
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of substituted (N,C) and (N,C,C) Au(III) complexes: the influence of sterics and electronics on cyclometalation reactions†
Knut T.
Hylland‡
 *ab,
Inga L.
Schmidtke‡
ab,
David S.
Wragg
ab,
Ainara
Nova
abcd and
Mats
Tilset
*abc
*ab,
Inga L.
Schmidtke‡
ab,
David S.
Wragg
ab,
Ainara
Nova
abcd and
Mats
Tilset
*abc
aDepartment of Chemistry, University of Oslo, P.O. Box 1033, Blindern, N-0315 Oslo, Norway. E-mail: k.t.hylland@smn.uio.no; mats.tilset@kjemi.uio.no
bCentre for Materials Science and Nanotechnology, University of Oslo, P.O. Box 1126 Blindern, N-0316 Oslo, Norway
cHylleraas Centre for Quantum Molecular Sciences, Department of Chemistry, University of Oslo, N-0315 Oslo, Norway
dUiT-The Arctic University of Norway, N-9037 Tromsø, Norway
First published on 4th March 2022
Abstract
Cyclometalated Au(III) complexes are of interest due to their catalytic, medicinal, and photophysical properties. Herein, we describe the synthesis of derivatives of the type (N,C)Au(OAcF)2 (OAcF = trifluoroacetate) and (N,C,C)AuOAcF by a cyclometalation route, where (N,C) and (N,C,C) are chelating 2-arylpyridine ligands. The scope of the synthesis is explored by substituting the 2-arylpyridine core with electron donor or acceptor substituents at one or both rings. Notably, a variety of functionalized Au(III) complexes can be obtained in one step from the corresponding ligand and Au(OAc)3, eliminating the need for organomercury intermediates, which is commonly reported for similar syntheses. The influence of substituents in the ligand backbone on the resulting complexes was assessed using DFT calculations, 15N NMR spectroscopy and single-crystal X-ray diffraction analysis. A correlation between the electronic properties of the (N,C) ligands and their ability to undergo cyclometalation was found from experimental studies combined with natural charge analysis, suggesting the cyclometalation at Au(III) to take place via an electrophilic aromatic substitution-type mechanism. The formation of Au(III) pincer complexes from tridentate (N,C,C) ligands was investigated by synthesis and DFT calculations, in order to assess the feasibility of C(sp3)–H bond activation as a synthetic pathway to (N,C,C) cyclometalated Au(III) complexes. It was found that C(sp3)–H bond activation is feasible for ligands containing different alkyl groups (isopropyl and ethyl), although the C–H activation is less energetically favored compared to a ligand containing tert-butyl groups.
Introduction
(N,C)-Cyclometalated Au(III) complexes1–4 have found application within catalysis5–9 and medicine10–15 and have also gained attention for their photophysical properties.3,4,16–18 While cyclometalated Au(III) dichloro complexes have been known since the 1980s,1 corresponding acetate and trifluoroacetate complexes are less studied. Due to the labile nature of carboxylate ligands, the reactivity of such complexes in e.g. ligand exchange reactions is higher than for their chloride analogues.2 Trifluoroacetate ligands are especially labile, making Au(III) trifluoroacetate complexes attractive catalyst candidates,5,7 as well as useful intermediates for the synthesis of other Au(III) complexes.19–22 In 2012 the first di(trifluoroacetate) complex of Au(III) with a chelating 2-arylpyridine ligand was reported by our group (2a-Au(OAcF)2, Fig. 1).19 This complex was conveniently synthesized by reacting tpy (2-(p-tolyl)-pyridine, 1a) with Au(OAc)3 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of HOAcF (trifluoroacetic acid) and water, using microwave-heating. This was a further development of a protocol for the synthesis of (N,C) Au(III) dichloro complexes, such as 2a-AuCl223 (Fig. 1). The reactivity of 2a-Au(OAcF)2 towards organolithium and Grignard reagents,19,20 ethylene,24,25 higher alkenes26 and acetylene7 has since then been explored. Subsequent to the initial findings, only few examples of trifluoroacetate complexes of Au(III) with 2-arylpyridine ligands have been reported.27–29 The scope of the microwave-assisted cyclometalation protocol has not been systematically investigated, although microwave-mediated synthesis of Au(III) complexes has had an increase in popularity in the last decade.3,30–36
:1 mixture of HOAcF (trifluoroacetic acid) and water, using microwave-heating. This was a further development of a protocol for the synthesis of (N,C) Au(III) dichloro complexes, such as 2a-AuCl223 (Fig. 1). The reactivity of 2a-Au(OAcF)2 towards organolithium and Grignard reagents,19,20 ethylene,24,25 higher alkenes26 and acetylene7 has since then been explored. Subsequent to the initial findings, only few examples of trifluoroacetate complexes of Au(III) with 2-arylpyridine ligands have been reported.27–29 The scope of the microwave-assisted cyclometalation protocol has not been systematically investigated, although microwave-mediated synthesis of Au(III) complexes has had an increase in popularity in the last decade.3,30–36
 | ||
| Fig. 1 Top: previously synthesized Au(III) complexes by our group: the (N,C) complexes 2a-AuCl223 and 2a-Au(OAcF)219 and the (N,C,C) complex 3b-AuOAcF.8 Bottom: (N,C) and (N,C,C) Au(III) trifluoroacetate complexes studied herein. | ||
(N,C,C) Au(III) pincer complexes have been reported to be more stable than (N,C) systems towards protolytic decomposition,8 and also possess interesting luminescence properties compared to (C,N,C) and (N,C) Au(III) complexes.16 For Au(III), the majority of reported (N,C,C) pincer ligands are derived from 2-([1,1′-biphenyl]-3-yl)pyridine (Fig. 2).2,16,18 While cyclometalation through C(sp3)–H bond activation is common for e.g. Pd(II),37–40 reports on C(sp3)–H bond activation in the synthesis of Au(III) complexes are scarce, and only a few examples have been reported.41–43 In 2018, we reported a rare example of C(sp3)–H bond activation for Au(III) in the synthesis of an (N,C,C)-cyclometalated Au(III) trifluoroacetate complex 3b-AuOAcF from 2-(3,5-di-tert-butylphenyl)pyridine (1b) (Fig. 1).8 The formation of complex 3b-AuOAcF from 1b was studied in detail by DFT calculations, showing that the steric bulk of the tert-butyl substituent promotes the C(sp3)–H activation by destabilising the intermediate (N,C) complex.
 | ||
| Fig. 2 Ligand for (N,C,C) Au(III) pincer formation derived from 2-([1,1′-biphenyl]-3-yl)pyridine (left) and the corresponding Au(III) complex that possess two Au–C(sp2) bonds (right). | ||
To understand and develop the chemistry of organometallic Au(III) complexes, robust synthesis protocols are of high importance. Varying the functionalization of the ligand backbone in coordination compounds is of interest as this can have a significant impact on their catalytic,44–47 photophysical,2,16–18,48–55 magnetic,56,57 electrochemical58–62 and biological63–65 properties. Furthermore, it allows for the evaluation of the robustness of the metalation protocol. Therefore, we wanted to explore the possibility to synthesize functionalized derivatives of 2a-Au(OAcF)2 and 3b-AuOAcF (see general structures in Fig. 1). Variation of the ancillary 2-arylpyridine ligand is easily implemented, particularly through cross-coupling reactions, and by this a series of new ligands for Au(III) is readily available. In addition, cyclometalation as a strategy for the synthesis of (N,C) and (N,C,C) Au(III) complexes remains a somewhat underdeveloped field. More traditional approaches, such as transmetalation of the corresponding organomercury compounds, are frequently being reported for the synthesis of cyclometalated Au(III) complexes.1,2,18,65–68 Although efficient, this method suffers from the toxicity of mercury, creating a need to investigate and further develop alternative synthesis methods. We herein present the synthesis and characterization of a series of (N,C) and (N,C,C) Au(III) complexes. All complexes were conveniently prepared by microwave-heating using Au(OAc)3 and the corresponding 2-arylpyridine ligand, with electron-donating or -withdrawing substituents at one or both rings. The formation and bonding properties of these complexes were assessed by DFT calculations, 15N NMR spectroscopy and single-crystal X-ray diffraction analysis, in order to address any substituent effects.
Results and discussion
Ligand synthesis
The ligands, substituted 2-arylpyridines (1c–1u), were readily available through the Suzuki–Miyaura cross-coupling of suitable 2-halogenated pyridine derivatives and arylboronic acids (Scheme 1A), using reaction conditions reported in the literature.8,69,70 Ligand 1r was prepared from 1q (Scheme 1B) according to a method developed by Fagnou and co-workers for the installation of pentafluorophenyl groups through Pd-catalysed C–H activation.71 | ||
| Scheme 1 (A) Synthesis of 2-arylpyridine ligands. Conditions I = Pd(OAc)2, PPh3, K3PO4, H2O, n-PrOH.8 Conditions II = Pd2dba3, HBF4·P(t-Bu)3, KF·2H2O, THF.69,70 (B) Synthesis of ligand 1r from 1q. | ||
Synthesis of (N,C)-cyclometalated Au(III) di(trifluoroacetate) complexes
Having in hand a wide variety of potential ligands, we investigated their ability to cyclometalate at Au(III) under the same reaction conditions as utilized for the synthesis of 2a-Au(OAcF)2 (Scheme 2).23 | ||
| Scheme 2 Microwave-mediated synthesis of (N,C)-cyclometalated Au(III) complexes. | ||
The Au(III) complexes were obtained in yields ranging from 27 to 95%, and both electron-withdrawing (nitro, trifluoromethyl) and electron-donating (methyl, methoxy) substituents were tolerated. They were characterized by multinuclear NMR spectroscopy (1H, 13C, 19F and 15N NMR), MS, elemental analysis and single-crystal X-ray diffraction analysis. For Au(III) complexes carrying substituents on the pyridine ring, no clear trends in yields were observed. For complexes with substituents on the phenyl ring, certain trends in yields were found. For evaluation of the experimental observations, the carbon that undergoes cyclometalation in the protonated ligands (C2′, see Scheme 3) was investigated by natural charge analysis. In the following section, the natural charge of C2′ in the ligands are discussed relative to the charge of C2′ in the 2-phenylpyridinium cation, ppy-H+ (ΔC2′ = 0) (for full details, see ESI†). The dimethyl-substituted complex 2h-Au(OAcF)2 was obtained in high yield (95%) like complex 2a-Au(OAcF)2 (94%).19 Complexes carrying either significantly electron-withdrawing (2e-Au(OAcF)2, 2o-Au(OAcF)2 and 2r-Au(OAcF)2) or electron-donating (2k-Au(OAcF)2) groups were obtained in lower yields. The difluoro-substituted complex 2c-Au(OAcF)2 was obtained in 85% yield, supporting an electrophilic aromatic substitution-type of cyclometalation mechanism (Scheme 3). This mechanism is generally preferred for the formation of cyclometalated complexes of late transition metals.72–76 Despite being inductively electron-withdrawing, fluorine groups are ortho/para-directing and activating substituents in aromatic electrophilic substitution reactions, causing the C(sp2)–H activation to proceed more easily at ligand 1c compared to e.g.1e, 1o and 1r.
 | ||
| Scheme 3 Postulated mechanism for the formation of cyclometalated Au(III) complexes. The carbon (C2′) that undergoes the C(sp2)–H bond activation and binds to gold in the final product is indicated. | ||
Neither di(trifluoromethyl)- nor dimethoxy-substituted ligands 1f and 1j provided the desired cyclometalated products. The reaction of 1j and Au(OAc)3 furnished a multitude of species as seen in the 1H NMR spectrum of the crude product (Fig. S89, ESI†), and no single, clean compound could be isolated. It was however possible to obtain crystals suitable for single-crystal XRD, showing an unusual dinuclear M2L3-type complex (2j3Au2(OAcF)2, see Fig. S146 and Fig. S147, ESI†). Having said this, we do not believe this to be the main reaction product, as no other characterization data can substantiate this. We surmise that the ligand is too electron rich and reactive to yield clean formation of a Au(III) complex under the investigated reaction conditions. This assumption is further supported by the calculated natural charge for C2′ in 1j-H+ (ΔC2′ = −0.120). The natural charge for C2′ in 1j-H+ is significantly larger than the ones found for ligands 1c-H+ (ΔC2′ = −0.084) and 1h-H+ (ΔC2′ = −0.008), which both have activating substituents in the 3′- and 5′-positions. For di(trifluoromethyl)-substituted 1f, the N-coordinated adduct 1f-Au(OAcF)3 (which is a likely precursor for the cyclometalation step) was isolated (Scheme 4).
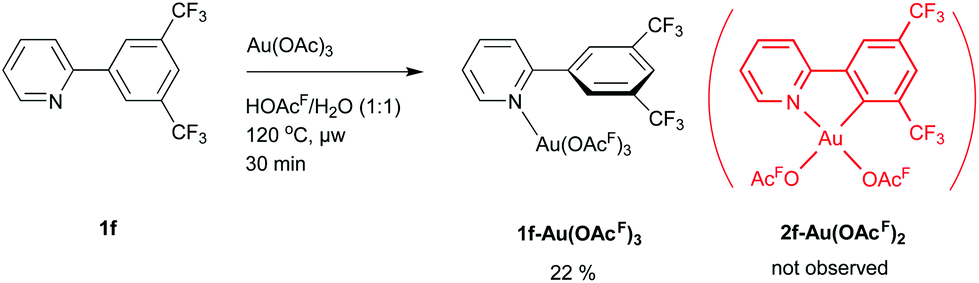 | ||
| Scheme 4 Synthesis of 1f-Au(OAcF)3. The corresponding (N,C) complex 2f-Au(OAcF)2 was not observed. | ||
The failure of obtaining cyclometalated 2f-Au(OAcF)2 can be related with the poor electrophilicity of 1f (ΔC2′ = +0.061 for 1f-H+). It was previously reported by our group that pincer complex 3b-AuOAcF (derived from the sterically encumbered and electron rich ligand 1b) proceeds via the corresponding (N,C)-cyclometalated complex 2b-Au(OAcF)2 (see below).8 As the trifluoromethyl group is smaller than the tert-butyl group,77,78 the formation of 2f-Au(OAcF)2 should be feasible from a steric point-of-view. If the Au–C bond formation takes place by electrophilic C(sp2)–H bond activation (Scheme 3), (a) strongly electron-withdrawing substituent(s) in the aryl ring of the ligand might impede the reaction. Reaction of the mono-trifluoromethyl-substituted ligand 1g with Au(OAc)3 gave a ca. 2:1 mixture of the two regioisomers 2g-p-Au(OAcF)2 and 2g-o-Au(OAcF)2 (Scheme 5). Thus, cyclometalation of Au(III) is compatible with the steric demands of an ortho-trifluoromethyl group. This result therefore suggests that the electronic influence is the main reason for the failure to produce 2f-Au(OAcF)2, as the di(trifluoromethyl)-substituted ligand 1f is more electron deficient than the corresponding mono-trifluoromethyl-substituted ligand 1g (ΔC2′ = +0.025 for 1g-p-H+ and ΔC2′ = +0.038 for 1g-o-H+).
 | ||
| Scheme 5 Reactivity of ligand 1g towards Au(OAc)3. | ||
Synthesis of (N,C,C)-cyclometalated Au(III) trifluoroacetate pincer complexes
Following the successful microwave-mediated synthesis of pincer complex 3b-AuOAcFvia C(sp2)–H and C(sp3)–H bond activation of ligand 1b (Fig. 3),8 we sought to investigate related tridentate ligands (1s–1u) in order to get more insight into the scope and limitations of the pincer formation. DFT calculations on the formation of 3b-AuOAcF suggested the directing effect of the bulky tert-butyl group in ligand 1b to be a key element in the successful synthesis of the complex.8 Therefore, ligands 1s and 1t with less sterically demanding substituents, but otherwise an identical substitution pattern to that of 1b were investigated as tridentate ligands for Au(III). Additionally, the mono-substituted analogue of 1t, ligand 1u, was investigated in order to probe the selectivity of pincer formation relative to (N,C) cyclometalation. | ||
| Fig. 3 Tridentate (N,C,C) ligands. | ||
Attempts of synthesizing 3s-AuOAcF in an analogous manner (microwave heating at 120 °C for 30 min) to 3b-AuOAcF failed, and the N-coordinated adduct of ligand 1s, 1s-Au(OAcF)3, was the main species observed in the 1H NMR spectrum of the crude product. The combination of lower reaction temperature (80 °C) and longer reaction time (3.5 h) did however furnish the pincer complex 3s-AuOAcF in moderate yields (38%) (Scheme 6). It is noteworthy that the C(sp3)–H bond activation of the isopropyl group introduces a chiral centre in close proximity to gold in 3s-AuOAcF, which has previously been observed for a structurally related Pt(IV) complex.79 We did not attempt to resolve the enantiomers, but the accessibility to chiral centres through cyclometalation is a topic that deserves further investigation, especially if an enantiopure complex can be obtained.80
 | ||
| Scheme 6 Synthesis of 3s-AuOAcF. The star indicates the chiral centre that is formed upon C(sp3)–H bond activation. | ||
By employing the reaction conditions in Scheme 6, the ethyl analogue 3t-AuOAcF was obtained in a good yield (65%) from ligand 1t. In addition to the synthesis of 3s-AuOAcF and 3t-AuOAcF, mono-ethyl-substituted 1u was explored as a tridentate ligand for Au(III). The reaction of 1u with Au(OAc)3 yielded the desired pincer complex 3u-AuOAcF as a mixture with the corresponding (N,C)-cyclometalated complex 2u-p-Au(OAcF)2 (Scheme 7). The other possible (N,C)-cyclometalated complex 2u-o-Au(OAcF)2 could not be observed, supporting the involvement of 2u-o-Au(OAcF)2 as an intermediate for the formation of 3u-AuOAcF (see ESI† for details). To summarize, the successful syntheses of 3s-AuOAcF and 3t-AuOAcF show that the sterically induced pre-orientation of the C–H bond that is activated (as seen in ligand 1b) is not a strict requirement for C(sp3)–H activation at Au(III), although the experimental observations suggest that the process is more feasible for 1b compared to 1s and 1t. Importantly, the results show that cyclometalation of Au(III) through C(sp3)–H bond activation takes place at a β position regardless of the nature of the alkyl group in the ligand.
 | ||
| Scheme 7 Reactivity of ligand 1u towards Au(OAc)3. | ||
Single-crystal X-ray diffraction analysis of (N,C)- and (N,C,C)-cyclometalated Au(III) complexes
Several of the (N,C)-cyclometalated Au(III) complexes were analysed by single-crystal X-ray diffraction (Fig. 4). In all cases, the complexes crystallized with the expected square planar geometry around Au(III). Deviations from ideal square planar geometries were evaluated by the τ′4 values (0 for square planar complexes, 1 for tetrahedral complexes)81,82 of the structures, which were found in the range from 0.02 to 0.09 (Table 1). All Au–ligand bond distances and angles are given in Table 1. The bond angles are similar to those previously reported for structurally related square planar Au(III) complexes.19,21,26,29 The Au1–N1 and Au1–C7 bond lengths are in the range of 1.9974(19)–2.0426(17) Å and 1.988(2)–2.02(2) Å, being comparable to those reported for 2a-Au(OAcF)2 (1.991(6) Å and 1.995(7) Å, respectively).19 The Au1–O3 (trans to N) bond lengths are in the range of 2.0032(16)–2.034(3) Å, all being slightly longer than the corresponding bond length in 2a-Au(OAcF)2 (1.993(5) Å). Larger variation is found in the Au1–O1 (trans to C) bond lengths. In 2c-Au(OAcF)2, the Au1–N1 and Au1–C7 bond lengths are comparable to the other complexes, but a slightly shorter Au1–O1 (trans to carbon) bond is noted (2.070(3) Å for 2c-Au(OAcF)2vs. 2.095(13)–2.1185(16) Å for the rest of the complexes). This may be attributed to the presence of the electron-withdrawing fluorine substituents in the ligand, making the phenyl ring a weaker trans influence ligand compared to the one in e.g.2a-Au(OAcF)2. Similar, but less pronounced, shortening of the Au1–O1 bond can be observed for other complexes having electron-withdrawing substituents (2e-Au(OAcF)2, 2o-Au(OAcF)2 and 2r-Au(OAcF)2; Au1–O1 = 2.1026(18) Å, 2.0984(16) Å and 2.095(13) Å, respectively). | ||
| Fig. 4 ORTEPs of 2c-Au(OAcF)2, 2d-Au(OAcF)2, 2e-Au(OAcF)2, 2h-Au(OAcF)2, 2i-Au(OAcF)2, 2l-Au(OAcF)2, 2m-Au(OAcF)2, 2n-Au(OAcF)2, 2o-Au(OAcF)2, 2p-Au(OAcF)2 and 2r-Au(OAcF)2. Ellipsoids are shown at 50% probability level. Hydrogen atoms and associated (disordered) solvent molecules (for 2d-Au(OAcF)2, 2i-Au(OAcF)2, 2m-Au(OAcF)2 and 2p-Au(OAcF)2) have been removed for clarity. For 2d-Au(OAcF)2 and 2i-Au(OAcF)2, only one of the two molecules in the asymmetric unit of each structure is shown. For 2d-Au(OAcF)2 and 2l-Au(OAcF)2, disorder in the trifluoroacetate ligands have been removed for clarity. For more details, see ESI.† | ||
| Complex | τ′4 | Au1–N1 | Au1–C7 | Au1–O1 | Au1–O3 | N1–Au1–C7 | N1–Au1–O1 | N1–Au1–O3 | O1–Au1–O3 | O1–Au1–C7 | O3–Au1–C7 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| The corresponding data for 2a-Au(OAcF)2 are included for reference purposes.19 For 2d-Au(OAcF)2 and 2i-Au(OAcF)2, metric data for one of the two molecules in the respective asymmetric unit are listed. See ESI (Fig. S140 and Fig. S145)† for metric data for both molecules in each asymmetric unit. | |||||||||||
| 2a-Au(OAcF)2 | 0.07 | 1.991(6) | 1.995(7) | 2.111(5) | 1.993(5) | 81.8(3) | 93.1(2) | 175.5(2) | 88.8(2) | 174.8(3) | 96.4(3) |
| 2c-Au(OAcF)2 | 0.08 | 2.003(3) | 2.004(4) | 2.070(3) | 1.999(3) | 81.86(14) | 91.37(12) | 175.45(12) | 87.60(11) | 172.50(13) | 98.89(14) |
| 2d-Au(OAcF)2 | 0.06 | 2.013(4) | 1.995(5) | 2.114(4) | 2.008(4) | 82.03(19) | 94.74(17) | 174.92(17) | 88.41(17) | 176.60(19) | 94.73(18) |
| 2e-Au(OAcF)2 | 0.04 | 2.007(2) | 1.991(2) | 2.1026(18) | 2.0065(18) | 81.83(9) | 95.48(8) | 175.98(8) | 87.67(7) | 177.24(9) | 95.05(9) |
| 2h-Au(OAcF)2 | 0.04 | 1.9996(19) | 2.022(2) | 2.1185(16) | 2.0085(16) | 81.84(8) | 93.69(7) | 178.23(7) | 84.76(7) | 175.38(8) | 99.68(8) |
| 2i-Au(OAcF)2 | 0.09 | 2.034(3) | 1.994(3) | 2.113(2) | 2.011(2) | 81.65(13) | 103.80(11) | 173.84(11) | 82.24(10) | 172.37(12) | 92.46(12) |
| 2l-Au(OAcF)2 | 0.05 | 1.999(4) | 2.000(5) | 2.101(4) | 2.005(4) | 81.77(17) | 96.02(17) | 176.75(16) | 86.37(18) | 176.63(17) | 95.74(18) |
| 2m-Au(OAcF)2 | 0.09 | 2.0426(17) | 1.995(2) | 2.1112(15) | 2.0119(15) | 81.23(8) | 104.62(6) | 171.95(6) | 83.31(6) | 173.98(7) | 90.81(7) |
| 2n-Au(OAcF)2 | 0.03 | 1.9974(19) | 1.994(2) | 2.1183(17) | 2.0032(16) | 81.43(9) | 99.00(8) | 173.40(7) | 86.39(7) | 179.55(8) | 93.19(8) |
| 2o-Au(OAcF)2 | 0.06 | 2.0047(18) | 1.988(2) | 2.0984(16) | 2.0097(16) | 81.62(8) | 95.05(7) | 175.35(7) | 89.37(7) | 176.23(7) | 94.01(8) |
| 2p-Au(OAcF)2 | 0.03 | 2.008(3) | 1.996(3) | 2.102(2) | 2.034(3) | 81.54(12) | 98.36(10) | 173.63(11) | 86.78(9) | 179.69(11) | 93.29(12) |
| 2r-Au(OAcF)2 | 0.02 | 1.999(12) | 2.020(19) | 2.095(13) | 1.984(11) | 82.3(7) | 95.7(5) | 178.9(5) | 84.0(5) | 177.0(6) | 98.0(6) |
For 3′,5′-disubstituted complexes 2c-Au(OAcF)2, 2h-Au(OAcF)2 and 2r-Au(OAcF)2 the O3–Au1–C7 cis angles (98.89(14)°, 99.68(8)° and 98.0(6)°, respectively) were slightly larger than in the other complexes (see Table 1). The angle was larger for 2h-Au(OAcF)2 than for 2r-Au(OAcF)2, being in accordance with literature reports of the similar effective steric bulk of the pentafluorophenyl group and the methyl group.83
The Au1–N1 bond lengths of 2i-Au(OAcF)2 and 2m-Au(OAcF)2 are 2.034(3) Å and 2.0426(17) Å, respectively, being longer than the corresponding Au1–N1 bond in the other complexes studied. Furthermore, the N1–Au1–O1 cis angles in 2i-Au(OAcF)2 and 2m-Au(OAcF)2 are also larger (103.80(11)° and 104.62(6)°, respectively) compared to the other complexes (91.37(12)°–99.00(8)°). Similar elongation of the Au1–N1 bond and widening of the corresponding cis angle have earlier been reported by Cinellu and co-workers for a structurally similar (N,C) Au(III) complex derived from 6,6′-dimethoxy-2,2′-bipyridine.84 The N1–C1–O5 angle in 2m-Au(OAcF)2 is 114.51(18)°, being slightly smaller than the corresponding N–C–O(alkoxy) angles in reported crystal structures of non-coordinated pyridines, bipyridines or phenanthrolines with alkoxy substituents α to nitrogen (typically around 120°).40,84–88 The angle is similar to the N–C–O(alkoxy) angle in (N,C) Au(III) and Pd(II) complexes derived from 6,6′-dimethoxy-2,2′-bipyridine reported by Cinellu and co-workers.40,84 The relatively small N–C–O(alkoxy) angle in 2m-Au(OAcF)2, together with a Au1–O5 distance of 3.13 Å may hint at a weak interaction between the gold centre and the oxygen atom in the methoxy group. This was further investigated by means of 15N NMR spectroscopy (see below). The N1–C1–C13 angle (120.9(3)°) in 6-methyl-substituted 2i-Au(OAcF)2 is slightly larger than the corresponding N–C–C(alkyl) angles in reported crystal structures of non-coordinated pyridines, bipyridines or phenanthrolines with alkyl substituents α to nitrogen (typically around 115°).89–93 This indicates that Au(III) coordination to 6-methyl-substituted pyridines results in a widening of the N–C–C(methyl) angle, contrary to the effect Au(III) coordination has on the N–C–O(methoxy) angle in 6-methoxy-substituted pyridines. The N–C–C(alkyl) angle in 2i-Au(OAcF)2 is similar to reported angles for related square planar Au(III)94 and Pd(II)58 complexes of 6-methyl-substituted (bi)pyridine ligands.
Au(III) pincer complexes 3s-AuOAcF and 3t-AuOAcF were also crystallographically characterized (Fig. 5). Both complexes crystallized with distorted square planar geometry around Au(III) (τ′4 = 0.09 for 3s-AuOAcF and τ′4 = 0.11 for 3t-AuOAcF). The bond lengths and angles of the complexes are similar to those reported for 3b-AuOAcF (Table 2). Interestingly, only one enantiomer of the racemic mixture of pincer complex 3s-AuOAcF could be modelled as the major component during refinement of the single-crystal X-ray structure.
 | ||
| Fig. 5 ORTEPs of 3s-AuOAcF and 3t-AuOAcF. Ellipsoids are shown at 50% probability level. Hydrogen atoms and disorder in the isopropyl group in 3s-AuOAcF have been omitted for clarity. Only one of the two molecules in the asymmetric unit is shown for 3s-AuOAcF, and only one of the four molecules in the asymmetric unit is shown for 3t-AuOAcF. For more details, see ESI.† | ||
| Complex | τ′4 | Au1–N1 | Au1–C7 | Au1–O1 | Au1–C13 | N1–Au1–C7 | N1–Au1–C13 | N1–Au1–O1 | C7–Au1–C13 | C7–Au1–O1 | C13–Au1–O1 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| The corresponding data for 3b-AuOAcF are included for reference purposes.8 For 3s-AuOAcF, metric data for one of the two molecules in the asymmetric unit are listed. See Fig. S160 (ESI)† for metric data for both molecules in the asymmetric unit. For 3t-AuOAcF, metric data for one of the four molecules in the asymmetric unit are listed. See Fig. S162 (ESI)† for metric data for all molecules in the asymmetric unit. | |||||||||||
| 3b-AuOAcF | 0.08 | 2.135(3) | 1.944(3) | 2.119(3) | 2.049(4) | 80.36(13) | 161.36(14) | 99.62(11) | 81.66(15) | 179.34(13) | 98.30(14) |
| 3s-AuOAcF | 0.09 | 2.144(3) | 1.954(3) | 2.108(3) | 2.055(4) | 80.59(13) | 161.67(14) | 98.52(11) | 81.33(15) | 177.53(13) | 99.67(14) |
| 3t-AuOAcF | 0.11 | 2.15(2) | 1.94(2) | 2.076(18) | 2.04(3) | 80.0(10) | 163.0(9) | 93.2(8) | 83.0(11) | 173.2(10) | 103.7(9) |
15N NMR spectroscopic studies of cyclometalated Au(III) complexes
Some of the complexes and ligands discussed herein were investigated by 15N NMR spectroscopy, and coordination shifts Δδ15N (δ15Ncomplex–δ15Nligand) were obtained in order to gain insight about the Au–N interactions. Furthermore, a selection of previously reported tpy-ligated Au(III) complexes8,14,19,22,25,95 with varying substituents cis and trans to pyridine-N was studied by 15N NMR spectroscopy to shed light on which factors influence the 15N NMR chemical shifts of N-ligated square planar d8 metal complexes (Fig. 6). | ||
| Fig. 6 Overview of (N,C) and (N,C,C) Au(III) complexes studied by 15N NMR spectroscopy herein. All data were collected in CD2Cl2 at either 600 or 800 MHz. See also Table S1, ESI.†δ15N for [2a-Au(C,N)]+[OAcF]− is from ref. 25. | ||
Δδ15N were found in the range of −88.2 ppm to −104.5 ppm for the (N,C) di(trifluoroacetate) Au(III) complexes (except for 2m-Au(OAcF)2; see discussion below). These shifts are similar to reported data for other pyridine-ligated Au(III) complexes with weak trans influence ligands trans to nitrogen.44,96–98 The 15N NMR data can be interpreted in a similar manner as the data from the single-crystal X-ray diffraction analysis of the complexes, where the Au–N bond lengths were found to be little dependent of the substituents in the ligand backbone. This reflects that other factors, such as the ligand trans to pyridine-N,99 affects Δδ15N stronger than the substituents on the pyridine ring. Similar observations have also been reported by Pazderski for square planar Pd(II) and Pt(II) complexes with substituted bipyridine and phenanthroline ligands.96
The coordination shift of the 6-methoxy-substituted complex 2m-Au(OAcF)2 (Δδ15N = −66.1 ppm) is significantly smaller than those of the other substituted di(trifluoroacetate) complexes. It seems likely that the relatively small Δδ15N found for 2m-Au(OAcF)2 is a result of the weak interaction between the methoxy-oxygen and gold. This potential interaction was also observed in the single-crystal structure of the Au(III) complex. The coordination shift for the 6-methyl-substituted complex 2i-Au(OAcF)2 (−88.2 ppm) was in the same range as those obtained for the other di(trifluoroacetate) complexes studied herein, although the Au–N bond length in the crystal structure of 2i-Au(OAcF)2 (2.034(3) Å) was similar to the one observed for 2m-Au(OAcF)2 (2.0426(17) Å). The very similar Au–N bond lengths in the two complexes strengthens the argument that the relatively small coordination shift obtained for 2m-Au(OAcF)2 is caused by an interaction between oxygen and gold, rather than steric repulsion between gold and the 6-substituent.84 If this was the case, it would be expected to yield a comparable coordination shift for 2i-Au(OAcF)2.
For complex 1f-Au(OAcF)3, Δδ15N was found to be −114.1 ppm, larger than what was observed for the cyclometalated complexes and also larger than what has been reported for pyridine-ligated AuCl3 complexes in the literature (ca. −80 ppm in CDCl3).100–102 This reflects that [OAcF]− is a weaker trans influence ligand than Cl−, being consistent with reported experimental and computational data for the trans influence of carboxylate ligands vs. chloride ligands in square planar complexes.103–105
To further evaluate the effect of the identity of the ligand trans to nitrogen on the coordination shift of pyridine-N, Δδ15N for 2a-Au(OAc)2 and 2a-Au(CH3)2 were obtained, and compared to the one found for 2a-Au(OAcF)2. For the three complexes, Δδ15N was found to decrease in the order 2a-Au(OAcF)2 ∼ 2a-Au(OAc)2 > 2a-Au(CH3)2, agreeing with the established trans influence of the corresponding ligands; [OAcF]− ∼ [OAc]− < CH3−.19,103,104 Coordination shifts were also obtained for the pincer complexes 3b-AuOAcF, 3s-AuOAcF and 3t-AuOAcF which all have an alkyl group trans to pyridine-N. As expected from the differences in relative trans influence strength of an alkyl ligand and a [OAcF]− ligand, the coordination shifts for the three pincers were significantly smaller (Δδ15N from −37.1 to −39.5 ppm) than those obtained for the di(trifluoroacetate) complexes. Surprisingly, they were also found to be smaller than the coordination shifts of 2a-Au(CH3)2, 2a-Au(CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)Br, 2a-Au(CH3)Br and [2a-Au(C,N)]+[OAcF]−, although it could be anticipated that the relative trans influence of the ligands trans to pyridine-N would be similar for all these complexes. The observations may be explained from differences in the relative cis influence of an alkyl ligand, a halide ligand and a carboxylate ligand. A smaller Δδ15N was found for 2a-Au(CH3)Br (Δδ15N = −46.7 ppm) compared to 2a-Au(CH3)2 (Δδ15N = −56.1 ppm) being in accordance with the reported higher cis influence of halide ligands compared to alkyl ligands.106,107 In summary, the findings from the 15N NMR spectroscopic studies show that functionalization of the ligand backbone has little effect on the interaction between pyridine-N and gold, whereas the nature of the ligands cis and trans to pyridine-N has a significantly larger effect.
CH2)Br, 2a-Au(CH3)Br and [2a-Au(C,N)]+[OAcF]−, although it could be anticipated that the relative trans influence of the ligands trans to pyridine-N would be similar for all these complexes. The observations may be explained from differences in the relative cis influence of an alkyl ligand, a halide ligand and a carboxylate ligand. A smaller Δδ15N was found for 2a-Au(CH3)Br (Δδ15N = −46.7 ppm) compared to 2a-Au(CH3)2 (Δδ15N = −56.1 ppm) being in accordance with the reported higher cis influence of halide ligands compared to alkyl ligands.106,107 In summary, the findings from the 15N NMR spectroscopic studies show that functionalization of the ligand backbone has little effect on the interaction between pyridine-N and gold, whereas the nature of the ligands cis and trans to pyridine-N has a significantly larger effect.
DFT calculations on the formation of (N,C,C)-cyclometalated Au(III) complexes 3s-AuOAcF and 3t-AuOAcF
In order to gain understanding of pincer formation for tridentate (N,C,C) ligands 1s and 1t, DFT calculations were performed. The formation of 3s-AuOAcF and 3t-AuOAcF starting from complexes 2s-Au(OAcF)2 and 2t-Au(OAcF)2via the same mechanism proposed for the formation of 3b-AuOAcF8 was explored (Fig. 7). As the formation of 2s-Au(OAcF)2 was found to be very similar to that of 2b-Au(OAcF)2, the energies for this first cyclometalation step are not included in the figure (see Table S17, ESI† for details), and was not calculated for 2t-Au(OAcF)2.
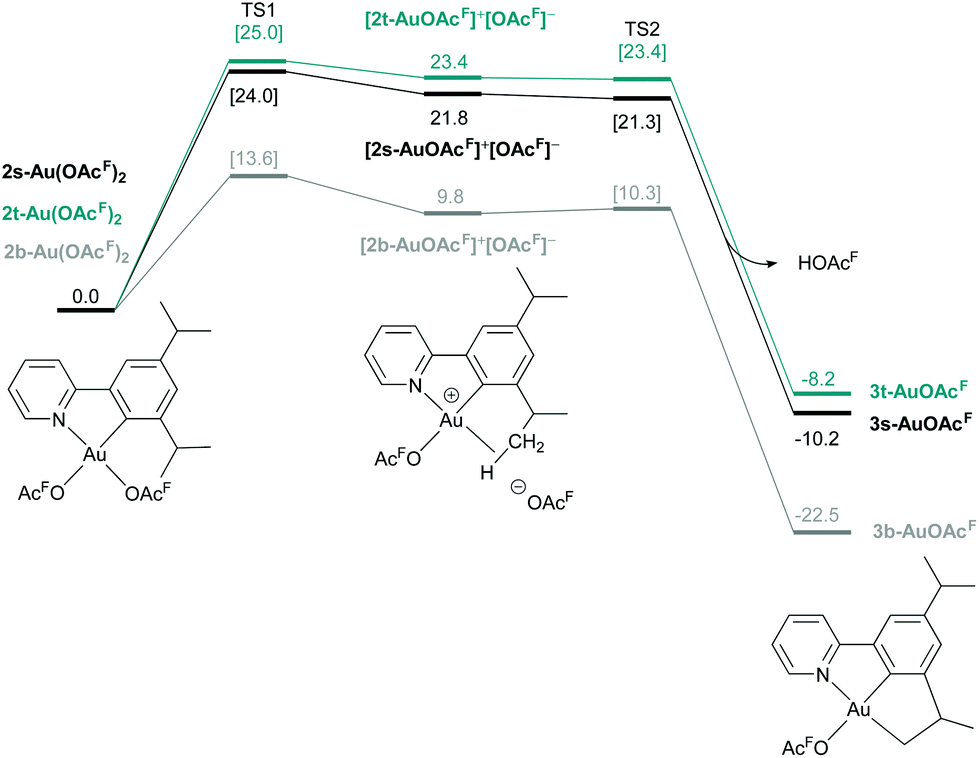 | ||
| Fig. 7 Free energy profile in kcal mol−1 for the formation of 3b-AuOAcF, 3s-AuOAcF and 3t-AuOAcF from the corresponding di(trifluoroacetate) complexes. In the figure, 2s-Au(OAcF)2, [2s-AuOAcF]+[OAcF]− and 3s-AuOAcF are displayed as structural examples. The energies of all minima and transition states in brackets are computed in CH2Cl2 (SMD) for 3s-AuOAcF and 3t-AuOAcF. The energies and transition states for 3b-AuOAcF were computed in HOAcF.8 | ||
Looking at the C–H activation step, a clear difference in energy for TS1 was observed for the three complexes, illustrating the effect of the substituent on the formation of the desired pincer complex. The endergonic dissociation step forming the agostic intermediates [2s-Au-OAcF]+[OAcF]− and [2t-Au-OAcF]+[OAcF]− is more than 10 kcal mol−1 higher in energy for 3s-AuOAcF and 3t-AuOAcF (24.0 kcal mol−1 and 25.0 kcal mol−1, respectively), compared to 3b-AuOAcF (13.6 kcal mol−1). The following proton abstractions by [OAcF]− that furnish 3s-AuOAcF and 3t-AuOAcF are barrier–free, which is in accordance with what was found for 3b-AuOAcF.
The energy barrier associated with the formation of the agostic intermediates is depending on the bulkiness of the alkyl substituent that undergoes C–H activation. The lower energy barrier for [2b-Au-OAcF]+[OAcF]− is due to the higher energy of 2b-Au(OAcF)2 relative to TS1 due to steric interaction with the tert-butyl group in the (N,C)-cyclometalated complex. In order to highlight the difference in stability of the (N,C)-cyclometalated complexes 2b-Au(OAcF)2 and e.g.2s-Au(OAcF)2, which is responsible for the significantly lower TS1 found for the tert-butyl system, an isodesmic reaction of a formal chelate ligand exchange on 2b-Au(OAcF)2 with 1s was investigated (Scheme 8). The formation of 2s-Au(OAcF)2 and 1b is favoured by 9.9 kcal mol−1 which illustrates the negative effect the large, bulky substituents have on the stability of di(trifluoroacetate) complexes. On the other hand, this characteristic of the tert-butyl substituent ultimately facilitates the C(sp3)–H bond activation step and subsequent pincer formation.
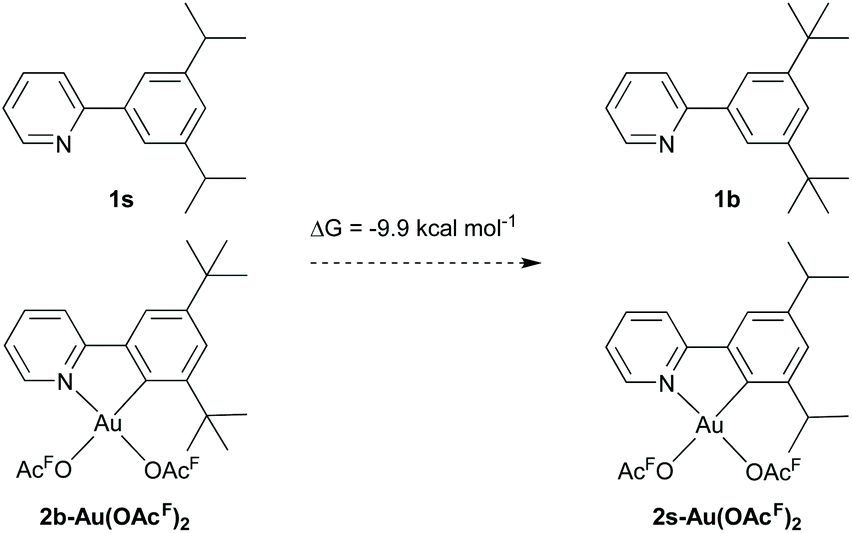 | ||
| Scheme 8 Isodesmic reaction for the formal chelate ligand exchange on 2b-Au(OAcF)2 furnishing 2s-Au(OAcF)2. Gibbs energy in CH2Cl2 (SMD) is given in kcal mol−1. | ||
Conclusions
In this work, a detailed experimental and computational study of a series of 2-arylpyridine-based (N,C)- and (N,C,C)-cyclometalated Au(III) complexes has been presented. For the (N,C) systems, it was found that the scope of microwave-mediated synthesis of cyclometalated Au(III) complexes is broad and that a large variety of different functional groups is tolerated. This makes it an attractive method for the synthesis of Au(III) complexes of substituted arylpyridine ligands without having to resort to any organomercury intermediates. The efficiency of the reaction is strongly dependent on the electronic features of the (N,C) ligand, being consistent with an electrophilic aromatic substitution-type mechanism for cyclometalation at Au(III). Natural charge analysis performed on the protonated (N,C) ligands was found to correlate with the experimental observations of their reactivity towards Au(OAc)3. Single-crystal X-ray diffraction and 15N NMR spectroscopy data suggest that the (N,C) Au(III) complexes are structurally similar species, meaning that the ligand scaffold is flexible to changes without drastically affecting the coordination sphere around Au. Detailed 15N NMR spectroscopic studies of different cyclometalated Au(III) complexes with varying ligands trans to the (N,C) backbone show that these ligands have a much stronger influence on the Au–N interaction than any substituents in the backbone, with the exception of the 6-methoxy-substituted complex 2m-Au(OAcF)2. For this complex, the relatively small coordination shift may be explained by a possible weak interaction between the methoxy-oxygen and gold. In addition to the studies of (N,C)-cyclometalated Au(III) complexes, C(sp3)–H bond activation as a synthetic feasible method to yield (N,C,C)-cyclometalated Au(III) complexes was expanded. Earlier we have shown that the ligand 2-(3,5-di-tert-butylphenyl)pyridine (1b) functions as a tridentate ligand for Au(III), yielding pincer complex 3b-AuOAcF. We have broadened the scope of Au(III) pincer formation, via C(sp3)–H bond activation to include less sterically encumbered ligands. These ligands contain either isopropyl (1s) or ethyl (1t and 1u) groups, where the C–H bond that is activated is able to rotate away from gold. In a combined experimental and computational effort we have shown that these ligands indeed undergo C(sp3)–H bond activation, but that the process is less facile than for the tert-butyl-substituted system. Further work will focus on broadening the scope of (N,C,C) pincer formation from alkyl-substituted 2-arylpyridines, as well as investigating their reactivity and optical properties.Experimental section
General considerations
2a-Au(OAcF)2,193b-AuOAcF,82a-Au(OAc)2,14,952a-Au(CH2CHCH2)Br,222a-Au(C6H5)Br,192a-Au(CH3)Br,192a-Au(CH3)2,19 3,5-diethylphenylboronic acid108,109 and 3,5-diisopropylphenylboronic acid108,109 were synthesized according to literature procedures. Au(OAc)3 was obtained from abcr. THF (unstabilized) and CH2Cl2 were dried using an MB SPS-800 solvent purifier system from MBraun. Hexanes and ethyl acetate were distilled before use. Deionized water was used. Other chemicals and solvents were used as received from commercial sources. TLC was performed using Merck 60 F254 plates. Flash chromatography was performed using silica gel from Merck (60, 0.040–0.063 mm). Microwave reactions were performed with a Milestone MicroSYNTH microwave reactor with a SK-10 rotor or, for reaction volumes smaller than 10 mL, in an Anton Paar GmbH Monowave 300 synthesis reactor equipped with an internal IR probe calibrated with a Ruby thermometer. NMR spectroscopy was performed using Bruker Avance DPX300, AVII400, AVIIIHD400, DRX500, AVI600, AVII600 or AVIIIHD800 operating at 300 MHz (1H NMR), or 400 MHz (1H NMR), 376 MHz (19F NMR), 101 MHz (13C NMR), or 500 MHz (1H NMR), or 600 MHz (1H NMR) and 151 MHz (13C NMR), or 800 MHz (1H NMR) and 201 MHz (13C NMR) respectively. All spectra were recorded at room temperature. 1H NMR and 13C NMR spectra have been referenced relative to the residual solvent signals, and the resonances are numbered according to Fig. 8. Chemical shifts in 19F NMR have been referenced to CFCl3 by using C6F6 or C6H5F (−164.9 ppm and −116.1 ppm with respect to CFCl3 at 0 ppm) as an internal standard, and are proton decoupled. Chemical shifts in 15N NMR have been calibrated against CH3NO2 as an external standard (0.0 ppm). All 15N NMR chemical shifts were obtained and assigned using 1H–15N HMBC experiments. The resonances in the 1H NMR and 13C NMR spectra were assigned using various 2D experiments (COSY, NOESY, HSQC and HMBC). MS (ESI and APPI) was recorded on a Bruker maXis II ETD spectrometer. All melting points are uncorrected and were obtained with a Stuart SMP10 melting point apparatus. Elemental analysis was performed by Mikroanalytisches Laboratorium Kolbe, Oberhausen, Germany. Single-crystal diffraction data were acquired on a Bruker D8 Venture equipped with a Photon 100 CMOS area detector, and using Mo Kα radiation (λ = 0.71073 Å) or Cu Kα radiation (λ = 1.5406 Å) from an Incoatec iμS microsource. Data reduction was performed with the Bruker Apex3 Suite, the structures were solved with ShelXT110 and refined with ShelXL.111 Olex2 was used as user interface.112 The cif files were edited with enCIFer v. 1.4.113 Full details of the data collection, structure solution and refinement for each compound are contained in the cif files, available from https://www.ccdc.cam.ac.uk/(CCDC 2115512 (2c-Au(OAcF)2), 2122527 (2d-Au(OAcF)2), 2085151 (2e-Au(OAcF)2), 2086346 (2h-Au(OAcF)2), 2126159 (2i-Au(OAcF)2), 2126283 (2j3Au2(OAcF)2), 2122284 (2l-Au(OAcF)2), 2111917 (2m-Au(OAcF)2), 2105655 (2n-Au(OAcF)2), 2086931 (2o-Au(OAcF)2), 2126114 (2p-Au(OAcF)2), 2130186 (2r-Au(OAcF)2), 2126097 (3s-AuOAcF) and 2114274 (3t-AuOAcF)). The data are summarized in Tables S2–S15, ESI.†
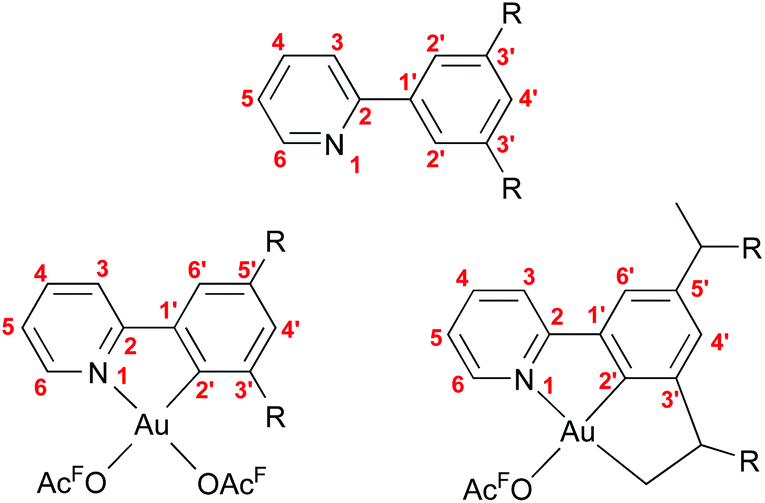 | ||
| Fig. 8 Numbering scheme used for reporting the NMR data. | ||
Experimental and analytical data for a selection of compounds described within the text are presented here, data for all compounds can be found in the ESI.†
General procedure for synthesis of arylpyridine ligands
2-Bromopyridine or substituted 2-bromopyridine (5.00 mmol, 1.0 equiv.) and arylboronic acid (4.75–5.50 mmol, 0.95–1.1 equiv.) were dissolved in n-PrOH (10 mL). A solution of K3PO4 (10.0–11.5 mmol, 2.0–2.3 equiv.) in water (10 mL) was added, and the resulting biphasic mixture was degassed for 10 min by bubbling Ar through it. Pd(OAc)2 (0.100 mmol, 2.0 mol%) and PPh3 (0.300 mmol, 6.0 mol%) were added, and the reaction mixture was heated at reflux temperature for 3 h under Ar. After cooling to rt, CH2Cl2 (50 mL) and water (50 mL) were added. The phases were separated, and the CH2Cl2 solution was washed with 2 M NaOH (aq) (2 × 50 mL), brine (50 mL), and was dried over Na2SO4. The solvent was removed under reduced pressure. The obtained residue was purified by flash column chromatography (hexanes/EtOAc, hexanes/CH2Cl2 or hexanes/EtOAc/CH2Cl2 mixtures), furnishing the arylpyridine ligand.:1 mixture of HOAcF and water (30 mL). The reaction mixture was heated at 120 °C for 30 min in a microwave. After cooling to room temperature, HOAcF (30 mL) was added to dissolve partially precipitated product, and the resulting solution was filtered. Water (50 mL) was added to the filtrate, resulting in the precipitation of a solid. After cooling on an ice-water bath for 15 min, the precipitate was filtered off, washed with water (3 × 5 mL) and Et2O (5 mL), and dried under a stream of air for ca. 3 h, furnishing 2c-Au(OAcF)2 as a colourless solid (0.527 g, 0.860 mmol, 85%). 1H NMR (800 MHz, CD2Cl2): δ 8.62 (dd, 3JH,H = 6.0 Hz, 4JH,H = 1.0 Hz, H6), 8.33 (ddd, 3JH,H = 7.9 Hz, 3JH,H = 7.8 Hz, 4JH,H = 1.4 Hz, H4), 7.95 (d, 3JH,H = 8.1 Hz, H3), 7.66 (ddd, 3JH,H = 7.6 Hz, 3JH,H = 6.0 Hz, 4JH,H = 1.3 Hz, H5), 7.28 (dd, 3JH,F = 7.8 Hz, 4JH,H = 2.4 Hz, H6′), 6.87 ppm (ddd, 3JH,F = 9.1 Hz, 3JH,F = 9.1 Hz, 4JH,H = 2.5 Hz, H4′). 13C NMR (201 MHz, CD2Cl2): δ 164.1 (C2), 164.0 (dd, 1JC,F = 251.5 Hz, 3JC,F = 12.0 Hz, C3′ or C5′), 162.8 (dd, 1JC,F = 253.4 Hz, 3JC,F = 12.1 Hz, C3′ or C5′), 161.1 (q, 2JC,F = 38.2 Hz, OCOCF3), 161.0 (q, 2JC,F = 39.5 Hz, OCOCF3), 148.2 (C6), 145.4 (C4), 145.1 (dd, 3JC,F = 9.9 Hz, 3JC,F = 9.6 Hz, C1′), 126.4 (C5), 122.9 (C3), 118.8 (dd, 2JC,F = 25.3 Hz, 4JC,F = 3.6 Hz, C2′), 118.0 (q, 1JC,F = 288.6 Hz, OCOCF3), 115.5 (q, 1JC,F = 287.8 Hz, OCOCF3), 109.2–109.5 ppm (m, C4′ + C6′). 19F NMR (188 MHz, CD2Cl2): δ −75.5 (d, 7JF,F = 4.0 Hz, 3F, OCOCF3 (trans-N)), −77.0 (s, 3F, OCOCF3 (cis-N)), −102.46 to −102.53 (m, 1F, ArF1), −109.2 ppm (d, 4JF,F = 10.0 Hz, 1F, ArF2). MS (ESI): m/z (rel. %): 499.998 (90) [M − OCOCF3]+. HRMS (ESI): Found: 499.9979. Calc. for C13H6AuF5NO2 [M − OCOCF3]+: 499.9979. Elemental analysis: Found: C, 29.4; H, 1.0; N, 2.3. Calc. for C15H6AuF8NO4: C, 29.4; H, 1.0; N, 2.3%.
:1 mixture of HOAcF and water (6 mL). The reaction mixture was heated at 100 °C for 60 min in a microwave. After cooling to room temperature, HOAcF (1 mL) was added, and the resulting solution was filtered. Water (8 mL) was added to the filtrate, resulting in the precipitation of a solid. After cooling on an ice-water bath for 10 min, the precipitate was filtered off, washed with water (3 × 5 mL) and Et2O (5 mL), and dried under a stream of air for ca. 3 h, furnishing 2m-Au(OAcF)2 as a pale yellow solid (0.0660 g, 0.106 mmol, 53%). 1H NMR (800 MHz, CD2Cl2): δ 8.09 (dd, 3JH,H = 8.2 Hz, 3JH,H = 7.8 Hz, 1H, H4), 7.42 (dd, 3JH,H = 7.7 Hz, 4JH,H = 0.9 Hz, 1H, H3), 7.35 (d, 3JH,H = 7.9 Hz, 1H, H6′), 7.23–7.25 (m, 1H, H5′), 6.82 (d, 4JH,H = 0.6 Hz, 1H, H3′), 6.81 (dd, 3JH,H = 8.5 Hz, 4JH,H = 0.8 Hz, 1H, H5), 4.01 (s, 3H, OCH3), 2.40 ppm (s, 3H, Ar–CH3). 13C NMR (201 MHz, CD2Cl2): δ 165.1 (C6), 163.7 (C2), 161.1 (q, 2JC,F = 36.6 Hz, OCOCF3), 160.4 (q, 2JC,F = 39.6 Hz, OCOCF3), 146.6 (C4), 143.8 (C4′), 141.7 (C2′), 139.5 (C1′), 131.3 (C5′), 128.7 (C3′), 125.9 (C6′), 118.7 (q, 1JC,F = 289.6 Hz, OCOCF3), 116.1 (q, 1JC,F = 288.1 Hz, OCOCF3), 113.3 (C3), 106.9 (C5), 58.0 (OCH3), 22.3 ppm (Ar–CH3). 19F NMR (376 MHz, CD2Cl2): δ −76.2 (s, 3F, OCOCF3), −76.5 ppm (broadened s, 3F, OCOCF3). 15N{1H} NMR (600 MHz, CD2Cl2): δ −187.7 ppm (N1). MS (ESI): m/z (rel. %): 426.076 (100) [M − 2OCOCF3 + OMe]+. HRMS (ESI): Found: 426.0761. Calc. for C14H15AuNO2 [M − 2OCOCF3 + OMe]+: 426.0763. Elemental analysis: Found: C, 32.8; H, 2.0; N, 2.3. Calc. for C17H12AuF6NO5: C, 32.9; H, 1.95; N, 2.25%.
:1 mixture of HOAcF and water (6 mL). The reaction mixture was heated at 120 °C for 60 min in a microwave. After cooling to room temperature, HOAcF (2 mL) was added, and the resulting solution was filtered. Water (20 mL) was added to the filtrate, resulting in the precipitation of a solid. After cooling on an ice-water bath for 15 min, and then overnight at 4–8 °C, the precipitate was filtered off, washed with water (3 × 5 mL) and Et2O (5 mL), and dried under a stream of air for ca. 3 h, furnishing 2r-Au(OAcF)2 as a colourless solid (0.0740 g, 0.0810 mmol, 41%). 1H NMR (800 MHz, CD2Cl2): δ 8.61 (dd, 3JH,H = 6.1 Hz, 4JH,H = 1.0 Hz, 1H, H6), 8.34 (ddd, 3JH,H = 8.1 Hz, 3JH,H = 7.7 Hz, 4JH,H = 1.4 Hz, 1H, H4), 8.05 (dd, 3JH,H = 8.2 Hz, 4JH,H = 1.2 Hz, 1H, H3), 7.80 (s, 1H, H6′), 7.65 (ddd, 3JH,H = 7.6 Hz, 3JH,H = 6.1 Hz, 4JH,H = 1.4 Hz, 1H, H5), 7.27 ppm (s, 1H, H4′). 13C NMR (201 MHz, CD2Cl2): δ 164.3 (C2), 161.2 (q, 2JC,F = 38.4 Hz, OCOCF3), 160.4 (q, 2JC,F = 39.6 Hz, OCOCF3), 148.4 (C6), 145.4 (C4), 144.6 (m, 2× Ar–C–F), 142.2 (d, 1JC,F = 254.8 Hz, Ar–C–F), 141.9 (d, 1JC,F = 256.2 Hz, Ar–C–F), 141.1 (C3′ + C5′), 138.6 (d, 1JC,F = 252.5 Hz, Ar–C–F), 137.6 (C4′), 137.2 (d, 1JC,F = 251.1 Hz, Ar–C–F), 130.8 (C1′ or C2′), 129.4 (C1′ or C2′), 128.3 (C6′), 126.3 (C5), 122.8 (C3), 117.9 (q, 1JC,F = 288.4 Hz, OCOCF3), 115.1 (q, 1JC,F = 288.1 Hz, OCOCF3), 113.0 ppm (m, 2× Ar–C–C–F). 19F NMR (470 MHz, CD2Cl2): δ −75.6 (s, 3F, OCOCF3), −77.1 (s, 3F, OCOCF3), −143.8 (dd, 3JF,F = 22.1 Hz, 4JF,F = 7.5 Hz, 2F, C6F5), −145.2 (dd, 3JF,F = 23.1 Hz, 4JF,F = 7.5 Hz, 2F, C6F5), −155.0 (dd, 3JF,F = 21.3 Hz, 3JF,F = 20.3 Hz, 1F, C6F5), −156.3 (dd, 3JF,F = 20.8 Hz, 3JF,F = 20.2 Hz, 1F, C6F5), −163.6 (m, 2F, C6F5), −164.7 ppm (m, 2F, C6F5). 15N{1H} NMR (800 MHz, CD2Cl2): δ −167.7 ppm (N1). MS (ESI): m/z (rel. %): 714.019 (15) [M − 2OCOCF3 + OMe]+, 768.027 (70) [M − 2OCOCF3 + 2OMe + Na]+, 800.053 (100) [M − 2OCOCF3 + 2OMe + MeOH + Na]+. HRMS (ESI): Found: 768.0269. Calc. for C25H12AuF10NNaO2 [M − 2OCOCF3 + 2OMe + Na]+: 768.0266. Elemental analysis: Found: C, 35.6; H, 0.7; N, 1.5. Calc. for C26H6AuF16NO4: C, 35.7; H, 0.7; N, 1.5%.
:1 mixture of HOAcF and water (6 mL). The reaction mixture was heated at 80 °C for 3.5 h in a microwave. The reaction mixture was kept at 4–8 °C overnight. Addition of HOAcF (4 mL), followed by water (6 mL), furnishing a white precipitate. The precipitate was collected by filtration, and washed with water (3 × 3 mL). The title compound was obtained as a colourless solid (0.0410 g, 0.0750 mmol, 38%). 1H NMR (600 MHz, CD2Cl2): δ 8.48 (ddd, 3JH,H = 5.4 Hz, 4JH,H = 1.4 Hz, 5JH,H = 0.8 Hz, 1H, H6), 7.99–8.03 (m, 1H, H4), 7.94 (d, 3JH,H = 8.1 Hz, 1H, H3), 7.50 (ddd, 3JH,H = 7.5 Hz, 3JH,H = 5.4 Hz, 4JH,H = 1.3 Hz, 1H, H5), 7.35 (s, 1H, H6′), 6.92 (s, 1H, H4′), 3.50 (sx, 3JH,H = 6.8 Hz, 1H, Ar–CH(CH2Au)CH3), 3.38 (dd, 2JH,H = 10.4 Hz, 3JH,H = 7.3 Hz, 1H, Ar–CH(CH2Au)CH3), 2.97 (dd, 2JH,H = 10.4 Hz, 3JH,H = 5.6 Hz, 1H, Ar–CH(CH2Au)CH3), 2.94 (sp, 3JH,H = 6.9 Hz, 1H, Ar–CH(CH3)2), 1.35 (d, 3JH,H = 7.0 Hz, 3H, Ar–CH(CH2Au)CH3), 1.290 (d, 3JH,H = 6.9 Hz, 3H, Ar–CH(CH3)2), 1.287 ppm (d, 3JH,H = 6.9 Hz, 3H, Ar–CH(CH3)2). 13C NMR (151 MHz, CD2Cl2): δ 162.2 (C2), 161.5 (q, 2JC,F = 36.5 Hz, OCOCF3), 161.2 (C3′), 149.8 (C5′), 148.1 (C6), 146.3 (C1′), 141.5 (C4), 139.7 (C2′), 126.1 (C4′), 125.1 (C5), 121.1 (C6′), 120.8 (C3), 118.5 (q, 1JC,F = 290.3 Hz, OCOCF3), 46.7 (Ar–CH(CH2Au)CH3), 41.4 (Ar–CH(CH2Au)CH3), 35.2 (Ar–CH(CH3)2), 24.5 (Ar–CH(CH3)2), 24.3 (Ar–CH(CH3)2), 23.0 ppm (Ar–CH(CH2Au)CH3). 19F NMR (376 MHz, CD2Cl2): δ −76.9 ppm (s, 3F, OCOCF3). 15N{1H} NMR (600 MHz, CD2Cl2): δ −111.7 ppm (N1). MS (ESI): m/z (rel. %): 434.118 (49) [M − OCOCF3]+, 452.128 (100) [M − OCOCF3 + H2O]+, 475.144 (82) [M − OCOCF3 + MeCN]+. HRMS (ESI): Found: 434.1176. Calc. for C17H19AuN [M − OCOCF3]+: 434.1178. Elemental analysis: Found: C, 41.6; H, 3.5; N, 2.5. Calc. for C17H15AuF3NO2: C, 41.7; H, 3.5; N, 2.6%.
:1 mixture of HOAcF and water (6 mL). The reaction mixture was heated at 80 °C for 3 h in a microwave. The reaction mixture was kept at 4–8 °C overnight. Addition of HOAcF (2 mL), followed by water (3 mL), furnishing a white precipitate. The precipitate was collected by filtration, and washed with water (3 × 3 mL) and pentane (5 mL). The title compound was obtained as a colourless solid (0.0680 g, 0.131 mmol, 65%). 1H NMR (600 MHz, CD2Cl2): δ 8.45–8.47 (m, 1H, H6), 7.99–8.01 (m, 1H, H4), 7.90 (d, 3JH,H = 8.0 Hz, 1H, H3), 7.47 (ddd, 3JH,H = 7.6, 3JH,H = 5.4, 4JH,H = 1.3 Hz, 1H, H5), 7.31 (s, 1H, H6′), 6.99 (s, 1H, H4′), 3.27–3.30 (m, 2H, Ar–CH2CH2Au), 3.18–3.20 (m, 2H, Ar–CH2CH2Au), 2.65 (q, 3JH,H = 7.6 Hz, 2H, Ar–CH2CH3), 1.25 ppm (t, 3JH,H = 7.6 Hz, 3H, Ar–CH2CH3). 13C NMR (151 MHz, CD2Cl2): δ 162.5 (C2), 161.5 (d (q expected), 2JC,F = 36.3 Hz, OCOCF3), 158.3 (C3′), 148.2 (C5′), 148.0 (C6), 145.0 (C1′), 141.5 (C4), 139.4 (C2′), 126.9 (C4′), 125.0 (C5), 122.0 (C6′), 120.9 (C3), 118.5 (d (q expected), 1JC,F = 290.5 Hz, OCOCF3), 40.3 (Ar–CH2CH2Au), 32.2 (Ar–CH2CH2Au), 29.7 (Ar–CH2CH3), 16.2 ppm (Ar–CH2CH3). 19F NMR (376 MHz, CD2Cl2): δ −76.9 ppm (s, 3F, OCOCF3). 15N{1H} NMR (600 MHz, CD2Cl2): δ −110.7 ppm (N1). MS (ESI): m/z (rel. %): 406.087 (63) [M − OCOCF3]+, 424.097 (100) [M − OCOCF3 + H2O]+, 447.113 (54) [M − OCOCF3 + MeCN]+. HRMS (ESI): Found: 406.0867. Calc. for C15H15AuN [M − OCOCF3]+: 406.0865. Elemental analysis: Found: C, 39.3; H, 2.9; N, 2.7. Calc. for C17H15AuF3NO2: C, 39.3; H, 2.9; N, 2.7%.
Computational details
Calculations were carried out at the DFT level as implemented in the Gaussian16 software package.116 The hybrid PBE0+GD3 functional117,118 including Grimme's model for dispersion forces was used to optimize all geometries. This methodology was selected based on previous studies which have proven its solid performance in the modelling of Au(III) complexes.7,24,25,119,120 C, H, F, N and O were described with the all-electron triple-ζ 6-311+G** basis set,121,122 whereas Au was described with the Stuttgart–Köln basis set including a small-core quasi-relativistic pseudopotential.123,124 NBO7 calculations were performed in order to analyse the natural charges.125 Geometries were fully optimized without any constraint. Vibrational frequencies were computed at the same level of theory to classify all stationary points as either saddle points (transition states, with a single imaginary frequency) or energy minima (reactants, intermediates and products, with only real frequencies). The Gibbs free energy used in the discussion includes both the thermochemistry and the refined energy. All optimizations were carried out in solvent (CH2Cl2 or HOAcF) using the SMD solvation model.126 HOAcF was defined as eps = 8.55, epsinf = 2.26 and rsolv = 13.7. In the bimolecular steps, the energies were corrected for the 1 M standard state.Author contributions
KTH: investigation, supervision, writing – original draft, writing – review and editing; ILS: investigation, writing – review and editing; DSW: investigation, supervision; AN: supervision, writing – review and editing; MT: supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Research Council of Norway through the Norwegian NMR Package in 1994, through the Norwegian NMR Platform, NNP (226244/F50) and through the Hylleraas Centre for Quantum Molecular Sciences (project number 262695). This work was also supported by the ERASMUS program of the European Union (exchange visit to Oslo for ILS), and the Norwegian Metacenter for Computational Science (NOTUR, nn4654k). Dr Marte S. M. Holmsen, Dr Sigurd Øien-Ødegaard, Lorena P. Escrivá, Michael Philipp and Sahra A. Ahmed are acknowledged for experimental help. We thank Osamu Sekiguchi, Lina Aarsbog and Sverre Løyland for performing the MS experiments, and Dr Richard H. Heyn (SINTEF Industry) for assistance with the elemental analyses. We thank Prof. Odile Eisenstein for helpful discussions. We acknowledge use of the Norwegian National Centre for X-ray Diffraction and Scattering (RECX).Notes and references
- W. Henderson, Adv. Organomet. Chem., 2006, 54, 207–265 CrossRef CAS.
- R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1994–2015 CrossRef CAS PubMed.
- R. Malmberg and K. Venkatesan, Coord. Chem. Rev., 2021, 449, 214182 CrossRef CAS.
- C. Bronner and O. S. Wenger, Dalton Trans., 2011, 40, 12409–12420 RSC.
- C. Blons, S. Mallet-Ladeira, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2018, 57, 11732–11736 CrossRef CAS PubMed.
- J. Rodriguez, N. Adet, N. Saffon-Merceron and D. Bourissou, Chem. Commun., 2020, 56, 94–97 RSC.
- M. S. M. Holmsen, A. Nova, D. Balcells, E. Langseth, S. Øien-Ødegaard, R. H. Heyn, M. Tilset and G. Laurenczy, ACS Catal., 2017, 7, 5023–5034 CrossRef CAS.
- M. S. M. Holmsen, A. Nova, K. Hylland, D. S. Wragg, S. Øien-Ødegaard, R. H. Heyn and M. Tilset, Chem. Commun., 2018, 54, 11104–11107 RSC.
- J. Segato, A. Del Zotto, L. Belpassi, P. Belanzoni and D. Zuccaccia, Catal. Sci. Technol., 2020, 10, 7757–7767 RSC.
- S. K. Fung, T. Zou, B. Cao, P.-Y. Lee, Y. M. E. Fung, D. Hu, C.-N. Lok and C.-M. Che, Angew. Chem., Int. Ed., 2017, 56, 3892–3896 CrossRef CAS PubMed.
- M. Williams, A. I. Green, J. Fernandez-Cestau, D. L. Hughes, M. A. O'Connell, M. Searcey, B. Bertrand and M. Bochmann, Dalton Trans., 2017, 46, 13397–13408 RSC.
- S. Gukathasan, S. Parkin and S. G. Awuah, Inorg. Chem., 2019, 58, 9326–9340 CrossRef CAS PubMed.
- B. Bertrand, M. R. M. Williams and M. Bochmann, Chem. – Eur. J., 2018, 24, 11840–11851 CrossRef CAS PubMed.
- E. Abás, M. Gómez-Bachiller, E. Colom, E. Pardina, A. Rodríguez-Diéguez, L. Grasa and M. Laguna, J. Organomet. Chem., 2020, 920, 121340 CrossRef.
- M. Frik, J. Fernández-Gallardo, O. Gonzalo, V. Mangas-Sanjuan, M. González-Alvarez, A. Serrano del Valle, C. Hu, I. González-Alvarez, M. Bermejo, I. Marzo and M. Contel, J. Med. Chem., 2015, 58, 5825–5841 CrossRef CAS PubMed.
- R. Kumar, A. Linden and C. Nevado, Angew. Chem., Int. Ed., 2015, 54, 14287–14290 CrossRef CAS PubMed.
- M. Bachmann, J. Terreni, O. Blacque and K. Venkatesan, Chem. – Eur. J., 2017, 23, 3837–3849 CrossRef CAS PubMed.
- D. Zhou, W.-P. To, G. S. M. Tong, G. Cheng, L. Du, D. L. Phillips and C.-M. Che, Angew. Chem., Int. Ed., 2020, 59, 6375–6382 CrossRef CAS PubMed.
- E. Langseth, C. H. Görbitz, R. H. Heyn and M. Tilset, Organometallics, 2012, 31, 6567–6571 CrossRef CAS.
- S. Witzel, M. S. M. Holmsen, M. Rudolph, M. C. Dietl, S. Øien-Ødegaard, F. Rominger, M. Tilset and A. S. K. Hashmi, Organometallics, 2020, 39, 2830–2837 CrossRef CAS.
- V. A. Levchenko, A. Nova, S. Øien-Ødegaard, D. Balcells and M. Tilset, Eur. J. Inorg. Chem., 2020, 3249–3258 CrossRef CAS.
- M. S. M. Holmsen, A. Nova, S. Øien-Ødegaard, R. H. Heyn and M. Tilset, Angew. Chem., Int. Ed., 2020, 59, 1516–1520 CrossRef CAS PubMed.
- A. P. Shaw, M. Tilset, R. H. Heyn and S. Jakobsen, J. Coord. Chem., 2011, 64, 38–47 CrossRef CAS.
- E. Langseth, A. Nova, E. A. Tråseth, F. Rise, S. Øien, R. H. Heyn and M. Tilset, J. Am. Chem. Soc., 2014, 136, 10104–10115 CrossRef CAS PubMed.
- M. S. M. Holmsen, A. Nova, D. Balcells, E. Langseth, S. Øien-Ødegaard, E. A. Tråseth, R. H. Heyn and M. Tilset, Dalton Trans., 2016, 45, 14719–14724 RSC.
- M. S. M. Holmsen, F. S. Ihlefeldt, S. Øien-Ødegaard, E. Langseth, Y. Wencke, R. H. Heyn and M. Tilset, Organometallics, 2018, 37, 1937–1947 CrossRef CAS.
- Q. Wu, C. Du, Y. Huang, X. Liu, Z. Long, F. Song and J. You, Chem. Sci., 2015, 6, 288–293 RSC.
- R. Kumar, A. Linden and C. Nevado, J. Am. Chem. Soc., 2016, 138, 13790–13793 CrossRef CAS PubMed.
- V. A. Levchenko, H.-S. M. Siah, S. Øien-Ødegaard, G. Kaur, A. Fiksdahl and M. Tilset, Mol. Catal., 2020, 492, 111009 CrossRef CAS.
- H. von Wachenfeldt, A. V. Polukeev, N. Loganathan, F. Paulsen, P. Röse, M. Garreau, O. F. Wendt and D. Strand, Dalton Trans., 2015, 44, 5347–5353 RSC.
- A. R. Browne, N. Deligonul, B. L. Anderson, M. Zeller, A. D. Hunter and T. G. Gray, Chem. Commun., 2015, 51, 15800–15803 RSC.
- M. Bachmann, R. Fessler, O. Blacque and K. Venkatesan, Dalton Trans., 2019, 48, 7320–7330 RSC.
- A. Beillard, X. Bantreil, T.-X. Métro, J. Martinez and F. Lamaty, Chem. Rev., 2019, 119, 7529–7609 CrossRef CAS PubMed.
- R. P. Herrera and M. C. Gimeno, Chem. Rev., 2021, 121, 8311–8363 CrossRef CAS PubMed.
- A. Szentkuti, J. A. Garg, O. Blacque and K. Venkatesan, Inorg. Chem., 2015, 54, 10748–10760 CrossRef CAS PubMed.
- M. Kondrashov, D. Provost and O. F. Wendt, Dalton Trans., 2016, 45, 525–531 RSC.
- G. C. Dickmu and I. P. Smoliakova, Coord. Chem. Rev., 2020, 409, 213203 CrossRef CAS.
- H. Tang, X.-R. Huang, J. Yao and H. Chen, J. Org. Chem., 2015, 80, 4672–4682 CrossRef CAS PubMed.
- I. P. Beletskaya and A. V. Cheprakov, J. Organomet. Chem., 2004, 689, 4055–4082 CrossRef CAS.
- F. Cocco, A. Zucca, S. Stoccoro, M. Serratrice, A. Guerri and M. A. Cinellu, Organometallics, 2014, 33, 3414–3424 CrossRef CAS.
- M. A. Cinellu, A. Zucca, S. Stoccoro, G. Minghetti, M. Manassero and M. Sansoni, J. Chem. Soc., Dalton Trans., 1996, 4217–4225 RSC.
- J. Vicente, M. T. Chicote, M. I. Lozano and S. Huertas, Organometallics, 1999, 18, 753–757 CrossRef CAS.
- D. Fan, E. Meléndez, J. D. Ranford, P. F. Lee and J. J. Vittal, J. Organomet. Chem., 2004, 689, 2969–2974 CrossRef CAS.
- A. C. Reiersølmoen, D. Csókás, S. Øien-Ødegaard, A. Vanderkooy, A. K. Gupta, A.-C. C. Carlsson, A. Orthaber, A. Fiksdahl, I. Pápai and M. Erdélyi, J. Am. Chem. Soc., 2020, 142, 6439–6446 CrossRef PubMed.
- L. Chiang, L. E. N. Allan, J. Alcantara, M. C. P. Wang, T. Storr and M. P. Shaver, Dalton Trans., 2014, 43, 4295–4304 RSC.
- D. J. Darensbourg, P. Rainey and J. Yarbrough, Inorg. Chem., 2001, 40, 986–993 CrossRef CAS.
- Y. Yang, G. Li, X. Mao and Y. She, Org. Process Res. Dev., 2019, 23, 1078–1086 CrossRef CAS.
- X.-J. Zhu, T. Zhang, S. Zhao, W.-K. Wong and W.-Y. Wong, Eur. J. Inorg. Chem., 2011, 3314–3320 CrossRef CAS.
- H. He, W.-K. Wong, J. Guo, K.-F. Li, W.-Y. Wong, W.-K. Lo and K.-W. Cheah, Inorg. Chim. Acta, 2004, 357, 4379–4388 CrossRef CAS.
- T. Usuki, H. Uchida, K. Omoto, Y. Yamanoi, A. Yamada, M. Iwamura, K. Nozaki and H. Nishihara, J. Org. Chem., 2019, 84, 10749–10756 CrossRef CAS PubMed.
- M.-C. Chen, D.-G. Chen and P.-T. Chou, ChemPlusChem, 2021, 86, 11–27 CrossRef CAS PubMed.
- P. G. Bomben, B. D. Koivisto and C. P. Berlinguette, Inorg. Chem., 2010, 49, 4960–4971 CrossRef CAS PubMed.
- L. Bergmann, C. Braun, M. Nieger and S. Bräse, Dalton Trans., 2018, 47, 608–621 RSC.
- T. Nyokong, Coord. Chem. Rev., 2007, 251, 1707–1722 CrossRef CAS.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- W. Klaeui, W. Eberspach and P. Guetlich, Inorg. Chem., 1987, 26, 3977–3982 CrossRef CAS.
- P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed. Engl., 1994, 33, 2024–2054 CrossRef.
- D. L. Bruns, D. G. Musaev and S. S. Stahl, J. Am. Chem. Soc., 2020, 142, 19678–19688 CrossRef CAS PubMed.
- M. H. Rønne, D. Cho, M. R. Madsen, J. B. Jakobsen, S. Eom, É. Escoudé, H. C. D. Hammershøj, D. U. Nielsen, S. U. Pedersen, M.-H. Baik, T. Skrydstrup and K. Daasbjerg, J. Am. Chem. Soc., 2020, 142, 4265–4275 CrossRef PubMed.
- F. Thomas, Eur. J. Inorg. Chem., 2007, 2379–2404 CrossRef CAS.
- M. Rentschler, M.-A. Schmid, W. Frey, S. Tschierlei and M. Karnahl, Inorg. Chem., 2020, 59, 14762–14771 CrossRef CAS PubMed.
- A. Giraudeau, H. J. Callot, J. Jordan, I. Ezhar and M. Gross, J. Am. Chem. Soc., 1979, 101, 3857–3862 CrossRef CAS.
- R. Gust, I. Ott, D. Posselt and K. Sommer, J. Med. Chem., 2004, 47, 5837–5846 CrossRef CAS PubMed.
- A. Erxleben, Inorg. Chim. Acta, 2018, 472, 40–57 CrossRef CAS.
- S. Jürgens, V. Scalcon, N. Estrada-Ortiz, A. Folda, F. Tonolo, C. Jandl, D. L. Browne, M. P. Rigobello, F. E. Kühn and A. Casini, Bioorg. Med. Chem., 2017, 25, 5452–5460 CrossRef PubMed.
- K. K.-Y. Kung, V. K.-Y. Lo, H.-M. Ko, G.-L. Li, P.-Y. Chan, K.-C. Leung, Z. Zhou, M.-Z. Wang, C.-M. Che and M.-K. Wong, Adv. Synth. Catal., 2013, 355, 2055–2070 CrossRef CAS.
- W.-P. To, G. S. M. Tong, C.-W. Cheung, C. Yang, D. Zhou and C.-M. Che, Inorg. Chem., 2017, 56, 5046–5059 CrossRef CAS PubMed.
- Z.-T. Yu, X.-L. Liu, Y.-J. Yuan, Y.-H. Li, G.-H. Chen and Z.-G. Zou, Dalton Trans., 2016, 45, 17223–17232 RSC.
- K. T. Hylland, S. Øien-Ødegaard and M. Tilset, Eur. J. Org. Chem., 2020, 4208–4226 CrossRef CAS.
- S. Lou and G. C. Fu, Adv. Synth. Catal., 2010, 352, 2081–2084 CrossRef CAS PubMed.
- M. Lafrance, D. Shore and K. Fagnou, Org. Lett., 2006, 8, 5097–5100 CrossRef CAS PubMed.
- G. W. Parshall, Acc. Chem. Res., 1970, 3, 139–144 CrossRef CAS.
- G. W. Parshall, Acc. Chem. Res., 1975, 8, 113–117 CrossRef CAS.
- M. I. Bruce, Angew. Chem., Int. Ed. Engl., 1977, 16, 73–86 CrossRef.
- A. D. Ryabov, Chem. Rev., 1990, 90, 403–424 CrossRef CAS.
- M. Albrecht, Chem. Rev., 2010, 110, 576–623 CrossRef CAS PubMed.
- G. Bott, L. D. Field and S. Sternhell, J. Am. Chem. Soc., 1980, 102, 5618–5626 CrossRef CAS.
- L. Lunazzi, M. Mancinelli, A. Mazzanti, S. Lepri, R. Ruzziconi and M. Schlosser, Org. Biomol. Chem., 2012, 10, 1847–1855 RSC.
- M. G. MacDonald, C. N. Kostelansky, P. S. White and J. L. Templeton, Organometallics, 2006, 25, 4560–4570 CrossRef CAS.
- J.-J. Jiang and M.-K. Wong, Chem. – Asian J., 2021, 16, 364–377 CrossRef CAS PubMed.
- A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Polyhedron, 2015, 90, 47–57 CrossRef CAS.
- D. Rosiak, A. Okuniewski and J. Chojnacki, Polyhedron, 2018, 146, 35–41 CrossRef CAS.
- R. Ruzziconi, S. Spizzichino, A. Mazzanti, L. Lunazzi and M. Schlosser, Org. Biomol. Chem., 2010, 8, 4463–4471 RSC.
- F. Cocco, M. A. Cinellu, G. Minghetti, A. Zucca, S. Stoccoro, L. Maiore and M. Manassero, Organometallics, 2010, 29, 1064–1066 CrossRef CAS.
- P. Kalaramna, D. Bhatt, H. Sharma and A. Goswami, Adv. Synth. Catal., 2019, 361, 4379–4385 CrossRef CAS.
- X.-Y. Wang, Y.-F. Ao, Q.-Q. Wang and D.-X. Wang, Inorg. Chem., 2018, 57, 13461–13469 CrossRef CAS PubMed.
- A. B. de Carvalho, G. M. Diogo, R. S. Correa and J. G. Taylor, J. Struct. Chem., 2020, 61, 763–768 CrossRef CAS.
- S. G. Zhang, L. M. Xie and H. Li, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, 2549 CrossRef PubMed.
- P. Biswal, S. K. Banjare, B. V. Pati, S. R. Mohanty and P. C. Ravikumar, J. Org. Chem., 2021, 86, 1108–1117 CrossRef CAS PubMed.
- N. Yoshikawa, S. Yamazaki, Y. Kakimoto, S. Eguchi, R. Yokoyama, N. Kanehisa, N. Tohnai, E. Nakata and H. Takashima, J. Mol. Struct., 2021, 1242, 130728 CrossRef CAS.
- M. Rok, M. Moskwa, P. Dopieralski, W. Medycki, M. Zamponi and G. Bator, CrystEngComm, 2020, 22, 6811–6821 RSC.
- J. E. Nycz, J. Wantulok, R. Sokolova, L. Pajchel, M. Stankevič, M. Szala, J. G. Malecki and D. Swoboda, Molecules, 2019, 24, 4102 CrossRef PubMed.
- W. Gong, Z. Zhou, J. Shi, B. Wu, B. Huang and W. Yi, Org. Lett., 2018, 20, 182–185 CrossRef CAS PubMed.
- A. P. Shaw, M. K. Ghosh, K. W. Törnroos, D. S. Wragg, M. Tilset, O. Swang, R. H. Heyn and S. Jakobsen, Organometallics, 2012, 31, 7093–7100 CrossRef CAS.
- R. V. Parish, J. P. Wright and R. G. Pritchard, J. Organomet. Chem., 2000, 596, 165–176 CrossRef CAS.
- L. Pazderski, Magn. Reson. Chem., 2008, 46, S3–S15 CrossRef PubMed.
- L. Pazderski, in Annu. Rep. NMR Spectrosc, ed. G. A. Webb, Academic Press, 2013, vol. 80, pp. 33–179 Search PubMed.
- L. Pazderski, in Annu. Rep. NMR Spectrosc, ed. R. Atta ur, Academic Press, 2020, vol. 101, pp. 151–284 Search PubMed.
- J. Mason, Chem. Rev., 1981, 81, 205–227 CrossRef CAS.
- L. Pazderski, J. Toušek, J. Sitkowski, L. Kozerski, R. Marek and E. Szłyk, Magn. Reson. Chem., 2007, 45, 24–36 CrossRef CAS PubMed.
- L. Pazderski, T. Pawlak, J. Sitkowski, L. Kozerski and E. Szłyk, Magn. Reson. Chem., 2010, 48, 417–426 CrossRef CAS PubMed.
- L. Pazderski, J. Toušek, J. Sitkowski, L. Kozerski and E. Szłyk, Magn. Reson. Chem., 2009, 47, 658–665 CrossRef CAS PubMed.
- T. G. Appleton, H. C. Clark and L. E. Manzer, Coord. Chem. Rev., 1973, 10, 335–422 CrossRef CAS.
- A. C. Tsipis, New J. Chem., 2020, 44, 7976–7986 RSC.
- L. Rocchigiani and M. Bochmann, Chem. Rev., 2020, 121, 8364–8451 CrossRef PubMed.
- L. Rigamonti, C. Manassero, M. Rusconi, M. Manassero and A. Pasini, Dalton Trans., 2009, 1206–1213 RSC.
- L. Rocchigiani, J. Fernandez-Cestau, I. Chambrier, P. Hrobárik and M. Bochmann, J. Am. Chem. Soc., 2018, 140, 8287–8302 CrossRef CAS PubMed.
- T. Wiedemann, G. Voit, A. Tchernook, P. Roesle, I. Göttker-Schnetmann and S. Mecking, J. Am. Chem. Soc., 2014, 136, 2078–2085 CrossRef CAS PubMed.
- V. Diemer, H. Chaumeil, A. Defoin, A. Fort, A. Boeglin and C. Carré, Eur. J. Org. Chem., 2006, 2727–2738 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- F. H. Allen, O. Johnson, G. P. Shields, B. R. Smith and M. Towler, J. Appl. Crystallogr., 2004, 37, 335–338 CrossRef CAS.
- P. Coppo, E. A. Plummer and L. De Cola, Chem. Commun., 2004, 1774–1775 RSC.
- C. A. Fleckenstein and H. Plenio, Green Chem., 2007, 9, 1287–1291 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- D. Balcells, O. Eisenstein, M. Tilset and A. Nova, Dalton Trans., 2016, 45, 5504–5513 RSC.
- E. Langseth, M. L. Scheuermann, D. Balcells, W. Kaminsky, K. I. Goldberg, O. Eisenstein, R. H. Heyn and M. Tilset, Angew. Chem., Int. Ed., 2013, 52, 1660–1663 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- D. Figgen, K. A. Peterson, M. Dolg and H. Stoll, J. Chem. Phys., 2009, 130, 164108 CrossRef PubMed.
- D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Chem. Phys., 2005, 311, 227–244 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2115512, 2122527, 2085151, 2086346, 2126159, 2126283, 2122284, 2111917, 2105655, 2086931, 2126114, 2130186, 2126097 and 2114274. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2dt00371f |
| ‡ Co-first authors. These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |