
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of halogenation of salicylaldehyde on the antiproliferative activities of {Δ/Λ-[Ru(bpy)2(X,Y-sal)]BF4} complexes†
Maryam
Taghizadeh Shool
a,
Hadi
Amiri Rudbari
 *a,
Tania
Gil-Antón
b,
José V.
Cuevas-Vicario
b,
Begoña
García
b,
Natalia
Busto
*bc,
Nakisa
Moini
d and
Olivier
Blacque
e
*a,
Tania
Gil-Antón
b,
José V.
Cuevas-Vicario
b,
Begoña
García
b,
Natalia
Busto
*bc,
Nakisa
Moini
d and
Olivier
Blacque
e
aDepartment of Chemistry, University of Isfahan, Isfahan 81746-73441, Iran. E-mail: h.a.rudbari@sci.ui.ac.ir; hamiri1358@gmail.com
bDepartamento de Química, Facultad de Ciencias, Universidad de Burgos, Plaza Misael Bañuelos s/n, 09001, Burgos, Spain. E-mail: nbusto@ubu.es
cDepartamento de Ciencias de la Salud, Facultad de Ciencias de la Salud, Universidad de Burgos, Hospital Militar, Paseo de los Comendadores, s/n, 09001 Burgos, Spain
dDepartment of Chemistry, Faculty of Physics and Chemistry Alzahra University, P.O. Box 1993891176, Vanak Tehran, Iran
eDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057, Zurich, Switzerland
First published on 20th April 2022
Abstract
Ru(II) polypyridyl complexes are widely used in biological fields, due to their physico-chemical and photophysical properties. In this paper, a series of new chiral Ru(II) polypyridyl complexes (1–5) with the general formula {Δ/Λ-[Ru(bpy)2(X,Y-sal)]BF4} (bpy = 2,2′-bipyridyl; X,Y-sal = 5-bromosalicylaldehyde (1), 3,5-dibromosalicylaldehyde (2), 5-chlorosalicylaldehyde (3), 3,5-dichlorosalicylaldehyde (4) and 3-bromo-5-chlorosalicylaldehy (5)) were synthesized and characterized by elemental analysis, FT-IR, and 1H/13C NMR spectroscopy. Also, the structures of complexes 1 and 5 were determined by X-ray crystallography; these results showed that the central Ru atom adopts a distorted octahedral coordination sphere with two bpy and one halogen-substituted salicylaldehyde. DFT and TD-DFT calculations have been performed to explain the excited states of these complexes. The singlet states with higher oscillator strength are correlated with the absorption signals and are mainly described as 1MLCT from the ruthenium centre to the bpy ligands. The lowest triplet states (T1) are described as 3MLCT from the ruthenium center to the salicylaldehyde ligand. The theoretical results are in good agreement with the observed unstructured band at around 520 nm for complexes 2, 4 and 5. Biological studies on human cancer cells revealed that dihalogenated ligands endow the Ru(II) complexes with enhanced cytotoxicity compared to monohalogenated ligands. In addition, as far as the type of halogen is concerned, bromine is the halogen that provides the highest cytotoxicity to the synthesized complexes. All complexes induce cell cycle arrest in G0/G1 and apoptosis, but only complexes bearing Br are able to provoke an increase in intracellular ROS levels and mitochondrial dysfunction.
Introduction
Chemotherapy is an effective and widespread way of cancer treatment in which one or more chemotherapeutic or alkylating agents are used. In the past few years, there has been great interest in the use of metal compounds for the treatment of diseases. Cisplatin (CDDP) is one of the best and first metal-based chemotherapeutic drugs.1,2 Nevertheless, cisplatin does not display its highest potential because of side effects, which include toxicity and drug resistance.3 Therefore, inorganic chemistry researchers have focused on the design, synthesis and investigation of the anticancer activity of complexes with other metal ions, such as Ru(II,III), Cu(II), Zn(II), Pd(II), Rh(III) etc.4–6In recent decades, ruthenium complexes have become an attractive option for biological application due to their distinct features such as (1) existence stable different oxidation states under biological conditions, (2) less toxicity due to higher selectivity of cancer cells toward healthy cells, and (3) ability to mimic iron in binding biomolecules such as transferrin and albumin because these proteins play a key role in the transport of metallodrugs and their receptors are largely overexpressed on the surface of malignant cells.7–10 Despite the synthesis of a large number of ruthenium complexes with anticancer properties, only a few of them, like NAMI-A and KP1019 (Fig. 1) have been employed in human clinical studies,11–13 probably due to their poor water solubility, insufficient effectiveness or ungovernable interaction with serum proteins.14 As a result, the synthesis of new Ru-based compounds is still necessary in order to improve the physicochemical properties (water solubility) and anticancer activity of complexes.15–17 The reactivities of transition metal coordination compounds often become controlled by the environment around the coordination sphere. Hence, polypyridines with multiple covalently bonded pyridine groups exhibit unique photophysical and redox properties.18,19 Bipyridine analogues not only function as supporting ligands stabilizing metal complexes, but also are utilized as photosensitizers20 and phosphorescent materials.21
 | ||
| Fig. 1 Chemical structures of NAMI-A, KP1019 and TLD-1433. | ||
For the first time, two Ru(II) polypyridyl complexes, [Ru(phen)3](ClO4)2 and [Ru(bpy)3](ClO4)2 have been biologically investigated in the 1950s.10 Ru(II) polypyridyl complexes have been widely investigated in cellular imaging, chemotherapy and photodynamic therapy due to their unique photochemical and photophysical properties, which can in turn be controlled by suitable variations of the auxiliary and primary ligands around the Ru(II) metal center.22,23 The relationship between the number of heteroatoms involved in the supporting ligand and the reactivity of the complex has been reported in a ruthenium complex containing bipyridine analogues.24 The investigation of the electronic properties of cyclometalated ruthenium polypyridyl has continued to be active for many years.25–27
Nowadays, the most attractive Ru(II) polypyridyl complex is the TLD-1433 compound (Fig. 1), that has recently entered phase II clinical studies for the treatment of nonmuscle invasive bladder cancer.14,28
To explore the structures of ruthenium complexes with salicylaldehyde derivatives, C. Chen et al. have studied the coordination modes of salicylaldehyde derivatives in the Ru(II) nitrosyl and Ru(II) bis(2,2′-bipyridine) complexes, with the cationic ruthenium complex [Ru(bpy)2(κ2-O,O-salCl)](PF6) being similar to ClByRu(3).29 The antitumoral and antimicrobial biological activity of some ruthenium carbonyl derivatives of the bis-(salicylaldehyde)phenylenediimine Schiff base ligand have been studied. The data showed that the complexes have the capacity of inhibiting the metabolic growth of the investigated bacteria to different extents, which may indicate their broad-spectrum properties, especially for the bipyridine derivative.30
Lastly, it is well-known that natural products contain halogens in their structures. Therefore, halogenation should be an invaluable approach for the structural modification of natural products for drug development.31 Thus, halogen atoms are widely used as substituents in medicinal chemistry which enhance the bioactivity and bioavailability of drugs through attractive intermolecular interaction (halogen bonding) between an electrophilic site on a halogen and a nucleophilic site of the molecule namely the lone pair of heteroatoms like N,O and S in proteins.32–34 Also, the introduction of halogens into the phenyl ring decreases drug metabolism.35–37 Therefore, halogen bonding is a powerful tool to design more effective medicinal compounds for medicinal chemistry. For instance, it has been recently described that in a series of Pd(II) complexes with halogen-substituted Schiff bases and 2-picolylamine, the number and types of halogens influence not only the chirality but also their cytotoxicity towards breast cancer cells.38 In contrast, Hartinger et al. found no significant differences as a function of halogens in the anticancer activity of piano stool Ru(II) complexes bearing 8-hydroxyquinoline.39
Recently we synthesized a series of Cu(diimine)(X,Y-sal)(NO3) complexes, where the diimine is either bpy or phen, sal is salicylaldehyde, and X and Y are Cl, Br, I and H. The data set showed the potential of these bpy derivatives for further in vivo studies.40
On the basis of these promising results, we synthesized such kinds of compounds with different metal ions. From the first attempt, based on the above description of the anticancer activity of the ruthenium compounds, we concluded on the one side that the ruthenium atom is the best alternative for the copper atom and, on the other side, the properties of the Ru(II)-bpy, Ru(II)-salicylaldehyde base derivatives and the introduction of halogens into the phenyl ring have prompted us to report the synthesis, structural characterization, and antibacterial and anticancer activity of this series of novel chiral {Δ/Λ-[Ru(bpy)2(X,Y-sal)]BF4} complexes (1–5), where X,Y-sal is monoanionic halogenated salicylaldehyde (X = Cl, Br and H; Y = Cl and Br).
Results and discussion
A series of chiral complexes Δ/Λ-[Ru(bpy)2(X,Y-sal)]BF4 were synthesized by reacting Ru(bpy)2Cl2 with halogen-substituted salicylaldehydes reported in the Experimental section. An outline of the synthesis route is presented in Scheme 1. The resulting brown-black complexes were analyzed by FT-IR, 1H NMR and 13C NMR spectroscopy, and elemental analysis and two of them, 1 and 5, were studied by X-ray diffraction analysis. Because of the geometrical arrangement of the bpy chelating ligands around the Ru(II) ion, the configuration at the Ru(II) metal center may be Δ or Λ (for more details see the X-ray structural analyses section). | ||
| Scheme 1 Synthetic route to the formation of Ru(bpy)2Cl2 and Δ/Λ-[Ru(bpy)2(X,Y-sal)]BF4 complexes. | ||
The IR spectra of the complexes exhibit common characteristic bands for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (aldehyde) and B–F (BF4). The stretching frequency for CO in compounds 1, 2, 3, 4 and 5 occurs at 1600.6, 1579.4, 1584.2, 1583.2 and 1578.4 cm−1, respectively.41 The main stretching frequency for the BF4 anion occurs at 1055.8, 1056.8, 1057.7, 1058.7 and 1054.8 cm−1 for compounds 1, 2, 3, 4 and 5, respectively.42
O (aldehyde) and B–F (BF4). The stretching frequency for CO in compounds 1, 2, 3, 4 and 5 occurs at 1600.6, 1579.4, 1584.2, 1583.2 and 1578.4 cm−1, respectively.41 The main stretching frequency for the BF4 anion occurs at 1055.8, 1056.8, 1057.7, 1058.7 and 1054.8 cm−1 for compounds 1, 2, 3, 4 and 5, respectively.42
All complexes showed well-defined 1H/13C NMR spectra (Fig. S1†), permitting the unambiguous identification and assessment of purity. In the 1H NMR spectra of the complexes, the aldehyde proton (CHO) from the halogen-substituted salicylaldehyde ligand gives rise to signals at 8.92, 9.04, 8.93, 9.05 and 9.04 ppm for 1, 2, 3, 4 and 5, respectively. In the range of 6.5–8.7 ppm the signals of the aromatic protons from the halogen-substituted salicylaldehyde ligand appear to be overlapped with those from the bpy ligand. The main modification observed in the 1H NMR spectra of the complexes in relation to that of the free salicylaldehyde ligand is the absence of a resonance at ∼10.90 ppm assigned to the proton of the phenol oxygen, indicating its deprotonation.43
The 13C NMR spectra show 27 signals for all complexes. The peak observed at ∼188.00 ppm is ascribed to the aldehyde carbon atom. The existence of this peak in the spectra of the complexes supports the presence of the salicylaldehyde ligand in the structure of the Ru(II) complexes. The peaks in the range of 104.6–169.9 ppm are assigned to the aromatic protons.
X-ray structural analyses
The formation of complexes 1 and 5 was also confirmed by single-crystal X-ray diffraction analysis (Fig. 2). Crystallographic data for these complexes are listed in Table 1, whereas selected bond lengths and angles are displayed in Table 2. | ||
Fig. 2 Molecular structure and atomic labeling scheme of the cationic part of compounds 1 (left) and 5 (right). Thermal ellipsoids are drawn at the 30% probability level, while the hydrogen size is arbitrary. Disordered water and ethanol molecules and the BF![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) counter ion are omitted for clarity. counter ion are omitted for clarity. | ||
| 1 | 5 | |
|---|---|---|
| Empirical formula | C54H42B2Br2F8N8O5Ru2 | C58H52B2Br2Cl2F8N8O7Ru2 |
| Formula weight | 1418.53 | 1579.55 |
| Temperature (K) | 298(2) | 160(1) |
| Wavelength (Å) | 0.71073 | 1.54184 |
| Crystal system | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a (Å) | 10.508(2) | 11.7796(7) |
| b (Å) | 12.625(3) | 12.0345(7) |
| c (Å) | 13.078(3) | 12.9612(8) |
| α (°) | 108.15(3) | 63.333(6) |
| β (°) | 107.82(3) | 70.416(6) |
| γ (°) | 94.36(3) | 76.745(5) |
| Volume (Å3) | 1541.1(8) | 1540.17(19) |
| Z/calculated density (g cm−3) | 1/1.529 | 1/1.703 |
| Absorption coefficient (mm−1) | 1.861 | 7.006 |
| F(000) | 702 | 786.0 |
| θ range (°) | 2.655 to 29.329 (θ) | 7.92 to 149.008 (2θ) |
| h; k; l ranges | −13,14; ±17; −18, 17 | ±14, −15, 12; −16, 15 |
| Reflections collected/unique | 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 927/8382 [R(int) = 0.0584] 927/8382 [R(int) = 0.0584] |
25964/6270 [Rint = 0.0855] |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 8382/0/379 | 6270/195/416 |
| Goodness-of-fit on F2 | 0.978 | 1.039 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0555/wR2 = 0.1611 | R 1 = 0.0583/wR2 = 0.1430 |
| R indices (all data) | R 1 = 0.0836/wR2 = 0.1795 | R 1 = 0.0960/wR2 = 0.1635 |
| Largest diff. peak and hole (e Å−3) | 1.091/−1.065 | 0.97/−0.93 |
| CCDC number | 2150468 | 2108989 |
| 1 | 5 | |||
|---|---|---|---|---|
| Experimental XRD | Calculated DFT | Experimental XRD | Calculated DFT | |
| Ru(1)–N(1) | 2.056(3) | 2.080 | 2.042(6) | 2.081 |
| Ru(1)–N(2) | 2.027(4) | 2.055 | 2.031(6) | 2.053 |
| Ru(1)–N(3) | 2.043(3) | 2.065 | 2.040(5) | 2.064 |
| Ru(1)–N(4) | 2.042(4) | 2.081 | 2.045(6) | 2.077 |
| Ru(1)–O(1) | 2.066(3) | 2.094 | 2.083(4) | 2.084 |
| Ru(1)–O(2) | 2.080(3) | 2.103 | 2.054(5) | 2.105 |
| N(1)–Ru(1)–N(4) | 177.08(15) | 175.92 | 175.2(2) | 176.20 |
| N(1)–Ru(1)–O(1) | 85.94(14) | 87.49 | 88.54(19) | 86.82 |
| N(1)–Ru(1)–O(2) | 95.05(13) | 95.79 | 95.0(2) | 95.50 |
| N(1)–Ru(1)–N(2) | 79.33(15) | 79.05 | 79.7(2) | 79.13 |
| N(3)–Ru(1)–N(2) | 89.21(15) | 91.69 | 88.8(2) | 93.05 |
| N(2)–Ru(1)–N(4) | 97.97(15) | 97.78 | 95.9(2) | 97.51 |
| N(2)–Ru(1)–O(1) | 88.17(14) | 88.95 | 91.87(18) | 87.77 |
| N(2)–Ru(1)–O(2) | 174.26(12) | 174.78 | 173.8(2) | 174.16 |
| N(3)–Ru(1)–N(1) | 99.66(14) | 98.53 | 98.4(2) | 99.37 |
| N(3)–Ru(1)–N(4) | 79.10(15) | 78.90 | 79.5(2) | 78.95 |
| N(3)–Ru(1)–O(1) | 173.27(13) | 173.96 | 173.1(2) | 173.80 |
| N(3)–Ru(1)–O(2) | 92.93(13) | 89.88 | 89.0(2) | 90.08 |
| N(4)–Ru(1)–O(1) | 95.12(15) | 95.05 | 93.5(2) | 94.85 |
| N(4)–Ru(1)–O(2) | 87.67(14) | 87.41 | 89.3(2) | 87.93 |
| O(1)–Ru(1)–O(2) | 90.26(13) | 90.01 | 91.10(17) | 89.66 |
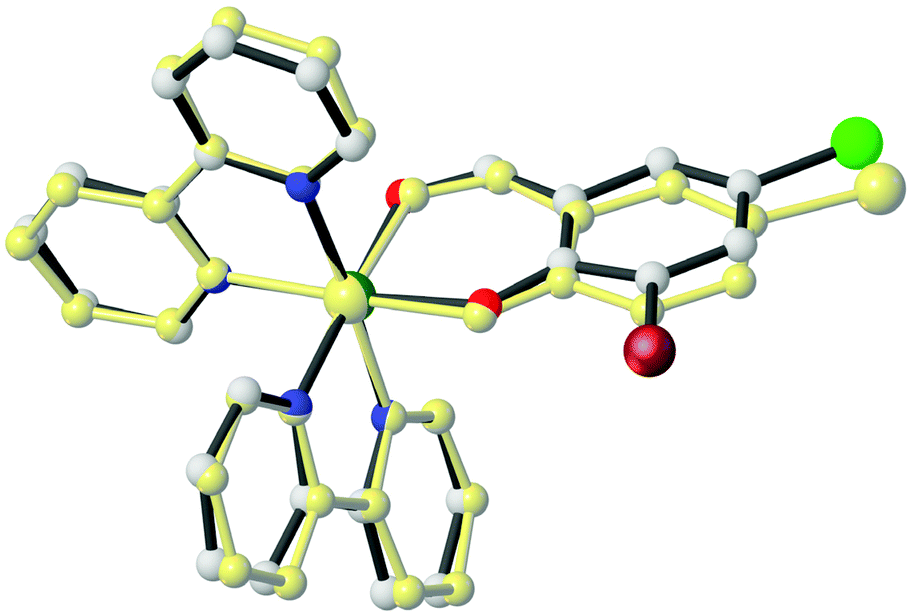
Both complexes, 1 and 5, have similar structures and crystallize in the space group P, with one enantiomer of the complex occupying the asymmetric unit.
In the cationic part of these complexes, the deprotonated aldehyde coordinates to the Ru(II) atom through the phenol-O and aldehyde-O atoms, forming a virtually planar six-membered chelate ring [maximum deviation from the least-squares plane = 0.053 Å (1) and 0.040 (5)], and two bidentate bpy co-ligands through their nitrogen atoms.44
The ruthenium atoms in both structures adopt a slightly distorted octahedral coordination geometry (Fig. 2). The Ru–Nbpy bond lengths are in the range of 2.027(4)–2.056(3) Å and 2.031(6)–2.045(6) Å for 1 and 5, respectively. The Ru–Oaldehyde bond lengths are 2.080(3) and 2.054(5) Å for 1 and 5, respectively, while the Ru–Ophenol bond lengths are 2.066(3) and 2.083(4) Å for 1 and 5, respectively. The Ru–N bond trans to the Ru–Ophenol bond (for 1: 2.043(3) Å; for 5: 2.040(5) Å) is longer than the Ru–N bond trans to the Ru–Oaldehyde bond (for 1: 2.027(4) Å; for 5: 2.031(6) Å). These results are consistent with the stronger trans influence of the Ophenol atom compared to that of the Oaldehyde atom.45
Because of the geometrical arrangement of the bpy chelating ligands and centrosymmetric space group (P for both complexes), the configuration at the Ru(II) metal center is Δ or Λ. Therefore, two enantiomers are possible such as Δ and Λ.46
The most noticeable distortion of the ideal octahedral geometry corresponds to the N–Ru–N bond angles, formed by the chelating bpy ligands, which are near 80° for both complexes (Table 2). These angles are shorter than ideal 90° found in a regular octahedron due to the geometrical requirements of the chelate rings formed by the bpy ligands.46
The similarity of both structures can be confirmed in the best way by overall conformation. As shown in Fig. 3, the dihedral angles between the three coordinating planes (N(1)–Ru(1)–N(2), N(3)–Ru(1)–N(4) and O(1)–Ru(1)–O(2)) in the two structures are slightly different. The dihedral angles are 93.956, 87.374 and 92.182° for complex 1 and 91.654, 89.335 and 91.172° for complex 5, respectively for planes O(1)–Ru(1)–O(2)/N(1)–Ru(1)–N(2), O(1)–Ru(1)–O(2)/N(3)–Ru(1)–N(4) and N(1)–Ru(1)–N(2)/N(3)–Ru(1)–N(4).
 | ||
| Fig. 3 A view of the structural overlap of cationic part of 1 and 5, having an RMSD deviation of 0.024 Å. Hydrogen atoms are omitted for clarity. | ||
Theoretical studies
Density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations for complexes 1–5 were performed to explain the photophysical properties of these complexes (see the Experimental section for details). As shown in Table 2, the comparison of the experimental XRD structures with the theoretically modeled complexes shows a good agreement between the bond distances, angles and torsional angles, validating the level of theory.Fig. 4 displays the energy levels and the isosurface contour plots of the selected frontier molecular orbitals for complex 1. The electronic structure of complexes 2–5 is very similar to the one calculated for complex 1 (see Fig. S3–S6†). In all of them the HOMO–LUMO gap is ranging between 3.10 and 3.13 eV (see Fig. S2†). In compound 1 (as a representative example) the HOMO is contributed by the orbitals of the Ruthenium atom (50.7%) and the salicylaldehyde ligand (39.3%) while the LUMO and LUMO+1 are mainly spread over the bpy ligands (see Table S1†) in a similar manner as it has been described for similar complexes47 or related complexes of ruthenium with bpy ligands and a chelating oxygen donor ligand.48,49
 | ||
| Fig. 4 Energy levels and isosurface contour plots (0.03 a.u.) for complex 1. | ||
TD-DFT calculations have been performed to explore the nature of the low-lying singlet and triplet states with the geometries of the ground state. Tables 3 and S2† summarize the calculated excited states. For complex 1, the absorption in the experimental spectra (Fig. 5A) appeared at 492 nm is assigned to the singlet excited state S8 (445.5 nm) and it is mainly a double transition from the HOMO−2 to the LUMO and to the LUMO+1 with a calculated oscillator strength of 0.1034 corresponding to a Metal-to-Ligand Charge Transfer (1MLCT) from the ruthenium center to the bpy ligands. Lower energy singlet excited states displayed very low values of the oscillator strength. In the same complex, the signal appeared at 363 nm is assigned to the singlet excited state S20 (343.7 nm, with a calculated oscillator strength of 0.0705), which is mainly a double transition from the HOMO−2 to the LUMO+3 and from the HOMO−1 to the LUMO+5. Both HOMO−2 and HOMO−1 show a high participation of atomic orbitals of the ruthenium center, and the LUMO+3 and the LUMO+5 are centered on the bpy ligands, therefore these transitions can be described as a Metal-to-Ligand charge Transfer (1MLCT). Similar results can be observed with complexes 2–5 as both the experimental absorption spectra and the calculated electronic structure of all of them are very close.
 | ||
| Fig. 5 (A) Absorption spectra of 20 μM of all complexes in DMSO. (B) Fluorescence spectra of all complexes (120 μM) in water at λex = 403 nm. | ||
| Complex | Estate | Energy (eV) | λ (nm) | f.osc. | Monoexcitations | Nature | Description |
|---|---|---|---|---|---|---|---|
| a Vertical excitation energies (E), dominant monoexcitations with contributions (within parentheses) of >15%, the nature of the electronic transition, and the description of the excited state are summarized. | |||||||
| 1 | S1 | 2.174 | 570.3 | 0.0067 | HOMO → LUMO (82) | dπ(Ru) + πsal → π*bpy | 1MLCT/1LLCT |
| S2 | 2.177 | 569.6 | 0.0041 | HOMO → LUMO+1 (74) | dπ(Ru) + πsal → π*bpy | 1MLCT/1LLCT | |
| S3 | 2.215 | 559.8 | 0.0168 | HOMO−1 → LUMO (81) | dπ(Ru) → π*bpy | 1MLCT | |
| S 8 | 2.782 | 445.5 | 0.1034 | HOMO−2 → LUMO (56) | dπ(Ru) → π*bpy | 1MLCT | |
| HOMO−2 → LUMO + 1 (27) | dπ(Ru) → π*bpy | 1MLCT | |||||
| S 20 | 3.607 | 343.7 | 0.0704 | HOMO−2 → LUMO + 3 (55) | dπ(Ru) → π*bpy | 1MLCT | |
| HOMO−1 → LUMO + 5 (27) | dπ(Ru) → π*bpy | 1MLCT | |||||
| T1 | 1.784 | 695.0 | HOMO−1 → LUMO + 2 (93) | dπ(Ru) → π*sal | 3MLCT | ||
| T2 | 1.796 | 690.2 | HOMO−1 → LUMO (54) | dπ(Ru) → π*bpy | 3MLCT | ||
| HOMO → LUMO (27) | dπ(Ru) + πsal → π*bpy | 3MLCT/3LLCT | |||||
| T3 | 1.842 | 673.0 | HOMO → LUMO + 1 (77) | dπ(Ru) + πsal → π*bpy | 3MLCT/3LLCT | ||
| T4 | 1.953 | 634.7 | HOMO → LUMO + 2 (73) | dπ(Ru) + πsal → π*sal | 3MLCT/3LC | ||
| T5 | 1.989 | 623.5 | HOMO−1 → LUMO (27) | dπ(Ru) → π*bpy | 3MLCT | ||
| HOMO−1 → LUMO + 1 (38) | dπ(Ru) → π*bpy | 3MLCT | |||||
| HOMO → LUMO (29) | dπ(Ru) + πsal → π*bpy | 3MLCT/3LLCT | |||||
The fluorescence emission spectra of all compounds are reported in Fig. 5B. The emission spectra of these complexes display important differences. Complexes bearing two halogen atoms on the salicylic ring (2, 4 and 5) feature a non-structured emission band at about 520 nm, while complexes with only one halogen atom (1 and 3) do not show any band in the same region. The first five calculated triplet excited states are reported in Tables 3 and S2.† In complexes 1 and 3, the triplet T1 lies 0.39 eV and 0.37 eV respectively lower than the corresponding singlet S1. This difference in the energy is bigger in the complexes featuring a second halogen atom on the salicylaldehyde ligand, with differences around 0.50 eV in these complexes. In complexes with only one halogen atom on the salicylaldehyde ligand (1 and 3) the triplets T1 and T3 are closer in energy (separated by 0.058 eV and 0.059 eV respectively) than those in the complexes with two halogen atoms in the salicylaldehyde ligand (with differences in the energy of 0.146 eV, 0.157 eV and 0.152 eV for 2, 4 and 5 respectively). In all cases, the first five triplet excited states are results of metal-to-ligand charge transfer from the ruthenium center to the bpy or to the salicylaldehyde ligands, along with some ligand–ligand charge transfer and ligand centered character. The excited states that display this ligand–ligand or ligand centered character are those in which there is an important participation of the HOMO as this molecular orbital is composed mainly of orbitals belonging to the ruthenium atom and to the salicylaldehyde ligand (see Table S2†). The lowest energy triplet state, T1, is described for all complexes as a Metal-to-Ligand Charge Transfer (3MLCT) from the ruthenium center to the salicylaldehyde ligand with the only one exception of complex 3 in which the same transition corresponds to the calculated exited state T3, with T1 for complex 3 being a combination of transitions from the HOMO to the LUMO and to the LUMO+1 (see Table S2†) that can be described as a 3MLCT from the ruthenium to the bpy ligands. The strong component of 3MLCT in the calculated triplet excited states is in good accordance with the observed unstructured band at around 520 nm observed for complexes 2, 4 and 5.
The geometries of the lowest triplet states T1 and T2 of complexes 1–5 were optimized using the spin-unrestricted DFT approach. After this geometry relaxation, the differences in the energy of each T1 and T2 state with their related S0 are calculated (adiabatic energy differences) and gathered in Table 4. In all cases, the calculated energy values are underestimated. The optimized triplet excited state of the lowest energy displays a spin-density distribution that supports the description of the TD-DFT calculations. For complexes 1, 2, 4 and 5, the spin-density distribution of the optimized T1 state is spread mainly over the salicylaldehyde ligand and the ruthenium center with values of spin densities for complex 1, as a representative example, of 0.77e for Ru and 1.20e for the salicylaldehyde ligand, in good agreement with the TD-DFT calculated T1 excited state (see Fig. 6 and Fig. S7† and Table 4 and Table S3†). Similarly, the second triplet state optimized is mainly spread over the bpy ligand and the ruthenium atom (0.84e for Ru and 1.01 for bpy ligand in complex 1 as example), in good agreement with the TD-DFT calculated T2 excited state. In the case of complex 3, the optimized T1 state displays an analogous spin-density distribution to the one described for the other complexes, but this state is represented by the excited state T3 in the TD-DFT calculations, as said above. As can be seen in Table 4 the difference in the energy between T1 and T3 for complex 3 is only 0.059 eV, and probably the apparent disorder of the triplet states in this complex arises from the fact that the triplet states calculated by TD-DFT are obtained over the geometry of the singlet ground state and the geometry of the triplet states is slightly modified.
 | ||
| Fig. 6 Spin-density contours (0.0008 a.u.) calculated for fully relaxed T1 (left) and T2 (right) states of complexes 1 (up) and 3 (down). | ||
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| Emission (experimental) | — | 2.38; 522 | — | 2.39; 518 | 2.37; 524 |
| Adiabatic T1–S0 | 1.68; 738 | 1.63; 761 | 1.67; 742 | 1.60; 775 | 1.58; 784 |
| Adiabatic T2–S0 | 1.82; 680 | 1.85; 669 | 1.81; 686 | 1.83; 676 | 1.84; 672 |
Stability in solution
The ligands and the Ru(II) complexes were firstly dissolved in DMSO. The stability of the synthesized complexes was evaluated in DMSO (Fig. S8†), in buffered aqueous solution (2.5 mM sodium cacodylate (NaCaC), pH = 7, 0.1% DMSO) (Fig. S9†) and under pseudophysiological conditions (2.5 mM NaCaC buffer with 0.1 M NaCl and pH = 7) by UV-vis measurements (Fig. S10†). All Ru(II) complexes were stable for 24 h in DMSO whereas some changes were observed in aqueous buffered solution for the dihalogenated complexes 2, 4 and 5. After 72 h of light-protection at room temperature, the bands around 360 and 480 nm decreased while a new band at 400 nm emerged. These changes occurred both in the absence and in the presence of 0.1 M NaCl. In order to shed some light on the possible cause of these changes, the absorption spectra of the dihalogenated ligands were recorded. All of them exhibited a band centered at 400 nm (Fig. S11†). Therefore, we hypothesized that the instability of the dihalogenated complexes in aqueous solution is due to the release of the salicylaldehyde ligand.To confirm this hypothesis, the evolution of complexes 1–5 in solution was monitored using NMR 1H spectroscopy. Initially, the stability of the complexes in DMSO was verified as no changes in the compounds were observed for 120 hours at room temperature (see Fig. S12A–E†). The influence of the presence of water in these solutions was studied by adding a small amount of deuterated water to the samples. As shown in Fig. S13A–E,† the complexes bearing two halogen atoms on the salicylaldehyde ligand (2, 4 and 5) undergo slight decomposition because we find the appearance of the signal of the free ligand at about 10 ppm. This decomposition process seems to be much slower in the complexes with only one halogen on the salicylaldehyde ligand (1 and 3), and can be attributed to the different steric hindrance of the halogen atom in comparison with the hydrogen atom.
Biological studies
Antibacterial activity
The antibacterial activity of the compounds was studied against pathogens of clinical interest endowed with a high rate of antibiotic resistance. The minimum inhibitory concentration (MIC), that is, the lowest concentration of the tested compounds which are able to inhibit bacterial growth, was determined against two Gram positive (vancomycin-resistant Enterococcus faecium and a methicillin – resistant Staphylococcus aureus) and two Gram negative (Acinetobacter baumannii and Pseudomonas aeruginosa) strains. The obtained results are collected in Table S4.† All the salicylaldehyde ligands and the metallic fragment ByRu are completely inactive. In contrast, all the complexes (with the exception of complex 3) are active only against Gram positive bacteria and, in general, they display a greater antibacterial activity against MRSA S. aureus than that against VR E. faecium. The lack of activity in Gram-negative strains may be related to the additional outer membrane that hinders their entrance into the cell.50 In comparison with the fluoroquinolone antibiotic Norfloxacin, the observed antimicrobial activities are low to deserve their study as antimicrobials.Cytotoxicity
The potential of the synthesized complexes as antitumoral agents is explored by evaluating their antiproliferative activity in a variety of cell lines: A549 (human lung adenocarcinoma), SW480 (colon adenocarcinoma) and A2780 (human ovarian carcinoma), and in the human embryonic kidney cell line (Hek293), after 72 h of treatment. The calculated IC50 values and the selectivity index (IC50healthy/IC50cancer) are collected in Table 5.| IC50 (μM) | SI = IC50, Hek293/IC50, A2780 | ||||
|---|---|---|---|---|---|
| A549 | SW480 | A2780 | Hek293 | ||
| CDDP | 3.5 ± 0.6 | 5.1 ± 0.6 | 4.0 ± 0.6 | 2.0 ± 0.3 | 0.5 |
| ClSal | >50 | >50 | >50 | >50 | — |
| BrSal | >50 | >50 | >50 | >50 | — |
| Cl 2 Sal | >50 | >50 | >50 | >50 | — |
| BrClSal | >50 | >50 | >50 | >50 | — |
| Br 2 Sal | >50 | >50 | >50 | >50 | — |
| ByRu | >50 | >50 | >50 | >50 | — |
| 1 | 6.5 ± 0.9 | 2.5 ± 0.5 | 2.7 ± 0.3 | 8.4 ± 0.4 | 3.1 |
| 2 | 1.3 ± 0.3 | 1.5 ± 0.2 | 0.8 ± 0.2 | 2.0 ± 0.1 | 2.5 |
| 3 | 7.9 ± 0.8 | 5.5 ± 0.5 | 3.3 ± 0.6 | 11.0 ± 1.0 | 3.3 |
| 4 | 2.8 ± 0.4 | 1.7 ± 0.3 | 0.7 ± 0.2 | 4.3 ± 0.6 | 6.1 |
| 5 | 2.3 ± 0.2 | 1.5 ± 0.2 | 0.8 ± 0.1 | 1.6 ± 0.5 | 2 |
Neither the ligands nor the metallic fragment (ByRu) are cytotoxic compounds. Interestingly, these Ru(II) complexes are less cytotoxic than previously studied Cu(bpy)(X-sal)(NO3) (X = Cl, Br, I or H).40 The cytotoxic potential of the Ru(II) complexes is influenced by both the number and the types of halogens in the salicylaldehyde ligand. Regarding the number of halogens, two exhibited higher cytotoxicity than one, and regarding the types of halogens, Br rendered the complex more cytotoxic than Cl. Interestingly, these results differ from those obtained for the Cu family where the monohalogenated complexes and those complexes bearing Cl as the halogen are the most cytotoxic derivatives.
On the other hand, if the selectivity index (SI) is considered, 4 is the most promising complex of the series since it displays the highest selectivity towards ovarian cancer cells (SI = 6.1). It seems that the increase in the cytotoxicity achieved with additional Br (in the position X of Scheme 1) is associated with a decrease in the selectivity of the Ru(II) complexes. Then, the cellular uptake of the Ru(II) metal complexes was studied by means of ICP-MS experiments. The collected results (Fig. 7) show that all the complexes are more internalized with cisplatin being the halogen key for the cellular accumulation since the complexes bearing Cl are less internalized than the Br-complexes.
 | ||
| Fig. 7 Cellular accumulation of the Ru(II) complexes in A549 cells treated with 2 μM of the studied complexes during 24 h. CDDP is included for comparison purposes. | ||
In order to shed some light on the mechanism of action of these Ru(II) complexes, images of the A549 cells treated with 10 μM of the Ru(II) complexes at different incubation times were recorded. After 17 h of treatment, important morphological changes compatible with apoptosis such as cell shrinkage, cytoplasmic vacuolization, membrane blebbing, and apoptotic body formation were observed (Fig. S14†).
Apoptosis studies
To properly confirm if these Ru(II) complexes are able to induce apoptosis, flow cytometry experiments with Annexin V-FITC/Propidium iodide double staining were performed. The collected results (Fig. 8) reveal that there is a greater percentage of cells in early apoptosis than in late apoptosis or necrosis for all the complexes except for the monohalogenated derivative 3. For this complex, the percentage of cells in early apoptosis (9.5%) is almost the same as the necrotic cells (10.1%). Anyway, all Ru(II) complexes induce apoptosis as a cell death mechanism in A549 cells.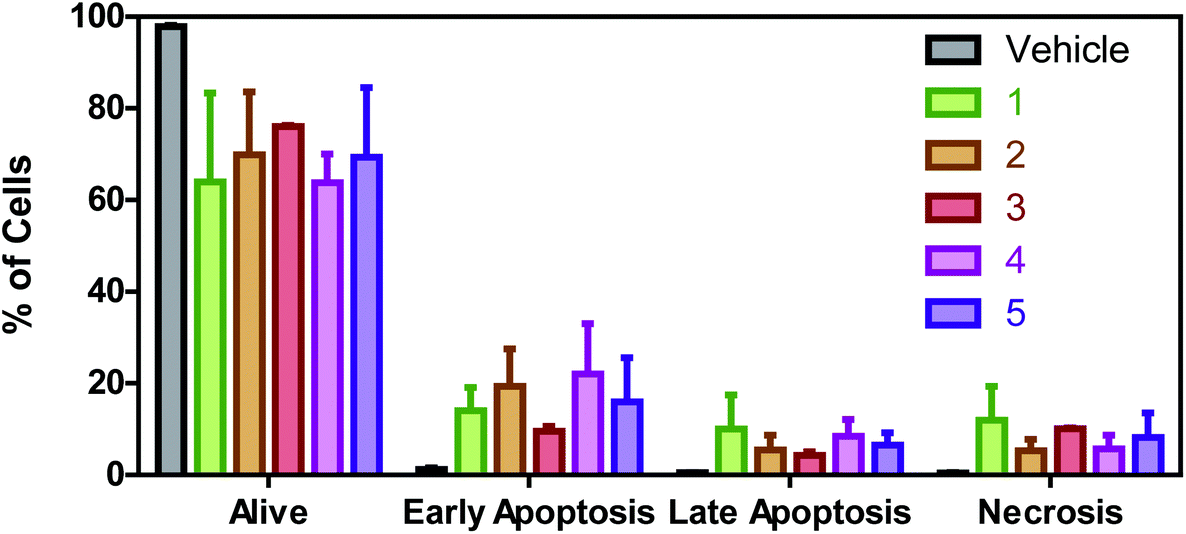 | ||
| Fig. 8 Percentage of A549 cells in alive, early apoptotic, late apoptotic and necrotic phases. Data were obtained from duplicates of two different flow cytometry experiments and plotted as mean ± SD (standard deviation). | ||
Once apoptosis induction is confirmed, we evaluate the cell cycle distribution of A549 cells treated with the Ru(II) complexes at their respective IC50 values by flow cytometry. An increase in the G0/G1 population along with a decrease in the percentage of cells in the S phase is observed due to the treatment with all the Ru(II) derivatives (Fig. 9). As for halogenation, it can be observed that Br induces a greater increase in the percentage of cells in G0/G1 than Cl. In addition, there is a reduction in the G2/M population for all the complexes except for monohalogenated 3.
 | ||
| Fig. 9 Cell cycle distribution of A549 cells treated with the Ru(II) complexes during 24 h. Data are expressed as mean ± SD (standard deviation). Statistical significance: ns – not significant, * – significant, ** – very significant and *** – extremely significant (ANOVA + Bonferroni). | ||
Since both apoptosis and cell cycle arrest at G0/G1 may be a consequence of an increase in reactive oxygen species (ROS) levels, the ability of these Ru(II) complexes to induce ROS production was investigated by fluorescence measurements with the probe 2′-7′-dichlorofluorescein diacetate (DCFH-DA).51 DCFH-DA gets into the cells by passive diffusion where it is hydrolyzed by esterases. Then, it is oxidized by ROS to yield fluorescent dichlorofluorescein. The fluorescence of the cells treated with the half maximal inhibitory concentration of the Ru(II) complexes was collected after 4 h of treatment. The variation in the emission intensity with respect to the untreated cells (corrected by the number of cells) is plotted in Fig. 10A and reflects the intracellular ROS levels. The halogen seems to influence ROS production since the presence of Br is essential for ROS generation as complexes without Br are not able to produce ROS. In addition, among the complexes bearing Br, the monohalogenated complex 1 is the least efficient as the ROS generator suggesting that the position of Br is a key issue for their biological activity.
 | ||
| Fig. 10 (A) Relative intracellular ROS levels with respect to untreated A549 cells after 4 h of treatment with the vehicle or the Ru(II) complexes at their IC50 values. (B) Relative MMP of A549 cells treated with the Ru(II) complexes with respect to untreated cells. Data were obtained from quadruplicates of two different experiments and plotted as mean ± SD. | ||
Mitochondrial membrane potential (MMP) is a key indicator of the mitochondrial bioenergetic state since it is the driving force for ATP production. On the other hand, mitochondrial activity is a prime source of endogenous ROS production and mitochondrial dysfunction may be responsible for the observed increase in ROS levels.52 Therefore, the effect of the Ru(II) complexes on the MMP was evaluated by means of the tetramethyl rhodamine methyl ester (TMRM) probe. The complexes bearing Br in the position X of Scheme 1 suffer mitochondrial depolarization since a substantial decrease in their MMP in comparison with untreated cells is observed (Fig. 10B).
Conclusions
To improve the anticancer activity of Ru(II) complexes, a series of new chiral Ru(II) polypyridyl complexes Δ/Λ-[Ru(bpy)2(X,Y-sal)]BF4 have been successfully synthesized and characterized. The excited states have also been characterized with the help of DFT and TD-DFT calculations. From the calculations it is found that the emission signals are described as Metal-to-Ligand Charge Transfers (3MLCT) from the ruthenium center to the salicylaldehyde ligand.The stability studies in solution have revealed that these complexes undergo decomposition with the release of the salicylaldehyde ligand being faster in dihalogenated than in monohalogenated complexes. In addition, it has been observed that halogenation is an important factor in the cytotoxicity of these complexes and in their mechanism of action. Indeed, dihalogenated complexes exhibit higher cytotoxicity than monohalogenated complexes and the type of halogen plays an important role. On the one side, complexes bearing chloride are more selective towards cancer cells than complexes bearing bromide. On the other side, bromide as an halogen is better than chloride in terms of half maximal inhibitory concentration in cancer cells. In fact, the presence of halogen is a decisive issue for their anticancer activity since complexes with Br are more internalized than complexes with Cl in cancer cells. Moreover, complexes bearing Br in the X position are not only the most cytotoxic complexes but also the most efficient derivatives in ROS generation along with an enhanced mitochondrion depolarization. In fact, oxidative stress and mitochondrial dysfunction seem to be the mechanism of action for this series of Ru(II) complexes.
Experimental
Chemicals and instrumentation
Ruthenium(III) chloride hydrate, lithium chloride, 2,2′-bipyridyl, silver tetrafluoroborate, triethylamine, 5-bromosalicylaldehyde, 3,5-dibromosalicylaldehyde, 5-chlorosalicylaldehyde, 3,5-dichlorosalicylaldehyde, and 3-bromo-5-chlorosalicylaldehyde were purchased from Sigma-Aldrich and used without purification. Ru(bpy)2Cl2 was prepared according to the literature procedure.53–55 The commercial solvents were distilled and then used for the preparation of ligands and complexes. The FT-IR spectrum was recorded on a JASCO, FT/IR-6300 spectrometer (4000–400 cm−1) in KBr pellets. Elemental analysis was performed on Leco, CHNS-932 and PerkinElmer 7300 DV elemental analyzers. 1H- and 13C-NMR spectra for the Ru(II) complexes were recorded on a Bruker Avance III 400 spectrometer using DMSO as the solvent at 20 °C.Synthesis of the complexes
A general synthetic route was used for all complexes, in which a solid sample of AgBF4 (2 mmol) was added to the solution of Ru(bpy)2Cl2 (1 mmol) in ethanol, and the mixture was stirred overnight at room temperature under an argon atmosphere, and then filtered to remove AgCl. Halogen-substituted salicylaldehyde (1 mmol) and one drop of Et3N in ethanol were slowly added to the filtered red solution and the solution was refluxed for 6 h under an argon atmosphere, and the solvent was removed by evaporation under reduced pressure. Finally, the dark red solid was dissolved in the lowest volume of chloroform and precipitated with n-hexane. The black crystals were obtained from ethanol solution in the refrigerator.O), 1055 (s, B–F). 1H-NMR data (CDCl3, 400 MHz): 8.92 (s, 1HAldehye), 8.67 (dd, 1HAr), 8.56 (dd, 1HAr), 8.48 (d, 1HAr), 8.43 (d, 1HAr), 8.35 (d, 1HAr), 8.29 (d, 1HAr), 8.09 (td, 1HAr), 8.03 (td, 1HAr), 7.78 (td, 1HAr), 7.74 (m, 2HAr), 7.62 (dd, 1HAr), 7.53 (m, 2HAr), 7.31 (d, 1HAr), 7.19 (dd, 1HAr), 7.13 (m, 2H), 6.50 (d, 1HAr). 13C-NMR data (100 MHz, CDCl3): 187.46 (CHO), 169.97, 159.13, 158.64, 157.87, 157.65, 153.31, 153.00, 150.06, 149.54, 138.74, 137.34, 136.79, 135.74, 135.38, 127.51, 126.57, 126.33, 125.76, 125.55, 123.45, 123.38, 123.30, 123.22, 122.74, 119.27, 105.58.
O), 1056 (s, B–F). 1H-NMR (CDCl3, 400 MHz): 9.04 (s, 1HAldehyde), 8.58 (td, 2HAr), 8.54 (d, 1HAr), 8.40 (d, 2HAr), 8.28 (d, 1HAr), 8.12 (td, 1HAr), 8.04 (td, 1HAr), 7.79 (m, 3HAr), 7.66 (d, 1HAr), 7.62 (d, 1HAr), 7.55 (m, 2HAr), 7.33 (d, 1HAr), 7.17 (td, 1HAr), 7.13 (td, 1HAr). 13C-NMR (100 MHz, CDCl3): 188.41 (CHO), 163.99, 159.13, 158.68, 157.88, 157.58, 153.53, 153.02, 149.78, 149.63, 140.15, 137.59, 137.21, 137.08, 135.95, 135.52, 126.72, 126.44, 125.83, 125.32, 123.63, 123.43, 123.27, 123.03, 122.95, 121.64, 104.62.
O), 1057 (s, B–F). 1H-NMR (CDCl3, 400 MHz): 8.93 (s, 1HAldehyde), 8.67 (d, 1HAr), 8.57 (d, 1HAr), 8.50 (d, 1HAr), 8.45 (d, 1HAr), 8.36 (d, 1HAr), 8.31 (d, 1HAr), 8.09 (td, 1HAr), 8.03 (td, 1HAr), 7.74 (m, 3HAr), 7.61 (d, 1HAr), 7.53 (m, 2HAr), 7.16 (d, 1HAr), 7.10 (m, 3HAr), 6.55 (d, 1HAr). 13C-NMR data (100 MHz, CDCl3): 187.44 (CHO), 169.78, 159.14, 158.66, 157.89, 157.67, 153.30, 152.98, 150.06, 149.54, 137.32, 136.77, 136.37, 135.73, 135.36, 133.92, 127.16, 126.54, 126.30, 125.75, 125.53, 123.45, 123.36, 123.29, 123.20, 122.43, 118.99.
O), 1058 (s, B–F). 1H-NMR data (CDCl3, 400 MHz): 9.05 (s, 1HAldehyde), 8.59 (t, 2HAr), 8.54 (d, 1HAr), 8.41 (dd, 2HAr), 8.27 (d, 1HAr), 8.12 (td, 1HAr), 8.04 (td, 1HAr), 7.79 (m, 3HAr), 7.66 (dd, 1HAr), 7.55 (m, 2HAr), 7.34 (d, 1HAr), 7.19 (m, 3HAr). 13C-NMR data (100 MHz, DMSO): 190.61 (CHO), 162.12, 158.97, 158.42, 157.33, 157.08, 153.67, 153.14, 149.98, 149.40, 137.58, 137.13, 135.77, 135.56, 134.19, 133.80, 129.17, 127.24, 126.62, 125.92, 125.67, 123.64, 123.58, 123.53, 123.33, 122.90, 116.01.
O), 1054 (s, B–F). 1H-NMR data (CDCl3, 400 MHz): 9.04 (s, 1HAldehyde), 8.58 (td, 2HAr), 8.54 (d, 1HAr), 8.40 (d, 2HAr), 8.28 (d, 1HAr), 8.12 (td, 1HAr), 8.04 (td, 1HAr), 7.79 (m, 3HAr), 7.66 (d, 1HAr), 7.55 (m, 3HAr), 7.19 (d, 1HAr), 7.15 (m, 2HAr). 13C-NMR data (100 MHz, CDCl3): 188.58 (CHO), 163.73, 159.18, 158.73, 157.86, 157.57, 153.48, 152.94, 149.78, 149.64, 137.90, 137.58, 137.11, 135.87, 135.54, 133.92, 126.73, 126.44, 125.76, 125.27, 123.55, 123.36, 123.27, 122.96, 122.02, 121.27, 118.43.
Single crystal X-ray details
X-ray data for 1 were collected on a STOE IPDS-II diffractometer with graphite monochromated Mo Kα radiation. Data were collected at 298(2) K in a series ω scans at 1° oscillations and integrated using the StöeX-AREA56 software package. A numerical absorption correction was applied using the X-RED and X-SHAPE57,58 software. The data were corrected for Lorentz and Polarizing effects. The structure was solved by direct methods using SIR2004.59 The non-hydrogen atoms were refined anisotropically by the full-matrix least-squares method on F2 using SHELXL.60Single-crystal X-ray diffraction data for 5 were collected at 160(1) K on a Rigaku OD XtaLAB Synergy, Dualflex, Pilatus 200 K diffractometer using a single wavelength X-ray source (Cu Kα radiation: l = 1.54184 Å) from a micro-focus sealed X-ray tube and an Oxford liquid-nitrogen Cryostream cooler. The selected suitable single crystal was mounted using polybutene oil on a flexible loop fixed on a goniometer head and immediately transferred to the diffractometer. Pre-experiments, data collection, data reduction and analytical absorption correction61 were performed with the program suite CrysAlisPro.62 Using Olex2,63 the structure was solved with the SHELXT64 small molecule structure solution program and refined with the SHELXL2018/3 program package65 by full-matrix least-squares minimization on F2. PLATON66 was used to check the result of the X-ray analysis. A solvent mask67 was used in Olex2 for structure 5 to take into account the residual electron density attributed to disordered solvent molecules of ethanol. Although they are not present in the final model, the formula moiety and the formula sum include the atoms of those molecules (two solvent molecules per cell) leading to many alerts in the checkCIF report.
For both structures, all hydrogen atoms were added at the ideal positions and constrained to ride on their parent atoms. Crystallographic data are listed in Table 1. Selected bond distances and angles are summarized in Table 2. More details concerning both crystal structures and their refinements can be found in the corresponding CIF files (ESI†).
Theoretical calculations
DFT and TD-DFT calculations were performed using Becke's three-parameter B3LYP exchange–correlation functional68,69 implemented ORCA 4.2.1.70,71 The basis sets used to define the atoms were LANL2DZ72 for Ru and def2-SVP73 for the other atoms. The empirical dispersion correction was taken into account using Grimme's dispersion with Becke–Johnson damping, D3BJ.74,75 The solvent (water) effects were considered within the self-consistent reaction field (SCRF) theory using the solvation model SMD of Trulhar et al.76 Time dependent DFT (TD-DFT)77–79 calculations of the lowest-lying 50 singlets were performed in the presence of the solvent for all complexes 1–5 with the minimum-energy geometry optimized for the ground state (S0).Stability studies
Fresh solutions of the synthesized complexes in DMSO were prepared. The absorbance spectra of 20 μM of the Ru(II) complexes were recorded at t = 0 and t = 24 h with an HP-8453 spectrophotometer (Agilent Technologies) fitted with a photodiode array detector and a Peltier thermostatic system at 25 °C. Similarly, the UV-vis spectra of 25 μM of the Ru(II) complexes in aqueous buffered solution (2.5 mM of sodium cacodylate, (CH3)2AsO2Na, named NaCaC, pH = 7.0) were also recorded as a function of time during 72 h. In order to mimic physiological conditions spectra were also recorded in a pseudo-physiological buffer (2.5 mM NaCaC, 0.1 M NaCl, pH = 7.0). Finally, the UV-spectra of 20 μM of the dihalogenated salicylaldehyde ligands in aqueous buffered solution were also recorded.For NMR 1H measurements, 5 mg of the compound was dissolved in 0.5 ml of DMSO-d6. The NMR 1H spectra were regularly registered in order to test the variations of the spectra. The influence of the presence of water was studied adding 0.1 ml of D2O to the samples and then, the evolution of the samples with time was monitored registering the NMR 1H spectra.
Antibacterial activity
S. aureus CECT 5190 (methicillin resistant, Gram positive bacteria), A. baumannii ATCC 17978 (Gram negative bacteria) and P. aeruginosa PAO1 (Gram negative bacteria) were maintained at 37 °C in Mueller-Hinton (MH) broth or agar while E. faecium CECT 5253 (vancomycin resistant, Gram positive bacteria) was maintained in Tryptic Soy. The broth microdilution plate method according to CLSI criteria80 was performed as previously described81 to evaluate the antibacterial activity of the synthesized complexes. The well-known fluoroquinolone antibiotic Norfloxacin was included as positive control for comparison purposes. The reported minimum inhibitory concentrations (MIC) are the mean values of three independent experiments with two replicates.Cell culture
The human lung carcinoma (A549), colon adenocarcinoma (SW480), ovarian carcinoma (A2780) and embryonic kidney (Hek293) cell lines were obtained from the European Collection of Cell Cultures (EACC). A549 and SW480 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM), whereas Hek293 cells were cultured in Eagle's Minimum Essential Medium (EMEM) supplemented with 1% of non-essential amino acids and A2780 (ovarian carcinoma) cells were cultured in RPMI-1640. All media (DMEM, EMEM and RPMI) were supplemented with 10% fetal bovine serum (FBS) and 1% amphotericin–penicillin–streptomycin solution. All reagents were purchased from Sigma Aldrich. The cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2.MTT antiproliferative assay
Approximately 3 × 103 A549, 5 × 103 SW480, 1 × 104 A2780 and Hek293 cells per well were seeded in 200 μL of their culture medium in 96-well plates and incubated for 24 h at 37 °C under a 5% CO2 atmosphere. Then, the cells were treated with different concentrations of the complexes under study for 72 h. Afterwards, the medium was removed and 5 mg ml−1 of MTT (3-(4,5-dimethylthiazol- 2-yl)-2,5-diphenyltetrazolium bromide) in culture medium was added. After 4 h of incubation, the formazan crystals were dissolved by overnight incubation with the solubilized solution (10% SDS and 0.01 M HCl). Finally, the absorbance was read at 590 nm on a microplate reader (Cytation 5 Cell Imaging Multi-Mode Reader-Biotek Instruments, USA). Four replicates per dose were included and at least two independent experiments were performed for the calculation of the half-maximal inhibitory concentration (IC50) values by means of the GraphPadPrism Software Inc. (version 6.01) (USA).In all the experiments, Cisplatin (CDDP) was included as positive control for comparison purposes.
Cellular uptake by ICP-MS
A549 cells were seeded in 6-well plates at a density of 1 × 106 cells per well and incubated at 37 °C with 5% CO2. After 24 h of incubation, the cells were treated with 2 μM of cisplatin and the synthesized complexes for 24 h. Then, the medium was removed and the cells were washed twice with DPBS (Dulbecco's Phosphate Buffered Saline) and harvested and centrifuged. The pellets were resuspended in 1 mL of DPBS and 10 μL of the resuspended cells were used to count cells in an automated cell counter (TC20 – Biorad). Then, the samples were digested with 65% HNO3 at room temperature for 24 h for ICP-MS. Finally, the diluted sample solutions (2% HNO3) were analyzed using a 8900 Triple Quadrupole ICP-MS (Agilent Technologies). Data are reported as the mean with the standard deviation of two independent experiment with two replicates per compound.Apoptosis studies
Apoptosis was evaluated by flow cytometry with an Annexin V:FITC Assay Kit (Biorad). According to the manufacturer's instructions, 2 × 105 A549 cells were seeded in 2 mL of cell culture medium in 12 well plates. After 24 h of incubation, the cells were treated with the half maximal inhibitory concentration of each Ru(II) complex for 24 h. Afterwards, the cells were washed with cold PBS, harvested, and resuspended in binding buffer. Then, the cells were doubly stained with the Annexin V:FITC conjugate and propidium iodide (PI). Immediately after PI addition, the cells were injected in a NovoCyte Flow cytometer (ACEA Biosciences, Inc., USA). 10000 events were counted and analyzed by using the NovoExpress 1.4.0 Software. Two replicates and two independent experiments were performed.
Cell cycle
A549 cells were seeded at a density of 1.5 × 105 cells per well in 6 well plates and incubated for 24 h. Then, the cells were treated with the half maximal inhibitory concentration of each Ru(II) complex for 24 h. Cells without any treatment and cells treated only with the vehicle (0.5% DMSO) were included as controls. After 24 h, the media were removed and the cells were harvested and resuspended in cold PBS for overnight fixation at 4 °C with 70% EtOH. Then, the samples were treated with the staining solution (0.1 mg mL−1 PI, 0.1 mM EDTA and 0.1% Triton- × 100 and 2 mg mL−1 of RNAse) and kept in ice and protected from light for 30 min. Finally, the cells were injected in a NovoCyte Flow cytometer (ACEA Biosciences, Inc., USA) and cell cycle distribution was evaluated by using the NovoExpress 1.4.0 Software. Two independent experiments with two replicates were performed.ROS generation
Intracellular ROS generation was evaluated by fluorescence measurements on a microplate reader (Cytation 5 Cell Imaging Multi-Mode Reader -Biotek Instruments, USA). A549 cells seeded at a density of 3 × 104 cells per well and incubated for 24 h in a clear bottom black side 96 well plate (Costar) were treated with 25 μM of H2DCFDA (2′-7′-Dichlorofluorescein diacetate) in DMEM without phenol-red and incubated for 30 min. Afterwards, the cells were treated with the vehicle, the Ru(II) complexes at their IC50 value and 20 μM of TBH (tertbutyl hydroperoxide) as positive control. After 4 h of treatment, the cells were washed twice with DPBS and emission was measured at λem = 530 nm and λexc = 490 nm. The collected results were corrected by the number of cells and the relationship between the treated and not treated cells was analysed. Two independent experiments with 4 replicates per treatment were performed.Mitochondrial membrane potential (MMP) assay by TMRM
Changes in the mitochondrial membrane potential of A549 cells treated with the synthesized complexes were evaluated by using the probe tetramethyl rhodamine methyl ester (TMRM) on a microplate reader (Cytation 5 Cell Imaging Multi-Mode Reader-Biotek Instruments, USA) according to a protocol previously described.82Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are grateful to the Research Council of the University of Isfahan (Iran) for financial support of this work. Financial support from La Caixa Foundation (LCF/PR/PR12/11070003), Ministerio de Ciencia, Innovación y Universidades (Grant PID2019-111215RB-I00 and RTI2018-102040-B-100) and Consejería de Educación, Junta de Castilla y León, FEDER (BU305P18) is gratefully acknowledged.We are indebted to M. Mansilla (PCT of the Universidad de Burgos) for the technical support.
This research has made use of the high-performance computing resources of the Castilla y León Supercomputing Center (SCAYLE, https://www.scayle.es), financed by FEDER (Fondo Europeo de Desarrollo Regional).
References
- B. Rosenberg, L. VanCamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385 CrossRef CAS PubMed.
- S. Ghosh, Bioorg. Chem., 2019, 88, 102925 CrossRef CAS PubMed.
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645 RSC.
- U. Das, B. Kar, S. Petea and P. Paira, Dalton Trans., 2021, 50, 11259 RSC.
- P. Jia, R. Ouyang, P. Cao, X. Tong, X. Zhou, T. Lei, Y. Zhao, N. Guo, H. Chang, Y. Miao and S. Zhou, J. Coord. Chem., 2017, 70, 2175 CrossRef CAS.
- M. Mital and Z. Ziora, Coord. Chem. Rev., 2018, 375, 434 CrossRef CAS.
- O. A. Lenis-Rojas, C. Roma-Rodrigues, A. R. Fernandes, F. Marques, D. Perez-Fernandez, J. Guerra-Varela, L. Sanchez, D. Vazquez-Garcia, M. Lopez-Torres, A. Fernandez and J. J. Fernandez, Inorg. Chem., 2017, 56, 7127 CrossRef CAS PubMed.
- D. K. S. Sales, L. M. T. Simplício, C. D. S. da Silva, C. M. B. Enju, V. B. Silva, T. de F. Paulo, I. P. Santos, H. C. Quadros, C. S. Meira, M. B. P. Soares, L. G. de F. Lopes, E. H. S. de Sousa and D. S. de Sá, Inorg. Chim. Acta, 2021, 516, 120125 CrossRef CAS.
- S. Parveen, Appl. Organomet. Chem., 2020, 34, e5687 CrossRef CAS.
- A. Notaro and G. Gasser, Chem. Soc. Rev., 2017, 46, 7317 RSC.
- S. Leijen, S. A. Burgers, P. Baas, D. Pluim, M. Tibben, E. van Werkhoven, E. Alessio, G. Sava, J. H. Beijenn and J. H. Schellens, Invest. New Drugs, 2015, 33, 201 CrossRef CAS PubMed.
- H. A. Burris, S. Bakewell, J. C. Bendell, J. Infante, S. F. Jones, D. R. Spigel, G. j. Weiss, R. K. Ramanathan, A. Ogden and D. Von Hoff, ESMO Open, 2016, 1, e000154 CrossRef PubMed.
- S. Monro, K. L. Colon, H. Yin, J. Roque III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2018, 119, 797 CrossRef PubMed.
- S. Thota, D. A. Rodrigues, D. C. Crans and E. J. Barreiro, J. Med. Chem., 2018, 61, 5805 CrossRef CAS PubMed.
- J. M. Rademaker-Lakhai, D. Van Den Bongard, D. Pluim, J. H. Beijnen and J. H. Schellens, Clin. Cancer Res., 2004, 10, 3717 CrossRef CAS PubMed.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodivers., 2008, 5, 2140 CrossRef CAS PubMed.
- E. Alessio and L. Messori, Molecules, 2019, 24, 1995 CrossRef CAS PubMed.
- K. Akatsuka, R. Abe, T. Takase and D. Oyama, Molecules, 2019, 25, 27 CrossRef PubMed.
- A. Winter, S. Hoeppener, G. R. Newkome and U. S. Schubert, Adv. Mater., 2011, 23, 3484 CrossRef CAS PubMed.
- A. Kumar, P. Kumar, M. S. Aathira, D. P. Singh, B. Behera and S. L. Jain, J. Ind. Eng. Chem., 2018, 61, 381 CrossRef CAS.
- A. Son, A. Kawasaki, D. Hara, T. Ito and K. Tanabe, Chem. – Eur. J., 2015, 21, 2527 CrossRef CAS PubMed.
- S. Mehanna, N. Mansour, H. Audi, K. Bodman-Smith, M. A. Mroueh, R. L. Taleb and R. S. Khnayzer, RSC Adv., 2019, 9, 17254 RSC.
- N. Soliman, G. Gasser and C. M. Thomas, Adv. Mater., 2020, 32, 2003294 CrossRef CAS PubMed.
- D. L. Ashford, C. R. Glasson, M. R. Norris, J. J. Concepcion, S. Keinan, M. K. Brennaman, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2014, 53, 5637 CrossRef CAS PubMed.
- K. Akatsuka, R. Abe, T. Takase and D. Oyama, Molecules, 2019, 25, 27 CrossRef PubMed.
- T. Funaki, H. Funakoshi, O. Kitao, N. Onozawa-Komatsuzaki, K. Kasuga, K. Sayama and H. Sugihara, Angew. Chem., Int. Ed., 2012, 51, 7528 CrossRef CAS PubMed.
- N. R. Palepu, S. L. Nongbri, J. R. Premkumar, A. K. Verma, K. Bhattacharjee, S. R. Joshi, S. Forbes, Y. Mozharivskyj, R. Thounaojam, K. Aguan and M. R. Kollipara, J. Biol. Inorg. Chem., 2015, 20, 619 CrossRef CAS PubMed.
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727 CrossRef CAS PubMed.
- C. Chen, J. Ji, C. J. Wang, A. Q. Jia and Q. F. Zhang, J. Coord. Chem., 2020, 73, 1306 CrossRef CAS.
- R. M. Ramadan, A. K. A. Al-Nasr and O. A. Ali, J. Mol. Struct., 2018, 1161, 100 CrossRef CAS.
- Z. Xu, Z. Yang, Y. Liu, Y. Lu, K. Chen and W. Zhu, J. Chem. Inf. Model., 2014, 54, 69 CrossRef CAS PubMed.
- J. Latham, E. Brandenburger, S. A. Shepherd, B. R. Menon and J. Micklefield, Chem. Rev., 2018, 118, 232 CrossRef CAS PubMed.
- R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, J. Med. Chem., 2013, 56, 1363 CrossRef CAS PubMed.
- M. R. Scholfield, C. M. V. Zanden, M. Carter and P. S. Ho, Protein Sci., 2013, 22, 139 CrossRef CAS PubMed.
- H. Sun, C. E. Keefer and D. O. Scott, Drug Metab. Lett., 2011, 5, 232 CrossRef CAS PubMed.
- P. F. Wang, A. Neiner, T. R. Lane, K. M. Zorn, S. Ekins and E. D. Kharasch, Mol. Pharm., 2018, 16, 898 CrossRef PubMed.
- N. Kordestani, H. A. Rudbari, Z. Fateminia, G. Caljon, L. Maes, P. G. Mineo and N. Micale, Appl. Organomet. Chem., 2021, 35, 6079 CrossRef.
- N. Kordestani, H. A. Rudbari, I. Correia, A. Valente, L. Côrte-Real, M. K. Islam, N. Micale, J. D. Braun, D. E. Herbert, N. Tumanov, J. Wouters and M. Enamullah, New J. Chem., 2021, 45, 9163 RSC.
- M. Kubanik, H. Holtkamp, T. Sohnel, S. M. Jamieson and C. G. Hartinger, Organometallics, 2015, 34, 5658 CrossRef CAS.
- N. Kordestani, H. A. Rudbari, A. R. Fernandes, L. R. Raposo, P. V. Baptista, D. Ferreira, G. Bruno, G. Bella, R. Scopelliti, J. D. Braun, D. E. Herbert and O. Blacque, ACS Comb. Sci., 2020, 22, 89 CrossRef CAS PubMed.
- X. S. Tai, P. F. Li, X. Wang and L. L. Liu, Bull. Chem. React. Eng. Catal., 2017, 12, 364 CrossRef CAS.
- D. M. Cropek, A. Metz, A. M. Müller, H. B. Gray, T. Horne, D. C. Horton, O. Poluektov, D. M. Tiede, R. T. Weber, W. L. Jarrett, J. D. Phillips and A. A. Holder, Dalton Trans., 2012, 41, 13060 RSC.
- A. C. de Melo, J. M. Santana, K. J. Nunes, B. L. Rodrigues, N. Castilho, P. Gabriel, A. H. Moraes, M. D. A. Marques, G. A. de Oliveira, Í. P. de Souza, H. Terenzi and E. C. Pereira-Maia, Molecules, 2019, 24, 2154 CrossRef CAS PubMed.
- A. Mori, T. Suzuki and K. Nakajima, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2015, 71, 142 CrossRef CAS PubMed.
- T. Suzuki, T. Kuchiyama, S. Kishi, S. Kaizaki, H. D. Takagi and M. Kato, Inorg. Chem., 2003, 42, 785 CrossRef CAS PubMed.
- O. A. Lenis-Rojas, C. Roma-Rodrigues, A. R. Fernandes, F. Marques, D. Perez-Fernandez, J. Guerra-Varela, L. Sanchez, D. Vazquez-Garcia, M. Lopez-Torres, A. Fernandez and J. J. Fernandez, Inorg. Chem., 2017, 56, 7127 CrossRef CAS PubMed.
- V. L. Constantino, H. Toma, L. C. de Oliveira, F. Rein, R. Rocha and D. O. Silva, J. Chem. Soc., Dalton Trans., 1999, 11, 1735 RSC.
- A. Das, T. M. Scherer, S. M. Mobin, W. Kaim and G. K. Lahiri, Inorg. Chem., 2012, 51, 4390 CrossRef CAS PubMed.
- L. Zeng, D. Sirbu, P. G. Waddell, N. V. Tkachenko, M. R. Probert and A. C. Benniston, Dalton Trans., 2020, 49, 13243 RSC.
- M. G. Page, Antibiot. Resist., 2012, 211, 67 CAS.
- A. Wojtala, M. Bonora, D. Malinska, P. Pinton, J. Duszynski and M. R. Wieckowski, Methods Enzymol., 2014, 542, 243 CAS.
- J. Ježek, K. F. Cooper and R. Strich, Antioxidants, 2018, 7, 13 CrossRef PubMed.
- B. P. Sullivan, D. J. Salmon and T. J. Meyer, Inorg. Chem., 1978, 17, 3334 CrossRef CAS.
- P. A. Lay, A. M. Sargeson, H. Taube, M. H. Chou and C. Creutz, Inorg. Synth., 1986, 24, 291 CAS.
- C. Viala and C. Coudret, Inorg. Chim. Acta, 2006, 359, 984 CrossRef CAS.
- S. Cie, Program for the Acquisition and Analysis of Data X-AREA, version1. 30, Stöe and Cie GmbH, Darmatadt, Germany, 2005 Search PubMed.
- X-RED, Program for Data Reduction and Absorption Correction, ed. S. C. GmbH, Darmatadt, Germanny, 2005 Search PubMed.
- C. Stoe, X-SHAPE, Version 2.05, Program for Crystal Optimization for Numerical Absorption Correction, Stoe & Cie GmbH, Darmatadt, Germany, 2004 Search PubMed.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2008, 64, 112 CrossRef CAS PubMed.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887 CrossRef.
- CrysAlisPro (version 1.171.41.113a), Rigaku Oxford Diffraction Ltd, Yarnton, Oxfordshire, England, 2021 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- F. Neese, The ORCA Program System, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CAS.
- F. Neese, Software Update: The ORCA Program System, Version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82(1), 299 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- S. Grimme, J. Antony, S. Ehrlic and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Phys. Chem., 1998, 108, 4439 CrossRef CAS.
- C. Jamorski, M. E. Casida and D. R. Salahub, J. Phys. Chem., 1996, 104, 5134 CrossRef CAS.
- M. Petersilka, U. J. Gossmann and E. K. U. Gross, Phys. Rev. Lett., 1996, 76, 1212 CrossRef CAS PubMed.
- P. Wayne, CLSI document M02-A11 and M07-A9, Clinical and Laboratory Standards Institute, 2012 Search PubMed.
- E. Ortega, C. Pérez-Arnaiz, V. Rodríguez, C. Janiak, N. Busto, B. García and J. Ruiz, Eur. J. Med. Chem, 2021, 222, 113600 CrossRef CAS PubMed.
- N. Fernández-Pampín, M. Vaquero, T. Gil, G. Espino, D. Fernández, B. García and N. Busto, J. Inorg. Biochem., 2022, 226, 111663 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2150468 and 2108989. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00401a |
| This journal is © The Royal Society of Chemistry 2022 |