
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the reactivity of Al-group 11 (Cu, Ag, Au) bonds†‡
Han-Ying
Liu
,
Samuel E.
Neale
 ,
Michael S.
Hill
*,
Mary F.
Mahon
* and
Claire L.
McMullin
*
,
Michael S.
Hill
*,
Mary F.
Mahon
* and
Claire L.
McMullin
*
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@bath.ac.ak; cm2025@bath.ac.uk
First published on 15th February 2022
Abstract
Reactions of the seven-membered heterocyclic potassium diamidoalumanyl, [K{Al(SiNDipp)}]2 (SiNDipp = {CH2SiMe2NDipp}2; Dipp = 2,6-di-isopropylphenyl), with a variety of Cu(I), Ag(I) and Au(I) chloride N-heterocyclic carbene (NHC) adducts are described. The resultant group 11-Al bonded derivatives have been characterised in solution by NMR spectroscopy and, in the case of [{SiNDipp}Al–Au(NHCiPr)] (NHCiPr = N,N′-di-isopropyl-4,5-dimethyl-2-ylidene), by single crystal X-ray diffraction. Although similar reactions of LAgCl and LAuCl, where L is a more basic cyclic alkyl amino carbene (CAAC), generally resulted in reduction of the group 11 cations to the base metals, X-ray analysis of [(CyCAAC)AgAl(SiNDipp)] (CyCAAC = 2-[2,6-bis(1-methylethyl)phenyl]-3,3-dimethyl-2-azaspiro[4.5]dec-1-ylidene) provides the first solid-state authentication of an Ag–Al σ bond. The reactivity of the NHC-supported Cu, Ag and Au alumanyl derivatives was assayed with the isoelectronic unsaturated small molecules, N,N′-di-isopropylcarbodiimide and CO2. While these reactions generally provided products consistent with nucleophilic attack of the group 11 atom at the electrophilic heteroallene carbon centre, treatment of the NHC-supported copper and silver alumanyls with N,N′-di-isopropylcarbodiimide yielded less symmetric Cu–C and Ag–C-bonded isomers. In contrast to the previously described copper and silver alumanyl derivatives, [(NON)Al(O2C)M(Pt-Bu3)] (M = Cu or Ag; NON = 4,5-bis(2,6-di-isopropylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene), which were prone to facile CO extrusion and formation of carbonate derivatives, the NHC-supported dioxocarbene species, [(NHCiPr)M(CO2)Al(SiNDipp)] (M = Cu, Ag, Au), are all stable at room and moderately elevated temperatures. The stabilising role of the NHC co-ligand was, thus, assessed by preparation of the t-Bu3P adducted copper-alumanyl, [(t-Bu3P)CuAl(SiNDipp)]. Treatment of this latter compound, which was also structurally characterised by X-ray analysis, with both N,N′-di-isopropylcarbodiimide and CO2 again provided smooth heteroallene insertion and formation of the relevant Cu–C-bonded products. Although both compounds were quite stable at room temperature, heating of [(t-Bu3P)Cu(CO2)Al(SiNDipp)] at 60 °C induced elimination of CO and formation of the analogous carbonate, [(t-Bu3P)Cu(OCO2)Al(SiNDipp)], which was identified by 13C and 31P NMR spectroscopy. Reflective of the more reliable nucleophilic behaviour of the gold centres in these group 11 alumanyls, computational (QTAIM and NBO) analysis highlighted a lower level of covalency of the Al–Au linkage in comparison to the analogous Al–Cu and Al–Ag interactions. Although substitution of the co-ligand significantly perturbs the charge distribution across the Cu–Al bond of [LCuAl(SiNDipp)] (L = NHCiPr or t-Bu3P), only a negligible difference is observed between the phosphine-coordinated copper systems derived from either the [SiNDipp]- or (NON)-based alumanyl ligands. Computational mapping of the reaction profiles arising from treatment of the various group 11 alumanyls with N,N′-di-isopropylcarbodiimide indicates that the observed formation of the Cu–N and Ag–N bound isomers do not provide the thermodynamic reaction outcome. In contrast, examination of the CO2-derived reactions, and their potential toward CO extrusion and subsequent carbonate formation, implies that the identity of the co-ligand exerts a greater influence on this aspect of reactivity than the architecture of the diamidoalumanyl anion.
Introduction
Progress in organometallic chemistry (i.e. M–C bonded compounds) during the latter half of the 20th Century was accompanied by the similar development of metallated derivatives of carbon's heavier group 14 congeners. While the study of heterometal-bonded silyl,1–5 germyl,6,7 stannyl8–10 and, to a lesser extent, plumbyl11 species continues as a pursuit of some significance, the first two decades of the 21st Century have seen the emergence of a broadly comparable chemistry derived from terminally-coordinated group 13-centred anions.12–15 In this latter regard, the most significant activity has centred on the exploitation of boron-centred (boryl) moieties as strongly σ-donating ligands to transition metal (TM) centres and their attendant applications in synthesis and C–B bond-forming catalysis.16–19Although the synthetic utility of organoboranes ensures the continuing prominence of TM–boryl complexes, the variations in TM–E bond strength and polarity resulting from comparable species featuring transition metals bonded to heavier group 13 elements (i.e. E = Al, Ga, In or Tl) promises to expand the scope of complementary or contrasting TM–E reactivity. A variety of gallium-,20–27 aluminium-28–38 and indium-centred39,40 anions suitable for the further development of TM-bonded species have now been described. Of most recent origin is an assortment of group 1 alumanyls exemplified by 1–3 (Fig. 1),28,31,32,41,42 the availability and reactivity of which as Al-centred nucleophiles enables the direct metathetical construction of terminal TM–Al σ bonds by their reaction with transition metal electrophiles.30,42,43 Although some variability of outcome was observed as a result of phosphine co-ligand identity, treatment of t-Bu3PAuI with Aldridge and Goicoechea's [K{Al(NON)}]2 (1, where NON is the chelating tridentate ligand 4,5-bis(2,6-di-isopropylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene) allowed the synthesis of the two-coordinate gold complex, [(NON)AlAu(Pt-Bu3)] (4-Au, Scheme 1a).30 The Auδ−–Alδ+ polarisation intrinsic to the intermetallic bond of compound 4-Au allowed it to function as an apparent source of nucleophilic gold in reactions with CO2 and N,N′-di-isopropylcarbodiimide.44 The observation that the resultant molecules exclusively comprise a carbenic (:CX2) (X = O, Ni-Pr) unit with a {Cu-μ-η1-:C-κ2-(X,X′)-Al} mode of heteroallene bridging inspired us to assay comparable reactions of the seven-membered heterocyclic potassium diamidoalumanyl, [K{Al(SiNDipp)}]2 (3, SiNDipp = {CH2SiMe2NDipp}2; Dipp = 2,6-di-isopropylphenyl) with the Cu(I) chloride carbene adducts, [(NHCiPr)CuCl] and [(Me2CAAC)CuCl] (NHCiPr = N,N′-di-isopropyl-4,5-dimethyl-2-ylidene; Me2CAAC = 1-(2,6-di-isopropylphenyl)-3,3,5,5-tetramethylpyrrolidin-2-ylidene).43 In contrast to the consistent behaviour of 4-Au as an Au-centred nucleophile, the resultant complexes, [LCu-Al(SiNDipp)] (7, L = NHCiPr; 8 L = Me2CAAC), exhibited divergent behaviour with carbodiimides and CO2, providing compounds 9–12, the structures of which imply the Cu–Al interaction displays an ambiphilicity modulated by the identities of the carbene co-ligand and the heteroallene electrophile (Scheme 1b).
 | ||
| Fig. 1 Structures of compounds 1–3. | ||
 | ||
Scheme 1 (a) Contrasting reactivity of [(NON)Al–M(Pt-Bu3)] (M = Cu, Ag, Au) toward CO2;30,45 (b) reactivity of compounds 7 and 8 with i-PrN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CNi-Pr and CO2.43 CNi-Pr and CO2.43 | ||
Aldridge, Goicoechea, Frenking and co-workers’ study of the reactivity of compound 1 was very recently extended to encompass the synthesis of the full range of group 11 derivatives, [(NON)Al–M(Pt-Bu3)] (M = Cu (4-Cu), Ag (4-Ag), Au (4-Au)).45 Although treatment of these compounds with carbodiimides also yielded species comprising stable {Cu-μ-η1-:C-κ2-(NR)2-Al} heteroallene bridging, the outcome of reactions with CO2 was found to be dependent on the identity of the group 11 metal. In contrast to the stability of [(NON)Al(O2C)Au(Pt-Bu3)] (5-Au), the analogous silver phosphine adduct (5-Ag) completely converted to the corresponding carbonate complex, [(NON)Al(O3C)Ag(Pt-Bu3)] (6-Ag), via the extrusion of CO at 80 °C (Scheme 1a). Although this process was slow at room temperature, the analogous copper system (4-Cu) was labile toward carbonate formation (6-Cu) such that the initial dioxocarbene (5-Cu) could not be observed, even at −78 °C (Scheme 1a). This behaviour was rationalised by a computational study to be consistent with a rate determining extrusion of CO from the initially formed dioxocarbene {M(CO2)Al} units, and subsequent reaction of the resultant M–O–Al bridged oxides with CO2. This chemistry provides a notable contrast to the ready isolation and stability of compounds 11 and 12 (Scheme 1b), an observation which implies that the reactivity of the M–Al bonds of these systems displays significant malleability contingent upon the electronic character of the alumanyl unit itself and/or the identity of the neutral co-ligand. In an attempt to further deconvolute these effects, therefore, we now describe a synthetic and computational study of the use of compound 3 to access a wider range of [LM-Al(SiNDipp)] (M = Cu, Ag, Au; L = carbene or phosphine donor) derivatives along with an assessment of the reactivity of the M–Al bonds toward a similar range of heteroallene small molecules.
Results and discussion
Synthesis and reactivity of carbene-supported copper, silver and gold alumanyl derivatives
To assess the impact of variations in the steric demands of the N-heterocyclic carbene (NHC) co-ligand, we first prepared a copper alumanyl species analogous to compounds 7 and 8 but supported by the more sterically encumbered NHC, (1,3-bis(2,6-di-isopropylphenyl)imidazol-2-ylidene) (IPr). A stoichiometric reaction of compound 3 with [(IPr)CuCl] in hexane provided [(IPr)CuAl(SiNDipp)] (13) as an off-white solid. In common with the syntheses of 7 and 8, but in contrast to the analogous reactions of compound 1 with [(t-Bu3P)CuI], which resulted in the bis-alumanyl cuprate K[Cu{Al(NON)}2],42 substitution of the chloride was straightforward. Although analysis of compound 13 was limited to the solution state, its formation was unambiguous and gave rise to a series of resonances in the 1H NMR spectrum consistent with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of carbene to SiNDipp-ligand environments. The corresponding 13C{1H} NMR spectrum displayed a resonance at δ 185.1 ppm assigned to the (IPr)C–Cu donor carbon nucleus that is, thus, symptomatic of a similar electronic environment at copper as that observed in compound 7 (δ 175.9 ppm).
:1 ratio of carbene to SiNDipp-ligand environments. The corresponding 13C{1H} NMR spectrum displayed a resonance at δ 185.1 ppm assigned to the (IPr)C–Cu donor carbon nucleus that is, thus, symptomatic of a similar electronic environment at copper as that observed in compound 7 (δ 175.9 ppm).
Consistent with this latter inference, reactions of compound 13 with N,N′-di-isopropylcarbodiimide and CO2 provided a similar outcome as previously observed for compound 7 (Scheme 2).43 The carbodiimide-based reaction resulted in the gradual consumption of both starting materials at 40 °C over the course of 3 days to provide clean access to compound 14. In common with our previous observations of compound 9 (Scheme 1b), the resultant 1H NMR spectrum was consistent with significant asymmetry and/or limited conformational flexibility such that the iso-propyl methine resonances of the carbodiimide fragment in 14 are separated into two distinct (1H by relative integration) multiplet signals at δ 3.38 and 2.99 ppm. Significantly, and in clear contrast to the low field resonance (δ 220.9 ppm) assigned to the copper-coordinated {Cu–C(Ni-Pr)2} carbon centre in compound 10, no signal could be observed for the formerly sp-hybridised carbodiimide carbon. These observations, therefore, suggest that the constitution of 14 is most likely reminiscent of that determined in the solid state for compound 9, in which the 2-coordinate copper centre is ligated by a molecule of IPr and a single nitrogen atom of the {CN2} fragment. These observations are also again consistent, therefore, with a course of reaction in which the aluminium centre has apparently maintained a level of nucleophilic behaviour.
 | ||
| Scheme 2 Reactivity of compound 13 with N,N′-di-isopropylcarbodiimide and CO2 and the synthesis of compounds 14 and 15. | ||
Additional evidence of the analogous nature of its Cu–Al bond with that of 7 was provided by a further reaction of compound 13 with 2 atm. of 13CO2 in benzene. In this case, complete conversion to a single new compound (15) was achieved within 30 minutes. Although the resultant 1H NMR spectrum was indicative of a symmetrical species, compound 15 was most clearly characterised by the emergence of a single new 13C-labelled resonance in its 13C{1H} NMR spectrum at δ 234.0 ppm. This chemical shift is closely comparable to signals observed in the corresponding spectra provided by both compound 11 (δ 236.2 ppm) and the copper CAAC adduct, compound 12 (δ 234.9 ppm).43 This latter compound was structurally characterised as a cupra-dioxocarbene derivative comprising a linear {NHCiPr-Cu-CO2} unit and the current data are, thus, strongly suggestive that a similar structure may be ascribed to compound 15 (Scheme 2). These observations again provide a significant contrast to the low temperature instability of [(NON)Al(O2C)Cu(Pt-Bu3)] (5-Cu),45 but infer that moderation of the steric demands associated with the N-heterocyclic carbene co-ligand exerts only a limited influence over the reactivity of the {(NHC)Cu–Al} bond.
With these results in hand, reactions of compound 3 were performed with NHCiPr adducts of both AgCl and AuCl (Scheme 3). In both cases, overnight reactions performed in hexane provided the respective silver and gold alumanyl derivatives, compounds 16 and 17 (Scheme 3), as colourless solids. Consistent with all three group 11 derivatives exhibiting isotypical structures, the 1H NMR spectra of both compounds 16 and 17 were strongly reminiscent of their previously reported copper analogue, compound 7.43 The (NHCiPr)-M donor carbon atoms of the heavier group 11 congeners, however, were observed to resonate at notably lower field (δ16: 230.5: 17: 216.1 ppm) in their 13C{1H} NMR spectra than the C-donor environment of 7 (δ 175.9 ppm). These chemical shift values are also significantly downfield of those reported for the relevant chloride starting materials [(NHCiPr)MCl] (δ M = Ag: 172.3; M = Au: 166.0 ppm).46,47 Similar observations have been reported, but not rationalised, for several borylsilver and -gold(I) NHC complexes and,48,49 thus, evidence comparable modulation of the electronic environments of the second and third row transition metals by the introduction of the alumanyl ligand.
 | ||
| Scheme 3 Synthesis of compounds 16 and 17. | ||
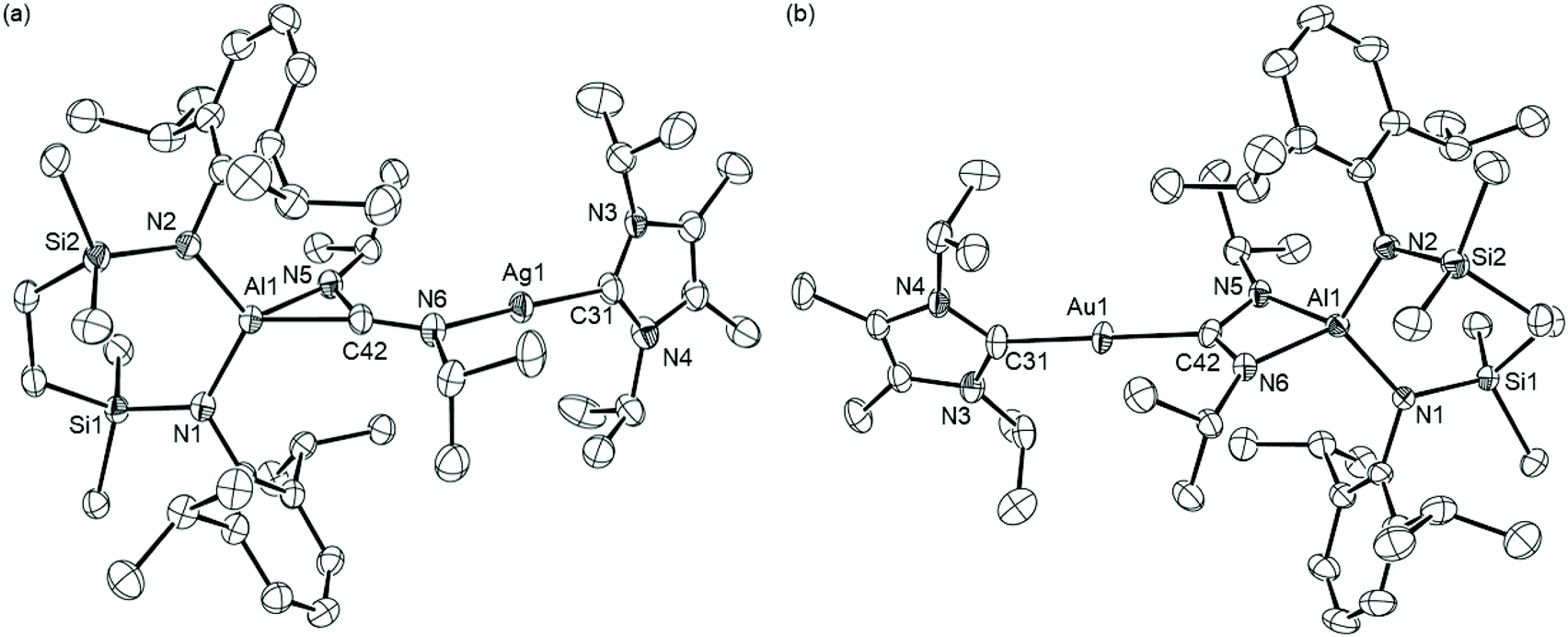
Although analytically pure bulk samples of compound 16 could be obtained for further studies of its reactivity (vide infra), all attempts to obtain single crystals were unsuccessful. Slow evaporation of a methylcyclohexane solution of compound 17, however, yielded colourless single crystals suitable for X-ray diffraction analysis. The result of this analysis is illustrated in Fig. 2a, while selected bond length and angle data are provided in Table 1. The gold coordination environment of compound 17 is provided solely by its interaction with the NHCiPr and alumanyl donor ligands such that the C31–Au1–Al1 angle approaches linearity [178.3(2)°]. The unsupported Au1–Al1 bond [2.369(2) Å] is significantly contracted in comparison to the corresponding distance in [(NON)AlAu(Pt-Bu3)] (4-Au: 2.402(3) Å), which previously represented the shortest reported Au–Al distance.30 The Al–Au interaction may, thus, be anticipated to exert a significant trans influence on the almost colinear NHCiPr ligand. The C31–Au1 bond [2.094(7) Å], however, is only marginally elongated in comparison to the C–Au distance observed in [(NHCiPr)AuCl] [1.996(9) Å]. Although no comparable examples have been described with the identical NHCiPr ligand, the bond length observed in 17 is closely comparable to the Au–C distances reported [range: 2.048(1)–2.109(16) Å] for the handful of 2-coordinate gold species which have been shown to feature a similarly pseudo-linear orientation of a NHC and boryl donor ligands.48,49
 | ||
| Fig. 2 ORTEPs (30% probability ellipsoids) of (a) compound 17 and (b) compound 18. Hydrogen atoms and an occluded molecule of methylcyclohexane solvent (17) have been omitted for clarity. | ||
| 17 | 18 | 23 | |
|---|---|---|---|
| a M = Au. b M = Ag. c M = Cu. d Cu1–P1. e P1–Cu1–Al1. | |||
| M1–Al1 | 2.369(2) | 2.4694(6) | 2.3755(3) |
| M1–C31 | 2.094(7) | 2.182(2) | 2.2807(3)d |
| Al1–N1 | 1.827(6) | 1.8526(18) | 1.8457(10) |
| Al1–N2 | 1.828(6) | 1.8484(18) | 1.8490(10) |
| C31–M1–Al1 | 178.3(2) | 171.13(6) | 175.879(14)e |
| N1–Al1–M1 | 122.8(2) | 124.91(6) | 120.28(3) |
| N2–Al1–M1 | 122.2(2) | 121.48(6) | 127.11(3) |
In contrast to the smooth access to compounds 16 and 17 provided by silver and gold adducts of NHCiPr, attempts to extend this reactivity to the more basic Me2CAAC ligand utilised in the synthesis of the copper alumanyl, 8, were unsuccessful. In all cases, attempted reactions of either [(Me2CAAC)AgCl] or [(Me2CAAC)AuCl] with compound 3 led to immediate decomposition and a complex mix of products by apparent reduction and precipitation of the group 11 metals. Analogous reactions performed with the more sterically encumbered cyclic (alkyl)(amino)carbene, CyCAAC (CyCAAC = 2-[2,6-bis(1-methylethyl)phenyl]-3,3-dimethyl-2-azaspiro[4.5]dec-1-ylidene), provided similar observations. In one case, however, reaction of [(CyCAAC)AgCl] with 3 in C6D6 enabled the identification of the silver alumanyl derivative, [(CyCAAC)AgAl(SiNDipp)] (18), by X-ray diffraction analysis performed on a colourless single crystal, which was isolated by its mechanical separation from the mixture of products formed. Although a bulk sample for further analysis could not be obtained, the resultant structure (Fig. 2b) provides the first solid-state authentication of any silver–aluminium bond. In a manner reminiscent of the copper and gold centres of compounds 7, 8 and 17, the silver atom occupies a pseudo-linear 2-coordinate environment provided by the CAAC and alumanyl donor atoms [C31–Ag1–Al1 171.13(6)°]. While the Ag1–Al1 bond [2.4694(6) Å] is unique, the Ag1–C31 separation [2.182(2) Å] is only slightly longer than the Ag–C distances observed in a variety of previously reported silver(I) adducts of CAAC donors (e.g. [(Me2CCAC)AgCl] 2.0769(18) Å).50–54
Our earlier communication documented the reactivity of compounds 7 and 8 toward N,N′-di-isopropylcarbodiimide and CO2 (Fig. 1b) and highlighted that the regiochemistry of the carbodiimide insertion was dictated by the nature of the carbene co-ligand.43 This apparent ambiphilicity of the Al–Cu bond was mirrored by the behaviour of compound 13 (Scheme 2). Although a study of similar scope was precluded by the instability of comparable alumanylsilver and -gold CAAC adducts, the availability of compounds 16 and 17, and the point of comparison provided by Aldridge and co-workers’ recent studies of the reactivity of [(NON)AlM(Pt-Bu3)] (Scheme 1, M = Cu, Ag, Au),30,45 allowed an assay of their reactivity toward the same heteroallene reagents to be performed (Scheme 4).
 | ||
| Scheme 4 Reactivity of the silver and gold alumanyl complexes, 16 and 17, with N,N′-di-isopropylcarbodiimide and CO2. | ||
Treatment of the silver and gold alumanyl derivatives, 16 and 17, with N,N′-di-isopropylcarbodiimide in benzene provided the respective compounds 19 and 20 within a few hours at room temperature. The 1H and 13C{1H} NMR spectra of compound 19 were reminiscent of those arising from the unsymmetrical insertion regiochemistry of compound 9.43 The Ni-Pr methine resonances of the former carbodiimide were separated into two distinct (1H by relative integration) multiplet signals at δ 3.46 and 4.34 ppm. In contrast, compound 20 exhibited only a single set of NCH(CH3)2 environments in its 1H NMR spectrum. These data are strongly suggestive, therefore, that compounds 19 and 20 are the respective products of a contrasting carbodiimide insertion regiochemistry. This deduction was confirmed by X-ray diffraction analysis of colourless single crystals of compounds 19 and 20, which were isolated by slow evaporation of methylcyclohexane solutions. The results of these analyses are shown in Fig. 3, while selected bond length and angle data are presented in Table 2. In both cases, the asymmetric units comprise two molecules, which are, in each instance, very similar. Discussion, therefore, is restricted to the Ag1- and Au1-containing molecules. The structure of 19 features a two-coordinate silver centre, ligated by NHCiPr and a single nitrogen atom of the {CN2} fragment [Ag1–C31 2.078(5) Å; Ag1–N6 2.087(4) Å]. The coordination sphere of the aluminium is satisfied by a side-on η2-interaction with the C42–N5 bond of the {CN2} unit, resulting in the formation of a constrained three-membered AlCN metallacycle with Al1–C42, Al1–N5 and C–N distances of 1.948(5), 1.853(4) and 1.362(6) Å, respectively. In contrast, compound 20 crystallises as an aura-amidinate structure in which the NHCiPr-ligated gold centre is bound to the {CN2} fragment through the central carbenic carbon atom. Despite the introduction of an alternative NHC co-ligand, the Au1–C42 distance of 20 [2.046(3) Å] is closely comparable to that reported for [(NON)Al{(Ni-Pr)2C}Au(Pt-Bu3)], [2.058(10) Å].30 Consistent with its assignment as a delocalised aura-amidinate, the {(Ni-Pr)2C} unit coordinates the aluminium centre in a N,N′-bidentate fashion, with essentially identical Al–N distances [Al1–N5 1.913(2); Al1–N6 1.904(2) Å].
 | ||
| Fig. 3 ORTEPs (30% probability ellipsoids) of (a) the Ag1-containing molecule of compound 19 and (b) the Au1-containing molecule of compound 20. H atoms and disordered atoms have been omitted for clarity. | ||
| 19 | 20 | 24 | |
|---|---|---|---|
| a M = Ag. b M = Au. c M = Cu. d Ag1–N6. e Al1–C42. f C31–Ag1–N6. g N1–Al1–C42. h N2–Al1–C42. i Cu1–C43. j Cu1–P1. k Al1–N3. l Al1–N4. m C43–N3. n C43–N4. o P1–Cu1–C43. p N1–Al1–N3. q N1–Al1–N4. r N2–Al1–N3. s N2–Al1–N4. t N3–C43–N4. | |||
| M1–C42 | 2.087(4)d | 2.046(3) | 1.9383(15)i |
| M1–C31 | 2.078(5) | 2.058(3) | 2.2095(4)j |
| Al1–N1 | 1.842(5) | 1.849(2) | 1.8654(13) |
| Al1–N2 | 1.854(4) | 1.839(2) | 1.8620(13) |
| Al1–N5 | 1.853(4) | 1.913(2) | 1.9091(13)k |
| Al1–N6 | 1.948(5)e | 1.904(2) | 1.9096(13)l |
| C42–N5 | 1.362(6) | 1.364(4) | 1.346(2)m |
| C42–N6 | 1.315(6) | 1.335(3) | 1.343(2)n |
| C31–M1–C42 | 174.9(2)f | 179.41(12) | 177.73(4)o |
| N1–Al1–N2 | 114.80(19) | 110.37(10) | 113.32(6) |
| N1–Al1–N5 | 121.54(19) | 123.61(10) | 122.64(6)p |
| N1–Al1–N6 | 118.8(2)g | 111.62(10) | 112.16(6)q |
| N2–Al1–N5 | 121.54(19) | 113.24(10) | 110.72(6)r |
| N2–Al1–N6 | 124.22(19)h | 123.65(10) | 122.14(6)s |
| N5–C42–N6 | 127.7(5) | 108.4(2) | 108.75(13)t |
Exposure of samples of compounds 16 and 17 to 2 atm. of 13CO2 provided clean conversion to the respective new species, 21 and 22, within 30 minutes at room temperature. Although the solid-state structure of neither compound could be crystallographically corroborated, the assignments of their solution-state constitutions were unambiguous. As discussed above for the copper derivative 15, the emergence of low field 13C-labelled resonances in the 13C{1H} NMR spectra at δ 240.4 (21) and 239.1 (22) ppm is characteristic of the formation of the respective silver and gold dioxocarbene derivatives, [(NHCiPr)M{CO2}Al(SiNDipp)] (21, M = Ag; 22, M = Au; Scheme 4). These 13C NMR data are, thus, reminiscent of those reported for the analogous group 11-bonded {13CO2} environments of [(NON)Al{O2C}M(Pt-Bu3)] (5-Ag, δ 220.2;455-Au, δ 242.3 ppm).30 In addition, and like the former of these two previously reported signals, the resonance arising from compound 21 is observed as two doublets resulting from coupling to the high abundance I = ½ 109Ag (48.2%) and 107Ag (51.8) isotopes (1J109Ag–C = 252.6, 1J107Ag–C = 218.9 Hz, Fig. S34‡).
Stability of copper dioxocarbene species
Although the previously reported silver complex, [(NON)Al{O2C}AgPt-Bu3] (5-Ag, Scheme 1a), could be characterised, it underwent slow conversion to the corresponding silver carbonate (6-Ag) at room temperature or over 24 h when heated at 80 °C under CO2 (Scheme 1).45 Notably, the putative copper species, [(NON)Al{O2C}Cu(Pt-Bu3)] (5-Cu), was prone to elimination of CO and sequestration of a molecule of CO2 even at −78 °C. This latter behaviour is in significant contrast to our isolation of the copper carbene adducts 11, 12 and 15, which appear to be indefinitely stable at room temperature and even when heated to 60 °C.To begin to assess the relative impact of alumanyl and co-ligand identity, therefore, an equimolar reaction of compound 3 and the copper(I) phosphine adduct [(t-Bu3P)CuCl] was carried out. In contrast to the synthesis of [(NON)AlCu(Pt-Bu3)] (4-Cu), which had to be performed with a twofold stoichiometric excess of the copper halide reagent to suppress the formation of the cuprate K[Cu{Al(NON)}2],45 this procedure provided clean access to the phosphine-supported copper alumanyl, [(t-Bu3P)CuAl(SiNDipp)] (23) (Scheme 5). The resultant 1H NMR spectrum confirmed a ratio of one phosphine per {SiNDipp} unit, while the 31P NMR resonance was recorded at δ 43.9 ppm. Consistent with only a marginal perturbation to the phosphorus and, by extension, the copper environments, this latter value is only slightly downfield of that reported for [(NON)AlCu(Pt-Bu3)] (4-Cu, δ 38.3 ppm).45 The constitution of compound 23 was confirmed by a further X-ray diffraction analysis performed on a single crystal isolated from hexane solution (Fig. 4a and Table 1). While the solid-state structure of 4-Cu is not available for comparison, the Cu1–Al1 bond length [2.3755(3) Å] of 23 lies between the corresponding measurements provided by compound 7 [2.3449(4) Å] and compound 8 [2.4028(7) Å].43 As these latter derivatives provided a contrasting outcome in their reactions with N,N′-di-isopropylcarbodiimide (Fig. 1), which was tentatively attributed to the relative basicity of the NHC and CAAC co-ligands, compound 23 was treated with the same heteroallene in benzene at room temperature. Inspection of the resultant 1H spectrum confirmed the ready insertion of the carbodiimide and the formation of a single symmetrical product (24) comprising a 1:1:1 ratio of {SiNDipp}, phosphine and carbodiimide resonances (Scheme 5). The maintenance of the nucleophilic character of the copper centre of 24 through the introduction of the strongly σ-donating phosphine was confirmed by the appearance of a low field signal at δ 218.0 ppm in the 13C{1H} NMR spectrum. This chemical shift is, thus, comparable with signals attributed to the Cu-bonded carbon nuclei of compound 10 (δ 220.9 ppm)43 and the data arising from Aldridge and co-workers’ [(NON)Al{(CyN)2C}Cu(Pt-Bu3)] (δ 215.5 ppm).45 Indicative of closely related electronic environments, the 31P{1H} NMR spectrum of 24 displayed a single resonance at δ 59.8 ppm, which is also effectively identical to that reported for [(NON)Al{(CyN)2C}Cu(Pt-Bu3)] (δ 59.6 ppm). These solution state observations were supported by an X-ray diffraction analysis of 24 performed on a single crystal isolated from hexane at room temperature (Fig. 4b and Table 2). The copper coordination geometry of compound 24 is close to linear [C43–Cu1–P1 177.73(4)°] and the C–Cu–P unit is broadly comparable to that reported for [(NON)Al{(CyN)2C}Cu(Pt-Bu3)]. Despite the variation in the carbodiimide substituents and the diamidoaluminium supporting environments employed in their synthesis, the Cu1–P1 [2.2095(4) Å] and Cu1–C43 [1.9383(15) Å] bond lengths of 24 are also only marginally perturbed in comparison to the analogous distances in Aldridge and co-workers’ previously reported phosphine adduct [Cu–P 2.2163(13); Cu–C 1.952(4) Å].45
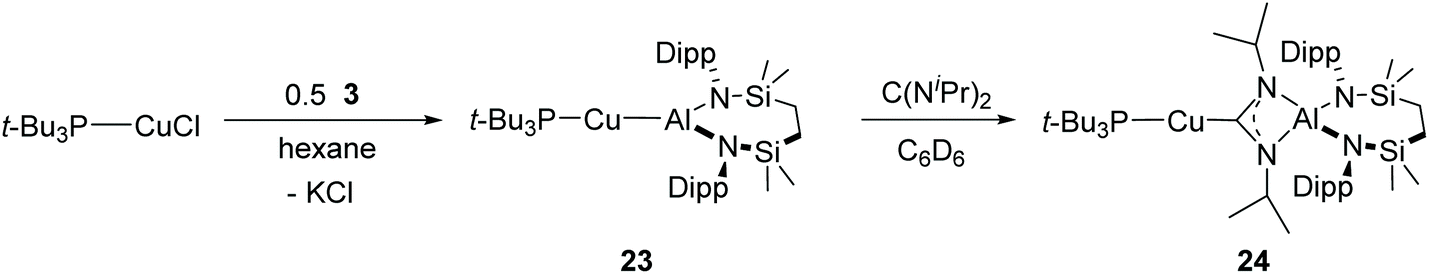 | ||
| Scheme 5 Synthesis of compounds 23 and 24. | ||
 | ||
| Fig. 4 ORTEPs (30% probability ellipsoids) of (a) compound 23 and (b) compound 24. H atoms and disordered atoms have been omitted for clarity. | ||
Compound 23 was also found to react rapidly with 13CO2 to provide compound 25, which was readily identified as a dioxocarbene species analogous to compounds 11, 12 and 15 (Scheme 5). Most characteristically, the 13C{1H} NMR spectrum displayed a low field doublet resonance, the chemical shift of which (δ 233.1 ppm, 2JPC = 73.9 Hz) was comparable to the {CuCO2} environments of the carbene-supported variants.43 Furthermore, a near equivalence of the copper centres within 25 and its structurally characterised diaminocarbene analogue, compound 24, was evidenced by the similarity of their phosphorus environments, both of which resonated at δ 59.8 ppm in their respective 31P{1H} NMR spectra. Single crystals of compound 25 suitable for X-ray diffraction analysis could not be obtained due to its slow solution decomposition at room temperature, which manifested most clearly in its 31P{1H} NMR spectrum through the appearance of further signals at 62.5 and 66.0 ppm. Heating of this solution with the maintenance of the atmosphere of CO2 overnight at 60 °C resulted in the complete disappearance of the 31P NMR signal assigned to compound 25. This process was accompanied by the emergence of a single copper phosphine species (27) observed as a resonance at δ 62.1 ppm, a chemical shift that is almost identical to that observed in Aldridge and co-workers’ spontaneously formed carbonate, [(NON)Al(O3C)Cu(Pt-Bu3)] (6-Cu, δ 62.5 ppm).45 Similarly, the disappearance after heating of 25 of the {CuCO2} signal at 233.1 ppm and the emergence of new sharp resonances at δ 166.7, 168.4 and 176.7 ppm in the corresponding 13C{1H} NMR spectrum are reminiscent of that arising from the {O3C} environment of 6-Cu (δ 170 ppm). Although we have not succeeded in isolating a pure bulk sample, we, therefore, assign compound 27 as the analogous copper carbonate, [(SiNDipp)Al(O3C)Cu(Pt-Bu3)], while the further species apparent at δ 66.0 ppm in the room temperature 31P NMR spectrum of 25 is tentatively attributed to the oxo derivative, [(SiNDipp)AlOCu(Pt-Bu3)] (26). This latter compound is analogous to the (Al–O–Cu)-bridged intermediate implicated computationally, but not spectroscopically identified, in Aldridge, Frenking and co-workers’ analysis of the formation of [(NON)Al(O2C)Cu(Pt-Bu3)] (6-Cu).45 The balance of probability, therefore, suggests that compound 27 is produced by an analogous route (Scheme 6).
 | ||
| Scheme 6 Synthesis of compound 25 and the proposed route to compound 27via compound 26. | ||
Quantum chemical studies
Characterisation of the E–M–Al (E = C, P; M = Cu, Ag, Au) interaction
With these synthetic insights in hand, the electronic structures of 7, 8, 16, 17, 23 and, for comparison, Aldridge co-workers’ 4-Cu, were initially probed with QTAIM analysis to discern how the identities of both co-ligand and alumanyl backbone perturb the nature of the central M–Al bond and, in turn, the respective nucleo- and electrophilicity of each centre (see the ESI‡ for full computational details). Table 3 details relevant bond critical point (BCP) data and atomic charges for the atoms involved in the M–E bonds and the M–Al bonds in these complexes.| Complex | BCP (A–B) | ρ(r) | ∇ 2 ρ(r) | H(r) | DI(A|B) | q A | q B | Δq (A–B) |
|---|---|---|---|---|---|---|---|---|
| 7 | Cu–Al | 0.061 | −0.045 | −0.030 | 0.752 | −0.323 | +1.817 | −2.140 |
| Cu–C | 0.105 | 0.275 | −0.039 | 0.734 | +0.622 | −0.9445 | ||
| 16 | Ag–Al | 0.060 | −0.028 | −0.029 | 0.719 | −0.428 | +1.874 | −2.302 |
| Ag–C | 0.086 | 0.214 | −0.025 | 0.665 | +0.670 | −1.098 | ||
| 17 | Au–Al | 0.066 | 0.049 | −0.029 | 0.618 | −0.720 | +2.080 | −2.800 |
| Au–C | 0.105 | 0.209 | −0.037 | 0.799 | +0.702 | −1.422 | ||
| 8 | Cu–Al | 0.059 | −0.061 | −0.028 | 0.762 | −0.212 | +1.735 | −1.947 |
| Cu–C | 0.106 | 0.272 | −0.040 | 0.812 | +0.325 | −0.537 | ||
| 23 | Cu–Al | 0.060 | −0.041 | −0.029 | 0.725 | −0.403 | +1.851 | −2.254 |
| Cu–P | 0.072 | 0.119 | −0.022 | 0.688 | +0.667 | −1.070 | ||
| 4-Cu | Al–O | 0.036 | 0.159 | −0.002 | 0.134 | +1.887 | −1.117 | +3.004 |
| Cu–Al | 0.060 | −0.044 | −0.030 | 0.723 | −0.406 | +1.887 | −2.292 | |
| Cu–P | 0.072 | 0.119 | −0.022 | 0.688 | +0.668 | −1.074 |
The electron densities associated with the M–Al BCPs are generally consistent across the trio of group 11 NHCiPr–M–Al(SiNDipp) complexes [ρ(r) = 0.061 (7), 0.060 (16), 0.066 (17) e bohr−3 ]. Moreover, the total energy density, H(r), is negative in each case, indicating a similarly stabilising M–Al interaction. The associated Laplacians [∇2ρ(r) = −0.045 (7), −0.028 (16) and +0.049 (17) e bohr−3], however, suggest that, while the Cu–Al and Ag–Al interactions are covalent in nature, the Au–Al interaction is non-covalent with a contraction of ρ(r) towards each nucleus. This latter inference is supported by the both the diagnostic V(r) < 2G(r),55 for which the Au–Al BCP of 17 meets the criterion to classify it as a non-covalent interaction, and the delocalisation indices, DI(A|B), which are indicative of a comparably lower bond order for the Au–Al [+0.618 (17)] linkage in comparison to the Ag–Al [+0.719 (16)] and Cu–Al [+0.752 (7)] interactions.56 The atomic charges, qCu = −0.323 (7), qAg = −0.428 (16) and qAu −0.720 (15), identify that Au is more electronegative in nature and thus lends itself to enhanced nucleophilic reactivity, a supposition that is further reinforced by inspection of Natural Localised Molecular Orbitals via NBO analysis, and the respective atomic contributions to each M–Al bonding orbital (see Table S6 in the ESI‡ for further details). While 7 [51.0% on Al and 45.5% on Cu] and 16 [53.1% on Al and 43.2% on Ag] have majority contributions arising from the Al centre, this is reversed in 17 [41.8% on Al and 55.5% on Au] where Au is the more significant contributor to the NLMO and so indicative of a greater degree of Mδ−–Alδ+ polarization across the trio of complexes.
Turning to a comparison of the four E–Cu–[Al] complexes (7, 8, 23 and 4-Cu), the identity of both the co-ligand and the alumanyl backbone can be seen to influence the electronic behaviour of the Cu centre, albeit to differing degrees. While all Cu–Al BCPs possess similar electron densities and Laplacians, subtle changes to the computed atomic charges arise across these complexes. Whereas the Cu atom of 8 bears the least negative charge (qCu = −0.202), the effect of co-ligand identity is highlighted by the marked step-change in qCu observed upon changing from NHCiPr in 7 (qCu = −0.323) to t-Bu3P in 23 (qCu = −0.403). Conversely, a negligible difference is observed in qCu between 23 and 4-Cu (qCu = −0.406), in which the [SiNDipp] alumanyl backbone is exchanged for [NON]. This latter observation infers that the identity of the co-ligand plays a much larger role in determining the Cu centre's electronic structure and potential nucleophilicity than the nature of the diamido backbone of the alumanyl ligand. Conversely, the adjustment in qAl between 23 (+1.887) and 4-Cu (+1.851) is more marked than the change in qCu, indicating that the Al(NON)-bound Cu centre is likely to display a higher degree of nucleophilic behaviour.
Reactivity of [(NHCiPr)MAl(SiNDipp)] (M = Cu, Ag, Au) with i-PrN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CNi-Pr and CO2
CNi-Pr and CO2
An initial thermodynamic analysis was undertaken via DFT at the BP86-D3BJ,C6H6/BS2//BP86/BS1 level of the reactions of the NHCiPr-supported group 11 alumanyl complexes, 7, 16 and 17, with CO2 and i-PrNCNi-Pr (Table 4). In all cases for the reactions with CO2, formation of the experimentally-observed “symmetric” dioxocarbene adducts (identified as “S” in Table 4) was calculated to be thermodynamically favoured over the asymmetric adduct “A”, [where ΔGS − ΔGA = −22.9, −23.5 and −33.6 for Cu (7), Ag (16) and Au (17), respectively]. This is in line with the observation of the dioxocarbene species 11, 21 and 22 and supports a uniform mode of CO2 activation irrespective of group 11 element identity. The exergonicity of dioxocarbene formation is larger with copper complex 7 over the Ag and Au analogues, with a smaller difference calculated between 16 and 17 (where ΔG = −19.3 < −11.4 < −10.8 kcal mol−1 for SCu, SAg and SAu, respectively). For i-PrNCNi-Pr, the symmetric insertion product was also thermodynamically favoured in all cases (where ΔG = −40.0, −31.6 and −32.1 kcal mol−1 for Cu, Ag and Au, respectively), even for Cu and Ag, where the characterisation of 9 and 19 implicated the alumanyl group as the nucleophilic reaction partner. This indicates that either the observed “A” product formation for Cu and Ag is not under thermodynamic control, where onwards exergonic A to S isomerisation is only kinetically accessible for [(NHCiPr)AuAl(SiNDipp)] (17), or A and S are formed through two regiodivergent pathways, whereby the kinetic dominance of pathways switches to S formation for 17. Consistent with the trend identified for CO2 insertion, i-PrNCNi-Pr activation is notably more exergonic for 7 over 16 and 17, (where ΔG = −40.0 < −31.6 < −32.1 kcal mol−1 for SCu, SAu and SAg, respectively).
CNi-Pr activation with compounds 7, 16 and 17 to form either the symmetric (“S” featuring a μ-η1(C):κ2(X,X′) binding mode) or asymmetric (“A” featuring a μ-η1(C):κ1(X):κ1(X′) binding mode) insertion products. Bold numbers indicate the experimentally observed product
| L | X (in XCX) |
Product | ΔG (kcal mol−1) | ||
|---|---|---|---|---|---|
| Cu (7) | Ag (16) | Au (17) | |||
| NHCiPr | O | A | +3.6 | +12.1 | +22.8 |
| S | −19.3 | −11.4 | −10.8 | ||
| i-PrN | A | −24.0 | −13.6 | −7.5 | |
| S | −40.0 | −31.6 | −32.1 | ||
Although profiles have also been computed for the carbodiimide-based processes (see Fig. S56 in the ESI‡), we here concentrate on the reactions with carbon dioxide. The computed free energy profile for the activation of CO2 at 7, which for comparison with the comparable Ag- and Au-derived reactions is relabelled as “ICu”, is shown in Fig. 5. From ICu, initial rotation of one of the i-Pr groups of the NHCiPr co-ligand is assumed to take place viaTS(I–II)Cu (+8.5 kcal mol−1) to yield conformer IICu (+1.5 kcal mol−1).57 Subsequent rate-limiting addition of CO2 can take place viaTS(II–III)Cu (ΔG = +14.8 kcal mol−1, relative to ICu and reactants) to yield IIICu (−4.2 kcal mol−1). Two divergent mechanistic pathways emerging from this intermediate were characterised. The facile and kinetically dominant pathway in which the “symmetric” and experimentally observed dioxocarbene species 11 is formed (relabelled “SCu” in the computed profile) proceeds via Cu–Al cleavage with concomitant reorientation viaTS(III–S)Cu (+4.5 kcal mol−1), and via an overall energetic span relative to IIIM of ΔG‡ = +8.7 kcal mol−1, to afford SCu (−19.3 kcal mol−1). Conversely, the thermodynamically disfavoured asymmetric adduct ACu (+3.6 kcal mol−1) forms via transition state TS(III–IV)Cu (+14.1 kcal mol−1) and subsequent conformational rearrangement of the resulting adduct, IVCu (+6.7 kcal mol−1), viaTS(IV–A)Cu (+10.8 kcal mol−1). This, therefore, indicates that the formation of dioxocarbene complex 11 is both kinetically and thermodynamically favoured in the activation of CO2 at 7. A similar qualitative picture of rate limiting CO2 addition emerges from the characterisation of CO2 activation by the Ag and Au analogues, 16 and 17 (labelled as IAg and IAu, respectively, in Fig. 5). In both cases, subsequent formation of SM (M = Ag, Au) from IIIM is again both kinetically and thermodynamically favoured over the formation of AM, confirming that changing the group 11 element has little qualitative impact on the activation mode of CO2.
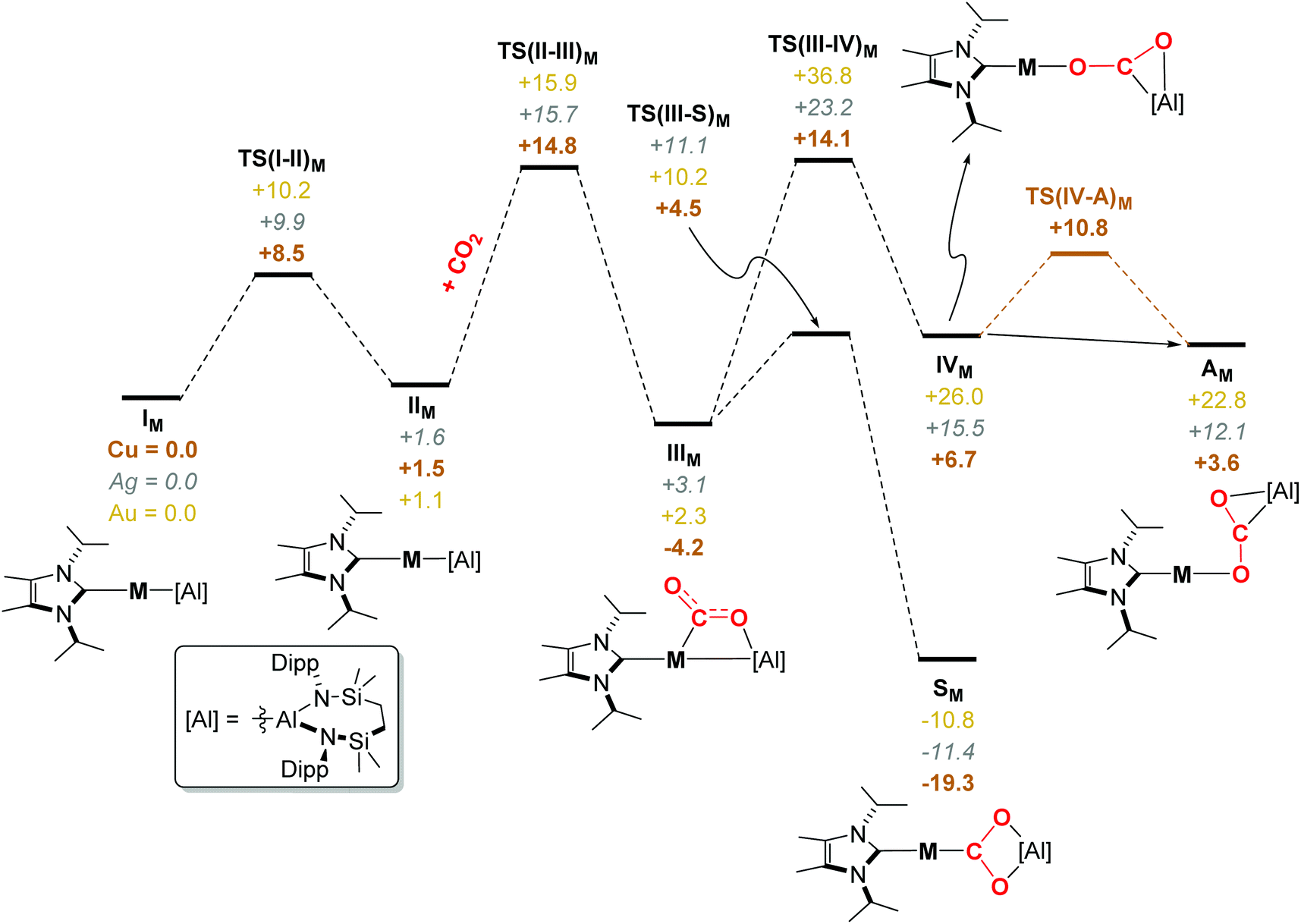 | ||
| Fig. 5 Computed free energy (kcal mol−1) profile of the addition and activation of CO2 at “IM”, [(NHCiPr)MAl(SiNDipp)], complexes, where M = Cu (7, copper brown, bold), Ag (16, silver, italics), Au (17, gold) at the BP86-D3BJ,C6H6/BS2//BP86/BS1 level. | ||
Computational analysis of CO extrusion
The observed uniformity of the spectral data across all the various copper, silver and gold alumanyl derivatives, indicate that only minor perturbations to the M–Al bonding are induced by changing the carbene or phosphine co-ligand. In contrast, the stability of the dioxocarbene derivatives resulting from the reactions of these compounds with CO2 appears to be subtly modulated by the identity of the (NON) and (SiNDipp) alumanyl environments. In order to further address these differences in observed CO extrusion reactivity between the various copper-alumanyl compounds, the thermodynamics of formation of the dioxocarbene derivatives, and the mechanisms of subsequent CO extrusion were computed for 7, 23, and 4-Cu.58Fig. 6 summarises the kinetics and thermodynamics of both CO2 addition and CO extrusion from the three corresponding dioxocarbene adducts. | ||
| Fig. 6 Free energy profile (kcal mol−1) of CO2 addition, dioxocarbene formation, and subsequent CO extrusion for L–Cu–[Al] complexes 7 (ICu, copper brown, bold), 23 (IP,H, green, italics) and 4-Cu (IP,A, gold). Energetic spans of CO extrusion reported in parenthesis are from the preceding adducts, SL,[Al]. | ||
While the disparity in the kinetic barriers toward CO2 addition between 23 (+12.1 kcal mol−1) and 7 (+14.8 kcal mol−1) is relatively minor, addition is kinetically more facile for 4-Cu with a barrier of +6.4 kcal mol−1. This observation infers that the nature of the alumanyl group has a larger influence on the kinetics of electrophile addition than the carbene or phosphine co-ligand. The previously discussed QTAIM analyses of compounds 7, 23 and 4-Cu, highlighted that the adjustment to the identity of the diamido backbone of the alumanyl group results in only subtle modulation of the Cu–Al BCP and qCu, and a modest change to qAl. On this basis, therefore, we suggest that the backbone influence could be more steric in nature, a deduction reinforced by inspection of the more accessible molecular surface of 4-Cu orthogonal to the Cu–Al bond vector in comparison to that of 23 (Fig. S55‡).
The energetic span of CO extrusion is notably higher for the system derived from 7 (+28.4 kcal mol−1 relative to SCu) than for 23 (+24.4 kcal mol−1, relative to SP,H). This is consistent with the observable CO extrusion reactivity from 25 at 60 °C and the contrasting stability of 11, highlighting the more significant influence exerted by the co-ligand in effecting this onwards reactivity from the dioxocarbene adducts. Indeed, a much smaller kinetic difference is characterised between the CO extrusion processes of 25 (where ΔΔG‡ = 0.2 kcal mol−1), which is qualitatively consistent with the observation that both resulting dioxocarbene adducts (SP,H and SP,A) are amenable to subsequent CO extrusion, and so confirms that the identity of the transition-metal's co-ligand exerts a greater influence on this aspect of reactivity than the backbone of the alumanyl anion.59
In conclusion, our observations suggest that variation in the steric demands of the supporting NHC impacts only marginally on the reactivity of the intermetallic {(NHC)Cu–Al} bond. In contrast to the previously reported copper-based chemistry, silver and gold alumanyl species supported by CAAC donors are more labile to reduction, albeit mechanical separation of a single crystal has allowed the first structural characterisation of an Ag–Al σ-bond. The ready accessibility of NHC-supported silver and gold alumanyl derivatives has enabled an assessment of the reactivity of the {(NHC)Ag–Al} and {(NHC)Au–Al} bonds with the representative heteroallenes, N,N′-di-isopropylcarbodiimide and carbon dioxide. In contrast to previously reported behaviour of [(NON)AlM(Pt-Bu3)] (M = Cu, Ag), the products of CO2 insertion are stable and isolable. Although NMR spectroscopic analysis argues that the introduction of a t-Bu3P co-ligand only induces a marginal perturbation to the {(SiNDipp)Al–Cu} interaction, the resultant product of CO2 insertion displays a resistance toward CO extrusion that is qualitatively intermediate between the NHC-supported {(SiNDipp)Al–C} and phosphine-coordinated {(NON)Al–Cu} species. These apparent subtleties are supported by our parallel computational study. The structures of both the neutral base and the alumanyl backbone impact upon the electronic character of all three group 11 metal centres. For copper in particular, however, the identity of the co-ligand exerts a more significant influence on the potential nucleophilicity of the group 11 atom than the structure of the alumanyl donor. Although these observations suggest that the influence of the diamidoalumanyl backbone is, thus, more kinetic/steric in nature, we opine that it is, as yet, imprudent to generalise too liberally. We are, thus, continuing to study the emergent behaviour of such metal-aluminium interactions with a wider variety of transition metal centres and complex types.
Data availability
See ESI‡ for complete synthetic, quantum chemical and crystallographic details. CCDC codes 2133123 (17), 2133122 (18), 2133127 (19), 2133126, (20), 2133125 (23) and 2133124 (24)‡ contain the supplementary crystallographic data for this paper.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the EPSRC (EP/R020752/1) for support of this research. This research made use of the Balena High Performance Computing (HPC) Service at the University of Bath.Notes and references
- C. Prasang and D. Scheschkewitz, Functional Molecular Silicon Compounds Ii: Low Oxidation States, 2014, vol. 156, pp. 1–47 Search PubMed.
- U. Schubert, Transition Met. Chem., 1991, 16, 136–144 CrossRef CAS.
- H. W. Lerner, Coord. Chem. Rev., 2005, 249, 781–798 CrossRef CAS.
- J. Y. Corey and J. Braddock-Wilking, Chem. Rev., 1999, 99, 175–292 CrossRef CAS PubMed.
- R. A. Gossage, J. Organomet. Chem., 2000, 608, 164–171 CrossRef CAS.
- K. Mochida, J. Synth. Org. Chem., Jpn., 1991, 49, 288–294 CrossRef CAS.
- G. L. Casty, T. D. Tilley, G. P. A. Yap and A. L. Rheingold, Organometallics, 1997, 16, 4746–4754 CrossRef CAS.
- G. M. de Lima, Quim. Nova, 2021, 44, 845–851 CAS.
- G. Albertin, S. Antoniutti, J. Castro, S. Garcia-Fontan and G. Zanardo, Organometallics, 2007, 26, 2918–2930 CrossRef CAS.
- G. Albertin, S. Antoniutti, A. Bacchi, G. Pelizzi and G. Zanardo, Organometallics, 2008, 27, 4407–4418 CrossRef CAS.
- H. F. Klein, J. Montag, U. Zucha, U. Florke and H. J. Haupt, Inorg. Chim. Acta, 1990, 177, 35–42 CrossRef CAS.
- M. Yamashita and K. Nozaki, Bull. Chem. Soc. Jpn., 2008, 81, 1377–1392 CrossRef CAS.
- M. Yamashita, Bull. Chem. Soc. Jpn., 2011, 84, 983–999 CrossRef CAS.
- G. J. Irvine, M. J. G. Lesley, T. B. Marder, N. C. Norman, C. R. Rice, E. G. Robins, W. R. Roper, G. R. Whittell and L. J. Wright, Chem. Rev., 1998, 98, 2685–2722 CrossRef CAS PubMed.
- Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113–115 CrossRef CAS PubMed.
- (a) I. A. I. Mkhalid, J. H. Bernard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (b) U. Kaur, K. Saha, S. Gayen and S. Ghosh, Coord. Chem. Rev., 2021, 446, 214106 CrossRef CAS.
- D. Hemming, R. Fritzemeier, S. A. Westcott, W. L. Santos and P. G. Steel, Chem. Soc. Rev., 2018, 47, 7477–7494 RSC.
- L. Dang, Z. Y. Lin and T. B. Marder, Chem. Commun., 2009, 3987–3995 RSC.
- J. F. Hu, M. Ferger, Z. Z. Shi and T. B. Marder, Chem. Soc. Rev., 2021, 50, 13129–13188 RSC.
- M. Brym, C. Jones, P. C. Junk and M. Kloth, Z. Anorg. Allg. Chem., 2006, 632, 1402–1404 CrossRef CAS.
- C. Jones, D. P. Mills, J. A. Platts and R. P. Rose, Inorg. Chem., 2006, 45, 3146–3148 CrossRef CAS PubMed.
- S. Aldridge, R. J. Baker, N. D. Coombs, C. Jones, R. P. Rose, A. Rossin and D. J. Willock, Dalton Trans., 2006, 3313–3320 RSC.
- S. P. Green, C. Jones, K. A. Lippert, D. P. Mills and A. Stasch, Inorg. Chem., 2006, 45, 7242–7251 CrossRef CAS PubMed.
- C. Jones, R. P. Rose and A. Stasch, Dalton Trans., 2007, 2997–2999 RSC.
- E. S. Schmidt, A. Jockisch and H. Schmidbaur, J. Am. Chem. Soc., 1999, 121, 9758–9759 CrossRef CAS.
- A. V. Protchenko, L. M. A. Saleh, D. Vidovic, D. Dange, C. Jones, P. Mountford and S. Aldridge, Chem. Commun., 2010, 46, 8546–8548 RSC.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed.
- N. Hara, T. Saito, K. Semba, N. Kuriakose, H. Zheng, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2018, 140, 7070–7073 CrossRef CAS PubMed.
- J. Hicks, A. Mansikkamaki, P. Vasko, J. M. Goicoechea and S. Aldridge, Nat. Chem., 2019, 11, 237–241 CrossRef CAS PubMed.
- R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489–1493 CrossRef CAS PubMed.
- R. J. Schwamm, M. P. Coles, M. S. Hill, M. F. Mahon, C. L. McMullin, N. A. Rajabi and A. S. S. Wilson, Angew. Chem., Int. Ed., 2020, 59, 3928–3932 CrossRef CAS PubMed.
- S. Kurumada, S. Takamori and M. Yamashita, Nat. Chem., 2020, 12, 36–39 CrossRef CAS PubMed.
- K. Sugita and M. Yamashita, Organometallics, 2020, 39, 2125–2129 CrossRef CAS.
- K. Sugita and M. Yamashita, Chem. – Eur. J., 2020, 26, 4520–4523 CrossRef CAS PubMed.
- K. Koshino and R. Kinjo, J. Am. Chem. Soc., 2020, 142, 9057–9062 CrossRef CAS PubMed.
- R. J. Schwamm, M. S. Hill, H. Y. Liu, M. F. Mahon, C. L. McMullin and N. A. Rajabi, Chem. – Eur. J., 2021, 27, 14971–14980 CrossRef CAS PubMed.
- M. J. Evans, M. D. Anker, C. L. McMullin, S. E. Neale and M. P. Coles, Angew. Chem., Int. Ed., 2021, 60, 22289–22292 CrossRef CAS PubMed.
- R. J. Schwamm, M. D. Anker, M. Lein, M. P. Coles and C. M. Fitchett, Angew. Chem., Int. Ed., 2018, 57, 5885–5887 CrossRef CAS PubMed.
- M. D. Anker, Y. Altaf, M. Lein and M. P. Coles, Dalton Trans., 2019, 48, 16588–16594 RSC.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CrossRef CAS.
- M. M. D. Roy, J. Hicks, P. Vasko, A. Heilmann, A. M. Baston, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 22301–22306 CrossRef CAS PubMed.
- H. Y. Liu, R. J. Schwamm, M. S. Hill, M. F. Mahon, C. L. McMullin and N. A. Rajabi, Angew. Chem., Int. Ed., 2021, 60, 14390–14393 CrossRef CAS PubMed.
- D. Sorbelli, L. Belpassi and P. Belanzoni, J. Am. Chem. Soc., 2021, 143, 14433–14437 CrossRef CAS PubMed.
- C. McManus, J. Hicks, X. L. Cui, L. L. Zhao, G. Frenking, J. M. Goicoechea and S. Aldridge, Chem. Sci., 2021, 12, 13458–13468 RSC.
- P. de Fremont, N. M. Scott, E. D. Stevens, T. Ramnial, O. C. Lightbody, C. L. B. Macdonald, J. A. C. Clyburne, C. D. Abernethy and S. P. Nolan, Organometallics, 2005, 24, 6301–6309 CrossRef CAS.
- P. de Fremont, N. M. Scott, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 2411–2418 CrossRef CAS.
- Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2007, 46, 6710–6713 CrossRef CAS.
- C. M. Zinser, F. Nahra, L. Falivene, M. Brill, D. B. Cordes, A. M. Z. Slawin, L. Cavallo, C. S. J. Cazin and S. P. Nolan, Chem. Commun., 2019, 55, 6799–6802 RSC.
- H. Braunschweig, W. C. Ewing, T. Kramer, J. D. Mattock, A. Vargas and C. Werner, Chem. – Eur. J., 2015, 21, 12347–12356 CrossRef CAS PubMed.
- B. Bertrand, A. S. Romanov, M. Brooks, J. Davis, C. Schmidt, I. Ott, M. O'Connell and M. Bochmann, Dalton Trans., 2017, 46, 15875–15887 RSC.
- A. S. Romanov and M. Bochmann, J. Organomet. Chem., 2017, 847, 114–120 CrossRef CAS.
- F. Chotard, A. S. Romanov, D. L. Hughes, M. Linnolahti and M. Bochmann, Eur. J. Inorg. Chem., 2019, 2019, 4234–4240 CrossRef CAS.
- R. Hamze, S. Shi, S. C. Kapper, D. S. M. Ravinson, L. Estergreen, M. C. Jung, A. C. Tadle, R. Haiges, P. I. Djurovich, J. L. Peltier, R. Jazzar, G. Bertrand, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2019, 141, 8616–8626 CrossRef CAS PubMed.
- P. S. V. Kumar, V. Raghavendra and V. Subramanian, J. Chem. Sci., 2016, 128, 1527–1536 CrossRef CAS.
- C. Outeiral, M. A. Vincent, Á. M. Pendás and P. L. A. Popelier, Chem. Sci., 2018, 9, 5517–5529 RSC.
- Isopropyl group rotation can potentially take place elsewhere in the profile, but characterisation of i-Pr rotation at all possible steps along the CO2 activation profile were deemed too time-consuming. For the sake of simplicity, this rotational process was assumed, and modelled to take place prior to CO2 addition, and can be seen to have a small enough barrier as to not interfere with the kinetics of CO2 addition and activation or infer that it is a rate-limiting process.
- Transition states for CO2 addition and CO extrusion of 4-Cu were adapted from ref. 45 and recomputed at the BP86-D3BJ,C6H6/BS2//BP86/BS1 level for better alignment with the computational methodology employed throughout.
- It must be noted that the energetic span for CO extrusion for 23 is marginally lower than for 4-Cu, which appears to be in conflict with the milder reaction conditions required to effect CO extrusion in 4-Cu (−78 °C). Functional testing the kinetics of this process indicates, that while most density functional approximations compute the difference barriers for these two CO extrusion processes to be small, the kinetically favoured process is inconsistent. See Table S7‡ for further details.
Footnotes |
| † Dedication: In celebration of the 70th birthday of our friend and colleague, Professor Paul Raithby. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2133122, 2133123, 2133124, 2133125, 2133126 and 2133127. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2dt00404f |
| This journal is © The Royal Society of Chemistry 2022 |