxidations at E1/2 = −0.11 V (ΔE = 540 mV) and E1/2 = +0.66 V (ΔE = 260 mV) and a further irreversible oxidation at Ean = +1.05 V vs. Fc+/0. Reversibility of the first oxidation is only maintained at faster scan rates, where ica/ian reaches 0.95. As expected, oxidations of 2 occur at more negative potentials than those of 1 due to the increased electron-donating ability of dcpf vs. dppf. The first oxidation of 2 was studied at different scan rates and when normalised for scan rate, slower scan rates resulted in comparably higher currents. This may indicate a geometric rearrangement occurring on the same timeframe as the CV measurement resulting in potential inversion, i.e., the potential of the second oxidation is lower than the first oxidation. We suggest that oxidation of 2 affords cation [2]+, which undergoes a significant geometric rearrangement to give [2*]+. The oxidation potential of the latter is lower than that for the formation of [2]+, leading to a spontaneous second oxidation occurring to give [2*]2+. In this scenario, while DFT calculations predict the lower energy mono-cation, [2*]+ over cation [2]+, the former will not be observed spectroscopically.
 | ||
| Fig. 7 CVs of 2 in CH2Cl2 at various scan rates (1 mM solution, supporting electrolyte [NBu4][PF6], glassy carbon electrode, potential vs. Fc+/Fc) scanning positive potential first (a) full scan, (b) first oxidation process. | ||
CVs of 4a–c (in MeCN) are different to 1 but similar to 3. We present data for 4a at scan rates ranging from 0.1–1.0 V s−1 (Fig. 8) and note that 4b shows almost identical behaviour. Oxidation occurs at +0.13 V but shows no signs of any reversibility, while a second oxidation at +0.78 V shows some reversibility at all scan rates (ΔE = 110 mV). We suggest the first process is an irreversible oxidation of the Fe2 centre, and the second the quasi-reversible oxidation of the ferrocene. CVs for amino-functionalised 4c were different (Fig. S4†), showing three separate oxidations at +0.04, +0.52 and +0.84 V, all of which are irreversible at all scan rates. The extra peak at +0.52 V is possibly due to the irreversible oxidation of the amino groups. Given the irreversible nature of the first oxidation process we did not further explore the oxidation chemistry of these bis(thiolate)-bridged complexes.
 | ||
| Fig. 8 CVs of 4a MeCN at various scan rates (1 mM solution, supporting electrolyte [NBu4][PF6], glassy carbon electrode, potential vs. Fc+/Fc) scanning positive potential first. | ||
IR-spectroelectrochemistry (IR-SEC) of 1 and 2
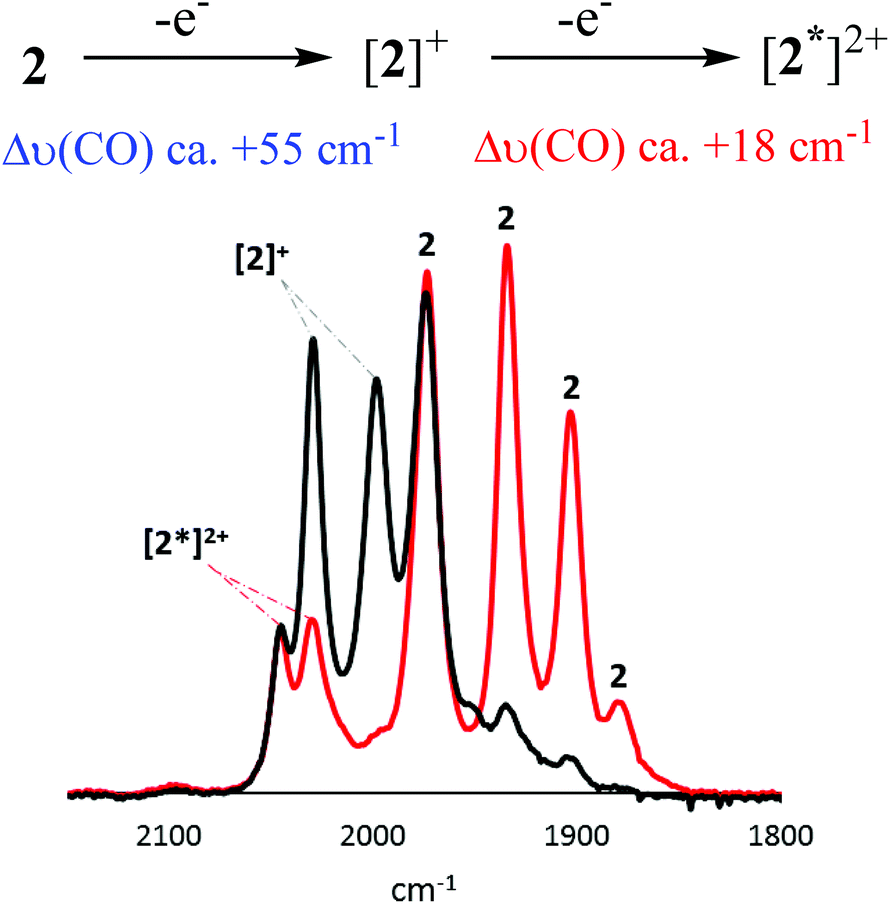
Oxidation of [FeFe]-H2ase biomimics results in a significant reduction in back-bonding, leading to a ca. 60 cm−1 hypsochromic shift in the highest frequency ν(CO) band,29 while oxidation of a ferrocene centre separated by a heteroatom from the diiron centre results in a ca. 15 cm−1 hypsochromic shift.30 In the event that oxidation results in a Δν(CO) that deviates significantly from these values, it is likely that some level of non-innocent behaviour is occurring. An important consideration when comparing CV and IR-SEC measurements is the timeframe of these experiments. The CV timeframe is significantly shorter than for the IR-SEC experiments, and the generated species are often different.We first consider 2, the results of which are concentration dependent (Fig. 9–11). Oxidation of a 3 mM solution results in initial formation of a new species characterised by a ca. +55 cm−1 hypsochromic shift of the ν(CO) bands being associated with formation of [2]+, and subsequently a small amount of this cation transforms to [2*]2+ (see later) accompanied by an additional ca. 18 cm−1 shift (Fig. 9). IR data for [2]+ is consistent with oxidation at the diiron centre, while the smaller second shift is consistent with oxidation at the ferrocenyl moiety. Together with our observation (above) that oxidation of 2 can result in potential inversion, this suggests that [2*]2+ is likely a dication, with one oxidation localised at the diiron centre, and the second at the ferrocenyl moiety.
 | ||
| Fig. 9 IR-SEC for oxidation of 2 (black) (3 mM) in CH2Cl2 at RT. Grey scan shows neutral and oxidised species during oxidation, and red shows two species after full conversion of 2. | ||
 | ||
| Fig. 10 IR-SEC showing oxidation of 2 (black) (0.5 mM) in CH2Cl2 at RT. Oxidation to give [2*]2+ is in red. In grey are intermediate scans showing conversion of 2 to [2*]2+ and the intermediate presence of [2]+. | ||
 | ||
| Fig. 11 IR-SEC of [2]+ (black) in CH2Cl2 at RT and its reduction to 2 leaving [2*]2+ in solution (red). | ||
Oxidation of a (less concentrated) 0.5 mM CH2Cl2 solution of 2 results in the near exclusive formation of [2*]2+ (Fig. 10). The apparent increased stability of [2]2+ at this concentration may due to disproportionation of [2]+. Furthermore, while [2]+ can be reduced back to 2, the product of the secondary transformation, [2*]2+, cannot be reduced to 2. Neither [2]+ nor [2*]2+ undergoes any further oxidative processes.
To better characterise [2]+, we carried out oxidation of 2 at 253 K in PrCN (Fig. 12). Under these conditions exclusive formation of [2]+ results, with no evidence of formation of [2*]2+, possibly since the activation energy for the structural rearrangement leading to potential inversion is now too high. Rather, scanning to higher potentials generates a dication, accompanied by a Δν(CO) of ca. +39 cm−1 shift, a value that does not fit hypsochromic shifts expected for localised oxidations at either diiron or ferrocenyl centres, and might be explained either by a second oxidation delocalised over the whole molecule, or by oxidation at the diiron centre followed by solvent coordination. Given the instability of [2]+ and DFT calculations (see later) which suggest the presence of a μ-CO in the cation (and thus a vacant coordination site) it seems most plausible that solvent binding to give [2(PrCN)]2+.
 | ||
| Fig. 12 IR-SEC of 2 (1 mM) in PrCN at 253 K showing first (purple) and second oxidation (orange) of a solution of 2 (black). | ||
As summarised (Fig. 13) CV and IR-SEC reveal that 2 undergoes a diiron-based oxidation at Ean = −0.10 V in CH2Cl2 to give [2]+. This cation is relatively unstable and undergoes a significant structural rearrangement to another species, [2*]+, which permits a ferrocene-based oxidation at a potential lower than −0.10 V (potential inversion). This event in turn yields the (triplet) dication, [2*]2+, in which one “hole” is localised on the diiron centre and the second at ferrocene. Although the (singlet) dication, [2]2+, is not observed spectroscopically, under the different time scales and reaction conditions of CV, formation of this species may be responsible for the oxidative wave at Ean = +0.66 V.
 | ||
| Fig. 13 Proposed oxidation-induced chemistry of 2 in (a) CH2Cl2, and (b) PrCN (S) with a table giving ν(CO) absorptions. | ||
We next turn our attention to SEC of 1, all experiments being performed in CH2Cl2. At room temperature, the behaviour of 1 is similar to 2. Oxidation of a 1 mM (or more concentrated) solution of [1] to [1]+ initially results in a hypsochromic shift of ca. +60 cm−1 (Fig. 14(i)). This transformation can also be affected chemically upon addition of [Cp2Fe][PF6] to CH2Cl2 solutions of 1. In the SEC experiment, the presence of a shoulder at 2056 cm−1 (red trace) and the larger than otherwise expected absorption at 1983 cm−1, is attributed on the basis of the chemistry of 2, to formation of [1*]2+. Unfortunately, overlap of the absorptions prohibited acquisition of information regarding ratios of the two species, and passivation of the electrode over time prevented further studies into the conversion of [1]+ to [1*]2+. Scanning [1]+ to higher potentials results in a second oxidation which, due to electrode passivation, is not well-resolved (Fig. 14(ii)). Nevertheless, a shift in the highest frequency ν(CO) band by ca. +60 cm−1 indicates that a second diiron-based oxidation seems likely and may correspond to formation of [1]2+.
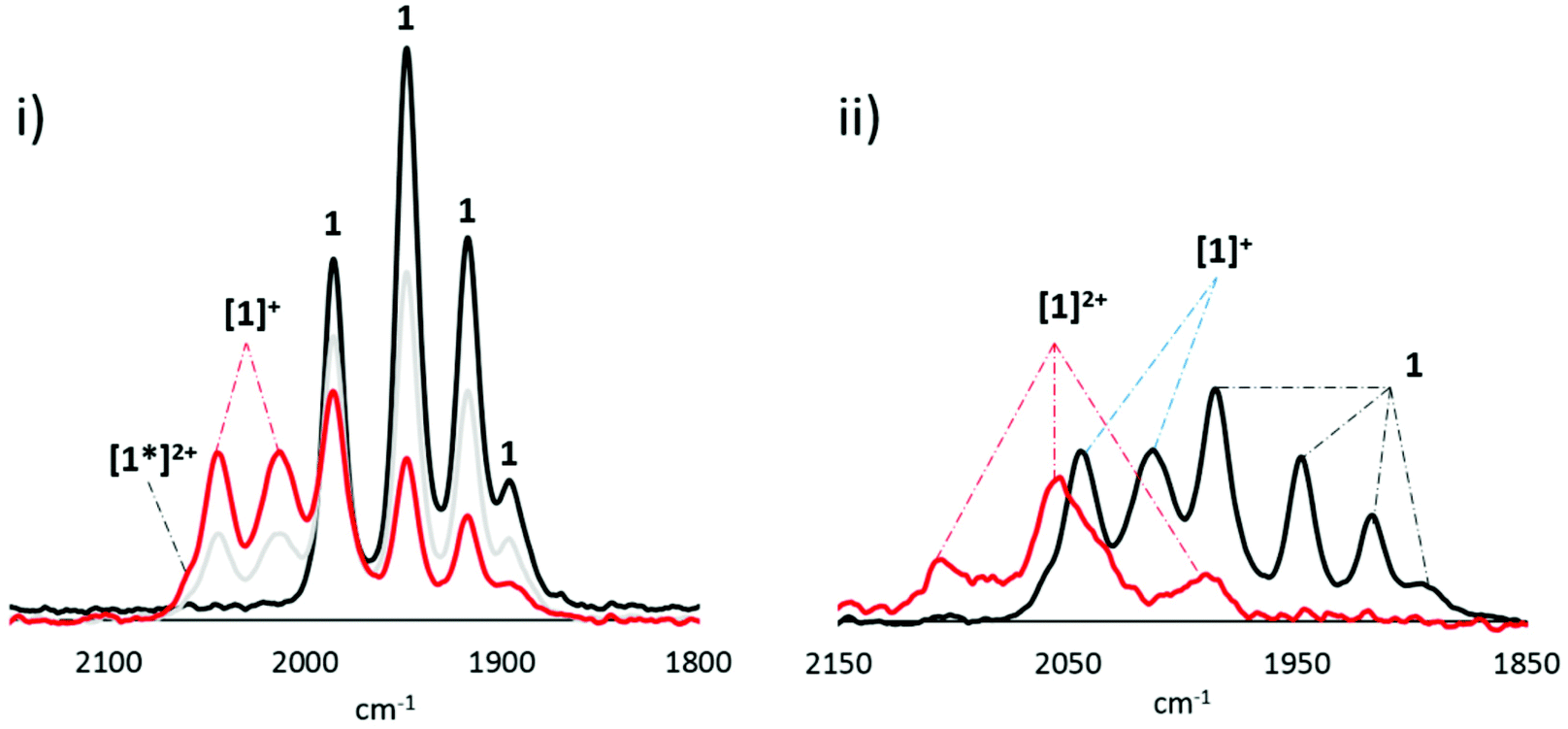 | ||
| Fig. 14 IR-SEC of 1 (1 mM) in 0.1 M [Bu4N][PF6]/CH2Cl2 showing (i) first oxidation and (ii) second oxidation. Red trace shows most oxidised species. | ||
Previously reported electrocatalytic H2 oxidation studies with 1 were conducted in MeCN.8 The activation barrier to the rearrangement of [1]+ and [2]+, which permits formation of [1*]2+ and [2*]2+ respectively, may be significantly different in the presence of a coordinating solvent. Unfortunately, due to the low solubility of 1 and 2 in MeCN leading to immediate complex adsorption onto the electrode, all attempts to record IR SEC in this solvent were unsuccessful. The lower solubility of 1vs.2 in CH2Cl2 also hampered our attempts to study SEC at low temperature. Only weak and broad new IR bands were observed which afforded little insight. We thus turned to DFT calculations (see below) for further clarification of the nature of the cations formed upon oxidation and to identify which species is/are responsible for the catalytic activity seen in MeCN.
DFT calculations on oxidised forms of Fe2(CO)4(μ-pdt)(Me2PC5H4FeC5H4PMe2) (A)
In order to better understand the nature of the oxidised species formed in experiments described above, we have carried out DFT calculations on oxidised forms of Fe2(CO)4(μ-pdt)(Me2PC5H4FeC5H4PMe2) (A), namely the radical cation B and triplet (3C) and singlet (1C) electronic configurations of dication C (Fig. 15). One electron oxidation of A results in a significant change to the geometry, resulting in formation of a semi-bridging carbonyl via a pronounced trigonal twist of one Fe(CO)2P centre leading to a significant lengthening of the Fe–Fe vector to 2.645 Å. Clearly a significant reduction of electron-density between the diiron centres results from this oxidation, suggesting that the hole is localised between these sites. The spin density plot of B (Fig. 16) confirms the Fe–Fe bond as the site of the first oxidation. Interestingly, we see no evidence of a semi-bridging carbonyl in the SEC of either 1 or 2. This suggests that there may be a significant activation energy for the trigonal twist of the Fe(CO)2P centre and thus in oxidation catalysis the ground state conformation B of the mono-cation may not be accessible. | ||
| Fig. 15 Radical cation B, and triplet (3C) and singlet (1C) electronic configurations of the dication C. | ||
 | ||
| Fig. 16 Spin density plots of the radical cation B (left) and the triplet dication 3C (right). The isovalue for each α-spin based contour plot is 0.02. | ||
The HOMO of radical cation B is largely based on the ferrocene, suggesting that the second oxidation would lead to electron loss from this centre. In support of this, we find that the most stable form of dication C is the triplet form 3C which has a highly rotated structure, being ca. 17.5 kcal mol−1 more stable than the singlet form. While this manuscript was in preparation, De Gioia, Zampella and co-workers reported related calculations on [1]2+ (phenyl groups included) and came to (broadly) similar conclusions.31 The energy order of singlet and triplet states was functional-dependent; with B3LYP functional the ground state is the triplet form, while with BP86, triplet and singlet states are nearly degenerate.31 This suggests that electron transfer from the Fe2 to Fc centres, resulting in conversion of 3C to 1C, may be facile, and this may go some way to explain the two spectroscopically identified dications. The spin density in 3C is largely delocalised over the three iron centres (Fig. 16) with smaller contributions for the Cp rings and the CO ligands. Cations [1]2+ and [2]2+ have a doubly oxidised diiron centre and likely correspond to the 1C dication configuration, while [1*]2+ and [2*]2+ would correspond to the 3C configuration. This also correlates to the observed preferential formation of the lower energy 3C configurations, [1*]2+ and [2*]2+, if the system has sufficient energy (298 K). Energy barriers to the formation of 3C and 1C have not been calculated, and intermolecular or solvent effects on the stability of these species (or indeed their intermediates) have not been considered. However, that formation of [1]2+ and [2]2+ occurs at low temperatures (253 K) suggests that formation of the more stable 3C configuration involves a large energy barrier. It is worth noting that we observe no conversion of [1]2+ and [2]2+ to their respective lower energy states [1*]2+ and [2*]2+.
Protonation of 1 and 2
Chelate complexes Fe2(CO)4(κ2-diphosphine)(μ-dithiolate) protonate readily to afford stable hydride-bridged cations, [Fe2(CO)4(μ-H)(κ2-diphosphine)(μ-dithiolate)]+,13,20,27,32 while in contrast related (sometimes isomeric) species with a bridging diphosphine are protonated only slowly by strong acids and (generally) form unstable cations.18 There are two exceptions to this chelate vs. bridge behaviour, namely bridging complexes with highly basic diiron centres or those with very flexible diphosphines. For example, protonation of Fe2(CO)4(μ-Cy2PCH2PCy2)(μ-pdt) by HBF4·Et2O readily affords crystallographically characterised [Fe2(CO)4(μ-H)(μ-Cy2PCH2PCy2)(μ-pdt)][BF4],18 while in contrast Fe2(CO)4(μ-Ph2PCH2PPh2)(μ-pdt) protonates only upon addition of excess HBF4·Et2O and the resulting cation shows limited stability.18,33 A second example relates to Fe2(CO)4{μ-Ph2P(CH2)4PPh2}(μ-pdt), which, unlike related species with fewer methylene groups in the backbone, reacts with HBF4·Et2O to form a relatively stable cationic hydride [20]. The reason(s) for the different protonation behaviour of Fe2(CO)4{μ-Ph2P(CH2)4PPh2}(μ-pdt) are less clear, as the diiron centre appears to have similar basicity to other diphosphine-bridged analogues. We have previously attributed this behaviour to the more flexible nature of this diphosphine, as shown by the relatively elongated iron–iron bond.Given the elongated nature of the iron–iron bond in 1, this seemed to be a good opportunity to test this hypothesis. Addition of a slight excess of HBF4·Et2O to a CH2Cl2 solution of 1 resulted in the rapid formation of [Fe2(CO)4(μ-H)(μ-dppf)(μ-pdt)][BF4], [1H][BF4] (Scheme 3). Interestingly, unlike related cationic-chelate complexes which generally do not easily deprotonate, addition of pyridine to 1H+ leads to regeneration of 1. Characterisation of 1H+ was straightforward, bands at 2058s, 2040s, 2002s cm−1 in the IR spectrum being shifted ca. +72–91 cm−1 to lower frequencies consistent with removal of significant electron-density from the diiron centre. The 31P{1H} NMR spectrum shows a singlet at 44.8 ppm, consistent with a bridging hydride and this is confirmed by the observation of a triplet at δ −12.40 (JPH 17.6 Hz) in the 1H NMR spectrum.
 | ||
| Scheme 3 Protonation of 1 to afford [1H]+. | ||
We have been unable to grow crystals of 1H+ suitable for X-ray analysis. Thus, we used DFT theory to calculate its ground state structure D and compare it with an isomer K with a terminal hydride (Fig. 17) finding that the former is ca. 22.4 kcal mol−1 lower in energy than the terminal hydride K. An Fe–Fe bond length of 2.642 Å is calculated for D suggesting that little overall structural rearrangement occurs to 1 upon protonation.
 | ||
| Fig. 17 Calculated molecular structures for isomers of 1H+ with bridging (D) and terminal (K) hydrides. | ||
Protonation of 2 with HBF4·Et2O in CD2Cl2 similarly affords [Fe2(CO)4(μ-H)(μ-dcpf)(μ-pdt)][BF4], [2H][BF4]. In the IR spectrum, peaks are moved to higher wavenumbers, and that at highest frequency is shifted by 82 cm−1. The spectral pattern is, however, a little different from that of 1H+, the lowest energy peak in 2H+ appearing at 1975 cm−1, as opposed to 2002 cm−1 in 1H+. We are not sure why this difference occurs. It may suggest a greater asymmetry in the molecule, as the low energy vibration is less shifted than might be expected. Both the hydride signal in the 1H NMR spectrum at δ −13.4, and the singlet in the 31P{1H} NMR spectrum at 53.3 ppm are broad, which may be due to a fluxional process or contamination by paramagnetic 2+, since oxidation of 2 is so facile (see below). Unlike 1H+, addition of pyridine to 2H+ did not result in the regeneration of 2, suggesting (as might be expected) that the hydride is more tightly bound than in 2H+.
Catalytic proton-reduction
Catalytic proton-reduction (the hydrogen evolution reaction) by [FeFe]-ase biomimics has been widely studied,34 We previously communicated that 1 catalyses proton-reduction in MeCN with HBF4·Et2O as the proton source.8Fig. 18 shows CVs upon addition of 1–10 equivalents of the acid. A new reduction wave appears at ca. −1.7 V which we associate with hydride-bridged cation 2H+ and this was confirmed in an independent experiment. The height of this peak grows with increasing acid addition consistent with proton-reduction and with higher amounts of acid (>7 equivalents) it splits into two distinct waves. The nature of the second of these (at ca. −1.8 V) is not known but could be associated with a terminal hydride species akin to K. A second catalytic wave is also observed at ca. −2.10 V which competes with the direct reduction of the acid at the glassy carbon electrode (>2.0 V).35 On the return scan a further reductive wave is seen at −1.55 V which also increases with acid concentration implying that a sufficiently stable species is generated in the depletion layer during catalysis on the forward scan which can release H2 by reducing at this potential.36 Thus it appears that 1 enters into the catalytic cycle via a CE mechanism to generate the neutral paramagnetic complex [Fe2(CO)4(μ-H)(μ-dppf)(μ-pdt)] (1H) upon reduction of 1H+, which can then either further protonate or undergo reduction. Peak heights of the oxidative process do not change during the experiment showing the robustness of 1 under the catalytic operating conditions.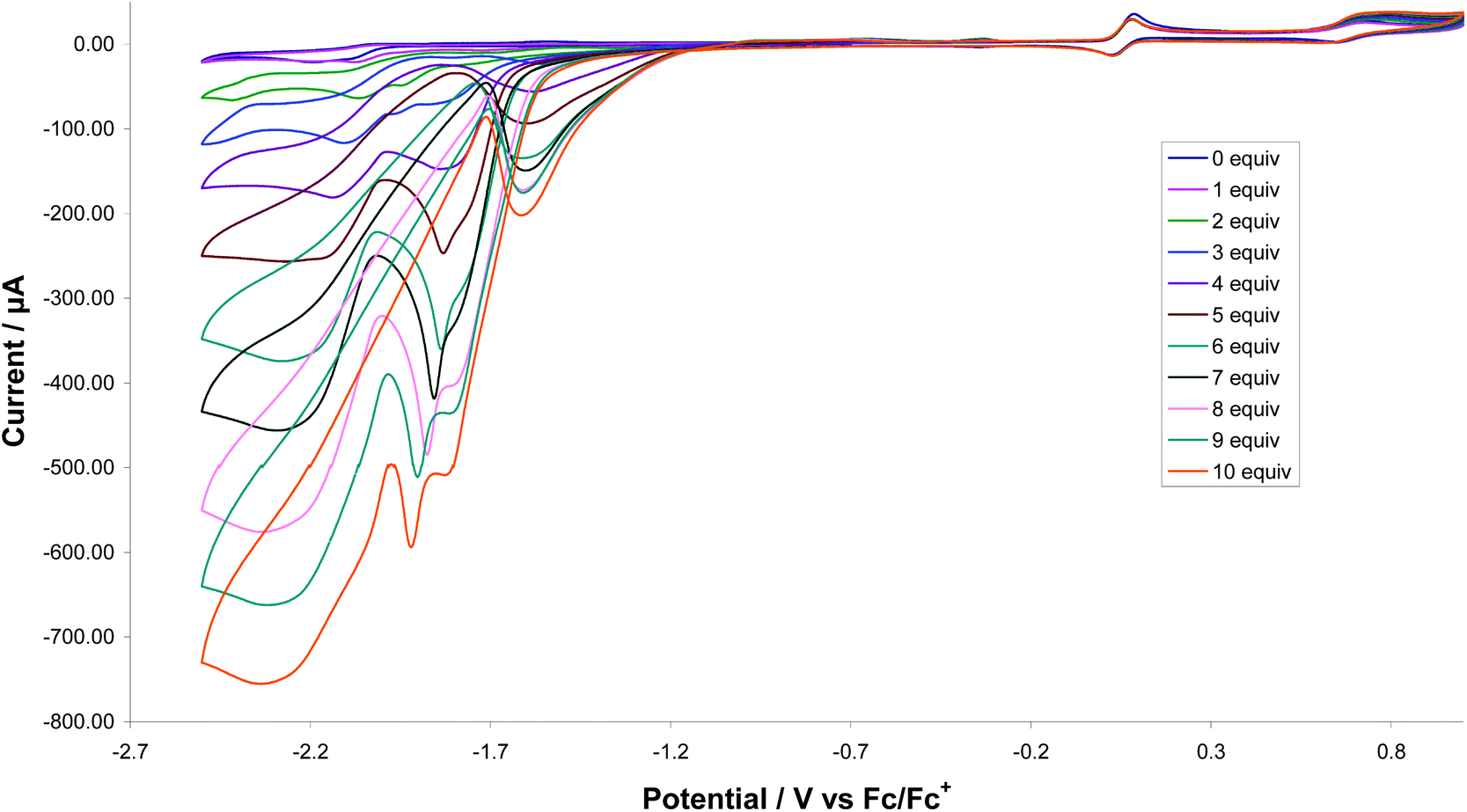 | ||
| Fig. 18 CVs of 1 in the absence of acid and in the presence of up to 10 molar equivalents of HBF4 (1 mM solution in acetonitrile, supporting electrolyte [NBu4][PF6], scan rate 0.1 V s−1, glassy carbon electrode, potential vs. Fc+/Fc). | ||
Proton-reduction studies of 2 were carried out in CH2Cl2, using CF3CO2H as a proton source, but under these conditions poor catalytic activity resulted (Fig. S5†). After addition of 3 equivalents of the acid, while it is clear from changes in the oxidation chemistry that 2 is no longer the major species in solution, somewhat surprisingly there is still no reductive chemistry visible within the potential window. After addition of 7 acid equivalents further changes are seen to the oxidation chemistry and a new reductive process is observed at ca. −1 V, but the extremely broad nature suggests that a single stable species is not generated. After 15 acid equivalents the oxidation chemistry again changes with a relatively sharp reductive process being seen at ca. −1.2 V. It is difficult to interpret these data and we will not speculate too much, apart from suggesting that perhaps at high acid concentrations (ca. 15 equivalents) a bis(hydride) dication [2HH]2+ results, which has a reduction potential within the spectral window.
Somewhat surprisingly, given the irreversible nature its first reduction potential, edt-analogue 3 shows similar catalytic behaviour to 1 with respect to electrocatalytic proton reduction. This provides further evidence that protonation occurs prior to reduction and presumably [1H]+ and [3H]+ have similar reduction potentials.
DFT studies probing the mechanism of catalytic proton-reduction by 1
In order to better understand the proton-reduction behaviour of 1 we have carried out a series of DFT calculations based on Fe2(CO)4(μ-pdt)(Me2PC5H4FeC5H4PMe2) (A) which allow an overall reaction scheme to be postulated (Scheme 4). | ||
| Scheme 4 Proposed reaction pathway for proton-reduction catalysed by 1. | ||
As established from the experimental studies, protonation affords cation D. This is not basic enough to add a second proton but reduces to afford the neutral 35-electron complex E (Fig. 19a). A small number of related 35-electron species including Fe2(CO)4(μ-H)(PMe3)2(μ-pdt)37 and Fe2(CO)2(κ2-dppv)2(μ-H)(μ-pdt)38 have been prepared and both experimental data and DFT studies suggest symmetrically bridging hydrides. Somewhat unexpectedly then, our DFT calculations for E suggest that upon reduction the hydride binds in an unsymmetrical manner to the diiron centre, with Fe–H distances of 1.973 and 1.577 Å. Zampella and co-workers have carried out DFT calculations on a range of cationic 34-electron diiron dithiolate complexes with both terminal (t-H) and bridging (μ-H) hydrides and their one-electron reduced products which has allowed some useful insights.39 While most μ-H cations reduce to give 35-electron species in which the hydride remains in this position, in one case the reduced product is calculated to be a t-H with a significantly elongated the Fe–Fe vector. Thus, while the semi-bridging nature of the hydride in E is unexpected it is not unprecedented. It is also shown that, while in μ-H complexes the spin density is delocalised across the Fe2 centre, in t-H analogues it is localised at the non-hydride bound iron atom, and thus such species are best considered as Fe(I)–Fe(II) mixed-valence complexes.39 Finally, the reduction potential of 35-electron radicals is very sensitive to the coordination nature of the hydride with t-H species reducing at consistently lower potentials than their μ-H analogues. Thus, while the structure of E is unexpected, it is also highly beneficial with respect to proton-reduction behaviour.
 | ||
| Fig. 19 Calculated molecular structures for 35-electron species (a) E and (b) I. | ||
As discussed in the previous section, E is a potential branching point, as it can be either protonated or further reduced. DFT calculations suggest that protonation leads to the mixed-valence dihydrogen complex I (Fig. 19b) in which H2 is bound to an Fe(II) centre, which also results in a further elongation of the Fe–Fe vector from 2.869 Å (in E) to 3.086 Å. Loss of H2 from I affords radical cation B or I can be reduced further, regenerating A upon H2 loss. As the reduction potential for t-H complexes are lower than those for μ-H analogues, we cannot discount possible reduction of E to generate 36-electron anion F. This is calculated to have a t-H and no direct Fe⋯Fe interaction (Fig. S6†).
H2 binding studies
Catalytic H2 oxidation by [FeFe]-ase biomimics remains a significant challenge with few authenticated examples of H2 activation.7,8,40 In the enzymes, the active catalytic H2 oxidation site (Hox) is a mixed-valence Fe(II)–Fe(I) state.41 Recent theoretical work by De Gioia, Zampella and co-workers has attempted to rationalise the ideal conditions needed for H2 oxidation31,41 (see discussion below). Their findings include: the mixed-valence diiron centre should not be too basic for H2 binding to be favorable and the presence of the Fe4S4 cluster (or surrogate) is essential as it allows electron delocalisation upon H2 binding.31 Molecular hydrogen oxidation is a two-electron process and we reasoned that the ability to access [1]2+ electrochemically (in MeCN) containing a mixed-valence Fe2 core that is not too electron-rich, and an oxidised dppf moiety, which can act as a surrogate of the Fe4S4 cluster, may provide a favorable centre for H2 oxidation.We first tried to detect H2 binding to oxidised forms of 1 and 2 electrochemically but CVs of 1 in MeCN in the absence and presence of H2 showed little change (Fig. S7†). We next turned to IR spectroscopy, generating oxidised forms of 1 and 2 in CH2Cl2 upon addition of an excess of the thianthrene (TA) radical cation, [TA][PF6]. For 2, on the basis of the IR spectrum (Fig. S8†), oxidation produces both [2]+ and [2*]2+, while for 1, it appears that [1]+ primarily results, however, due to overlap of IR signals for [1]+ and [1*]2+, the presence of small amounts of [1*]2+ cannot be excluded. H2 was then bubbled through these solutions (5 min) and a second IR spectrum recorded. In both cases, small changes were seen following H2 addition, but no compelling evidence for H2 binding could be found. For 1, a (very) small new absorption was seen at 2069 cm−1. If dication [1*]2+, at best present in small amounts, binds H2 then we might expect to see only a small change in IR spectra.
We next used DFT calculations to probe the likely nature of H2 binding and activation at the dication site. This has recently been independently addressed by De Gioia, Zampella and co-workers.31 They found two possible binding sites for H2 to 3C, at the apical site of the rotated Fe(CO)2P moiety and at the diiron bond, the latter being favorable. We focused exclusively on H2 binding to the apical site, which via a Kubas interaction (in which H2 acts as both a donor and acceptor) results in formation of singlet J (Fig. 20). Thus, upon H2 binding electron-transfer occurs leading to the effective formation of Fe(II)–Fe(II) diiron centre.
 | ||
| Fig. 20 Calculated structure of J generated from H2 binding to 3C. | ||
Catalytic H2 oxidation
We previously communicated that 1 catalyses H2 oxidation in the presence of pyridine which acts as an external base.8 Thus, while we have been unable to access [1]2+ in significant amounts to fully probe H2 binding, we can access it electrochemically. Upon addition of 1 equivalent of pyridine to 1 in MeCN, the oxidation wave at +0.05 V remains unchanged but the oxidative peak current of the second oxidation process, associated with generation of [1]2+ increased by ca. 10 mA, and upon sequential addition of up to 10 equivalents of pyridine the current increase was 22 mA (Fig. 21). No such catalytic wave was observed when the same experiment was carried out in absence of base. In contrast to the behaviour of 1, the edt-analogue 3 does not oxidise dihydrogen suggesting that the reversibility of the first (diiron-based) oxidation is a requirement for this type of diiron complexes to act as an electrocatalyst for dihydrogen oxidation. | ||
| Fig. 21 CVs of 1 under a H2 atmosphere in the absence of pyridine and in the presence of 1–10 molar equivalents (1 mM solution in MeCN, supporting electrolyte [NBu4][PF6], scan rate 0.1 V s−1, glassy carbon electrode, potential vs. Fc+/Fc). | ||
To probe a possible H2 activation pathway we returned to DFT. As discussed above, H2 binding to [1]2+ affords singlet J (Fig. 20) with diiron centre. Subsequent removal of H+ (by an external base) affords the terminal hydride K, which is unstable with respect to the bridging isomer D, the same species being formed upon protonation of A (Scheme 5). Interestingly, De Gioia, Zampella and co-workers found a different H2 activation pathway, whereby H2 scission led to formation of a dihydride in which one proton bridged the diiron centre and the other at the ferrocene.31 Thus, the dppf/dcpf ligands may be playing a role of both a redox and chemical centre, something that we had not previously considered. Presumably if this indeed occurs, then it is the ferrocene-bound hydride that is deprotonated by external base and thus the same cationic bridging hydride D results.
 | ||
| Scheme 5 Proposed reaction pathway(s) for the H2 oxidation catalysis by 1. | ||
We can thus propose an overall reaction pathway for the H2 oxidation catalysis by 1 based on these calculations (Scheme 5). Key steps are the formation of a triplet dication which is able to bind H2 and (importantly) this binding leads to an increase in electron-density at the diiron centre which in turn favors electron-transfer to the Fe(III) ferrocenium site; the overall conversion of the Fe2(I)/(II)–Fc(III) centre to an Fe2H2(II)/(II)–Fc(II) state. Abstraction of a proton from this complex results in formation of a cationic hydride, the most stable state of which is in the bridging site. [Fe2(CO)4(μ-H)(μ-pdt)(μ-dppf)]+ (1H+) results from the addition of strong acids to 1, a reaction that is reversed in the presence of strong bases, thus closing the catalytic cycle. This is undoubtedly a key step, as oxidation of 1H+ would occur only at high potentials and thus conversion of 1H+ (D) to 2 (A) is likely to be rate-limiting.
Summary and conclusions
In this contribution we have prepared and characterised a small series of new [FeFe]-ase biomimics which contain a ferrocenyl diphosphine as a surrogate for the Fe4S4 moiety in the H-cluster of the enzyme. In all, the diphosphine bridges the diiron centre and this likely results from the thermodynamic stability of these species over the initially targeted chelate isomers. The two redox active centres are ca. 4.5–4.7 Å apart in the solid-state and the flexibility of the diphosphine should allow them to get slightly closer in solution, to be comparable with the average Fe4S4⋯Feproximal distance in the enzyme of ca. 4 Å.42 To investigate cooperativity between the two redox centres, we carried out a series of CV experiments. While oxidation of the edt and SAr complexes was irreversible, the first oxidation of 1–2 showed (chemically) reversible or quasi-reversible character (in MeCN) and bathochromic shifts in IR-SEC (in CH2Cl2) allow us to correlate this with electron-loss from the Fe2 centre, while the second oxidation likely results from the ferrocenyl centre. The nature of the oxidised species was further probed by DFT calculations which suggest that the experimental assignment of oxidation centres is correct, and the most stable form of the dication is a triplet state with a highly rotated structure. The quasi-reversibility of the second oxidation in the pdt complexes suggests that chemically the removal and addition of two electrons is reversible but it is difficult to say unambiguously whether the two oxidised centres interact directly. Nevertheless, the dppf complex is able to oxidise H2 in the presence of added base at its second oxidation potential, although all others are inactive, being associated with the irreversible nature of the first oxidation. As H2 oxidation is a two-electron process, at some point an electron must be transferred from (bound) H2 to the Fe(III) ferrocenyl centre although it remains unclear as to when this occurs. All the ferrocenyl diphosphine complexes are also able to reduce protons to H2, a process which occurs via initial protonation of the diiron centre, and thus Fe2(CO)4(μ-dppf)(μ-pdt) is a true [FeFe]-ase biomimic in that it is able to catalyse the reaction in both directions.Experimental
General procedures
All reactions were carried out using standard Schlenk-line techniques under N2 and reaction solvents were purified on alumina columns. Work-up was done in air using standard bench reagents. Diiron hexacarbonyls were prepared by standard procedures43–46 and diphosphines were purchased from Aldrich and used as supplied. NMR spectra were recorded on a Bruker AMX400 spectrometer and referenced internally to the residual solvent peak (1H) or externally to P(OMe)3 (31P). Infrared spectra were recorded on a Nicolet 205 FT-IR spectrometer in a solution cell fitted with calcium fluoride plates, subtraction of the solvent absorptions being achieved by computation. Fast atom bombardment mass spectra were recorded on a VG ZAB-SE high resolution mass spectrometer and elemental analyses were performed at UCL.:2, v/v) developed five bands. The fastest moving band gave unreacted dppf. The second major band gave red crystals of Fe2(CO)4(μ-edt)(μ-dppf) (3) (93 mg, 20%) after recrystallisation from n-hexane/CH2Cl2 at 4 °C. The contents of other bands were too small for complete characterisation. Characterising data for 3: IR ν(CO)(CH2Cl2): 1989s, 1952vs, 1921s, 1900w cm−1. 1H NMR (CDCl3): δ 7.96 (t, J 8.0, 2H, Ph), 7.43–7.37 (m, 14H, Ph), 7.28 (s, 2H, Ph), 4.81 (s, 2H, CH), 4.45 (s, 2H, CH), 4.37 (s, 2H, CH), 3.98 (s, 2H, CH), 2.77 (br, s, 2H, CH2), 2.33 (br, s, 2H, CH2). 31P{H} NMR (CDCl3): δ 54.9 (s) ppm. ESI-MS: m/z 870.95 (M + H+, 10%), 841.94 (M − CO, 30%), 813.94 (M − 2CO, 30%), 757.96 (M − 4CO, 30%), 702.13 (100%). Elemental analysis. Calcd (found): C40H32Fe3O4P2S2·C6H14: C 57.78 (57.24), H 4.81 (4.24).
:1) developed two bands in addition to the starting material, the major band being Fe2(CO)4(μ-SPh)2(μ-dppf) (4a) (0.042 g, 0.04 mmol, 14% yield). Recrystallisation from a CH2Cl2–hexane mixture afforded crystals suitable for X-ray diffraction. Characterising data for 4a: IR ν(CO)(CH2Cl2) 1990m, 1956vs, 1927s, 1902w cm−1. 1H NMR (CDCl3) δ 8.19–6.99 (m, 30H, Ph), 4.99 (s, 2H, Cp), 4.52 (d, J 15.1, 4H, Cp), 4.06 (s, 2H, Cp). 31P{1H} NMR (CDCl3) δ 52.4 (s). Elemental analysis calc. for Fe3S2P2O4C50H38·C6H14 (found): C 62.11 (61.61), H 4.81 (4.67). Fe2(CO)4(μ-STol)2(μ-dppf) (4b) (54%) was prepared in a similar manner. Characterising data for 4b: IR ν(CO)(CH2Cl2) 1989m, 1964vs, 1925s, 1902w cm−1. 1H NMR (CDCl3) δ 8.17–6.80 (m, 28H, Ph), 4.99 (s, 2H, Cp), 4.52 (d, J 18.0, 4H, Cp), 4.06 (s, 2H, Cp), 2.36 (s, 3H, CH3), 2.14 (s, 3H, CH3). 31P{1H} NMR (CDCl3) δ 52.4 (s). Elemental analysis calc. for Fe3S2P2O4C52H42·C6H14 (found): C 62.70 (61.79), H 5.05 (4.85).
X-ray structure determinations
For 1, 4a and 4c, single crystals were mounted on a glass fiber and all geometric and intensity data were taken from this sample using a Bruker SMART APEX CCD diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) at 150 ± 2 K. Data reduction were carried out with SAINT PLUS47 and absorption corrections applied using the programme SADABS.48 Structures were solved by direct methods and developed using alternating cycles of least-squares refinement and difference-Fourier synthesis. All non-hydrogen atoms were refined anisotropically. Hydrogens were placed in calculated positions (riding model). Structure solution used SHELXTL PLUS V6.10 program package.49 For 3, a single crystal was mounted on a MiTeGen loop on an XtaLAB AFC11 (RCD3) quarter-chi single diffractometer. The crystal was kept at 100.00(11) K during data collection using Cu-Kα radiation (λ = 1.54184 Å). Using Olex2,50 the structure was solved with the olex2.solve50 structure solution program using Charge Flipping and refined with the SHELXL51 refinement package using Least Squares minimisation. Details of data collection and structure refinement are given in Table 1.52 For 4a voids in the initial structure solution were treated with the Olex implementation of the SQUEEZE function is Platon. Unfortunately it was not possible to model this as a disordered hexane although this is what we anticipate is present and is in accord with the elemental analysis results.Electrochemical and spectroelectrochemical studies
Electrochemistry was carried out in deoxygenated acetonitrile or dichloromethane solutions with 0.1 M TBAPF6 as the supporting electrolyte. The working electrode was a 3 mm diameter glassy carbon electrode that was polished with 0.3 μm alumina slurry prior to each scan. The counter electrode was a Pt wire and the quasi-reference electrode was a silver wire. All CVs were referenced to the Fc+/Fc redox couple. An Autolab potentiostat (EcoChemie, Netherlands) was used for all CV experiments and an EmStat3 (Palmsens, Netherlands) potentiostat for IR-SEC experiments. Catalysis studies were carried out by adding equivalents of HBF4·Et2O or CF3CO2H (Sigma-Aldrich) for proton reduction and pyridine (Sigma-Aldrich) for dihydrogen oxidation. Spectroelectrochemical (SEC) measurements were conducted within an optically transparent thin-layer electrochemical (OTTLE) cell (Spectroelectrochemistry Reading). The OTTLE cell was equipped with a Pt mini-grid working electrode, a Pt counter electrode, a Ag wire pseudo-reference electrode, and CaF2 windows. SEC samples contained 3 × 10−1 M TBAH as a supporting electrolyte. IR SEC (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) < 7500 cm−1) was run on a Bruker Vertex 70v Fourier transform infrared spectrometer equipped with a DTLaGS detector.
< 7500 cm−1) was run on a Bruker Vertex 70v Fourier transform infrared spectrometer equipped with a DTLaGS detector.
Computational methodology
The DFT calculations reported here were performed with the Gaussian 09 package of programs.53 The calculations were carried out with the B3LYP functional, which utilises the Becke three-parameter exchange functional (B3)54 combined with the correlation functional of Lee, Yang, and Parr (LYP).55 The iron atoms were described by Stuttgart-Dresden effective core potentials (ecp) and SDD basis set, while the 6-31G(d’) basis set was employed for the remaining atoms. The geometry-optimised structures reported here represent minima based on zero imaginary frequencies (positive eigenvalues) in the Hessian matrix. Unscaled vibrational frequencies were used to make zero-point and thermal corrections to the electronic energies. The geometry-optimised structures have been drawn with the JIMP2 molecular visualisation and manipulation program.56Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Commonwealth Scholarship Commission for the award of Commonwealth Scholarships (SG and JCS) and King's College London (GRFO) for PhD funding. GH thanks The Royal Society of Chemistry for an International Authors Grant which allowed this work to be developed during his visit to the University of North Texas and King's College London for funding. We thank Dr Nathan Patmore (University of Huddersfield) for some early IR-SEC on 1, Professor Katherine J. Holt (UCL) for the initial electrochemical study on 1, Dr Nathan Hollingsworth (ex-UCL) for part supervision of LA, and Kishan Muthu (KCL) for early attempts to prepare 2 while working under the supervision of GRFO. MGR acknowledges financial support from the Robert A. Welch Foundation (Grant B-1093-MGR). Computational resources through the High-Performance Computing Services and CASCaM at the University of North Texas are acknowledged. Dr David A. Hrovat is thanked for the generation of the spin density plots of B and 3C. The experimental work in Reading was conducted with the support from Spectroelectrochemistry Reading, a spinout company of the University.References
- C. Sommer, A. Adamska-Venkatesh, K. Pawlak, J. A. Birrell, O. Rűdiger, E. J. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2017, 129, 1440–1443 CrossRef PubMed.
- F. Wittkamp, M. Senger, S. T. Stripp and U.-P. Apfel, Chem. Commun., 2018, 54, 5934–5942 RSC.
- V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- For reviews of this area see: (a) I. P. Georgakaki, L. M. Thomson, E. J. Lyon, M. B. Hall and M. Y. Darensbourg, Coord. Chem. Rev., 2003, 238–239, 255–266 CrossRef CAS; (b) D. J. Evans and C. J. Pickett, Chem. Soc. Rev., 2003, 32, 268–287 RSC; (c) T. B. Rauchfuss, Inorg. Chem., 2004, 43, 14–26 CrossRef CAS PubMed; (d) L. Sun, B. Åkermark and S. Ott, Coord. Chem. Rev., 2005, 249, 1653–1663 CrossRef CAS; (e) X. Liu, S. K. Ibrahim, C. Tard and C. J. Pickett, Coord. Chem. Rev., 2005, 249, 1641–1652 CrossRef CAS; (f) C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed; (g) J.-F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, Coord. Chem. Rev., 2005, 249, 1664–1676 CrossRef CAS; (h) J.-F. Capon, F. Gloaguen, F. Y. Pétillon, P. Schollhammer and J. Talarmin, Eur. J. Inorg. Chem., 2008, 4671–4681 CrossRef CAS; (i) D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed; (j) J. T. Kleinhaus, F. Wittkamp, S. Yadav, D. Siegmund and U.-P. Apfel, Chem. Soc. Rev., 2021, 50, 1668–1784 RSC; (k) F. Wittkamp, M. Senger, S. T. Stripp and U.-P. Apfel, Chem. Commun., 2018, 54, 5934–5942 RSC; (l) F. Arrigoni, L. Bertini, R. Breglia, C. Greco, L. De Gioia and G. Zampella, New J. Chem., 2020, 44, 17596–17615 RSC.
- Y.-C. Liu, T.-H. Yen, K.-T. Chu and M.-H. Chiang, Comments Inorg. Chem., 2016, 36, 141–181 CrossRef CAS.
- C. Tard, X. Liu, S. K. Ibrahim, M. Bruschi, L. De Gioia, S. C. Davies, X. Yang, L.-S. Wang, G. Sawers and C. J. Pickett, Nature, 2005, 433, 610–613 CrossRef CAS PubMed.
- (a) J. M. Camara and T. B. Rauchfuss, Nat. Chem., 2012, 4, 26–30 CrossRef CAS PubMed; (b) J. C. Lansing, J. M. Camara, D. E. Gray and T. B. Rauchfuss, Organometallics, 2014, 33, 5897–5906 CrossRef CAS PubMed.
- S. Ghosh, G. Hogarth, N. Hollingsworth, K. B. Holt, S. E. Kabir and B. E. Sanchez, Chem. Commun., 2014, 50, 945–947 RSC . CCDC 956914 (1).
- (a) S. Ghosh, N. Hollingsworth, M. Warren, D. A. Hrovat, M. G. Richmond and G. Hogarth, Dalton Trans., 2019, 48, 6051–6060 RSC; (b) Y. Si, K. Charreteur, J.-F. Capon, F. Gloaguen, F. Y. Pétillon, P. Schollhammer and J. Talarmin, J. Inorg. Biochem., 2010, 104, 1038–1042 CrossRef CAS PubMed; (c) S. Ghosh, S. Rana, N. Hollingsworth, M. G. Richmond, S. E. Kabir and G. Hogarth, Inorganics, 2018, 6, 122 CrossRef CAS; (d) R. Becker, S. Amirjalayer, P. Li, S. Wouterse and J. N. H. Reek, Sci. Adv., 2016, 2, e1501014 CrossRef PubMed; (e) Y.-C. Liu, C.-H. Lee, G.-H. Lee and M.-H. Chiang, Eur. J. Inorg. Chem., 2011, 1155–1162 CrossRef.
- (a) S. Ghosh, A. Rahaman, K. B. Holt, E. Nordlander, M. G. Richmond, S. E. Kabir and G. Hogarth, Polyhedron, 2016, 116, 127–135 CrossRef CAS; (b) S. Roy, T. L. Groy and A. K. Jones, Dalton Trans., 2013, 42, 3843–3853 RSC; (c) S. Roy, J. A. Laureanti, T. L. Groy and A. K. Jones, Eur. J. Inorg. Chem., 2017, 2017, 2942–2950 CrossRef CAS; (d) P.-Y. Orain, J.-F. Capon, N. Kervarec, F. Gloaguen, F. Pétillon, R. Pichon, P. Schollhammer and J. Talarmin, Dalton Trans., 2007, 3754–3756 RSC; (e) J. Zhao, Z. Wei, X. Zeng and X. Liu, Dalton Trans., 2012, 41, 11125–11133 RSC; (f) Y.-C. Liu, T.-H. Yen, Y.-J. Tseng, C.-H. Hu, G.-H. Lee and M.-H. Chiang, Inorg. Chem., 2012, 51, 5997–5999 CrossRef CAS PubMed.
- C. Nataro, A. N. Campbell, M. A. Ferguson, C. D. Incarvito and A. L. Rheingold, J. Organomet. Chem., 2003, 673, 47–55 CrossRef CAS.
- J. W. Tye, M. B. Hall and M. Y. Darensbourg, Inorg. Chem., 2006, 45, 1552–1559 CrossRef CAS PubMed.
- (a) F. I. Adam, G. Hogarth, I. Richards and B. E. Sanchez, Dalton Trans., 2007, 2495–2498 RSC; (b) S. Ghosh, G. Hogarth, N. Hollingsworth, K. B. Holt, I. Richards, M. G. Richmond, B. E. Sanchez and D. Unwin, Dalton Trans., 2013, 42, 6775–6792 RSC.
- X.-F. Liu and B.-S. Yin, J. Coord. Chem., 2010, 63, 4061–4067 CrossRef CAS.
- C.-G. Li, F. Xue, M.-J. Cui, J.-Y. Shang and T.-J. Lou, Transition Met. Chem., 2015, 40, 47–52 CrossRef CAS.
- H. Li, Q. Gong, X. Chai, C. Yao and Q. Han, Asian J. Chem., 2015, 27, 1553–1554 CrossRef CAS.
- X.-F. Liu, Z.-Q. Jiang and Z.-J. Jia, Polyhedron, 2012, 33, 166–170 CrossRef CAS.
- F. I. Adam, G. Hogarth and I. Richards, J. Organomet. Chem., 2007, 692, 3957–3968 CrossRef CAS.
- S. Rana, S. Ghosh, Md. K. Hossain, A. Rahaman, G. Hogarth and S. E. Kabir, Transition Met. Chem., 2016, 41, 933–942 CrossRef CAS.
- F. I. Adam, G. Hogarth, S. E. Kabir and I. Richards, C. R. Chim., 2008, 11, 890–905 CrossRef CAS.
- G. Hogarth, M. O'Brien and D. A. Tocher, J. Organomet. Chem., 2003, 672, 29–33 CrossRef CAS.
- C.-G. Li, S.-L. Wang and J.-Y. Shang, J. Coord. Chem., 2016, 69, 2845–2854 CrossRef CAS.
- N. Begum, U. K. Das, M. Hassan, G. Hogarth, S. E. Kabir, E. Nordlander, M. A. Rahman and D. A. Tocher, Organometallics, 2007, 26, 6462–6472 CrossRef CAS.
- (a) X. L. Lu, S. Y. Ng, J. J. Vittal, G. K. Tan, L. Y. Goh and T. S. A. Hor, J. Organomet. Chem., 2003, 688, 100–111 CrossRef CAS; (b) D. Cauzzi, C. Graiff, C. Massera, G. Predieri, A. Tiripiccho and D. Acquotti, J. Chem. Soc., Dalton Trans., 1999, 3515–3521 RSC.
- (a) G. Hogarth and I. Richards, Inorg. Chem. Commun., 2007, 10, 66–70 CrossRef CAS; (b) G. M. Chambers, R. Angamuthu, D. L. Gray and T. B. Rauchfuss, Organometallics, 2013, 32, 6324–6329 CrossRef CAS; (c) D. Schilter, M. J. Nilges, M. Chakrabarti, P. A. Lindahl, T. B. Rauchfuss and M. Stein, Inorg. Chem., 2012, 51, 2338–2348 CrossRef CAS PubMed.
- G. Hogarth, S. E. Kabir and I. Richards, Organometallics, 2010, 29, 6559–6568 CrossRef CAS.
- S. Ghosh, B. E. Sanchez, I. Richards, M. N. Haque, K. B. Holt, M. G. Richmond and G. Hogarth, J. Organomet. Chem., 2016, 812, 247–258 CrossRef CAS.
- (a) A. C. Ohs, A. L. Rheingold, M. J. Shaw and C. Nataro, Organometallics, 2004, 23, 4655–4660 CrossRef CAS; (b) M. R. Ringenberg, M. Schwilk, F. Wittkamp, U.-P. Apfel and W. Kaim, Chem. – Eur. J., 2017, 23, 1770–1774 CrossRef CAS PubMed; (c) M. R. Ringenberg and F. Döttinger, Eur. J. Inorg. Chem., 2019, 2019, 2430–2435 CrossRef CAS.
- (a) M. E. Carroll, B. E. Barton, T. B. Rauchfuss, P. J. Carroll and J. Am, Chem. Soc., 2012, 134, 18843–18852 CrossRef CAS PubMed; (b) G. Eilers, L. Schwartz, M. Stein, G. Zampella, L. De Gioia, S. Ott and R. Lomoth, Chem. – Eur. J., 2007, 13, 7075–7084 CrossRef CAS PubMed.
- M. Häßner, J. Fiedler and M. R. Ringenberg, Inorg. Chem., 2019, 58, 1742–1745 CrossRef PubMed.
- F. Arrigoni, L. Bertini, M. Bruschi, C. Greco, L. De Gioia and G. Zampella, Chem. – Eur. J., 2019, 25, 1227–1241 CrossRef CAS PubMed.
- (a) S. Ezzaher, J.-F. Capon, F. Gloaguen, F. Y. Petillon, P. Schollhammer, J. Talarmin, R. Pichon and N. Kervarec, Inorg. Chem., 2007, 46, 3426–3428 CrossRef CAS PubMed; (b) S. Ezzaher, J.-F. Capon, N. Dumontet, F. Gloaguen, F. Y. Petillon, P. Schollhammer and J. Talarmin, J. Electroanal. Chem., 2009, 626, 161–170 CrossRef CAS; (c) C. Greco, P. Fantucci, L. De Gioia, R. Suarez-Bertoa, M. Bruschi, J. Talarmin and P. Schollhammer, Dalton Trans., 2010, 39, 7320–7329 RSC; (d) B. E. Barton, G. Zampella, A. K. Justice, L. De Gioia, T. B. Rauchfuss and S. R. Wilson, Dalton Trans., 2010, 39, 3011–3019 RSC.
- W. Gao, J. Ekström, J. Liu, C. Chen, L. Eriksson, L. Weng, B. Akermark and L. Sun, Inorg. Chem., 2007, 46, 1981–1991 CrossRef CAS PubMed.
- T. Agarwal and S. Kaur-Ghumaan, Coord. Chem. Rev., 2019, 397, 188–219 CrossRef CAS.
- G. A. N. Felton, R. S. Glass, D. L. Lichtenberger and D. H. Evans, Inorg. Chem., 2006, 45, 9181–9184 CrossRef CAS PubMed.
- R. Mejia-Rodriguez, D. Chong, J. H. Reibenspies, M. P. Soriaga and M. Y. Darensbourg, J. Am. Chem. Soc., 2004, 126, 12004–12014 CrossRef CAS PubMed.
- A. Jabloskyté, J. A. Wright, S. A. Fairhurst, J. N. T. Peck, S. K. Ibrahim, V. S. Oganesyan and C. J. Pickett, J. Am. Chem. Soc., 2011, 133, 18606–18609 CrossRef PubMed.
- W. Wang, M. J. Nilges, T. B. Rauchfuss and M. Stein, J. Am. Chem. Soc., 2103, 135, 3633–3639 CrossRef PubMed.
- G. Filippi, F. Arrigoni, L. Bertini, L. De Gioia and G. Zampella, Inorg. Chem., 2105, 54, 9529–9542 CrossRef PubMed.
- N. Wang, M. Wang, Y. Wang, D. Zheng, H. Han, M. S. G. Ahlquist and L. Sun, J. Am. Chem. Soc., 2013, 135, 13688–13691 CrossRef CAS PubMed.
- F. Arrigoni, L. Bertini, R. Breglia, C. Greco, L. De Gioia and G. Zampella, New J. Chem., 2020, 44, 17596–17615 RSC.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- A. Winter, L. Zsolnai and G. Huttner, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1982, 37, 1430–1436 CrossRef.
- P. C. Ellgen and J. N. Gerlach, Inorg. Chem., 1973, 12, 2526–2532 CrossRef CAS.
- A. L. Haley, L. N. Broadbent, L. S. McDaniel, S. T. Heckman, C. H. Hinkle, N. N. Gerasimchuk, J. C. Hershberger and C. A. Mebi, Polyhedron, 2016, 114, 218–224 CrossRef CAS.
- C. A. Mebi, D. S. Karr and R. Gao, J. Coord. Chem., 2011, 64, 4397–4407 CrossRef CAS.
- SAINT Version 6.36A, Bruker AXS, Inc., Analytical X-ray Systems, 5465 East Cheryl Parkway, Madison WI 53711-5373, 2002 Search PubMed.
- G. M. Sheldrick, SADABS Version 2.10, University of Göttingen, 2003 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 59–75 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 Search PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 37, 785–789 CrossRef PubMed.
- (a) JIMP2, version 0.091, a free program for the visualisation and manipulation of molecules Search PubMedM. B. Hall and R. F. Fenske, Inorg. Chem., 1972, 11, 768–775 CrossRef CAS; (b) J. Manson, C. E. Webster and M. B. Hall, Texas A&M University, College Station, TX, 2006, https://www.chem.tamu.edu/jimp2/index.html.
Footnote |
| † Electronic supplementary information (ESI) available: Text, tables and cif files giving details of the X-ray crystallographic structure determinations; atomic coordinates and energies of all optimised stationary points. Some additional electrochemical information and experiments, crystallographic data and computational details. CCDC 2151071 (3), 2151142 (4a) and 2151143 (4c). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00419d |
| This journal is © The Royal Society of Chemistry 2022 |